Embed Size (px)
Citation preview

Gabarito TVQ 2017 – Segunda Fase
Sumário
I. Síntese Polimérica: entendimento a nível molecular _________________________ 1
II. Regras de Baldwin ____________________________________________________ 8
III. Química de Coordenação: um ensaio sobre a Teoria do Orbital Molecular e
Espectroscopia Vibracional ________________________________________________ 11
IV. Sólidos iônicos: de uma fase à outra ___________________________________ 16
V. Ácido Maleico e Ácido Fumárico ________________________________________ 18
VI. Titulação de Ni com dimetilglioxima ___________________________________ 24
VII. Espectroscopia no infravermelho ______________________________________ 26
VIII. Dinâmica de colisões reacionais _______________________________________ 28

1
I. Síntese Polimérica: entendimento a nível molecular
a) A reação representada abaixo corresponde a uma reação de polimerização
por condensação, mais especificamente, trata-se de uma reação de esterificação.
O mecanismo está representado abaixo. No qual observa-se (i) a ativação
da carbonila com meio ácido, seguida da (ii) adição nucleofílica do etilenoglicol com
a (iii) subsequente eliminação de água, como é esperado para reações de
condensação.
O meio ácido é utilizado para catalisar a reação. De modo que a utilização
de ácido permite que a reação corra por uma via de menor energia de ativação,
favorecendo cineticamente a reação. Além disso, é importante salientar que
carbonila de ácidos carboxílicos não são tão eletrofílicas quanto carbonilas de
aldeídos, cetonas, cloreto de ácidos e anidridos de ácidos carboxílicos. Tal
característica pode ser explicada pelas possíveis estruturas de ressonância, nas
quais fica evidente a deslocalização dos pares de elétrons não ligantes do grupo –
OH.

2
No esquema acima, fica evidente que em meio ácido a estrutura de
ressonância estabiliza garante maior eletrofilicidade para o carbono do grupo
carbonila.
O raciocínio desenvolvido acima é dirigido pela Teoria de Ligação de
Valência, mas também poderia ser explicado pela Teoria de Orbital Molecular, a
qual sugere que a catálise ácida promove a diminuição entre a diferença dos níveis
energéticos do orbital molecular LUMO (lowest unocoppied molecular orbital) do
eletrófilo e a energia do orbital molecular HOMO (highest ocuppied molecular orbital)
do nucleófilo, permitindo portanto que a adição nucleofílica ocorra.
b) A estratégia apresentada, faz sentido do ponto de vista teórico e tem sido
explorada em escala industrial. O polímero inicial (PET), possui grupos carbonilicos
pouco eletrofílicos, tal característica pode ser entendida pela Teoria de Ligação de
Valência e especialmente pela representação das principais estruturas de
ressonância possíveis para grupos estéres ressonantes com anel aromático.
As estruturas canônicas representadas acima, evidenciam que a deficiência
de elétrons do carbono do grupo C=O é parcialmente suprida pela ressonância dos
elétrons do sistema aromático e pelos pares de elétrons não ligante do oxigênio
procedente do álcool. Em resumo, pode-se afirmar que grupos carbonilicos de
ésteres adjacentes a sistemas aromáticos são menos eletrofílicos e

3
consequentemente menos susceptíveis aos ataques de nucleófilos. Além disso vale
ressaltar que a degradação é catalisada por enzimas, de modo que sistemas mais
rígidos como os aromáticos serão menos propensos a se ajustar nos sítios
catalíticos das enzimas, dificultando a hidrólise do polímero.
Desse modo, a adição de um segmento alifático com grupos carbonílicos
mais eletrofílicos favorecerá enormemente a biodegradação do material.
c) Os sais de sulfato em questão, quando são utilizados em suas formas anidras
atuam como agentes secantes – retirando pequenos volumes de água do ambiente.
Na reação de polimerização por condensação ocorre a formação de água como um
subproduto reacional. Desse modo, utilizando os sais de sulfato, pode-se remover
a água do meio reacional perturbando a reação no sentido de formação dos
produtos, tal como é proposto pelo Princípio de Le Chatelier.
d) Como é esperado a partir da interpretação do enunciado o fragmento
metálico de Zn2+ está diretamente relacionado com a atividade do catalisador. De
modo que, a coordenação da espécie catiônica de zinco à carbonila deve tornar o
grupo C=O significativamente mais eletrofílico, facilitando a adição de nucleófilos
como a água, por exemplo. Nesse contexto, é importante enfatizar que a ativação
da carbonila é substancial para que a molécula de água adicione-se a carbonila do
grupo éster.
Duas abordagens são possíveis para entender fenomenologicamente a
atividade do catalisador. A primeira seria a interpretação a partir da Teoria de
Ligação de Valência, que permite afirmar que a espécie de Zn2+ estabiliza a forma
canônica cujo átomo de carbono possui maior eletrofilicidade, favorecendo portanto,
reações de adição nucleofílica.

4
A segunda alternativa seria interpretar o aumento da reatividade do grupo
carbonílico pela Teoria de Orbital Molecular (TOM). Nesse caso é esperado que a
coordenação do fragmento metálico de Zinco à carbonila promova diminuição
energética dos orbitais moleculares HOMO e LUMO, cuja denominação deriva do
inglês highest ocuppied molecular orbital e lowest unocuppied molecular orbital,
respectivamente.
Para entender as reações de adição nucleofílica pela TOM, é importante
destacar os seguintes aspectos:
› Orbitais moleculares LUMO geralmente possuem maior energia que
os orbitais moleculares HOMO;
› A adição nucleofílica ocorre pela interação orbitalar do HOMO do
nucleofílo com o LUMO do eletrófílo;
› Além disso, quanto menor for a diferença energética entre o HOMO
do nucleofílo e o LUMO do eletrófilo mais favorável será a reação de
adição nucleofilica.
Nesse sentido, analisando qualitativamente os orbitais moleculares
envolvidos na adição nucleofílica, antes e após a coordenação do Zinco, temos:

5
O esquema acima, representa a diminuição energética após a coordenação
do Zn2+ promovendo, portanto, a diminuição da diferença energética entre os
orbitais HOMO e LUMO do nucleófilo e do eletrófilo, respectivamente, possibilitando
a adição nucleofilica ao grupo C=O do éster.
e) A partir das proporções mássicas em porcentagem, é possível se determinar
a fórmula mínima em termos das proporções mássicas:
Zn41,394%C22,813%H2,872% O32,921%
Considerando 100g do composto, temos:
Zn41,394gC22,813gH2,872g O32,921g
Em seguida, dividindo-se as respectivas quantidades em massa pelas
massas molares dos elementos em questão ( Zn = 65,38 g mol-1, O = 16 g mol-1, H
= 1,008 g mol-1, C = 12,011 g mol-1).
Zn0,63C1,90H2,85 O2,06
Dividindo todos pelo menor valor (0,63) é possível encontrar fórmula mínima
do complexo de zinco:
ZnC3H4,5 O3,25
Para se obter a fórmula molecular do complexo é necessário multiplicar por
um valor para obter índices estequiométricos inteiros. Logo, multiplicando cada um
dos índices por 4, produziremos
Zn4C12H18O13
Analisando o material de partida e assumindo que a estrutura do ligante
acetato tenha sido preservada é possível escrever a seguinte formula molecular
para complexo.

6
Zn4(OCOCH3)6O
Logo, a partir da fórmula e considerando a existência de um ligante oxo
fazendo quatro ligações (μ4 − O) com os núcleos metálicos de Zinco é possível
chegar na seguinte estrutura do complexo.
A ligação μ4 − O é possível devido ao fato do oxigênio central possuir
valência 2-, e consequentemente possuir oito elétrons de valência, alocados em
orbitais hibridizados sp3, permitindo portanto que os quatro pares de elétrons sejam
utilizados para fazer ligações com o centro metálico de Zinco.
f)

7
A análise das curvas para as amostras A, B e C de PET permite afirmar que
a Amostra C apresenta menor polidispersidade, pois esta curva apresenta
claramente a menor largura a meia altura. Além disso, a amplitude de massa molar
do polímero que compõem a amostra C é menor, de modo a reforçar que a Amostra
C corresponde a amostra mais homogênea.
Considerando a massa molar cuja fração de polímero maior é igual a 61.000
g mol-1 e que o polímero é composto por (C10H8O4)n onde a massa molar da unidade
monomérica é 192 g mol-1. É possível determinar o valor de n:
192n g mol−1 = 61.000 g mol−1
Onde n = 318 unidades.
Observação: O número de unidades monoméricas depende essencialmente da
massa molar do polímero observada no gráfico acima, como a resolução da
abscissa não permite a escolha de um valor exato, caso o estudante encontre valor
próximo de 300 unidades, utilize a massa molar do monômero correta e evidencie
o raciocínio corretamente – a resposta será considerada correta.

8
II. Regras de Baldwin
a) Orbitais moleculares do fragmento C=C. Note que os orbitais HOMO podiam
ser apresentados de duas formas distintas, como representado abaixo
b) O ângulo de Burgi-Dunitz podem ser obtido qualitativamente através da
racionalização de interações eletronicamente favoráveis entre o orbital HOMO de
um nucleófilo com o LUMO do framento C=C em conjunto com fenômenos de
repulsão entre o HOMO do nucleófilo com o HOMO do fragmento C=C.
No caso a trajetória que apresentaria maior sobreposição entre o orbital
HOMO do nucleófilo com o LUMO do fragmento C=C também apresentaria grande
repulsão eletrônica entre a sobreposição dos orbitais ocupados HOMO do nuleófilo
com HOMO do framento C=C, como pode ser visualizado acima.

9
Portanto a adoção de uma trajetória lateral como observada por Bürgi e
Dunitz permite a interação do nucleófilo com o LUMO enquanto mantem reduzida
a repulsão entre os orbitais ocupados do fragmento e do nucleófilo.
c) O mecanismo para a formação do produto 2a corresponde inicialmente a
desprotonação da hidroxíla fenólica, seguida da adição conjugada 1,4. Note que
nessa reação o metóxido de sódio atua como base.
d) No caso da formação dos produtos 2a e 2b, sendo ciclizações 6-endo-trig e
5-exo-trig , respectivamente, portanto permitidas pelas regras de Baldwin.
Considerando os orbitais envolvidos nos reagentes a aproximação esperadas entre
os orbitais HOMO e LUMO correspondentes podem ser observadas abaixo.
Percebe-se que nas conformações apresentadas a interação orbitalar necessária
para ocorrência da reação química ocorre sem grandes desvios da geometria
molecular.

10
No entanto no caso da ciclização do produto 3 ocorre facil sobreposição
adequada de orbitais para obtenção do produto 4, porém não temos sobreposição
adequada para a interação com o outro carbono da dupla ligação
consequentemente impossibilitando a formação de outro produto em quantidades
relevantes. Do ponto de vista da regra de Baldwin a reação observada corresponde
a uma ciclização 4-exo-trig que é permitida enquanto o produto não observado
corrresponde a uma ciclização 5-endo-trig que como esperado não é permitida
pelas regras de Baldwin.
e) Observando comparativamente as estruturas dos compostos 5 e 6 pode-se
concluir que ocorreu uma reação de 5-endo-trig. No entanto pelas regras de Baldwin
esse tipo de reação de ciclização não seria permitida. Adicionalmente é indicado
que tal reação só ocorre em condições ácidas , porém não em condições básicas.
A reação do composto 6 apenas em condições ácidas, como apresentado no
enunciado, ocorre devido a estabilização de diferentes formas de ressonância
nessas condições, indicandando assim que ocorrem alterações significantes na
distribuição de densidade eletrônica pela molécula de forma que o mecanismo de
ciclização se torna favorável.
No caso nas condições ácidas ocorre o favorecimento da protonação da
carbonila, como demonstrado abaixo, assim permitindo a formação de uma nova
espécie positivamente carregada que pode ser representada pelas formas de
ressonância I, II e III. Especificamente as espécies II e III permitem a explicação do
fenômeno observado, pois pela forma de ressonância II nota-se que existe um
caráter carbocatiônico, no entanto as regras de Baldwin não são diretamente
aplicáveis a sistemas carbocatiônicos, então mesmo sendo uma ciclização 5-endo-

11
trig, portanto não permitida, ela ocorre. Analogamente pode-se obervar que a forma
de ressonância III indica a ocorrência de uma ciclização 6-exo-trig e portanto
permitida pelas regras de Baldwin.
Ambas as racionalizações atuam na explicação da ciclização aparente 5-
endo-trig observada. Como em condições básicas esse equílibrio com a forma
protonada da carbonila é impossibilitado, a reação não ocorre, assim como
esperado pelas regras de Baldwin.
III. Química de Coordenação: um ensaio sobre a Teoria do Orbital
Molecular e Espectroscopia Vibracional
a) Seguindo a representação exemplificada para o O2 temos o seguinte
diagrama de orbitais moleculares para a molécula de CO. Note que diferentemente
do caso do oxigênio molecular existe uma inversão de energia entre os orbitais 1𝜋
e 3𝜎 devido a interação orbitalar entre os orbitais 2s e 2p, como representado
abaixo.

12
Adicionalmente está exemplificado abaixo a representação espacial dos orbitais
HOMO e LUMO para esta diatômica.
b) Como informado no enunciado, considerando a teoria do campo cristalino a
aproximação de seis ligantes em um arranjo octaédrico resultará no seguinte
desdobramento dos orbitais d do metal.

13
Como se trata de uma espécie de Fe(II) de campo forte, os seis elétrons
presentes no metal ocuparão os três orbitais degenerados de baixa energia (dxy, dxz
e dyz). Consequemente apenas or orbitais vazios dx2-y2 e dz2 podem interagir
preferencialmente com o HOMO do ligante. Analogamente os orbitais dxy, dxz e dyz
são apresentam capacidade de interagir com o LUMO dos ligantes, assim
realizando retrodoação.
No caso dos ligantes O2 ou CO, seus respectivos orbitais HOMO (ou SOMO)
como representado no exercício anterior serão responsáveis pela doação de
densidade eletrônica para os orbitais degenerados eg enquanto os orbitais t2g
estarão envolvidos na retrodoação para o orbital LUMO (ou SOMO) dos ligantes.
c) Do ponto de vista orbitalar pode-se notar que no caso do CO o orbital HOMO
se apresenta paralelo ao eixo da ligação na diatômica enquanto o respectivo orbital
LUMO está perpendicular a este mesmo eixo. De forma que a aproximação com

14
ângulo de 180° com a plano do grupo heme permite a interação entre os orbitais
citados anteriormente.
No entanto o orbital SOMO da molécula de O2 apresentá certa angulação em
relação ao eixo da ligação O=O, consequemente a aproximação adequada para
favorecer a sobreposição entre os orbitais eg e t2g do metal com o SOMO exige a
formação de um angulo de aproximadamente 120° como observado
experimentalmente.
d) No processo de retrodoação temos usualmente a doação de densidade
eletrônica para orbitais de caráter antiligante nos ligantes, consequentemente tal
transferência de carga atua reduzindo a ordem de ligação no ligante. Considerando
que o parâmetro k , correspondente à constante de força da ligação, é proporcional
a força da ligação na diatômica então a redução da ordem de ligação como
resultado da retrodoação tem como consequência a redução do parâmetro k.

15
Os demais termos da equação permanecem invariantes, portanto a redução
do k resulta em uma diminuição da frequência de estiramento observada.
e) Como discutido no item anterior, é esperado que com aumento da
retrodoação ocorra uma redução na frequência de estiramento observada. Portanto
observando os dados experimentais apresentados no exercício espera-se que o
complexo com ligante thiofenoláto axial apresente maior retrodoação que o
correspondente com o ligante derivado do imidazol.
Em relação às enzimas podemos correlacionar os grupos axiais histidina e
cisteína com os dos complexos modelo apresentados no enunciado. Nota-se que o
resíduo de histidina apresenta grande semelhança estrutural com o ligante 1-metil-
imidazol enquanto que o resíduo de cisteína é semelhante ao ligante thiofenol. De
forma que podemos esperar que assim como no caso dos complexos modelo, o
complexo com resíduo de cistéina possibilite maior retrodoação ao ligante
coordenado que o correspondente com o resíduo de histidina.
Nas condições biológicas em ambas as enzimas, nas cyps 450 e na
hemoglobina, o ligante coordenado é usualmente uma molécula de oxigênio.
Consequentemente com a maior retrodoção do resíduo axial de cistéina nas CYPs
450 ocorre um maior enfraquecimento da ligação O=O , assim facilitando a clivagem
da ligação. De forma contrária, na hemoglobina, esse tipo de fenômeno não é
observado devido a menor retrodoação no complexo com ligante axial de histidina.

16
IV. Sólidos iônicos: de uma fase à outra
a) Temos que, a partir da termodinâmica, o composto β -AgI é o mais estável
na temperatura e pressão ambiente. Sendo assim, ao ser formado o composto α -
AgI espera-se que esse seja convertido no composto mais estável, o β -AgI.
Entretanto, isso não é observado na experiência realizada na solução com excesso
de Ag+. Essa aparente contradição pode ser elucidada a partir da cinética da reação:
ao ser formado o composto em sua forma alfa, a conversão para a estrutura mais
estável ocorre de forma muito lenta por se tratar de fases sólidas, não podendo ser
observada na escala de tempo da realização do experimento.
b) O gráfico mostra uma brusca mudança do coeficiente de condutividade do
AgI em aproximadamente 420 K, coincidente com a temperatura da transição de
fase do composto, passando de mais estável a fase β para α. Sabendo que a
estrutura do sólido é um fator determinante para sua capacidade condutora, visto
que determina a menor ou maior mobilidade dos íons, temos que a mudança na
estrutura cristalina que ocorre na transição de fases é a responsável pela mudança
do coeficiente observada no gráfico.
c) Na curva do cloreto de sódio, a variação abrupta do coeficiente de
condutividade ocorre em seu ponto de fusão, ou seja, a mudança de estado físico
proporciona grande alteração na sua condutividade iônica.
Na fase líquida, a movimentação das cargas é muito mais eficiente do que
na estrutura sólida e, consequentemente, a condutividade também.
d) De forma também generalizada, podemos dizer que os tamanhos dos cátions
são menores do que os tamanhos dos ânions. Os cátions são átomos que doaram
um ou mais elétrons a ânions, fazendo com que os eletróns restantes no átomo
sentissem uma carga nuclear efetiva maior, se aproximando do núcleo e diminuindo

17
assim o seu raio iônico. De forma similar, os ânions, que recebem elétrons, têm seu
raio aumentado, pois os elétrons adicionados à sua eletrosfera resultam na
diminuição da carga nuclear efetiva na camada de valência, permitindo, assim, o
aumento do tamanho do átomo. Essa racionalização está de acordo com a
generalização proposta, de forma que os cátions, como menores íons, podem ser
considerados os portadores de carga, visto que a mobilidade é inversamente
proporcional ao tamanho do íon.
e) Como mostra o gráfico, a condutividade da fase α é muito superior àquela
observada para a fase β, ou seja, pode-se inferir que a mobilidade dos portadores
de carga na estrutura cristalina do α -AgI é maior. Tendo em vista que quanto maior
o fator de empacotamento (razão entre o volume ocupado pelos átomos e o volume
da célula unitária), menor o espaço vazio nas células unitárias que compõe a
estrutura cristalina, espera-se que o fator de empacotamento para a estrutura α-
AgI seja menor do que para β-AgI, garantinda que a mobilidade dos portadores de
carga seja favorecida na estrutura α.
f) O seguinte diagrama mostra como pode ser obtido o ΔHtransição a partir das
respectivas energias de rede das fases α e β:
Sendo assim, o ΔHtransição pode ser escrito segundo a seguinte equação:
ΔHrede α-AgI – ΔHrede β-AgI = ΔHtransição .

18
g) Como temos que nas transições de fase ΔG = 0, podemos reorganizar a
seguinte expressão para obter a entropia de transição:
ΔG = ΔH – T ΔS.
ΔStransição = 𝚫𝐇 𝐭𝐫𝐚𝐧𝐬𝐢çã𝐨
𝑻
V. Ácido Maleico e Ácido Fumárico
a) A interconversão direta do ácido maleico para o ácido fumárico envolveria
uma rotação da ligação dupla. Este fenômeno possui uma grande barreira
energética, já que a rotação prejudica o alinhamento dos orbitais π e
consequentemente, enfraquece a ligação química. Em outras palavras, a reação
não ocorre porque a ligação dupla é rígida.
Com a protonação, esta ligação adquire um maior caráter de ligação simples
e, desta forma, a barreira energética da rotação é diminuída drasticamente.
b) Ponto de fusão: O ácido fumárico possui uma geometria que permite a
formação de uma rede de moléculas interagindo por meio de ligações de hidrogênio,
levando a uma estrutura cristalina com um bom empacotamento. Já no ácido
maleico, parte das ligações de hidrogênio serão intermoleculares e sua geometria
não permite a formação de uma rede cristalina tão favorável, o que resulta em

19
interações intermoleculares mais fracas. Portanto, o ácido fumárico possui maior
ponto de fusão.
Solubilidade: No ácido maleico, os momentos de dipolo resultantes dos
grupos carboxila de adicionam, fazendo com que a molécula tenha um momento de
dipolo diferente de zero – esta polaridade favorece o processo de hidratação. No
ácido fumárico, os momentos de dipolo das duas carboxilas se cancelam, fazendo
com que o momento de dipolo da molécula seja igual a zero. Desta forma, o ácido
maleico terá uma solubilidade maior que a do ácido fumárico.
c) A diferença do primeiro pKa do ácido maleico e do ácido fumárico está
relacionada com a estabilidade da base conjugada. A remoção de um próton no
ácido maleico resulta em um ânion estabilizado por uma ligação de hidrogênio
intramolecular. No ácido fumárico, a geometria da molécula não permite essa
ligação de hidrogênio intramolecular. Desta forma, a desprotonação é mais
favorável para o ácido maleico, fazendo com que este tenha um pKa1 menor quando
comparado ao ácido fumárico.

20
Essa estabilização resultante da ligação de hidrogênio intramolecular precisa sem
superada para que ocorra a segunda desprotonação do ácido maleico, o que
dificulta o processo. Desta forma, o ácido maleico possui pKa2 maior que o do ácido
fumárico.
d)
Espécie predominante após adição de titulante:
(1) 10 mL – H2B
(2) 30 mL – HB-
(3) 50 mL – B2-
e) Primeiramente, devemos escrever os equilíbrios químicos envolvidos e suas
respectivas constantes
- Primeira ionização do ácido maleico (H2Ma)
𝐻2𝑀𝑎 ⇌ HMa− + 𝐻+

21
𝐾𝑎1 =[HMa−][𝐻+]
[𝐻2𝑀𝑎]
𝐾𝑎1 = 10−𝑝𝐾𝑎1 = 1,259 × 10−2
- Segunda ionização do ácido maleico (H2Ma)
HMa− ⇌ Ma2− + 𝐻+
𝐾𝑎2 =[Ma2−][𝐻+]
[HMa−]
𝐾𝑎2 = 10−𝑝𝐾𝑎2 = 8,511 × 10−7
- Autoionização da água
𝐻2O ⇌ OH− + 𝐻+
𝐾𝑤 = [OH−][𝐻+]
𝐾𝑤 = 1,00 × 10−14
- Solubilização do sal
𝑁𝑎2Ma ⇌ Ma2− + 2𝑁𝑎+
Em seguida, fazemos os balanços de massa correspondente ao ácido e ao sal
adicionados
𝐶á𝑐𝑖𝑑𝑜 + 𝐶𝑠𝑎𝑙 = [Ma2−] + [HMa−] + [𝐻2𝑀𝑎]
2𝐶𝑠𝑎𝑙 = [Na+]
Por fim, fazemos o balanço de cargas

22
[𝐻+] + [Na+] = 2[Ma2−] + [HMa−] + [OH−]
Para facilitar os cálculos, vamos considerar as seguintes aproximações
- Como estamos em meio ácido
[OH−] ≪ [𝐻+]
- Como Ka1 é muito maior que Ka2
[Ma2−] ≪ [HMa−]
Desta forma, as equações do balanço de carga e do balanço de massa podem ser
escritas como
[𝐻+] + [Na+] = [HMa−]
𝐶á𝑐𝑖𝑑𝑜 + 𝐶𝑠𝑎𝑙 = [HMa−] + [𝐻2𝑀𝑎]
2𝐶𝑠𝑎𝑙 = [Na+]
Somando [H2Ma] dos dois lados da equação do balanço de carga
[𝐻+] + [Na+] + [𝐻2𝑀𝑎] = [HMa−] + [𝐻2𝑀𝑎]
Substituindo as equações do balanço de massa na equação anterior
[𝐻+] + 2𝐶𝑠𝑎𝑙 + [𝐻2𝑀𝑎] = 𝐶á𝑐𝑖𝑑𝑜 + 𝐶𝑠𝑎𝑙
[𝐻2𝑀𝑎] = 𝐶á𝑐𝑖𝑑𝑜 − 𝐶𝑠𝑎𝑙 − [𝐻+]
Como Cácido= 0,005 mol/L e Csal= 0,001 mol/L
[𝐻2𝑀𝑎] = 0,004 − [𝐻+]

23
Desta forma, encontramos a concentração de H2Ma em função apenas da
concentração de H+ do meio. Agora, podemos substituir [H2Ma] na equação de Ka1
para encontrar [HMa-]
[HMa−] =[𝐻2𝑀𝑎]𝐾𝑎1
[𝐻+]
[HMa−] =(0,004 − [𝐻+]) × 0,01259
[𝐻+]
[HMa−] =(5,036 × 10−5) − 0,01259[𝐻+]
[𝐻+]
Com isso, podemos substituir esta concentração na equação do balanço de carga
[𝐻+] + [Na+] = [HMa−]
[𝐻+] + 2𝐶𝑠𝑎𝑙 =(5,036 × 10−5) − 0,01259[𝐻+]
[𝐻+]
Organizando os termos, chegamos a um polinômio de segundo grau
[𝐻+]2 + 0,002[𝐻+] = (5,036 × 10−5) − 0,01259[𝐻+]
[𝐻+]2 + 0,01459[𝐻+] − (5,036 × 10−5) = 0
Substituindo [H+] por x, podemos usar softwares ou alguns sites para encontrar mais
facilmente as raízes desta equação. Deste modo, obtemos
𝑥1 = −0,01747
𝑥2 = 0,002882
Como a concentração de H+ é um valor positivo, temos que
[𝐻+] = 2,882 × 10−3 𝑚𝑜𝑙/𝐿

24
Finalmente
𝑝𝐻 = −log[𝐻+]
𝑝𝐻 = −log[𝐻+]
𝑝𝐻 = 2,54
VI. Titulação de Ni com dimetilglioxima
a) Como o teor de Ni na liga está em torno de 2%, 1,0g da liga metálica conterá
cerca de 0,02g de Ni, o que corresponde a
0,02 𝑔 𝑑𝑒 𝑁𝑖
58,69 𝑔/𝑚𝑜𝑙= 3,41 × 10−4 𝑚𝑜𝑙 𝑑𝑒 𝑁𝑖
Essa quantidade de metal requer
2 × (3,41 × 10−4 𝑚𝑜𝑙 𝑑𝑒 𝑁𝑖)(116,12𝑔 𝑑𝑒 𝐷𝑀𝐺/𝑚𝑜𝑙 𝑑𝑒 𝑁𝑖) = 0,0792 𝑔 𝑑𝑒 𝐷𝑀𝐺
Pois 1 mol de Ni2+ necessita de 2 mol de DMG. Um excesso de 50% de DMG seria
(1,5 x 0,0792g) = (0,119g). Essa quantidade de DMG está contida em
0,119𝑔 𝑑𝑒 𝐷𝑀𝐺
0,01𝑔 𝑑𝑒 𝐷𝑀𝐺/𝑔 𝑑𝑒 𝑠𝑜𝑙𝑢çã𝑜= 11,9𝑔 𝑑𝑒 𝑠𝑜𝑙𝑢çã𝑜
Que ocupa um volume de
11,9𝑔 𝑑𝑒 𝑠𝑜𝑙𝑢çã𝑜
0,79 𝑔 𝑑𝑒 𝑠𝑜𝑙𝑢çã𝑜/𝑚𝐿= 15 𝑚𝐿
b) Para cada mol de Ni existente no aço, será formado 1 mol de precipitado.
Portanto, 0,1340 g de precipitado corresponde a

25
0,1340 𝑔 𝑑𝑒 𝑁𝑖(𝐷𝑀𝐺)2
288,91 𝑔 𝑑𝑒 𝑁𝑖(𝐷𝑀𝐺)2/𝑚𝑜𝑙 𝑑𝑒 𝑁𝑖(𝐷𝑀𝐺)2 = 4,638 × 10−4 𝑚𝑜𝑙 𝑑𝑒 𝑁𝑖(𝐷𝑀𝐺)2
A massa de Ni no aço é
(4,638 × 10−4 𝑚𝑜𝑙 𝑑𝑒 𝑁𝑖)(58,69 𝑔/𝑚𝑜𝑙) = 0,02722 𝑔 𝑑𝑒 𝑁𝑖
E a porcentagem em massa de Ni presente no aço é
0,02722 𝑔 𝑑𝑒 𝑁𝑖
1,1634 𝑑 𝑑𝑒 𝑎ç𝑜× 100 = 2,34%
c) Em pH ácido, a dimetilglioxima (que neste caso pode ser considerada um
ácido) estará na sua forma protonada. Desta forma, a presença desses hidrogênios
poderia impedir a formação do complexo devido a repulsão estérica ou, caso
ocorresse formação de um complexo, o composto de coordenação formado,
Ni(DMG)2 2+, seria solúvel em por ser um cátion, não ocorrendo precipitação.
d) A configuração eletrônica do metal (mais especificamente o preenchimento
dos orbitais d) é um dos principais fatores que indicam quando o metal formará um
complexo estável com determinados ligantes. O cátion Ni2+ possui configuração
eletrônica 1s2 2s2 2p6 3s2 3p6 3d8. Assim como o Ni2+, os cátions Au3+ e Pd2+
também possuem oito elétrons em seus orbitais d mais externos – em outras
palavras, são do tipo d8. Desta forma, é justificável que estes metais também
formem complexos estáveis com DMG. Já os cátions Zr4+ e Ca2+ são do tipo d0 e,
por isso, é compreensível que eles não formem complexos estáveis com DMG.
e) Em meio básico, se os cátions Fe(III), Al(III) e Cr(III) estiverem presentes,
pode ocorrer precipitação dos hidróxidos metálicos Fe(OH)3, Al(OH)3 e Cr(OH)3. A
massa desses hidróxidos se somaria à massa de Ni(DMG)2 na análise, fazendo com
que a concentração de níquel calculada seja maior do que a concentração real.
Citrato e tartarato formam complexos solúveis e estáveis com Fe(III), Al(III) e Cr(III);

26
desta forma, os cátions complexados ficam indisponíveis para reagir com os íons
hidróxido, impedindo a precipitação indesejada.
VII. Espectroscopia no infravermelho
a) As geometrias e o grupo pontual das moléculas em questão encontram-se
abaixo:
Os grupos pontuais são determinados seguindo o fluxograma. Assim, temos os
grupos C2v para a água, D∞h para o gás carbônico e Td para o metano.

27
b) Todos os espectros apresentam apenas duas bandas vibracionais, desse
modo, o único jeito de determinar a qual molécula pertence cada espectro é
avaliando a freqüência de vibrações. Considere a equação abaixo:
k
c2
1
Onde é a freqüência em cm-1, c é a velocidade da luz, k é a constante de
força e é a massa reduzida.
Apesar de ser uma aproximação rudimentar, pois o calculo da freqüência não
é tão simples para um modo vibracional que envolva o movimento de vários átomos,
podemos utilizar essa equação quantitativamente para atribuir os espectros.
Dessa forma, como o CO2 possui maior massa reduzida, espera-se que suas
bandas se apresentem com menor freqüência, portanto, corresponde ao espectro
1.
A água e o metano possuem modos vibracionais com massa reduzida
parecida, porém, a constante de força das ligações OH é maior do que as ligações
CH, assim, OH deve possuir modos vibracionais mais energéticos (maior número
de onda). Portanto o espectro 2 corresponde à molécula de água e o espectro 3
corresponde ao metano.
c) Ao substituir o hidrogênio por flúor, aumenta-se consideravelmente a massa
reduzida do sistema. Dessa forma, segundo a equação do item B, esperar-se-á um
deslocamento das bandas para menores números de onda. Quanto ao número de
bandas, vemos que este permanece constante, pois não há mudança de simetria.
Em relação à intensidade das bandas nada se pode afirmar, uma vez que a
intensidade é determinada pela variação do momento de dipolo com o movimento

28
dos átomos, fator que não é possível quantificar considerando apenas a estrutura
da molécula.
d) A regra de seleção para espectroscopia no infravermelho estabelece que,
para que um modo seja ativo, precisa haver alteração no momento de dipolo da
molécula com o movimento dos átomos. A molécula Br2 livre não é ativa no
infravermelho pois o seu momento de dipolo não se altera quando a ligação Br-Br
estica no movimento vibracional.
Quando o Br2 está posicionado paralelamente ao anel, graças à simetria do sistema,
o movimento vibracional dos átomos de Br não irá causar alteração no momento de
dipolo, dessa forma, não será ativo no infravermelho e não teremos a banda
presente no espectro do enunciado. Porém, quando o Br2 se posiciona
perpendicularmente ao anel, o movimento vibracional dos átomos de bromo
provoca alteração no momento de dipolo e, portanto, o modo será ativo no
infravermelho, correspondendo à banda do espectro apresentado.
Assim, pode-se afirmar, com base no espectro dado, que a conformação mais
estável é aquela na qual a molécula de bromo se apresenta perpendicular ao anel
benzênico.
VIII. Dinâmica de colisões reacionais
a) Na figura abaixo está representada a curva do potencial de morse para o
estado fundamental da molécula de H2, onde D0 corresponde a energia de
dissociação da molécula em questão. A análise da curva sugere que a magnitude
da energia de dissociação é proporcional a profundidade da curva de potencial.
Desse modo, pode-se tirar conclusões sobre a força da ligação das moléculas, a
partir das curvas de potencial de morse.

29
A imagem fornecida no enunciado do exercício, permite afirmar que as forças
de ligação das moléculas de hidrogênio no estado fundamental e estado excitado
são diferentes.
A partir dos diagramas de orbitais moleculares da molécula H2 (estado
fundamental) e H2∗ (estado excitado) verifica-se que quando a molécula de
hidrogênio é excitada eletronicamente ocorre a ocupação de orbitais moleculares
com caráter antiligante o que promove a diminuição da ordem de ligação e
consequentemente a diminuição da força de ligação/magnitude da energia de
dissociação.
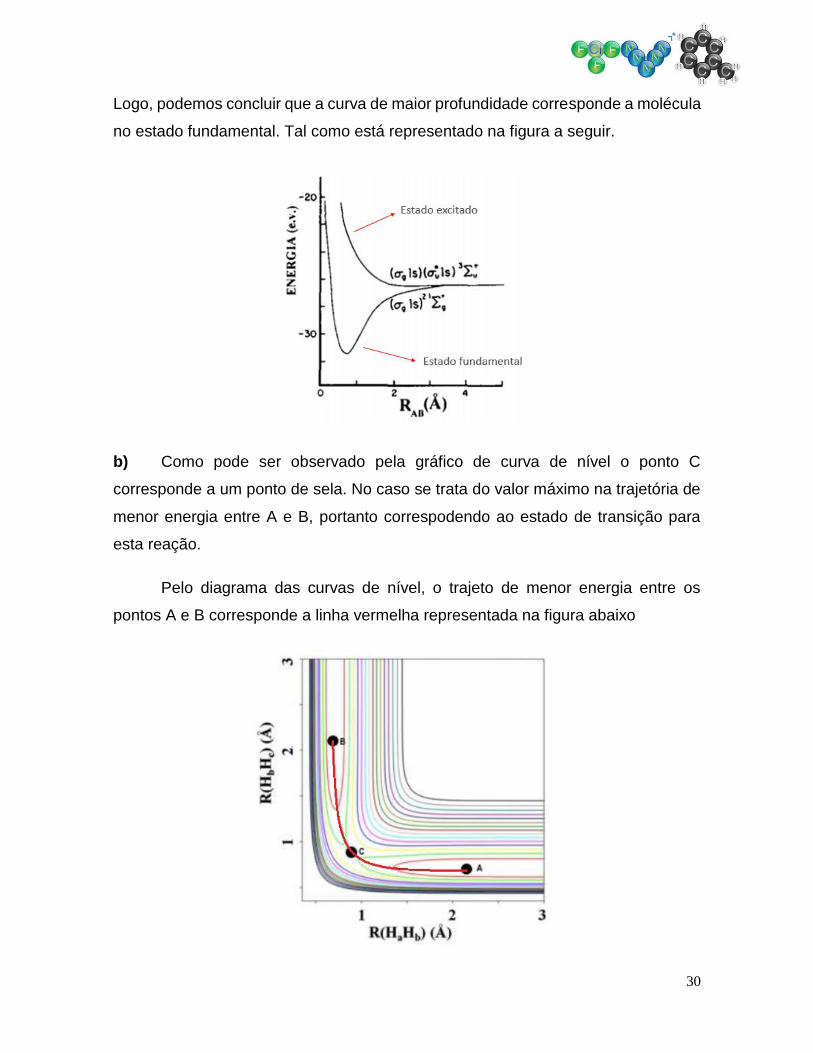
30
Logo, podemos concluir que a curva de maior profundidade corresponde a molécula
no estado fundamental. Tal como está representado na figura a seguir.
b) Como pode ser observado pela gráfico de curva de nível o ponto C
corresponde a um ponto de sela. No caso se trata do valor máximo na trajetória de
menor energia entre A e B, portanto correspodendo ao estado de transição para
esta reação.
Pelo diagrama das curvas de nível, o trajeto de menor energia entre os
pontos A e B corresponde a linha vermelha representada na figura abaixo
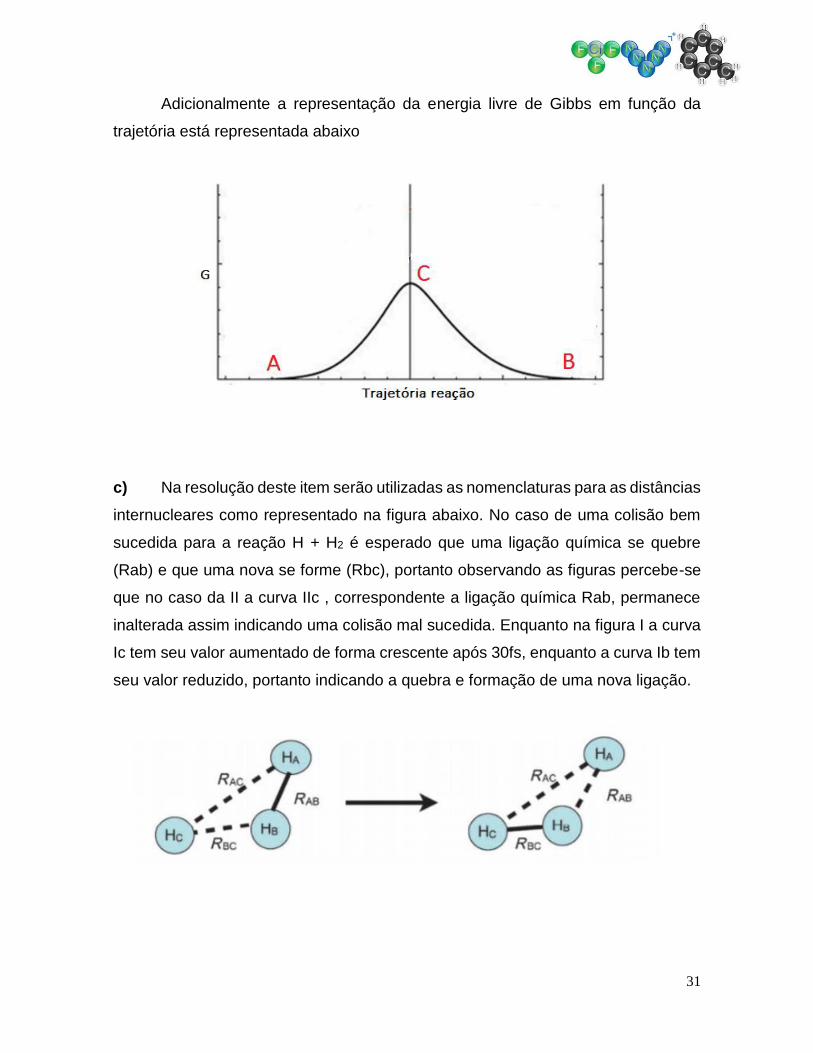
31
Adicionalmente a representação da energia livre de Gibbs em função da
trajetória está representada abaixo
c) Na resolução deste item serão utilizadas as nomenclaturas para as distâncias
internucleares como representado na figura abaixo. No caso de uma colisão bem
sucedida para a reação H + H2 é esperado que uma ligação química se quebre
(Rab) e que uma nova se forme (Rbc), portanto observando as figuras percebe-se
que no caso da II a curva IIc , correspondente a ligação química Rab, permanece
inalterada assim indicando uma colisão mal sucedida. Enquanto na figura I a curva
Ic tem seu valor aumentado de forma crescente após 30fs, enquanto a curva Ib tem
seu valor reduzido, portanto indicando a quebra e formação de uma nova ligação.

32
Utilizando este racioncínio podemos classificar a figura I como a
correspondente a uma colisão bem sucedida enquanto a figura II corresponde a
uma colisão mal sucedida
Analisando inicialmente a curva I, percebe-se que a curva Ic apresenta o
menor valor no início da trajetória consequentemente indicando que esta curva se
trata da ligação inicial H-H , portanto Rab de acordo com a nomenclatura escolhida.
Esta escolha é confirmada pelo caráter oscilatório da curva Ic devido à vibração
molecular. Sabendo que a figura I corresponde a uma colisão bem sucedida a curva
Ib que assume o comportamento da curva Ic após os 30fs possivelmente
corresponde a nova ligação formada (Rbc). Por fim a curva restante, Ia, representa
a distância Rac.
De forma análoga, no caso da figura II , na qual a colisão é mal sucedida,
pode-se identificar que a curva inalterada de caráter oscilatório, IIc, representa a
distância da ligação química Rab. Enquanto que pela figura não é possível
identificar se Ia e Ib corresponde a respectivamente Rac e Rbc ou vice versa.
Distâncias
Internucleares
Figura I Figura II
Rac Ia IIa/IIb
Rbc Ib IIb/IIa
Rab Ic IIc
d) Note que entre as duas reações comparadas, representadas abaixo, o único
parâmetro da equação Z12 que distinto entre elas é a massa reduzida.
H + D2 → HD + D
D + H2 → HD + H

33
Sendo assim podemos facilmente utilizar a seguinte razão das massas reduzidas
para deteminar a razão entre as densidades de colisão para cada uma das reações.
Nas contas serão utilizadas as seguintes massas aproximadas mH=1 e mD=2.
𝜇𝐻+𝐷2=
1 ∗ 4
1 + 4= 0,8
𝜇𝐷+𝐻2=
2 ∗ 2
2 + 2= 1
Portanto a razão entre as densidades de colisão será
𝑍𝐷+𝐻2
𝑍𝐻+𝐷2
=
√1
𝜇𝐷+𝐻2
√1
𝜇𝐻+𝐷2
= √𝜇𝐻+𝐷2
𝜇𝐷+𝐻2
= √0,8 = 0,89
A razão entre os fatores pré-exponenciais será
𝐴𝐷+𝐻2
𝐴𝐻+𝐷2
=3,17 × 10−10
2,67 × 10−10= 1,19
O resultado pode ser interpretado de duas formas. Caso você esperasse a
ocorrência de outros fatores dinâmicos não presentes no simples modelo de colisão
de esferas rígidas, a pequena diferença da razão experimental com o valor obtido
pela razão das densidades de colisão poderia ser esperada. No entanto, a
consideração da validade do modelo de esferas rígidas para esta reação simples
leva a uma discordância entre o esperado e o calculado.





























