Embed Size (px)
Citation preview

Um químico analítico pode se defrontar com dois tipos de problemas:
Qualitativo:
•Esta água destilada contém boro?
•Estes dois solos são do mesmo lugar?
Quantitativo:
•Quanta albumina existe nesta amostra de soro sanguíneo?
•Quanto chumbo existe na água da torneira?
•Esta amostra de aço contém pequenas quantidades de cromo, tungstênio e manganês,
quanto de cada um?
Química Analítica moderna tem um caráter essencialmente quantitativo.
Uma resposta quantitativa, a qualquer das perguntas anteriores é mais indicada que uma
qualitativa.
Problemas em Química Analítica:

A pessoa que precisou da análise pode, com os resultados quantitativos, julgar se o
conteúdo do analito é nocivo e exige alguma providência ou não.
Em alguns casos, apenas uma resposta quantitativa tem algum valor. Por exemplo,
virtualmente todo o soro sanguíneo humano tem albumina, a dúvida só poderia ser quanto.
É importante considerar que, mesmo quando uma resposta qualitativa é solicitada, métodos
quantitativos têm de ser usados para obtê-la.
Na realidade, um químico analítico nunca pode dizer simplesmente que encontrou ou não
encontrou boro na amostra de água.
Ele deve empregar um método quantitativo, capaz de detectar, digamos, 1 μg mL-1 de boro.

Como devo expressar o resultado final?
Qual a densidade de um mineral que apresenta uma massa 4,635 (± 0,002) g e um
volume de 1,13 (± 0,05) mL?
a) Qual a incerteza da densidade calculada?
b) Quantos algarismos significativos devem ser usados para expressar a densidade?

Algarismos significativos
O número de algarismos significativos de uma medida é o número de dígitos que
representam um resultado experimental, de modo que apenas o último algarismo seja
duvidoso.
Pode ser obtido de duas formas:
-Diretamente ou
-Indiretamente

Algarismos significativos
Diretamente
Ex 1. Determinação da massa de uma substância em uma balança.
Balança analítica com incerteza ± 0,1 mg: 0,0402 g de NaCl; 1,0056 g de CaCO3
Ex 2. Medida do volume de uma solução com uma pipeta.
Pipeta volumétrica capacidade de 5mL (classe A) tem incerteza de ± 0,01 mL: 5,00 mL
Ex 3. Medida do volume de uma solução com uma bureta.
Bureta capacidade 10 mL (classe A) tem incerteza de ± 0,02 mL: 8,00 mL
Indiretamente
A partir dos valores de outras grandezas medidas.
Ex 4. Cálculo da concentração de uma solução a partir da massa do soluto e do volume
da solução. Solubilização de 0,0402 g de NaCl pesado em balança analítica em um
balão volumétrico de 100 mL.
Ex 5. Cálculo da densidade do mineral.

Como devo expressar o resultado final?
Qual a densidade de um mineral que apresenta uma massa 4,635 (± 0,002) g e um
volume de 1,13 (± 0,05) mL?
a) Qual a incerteza da densidade calculada? PROPAGAÇÃO DAS INCERTEZAS!!!!
mLgmL
gd /1018,4
)05,0(13,1
)002,0(635,4
mLg /)2,0(1,4

Como devo expressar o resultado final?
Qual a densidade de um mineral que apresenta uma massa 4,635 (± 0,002) g e um
volume de 1,13 (± 0,05) mL?
b) Quantos algarismos significativos devem ser usados para expressar a densidade?
REGRA DOS ALGARISMOS SIGNIFICATIVOS!!!!
mLgmL
gd /10,41018,4
)05,0(13,1
)002,0(635,4
Zero é significativo:
(a) Entre 2 algarismos significativos;
(b) Depois da vírgula e a direita de outro dígito significativo.
Zero não é significativo antes da vírgula e a esquerda de um número significativo.
O arredondamento deve ser
feito somente na resposta final
(não em resultados
intermediários).

Adição e subtração
-se os números a serem adicionados ou subtraídos têm igual número de algarismos
significativos: resposta deve ficar com o mesmo número de casas decimais do
número individual
Ex: 5,345 + 6,728 = 12,073
-se os números a serem adicionados ou subtraídos não possuírem o mesmo número
de algarismos significativos: limita-se pelo de menor número
Ex: 18,9984032 + 83,80 = 102,7984032 102,80
Ex: 1,632 x 105 + 4,107 x 103 + 0,984 x 106 = ?????
Algarismos significativos em aritmética

Algarismos significativos em aritmética
Multiplicação e divisão
-limita-se ao número de dígitos contidos no número com menos algarismos
significativos
Ex: 3,26 x 10-5 x 1,78 = 5,80 x 10-5
Ex: 4,3179 x 1012 x 3,6 x 10-19 = 1,6 x 10-6
Ex: 34,60 / 2,46287 = 14,05
A potência de 10 não influencia em nada o número de algarismos significativos que
devem ser mantidos.
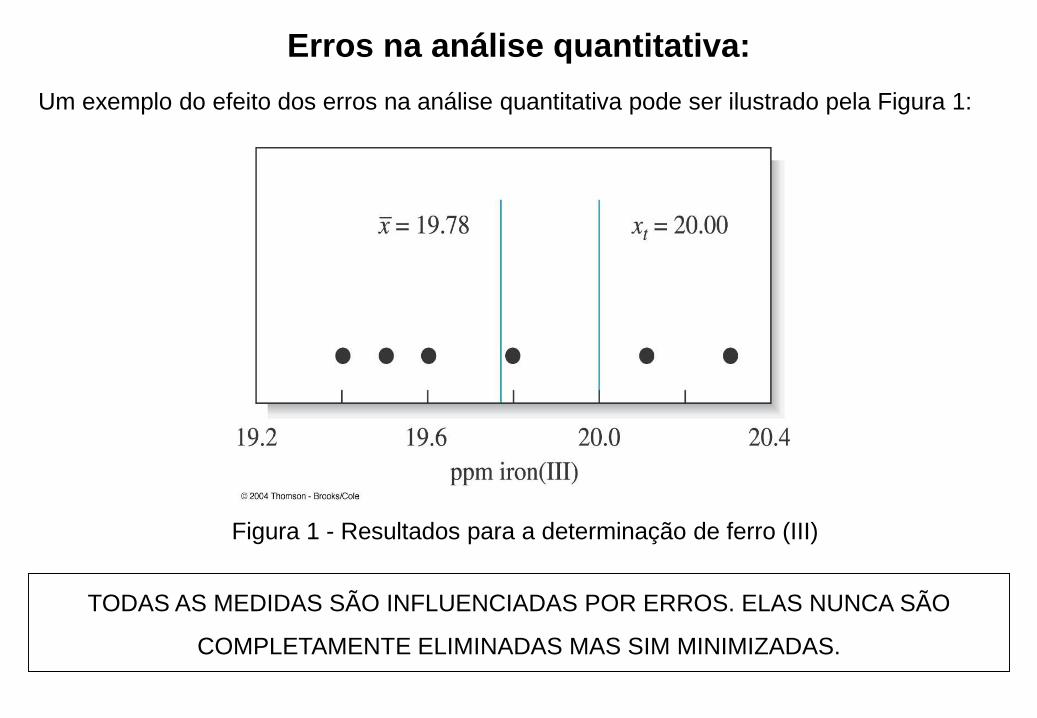
Erros na análise quantitativa:
Um exemplo do efeito dos erros na análise quantitativa pode ser ilustrado pela Figura 1:
Figura 1 - Resultados para a determinação de ferro (III)
TODAS AS MEDIDAS SÃO INFLUENCIADAS POR ERROS. ELAS NUNCA SÃO
COMPLETAMENTE ELIMINADAS MAS SIM MINIMIZADAS.

A primeira pergunta a ser respondida antes de começar uma análise é “QUAL É O
ERRO MÁXIMO QUE EU POSSO TOLERAR NO RESULTADO?”
A resposta para essa pergunta determina o quanto tempo você gastará na análise.
Definições:
MÉDIA:
N
x
x
N
ii
1
xi = valor individual
N = número de replicatas

PRECISÃO: concordância entre duas ou mais medidas realizadas exatamente do
mesmo jeito.
DESVIO PADRÃO:
1
1
2
N
xx
s
N
ii

VARIÂNCIA:
DESVIO PADRÃO RELATIVO:
COEFICIENTE DE VARIAÇÃO:
EXATIDÃO: proximidade do resultado do valor verdadeiro
1000xx
sRSD
%100xx
sCV
1
1
2
2
N
xx
s
N
ii

Exatidão e precisão:

Tipos de erros:
O ato de medir é, em essência, um ato de comparar, e essa comparação envolve erros
de diversas origens (dos instrumentos, do operador, do processo de medida etc.).
Quando se pretende medir o valor de uma grandeza, pode-se realizar apenas uma ou
várias medidas repetidas, dependendo das condições experimentais particulares ou
ainda da postura adotada frente ao experimento.
Todos os tipos de erro podem ser expressos como "erro absoluto" ou como "erro
relativo".

Tipos de erros:
Erro absoluto:
Erro relativo:
verdadeiroixxE
%100xx
xxE
verdadeiro
verdadeiroi
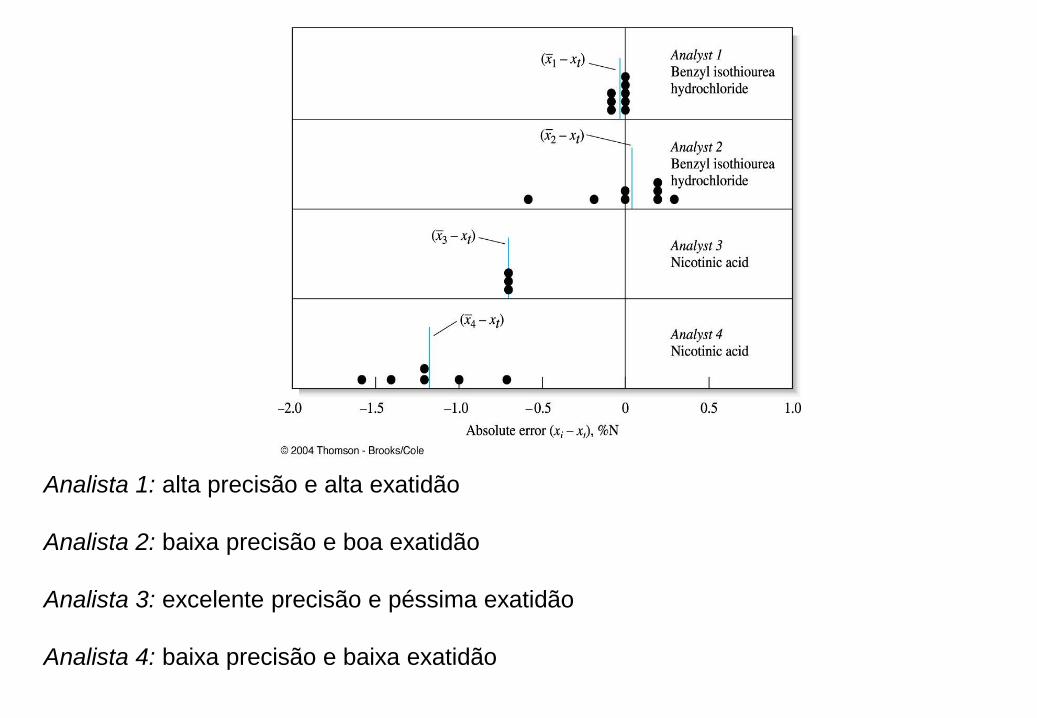
Analista 1: alta precisão e alta exatidão
Analista 2: baixa precisão e boa exatidão
Analista 3: excelente precisão e péssima exatidão
Analista 4: baixa precisão e baixa exatidão

ERROS GROSSEIROS: são facilmente reconhecidos. Eles são erros tão sérios que não
deixam alternativas a não ser refazer todo o experimento.
Exemplos incluem a quebra do equipamento, contaminação de reagentes, erros na
adição de alíquotas, etc.
ERROS RANDÔMICOS OU ALEATÓRIOS (indeterminados): estes erros se
manifestam na forma de pequenas variações nas medidas de uma amostra, feitas em
sucessão pelo mesmo analista, com todas as precauções necessárias e em condições
de análise praticamente idênticas. Não podem ser controlados.
ERROS SISTEMÁTICOS (determinados) : são erros em que se pode conhecer a sua
fonte. São independentes das leis do acaso e produzem-se sempre no mesmo sentido,
podendo ser anulados ou corrigidos.
Exemplos incluem balança mal calibrada, deficiência de funcionamento, erros de
operação, etc.

Como reduzir os erros sistemáticos?
•Calibração de instrumentos e sua correção
•Determinação do branco de uma amostra: consiste na execução de uma análise nas
mesmas condições experimentais usadas na análise da amostra, porém na ausência do
constituinte de interesse.
•Análise de uma substância padrão nas mesmas condições experimentais usadas na
análise da amostra.
•Uso de métodos de análise independentes
•Determinações paralelas
•Adição de padrão: adiciona-se à amostra uma quantidade conhecida de constituinte a
ser determinado.
•Padrões internos: adição de uma quantidade fixa de um material de referência a uma
série de amostras de concentrações conhecidas da substância a ser determinada.

Distribuição dos erros aleatórios
•A dispersão dos resultados de um conjunto de medidas pode ser estimada pelo desvio
padrão.
•Quando se faz um número elevado de leituras, pelo menos 50, de uma variável
contínua, por exemplo, o ponto final de uma titulação, os resultados se distribuem, em
geral de forma aproximadamente simétrica em torno da média.
68,26% - um intervalo
95,44% - 2 intervalos
99,72% - 3 intervalos
Distribuição Normal (ou Gaussiana)
média

Limite de confiança

Confiabilidade dos resultados
Um ponto muito importante é poder rejeitar certos resultados de forma sensata.
Aplicação do teste Q:
valormenorvalormaior
próximomaisvalorsuspeitovalorQ
Intervalo de confiança
Permite estimar a faixa na qual a média verdadeira poderá ser encontrada.
Aplicação tabela distribuição de t:
n
tsx
Limite de confiança de
para n análises repetidas

Exercício
A análise de uma amostra de calcita gerou porcentagens de CaO de 55,95; 56,00;
56,04; 56,08 e 56,23. O último valor parece anômalo; deve ser mantido ou rejeitado
em nível de confiança de 95%?
valormenorvalormaior
próximomaisvalorsuspeitovalorQ

Intervalo de confiança
Permite estimar a faixa na qual a média verdadeira poderá ser encontrada.
Aplicação tabela distribuição de t:
n
tsx
Limite de confiança de
para n análises repetidas

Exercício
Um químico obteve os seguintes dados para o teor alcoólico de uma amostra de
sangue: % de C2H5OH: 0,084; 0,089 e 0,079. Calcule o intervalo de confiança a
95%, sabendo que o desvio padrão do método é 0,005%.
Grau de liberdade é, em estatística, o número de determinações independentes (dimensão da amostra)
menos o número de parâmetros estatísticos a serem avaliados na população.

Comparação de resultados
A comparação dos valores de um conjunto de resultados com o valor verdadeiro ou
com os valores de outros conjuntos de resultados permite verificar a exatidão e
precisão do método analítico, ou se ele é melhor do que outro.
Existem 2 métodos muito usados para comparar resultados:
-teste t de Student
-teste da razão de variâncias (teste F)
Estes métodos utilizam o número de GRAUS DE LIBERDADE, em termos
estatísticos, o número de determinações independentes (dimensão da amostra)
menos o número de parâmetros estatísticos a serem avaliados na população.

Teste t de Student
-Usado para amostras pequenas
-Comparar a média de uma série de resultados com um valor de referência e
exprimir o nível de confiança associado ao significado de comparação
-Também usado para testar a diferença entre as médias de dois conjuntos de
resultados
s
nxt
)( = valor verdadeiro

Probabilidade do valor de t estar dentro de certos limites

Caso 1 – Comparando um resultado medido com um valor conhecido
Uma amostra de carvão foi adquirida como sendo um Material Padrão de Referência
certificado pelo Instituto Nacional de Padrões e Tecnologia (NIST) dos Estados Unidos,
contendo 3,18%pp de enxofre. Está se testando um novo método analítico para verificar
se o valor conhecido pode ser produzido ou não. Os valores medidos são 3,29; 3,22;
3,30 e 3,23%pp de enxofre, dando uma média de 3,26 e um desvio-padrão de 0,04. Esta
resposta concorda com o valor fornecido pelo NIST?
n
tsxμ
Valores necessários: t (Tabela t student), média (valor dado), desvio-padrão (valor dado) e
número de medidas (valor dado)

Lembrar que graus de liberdade nesse caso = n - 1

Caso 1 – Comparando um resultado medido com um valor conhecido
Uma amostra de carvão foi adquirida como sendo um Material Padrão de Referência
certificado pelo Instituto Nacional de Padrões e Tecnologia (NIST) dos Estados Unidos,
contendo 3,18%pp de enxofre. Está se testando um novo método analítico para verificar
se o valor conhecido pode ser produzido ou não. Os valores medidos são 3,29; 3,22;
3,30 e 3,23%pp de enxofre, dando uma média de 3,26 e um desvio-padrão de 0,04. Esta
resposta concorda com o valor fornecido pelo NIST?
0602634
0401823263 ,,
),)(,(,
n
tsxμ
Intervalo de confiança de 95% = 3,20 até 3,32%pp
O valor conhecido está pouco fora do intervalo de confiança de 95% Há menos do
que uma chance de 5% de que nosso método concorde com a resposta conhecida.

Caso 2 - Comparação entre as médias de duas amostragens
Quando um novo método analítico está sendo desenvolvido é comum comparar-se a
média e precisão do novo método com as do método de referência.
21
2121
nn
nn
s
xxt
p
2
1
2
1
médiax
médiax
sP = desvio padrão agrupado
2
)1()1(
21
2
22
2
11
nn
snsns
p
É necessário que não haja uma diferença significativa entre as precisões dos
métodos aplica o teste F antes de usar o teste t.
t calculado > t tabelado (95%)
diferença significativa resultados são
considerados diferentes

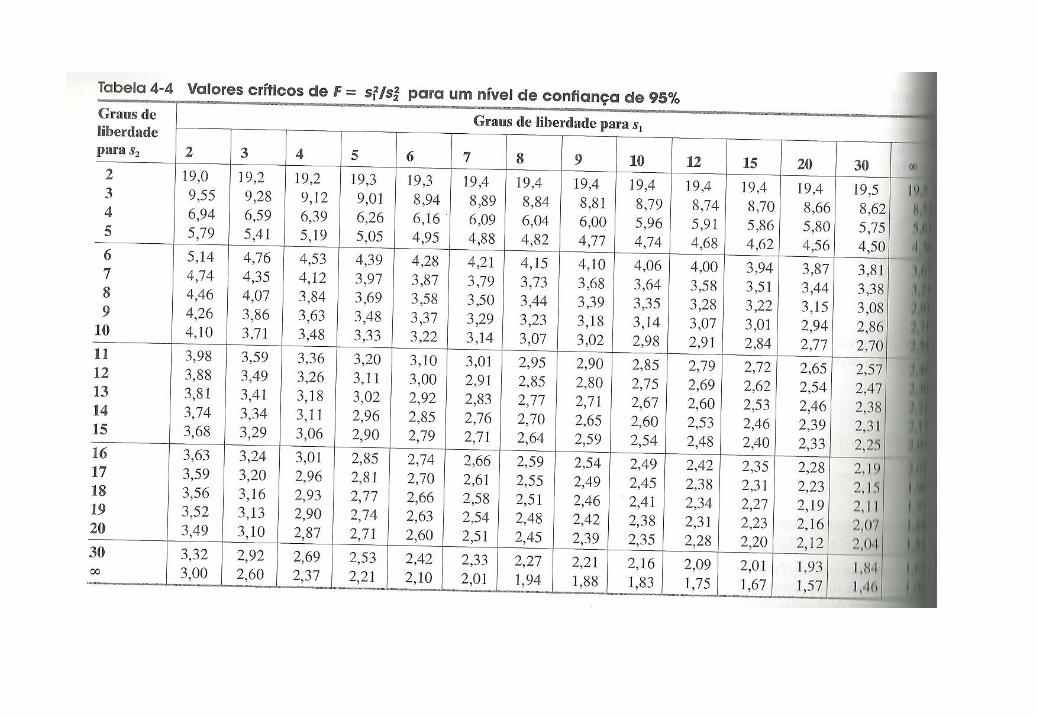
Teste F
-Usado para comparar as precisões de dois grupos de dados, como, por exemplo, os
resultados de dois métodos de análise diferentes ou resultados de dois laboratórios
diferentes.
-O maior valor de s é sempre colocado no numerador, o que faz com que o valor de F seja
sempre maior do que a unidade.
2
2
B
A
s
sF
F calculado > F tabelado a diferença é significativa

Caso 3 - Comparação diferenças individuais
-Usamos dois métodos diferentes para fazer medidas simples em várias amostras
diferentes.
-Os dois métodos fornecem a mesma resposta “dentro do erro experimental”?
Para cada amostra, ambos os resultados
são similares, porém não são idênticos.
Para verificar se existe uma diferença
significativa entre os dois métodos
realizaremos o teste t.

Caso 3 - Comparação diferenças individuais
-Usamos dois métodos diferentes para fazer medidas simples em várias amostras
diferentes.
-Os dois métodos fornecem a mesma resposta “dentro do erro experimental”?
1
)( 2
n
dds i
d
ns
dt
d
calculado
t tabelado = 2,228 há menos do que
95% de chance de que os dois resultados
sejam diferentes

Amostragem, padronização e calibração
Dimensão da amostra
Dimensão da amostra Tipo de análise
> 0,1 g Macro
0,01 a 0,1 g Semimicro
0,0001a 0,01 g Micro
<10-4 g Ultramicro
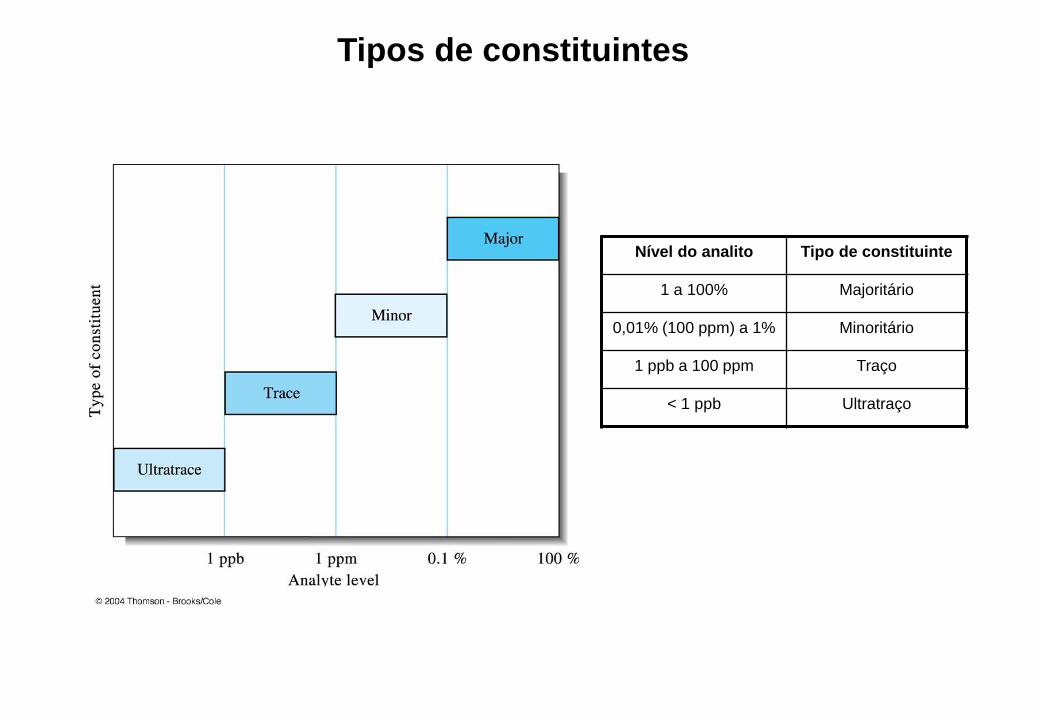
Tipos de constituintes
Nível do analito Tipo de constituinte
1 a 100% Majoritário
0,01% (100 ppm) a 1% Minoritário
1 ppb a 100 ppm Traço
< 1 ppb Ultratraço

Erros interlaboratoriais em função da concentração

Etapas envolvidas no processo de amostragem
Identificação da população da qual a amostra vai ser obtida
-Amostra líquida grande e heterogênea: água de um lago
-Amostra sólida grande e heterogênea: amostra de solo ou uma amostra de
minério
-Amostra de origem biológica: pedaço de tecido animal
Amostragem:
-Pequena fração representativa do material cuja composição se deseja determinar
-Composição deve refletir a composição média do todo
É a etapa mais difícil do processo analítico global e o fator determinante da
exatidão da análise

Coleta da amostra bruta que é representativa da população que está sendo
amostrada
Erros sistemáticos: eliminados com trabalho cuidadoso, uso adequado de padrões,
calibrações, brancos e materiais de referência.
Erros aleatórios podem ser mantidos em níveis aceitáveis, controlando as
variáveis que afetam as medições
Redução da amostra bruta a poucas centenas de grama de uma amostra
laboratorial homogênea, conveniente para análise.
AMOSTRAGEM
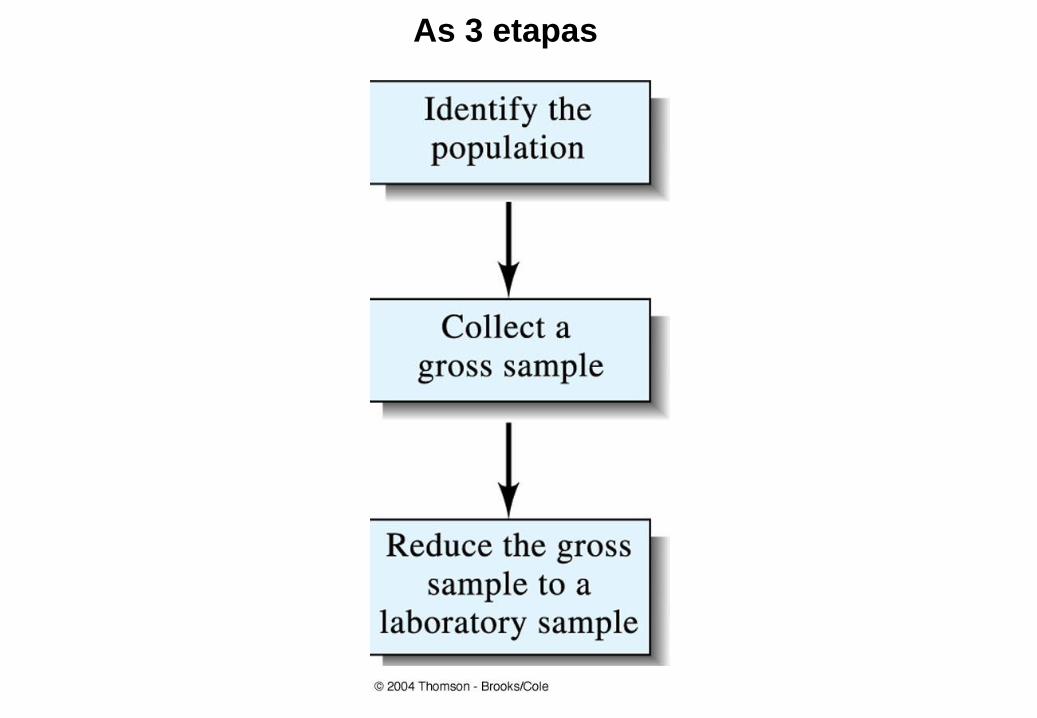
As 3 etapas

Objetivos do processo de amostragem
-Obter um valor médio que seja uma estimativa sem tendências da média da
população. Esse objetivo pode ser atingido apenas se todos os membros da
população tiverem uma probabilidade igual de estarem incluídos na amostra.
-Obter uma variância que seja uma estimativa sem erros sistemáticos da
variância da população, para que limites de confiança válidos para a média
possam ser encontrados e vários testes de hipóteses possam ser aplicados. Esse
objetivo pode ser alcançado apenas se toda amostra possível puder ser
igualmente coletada.
Ambos os objetivos requerem a obtenção de uma AMOSTRA ALEATÓRIA.

Incertezas na amostragem
Desvio padrão global para uma medida analítica:
222
mag sss sa = desvio padrão do processo de amostragem
sm = desvio padrão do método
Determinação do valor de sa:
-Retirada de uma amostra laboratorial (material bruto inicialmente coletado);
-Divisão dessa amostra em 6 partes iguais;
-Análises em replicata para cada uma das 6 partes.
Determinação do valor de sm:
-Diferentes porções da amostra laboratorial

Incertezas na amostragem
Desvio padrão global para uma medida analítica:
222
mag sss sa = desvio padrão do processo de amostragem
sm = desvio padrão do método
Youden sm < sa/3 melhorias adicionais associada à medida são infrutíferas
Se a incerteza da amostragem for muito elevada e não puder ser melhorada,
muitas vezes é interessante mudar para um método de análise menos preciso,
porém mais rápido, assim mais amostras podem ser analisadas em um dado
intervalo de tempo e com isso aumentar a precisão da medida analítica da
espécie de interesse.

Coleta da amostra bruta que é representativa da população que está sendo
amostrada
Idealmente, a amostra bruta é uma réplica em miniatura da massa inteira do
material a ser analisado. Deve corresponder ao todo do material em sua
composição química e, se composto por partículas, na distribuição do tamanho das
partículas.
Diretrizes:
1.Massa da porção amostrada não deve ser maior que a necessária;
2.Levar em consideração os fatores que determinam a massa da porção a ser
amostrada.
Quais são esses fatores?

Fatores que determinam a massa da porção a ser amostrada
1.Incerteza que pode ser tolerada entre a composição da amostra coletada frente a
amostra como um todo;
2.Grau de heterogeneidade do material;
3.Tamanho de partícula para a qual a heterogeneidade se inicia.
Sólidos: heterogeneidade em nível macroscópico

Dispõe-se de uma carroça contendo minério de chumbo constituído de partículas
esféricas com raio de 5 mm. Aproximadamente 4% das partículas são de galena,
têm densidade de 7,6 g cm-3 e contém 70% de chumbo. As partículas
remanescentes têm densidade de 3,5 g cm-3 e não contém ou contém muito pouco
chumbo.
Questão formulada: Quantas gramas de minério deveriam ser amostradas para
que a incerteza relativa da amostragem se mantivesse na ordem de 0,5%?

22
2)1(
P
PP
d
ddppN
r
BABA
N = número de partículas que devem ser amostradas
p = fração de partículas que contém chumbo
(1-p) = fração de partículas que não contém chumbo
dA = densidade do componente A, partículas de galena
dB = densidade do componente B, partículas que não são de galena
PA = porcentagem do analito do componente A
PB = porcentagem do analito do componente B
r = incerteza relativa de amostragem desejada
d = densidade média das partículas
P = porcentagem média do analito

(PA - PB) Grau de heterogeneidade da amostra
N é (1/r2)
N é (1/P2)
Se (PA - PB) aumenta então N aumenta
d = densidade média = (0,04 x 7,6) + (0,96 x 3,5) = 3,7 g cm-3
P = % média de Pb = (0,04 x 0,70 x 7,6 x 100) / 3,7 = 5,8%
N = 8,45 x 105 partículas
Massa = V x d = 8,45 x 105 x 4/3 r3 x 3,7 = 1,6 x 106 g = 1,6 toneladas

Redução da amostra bruta a poucas centenas de grama de uma amostra
laboratorial homogênea, conveniente para análise
Amostra laboratorial = 454 g
É necessário manter o número de 8,45x105 partículas
A que tamanho as partículas devem se reduzidas?
Passo 1: Calcular a massa de uma partícula
mpartícula = 454 / (8,45 x 105) = 5,37 x 10-4 g / partícula
Passo 2: Calcular o raio que as partículas devem possuir para preencher estes
requisitos. Usar densidade média e volume da partícula esférica.
r3 = m x 3/4d
r = 0,3 mm
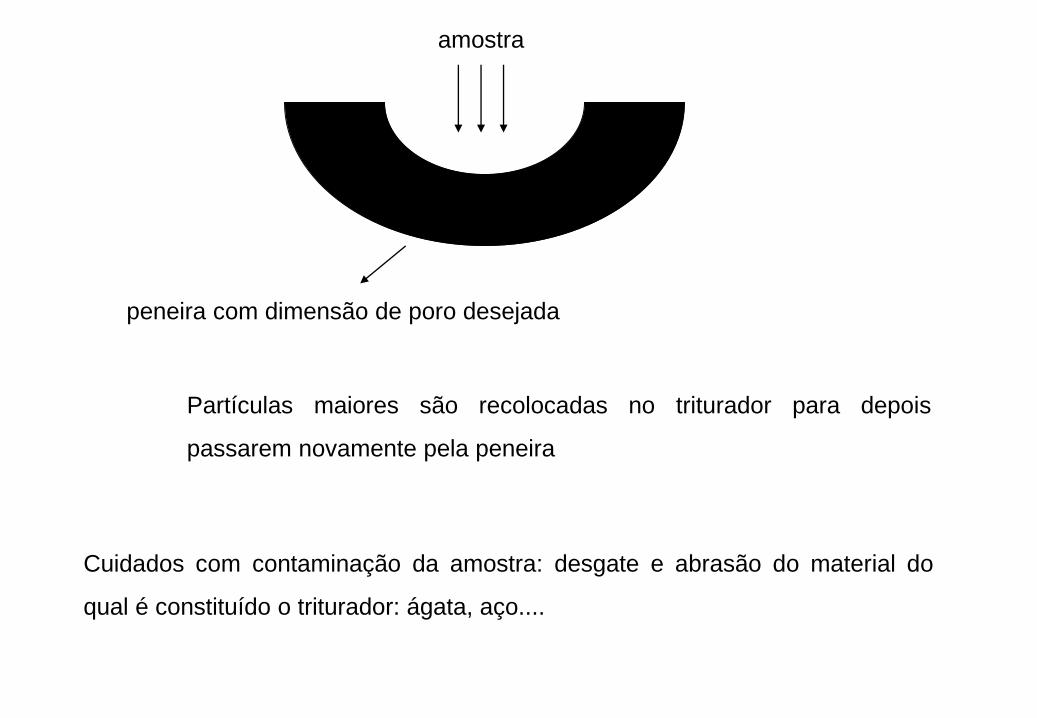
amostra
peneira com dimensão de poro desejada
Partículas maiores são recolocadas no triturador para depois
passarem novamente pela peneira
Cuidados com contaminação da amostra: desgate e abrasão do material do
qual é constituído o triturador: ágata, aço....

Cuidados para que durante a trituração da amostra não ocorra
alteração de composição
1.Calor: perda de componentes voláteis e perda de água de cristalização
2.Trituração: diminuição do tamanho de partícula, aumento da área superficial
(aumento da reação com atmosfera) e aumento da água de absorção
Ex: Cerâmica na forma de pó fino absorve cerca de 0,6% de água por grama de
material

Secagem da amostra e determinação do teor de umidade
H2O (amostra) H2O (atmosfera)
A umidade da atmosfera dependerá da umidade relativa do ar e da
temperatura do local de armazenamento e análise.
UR = Pressão de vapor H2O atmosfera / Pressão de vapor em atm. sat. com ar
Teor de H2O aumenta se T diminui e UR aumenta
Pressão do vapor em atm. sat. com ar = 23,76 torr

Formas de água em sólidos
Água essencial
1. Água de cristalização: CaSO4.2H2O
2. Água de constituição: Ca(OH)2 (s) / aquecimento = CaO + H2O (g)
Água não essencial
• Água adsorvida
• Água “sorvida”: amido, proteínas, zeólitas, sílica gel, carvão
Água ocluída

Processos de eliminação de água
Aquecimento da amostra em forno convencional
Aquecimento da amostra em forno à vácuo
Aquecimento da amostra em forno de microondas
Determinação do teor de água
Pesagem
Karl Fisher

Amostragem de metais e ligas
As amostra de metais e ligas são obtidas por meio de limalhas, moagem ou
perfuração.
Pedaços de metais removidos da superfície não são representativos do interior
do sólido.
Parte interna também deve ser amostrada!!!
Pedaços serrados transversalmente.
Perfuração com brocas de lado a lado. Coleta do material interno para análise!!!

Amostragem de gases e líquidos industriais
Efetuar amostragem contínua!!!
Garantir que a amostra coletada represente uma fração constante do fluxo

Amostragem de gases e soluções homogêneas
A amostra laboratorial pode ser relativamente pequena, porque a
heterogeneidade se inicia em nível molecular!!!
Assim pequeno volume de amostra contém uma quantidade apreciável de
partículas!!!
O líquido ou gás a ser amostrado deve ser bem homogeneizado antes da
amostragem. Se o volume é grande o procedimento de homogeneização pode
ser eficiente.
Várias amostragens consecutivas pode ser mais eficiente.
Ex: Determinação de O2 dissolvido na água de um lago!!!

Padronização e calibração
Uma parte importante de todos os procedimentos analíticos é o processo de
calibração e padronização.
Calibração: determina a relação entre a resposta analítica e a concentração do
analito (uso de padrões químicos)
•Quase todos os métodos analíticos requerem algum tipo de calibração com
padrões químicos.
•Os métodos gravimétricos e alguns métodos coulométricos estão entre os
poucos métodos absolutos que não dependem da calibração com padrões
químicos.
Diversos tipos de procedimentos de calibração

Comparação com padrões
2 tipos:
-Comparação direta
-Procedimento de titulação
Comparação direta:
Comparação de uma propriedade do analito (ou o produto de uma reação com o
analito)
Ex: a cor produzida como resultado de uma reação química do analito x aquela
produzida pela reação de padrões
Titulações:
Analito reage com um reagente padronizado (titulante) em uma reação de
estequiometria conhecida

MÉTODOS DE CALIBRAÇÃO
Modos de calibração:
a) Calibração Pontual – determina-se o valor de uma constante K com um único
padrão, a qual expressa a relação entre a medida instrumental e a concentração do
analito de interesse. Esta hipótese deve ser testada experimentalmente.
b) Calibração Multipontual – calibração com mais de dois padrões.
*O método mais empregado consiste na calibração multipontual com até 5 níveis de
concentração, podendo apresentar uma relação linear (sensibilidade constante na faixa
de concentração de trabalho) ou não-linear (sensibilidade é função da concentração do
analito).

Para muitos tipos de análises químicas, a resposta para o procedimento analítico deve
ser avaliado para quantidades conhecidas de constituintes (chamados padrões), de
forma que a resposta para uma quantidade desconhecida possa ser interpretada.
1. Curva de calibração externa ou curva analítica
2. Curva de adição de padrão
3. Padrão interno
MÉTODOS DE CALIBRAÇÃO

AMOSTRA PADRÃO
É uma amostra de referência que contém o analito de interesse.
Padrão externo:
Amostra e padrão são injetados separadamente e a identificação do composto
desejado é feita através da comparação de alguma característica, por exemplo, em
cromatografia, tempo de retenção.
Padrão interno:
Adição de quantidade conhecida de elemento de referência nos padrões e na
amostra.

BRANCO
Os brancos indicam a interferência de outras espécies na amostra e os traços de
analito encontrados nos reagentes usados na preservação, preparação e análise.
Medidas frequentes de brancos também permitem detectar se analitos provenientes de
amostras previamente analisadas estão contaminando as novas análises, por estarem
aderidos aos recipientes ou aos instrumentos.
1.Branco do método
2.Branco para reagentes
3.Branco de campo

BRANCO
Branco de método: é uma amostra que contém todos os constituintes exceto o analito,
e deve ser usada durante todas as etapas do procedimento analítico.
Branco para reagente: é semelhante ao branco de método, mas ele não foi submetido a
todos os procedimentos de preparo de amostra.
Branco de campo: é semelhante a um branco de método, mas ele foi exposto ao local
de amostragem.
Obs.: O branco de método é a estimativa mais completa da contribuição do branco para
a resposta analítica, sendo que sua resposta deve ser subtraída da resposta de uma
amostra real antes de calcularmos a quantidade de analito na amostra.

Determinação de ácido ascórbico (vitamina C) em suco de laranja:
Branco: sinal = 0,10 unidade
Padrão: [AA] = 2 μmol L-1; sinal = 2,39 unidade
sinal = K [AA]padrão
(2,39-0,10) = K (2)
K = 1,145 unidade L mol-1
Amostra: [AA] = x μmol L-1; sinal = 6,11 unidade
sinal = K [AA]padrão
(6,11-0,10) = 1,145 [AA]
[AA] = 5,24 μmol L-1
CALIBRAÇÃO PONTUAL

Correlação e regressão
CONCENTRAÇÃO DO PADRÃO
SINAL
Quando se usam métodos instrumentais, é necessário calibrar, freqüentemente, os
instrumentos usando uma série de amostras (padrões), cada uma em uma concentração
diferente e conhecida do analito CURVA DE CALIBRAÇÃO EXTERNA
Dois procedimentos estatísticos devem ser aplicados à curva de calibração:
a) Verificar se o gráfico é linear ou não
b) Encontrar a melhor reta (ou melhor curva) que passa pelos pontos.

Coeficiente de correlação
Para verificar se existe uma relação linear entre duas variáveis x1 e y1, usa-se o
coeficiente de correlação de Pearson, r:
n = número de pontos experimentais
O valor de r deve estar entre -1 e +1.
2
1
2
1
2
1
2
1
1111
yynxxn
yxyxnr

r = +1,0
x
y
r = 0,0
x
y
r = -1,0
x
y
r = 0,0
x
y
Quanto mais próximo de ±1, maior a probabilidade de que exista uma relação linear
entre as variáveis x e y.
Valores de r que tendem a zero indicam que x e y não estão linearmente
correlacionados.

Regressão linear
Determinar a melhor reta que passa pelos pontos experimentais REGRESSÃO
LINEAR OU MÉTODO DOS MÍNIMOS QUADRADOS.
A equação de uma linha reta é: y = a + bx
y = variável dependente
x = variável independente
b = inclinação da reta
a = intersecção no eixo dos y
2
1
2
1
1111
xxn
yxyxnb
xbya
1
1
xdevaloresostodosdemédiax
ydevaloresostodosdemédiay

Pela curva de calibração
a = intersecção
b = coeficiente angular = y/x
0,0 0,1 0,2 0,3 0,4 0,50
5
10
15
20
25
Intersecçãox
y
y
x
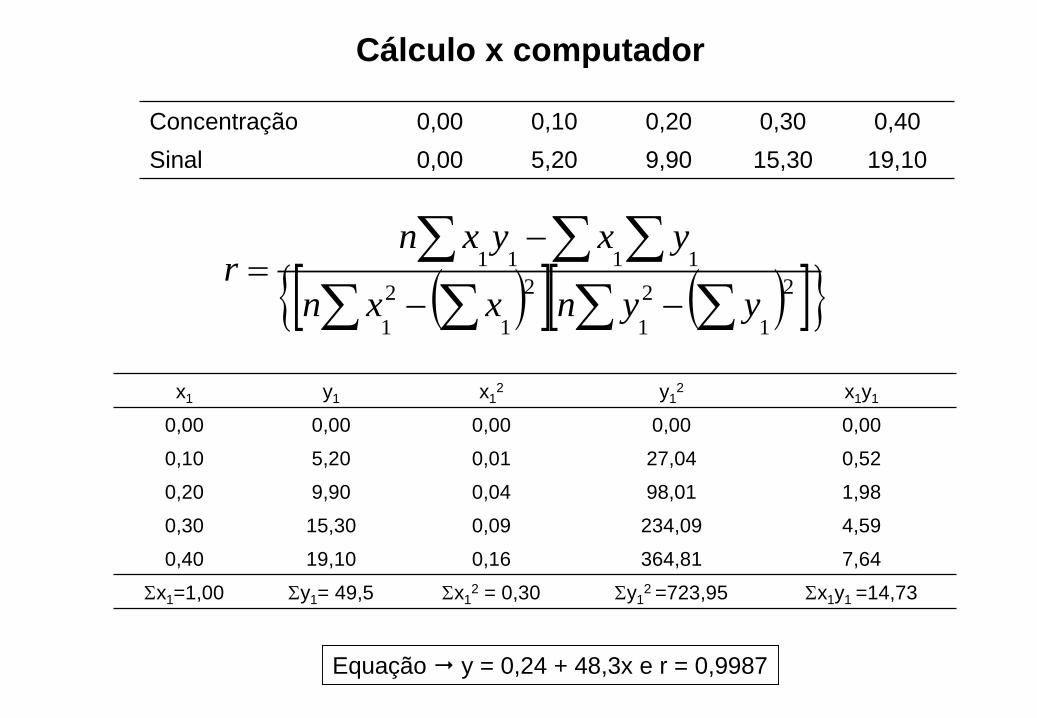
Cálculo x computador
Concentração 0,00 0,10 0,20 0,30 0,40
Sinal 0,00 5,20 9,90 15,30 19,10
x1 y1 x12 y1
2 x1y1
0,00 0,00 0,00 0,00 0,00
0,10 5,20 0,01 27,04 0,52
0,20 9,90 0,04 98,01 1,98
0,30 15,30 0,09 234,09 4,59
0,40 19,10 0,16 364,81 7,64
x1=1,00 y1= 49,5 x12 = 0,30 y1
2 =723,95 x1y1 =14,73
Equação y = 0,24 + 48,3x e r = 0,9987
2
1
2
1
2
1
2
1
1111
yynxxn
yxyxnr

Cálculo x computador
0,0 0,1 0,2 0,3 0,4 0,50
5
10
15
20
25
Sin
al
Concentração Computador
Equação y = 0,24 + 48,3x e r = 0,9987

Erros na inclinação e intersecção da reta
2/
^
1/nyyS
xyregressãoderetadapartiravaloresy
^
2
1/)(/ xxSS
xyb
2
1
2
1/)(/ xxnxSS
xya
Desvio padrão da inclinação:
Desvio padrão da intersecção:
RESULTADO: b ± ts e a ± ts
Erro na estimativa da concentração
2/1
22
2
0/ 11
xxb
yy
nb
SS
xy
xc

Exemplo
Calcule os desvios padrões da inclinação e da intersecção da reta y = 48,3x + 0,24 para
intervalo de confiança a 95% .
Concentração (g mL-1) 0,00 0,10 0,20 0,30 0,40
Resposta 0,00 5,20 9,90 15,30 19,10
2/
^
1/nyyS
xy 2
1/)(/ xxSS
xyb
2
1
2
1/)(/ xxnxSS
xya
Desvio padrão da inclinação:
Desvio padrão da intersecção:

•O sucesso do método da curva de calibração é muito dependente da exatidão com
que são conhecidas as concentrações dos padrões e quão próxima a matriz dos
padrões está da matriz das amostras a serem analisadas.
•Infelizmente, estabelecer esta similaridade de matriz entre amostras complexas e
padrões geralmente é difícil ou impossível de ser feita, e os efeitos da matriz levam a
erros de interferência.
•Para minimizar os efeitos da matriz, normalmente é necessário separar o analito do
interferente antes de obter a resposta medida do instrumento.

Método da adição de padrão
-Adições de quantidades conhecidas do analito na amostra (spiking)
-Elimina ou minimiza interferências introduzidas pela matriz de amostras complexas
-A matriz permanece quase inalterada após cada adição, a única diferença é
concentração do analito.
t
xxss
V
ckVckVS
S = resposta instrumental
k = constante de proporcionalidade
Vs = volume da solução padrão
cs = concentração da solução padrão
Vx = volume da solução problema
cx = concentração da solução problema
Vt = volume total

A equação da reta é: y = a + bx
y = resposta do instrumento
x = concentração da solução padrão
b = inclinação da reta
a = intersecção no eixo dos y
t
s
V
kVb
t
xx
V
ckVa
Determinação por extrapolação

2/1
2
1
2
2
/ 1
xxb
y
nb
SS
xy
xE
Desvio padrão do valor extrapolado
Determinação da concentração por extrapolação
0
t
xxss
V
ckVckVS
x
ss
x V
cVc 0
)(
Via gráfico
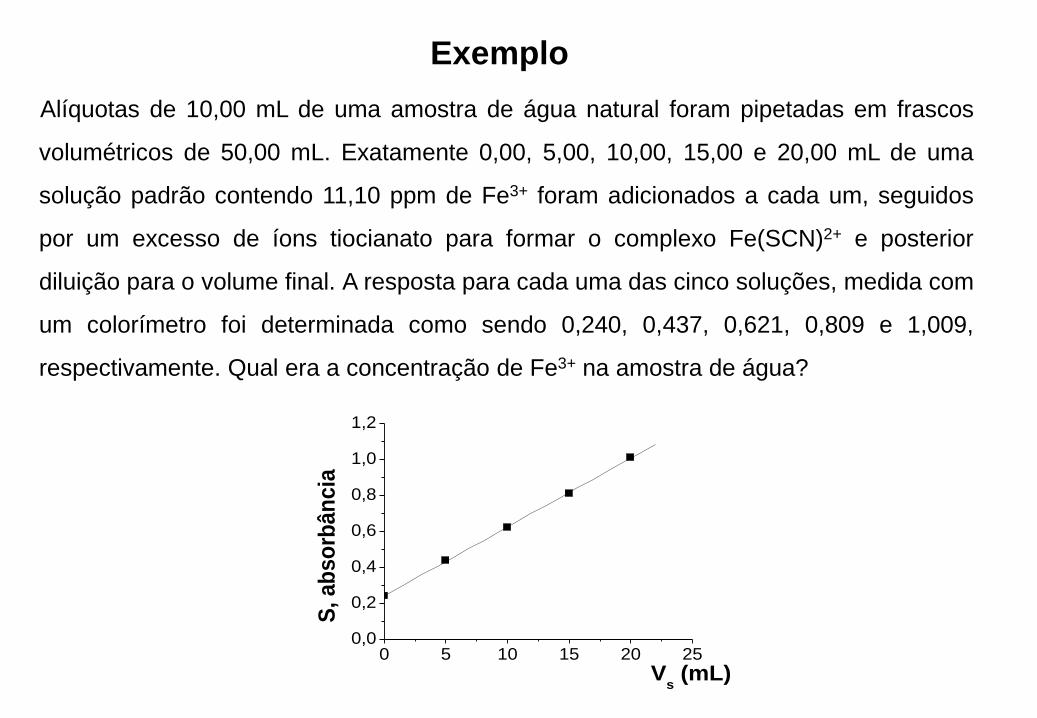
Exemplo
Alíquotas de 10,00 mL de uma amostra de água natural foram pipetadas em frascos
volumétricos de 50,00 mL. Exatamente 0,00, 5,00, 10,00, 15,00 e 20,00 mL de uma
solução padrão contendo 11,10 ppm de Fe3+ foram adicionados a cada um, seguidos
por um excesso de íons tiocianato para formar o complexo Fe(SCN)2+ e posterior
diluição para o volume final. A resposta para cada uma das cinco soluções, medida com
um colorímetro foi determinada como sendo 0,240, 0,437, 0,621, 0,809 e 1,009,
respectivamente. Qual era a concentração de Fe3+ na amostra de água?
0 5 10 15 20 250,0
0,2
0,4
0,6
0,8
1,0
1,2
S, ab
so
rbân
cia
Vs (mL)
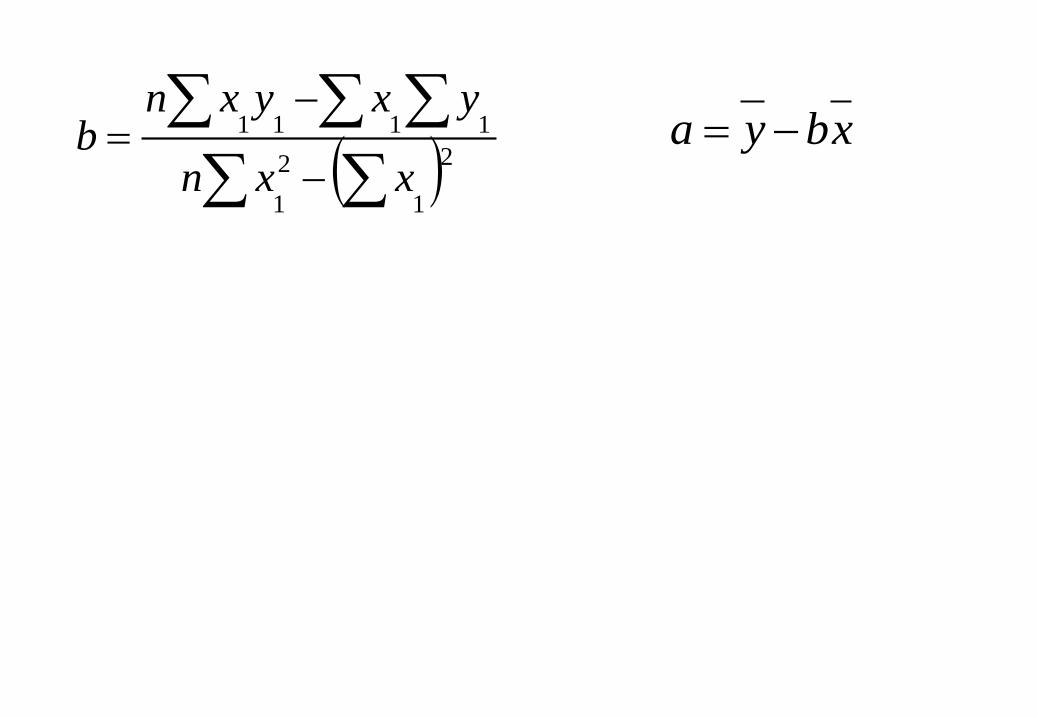
2
1
2
1
1111
xxn
yxyxnb xbya

x1 y1 x12 y1
2 x1y1
0,00 0,240 0,00 0,0576 0,00
1,11 0,437 1,23 0,1910 0,48
2,22 0,621 4,93 0,3856 1,38
3,33 0,809 11,09 0,6545 2,69
4,44 1,009 19,71 1,0181 4,48
x1=11,1 y1= 3,116 x12 = 36,96 y1
2 = 2,3068 x1y1 = 9,03
2
1
2
1
1111
xxn
yxyxnb xbya

x1 y1 x12 y1
2 x1y1
0,00 0,240 0,00 0,0576 0,00
1,11 0,437 1,23 0,1910 0,48
2,22 0,621 4,93 0,3856 1,38
3,33 0,809 11,09 0,6545 2,69
4,44 1,009 19,71 1,0181 4,48
x1=11,1 y1= 3,116 x12 = 36,96 y1
2 = 2,3068 x1y1 = 9,03
Equação S = 0,24 + 0,171 [Fe3+]
2
1
2
1
1111
xxn
yxyxnb xbya

Determinação da concentração por extrapolação
0 = 0,24 + 0,171 [Fe3+] [Fe3+] = 1,404 ppm
10 mL de amostra em um balão de 50,00 mL
[Fe3+] = 7,018 ppm

Método do padrão interno
-Adição de quantidade conhecida de elemento de referência nos padrões e na amostra
-Corrige variações no sinal analítico devido a mudanças nas condições de análise.
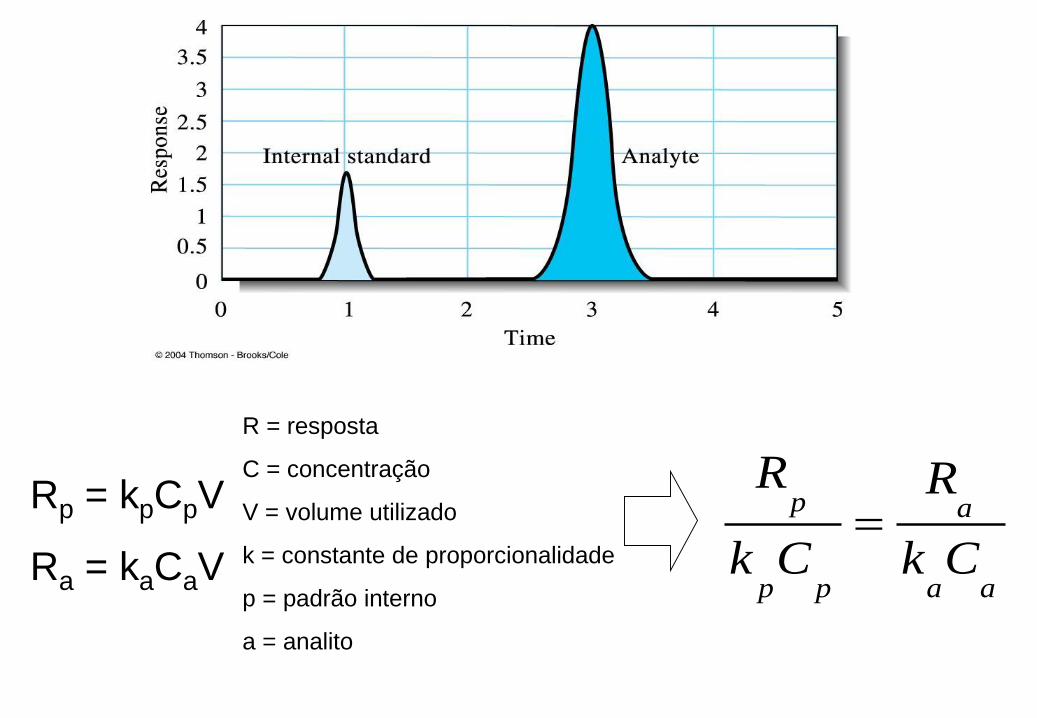
Rp = kpCpV
Ra = kaCaV
R = resposta
C = concentração
V = volume utilizado
k = constante de proporcionalidade
p = padrão interno
a = analito
aa
a
pp
p
Ck
R
Ck
R

Uma solução padrão contendo iodoacetona 6,3x10-8 mol L-1 e p-diclorobenzeno 2,0x10-7
mol L-1 (um padrão interno) deu áreas de pico de 395 e 787, respectivamente; 3,00 mL
de uma solução desconhecida de iodoacetona foram tratados com 0,100 mL de p-
diclorobenzeno 1,6x10-5 mol L-1 e a mistura foi diluída para 10,00 mL. As áreas dos picos
foram de 633 e 520 para a iodoacetona e p-diclorobenzeno, respectivamente. Encontre
a concentração de iodoacetona nos 3,00 mL da solução desconhecida original.

Seleção de um método analítico
Definindo o problema:
1. Que exatidão é necessária? TEMPO GASTO
2. Qual é a quantidade de amostra disponível? SENSIBILIDADE
3. Qual é o intervalo de concentração do analito? AMPLITUDE DE INTERVALO
4. Que componentes da amostra causarão interferência? SELETIVIDADE
5. Quais são as propriedades físicas e químicas da matriz da amostra?
ALGUNS MÉTODOS SÃO APLICADOS A SOLUÇÕES AQUOSAS E OUTROS
MAIS APROPRIADOS PARA ANÁLISE DIRETA DE SÓLIDOS
6. Quantas amostras serão analisadas? PONTO DE VISTA ECONÔMICO

Outras características
• Velocidade
• Facilidade e conveniência
• Habilidade requerida do operador
• Custo e disponibilidade do equipamento
• Custo por amostra

Parâmetros de avaliação dos métodos
Faixa dinâmica linear
Para qualquer método quantitativo, existe uma faixa de concentrações do analito
ou valores da propriedade no qual o método pode ser aplicado.
No limite inferior da faixa de concentração, os fatores limitantes são os valores
dos limites de detecção e de quantificação. No limite superior, os fatores limitantes
dependem do sistema de resposta do equipamento de medição.
Dentro da faixa de trabalho pode existir uma faixa de resposta linear e dentro
desta, a resposta do sinal terá uma relação linear com o analito ou valor da
propriedade.
A faixa linear de trabalho de um método de ensaio é o intervalo entre os níveis
inferior e superior de concentração do analito no qual foi demonstrado ser possível
a determinação com a precisão, exatidão e linearidade exigidas, sob as condições
especificadas para o ensaio.

Sensibilidade de calibração
-É uma medida de sua habilidade em discriminar entre pequenas diferenças na
concentração de um analito.
-Dois fatores limitam a sensibilidade: a inclinação da curva de calibração e a
reprodutibilidade ou precisão do dispositivo de medida.
-Para dois métodos que tenham a mesma precisão, aquele que tem a curva de
calibração mais inclinada será o mais sensível.
-Definição: INCLINAÇÃO DA CURVA DE CALIBRAÇÃO A UMA DADA
CONCENTRAÇÃO DE INTERESSE (m).
Sensibilidade analítica: = m/sS
m = inclinação da curva de calibração
sS = desvio padrão da medida
0,0 0,1 0,2 0,3 0,4 0,50
5
10
15
20
25
m
y
x

brbr
mksSS
calibração de curva da inclinação m
detecção de limite LD
branco do medidas das padrão desvio s
3constantek
branco do médiosinalS
eldistinguív mínimo analíticosinalS
br
br
m
m
s
m
SSLD br
brm
3
Limite de detecção
-Pode ser definido como a concentração mais baixa de um analito que pode ser
distinguida com confiança razoável do branco operacional (uma amostra que contém
o analito em concentração zero)
-Este limite depende da razão entre a magnitude do sinal analítico e o tamanho das
flutuações estatísticas no sinal do branco.
Sinal < LD
Espécie não detectada ao limite de
detecção da concentração x,
porém há presença de sinal
analítico não presente no branco.

Limite de quantificação
-É a menor concentração do analito que pode ser determinada com um nível
aceitável de precisão e veracidade.
-Pode ser considerado como sendo a concentração do analito correspondente
ao valor da média do branco mais 5, 6 ou 10 desvios padrão.
Sinal < LQ
Espécie não detectada ao limite de
determinação ou quantificação da
concentração x, porém há presença
de sinal analítico não presente no
branco.
O cálculo do desvio padrão do branco pode ser feito com base na variação das
medidas do branco analítico, da linha de base ou de um padrão de concentração
muito baixa da(s) espécie(s) analisada(s). A escolha depende da técnica e/ou
instrumentação analítica, sendo função do parâmetro que está sendo medido.

Especificidade e seletividade
-A especificidade e a seletividade estão relacionadas ao evento da detecção.
-Um método que produz resposta para apenas um analito é chamado
específico e respostas para vários analitos, mas que pode distinguir a resposta
de um analito da de outros, é chamado seletivo (livre de interferência).

Recuperação ou fortificação:
-Consiste na adição de uma quantidade conhecida de analito à amostra para testar se a
resposta da amostra corresponde ao esperado a partir da curva de calibração. As
amostras fortificadas são analisadas da mesma forma que as desconhecidas.
-Deve-se adicionar pequenos volumes de um padrão concentrado para evitar mudança
significativa no volume de amostra.
Exemplo: Sabe-se que em uma amostra desconhecida existem 10,0 μg de um analito por
litro. Uma adição intencional de 5,0 μg L-1 foi feita numa porção idêntica da amostra
desconhecida. A análise da amostra modificada forneceu uma concentração de 14,6 μg L-
1. Determine o percentual de recuperação da substância intencionalmente adicionada.
100% xC
CCorecuperaçã
adicionada
afortificadnãoamostraafortificadamostra

Repetibilidade ou repetitividade:
Máxima diferença aceitável entre duas repetições, vale dizer dois resultados
independentes, do mesmo ensaio, no mesmo laboratório e sob as mesmas condições.
a) Mesma amostra;
b) Mesmo analista;
c) Mesmo equipamento;
d) Mesmo momento;
e) Mesmo ajuste;
f) Mesma calibração

Reprodutibilidade:
Máxima diferença aceitável entre dois resultados individuais para um mesmo processo e
com demais condições como especificado.
a) Amostras diferentes do mesmo ponto amostral, ou
b) Diferentes analistas, ou
c) Diferentes equipamentos, ou
d) Diferentes técnicas, ou
e) Diferentes calibrações, ou ajustes.

Exatidão:
1.Testes de calibração: a cada dez análises realizadas um padrão de concentração
conhecida e diferentes dos usados para construir a curva de calibração deve ser
analisado.
2. Recuperação da substância fortificada.
3.Amostra de controle de qualidade: são medidas do controle de qualidade que ajuda a
eliminar vícios introduzidos pelo analista, que sabe a concentração das amostras de
verificação de calibração. Amostras de composição conhecida são fornecidas ao analista
como se fossem desconhecida.
4.Brancos.
Precisão:
1.Amostras repetidas (repetibilidade).
2.Porções repetidas da mesma amostra (reprodutibilidade).

Planejamento fatorial
Conhecer como os diferentes parâmetros experimentais afetam o resultado do
experimento (ou análise).
Ex: Reação química – rendimento – temperatura e catalisador (tipo)
Resposta (Rendimento)
Fatores (Temperatura e catalisador)
Sistema
Fator 1
Fator 2
Fator k
Resposta 1
Resposta 2
Resposta k
Benício de Barros Neto et al. ,Como fazer experimentos, 4a ed.

Planejamento fatorial
O sistema atua como função – desconhecida, em princípio, senão precisaríamos de
experimentos – que opera sobre as variáveis de entrada (fatores) e produz como saída
as respostas observadas.
O objetivo da pessoa que realiza os experimentos é descobrir essa função, ou pelo
menos obter uma aproximação satisfatória para ela.
Com esse conhecimento, ela poderá entender melhor a natureza da reação em estudo,
e assim escolher as melhores condições de operação do sistema.
Benício de Barros Neto et al. ,Como fazer experimentos, 4a ed.

Planejamento fatorial
Decidir quais são os fatores e as respostas de interesse.
Fatores: variáveis que o experimentador tem condições de controlar. Podem ser
qualitativos (catalisador) ou quantitativos (temperatura).
Respostas: variáveis de saída do sistema e que serão ou não afetadas por
modificações provocadas nos fatores.
Próximo passo: “O OBJETIVO”
Benício de Barros Neto et al. ,Como fazer experimentos, 4a ed.

Planejamento fatorial
“O OBJETIVO”
Nosso químico pode estar só querendo saber se trocar o catalisador por um mais
barato não vai diminuir o rendimento da reação;
Ou então, pode querer descobrir que temperatura deve ser usada para se obter o
rendimento máximo;
Ou ainda, até quando ele pode variar os fatores sem alterar o rendimento ou a
qualidade do produto final, e assim por diante.
Benício de Barros Neto et al. ,Como fazer experimentos, 4a ed.

Planejamento fatorial
O planejamento de experimentos, isto é, a especificação detalhada de todas as
operações experimentais que devem ser realizadas, vai depender do objetivo particular
que ele quiser atingir. Objetivos diferentes precisarão de planejamentos diferentes.
Pense num experimento, de preferência numa área de seu interesse, cuja resposta
seja quantitativa. Que fatores você gostaria de examinar para determinar a possível
influência deles sobre a resposta?
Benício de Barros Neto et al. ,Como fazer experimentos, 4a ed.

Calibração multivariada
A calibração multivariada é uma área do que hoje se entende como quimiometria, que
por sua vez, já é considerada uma parte da química analítica.
O termo quimiometria (do inglês chemometrics) foi proposto no final dos anos 70, para
descrever as técnicas e operações associadas com a manipulação matemática e
interpretação de dados químicos.
Com o avanço da instrumentação e automação dentro dos laboratórios de análise, uma
enorme quantidade dados, tabelas e gráficos começaram a ser gerados muito
rapidamente. A identificação, classificação e interpretação desses dados podem ser
fatores limitantes na eficiência e efetiva operação das análises, principalmente sem a
utilização de um adequado tratamento dos dados.

Calibração multivariada
A calibração multivariada é empregada de forma bastante efetiva justamente nos casos
onde existe o problema da superposição de sinais analíticos e para determinações
simultâneas. Um modelo é produzido, baseado em todas as informações disponíveis,
que consegue fazer uma relação entre todo o sinal analítico e a propriedade de
interesse (concentração em muitos casos).

Aplicações

Aplicações





























