Embed Size (px)
Citation preview

Universidade de Aveiro 2011
Departamento de Biologia
Sónia Raquel Neiva Santos
Aspectos Bioquímicos e Moleculares da Coenzima Q10

Universidade de Aveiro 2011
Departamento de Biologia
Sónia Raquel Neiva Santos
Aspectos Bioquímicos e Moleculares da Coenzima Q10
Dissertação apresentada à Universidade de Aveiro para cumprimento dosrequisitos necessários à obtenção do grau de Mestre em Biologia Molecular eCelular, realizada sob a orientação científica da Doutora Laura Vilarinho, Investigadora Auxiliar do Centro de Genética Médica Jacinto Magalhães,Instituto Nacional de Saúde Dr. Ricardo Jorge, INSA e da Professora Paula Gonçalves, Professora Associada do Departamento de Biologia da Universidade de Aveiro.

Dedico este trabalho às famílias afectadas por esta doença, esperando contribuir para a melhoria da sua qualidade de vida.

o júri
presidente Professora Maria do Céu Gomes dos Santos Professora Auxiliar Convidada do Departamento de Biologia da Universidade de Aveiro
Professora Doutora Luísa Cristina da Costa Azevedo Investigadora do Instituto de Patologia e Imunologia Molecular da Universidade do Porto - IPATIMUP
Doutora Laura Vilarinho Investigadora Auxiliar do Departamento de Genética do Centro de Genética Médica Jacinto Magalhães – CGMJM -INSA
Professora Doutora Maria Paula Polónia Gonçalves Professora Associada do Departamento de Biologia da Universidade de Aveiro

agradecimentos
Os meus agradecimentos a todas as pessoas que me ajudaram e apoiaram narealização deste trabalho. À Doutora Laura Vilarinho pela orientação deste trabalho, disponibilidade,confiança nas minhas capacidades e apoio prestado, que me permitiramconcluir este trabalho. À Dr.ª Dulce Quelhas e Doutora Lúcia Lacerda por me terem permitidodesenvolver este trabalho sem restrições no Serviço de Bioquímica Genéticado Centro de Genética Médica Jacinto Magalhães. A todos os meus colegas da Unidade de Bioquímica Genética que de algummodo contribuíram para este trabalho. À minha colega de mestrado e amiga Sónia que me acompanhou e apoiounos momentos difíceis e nos bons momentos que surgiram durante arealização deste trabalho. Aos meus amigos de longa data e aos mais recentes, mas muito especiais,agradeço todo o apoio, amizade e cumplicidade. Aos meus pais e família, em especial à minha mãe, por todo o carinho e apoioincondicional desde sempre.

palavras-chave
Coenzima Q10; Citopatias Mitocondriais
resumo
A ubiquinona (também chamada de Coenzima Q10, CoQ10) é umabenzoquinona presente em praticamente todas as células do organismo queparticipam dos processos de produção de ATP e é sintetisada na membranainterna da mitocondria. A principal fonte de CoQ10 é obtida por sínteseendógena e uma pequena parte é adquirida através da dieta. A CoQ10 desempenha diversas funções biológicas nas células, entre as que sedestacam a de transportador de electrões na cadeia respiratória mitocondrial ea participação no sistema anti-oxidante do organismo. Como tal, umadeficiência de ubiquinona pode causar efeitos nocivos importantes noorganismo, uma vez que se veriam implicadas estas funções chave dometabolismo celular. A deficiência primária de CoQ10 (MIM 607426) é uma doença rara, detransmissão autossómica recessiva e com uma apresentação clínica muitovariável, estando associada a quatro principais fenótipos clínicos: 1) encefalopatia caracterizada por mioglobinúria mas também comenvolvimento do sistema nervoso central; (2) doença predominantementeencefalopática com ataxia e atrofia cerebelar; (3) miopatia isolada com RRFs earmazenamento lipídico; (4) doença multissistémica tipicamente infantil eencefalopatia; (5) nefropatia isolada ou associada a encefalopatia. Se as mutações encontradas, nestes doentes, estiverem localizadas nosgenes envolvidos na biossíntese da CoQ10, então classifica-se estessíndromes como primários, se pelo contrário, as mutações estão presentesnoutros genes, estamos perante a formas secundárias. O objectivo deste estudo é identificar e caracterizar sob o ponto de vistabioquímico e molecular as formas primárias com alteração da cadeiarespiratória mitocondrial (CRM), patologia ainda não diagnosticada no nossopaís. Inicialmente será implementado o doseamento da CoQ10 no músculo, plasmae eventualmente em fibroblastos por HPLC-reversa por detecçãoelectroquímica. Nos doentes com os valores baixos de CoQ10, vai-se procederao estudo molecular dos genes envolvidos na biossíntese, em que já existemalterações descritas, no sentido de estabelecer uma relação fenótipo-genótipo. Os doentes vão ser seleccionados pelo diagnóstico clínico suspeito e/ou comalterações da CRM compatíveis com deficiência da ubiquinona. Este trabalho vai permitir a identificação das deficiências de CoQ10 que são deextrema importância visto serem as únicas Citopatias Mitocondriaispotencialmente tratáveis. Para além disso, a identificação das mutaçõesresponsáveis por esta deficiência, vai permitir a possibilidade de diagnósticopré-natal e implementação de terapêutica precoce.

keywords
Coenzyme Q10, mitochondrial cytopathies
abstract
The ubiquinone (also known as Coenzyme Q10, CoQ10) is a benzoquinonepresent in virtually all body cells that participate in production of ATP, and issynthesized in the inner membrane of mitochondria. The main source ofCoQ10 is the endogenous synthesis and only a small part is acquired throughthe diet. CoQ10 has several biological functions in cells, among which stand out theelectron carrier function in the mitochondrial respiratory chain and theparticipation in the body’s anti-oxidant system. Thus, a ubiquinone deficiencycan cause significant adverse effects on the body, since key functions involvedin cellular metabolism are affected. The primary deficiency of CoQ10 (MIM 607426) is a rare autosomal recessivedisease with a highly variable clinical presentation, and which is associatedwith four major clinical phenotypes: (1) encephalomyopathy characterized by mioglobinúria and brain involvement;(2) predominantly encephalopathic illness with ataxia and cerebellar atrophy,(3) isolated myopathy with ragged-red fibers (RRF's); (4) typical infantilemultisystemic disease with encephalopathy and (5) isolated nephropathy orassociated with encephalopathy. If the mutations found in these patients are located in genes involved in thebiosynthesis of CoQ10, these syndromes are classified as primary, on thecontrary, if mutations are present in other genes, these are secondary forms. The aim of this study is to identify and characterize, from the point of view ofbiochemical and molecular changes, the primary forms of CoQ10 deficiencywith mitochondrial respiratory chain (CRM) alterations, a disease not yetdiagnosed in our country. Initially it will be implemented the determination of CoQ10 in muscle, plasmaand possibly in fibroblasts by reverse HPLC-electrochemical detection. Inpatients with low levels of CoQ10, will be carried out the molecular study of thegenes involved in biosynthesis, for which there are already changes described,to establish a phenotype-genotype relationship. Patients will be selected by the suspected clinical diagnosis and / or changes inCRM compatible with ubiquinone deficiency. The work will allow the identification of the deficiencies of CoQ10 which areextremely important because they are the only potentially treatablemitochondrial cytopathies. In addition, the identification of mutationsresponsible for this deficiency will allow the possibility of prenatal diagnosis andimplementation of early treatment.

i
ÍNDICE
1. INTRODUÇÃO ..................................................................................................................... 3
1.1. A Mitocôndria ........................................................................................................................ 3
1.2. Cadeia Respiratória Mitocondrial e Fosforilação Oxidativa .................................................. 5
1.3. Doenças Mitocondriais .......................................................................................................... 7
1.4. Défices da Coenzima Q10 ........................................................................................................ 9
1.5. Correlação fenótipo/genótipo ............................................................................................. 27
1.6. Tratamento ........................................................................................................................... 29
1.7. Novas abordagens terapêuticas .. ........................................................................................ 31
1.8. Aconselhamento Genético e Diagnóstico Pré‐Natal ........................................................... 32
2. OBJECTIVOS ..................................................................................................................... 35
3. MATERIAL E MÉTODOS .................................................................................................... 39
3.1. Amostra seleccionada .......................................................................................................... 39
3.2. Material biológico ................................................................................................................ 41
3.3. Extracção de DNA ................................................................................................................ 41
3.4. Estudo dos genes PDSS1 e PDSS2, COQ2, COQ9, ADCK3 .................................................... 41
3.5. Análise bioinformática ........................................................................................................ 49
3.6. Nomenclatura das mutações e base de dados ................................................................... 50
4. RESULTADOS E DISCUSSÃO .............................................................................................. 53
4.1. Variantes missense .............................................................................................................. 58
4.2. Variante nonsense ............................................................................................................... 67
4.3. Variantes intrónicas .............................................................................................................. 68
5. CONCLUSÃO ..................................................................................................................... 77
6. PERSPECTIVAS FUTURAS .................................................................................................. 79
7. REFERÊNCIAS BIBLIOGRÁFICAS .......................................................................................... 83
8. ANEXOS ............................................................................................................................ 95

ii
ÍNDICE DE FIGURAS
Figura 1‐ Representação esquemática da mitocôndria, e da localização das enzimas da cadeia
respiratória mitocondrial (CRM) e outras vias metabólicas importantes ......................................... 4
Figura 2‐ Representação esquemática da Cadeia Respiratória Mitocondrial/OXPHOS e os
transportadores móveis de electrões (CoQ e Cit c) .......................................................................... 6
Figura 3‐ Heterogeneidade dos órgãos afectados por citopatias mitocondriais .............................. 8
Figura 4‐ Corte histológico onde podem ser visualizadas RRFs, coradas com o corante de tricrómio
de Gomori .......................................................................................................................................... 9
Figura 5‐ Estrutura química da ubiquinona ..................................................................................... 10
Figura 6‐ Via da biossíntese da CoQ10 humana ............................................................................... 12
Figura 7‐ Representação do cromossoma 10 e locus do gene PDSS1 ............................................. 20
Figura 8‐ Representação do cromossoma 6 e do locus do gene PDSS2 .......................................... 21
Figura 9‐ Representação do cromossoma 4 e do locus do gene COQ2 .......................................... 22
Figura 10‐ Representação do cromossoma 16 e do locus do gene COQ9 ...................................... 23
Figura 11‐ Representação do cromossoma 1 e do locus do gene ADCK3/CABC1 ........................... 24
Figura 12‐ Representação parcial da sequência dos casos 21 e 25, onde se evidência a alteração
c.255T>G, comparada com um controlo normal ............................................................................. 59
Figura 13‐ Estrutura linear dos aminoácidos glutamina (A) e histidina (B), com os radicais
assinalados . ..................................................................................................................................... 59
Figura 14‐ Alinhamento entre espécies de parte da sequência proteica do gene ADCK3............... 60
Figura 15‐ Previsão da patogenicidade da alteração c.255T>G no gene ADCK3 através do
programa Polyphen ......................................................................................................................... 60
Figura 16‐ Frequência alélica do polimorfismo rs2297411 .............................................................. 61
Figura 17‐ Representação parcial da sequência de um dos casos, onde se evidência a alteração
c.196G>T, em homozigotia e em heterozigotia .............................................................................. 62
Figura 18‐ Estrutura linear dos aminoácidos valina (A) e leucina (B), com os radicais assinalados. 62

iii
Figura 19‐ Alinhamento entre espécies de parte da sequência proteica do gene COQ2 ................ 63
Figura 20‐ Previsão da patogenicidade da alteração c.196G>T no gene COQ2 através do programa
mmb . ............................................................................................................................................... 64
Figura 21‐ Representação parcial da sequência dos casos 8 e 9, onde se evidência a alteração
c.320G>C, comparada com um controlo normal . ........................................................................... 65
Figura 22‐ Estrutura linear dos aminoácidos serina (A) e treonina (B), com os radicais assinalados.
(Nelson e Cox, 2004) ........................................................................................................................ 65
Figura 23‐ Alinhamento entre espécies de parte da sequência proteica do gene COQ2 ................ 66
Figura 24‐ Previsão da patogenicidade da alteração c.320G>C no gene COQ2 através do programa
Polyphen ........................................................................................................................................... 66
Figura 25‐ Previsão da patogenecidade da alteração c.320G>C no gene COQ2 através do programa
mmb ................................................................................................................................................. 67
Figura 26‐ Representação parcial da sequência dos casos 20 e 24, onde se evidência a alteração
c.64A>T, comparada com um controlo normal. .............................................................................. 68
Figura 27‐ Representação parcial da sequência do caso 16, onde se evidência a alteração c.1‐
29C>T, comparada com um controlo normal .................................................................................. 69
Figura 28‐ Representação parcial da sequência do caso 12, onde se evidência a alteração
c.721+4C>T, comparada com um controlo normal .......................................................................... 70
Figura 29‐ Representação parcial da sequência dos casos 10 e 16, onde se evidência a alteração
c.693‐136A>G, comparada com um controlo normal. .................................................................... 71
Figura 30‐ Representação parcial da sequência do caso 6, onde se evidência a alteração c.1660‐
81C>G, comparada com um controlo normal . ................................................................................ 72
Figura 31‐ Previsão dos locais de splicing para a região 5`UTR do gene PDSS1 normal (A) e com a
alteração c.1‐29C>T (B) através do programa NetGene 2 ............................................................. 104
Figura 32‐ Previsão dos locais de splicing para a região 5`UTR do gene PDSS1 normal (A) e com a
alteração c.1‐29C>T (B) através do programa Splice Site Prediction ............................................. 105
Figura 33‐ Previsão dos locais de splicing para a região exão7 – intrão7 do gene PDSS1 normal (A)
e com a alteração c.721+4C>T (B) através do programa NetGene 2 ............................................. 106

iv
Figura 34‐ Previsão dos locais de splicing para a região exão7 – intrão7 do gene PDSS1 normal (A)
e com a teração c.721+4C>T (B) através do programa Splice Site Prediction ................................ 106
Figura 35‐ Previsão dos locais de splicing para a região exão4 – intrão4 do gene COQ2 normal (A) e
com a alteração c.693‐136A>G (B) através do programa NetGene 2 . .......................................... 107
Figura 36‐ Previsão dos locais de splicing para a região exão4 – intrão4 do gene COQ2 normal (A) e
com a alteração c.693‐136A>G (B) através do programa Splice Site Prediction . .......................... 108
Figura 37‐ Previsão dos locais de splicing para a região exão15 – intrão15 do gene ADCK3/CABC1
normal (A) e com a alteração c.1660‐81C>G (B) através do programa NetGene 2 ....................... 109
Figura 38‐ Previsão dos locais de splicing para a região exão15 – intrão15 do gene ADCK3/CABC1
normal (A) e com a alteração c.1660‐81C>G (B) através do programa Splice Site Prediction . ..... 110

v
ÍNDICE DE TABELAS
Tabela 1‐ Descrição das mutações identificadas no gene PDSS1 ................................................... 20
Tabela 2‐ Descrição das mutações identificadas no gene PDSS2 ................................................... 21
Tabela 3‐ Descrição das mutações identificadas no gene COQ2 ..................................................... 23
Tabela 4‐ Descrição das mutações identificadas no gene COQ9 ..................................................... 24
Tabela 5‐ Descrição das mutações identificadas no gene ADCK3/CABC1 ...................................... 26
Tabela 6‐ Resumo dos resultados bioquímicos dos casos seleccionados para este estudo ............ 40
Tabela 7‐ Descrição dos primers utilizados para o estudo do gene PDSS1...................................... 42
Tabela 8‐ Descrição dos primers desenhados para o estudo do gene PDSS1 ................................. 43
Tabela 9‐ Descrição dos primers utilizados para o estudo do gene PDSS2...................................... 43
Tabela 10‐ Descrição dos primers desenhados para o estudo do gene PDSS2 ............................... 44
Tabela 11‐ Descrição dos primers desenhados para o estudo do gene COQ2 ................................ 44
Tabela 12‐ Descrição dos primers utilizados para o estudo do gene COQ9 .................................... 45
Tabela 13‐ Descrição dos primers desenhados para o estudo do gene COQ9 ................................ 45
Tabela 14‐ Descrição dos primers utilizados para o estudo do gene ADCK3/CABC1 ...................... 46
Tabela 15‐ Descrição dos primers desenhados para o estudo do gene ADCK3/CABC .................... 46
Tabela 16‐ Referência das sequências de cDNA e proteína ............................................................. 50
Tabela 17‐ Variantes descritas no gene PDSS2 encontradas neste estudo .................................... 53
Tabela 18‐ Variantes descritas no gene COQ9 encontradas neste estudo ...................................... 53
Tabela 19‐ Variantes descritas no gene PDSS1 encontradas neste estudo .................................... 54
Tabela 20‐ Variantes descritas no gene COQ2 encontradas neste estudo ...................................... 55
Tabela 21‐ Variantes descritas no gene ADCK3 encontradas neste estudo ................................... 56
Tabela 22‐ Variantes não descritas encontradas neste estudo ....................................................... 57
Tabela 23‐ Descrição da variante missense encontrada neste estudo ........................................... 64
Tabela 24‐ Descrição da variante nonsense encontrada neste estudo .......................................... 67
Tabela 25‐ Descrição das variantes intrónias encontradas neste estudo ....................................... 68

vi
ABREVIATURAS E SÍMBOLOS
ADP Adenosina‐5’‐difosfato
ATP Adenosina trifosfato
ATPase 6 e 8 ATP sintetase 6 e 8
cDNA DNA complementar ao mRNA
CGMJM Centro de Genética Médica Jacinto Magalhães
CI a IV Complexo I a IV
CoA Coenzima A
CoQ10 Coenzima Q10
COX I a III Citocromo c oxidase subunidade I a III
CRM Cadeia respiratória mitocondrial
ddNTPs 2,3 ‐ didesoxiribonucleótidos
DM Doença mitocondrial
DNA Ácido desoxirribonucleico
dNTP Desoxiribonucleótido trifosfato
FADH2 Dinucleótido de flavina e adenina
HGMD Human Gene Mutation Database
KDa
CK
Kilo Dalton
Creatina quinase
MgCl2 Cloreto de magnésio
MIM
mRNA
Mendelian Inheritance in Man
Ácido ribonucleico mensageiro
mmb Molecular Modeling e Bioinformatics
mtDNA DNA mitocondrial
NADH Dinucleótido de nicotinamida‐adenina
NCBI National Center for Biotechnology Information
ND 1 a 6 NADH desidrogenase subunidade 1 a 6
nDNA DNA nuclear
OXPHOS Fosforilação oxidativa
pb pares de bases

vii
PCR Reacção da polimerase em cadeia (Polymerase Chain Reaction)
PTC Codão de terminação prematuro
RNA Ácido ribonucleico
ROS Espécies reactivas de oxigénio
rpm Rotações por minuto
RRFs Fibras rotas e vermelhas
rRNA RNA ribossómico
SNC Sistema Nervoso Central
TAE Tampão Tris‐Acetato‐EDTA
tRNA RNA de transferência


Introdução


3
1. Introdução
1.1. A Mitocôndria
As mitocôndrias são organelos presentes em todas as células eucarióticas, excepto nos
glóbulos vermelhos, tendo como função principal transformar a energia química dos
substratos orgânicos em energia facilmente acessível à célula (Wallace et al., 1997). São
organelos esféricos ou alongados, medindo entre 0,5 a 1,0 µm de largura e até 10 µm de
comprimento. A sua distribuição na célula é variável, estando localizadas no citoplasma
onde o gasto de energia é mais intenso. Ao microscópio electrónico, as mitocôndrias
apresentam uma estrutura característica. São constituídas por duas membranas, a
externa e a interna (fracção insolúvel), matriz e espaço intermembranar (fracção solúvel).
A membrana externa é lisa e permeável a pequenas moléculas e iões. Possui inúmeras
enzimas importantes para o metabolismo energético da célula, receptores e outros
componentes chave do sistema de transporte transmembranar das proteínas. A
membrana interna é constituída por grande quantidade de proteínas e fosfolípidos assim
como transportadores específicos, ou translocases, responsáveis pelo transporte de
metabolitos hidrófilos e ionizados da matriz para o exterior. Esta membrana apresenta
cristas que aumentam consideravelmente a sua superfície, sendo o local onde se
encontram as subunidades dos complexos multienzimáticos, transportadores de
electrões móveis da cadeia respiratória mitocondrial (CRM), e da fosforilação oxidativa
(OXPHOS) (Figura 1).
A mitocôndria está envolvida na homeostasia celular, tendo um importante papel na
sinalização intracelular, apoptose, metabolismo de aminoácidos, lípidos, colesterol,
esteróides e nucleótidos. Contudo, a sua principal função reporta‐se ao nível do
metabolismo energético, isto é, na β‐oxidação dos ácidos gordos, no ciclo da ureia e na
via final comum de produção de ATP – cadeia respiratória.

4
Figura 1. Representação esquemática da mitocôndria, e da localização das enzimas da cadeia respiratória mitocondrial (CRM) e outras vias metabólicas importantes. (http://www.awl.com/mathews/ch01/frames.htm)
Possui um DNA próprio (mtDNA) (Nass e Nass, 1963), tendo várias moléculas de DNA e
múltiplas cópias do mesmo gene.
O genoma mitocondrial humano é uma molécula circular, constituída por DNA de cadeia
dupla, com cerca de 16.569 pb e a sua transcrição ocorre nas mitocôndrias,
independentemente do núcleo. O mtDNA humano contém 37 genes que codificam 2
RNAs ribossómicos (rRNAs) – 12S e 16S, 22 RNAs de transferência (tRNAs – designados
por letras maiúsculas correspondentes ao aminoácido que transferem) e 13 RNAs
mensageiros (mRNAs) que codificam 13 polipeptídeos componentes da CRM‐OXPHOS: 7
subunidades do complexo I (ND1‐6, ND4L); 1 subunidade do complexo III (citocromo b); 3
subunidades do complexo IV (COX I, COX II e COX III) e 2 subunidades do complexo V
(ATPase 6 e 8) (Zeviani et al., 1989). Os restantes polipeptídeos, constituintes essenciais
dos complexos da CRM, são codificados por cerca de 90 genes do DNA nuclear (nDNA) e
posteriormente transportados para as mitocôndrias para participar na OXPHOS. Todas as
subunidades do complexo II são codificadas pelo nDNA.
O nDNA é responsável pela síntese de proteínas que terão funções diversas na
mitocôndria, desde a participação na estrutura da mitocôndria até o controlo da

5
replicação e da transcrição do mtDNA. Assim, o funcionamento perfeito da mitocôndria
depende da interacção adequada entre os dois genomas. Podendo ter qualquer tipo de
hereditariedade, seja ela autossómica dominante, recessiva, ligada ao X ou ainda materna,
ocorrendo também casos esporádicos. A hereditariedade materna é altamente sugestiva
de um defeito no mtDNA.
Apenas uma parte mínima dos componentes da mitocôndria é codificada neste organelo,
no entanto assume uma grande importância em processos metabólicos e na produção de
energia, daí que as doenças mitocondriais se caracterizem essencialmente por uma
deficiente produção energética.
1.2. Cadeia Respiratória Mitocondrial e Fosforilação Oxidativa
Na mitocôndria ocorrem numerosos processos bioquímicos complexos que culminam
com a produção de ATP. Indissociáveis desta função mitocondrial encontram‐se a Cadeia
Respiratória Mitocondrial e a Fosforilação Oxidativa.
O sistema de fosforilação oxidativa é composto por 4 complexos enzimáticos
constituintes da CRM: o complexo I ‐ NADH‐ubiquinona oxidoredutase (EC‐1.6.5.3); o
complexo II ‐ Succinato‐ubiquinona oxidoredutase (EC‐1.3.5.1); o complexo III ‐ ubiquinol‐
citocromo c oxidoredutase (EC‐1.10.2.2) e complexo IV ‐ citocromo c oxidase ou COX (EC‐
1.9.3.1); e pelo Complexo V – ATPsintetase (EC‐3.6.1.34) que usa a energia gerada pelo
transporte de electrões, ao longo da cadeia respiratória, para formar o ATP. Cada
complexo é composto por subunidades envolvidas no transporte de electrões através da
membrana, e no conjunto são responsáveis pela fosforilação oxidativa mitocondrial. A
CRM possui ainda moléculas transportadoras de electrões, a ubiquinona e o citocromo c
(Figura 2).
A estrutura membranar da mitocôndria permite fixar os componentes da CRM segundo
uma ordem sequencial que facilita a transferência de electrões entre eles e determina
uma alta velocidade e eficiência do sistema. Todos os componentes estão de tal forma

interliga
evitand
diferent
seguida
aceitado
Figura 2transpor
Enquan
protões
complex
armaze
ATP, for
(Cooper
ados que
o‐se reacçõ
tes desidro
a, os electrõ
or final, o o
2‐ Represenrtadores móv
to os elect
s são bom
xos I, III e I
nada no gr
rmando‐se
r et al, 1994
o transpor
ões laterais
ogenases e
ões são env
oxigénio, for
ntação esquveis de elect
trões se m
mbeados at
IV produzin
radiente de
três moléc
4).
rte de ele
s. Os comp
transferem
iados para
rmando águ
uemática datrões (CoQ e
movem atra
través da
ndo um gra
e protões p
ulas de ATP
6
ctrões se
lexos I e II
m‐nos para
os complex
ua.
a Cadeia RCit c). Adapt
vés desta
membrana
diente. O c
ara conden
P por cada
realiza co
recebem e
um comp
xos III e IV e
Respiratória tado de (Zevi
cadeia de
interna m
complexo V
nsar o ADP
NADH oxida
m elevada
electrões p
osto quinó
e finalment
Mitocondriiani e Di Dona
transporte
mitocondria
V utiliza a e
e o fosfato
ado e duas
especificid
proveniente
óide (CoQ10
e reagem c
al/OXPHOS ato, 2004)
electrónic
al ao nível
nergia pote
o inorgânic
por cada F
dade,
s das
0). De
com o
e os
co, os
l dos
encial
o em
ADH2

7
1.3. Doenças Mitocondriais
As Doenças Mitocondriais (DM) foram descritas pela primeira vez em 1959 (Ernster et al.,
1959) num paciente com sintomas neurológicos relacionados com um estado
permanente de hipermetabolismo, apresentando alterações morfológicas e bioquímicas
da mitocôndria. A patologia foi apenas reconhecida em 1962 sendo atribuído o nome de
doença de Luft (Luft et al., 1962). Somente no início da década 70 é que outras doenças
mitocondriais começaram a ser descritas, tendo‐se constatado que as aberrações na
cadeia respiratória, com ou sem as alterações estruturais na mitocôndria, encontradas na
doença de Luft, também ocorriam em outras miopatias (Shy e Gonatas, 1964; Shy et al.,
1966). Durante essa década foram associadas doenças envolvendo o sistema nervoso
central (SNC) e o músculo esquelético à deficiência na cadeia respiratória mitocondrial,
surgindo, pela primeira vez, o termo “encefalomiopatia mitocondrial” (Shapira et al.,
1977).
A classificação bioquímica destas patologias baseia‐se nas cinco principais funções do
metabolismo da mitocôndria (DiMauro et al., 1985): defeitos no transporte do substrato
(deficiência do transportador da carnitina‐CPT); defeitos na utilização do substrato
(deficiência da piruvato desidrogenase‐PDH); defeitos no ciclo de krebs (deficiência da
fumarase); defeitos da CRM (deficiência da citocromo c oxidase) e os defeitos no
acopolamento oxidação/fosforilação (doença de Luft).
Apesar de ser antigo o conhecimento de que a mitocôndria possui o seu próprio DNA
(mtDNA) (Nass e Nass, 1963), a sequenciação completa do mtDNA humano só surge em
1981 (Anderson et al., 1981) e a primeira mutação foi identificada em 1988 (Holt et al.,
1988; Wallace et al., 1988). Esta descoberta tornou possível estabelecer uma ligação
entre as alterações estruturais, bioquímicas, histopatológicas e moleculares das doenças
mitocondriais.
Estas doenças constituem um grupo de doenças de expressão clínica heterogénea que
têm na sua origem alterações do metabolismo energético celular. As disfunções
mitocondriais hereditárias podem ser resultantes quer de mutações do nDNA quer do
mtDNA. Estas doenças envolvem essencialmente, o SNC, o músculo‐esquelético ou ambos.

Outros
levando
Possui u
Figura 3
O apare
ser fata
Actualm
contudo
(DiMau
patolog
dos sint
A marca
fibers) d
(Figura
sarcolém
indiscut
mitocon
órgãos, co
o a um com
uma incidên
‐ Heterogene
ecimento do
ais, progres
mente não
o já foram
ro e Manc
gias mitocon
tomas na m
a histológic
demonstrad
4). A alte
mica de mit
tível, é ago
ndrial.
mo o coraç
plexo espec
ncia de 1 em
eidade dos ó
os primeiro
ssivamente
existe um
descritos a
uso, 2007)
ndriais poss
maioria dos c
ca destas d
das com col
ração das
tocôndrias a
ora claro qu
ção, pâncre
ctro de man
m cada 5000
órgãos afecta
os sintomas
lentos ou
tratamento
alguns efeit
. A deficiên
sui um trata
casos (Quin
oenças são
oração de T
fibras mus
anormais. A
ue a ausênc
8
eas, olhos e
nifestações
0 nados‐viv
ados por cito
pode ocorr
rápidos o
o geral com
tos benéfico
ncia em ub
amento efic
zii e Hirano
o as fibras
Tricrómio d
sculares em
Apesar da im
cia de RRFs
e rins pode
clínicas (Fig
os (Schaefe
opatias mitoc
rer em qua
u até mes
m sucesso p
os de terap
biquinona,
caz, havend
o, 2011).
rotas e ver
de Gomori (
m RRFs é d
mportância
s não exclu
em também
gura 3).
er et al., 200
condriais. (Jo
lquer idade
mo regred
para este t
pêutica em
ao contrári
do uma mel
rmelhas ‐ R
Engel e Cun
devida à a
do diagnós
ui o diagnó
m ser afect
04).
ohns, 1995)
e, podendo
ir com a id
ipo de doe
alguns doe
io das rest
horia acent
RRFs (red ra
nningham, 1
cumulação
stico de RRF
stico de do
ados,
estes
dade.
enças,
entes
antes
tuada
agged
1963)
sub‐
Fs ser
oença

Figurde Go
Os e
CRM
hete
mito
muta
1.4.
1.4.1
A ub
Fred
este
trans
et al
rato,
(Mor
quím
et al.
Em m
ra 4‐ Corte hiomori. (http:
studos hist
, contudo,
rogéneo. O
condrial é
ação, quer n
Défices
1. Via Meta
biquinona f
erick Crane
composto
sporte de e
., 1957). No
, a partir d
rton, 1958)
mica precisa
., 1958) (Fig
meados dos
istológico on://www.neuro
toquímicos
, não são
O diagnós
realizado
no genoma
s da Coenz
abólica da S
foi isolada
e em 1957 q
era parte
lectrões en
o mesmo a
da vitamina
. No final d
a da ubiquin
gura 5), sen
s anos 60, p
nde podem so.wustl.edu/n
específicos
o suficiente
tico defini
através do
mitocondri
zima Q10
Síntese da U
pela prime
que lhe atrib
integrante
tre as desid
no, o profe
A, e atrib
da década
nona, 2,3 d
ndo pela pri
professor Ya
9
er visualizadneuromuscula
s e os bioqu
es para cl
itivo dos
o estudo m
ial quer no
Ubiquinona
eira vez de
bui o nome
e da cadeia
drogenases
essor Morto
uiu‐lhe, pe
de 50, Folk
imetoxi‐5‐m
imeira vez s
amamura do
das RRFs, corr/pathol/mito
uímicos per
lassificar e
doentes s
molecular c
genoma nu
a
e mitocônd
de coenzim
a respirató
(NADH e su
on isolou es
ela primeira
kers e coleg
metil‐6‐deca
sintetizada
o Japão usa
radas com o ochondrial.htm
rmitem ide
este grupo
suspeitos d
com a ide
uclear (Won
rias de co
ma Q10 (CoQ
ria mitocon
uccinato) e
ste compost
a vez, o no
gas determ
aprenil‐1,4‐
e produzid
a pela prime
corante de tm)
ntificar def
de doen
de uma c
ntificação
g e Boles, 2
ração de v
Q10), e verifi
ndrial med
o citocromo
to de um fí
ome de ubi
inaram a e
‐benzoquino
a por ferme
eira vez a co
tricrómio
feitos da
ças tão
citopatia
de uma
2005).
vaca por
icou que
diando o
o (Crane
ígado de
quinona
estrutura
ona (Ott
entação.
oenzima

Q7 (um
cardíaca
antioxid
juntame
de cora
aperfeiç
suficien
pela sua
da form
de trans
A CoQ1
process
desemp
respirat
uma mo
contribu
encontr
coração
composto
a congestiv
dante efica
ente com o
ação human
çoaram a
ntes para en
a contribuiç
mulação da t
sferência de
Fi
10 apresenta
so de resp
penhando
tória mitoco
olécula mó
uindo para
ra‐se em ma
o, o fígado e
o relacionad
a. Em 1966
z (Mellors
o professor
no (Littarru
tecnologia
nsaios clínic
ção para a
teoria quim
e energia.
igura 5‐ Estru
a particula
piração cel
um papel
ondrial. Ape
óvel que pa
a biossínte
aior quantid
e os rins (Lit
do) no trat
6, Mellors e
e Tappel,
Karl Folker
et al., 197
industrial
os maiores
compreens
miosmótica,
utura químic
r relevânci
lular, prod
crucial, c
esar de não
articipa no t
ese de ATP
dade nos ó
ttarru e Lam
10
tamento de
e Tappel mo
1966). Em
rs documen
72). Por me
l para pro
. Peter Mitc
são da tran
que inclui
ca da ubiquin
a em euca
ução de A
como tran
o fazer parte
transporte
(Ernster e
rgãos com m
mbrechts, 20
e doenças
ostraram qu
m 1972, Gia
ntaram um
eados da d
oduzir CoQ
chell recebe
sferência d
o papel vita
nona. (Jeya e
ariotas, um
ATP, que
nsportador
e de nenhum
de electrõ
Dallner, 19
maior nece
011).
humanas,
ue a CoQ6 r
an Paolo L
défice de C
écada de 7
Q10 pura e
eu o Prémio
e energia b
al dos protõ
et al., 2010)
ma vez que
ocorre na
de electr
m dos comp
es dos CI e
995). Assim
ssidade ene
na insufici
reduzida er
Littarru de
CoQ10 na do
70, os japon
em quantid
o Nobel em
biológica at
ões nos sist
é essencia
s mitocôn
rões na c
plexos da C
e CII para o
m, esta mol
ergética, co
ência
ra um
Itália
oença
neses
dades
1978
través
emas
al no
drias,
adeia
CRM é
o CIII,
écula
omo o

11
1.4.2. Patofisiologia
O conhecimento actual da via da biossíntese da CoQ10 nos eucariotas deve‐se sobretudo,
aos estudos realizados na caracterização dos mutantes deficientes em CoQ10 de
Saccharomyces cerevisiae (Kawamukai, 2009).
A CoQ10 é a forma humana predominante de ubiquinona endógena e é sintetizada
predominantemente na membrana mitocondrial interna. A sua biossíntese envolve uma
interacção entre duas vias metabólicas sendo composta por uma benzoquinona e por
uma cadeia lateral de decaprenilo. A porção quinona é derivada predominantemente da
tirosina (em alguns casos a partir da fenilalanina), que é convertida em 4‐hidroxibenzoato
(4‐HBA). A cadeia lateral poliprenil é sintetizada a partir de acetil‐CoA através da via do
mevalonato (Grunler et al., 1994), havendo a formação de farnesil‐difostafo seguida de
uma conversão em decaprenil‐difosfato que condensa com ácido 4‐hidroxibenzóico
dando origem ao decaprenoil‐4‐hidroxibenzoato. O comprimento da cadeia lateral é
determinado pela natureza dos isoprenóides e não pela especificidade do decaprenoil‐4‐
hidroxibenzoato. O comprimento desta cadeia é constante e específico de cada
organismo, sendo determinada pela disponibilidade do prenil‐difosfato na célula.
O composto formado, o decaprenoil‐4‐hidroxibenzoato, entra na via da biossíntese da
ubiquinona sofrendo uma série de reacções de descarboxilação, hidroxilação e de
metilação, até à síntese da ubiquinona (Turunen et al., 2004)(Figura 6).

Figura 6
A bioss
membra
diferent
complex
membra
mitocon
CoQ10 p
(Turune
antioxid
também
proteína
reconhe
deficiên
apresen
‐ Via da bios
síntese da
anas do co
tes sítios n
xo de Golg
ana plasmá
ndrial como
participa em
en et al.,
dantes lipof
m necessár
a mitocond
ecido o seu
ncia de C
ntações clín
síntese da C
CoQ10 co
omplexo de
na célula. A
i, no reticu
ática (Crane
o a transpor
m outras fu
2004). Na
fílicos em
ria na bios
drial e pod
efeito na e
CoQ10 pod
icas variáve
oQ10 human
meça no
e Golgi, a p
A ubiquino
ulo endopla
e, 2008). A
rtadora de e
nções celu
forma re
todas as m
ssíntese da
de modula
expressão d
e ter vá
eis.
12
a. (Mollet et
retículo en
partir das q
ona pode s
smático, no
lém de seu
electrões do
lares, tais c
eduzida (ub
membranas
a pirimidin
ar a apopt
de genes (G
rios efeito
t al., 2007)
ndoplasmát
quais a qu
ser encontr
os lisossom
u papel cen
o complexo
como, na e
biquinol), é
celulares
a, particip
ose (Turun
Groneberg e
os bioquím
tico e é c
inona é tra
rada nas m
mas, nos pe
ntral na cad
o I e II para
liminação d
é um dos
(Bentinger
a no desa
nen et al;
et al., 2005)
micos, pod
completada
ansportada
mitocôndria
roxissomas
deia respira
o complexo
de radicais
mais pot
et al., 200
acoplament
2004). É
). Deste mo
dendo pro
a nas
para
s, no
e na
atória
o III, a
livres
entes
07). É
to da
ainda
odo, a
oduzir

13
A sua obtenção pode ser por síntese endógena, como também por via exógena, através
da alimentação ou toma de suplementos orais (Kwong et al., 2002). A quantidade de
CoQ10 no organismo aumenta até aos 20 anos e posteriormente decai durante o resto da
vida (Turunen et al., 2004).
1.4.3. Aspectos Clínicos
Os défices de CoQ10 são classificados como primários e secundários: se o défice é devido
a uma alteração da biossíntese da CoQ10 ou a uma inibição desta via metabólica devido a
outros metabolitos, respectivamente. Se as mutações encontradas, nestes doentes,
estiverem localizadas nos genes envolvidos na biossíntese da CoQ10, então classificam‐se
estes síndromes como primários, se pelo contrário, as mutações estão presentes noutros
genes, estamos perante formas secundárias (DiMauro et al., 2007). Neste trabalho vamos
apenas focar as formas primárias da deficiência da CoQ10.
A deficiência primária de CoQ10 (MIM 607426) tem sido relatada em doentes
apresentando uma clínica heterogénea e associada a cinco principais fenótipos clínicos: (1)
encefalomiopatia caracterizada por mioglobinúria mas também com envolvimento do
sistema nervoso central; (2) doença predominantemente encefalopática com ataxia e
atrofia cerebelar; (3) miopatia isolada com RRFs e armazenamento lipídico; (4) doença
multissistémica tipicamente infantil e encefalopatia; (5) nefropatia isolada ou associada a
encefalopatia (DiMauro, 2011).
O fenótipo clínico de encefalomiopatia foi descrito pela primeira vez em 1989, relatando
o primeiro doente com deficiência em CoQ10. Este fenótipo é caracterizado por miopatia
mitocondrial, mioglobinúria recorrente e com sinais neurológicos, sendo associada a uma
diminuição das actividades dos complexos I‐III e II‐III da CRM e ao défice marcado de
CoQ10 no músculo (Ogasahara et al., 1989). Desde então, vários doentes com
encefalomiopatia com as mesmas manifestações clínicas têm vindo a ser descritos
(Sobreira et al., 1997; Boitier et al., 1998; Di Giovanni et al., 2001; Aure et al., 2004). Os
achados histológicos na biópsia muscular destes doentes eram sugestivos de disfunção

14
mitocondrial, em que as actividades dos complexos da CRM eram normais excepto
aquelas dependentes da ubiquinona, CI‐III e CII‐III, que estavam significativamente
diminuídas. O conteúdo em CoQ10 foi consistentemente diminuído em todos os doentes.
Neste grupo, apenas num doente foi caracterizado molecularmente, identificando‐se uma
alteração no gene ADCK3/CABC1 (Aure et al., 2004; Mollet et al., 2008).
A apresentação cerebelar, com ataxia e atrofia cerebelar, é o fenótipo mais comum da
deficiência em CoQ10 e foi descrita, pela primeira vez, por Musumeci em 2001. Esta forma
clínica caracteriza‐se por ter início na infância e ser acompanhada por outras
manifestações como a neuropatia, convulsões, atraso mental, hipogonadismo e fraqueza
muscular (Artuch et al., 2006; Lagier‐Tourenne et al., 2008; Mollet et al., 2008). O défice
de CoQ10 foi comprovado no músculo e nos fibroblastos, contudo nem todas as biópsias
musculares revelaram proliferação mitocondrial anormal ou presença de depósitos de
lípidos, como foi apresentado nos primeiros casos descritos (Musumeci et al., 2001;
Lamperti et al., 2003). Um pequeno grupo de doentes com início da ataxia cerebelar na
idade juvenil revelou uma deficiência primária da CoQ10 com alterações moleculares no
gene ADCK3/CABC1 (Lagier‐Tourenne et al., 2008; Mollet et al., 2008).
Este fenótipo está também associado a formas secundárias da deficiência de CoQ10 já
com alterações encontradas noutros genes, como é o caso do gene APTX, que codifica a
aprataxina responsável pela ataxia com apraxia oculomotora (AOA1) (Quinzii et al., 2005).
A forma miopática é caracterizada pela associação de miopatia, fraqueza muscular e
hiperlactacidemia (Di Giovanni et al., 2001; Aure et al., 2004). Todos os doentes
apresentaram RRFs e depósitos de lípidos no músculo, bem como um aumento dos
valores de lactato e da creatina quinase (CK). A idade de aparecimento da doença é
relativamente mais elevada do que nas outras formas de deficiência de ubiquinona. É
também observado uma disfunção da CRM, nomeadamente das actividades dependentes
de ubiquinona e um decréscimo do conteúdo de ubiquinona no músculo (Lalani et al.,
2005; Horvath et al., 2006). Estes doentes apresentam melhorias significativas com a
suplementação oral de CoQ10 (Quinzii et al., 2007).

15
A apresentação miopática é também característica dos défices secundários da CoQ10
como é o caso da Acidúria Glutárica tipo II (AG II) causada por alterações no gene ETFDH
(Gempel et al., 2007). Nestas patologias o conteúdo de CoQ10 no músculo é normal (Liang
et al., 2009; Ohkuma et al., 2009).
Os doentes com a forma multissistémica infantil têm na sua maioria a confirmação
genética de deficiência de CoQ10. Este fenótipo foi descrito, pela primeira vez, em dois
irmãos que apresentavam após o nascimento sintomas neurológicos, incluindo
nistagmos, atrofia óptica, surdez neurosensorial, ataxia, distonia, fraqueza e uma
nefropatia rapidamente progressiva, contudo a alteração genética nesta família nunca foi
encontrada (Rötig et al., 2000). A primeira mutação associada a este fenótipo, foi descrita
no gene COQ2, em 2006, quando observados dois irmãos com doença multissistémica
infantil (Quinzii et al., 2006). Desde então, têm sido descritas mutações noutros genes da
biossíntese da ubiquinona associados a este fenótipo. No gene PDSS2 foi descrito um caso
combinado de síndrome nefrótico e de síndrome de Leigh (Lopez et al., 2006). Numa
forma da doença com aparecimento neonatal e com atingimento cardíaco foi encontrada
uma alteração no gene PDSS1 (Mollet et al., 2007). Recentemente, foi também associado
a este fenótipo alterações noutro gene necessário à biossíntese, o COQ9, acompanhado
por uma atrofia cerebral e cerebelar (Duncan et al., 2009).
Em todos os síndromes multissistémicos infantis descritos houve uma diminuição
acentuada dos níveis de CoQ10 no músculo e nos fibroblastos. A histologia não revelou
alterações mitocondriais sugestivas. Contudo, o estudo enzimático da CRM realizado no
músculo e no fígado revelou uma deficiência nas actividades dos complexos I‐III e II‐III.
Este défice das actividades da CRM foi também observado nos fibroblastos e nos
linfócitos, sugerindo um defeito de CoQ10 generalizado. É também de referir, que em
todos os doentes relatados que apresentavam mutações nos genes PDSS1, PDSS2, COQ2
e COQ9 tinham doença renal.
A apresentação nefrótica foi descrita por Diomedi‐Camassei num paciente com
glomerulopatia sem manifestações extra‐renais desenvolvendo rapidamente para um
síndrome nefrótico aos 18 meses de idade. Um segundo paciente com oligúria aos 5 dias
de vida, que evolui rapidamente para doença renal terminal. Esta criança morre aos 6

16
meses de idade por uma complicação da encefalopatia epiléptica progressiva (Diomedi‐
Camassei et al., 2007). As biópsias renais e musculares revelaram uma deficiência
combinada das actividades do CII‐III e do conteúdo de CoQ10. Foi também possível
identificar a alteração no gene COQ2, responsável por este fenótipo (Diomedi‐Camassei
et al., 2007).
Na maioria dos fenótipos a história familiar sugere um modo de transmissão autossómica
recessiva, pois os irmãos são frequentemente afectados, enquanto os pais não são
normalmente afectados e por vezes consanguíneos.
O síndrome da deficiência em CoQ10 é caracterizado por uma diminuição na
concentração de CoQ10 no músculo e/ou nos fibroblastos. Os doentes
relatados mostraram um grau variável da deficiência de CoQ10 no músculo
e/ou fibroblastos causando uma diminuição da actividade da NADH: citocromo c
oxidorredutase (CI + CoQ10 + CIII) e succinato: citocromo c oxidorredutase (CII + CoQ10 +
CIII) da cadeia respiratória mitocondrial. Tem sido observado uma correlação positiva
entre as actividades enzimáticas e o conteúdo de CoQ10 no músculo (Montero et al.,
2005). Os valores podem variar de concentrações no músculo extremamente baixas até
deficiências mais leves (Montero et al., 2007). No entanto, o diagnóstico bioquímico
apresenta algumas dificuldades, já que há doentes com comprovada deficiência de CoQ10
nos fibroblastos, mas que podem apresentar actividades da CRM e concentração de
CoQ10 no músculo normais (Artuch et al., 2006).
1.4.4. Diagnóstico Bioquímico/Histopatológico
O diagnóstico de deficiência primária de CoQ10 requer uma forte suspeita clínica e é um
diagnóstico extremamente importante no sentido de parar a progressão dos sintomas e
em muitos casos, levar à regressão total da doença. A história clínica deve ser detalhada e
pormenorizada.

17
A abordagem bioquímica deve iniciar‐se por um estudo metabólico completo. Deve
englobar a determinação de lactato, piruvato e dos corpos cetónicos sanguíneos e
respectivas razões molares (L/P e 3OH/AA). Pode ser também efectuado o estudo dos
ácidos orgânicos urinários, realizado por cromatografia gasosa/espectrometria de massa
(GC/MS), que permite identificar um aumento dos metabolitos do ciclo de krebs (ácido
fumárico, succínico e málico) e dos corpos cetónicos. O estudo do perfil dos aminoácidos
pode também revelar‐se de alguma importância, pela alteração do perfil plasmático,
quando se verifica um aumento da alanina (hiperalaninemia) e/ou urinário revelando
uma hiperaminoacidúria generalizada (Rustin et al., 1994). Estas alterações podem ser
sugestivas de uma citopatia mitocondrial.
Se houver uma forte suspeita clínica de deficiência primária de CoQ10 deve ser realizado,
numa primeira fase, a avaliação do conteúdo de CoQ10 no plasma.
A biópsia muscular é o tecido de eleição para realizar os estudos histopatológicos e
bioquímicos. A análise histológica e a microscopia electrónica revelam um aumento de
lípidos, fibras RRFs e acumulação sub‐sarcolémica de mitocôndrias na apresentação
miopática (Ogasahara et al., 1989; Lalani et al., 2005). No fenótipo puramente atáxico as
alterações musculares são muito menos evidentes (Quinzii et al., 2005).
A análise das enzimas da CRM é realizada por espectrofotometria e consiste na
determinação das actividades isoladas e combinadas dos diversos complexos. Estes
ensaios baseiam‐se na transferência dos equivalentes reduzidos obtidos por substratos
naturais ou artificiais (NADH, CoQ, citocromo c, ascorbato). Não requerem o isolamento
de fracções mitocondriais e podem ser realizados em homogeneizados de tecidos
criopreservados. Por esta razão, a quantidade de material requerida é muito pequena e
pode ser facilmente derivada de uma amostra de biópsia de músculo (50 – 100 mg),
fígado, rim e miocárdio ou de um pellet de linfócitos. No entanto este estudo deve ser
efectuado preferencialmente em biópsia de músculo.
Nesta avaliação é efectuada a determinação individualizada das actividades dos
complexos I, II, III e IV e conjunta dos complexos II e III da CRM. Através da utilização de
inibidores específicos adequados para cada um dos complexos, são eliminadas as

18
interferências devido às actividades inespecíficas. A actividade da enzima citrato sintetase
(CS), enzima do ciclo de Krebs, é determinada simultaneamente com os restantes
doseamentos, sendo utilizada como controlo de qualidade da amostra e do seu estado de
conservação. Os resultados são calculados em função da quantidade das proteínas,
determinadas pelo método de Lowry modificado (Lowry et al., 1951), presentes no
músculo e referidos à actividade da enzima de referência, CS.
O doseamento das enzimas da CRM é de extrema importância pois pode sugerir uma
deficiência de CoQ10, mas requer algum cuidado na compreensão das condições exactas
de análise. A determinação da actividade do complexo I baseia‐se na redução da
coenzima Q10 pelo NADH, sendo feita na presença e ausência de rotenona, que é um
inibidor deste complexo. Se nesta determinação for utilizada CoQ10 como último
aceitador de electrões não se observam valores baixos da actividade deste complexo. No
entanto, se forem utilizados outros métodos que não utilizam a CoQ artificial, mas apenas
utilizam a CoQ endógena como receptor final de electrões, resulta num défice aparente
da actividade deste complexo, o que leva a um desvio na investigação, atrasando o
diagnóstico e eventual início do tratamento (Land et al., 2007).
O doseamento da CoQ10 nos diversos tecidos pode ser realizado por várias metodologias,
mas a mais utilizada é a determinação por HPLC com detecção ultra‐violeta (UV) ou
electroquímica. A detecção UV foi utilizada durante algum tempo mas possui
desvantagens relativamente à detecção electroquímica e espectrometria de massa em
tandem. A extracção da coenzima Q10 é realizada através da utilização de vários solventes
e muitos autores utilizam a CoQ9 como padrão interno com amostras de sangue, não
tendo em conta os níveis das lipoproteínas. Assim, os valores dos intervalos de referência
calculados para o conteúdo de CoQ10 nos diferentes tecidos são extremamente variáveis
e de valor questionável. O que se tem observado é que os valores sanguíneos não são um
bom indicador da deficiência desta coenzima, podendo estar perfeitamente normal em
casos que, posteriormente, é demonstrada uma deficiência ao nível muscular e dos
fibroblastos (Artuch et al., 2006; Hirano et al., 2006). A concentração CoQ10 no plasma e o
seu significado não é claro, pois nem sempre existe uma correlação directa com a doença
e com a resposta terapêutica.

19
Na tentativa de contornar este problema, alguns autores utilizam a diproxi‐ CoQ10,
sintetizada artificialmente, como padrão interno e confirmam a sua estrutura,
posteriormente, por espectrometria de massa em tandem. A avaliação do conteúdo de
CoQ10, sempre que possível, é realizada em vários tecidos tais como plasma, células
mononucleares e músculo esquelético (Duncan et al., 2005).
A quantificação exacta de CoQ10 é de extrema importância no diagnóstico, avaliação dos
estados da doença e na interpretação dos ensaios clínicos.
1.4.5. Diagnóstico Molecular
A heterogeneidade clínica da deficiência da CoQ10 é sugestiva de uma heterogeneidade
genética que está relacionada ao grande número de enzimas e de genes envolvidos na
sua biossíntese até agora já identificados. Os genes têm vindo a ser estudados e
caracterizados em várias espécies incluindo bactérias, leveduras e nos nemátodos.
A via da biossíntese da CoQ10 envolve nove etapas e várias enzimas tendo sido
identificadas e sequenciados alguns dos genes que codificam para essas enzimas. Têm
vindo a ser descritas algumas mutações em cinco destes genes, PDSS1, PDSS2, COQ2,
COQ9 e ADCK3, e recentemente foram descritas pela, primeira vez, mutações no gene
COQ6 (Heeringa et al., 2011).
PDSS1
O gene estrutural do PDSS1 (decaprenil‐difosfato sintetase subunidade 1) (MIM 607429)
está localizado no cromossoma 10p12.1 (Figura 7) e abrange cerca de 49 kb do DNA
genómico e compreende 12 exões (Saiki et al., 2005). O cDNA correspondente
(NM_014317) possui 1679 pb e uma Open Reading Frame de 47 a 1294 pb que codifica
uma proteína de 415 aminoácidos (subunidade 1) com massa molecular de
aproximadamente de 46 kDa.

O PDSS
alonga a
et al., 2
Figura http://ww
A funçã
pode se
pirofosf
muito b
devidos
activida
apesar
que o m
síntese
para ma
al., 2007
As mut
Tabela 1
Alteraç
cDNA
c.924T
Adaptado
S1 codifica
a cadeia lat
004).
7‐ Represeww.genecards
o exacta da
er equacion
fato para fo
baixos de Co
s à falha na
ades da CRM
da deficiên
mutante de
de quantid
anter uma f
7).
tações já de
1‐ Descrição
ção
A
T>G
o da base de d
a subunida
teral de pre
entação do s.org/)
a proteína P
nada, por an
ormar o dec
oQ10 encon
a adição do
M dependen
cia severa
PDSS1 man
ades vestig
função da c
escritas nest
das mutaçõe
Efeito
Proteína
p.D308E
dados HGMD®
de 1 da de
nil da coenz
cromossom
PDSS1 nos h
nalogia com
caprenil pir
ntrados nos
o décimo p
ntes da ubiq
de CoQ10 n
ntém uma a
giais de CoQ
cadeia respi
te gene enc
es identificad
Ref
Mo
® ( http://www
20
ecaprenil‐di
zima Q na v
ma 10 e
humanos nã
m as das lev
ofosfato. C
doentes co
prenil à cad
quinona est
nos fibrobla
ctividade re
Q10 e/ou qua
iratória resi
contram‐se
das no gene
ferência
llet (2007) J
w.biobase‐int
ifosfato sin
via da biossí
locus do
ão está perf
veduras, qu
ontudo, os
om mutaçõ
deia de pol
tavam apen
stos. Uma
esidual, ma
antidades n
idual compa
enumerada
PDSS1
Clin Invest 1
ernational.com
tetase (EC‐
íntese da qu
gene PDSS
feitamente
ue apenas a
valores no
es neste ge
iprenil. É d
nas diminuí
hipótese pa
s significati
normais de
atível com a
as na tabela
117, 765
m/)
‐2.5.1.91) a
uinona (Tur
S1. (adaptad
esclarecida
alonga o ge
rmais de Co
ene parecem
de notar, qu
ídas nos do
ara este fac
va, permitin
CoQ9 sufici
a vida (Mol
a 1.
qual
runen
do de
, mas
eranil‐
oQ9 e
m ser
ue as
entes
cto, é
ndo a
entes
let et

PDSS
O ge
está
genó
(NM_
codif
apro
Figurhttp:/
O PD
along
et al.
Saiki
Schiz
subu
codif
dos g
al., 2
As m
Tabe
Alte
c
p.Q
p.S
Adapt
S2
ene estrutur
localizado
ómico e c
_020381) é
fica uma p
ximadamen
ra 8‐ Repre//www.genec
DSS2 codific
ga a cadeia
., 2004).
e colega
zosaccharom
unidades, co
fica a subun
genes refer
2005).
mutações já
la 2‐ Descriç
eração
DNA
Q322X
S382L
tado da base d
ral do PDSS
no cromos
ompreende
é extenso co
proteína de
nte 44 kDa.
esentação dards.org/)
ca a subun
lateral de p
as demon
myces pomb
odificadas p
nidade 2). N
ridos, a enz
descritas ne
ção das muta
Efeito
Proteí
c.964C
c.1145C
de dados HGM
S2 (decapre
ssoma 6p2
e 8 exões
om 3568 pb
e 399 amin
do cromoss
idade 2 da
prenil da co
straram q
be tem uma
por genes d
Na ausência
ima não é f
este gene e
ações identif
o
na
C>T C
C>T C
MD® ( http://w
21
enil‐difosfat
.1 (Figura
s (Saiki et
b e uma Op
noácidos (su
soma 6 e
decapreni
oenzima Q n
que esta
a configuraç
diferentes (P
a de uma s
funcional, n
encontram‐s
icadas no ge
Referência
Carlos Lopez
Carlos Lopez
www.biobase
o sintetase
8) e abran
t al., 2005
pen Reading
ubunidade
do locus
l difosfato
na via da bio
enzima no
ção heterot
PDSS1 codif
ubunidade,
não havend
se enumera
ene PDSS2
z (2006) Am J
z (2006) Am J
‐internationa
subunidad
nge cerca d
5). O cDN
g Frame de
2) com m
do gene P
sintetase (
ossíntese da
o homem,
tetramérica
fica a subun
, devido a u
o formação
adas na tabe
J Hum Genet
J Hum Genet
l.com/)
e 2) (MIM
de 307 kb
A correspo
291 a 1490
massa molec
PDSS2. (adap
EC‐2.5.1.91
a quinona (T
, no rato
a, formada p
nidade 1 e
uma alteraç
o de CoQ10
ela 2.
t 79, 1125
t 79, 1125
610564)
do DNA
ondente
0 pb que
cular de
ptado de
1) a qual
Turunen
o e na
por duas
o PDSS2
ção num
(Saiki et

COQ2
O gene
localizad
genómi
(NM_01
uma pro
(Forsgre
Figura 9http://ww
O COQ
envolvid
final da
polipren
conden
A análi
forteme
As muta
e estrutural
do no crom
co e comp
15697) poss
oteína de 4
en et al., 20
9‐ Represenww.genecards
Q2 codifica
da na biossí
a biossíntes
nil (Forsgre
sação da ca
se em mú
ente expres
ações já des
l COQ2 (4‐
mossoma 4
preende 7
sui 1657 pb
421 aminoác
004).
ntação do s.org/)
a enzima
íntese da u
se, a preni
n et al., 200
adeia latera
últiplos teci
ssa no músc
scritas neste
hidroxibenz
4q21.23 (F
exões (Fo
b e uma Op
cidos com m
cromossom
para‐hidro
biquinona.
lação do p
04) e mede
l do poli‐iso
idos huma
culo esquelé
e gene enco
22
zoato polip
igura 9), a
orsgren et
pen Readin
massa mole
a 4 e do
oxibenzoato
Esta enzim
para‐hidroxi
eia a segund
oprenóide c
nos, adulto
ético, coraç
ontram‐se e
preniltransfe
brangendo
al., 2004).
g Frame de
ecular de ap
locus do
o polipreni
a catalisa o
ibenzoato
da etapa da
com o PHB.
os e fetais
ão e glându
enumerada
erase) (MIM
cerca de
O cDNA
e 1 a 1266
proximadam
gene COQ2
l‐transferas
o segundo p
(PHB) com
a biossíntese
, permitiu
ula adrenal.
s na tabela
M 609825)
21 kb do
correspond
pb que co
mente de 46
2. (adaptad
se (EC‐2.5.1
passo da rea
o grupo t
e promoven
concluir q
3.
está
DNA
dente
difica
6 kDa
do de
1.39),
acção
trans‐
ndo a
que é

Tabe
Al
c.4
c.5
c.6
c.8
c.1
Adapt
COQ
O ge
6128
DNA
(NM_
uma
Figurhttp:/
A fu
leved
CoQ1
estar
la 3‐ Descriç
lteração
cDNA
437G>A
590G>A
683A>G
890A>G
1197delT G
tado da base d
9
ene estrutu
837) está lo
genómico
_020312) p
proteína de
ra 10‐ Repr//www.genec
nção deste
dura, pensa
10 (Hsieh et
r envolvido
ção das muta
E
Pr
p
p
p
p
GGATT^398GT
de dados HGM
ural COQ9
ocalizado no
e compree
possui 2580
e 381 amino
resentação ards.org/)
e gene aind
a‐se que fa
t al., 2007).
no passo d
ações identif
Efeito
roteína
.S146N
.R197H
.N228S
.Y297C
TCCTtGGGAA
MD® ( http://w
(proteína d
o cromosso
ende 9 exõ
0 pb e uma
oácidos com
do cromos
da perman
az parte de
Os estudos
a modificaç
23
icadas no ge
Ref
Dio18,
Dio18,
Dio18,
Qu
Salv
ATTTGT Mo
Bel
www.biobase
da biossínt
ma 16q21
ões (Duncan
Open Read
m massa mo
soma 16 e
ece obscur
e um comp
s realizados
ção do anel
ene COQ2
ferência
omedi‐Camas, 2773
omedi‐Camas, 2773
omedi‐Camas, 2773
inzii (2006) A
viati (2007) H
ollet (2007) J
ll (2011) Sci T
‐internationa
ese da ubi
(Figura 10),
n et al., 20
ding Frame
olecular de
e do locus
ra, mas po
plexo multi
s suportam
de benzoq
ssei (2007) J
ssei (2007) J
ssei (2007) J
Am J Hum Ge
Hum Mol Ge
Clin Invest 1
Transl Med 3
l.com/)
iquinona m
, abrange c
009). O cDN
de 82 a 10
aproximada
do gene
or homolog
enzimático
a ideia de
quinona, po
Am Soc Nep
Am Soc Nep
Am Soc Nep
enet 78, 345
enet 16: 1091
117, 765
3: 65ra4
mitocondrial
cerca de 13
NA correspo
038 pb que
amente de
COQ9. (ada
ia com o g
da biossín
que o COQ
rque na aná
phrol
phrol
phrol
5
1
l,) (MIM
.9 kb do
ondente
codifica
36 kDa.
ptado de
gene da
ntese da
Q9 parece
álise por

HPLC do
compos
As muta
Tabela 4
Alteraç
cDNA
c.730C
Adaptado
ADCK3/
O gene
no crom
compre
2932 pb
aminoá
2001).
Figura 1http://ww
O gene
na via d
envolvid
membra
os fibroblas
sto intermé
ações já des
4‐ Descrição
ção
A
C>T
o da base de d
/CABC1
estrutural
mossoma 1
eende 15 ex
b e uma Op
cidos com
1‐ Representww.genecards
ADCK3/CAB
de apoptos
da na bios
ana. Esta
stos dos do
dio acumul
scritas neste
das mutaçõe
Efeito
Proteína
p.R244X
dados HGMD®
do ADCK3/
1q42.13 (Fig
xões (Iiizum
pen Reading
massa mo
tação do cros.org/)
BC1 é um d
se. Este gen
ssíntese da
proteína é
oentes, apa
ado da bios
e gene enco
es identificad
Ref
Dun
® ( http://www
/CABC1 (pro
gura 11) e
i et al., 200
g Frame de
olecular de
omossoma 1
dos vários g
ne, homólo
a CoQ10, q
é essencia
24
arece um p
ssíntese.
ontram‐se e
das no gene
ferência
ncan (2009) A
w.biobase‐int
oteína chap
abrange ce
02). O cDNA
173 a 2116
aproximad
e do locus d
genes induz
ogo do ABC
que funcion
l para a
ico extra q
enumerada
COQ9
Am J Hum G
ernational.com
perone) (M
erca de 47.
A correspon
6 pb que co
damente de
o gene ADCK
zidos pela e
C1 da leved
na num co
conformaçã
ue poderá
s na tabela
enet 84, 558
m/)
IM 606980
.3 kb do D
dente (NM_
odifica uma
e 72 kDa (H
K3/CABC1. (a
expressão d
ura, codific
omplexo d
ão e func
representa
4.
8
) está local
DNA genóm
_020247) p
proteína de
Holzinger e
adaptado de
o p53 envo
ca uma pro
de proteína
cionamento
ar um
izado
mico e
possui
e 647
et al.,
olvido
oteína
as de
o dos

25
complexos das proteínas da cadeia respiratória mitocondrial (Iiizumi et al., 2002).
Pertence à superfamília das proteínas cinases e possui um local conservado necessário à
ligação ao ATP (Lagier‐Tourenne et al., 2008). A função exacta desta proteína ainda não
está totalmente esclarecida, mas a sua homológa da levedura (CoQ8) foi recentemente
publicada como tendo funções de uma cinase (Tauche et al., 2008). A proteína humana
possui vários “motivos” que indicam que também actua como uma cinase (Lagier‐
Tourenne et al., 2008).
A falta de CoQ8 na levedura não é letal, mas leva à desestabilização do CoQ3 no complexo
da biossíntese CoQ10 (Tauche et al., 2008) desempenhando uma função essencial na
montagem deste complexo. No Homem a supressão desta proteína também não se
revela letal, como se tem verificado nos doentes com alterações neste gene. Estes
doentes apresentam formas clínicas mais suaves em comparação com os que possuem
um bloqueio enzimático de síntese de CoQ10. Este facto sugere que esta enzima possui
uma função reguladora indirecta na síntese de CoQ10 (Lagier‐Tourenne et al., 2008).
As mutações já descritas neste gene encontram‐se enumeradas na tabela 5.

26
Tabela 5‐ Descrição das mutações identificadas no gene ADCK3/CABC1
Alteração
cDNA
Efeito
Proteína Referência
c.637C>T p.R213W Mollet (2008) Am J Hum Genet 82, 623
c.815G>A p.G272D Mollet (2008) Am J Hum Genet 82, 623
c.815G>T p.G272V Mollet (2008) Am J Hum Genet 82, 623
c.1541A>G p.Y514C Lagier‐Tourenne (2008) Am J Hum Genet 82, 661
c.1645G>A p.G549S Lagier‐Tourenne (2008) Am J Hum Genet 82, 661
c.637C>T p.R213W Mollet (2008) Am J Hum Genet 82, 623
c.815G>A p.G272D Mollet (2008) Am J Hum Genet 82, 623
c.815G>T p.G272V Mollet (2008) Am J Hum Genet 82, 623
c.1541A>G p.Y514C Lagier‐Tourenne (2008) Am J Hum Genet 82, 661
c.1645G>A p.G549S Lagier‐Tourenne (2008) Am J Hum Genet 82, 661
c.637C>T p.R213W Mollet (2008) Am J Hum Genet 82, 623
c.815G>A p.G272D Mollet (2008) Am J Hum Genet 82, 623
c.815G>T p.G272V Mollet (2008) Am J Hum Genet 82, 623
c.1541A>G p.Y514C Lagier‐Tourenne (2008) Am J Hum Genet 82, 661
c.1651G>A p.E551K Mollet (2008) Am J Hum Genet 82, 623
c.1750_1752delACC
TCAGAGC^583ACCaccGAGAAGATCC
Lagier‐Tourenne (2008) Am J Hum Genet 82, 661
c.1813dupG CCCACCC^604GAGgGAAA
CCTACT Mollet (2008) Am J Hum Genet 82, 623
c.993+54C>T (1) Lagier‐Tourenne (2008) Am J Hum Genet 82, 661
c.1398+2T>C (1) Lagier‐Tourenne (2008) Am J Hum Genet 82, 661
c.500_521delinsTTG
(2) Lagier‐Tourenne (2008) Am J Hum Genet 82, 661
Adaptado da base de dados HGMD® ( http://www.biobase‐international.com/).
(1) alteração de splicing; (2) alterações de rearranjo

27
1.5. Correlação fenótipo/genótipo
Os défices primários da CoQ10 já foram confirmados molecularmente em 8 doentes com
apresentação infantil (DiMauro et al., 2007) e em 11 com ataxia cerebelar (Lagier‐
Tourenne et al., 2008; Mollet et al., 2008).
Em 2006 foi descrita a primeira mutação missense no gene COQ2, encontrada em dois
irmãos, de pais consanguíneos, com nefropatia infantil resistente aos esteróides, e
encefalopatia na criança mais velha. Apresentavam uma deficiência de CoQ10 muscular e
nos fibroblastos (Quinzii et al., 2005). Em 2007, Mollet e os colegas reportaram uma
delecção no exão 7 presente em homozigotia no gene COQ2, num doente com stress
neurológico neonatal, síndrome nefrótico, hepatopatia, pancitopenia, diabetes,
convulsões e acidose láctica. Este doente faleceu aos 12 dias de vida por falência
multiorgânica (Mollet et al., 2007).
No mesmo ano, Diomedi‐Cassadei e colegas descreveram dois doentes com lesões
glomerulares com instalação precoce e com mutações no gene COQ2 (Diomedi‐Camassei
et al., 2007). O primeiro paciente apresentava aos 18 meses de idade, um síndrome
nefrótico resistente aos esteróides como resultado do colapso da glomerulopatia, sem
sintomas extra‐renais. O segundo paciente apresentava aos cinco dias de vida uma
oligúria, que na biópsia renal apresentou uma proliferação extra‐capilar grave.
Rapidamente evoluiu para uma doença renal terminal tendo falecido aos 6 meses de
idade, com encefalopatia epiléptica progressiva. As biópsias renais e musculares
revelaram uma diminuição da CoQ10 e do complexo II‐III (Diomedi‐Camassei et al., 2007).
Foram também já descritas mutações nas duas subunidades do PDSS, que codificam a
decaprenil difosfato sintase, primeira enzima da biossíntese da CoQ10. Foram encontradas
duas alterações não sinónimas no gene PDSS2, num rapaz com síndrome nefrótico e
síndrome de Leigh, que faleceu aos dois meses de vida devido a um estado epiléptico
refractário focal (Lopez et al., 2006). No gene PDSS1 foi descrita uma mutação missense
em homozigotia numa família consanguínea com deficiência de CoQ10, manifestando‐se
como doença multissistémica com surdez de instalação precoce, encefaloneuropatia,
obesidade, livedo reticularis e valvulopatia (Mollet et al., 2007). Em todas as formas

28
multissistémicas infantis descritas, os níveis de CoQ10 estavam diminuídos no músculo e
nos fibroblastos.
No gene ADCK3 já foram descritas mutações em 11 doentes de sete famílias com fenótipo
cerebelar. Todos os doentes apresentaram no início da infância ataxia cerebelar
variavelmente associada à intolerância ao exercício que melhorou com os anos, atraso
psicomotor leve e neuropatia. A deficiência de CoQ10 foi documentada no músculo e nos
fibroblastos apenas em alguns dos doentes, contudo em nenhum destes se manifestou
doença renal. Este facto parece sugerir, não haver uma correlação entre doença renal e
alterações no gene (Lagier‐Tourenne et al., 2008; Mollet et al., 2008).
Em 2009, Duncan e colegas descreveram pela primeira vez uma mutação no gene COQ9,
necessário na biossíntese da CoQ10 no Homem, num paciente que às 6 horas de vida
apresentava acidose láctica grave e hipertermia que evolui para uma doença
multissistémica severa que se manifesta com convulsões, atraso de desenvolvimento
psicomotor com atrofia cerebral e cerebelar, hipertrofia ventricular e disfunção renal
tubular. A biópsia muscular apresentava atrofia com acumulação de lípidos e um défice
severo do CII‐III e de CoQ10. Este paciente teve uma irmã mais velha que faleceu com 1
dia de vida com convulsões, acidose e aminoacidúria, que não foi possível estabelecer um
diagnóstico (Duncan et al., 2009).
Recentemente foram identificadas, pela primeira vez, 6 mutações no gene COQ6, que
codifica a mono‐oxigenase 6 que participa na biossíntese da CoQ10, em 13 indivíduos de 7
famílias, com alterações da substância branca, acabando por falecer por sepsis e falência
multiorgânica. Os outros dois doentes com mutações identificadas apresentaram ataxia e
dismorfias faciais (Heeringa et al., 2011).
O fenótipo clínico e a progressão da doença devido a alterações no gene ADCK3 parecem
ser de apresentação mais moderada do que a observada no doente com alterações no
gene COQ9, evidenciando que estas duas proteínas têm funções muito distintas.
É interessante verificar que a atrofia cerebelar é partilhada por doentes com mutações
nos genes ADCK3 e COQ9, enquanto a ataxia parece ser comum a várias formas de
deficiência da coenzima Q10, tanto primárias como secundárias.

29
Actualmente, nenhum estudo conseguiu estabelecer uma relação entre as alterações
genotípicas com os fenótipos específicos, uma vez que apenas um número reduzido de
famílias afectadas têm um diagnóstico molecular definitivo.
A identificação de mutações causadoras de doença é importante e necessária para ajudar
a elucidar a patogénese das deficiências de CoQ10 e levar a uma melhor compreensão da
biossíntese e regulação da CoQ10.
1.6. Tratamento
A deficiência primária de CoQ10 é uma doença mitocondrial com tratamento eficaz. Por
isso é importante que os clínicos, especialmente pediatras e neurologistas, conheçam
bem esta patologia de forma a estabelecer um diagnóstico o mais precoce possível. Pois,
um tratamento iniciado quando os sintomas ainda não estão totalmente instalados, muda
radicalmente o curso da doença (Artuch et al., 2006; DiMauro e Mancuso, 2007; Montini
et al., 2008).
Todos os doentes com deficiência primária de CoQ10 beneficiam com a suplementação
oral de CoQ10 contudo a resposta clínica varia mesmo entre os casos com fenótipos
semelhantes, que podem apresentar uma excelente resposta à CoQ10 ou apenas uma
melhoria parcial de alguns sinais ou sintomas (Montero et al., 2007). Tem sido verificado
que as formas miopáticas respondem melhor que os fenótipos de ataxia (Ogasahara et al.,
1989; Musumeci et al., 2001; Rahman, 2001).
Uma vez que a biodisponibilidade oral é pobre, devido à sua extrema afinidade lipídica,
apenas uma pequena fracção de CoQ10 administrada atinge o sistema circulatório e os
seus efeitos ao nível da mitocôndria são ainda menores. Na tentativa de contornar este
problema são administradas doses altas de CoQ10 e por um longo período de tempo
(Bhagavan e Chopra, 2006).

30
Não existe consenso na dose oral para o tratamento desta patologia. Embora existam
ensaios com uma gama de concentrações muito ampla, 300‐3000 mg/dia, alguns autores
relataram uma melhoria clara dos sintomas com doses de 1000 mg/dia .
Os efeitos benéficos da suplementação de CoQ10 são suportados ao nível da correcção
das alterações bioquímicas e histológicas (Di Giovanni et al., 2001; Artuch et al., 2006;
Rotig et al., 2007). No entanto, tem sido verificado uma melhoria significativa dos
sintomas musculares em geral, mas os cerebrais são apenas parcialmente atenuados. Esta
situação pode ser explicada pela presença de danos cerebrais irreversíveis já instalados
ao início do tratamento ou pela dificuldade que a CoQ10 tem em atravessar a barreira
hemato‐encefálica (Rotig et al., 2007).
Vários estudos têm demonstrado que altas doses de CoQ10 exógena administradas podem
produzir aumentos significativos dos níveis de CoQ10 em algumas regiões do cérebro
(Matthews et al., 1998). Mas um estudo recente sobre a distribuição da CoQ10 no cérebro
de ratos demonstrou que, após várias semanas com suplementação de CoQ10, o cerebelo
é a região que mantém os níveis mais baixos de CoQ10. Se o cérebro humano tiver a
mesma distribuição por região como o de cérebro do rato, o cerebelo pode ter a margem
mais estreita de segurança e, portanto, será o primeiro a sofrer com a falta patológica de
CoQ10 (Naini et al., 2003).
Contudo, a CoQ10 para funcionar como um antioxidante deve estar na forma reduzida,
mas apenas só 20% dos lípidos encontram‐se nesta forma no cérebro (Naini et al., 2003).
Por isso a alta proporção de CoQ10 oxidada no cérebro pode ser um reflexo do alto
consumo de oxigénio no tecido, causando um aumento na necessidade de anti‐oxidantes
(Turunen et al., 2004).
Durante a suplementação oral, são monitorizados os níveis de CoQ10 plasmáticos no
sentido de garantir a eficácia do tratamento, uma vez que a biodisponibilidade da
molécula é variável nas diferentes fórmulas comerciais, e existe uma grande variação
inter‐individual na absorção da CoQ10. No entanto, os níveis plasmáticos podem não
reflectir os níveis celulares, daí que seria mais apropriado, sempre que possível, utilizar as
células mononucleares do sangue (Pineda et al., 2010).

31
A CoQ10 é também utilizada com sucesso, no tratamento de outras condições clínicas, tais
como doenças neurodegenerativas, diabetes, doenças musculares e cardiovasculares e
em alguns cancros. Os seus efeitos bioenergéticos e as suas propriedades antioxidantes
também têm sido estudados ao nível da pele, com resultados muito importantes no
rejuvenescimento da pele enrugada (Littarru e Lambrechts, 2011).
1.7. Novas abordagens terapêuticas
Diversos avanços têm sido realizados na melhoraria da biodisponibilidade de CoQ10
utilizando várias abordagens como a redução de tamanho da molécula, o realce da sua
solubilidade e o uso combinado com outras moléculas, tais como lipossomas que
melhoram o seu transporte (Beg et al., 2010).
Uma das abordagens alternativas, para evitar a toma de doses terapêuticas elevadas de
coenzima Q10, é a utilização de agentes que aumentam a síntese endógena. Para isso
foram desenvolvidos análogos da coenzima Q, como a idebenona e o mitoQ, que são
absorvidos de uma forma mais eficiente, melhorando a eficácia antioxidante nos tecidos
(Mariotti et al., 2003). Estes compostos têm vindo a ser avaliados em ensaios clínicos de
forma a estudar a sua toxicidade, segurança e efeitos terapêuticos nas diferentes
patologias (Villalba, 2010).
A idebenona actua como um eliminador de radicais livres protegendo a membrana
mitocondrial contra a peroxidação dos lípidos e interage com outros transportadores de
electrões na CRM. Assim este composto pode ser utilizado tanto no transporte de
electrões como um antioxidante. Este análogo do CoQ10 é rapidamente absorvido e
metabolizado. As concentrações plasmáticas aumentam em proporção com a dose
administrada, quando acompanhada pela alimentação a sua concentração aumenta
exponencialmente (Villalba et al., 2010). Estudos realizados em ratos e um tratamento de
sucesso num doente de Leigh, descrito em 2009, parecem sugerir que a idebenona atinge
facilmente o cérebro e melhora o estado energético do sistema nervoso (Haginoya et al.,
2009). Esta descoberta, permitiu a realização de ensaios clínicos em doentes com outras

32
apresentações mitocondrias, como a encefalomiopatia mitocondrial com acidose láctica e
com episódios de stroke‐like (MELAS) e na neuropatia óptica de Leber (LHON) (Villalba et
al., 2010).
O mitoQ é um análogo da idebenona, e também actua como um antioxidante mas é
bastante mais potente na prevenção do stress oxidativo endógeno. É administrado por via
oral, semelhante à idebenona, mas possui um grupo terminal diferente que lhe confere
um maior poder oxidativo. Este composto atravessa com facilidade todas membranas
biológicas incluindo as das células musculares, e devido à sua carga positiva, acumula‐se
em grande quantidade no interior das mitocôndrias protegendo‐a dos danos oxidativos.
Os estudos pré‐clínicos demonstram a eficácia na redução do estado oxidativo celular
inibindo o processo da apoptose (Villalba et al., 2010).
1.8. Aconselhamento Genético e Diagnóstico Pré‐Natal
A identificação das mutações responsáveis por esta deficiência é de extrema importância
pois permite a implementação de terapêutica precoce e a possibilidade de um
diagnóstico pré‐natal.
Apesar de em muitas situações o tratamento ser eficaz, existem muitas outras em que há
apenas uma melhoria de alguns sintomas mas não uma cura. Perante isto, o diagnóstico
pré‐natal é importante para estas situações. Para isso é necessária a identificação da
mutação responsável pela doença, o que se revela complicado pois apenas ainda só são
conhecidas mutações em seis dos genes da biossíntese da CoQ10.

Objectivos


Objectivos
35
2. Objectivos
O objectivo deste estudo é identificar e caracterizar molecularmente as formas primárias
com alteração da cadeia respiratória mitocondrial (CRM), patologia ainda não
diagnosticada no nosso país.
Os doentes vão ser seleccionados pelo diagnóstico clínico suspeito e/ou com alterações
da CRM compatíveis com deficiência da ubiquinona. Nestes doentes vai‐se proceder ao
estudo molecular dos genes envolvidos na biossíntese, em que já existem mutações
descritas, no sentido de estabelecer uma relação fenótipo‐genótipo.
Este trabalho vai permitir a identificação das deficiências de CoQ10 que são de extrema
importância visto ser as únicas citopatias mitocondriais potencialmente tratáveis. Para
além disso, a identificação das mutações responsáveis por esta deficiência, vai permitir a
implementação de terapêutica precoce e aconselhamento genético e diagnóstico pré‐
natal nos casais de risco.


Material e Métodos


Material e Métodos
39
3. Material e Métodos
Este capítulo descreve o processo de selecção da amostra, e os métodos utilizados, assim
como os equipamentos, os processos e ferramentas informáticas usados na análise de
dados.
O estudo dos genes PDSS1, PDSS2, COQ2, COQ9 e ADCK3 foi implementado no nosso
laboratório disponibilizando este estudo genético a todos os doentes do nosso país
suspeitos de défice de CoQ10.
3.1. Amostra seleccionada
O estudo baseou‐se numa população de 25 doentes, em que 24 foram seleccionados pelo
resultado suspeito das actividades das enzimas da CRM em biópsia de músculo, cujo o
estudo foi realizado na Unidade de Bioquímica Genética do CGMJM. Apenas um paciente
foi estudado pela suspeita clínica. Não foi possível incluir mais doentes com défice da
CRM devido a uma quantidade insuficiente de amostra desses doentes para estudo.
Para efeitos de estudo, foram seleccionados os doentes que apresentaram uma
diminuição na actividade da enzima dependente da ubiquinona (CII‐III) o que é sugestivo
de uma deficiência de CoQ10. Os resultados das actividades enzimáticas da CRM
encontram‐se descritos em anexo (anexo 8.1).
A metodologia utilizada na determinação das actividades das enzimas da CRM, desde a
preparação da amostra até à sua análise encontra‐se descrita em anexo (anexo 8.2).
Os 24 doentes que apresentaram alterações das actividades da citrato sintetase e do
complexo II‐III da CRM, apresentavam idades compreendidas entre os 6 meses e os 75
anos e eram provenientes de hospitais de todo o país.
O caso 25 apresentou o estudo das actividades enzimáticas da CRM no músculo normal
mas foi seleccionado para o estudo pelo fenótipo suspeito. Na tabela 6 encontram‐se
resumidos os dados biográficos, clínicos e resultados dos estudos bioquímicos e
histopatológicos dos doentes incluídos neste estudo.

Tabela 6‐ Resumo dos resultados bioquímicos dos casos seleccionados para este estudo
Caso Sexo Idade (anos) Clínica Parâmetros Analíticos Histologia
1 F 3 Hiperlactacidemia; Diarreia crónica; AD Ala‐↑ Predomínio fibras tipo I 2 F 12 Fraqueza muscular; Mialgias Ala– N; AO– N n.d.3 M 2 Hipotonia n.d. Predomínio fibras tipo I 4 F 1 Doença mitocondrial L/P– N; Ala‐ N; AO‐Alt. n.d.5 M 4 Doença mitocondrial Lac‐ ↑; Ala– N; AO– N Predomínio fibras tipo I
6 M 75 Demência; parkinsonismo; RMN com alterações sugestivas de CM L/P– ↑; n.d.
7 M 45 Doença mitocondrial n.d. n.d.
8 M 6 Hipotonia; Fraqueza muscular n.d. Compatível com atrofia muscular espinal
9 F 10 Doença mitocondrial n.d. Alterações miopáticas inespecíficas 10 M 8 Doença mitocondrial Lac‐ ↑; L/P– ↑; AO– N n.d.11 F 10 Insuficiência renal aguda; insuficiência hepática crónica; ADPM; obesidade Ala‐ N; AO‐Alt. n.d.
12 M 3 Doença mitocondrial Lac‐ ↑; L/P– ↑; Ala‐ N; AO– N n.d.
13 M 1 Hipotonia grave; convulsões; dismorfia; má evolução estato‐ ponderal; polineuropatia sensitivomotora; Ala‐ N; AO‐Alt. n.d.
14 M 2 Hiperlactacidemia; hipotonia n.d Músculo sem alterações
15 F 44 Miopatia n.d Alterações discretas e inespecíficas (ligeira acumulação de glicogénio)
16 M 44 hipoacusia desde infância, AM; enfarte cerebral e episódios stroke‐like; RM c/ múltiplas lesões substância branca; LCR c/ proteinorráquia aumentada n.d n.d.
17 M 54 Doença mitocondrial n.d n.d.18 F 47 Doença mitocondrial n.d.19 F 0,5 Hipotonia grave; convulsões; AD; RMN sugestiva de LEIGH Lac‐↑; Ala‐ N; AO– Alt. n.d.
20 F 15 Miopatia metabólica; fraqueza muscular; insuficiência pulmonar recorrente; AD Lac‐↑; Ala‐ N; AO– N Fibras RRFs e infiltrações lipídicas
21 M 6 Artogripose; distrofia muscular congénita e ortopédicas n.d Músculo sem alterações 22 F 9 Sindrome Reye‐like Lac‐↑; Ala‐ N; AO–Alt. n.d.23 M 55 Ptose palpebral bilateral RRFs com sobrecarga de glicogénio 24 F 4 Miopatia mitocondrial; hipotonia; ADPM; mamilos invertidos Ala‐ N Alterações discretas inespecíficas 25 M Ataxia cerebelosa com atrofia do cerebelo Lac‐N; Ala‐N Alterações miopáticas inespecíficas
n.d.‐não disponível; F‐feminino; M‐ masculino; ADPM‐ atraso de desenvolvimento psicomotor; AD‐ atraso de desenvolvimento; Ala‐ alanina; AO‐ ácidos orgânicos; N‐normal; Alt.‐ alterações no perfil dos AO.
40

Material e Métodos
41
3.2. Material biológico
Para a realização do estudo molecular utilizou‐se sangue periférico ou homogeneizado de
biópsia muscular criopreservada como material biológico.
3.3. Extracção de DNA
A extracção de DNA foi feita a partir de sangue total por extracção automática e a partir
de biópsia muscular, através de um kit comercial da QIAGEN (Cat. No. 158622). A
determinação da concentração (ng/μL) e da pureza do DNA total foi obtida por
quantificação no aparelho NanoDrop® ND‐1000 UV‐Vis Spectrophotometer (a razão
Abs260/Abs280 deverá ser 1.8).
O DNA genómico foi obtido por extracção automática no aparelho BioRobot EZ1
(QIAGEN), utilizando o kit EZ1 DNA Blood 350 μL (QIAGEN), específico para a extracção de
DNA a partir de sangue total. Na utilização deste kit foi seguido o protocolo indicado pelo
fabricante.
O isolamento de DNA a partir de biópsia muscular é efectuado mediante a lise celular
pela proteinase K a 55 ⁰C, até a solução se tornar homogénea. Segue‐se um tratamento
com RNase e precipitação de proteínas. O DNA é seguidamente, precipitado pelo
isopropanol, seguido de lavagem com etanol a 70%. Posteriormente é dissolvido e
hidratado em soluções fornecidas pelo kit (anexo 8.3.).
3.4. Estudo dos genes PDSS1 e PDSS2, COQ2, COQ9, ADCK3
Na abordagem de sequenciação do DNA genómico, a amplificação dos exões e junção
exão‐intrão dos genes PDSS1, PDSS2, COQ2, COQ9 e ADCK3, foi efectuada utilizando
primers específicos descritos por Mollet, Lopez e Duncan, com ligeiras alterações. Outros

Material e Métodos
42
foram desenhados no nosso laboratório com o auxílio do programa Primer3 v.0.4.0 e
FastPCR©, representados nas tabelas 7 a 15.
A estes primers foi adicionada uma sequência de 18 nucleotídos (M13F e M13R) que
permitem a utilização de um primer universal para todos os fragmentos na realização do
PCR de sequenciação. O primer M13F possui a sequência TGTAAAACGACGGCCAGT,
enquanto o primer M13R apresenta a sequência CAGGAAACAGCTATGACC (direcções 5´‐3´).
Antes de se proceder ao estudo dos doentes foram efectuados gradientes de oito
temperaturas, de 45 °C a 65 °C, para todos os exões em DNA controlo, de forma a
optimizar as condições de PCR e assim obter uma melhor amplificação.
Tabela 7‐ Descrição dos primers utilizados para o estudo do gene PDSS1
(Mollet et al., 2008)
RReeggiiããoo TTaammaannhhoo ddoo ffrraaggmmeennttoo
((ppbb))
TTeemmppeerraattuurraa ddee aannnneeaalliinngg
((°°CC)) SSeeqquuêênncciiaa ddoo pprriimmeerr ((55’’‐‐>> 33’’))
Exão 5 339 61 F tgtaaaacgacggccagtCAACAAGAGCGAAACTCCGT
R caggaaacagctatgaccCCCAGCCGGTATATGCTAAT
Exão 7/8 515 61 F tgtaaaacgacggccagtCCTTCCAGTCAGTTTCATCA
R caggaaacagctatgaccCTCTTGGGATTTTTGGGTAC
Exão 12 595 61 F tgtaaaacgacggccagtGCAGGAAGTTAAAACGCTGA
R caggaaacagctatgaccGACTTCAATGCAGACTTCAC
pb – pares de bases °C‐ graus centígrados

Material e Métodos
43
Tabela 8‐ Descrição dos primers desenhados para o estudo do gene PDSS1
RReeggiiããoo TTaammaannhhoo ddoo ffrraaggmmeennttoo
((ppbb))
TTeemmppeerraattuurraa ddee aannnneeaalliinngg
((°°CC)) SSeeqquuêênncciiaa ddoo pprriimmeerr ((55’’‐‐>> 33’’))
Exão 1 330 61 F tgtaaaacgacggccagtCAAGCGAGGAAGAGCGAAC
R caggaaacagctatgaccCCCACTGCTGTCATTGGAG
Exão 2 270 61 F TgtaaaacgacggccagtTGAGGCCTGTAAGCCCCTTC
R caggaaacagctatgaccCTTGCTCCTCCCAAATCTCA
Exão 3 315 61 F tgtaaaacgacggccagtTGGGGGTTTTATCATCCAAT
R caggaaacagctatgaccGTCTGCAATCCCAGCACTTT
Exão 4 322 58 F tgtaaaacgacggccagtTGGGCTGAATTTTCCGTATC
R caggaaacagctatgaccGGCAACAGAGTGAGACTCC
Exão 6 280 61 F tgtaaaacgacggccagtTCCGATGTCCAGTTTTCAC
R caggaaacagctatgaccGTCCGAAAAGTGTGTGATTC
Exão 9/10 525 61 F tgtaaaacgacggccagtCCGCTGCCTCCTATTTTAAG
R caggaaacagctatgaccTTATGCAATCTTCCCATCAGC
Exão 11 346 61 F tgtaaaacgacggccagtCCCCCACATCATCTAGACACT
R caggaaacagctatgaccCACATCAAGAGTTGACCAAAC
pb – pares de bases °C‐ graus centígrados
Tabela 9 – Descrição dos primers utilizados para o estudo do gene PDSS2 (Lopez et al., 2006)
RReeggiiããoo TTaammaannhhoo ddoo ffrraaggmmeennttoo
((ppbb))
TTeemmppeerraattuurraa ddee aannnneeaalliinngg
((°°CC)) SSeeqquuêênncciiaa ddoo pprriimmeerr ((55’’‐‐>> 33’’))
Exão 2 349 57 F tgtaaaacgacggccagtATTTTCCTCCCTGACCCTGT
R caggaaacagctatgaccTTTCCACTGACCTCTGTCCA
Exão 3 368 57 F tgtaaaacgacggccagtGGGGGCAACCTATGGAATA
R caggaaacagctatgaccATTAGTTGGCTGCTGGAGCT
Exão 5 426 57 F tgtaaaacgacggccagtGCAGTTTTCCCACCACATTC
R caggaaacagctatgaccGGCAAAAGGTTTCTTGTGTG
Exão 8 550 57 F tgtaaaacgacggccagtGCCTCAAGATCACTGGGAAA
R caggaaacagctatgaccCTTCTGGCGTGACAAGTGAA
pb – pares de bases °C‐ graus centígrados

Material e Métodos
44
Tabela 10 – Descrição dos primers desenhados para o estudo do gene PDSS2
pb – pares de bases °C‐ graus centígrados
Tabela 11 – Descrição dos primers desenhados para o estudo do gene COQ2
pb – pares de bases °C‐ graus centígrados
RReeggiiããoo TTaammaannhhoo ddoo ffrraaggmmeennttoo
((ppbb))
TTeemmppeerraattuurraa ddee aannnneeaalliinngg
((°°CC)) SSeeqquuêênncciiaa ddoo pprriimmeerr ((55’’‐‐>> 33’’))
Exão 1 527 57 F tgtaaaacgacggccagtCCAGGAAGCCACCTTACTC
R caggaaacagctatgaccAGGAGCCAGTCTGAGAGAGC
Exão 4 258 57 F tgtaaaacgacggccagtCCACTGAGCCTTCTGAAAC
R caggaaacagctatgaccGCCATGTACAATTTAAACACA
Exão 6 334 57 F tgtaaaacgacggccagtGCATTAACTGGTACTTTCTG
R caggaaacagctatgaccGTTACGTGAATATGCCTAAG
Exão 7 303 57 F tgtaaaacgacggccagtAGAGGGAAAGGGGGACAGAG
R caggaaacagctatgaccGAATGAACCTTATTCCACAT
RReeggiiããoo TTaammaannhhoo ddoo ffrraaggmmeennttoo
((ppbb))
TTeemmppeerraattuurraa ddee aannnneeaalliinngg
((°°CC)) SSeeqquuêênncciiaa ddoo pprriimmeerr ((55’’‐‐>> 33’’))
Exão 1 573 61 F tgtaaaacgacggccagtGCCTTTTGCCAATAGAAATCC
R caggaaacagctatgaccCAGGTTCTCATTTCCATCACG
Exão 2 392 61 F tgtaaaacgacggccagtTGTTCTTTATTTCTTTGGTAT
R caggaaacagctatgaccGCATTTTCCATGCTGGA
Exão 3 357 61 F tgtaaaacgacggccagtTACCATGGGCCAGTCTCTTC
R caggaaacagctatgaccTGTGTGGTGAGTTACTTACAC
Exão 4 342 55 F tgtaaaacgacggccagtAGGGAAAGTCGAAGGCTAGG
R caggaaacagctatgaccGACTGATTTGTCATTTCATCT
Exão 5 317 61 F tgtaaaacgacggccagtGAAGGAACCCACAGAAAGCA
R caggaaacagctatgaccAAAAACAGCAACAACTAAACC
Exão 6 317 61 F tgtaaaacgacggccagtTGGTGTGTGAACTAAATCTCA
R caggaaacagctatgaccTTCCTTGCCAGGTAAACACAG
Exão 7
7A_ 337
7B_500
55
57
F tgtaaaacgacggccagtTGACCCAAAGTCCAGACTAGG
R caggaaacagctatgaccTGCTCTAAATCTTCATCTTCA
F tgtaaaacgacggccagtGTCCTTGGGAATTTGTGGAA
R caggaaacagctatgaccCAGTTTTTGGCAGTGACTAA

Material e Métodos
45
Tabela 12 – Descrição dos primers utilizados para o estudo do gene COQ9 (Duncan et al., 2009)
pb – pares de bases °C‐ graus centígrados
Tabela 13‐ Descrição dos primers desenhados para o estudo do gene COQ9
pb – pares de bases °C‐ graus centígrados
RReeggiiããoo TTaammaannhhoo ddoo ffrraaggmmeennttoo
((ppbb))
TTeemmppeerraattuurraa ddee aannnneeaalliinngg
((°°CC)) SSeeqquuêênncciiaa ddoo pprriimmeerr ((55’’‐‐>> 33’’))
Exão 1 376 61 F tgtaaaacgacggccagtCCCCACCACTCTCACCTG
R caggaaacagctatgaccCTTCACCTCACCTTGCTCGT
Exão 2 393 61 F tgtaaaacgacggccagtGGGACCCAACACTTGGATAA
R caggaaacagctatgaccAAAAGGGGATGGAGAGGAGA
Exão 3 284 61 F tgtaaaacgacggccagtTGCTTAGAGGAGATCCAGAGC
R caggaaacagctatgaCCGTGACTCCATCCTGGCTCA
Exão 6 283 61 F tgtaaaacgacggccagtCCCCTCATACACCTCTTGGA
R caggaaacagctatgaccTGATCAAAGCAATTTCACAAGC
RReeggiiããoo TTaammaannhhoo ddoo ffrraaggmmeennttoo
((ppbb))
TTeemmppeerraattuurraa ddee aannnneeaalliinngg
((°°CC)) SSeeqquuêênncciiaa ddoo pprriimmeerr ((55’’‐‐>> 33’’))
Exão 4 415 61 F tgtaaaacgacggccagtCAAAATCACATGGACCCAGA
R caggaaacagctatgaccTTTCAGTCCCATCACAGCAG
Exão 5 323 61 F tgtaaaacgacggccagtCTGCTGTGATGGGACTGAAA
R caggaaacagctatgaccCATCTGAGTGAAAGGTGCTGA
Exão 7 364 61 F tgtaaaacgacggccagtGTTGTCCAGAGGGCCAGAT
R caggaaacagctatgaccCAAGACCAGCTGTGACCTGA
Exão 8 257 61 F tgtaaaacgacggccagtTGTGTTGTCCAAAGCTCTCC
R caggaaacagctatgaccTGTATCACAGAGCCGGAATG
Exão 9 343 61 F tgtaaaacgacggccagtAGTCTGGCCAACATAGCAA
R caggaaacagctatgaccAGCATCAAATGCCTTGTGG

Material e Métodos
46
Tabela 14 – Descrição dos primers utilizados para o estudo do gene ADCK3/CABC1 (Mollet et al., 2008)
pb – pares de bases °C‐ graus centígrados
Tabela 15‐ Descrição dos primers desenhados para o estudo do gene ADCK3/CABC1
pb – pares de bases °C‐ graus centígrados
RReeggiiããoo TTaammaannhhoo ddoo ffrraaggmmeennttoo
((ppbb))
TTeemmppeerraattuurraa ddee aannnneeaalliinngg
((°°CC)) SSeeqquuêênncciiaa ddoo pprriimmeerr ((55’’‐‐>> 33’’))
Exão 3 528 61 F tgtaaaacgacggccagtACAGGCAGGGAGGAGC
R caggaaacagctatgaccATCACTGCTGTGGGTGG
Exão 6 252 61 F tgtaaaacgacggccagtCTTGCCATCCCACTCCC
R caggaaacagctatgaccCGGCCCTGGAAGTTGAC
Exão 12/13 526 61 F tgtaaaacgacggccagtCTTCTCTGGCCCCAGTCTC
R caggaaacagctatgaccCATCCTGTCCCAGCCTCAG
Exão 14 252 61 F tgtaaaacgacggccagtCTCGGGTGTGGCACTTG
R caggaaacagctatgaccCCCTGTGCTTTCATCCATTC
RReeggiiããoo
TTaammaannhhoo ddoo
ffrraaggmmeennttoo ((ppbb))
TTeemmppeerraattuurraa ddee aannnneeaalliinngg
((°°CC)) SSeeqquuêênncciiaa ddoo pprriimmeerr ((55’’‐‐>> 33’’))
Exão 2 311 61 F tgtaaaacgacggccagtGTTGGTAGTGTGGGTTTGGC
R caggaaacagctatgaccAGAGCTCCAGGTCCCACC
Exão 4 240 61 F tgtaaaacgacggccagtTCTCTTGGTGGAGTGTGCTG
R caggaaacagctatgaccGCAGGTGAGGCAGTCCAG
Exão 5 318 61 F tgtaaaacgacggccagtCCTTGTTCCCCACTTTTCAA
R caggaaacagctatgaccGACACTCCCAGAGCACAGGT
Exão 7/8 480 61 F tgtaaaacgacggccagtAGGGCATCCCAGGAGTGT
R caggaaacagctatgaccCCCCATAGGGACAGGCACT
Exão 9/10
583 61 F tgtaaaacgacggccagtCTCTGGTTCTCCAGGGTGTG
R caggaaacagctatgaccCCTTCACCCCAGGCAGGATA
Exão 11 313 61 F tgtaaaacgacggccagtTATCCTGCCTGGGGTGAA
R caggaaacagctatgaccTCACCAAACAGCCACACAG
Exão 15 497 61 F tgtaaaacgacggccagtCCCCTGTGGGTTTGAGTTTA
R caggaaacagctatgaccGCCTGTCTGAGGGAGAGAGTT

Material e Métodos
47
3.4.1. Amplificação do gDNA
A amplificação dos exões foi efectuada utilizando uma mix, ImmoMix™ Red (Bioline‐
Biocompare®). A 12,5 μL desta mix (que inclui IMMOLASE Taq DNA Polymerase, tampão
de reacção, dNTPs e cloreto de magnésio (MgCl2)), adicionou‐se 0,25 μL de primers (F/R)
(50 pmol) correspondentes a cada exão ou fragmento a amplificar, 0,5 – 1 μL gDNA alvo e
água bidestilada para um volume final de 25 μL. Submeteu‐se a mistura final ao seguinte
ciclo de temperaturas: uma desnaturação inicial de 5 minutos a 94 °C, seguida de 35
ciclos de 30 segundos a 94 °C, 45 segundos a 55‐61 °C e 60 segundos a 72 °C, e uma
extensão final de 7 minutos de 72 °C, no termociclador Thermal Cycler (BioRad).
A verificação da amplificação por PCR foi efectuada por electroforese num gel de agarose
1%/TAE (Invitrogen) (m/v), onde se adiciona 6 μL de GelRed (Biotium) e se aplica 6 μL de
cada amostra. O GelRed é um corante fluorescente altamente sensível para detecção de
cadeias de DNA e oligonucleotídos em géis de agarose e poliacrilamida.
As amostras correm em agarose numa tina de electroforese a 125 volts durante 15
minutos. O princípio da eletroforese utilizada para separação de DNA baseia‐se na carga
negativa do DNA dada pelos átomos de oxigénio dos grupos fosfato. No final do processo,
as cadeias de DNA estão próximas ao pólo positivo. Moléculas de menor peso molecular
terão mais facilidade de migrar pelo gel do que as de maior peso molecular, e percorrerão
uma distância maior ficando mais próximas do pólo positivo. A visualização das bandas de
amplificação é possível devido ao corante usado, GelRed que intercala as cadeias de DNA
emitindo fluorescência quando excitado por luz ultravioleta num transiluminador,
permitindo assim a visualização das bandas.
3.4.2. Purificação dos produtos PCR
Os produtos de PCR foram purificados pelo método enzimático utilizando o kit Exosap‐IT®,
de forma a eliminar os primers não incorporados e/ou amplificações inespecíficas. Para
tal, adicionou‐se 1 μL de ExoSap‐IT® a cada 2,5 μL de produto PCR, e incubou‐se no

Material e Métodos
48
termociclador a 37 °C durante 30 minutos, temperatura óptima para a actividade da
enzima, seguida de uma incubação a 80 °C durante 15 minutos, para inactivação da
enzima.
3.4.3. Reacção de sequenciação
Para a reacção de sequenciação, utilizou‐se 1 μL Big Dye® Terminator v1.1 Cycle
Sequencing Mix (que contém MgCl2, dNTPs, ddNTPs marcados com fluorocromos e
AmpliTaq DNA polimerase), 0,5 μL de primer M13 (F ou R) a 5 pmol/μL e 3,5 μL de
produto PCR purificado e água para um volume final de 10 μL.
O programa incluiu uma desnaturação inicial de 2 minutos a 94 oC, seguida de 25 ciclos de
10 segundos a 94 oC, 6 segundos a 50 oC e 4 segundos a 60 oC, no termociclador Thermal
Cycler (BioRad).
3.4.4. Purificação dos produtos de sequenciação
Os produtos de sequenciação foram purificados utilizando o kit DyeEX 96 (Quiagen), de
forma a eliminar os dNTPs e ddNTPs não incorporados e sais que pudessem interferir com
a electroforese capilar.
3.4.5. Electroforese capilar
Os produtos purificados foram ressuspendidos, por agitação no vortex, em 15‐25 μL de
formamida desionizada (formamida Hi‐DiTM Applied Biosystems), substância
desnaturante e de elevada viscosidade, e incubar à temperatura ambiente durante 10
minutos.

Material e Métodos
49
A separação dos fragmentos foi realizada por electroforese capilar no sequenciador
automático ABI3130xl Genetic Analyser (Applied Biosystems) de 16 capilares de 36 cm.
Este tipo de sequenciação baseia‐se no método desenvolvido por Sanger (Sanger et al.,
1977) e recorre a nucleótidos terminadores da cadeia que são didesoxinucleótidos
trifosfatados marcados com um fluorocromo na extremidade 3’ (ddNTP). O sequenciador
detecta a fluorescência dos 4 didesoxinucleótidos, através dos diferentes comprimentos
de onda de luz emitida por radiação da amostra. Os resultados foram analisados no
programa SeqScapeTM v.2.5 (Applied Biosystems).
3.5. Análise bioinformática
O desenho de primers específicos foi efectuado no programa Primer3
(http://primer3.sourceforge.net/). Para a verificação da qualidade dos primers, no que diz
respeito à temperatura de melting, formação de estruturas secundárias e formação de
dímeros de primers foi utilizado o software FastPCR
(http://www.biocenter.helsinki.fi/bi/Programs/).
A análise dos resultados de sequenciação foi efectuada no programa SeqScapeTM v.2.5
(Applied Biosystems).
As sequências aminoacídicas utilizadas para efectuar o alinhamento fazem parte da base
de dados National Center for Biotechnology Information (NCBI)
(http://www.ncbi.nlm.nih.gov/) e do Ensembl (http://www.ensembl.org/index.html) e o
alinhamento das sequências seleccionadas foi realizado no programa ClustalW2
(http://www.ebi.ac.uk/clustalw). Este programa realiza alinhamentos com outras
espécies previamente definidas e através de um algoritmo de alinhamento progressivo,
baseado na similaridade das sequências, alinha as sequências de aminoácidos. Com o
alinhamento é possível identificar motivos com elevado grau de conservação e permite
observar a conservação da zona mutada, sendo possível concluir que quanto maior o grau
de conservação maior é a probabilidade de a alteração ser patogénica.

Material e Métodos
50
Para a previsão das alterações de splicing recorreu‐se aos programas de análise Splice Site
Prediction by Neural Network at BDGP (http://www.fruitfly.org/) e Netgene 2
(http://www.cbs.dtu.dk/services/NetGene2/). Estas ferramentas prevêem os efeitos das
mutações nos sinais de splicing ou identificam os locais de splicing em qualquer sequência
humana (Desmet et al., 2009).
3.6. Nomenclatura das mutações e base de dados
As variações de sequência detectadas foram descritas de acordo com as recomendações
de nomenclatura de mutações da Human Gene Variation Society (HGVS) (den Dunnen e
Antonarakis, 2001).
Foram utilizadas as sequências de referência de cDNA e proteicas para os genes
estudados descritas no NCBI (tabela 16).
Tabela 16‐ Referência das sequências de cDNA e proteína
Gene cDNA_RefSeq NCBI proteína_RefSeq NCBI
PDSS1 NM_014317.3 NP_055132.2
PDSS2 NM_020381.3 NP_065114.3
COQ2 NM_015697.7 NP_056512.5
COQ9 NM_020312.3 NP_064708.1
ADCK3/CABC1 NM_020247.4 NP_064632.2
Adaptado do NCBI
Na pesquisa de variações já descritas foi consultada a literatura e as bases de dados
HGMD®‐ Human Gene Mutation Database Professional, disponível na página
http://www.biobase‐international.com/, NCBI (http://www.ncbi.nlm.nih.gov/) e do
Ensembl (http://www.ensembl.org/index.html).

Resultados e Discussão


Resultados e Discussão
53
4. Resultados e Discussão
Nos 25 casos seleccionados para o estudo, foram detectadas 48 variantes em que 5 não
se encontram descritas na literatura nem na base de dados Human Gene Mutation
Database Professional ‐ HGMD®. As variantes não descritas distribuem‐se da seguinte
forma: 1 variante missense e 4 variantes intrónicas. Estes resultados estão
esquematizados nas tabelas 17 a 22.
Tabela 17‐ Variantes descritas no gene PDSS2 encontradas neste estudo
Caso Variantes descritas encontradas Referência
1 c.876+48C>T rs3734677
6 c.876+48C>T rs3734677
24 c.876+48C>T rs3734677
25 c.876+48C>T rs3734677
Tabela 18 – Variantes descritas no gene COQ9 encontradas neste estudo
Caso Variantes descritas encontradas Referência
1 c.‐107A>G rs178602
5 c.‐107A>G rs178602
8 c.‐107A>G; c.74‐35G>A rs3734677; rs223863
11 c.‐107A>G rs178602
12 c.‐107A>G rs178602
13 c.‐107A>G rs178602
14 c.‐107A>G rs178602
17 c.‐107A>G rs178602
19 c.‐107A>G rs178602
21 c.‐107A>G rs178602
24 c.‐107A>G rs178602
25 c.‐107A>G rs178602

Resultados e Discussão
54
Tabela 19 – Variantes descritas no gene PDSS1 encontradas neste estudo
A azul estão representadas as alterações descritas que foram detectadas no gene PDSS1 em apenas num doente.
Caso Variantes descritas encontradas Referência
1 c.130‐46T>C; c.130‐23delG; c.130‐54A>G rs1780186; rs71403882; rs1677725
2 c.227+55G>A; c.227+67A>T; c.*76C>T rs1677730; rs1780193; rs12776877
3 c.162+56G>A rs1780185
4 c.130‐46T>C; c.130‐23delG; c.130‐54A>G; c.162+56G>A rs1780186; rs71403882; rs1677725; rs1780185
5 c.130‐54A>G; c.130‐46T>C; c.130‐23delG; c.162+56G>A; c.163‐5delT; c.227+55G>A; c.227+67A>T; c.467+42G>C
rs1677725; rs1780186; rs71403882; rs1780185; rs71403883; rs1677730; rs1780193; rs2368183
6 c.130‐54A>G; c.130‐46T>C; c.130‐23delG; c.162+56G>A; c.163‐5delT; c.227+55G>A; c.227+67A>T
rs1677725; rs1780186; rs71403882; rs1780185; rs71403883; rs1677730; rs1780193
7 c.130‐54A>G; c.130‐46T>C; c.130‐23delG; c.162+56G>A; c.163‐5delT; c.227+55G>A; c.227+67A>T
rs1677725; rs1780186; rs71403882; rs1780185; rs71403883; rs1677730; rs1780193
8 c.162+56G>A; c.467+42G>C; c.*76C>T rs1780185; rs2368183; rs12776877
9 c.*76C>T rs12776877
10 c.162+56G>A; c.*76C>T rs1780185; rs12776877
11 c.130‐54A>G; c.130‐46T>C; c.130‐23delG; c.162+56G>A; c.336+22G>A; c.467+42G>C; c.*76C>T
s1677725; rs1780186; rs71403882; rs1780185; rs1748356; rs2368183; rs12776877
12 c.227+55G>A; c.227+58G>A; c.227+67A>T rs1677730; rs6482571; rs1780193
13 c.130‐54A>G; c.130‐46T>C; c.130‐23delG; c.162+56G>A; c.163‐5delT; c.227+55G>A; c.227+67A>T; c.467+42G>C
rs1677725; rs1780186; rs71403882; rs1780185; rs71403883; rs1677730; rs1780193; rs2368183
14 c.162+56G>A; c.227+55G>A; c.227+58G>A; c.227+67A>T rs1780185; rs1677730; rs6482571; rs1780193
15 c.162+56G>A rs1780185
16 c.162+56G>A; c.467+42G>C rs1780185; rs2368183
17 c.162+56G>A; c.467+42G>C; c.*76C>T rs1780185; rs2368183; rs12776877
18 c.130‐54A>G; c.130‐46T>C; c.130‐23delG; c.162+56G>A; c.163‐5delT; c.227+55G>A; c.227+67A>T
rs1677725; rs1780186; rs71403882; rs1780185; rs71403883; rs1677730; rs1780193
19 c.162+56G>A; c.227+55G>A; c.227+67A>T; c.467+42G>C rs1780185; rs1677730; rs1780193; rs2368183
20 c.467+42G>C rs2368183
21 c.162+56G>A rs71403882
22 c.130‐54A>G; c.130‐46T>C; c.130‐23delG; c.162+56G>A; c.467+42G>C; c.*76C>T
rs1677725; rs1780186; rs71403882; rs1780185; rs2368183; rs12776877
23 c.130‐54A>G; c.130‐46T>C; c.130‐23delG; c.162+56G>A; c.1027‐33_1027‐30delGATT
rs1677725; rs1780186; rs71403882; rs1780185; rs34974619
24 c.227+55G>A; c.227+67A>T; c.*76C>T rs1677730; rs1780193; rs12776877
25 c.227+55G>A; c.227+58G>A; c.227+67A>T; c.467+42G>C rs1677730; rs6482571; rs1780193; rs2368183

Resultados e Discussão
55
Tabela 20 – Variantes descritas no gene COQ2 encontradas neste estudo
A vermelho estão representadas as alterações descritas que foram detectadas nas regiões codificantes do gene COQ2.
Caso Variantes descritas encontradas Referência
1 c.196G>T; c.292+47T>G; c.693‐41G>A; c.894T>C rs6818847; rs13113787; rs6841889; rs6535454
2 c.196G>T; c.292+47T>G; c.693‐41G>A; c.894T>C rs6818847; rs13113787; rs6841889; rs6535454
3 c.196G>T; c.292+47T>G; c.693‐41G>A; c.894T>C; c.990C>T rs6818847; rs13113787; rs6841889; rs6535454; rs1129617
4 c.196G>T; c.292+47T>G; c.693‐41G>A; c.894T>C; c.990C>T rs6818847; rs13113787; rs6841889; rs6535454; rs1129617
5 c.292+47T>G; c.693‐41G>A rs13113787; rs6841889
6 c.693‐41G>A rs6841889
7 c.196G>T; c.292+47T>G; c.894T>C rs6818847; rs13113787; rs6535454
8 c.292+47T>G; c.693‐41G>A rs13113787; rs6841889
9 c.196G>T; c.292+47T>G; c.693‐41G>A; c.894T>C; c.990C>T rs6818847; rs13113787; rs6841889; rs6535454; rs1129617
10 c.693‐41G>A; c.894T>C rs6841889; rs6535454
11 c.196G>T; c.292+47T>G; c.693‐41G>A; c.894T>C rs6818847; rs13113787; rs6841889; rs6535454
12 c.693‐41G>A; c.894T>C; c.990C>T rs6841889; rs6535454; rs1129617
13 c.196G>T; c.292+47T>G; c.693‐41G>A; c.894T>C; c.990C>T rs6818847; rs13113787; rs6841889; rs6535454; rs1129617
14 c.196G>T; c.292+47T>G; c.693‐41G>A; c.894T>C rs6818847; rs13113787; rs6841889; rs6535454
15 c.693‐41G>A; c.894T>C rs6841889; rs6535454
16 c.894T>C rs6535454
17 c.196G>T; c.292+47T>G; c.693‐41G>A; c.894T>C; c.990C>T rs6818847; rs13113787; rs6841889; rs6535454; rs1129617
19 c.196G>T; c.292+47T>G; c.693‐41G>A; c.894T>C rs6818847; rs13113787; rs6841889; rs6535454
20 c.64A>T; c.196G>T; c.292+47T>G; c.693‐41G>A; c.894T>C rs112033303; rs6818847; rs13113787; rs6841889; rs6535454
21 c.196G>T; c.292+47T>G(homo); c.404‐50A>T; c.693‐41G>A; c.894T>C; c.990C>T
rs6818847; rs13113787; rs116687075; rs6841889; rs6535454; rs1129617
22 c.196G>T; c.292+47T>G; c.693‐41G>A; c.894T>C rs6818847; rs13113787; rs6841889; rs6535454
23 c.196G>T; c.292+47T>G; c.693‐41G>A; c.894T>C rs6818847; rs13113787; rs6841889; rs6535454
24 c.64A>T; c.196G>T; c.292+47T>G; c.693‐41G>A; c.894T>C rs112033303; rs6818847; rs13113787; rs6841889; rs6535454
25 c.196G>T; c.292+47T>G; c.693‐41G>A; c.894T>C rs6818847; rs13113787; rs6841889; rs6535454
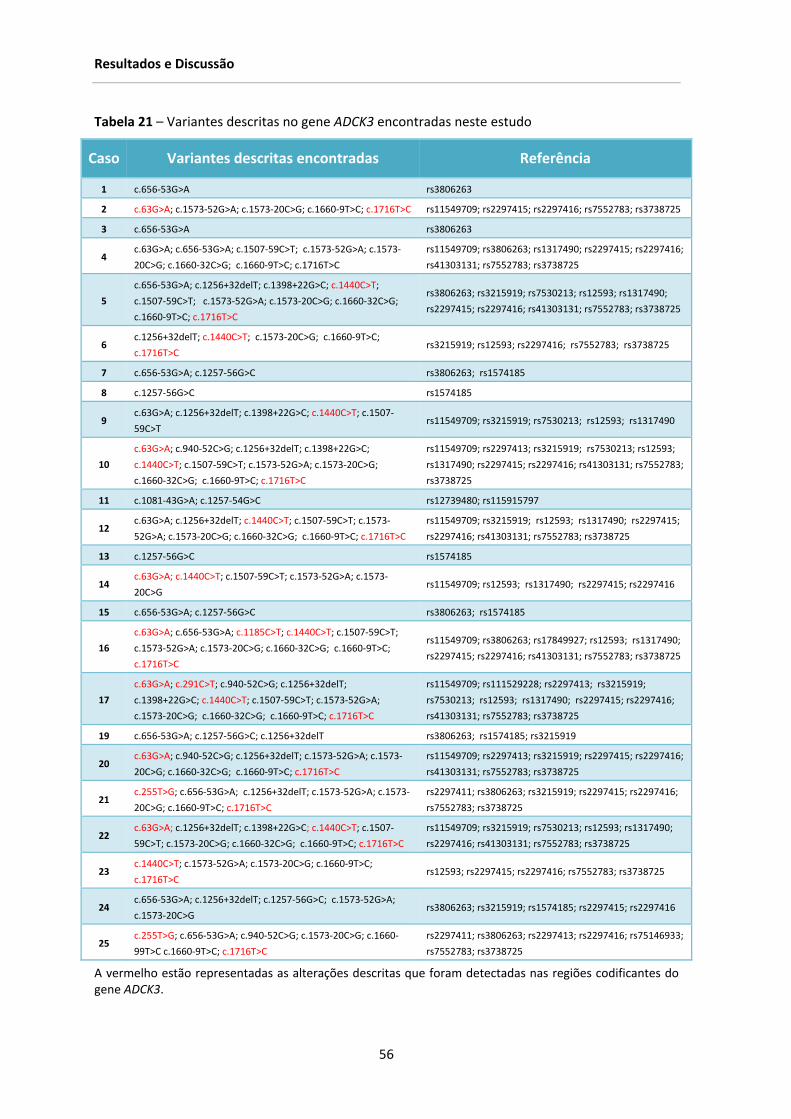
Resultados e Discussão
56
Tabela 21 – Variantes descritas no gene ADCK3 encontradas neste estudo
A vermelho estão representadas as alterações descritas que foram detectadas nas regiões codificantes do gene ADCK3.
Caso Variantes descritas encontradas Referência
1 c.656‐53G>A rs3806263
2 c.63G>A; c.1573‐52G>A; c.1573‐20C>G; c.1660‐9T>C; c.1716T>C rs11549709; rs2297415; rs2297416; rs7552783; rs3738725
3 c.656‐53G>A rs3806263
4 c.63G>A; c.656‐53G>A; c.1507‐59C>T; c.1573‐52G>A; c.1573‐
20C>G; c.1660‐32C>G; c.1660‐9T>C; c.1716T>C
rs11549709; rs3806263; rs1317490; rs2297415; rs2297416;
rs41303131; rs7552783; rs3738725
5
c.656‐53G>A; c.1256+32delT; c.1398+22G>C; c.1440C>T;
c.1507‐59C>T; c.1573‐52G>A; c.1573‐20C>G; c.1660‐32C>G;
c.1660‐9T>C; c.1716T>C
rs3806263; rs3215919; rs7530213; rs12593; rs1317490;
rs2297415; rs2297416; rs41303131; rs7552783; rs3738725
6 c.1256+32delT; c.1440C>T; c.1573‐20C>G; c.1660‐9T>C;
c.1716T>C rs3215919; rs12593; rs2297416; rs7552783; rs3738725
7 c.656‐53G>A; c.1257‐56G>C rs3806263; rs1574185
8 c.1257‐56G>C rs1574185
9 c.63G>A; c.1256+32delT; c.1398+22G>C; c.1440C>T; c.1507‐
59C>T rs11549709; rs3215919; rs7530213; rs12593; rs1317490
10
c.63G>A; c.940‐52C>G; c.1256+32delT; c.1398+22G>C;
c.1440C>T; c.1507‐59C>T; c.1573‐52G>A; c.1573‐20C>G;
c.1660‐32C>G; c.1660‐9T>C; c.1716T>C
rs11549709; rs2297413; rs3215919; rs7530213; rs12593;
rs1317490; rs2297415; rs2297416; rs41303131; rs7552783;
rs3738725
11 c.1081‐43G>A; c.1257‐54G>C rs12739480; rs115915797
12 c.63G>A; c.1256+32delT; c.1440C>T; c.1507‐59C>T; c.1573‐
52G>A; c.1573‐20C>G; c.1660‐32C>G; c.1660‐9T>C; c.1716T>C
rs11549709; rs3215919; rs12593; rs1317490; rs2297415;
rs2297416; rs41303131; rs7552783; rs3738725
13 c.1257‐56G>C rs1574185
14 c.63G>A; c.1440C>T; c.1507‐59C>T; c.1573‐52G>A; c.1573‐
20C>G rs11549709; rs12593; rs1317490; rs2297415; rs2297416
15 c.656‐53G>A; c.1257‐56G>C rs3806263; rs1574185
16
c.63G>A; c.656‐53G>A; c.1185C>T; c.1440C>T; c.1507‐59C>T;
c.1573‐52G>A; c.1573‐20C>G; c.1660‐32C>G; c.1660‐9T>C;
c.1716T>C
rs11549709; rs3806263; rs17849927; rs12593; rs1317490;
rs2297415; rs2297416; rs41303131; rs7552783; rs3738725
17
c.63G>A; c.291C>T; c.940‐52C>G; c.1256+32delT;
c.1398+22G>C; c.1440C>T; c.1507‐59C>T; c.1573‐52G>A;
c.1573‐20C>G; c.1660‐32C>G; c.1660‐9T>C; c.1716T>C
rs11549709; rs111529228; rs2297413; rs3215919;
rs7530213; rs12593; rs1317490; rs2297415; rs2297416;
rs41303131; rs7552783; rs3738725
19 c.656‐53G>A; c.1257‐56G>C; c.1256+32delT rs3806263; rs1574185; rs3215919
20 c.63G>A; c.940‐52C>G; c.1256+32delT; c.1573‐52G>A; c.1573‐
20C>G; c.1660‐32C>G; c.1660‐9T>C; c.1716T>C
rs11549709; rs2297413; rs3215919; rs2297415; rs2297416;
rs41303131; rs7552783; rs3738725
21 c.255T>G; c.656‐53G>A; c.1256+32delT; c.1573‐52G>A; c.1573‐
20C>G; c.1660‐9T>C; c.1716T>C
rs2297411; rs3806263; rs3215919; rs2297415; rs2297416;
rs7552783; rs3738725
22 c.63G>A; c.1256+32delT; c.1398+22G>C; c.1440C>T; c.1507‐
59C>T; c.1573‐20C>G; c.1660‐32C>G; c.1660‐9T>C; c.1716T>C
rs11549709; rs3215919; rs7530213; rs12593; rs1317490;
rs2297416; rs41303131; rs7552783; rs3738725
23 c.1440C>T; c.1573‐52G>A; c.1573‐20C>G; c.1660‐9T>C;
c.1716T>C rs12593; rs2297415; rs2297416; rs7552783; rs3738725
24 c.656‐53G>A; c.1256+32delT; c.1257‐56G>C; c.1573‐52G>A;
c.1573‐20C>G rs3806263; rs3215919; rs1574185; rs2297415; rs2297416
25 c.255T>G; c.656‐53G>A; c.940‐52C>G; c.1573‐20C>G; c.1660‐
99T>C c.1660‐9T>C; c.1716T>C
rs2297411; rs3806263; rs2297413; rs2297416; rs75146933;
rs7552783; rs3738725

Resultados e Discussão
57
Tabela 22‐ Variantes não descritas encontradas neste estudo
Pela análise das tabelas, verifica‐se que os genes PDSS1, COQ2 e ADCK3 são bastante
polimórficos e que as variantes descritas aparecem de uma forma geral distribuídas pela
maioria dos doentes. Enquanto no PDSS1 as alterações encontradas estão localizadas nas
regiões intrónicas do gene, nos COQ2 e ADCK3 foram detectadas também alterações nas
regiões codificantes (representadas a vermelho).
No gene PDSS1 foram detectadas duas variantes que apareceram apenas uma vez. A
variante c.336+22G>A foi detectada no caso 11 em heterozigotia e a c.1027‐33_1027‐
30delGATT no caso 23, igualmente em heterozigotia (representadas a azul). Neste gene
foram também encontradas duas alterações não descritas nas regiões flanqueadoras dos
exões 1 e 7 em heterozigotia, a c.1‐29C>T e c.721+4C>T respectivamente, que vão ser
apresentadas mais à frente (Tabela 19).
No gene PDSS2 apesar de já existirem alterações descritas, patogénicas e benignas,
apenas só foi detectada uma alteração benigna descrita, em 3 doentes, a c.876+48C>T em
heterozigotia localizada na região flanqueadora do exão 5 (Tabela 17).
No COQ2 foram detectados quatro alterações benignas já descritas nas regiões
codificantes em que em numa delas, a variante c.196G>T, há troca de aminoácido e a
outra, a c.64A>T origina um codão de terminação prematuro (PTC) (Tabela 20). Por este
motivo, foi verificado o impacto destas alterações.
Neste gene foram também detectadas 2 alterações não descritas na literatura, a variante
c.320G>C foi encontrada em heterozigotia e localiza‐se na região codificante do exão 1 e
Caso Gene Exão Alteração cDNA Efeito na proteína
Referência
16 PDSS1 1 c.1‐29C>T/N ? Não descrita
12 PDSS1 7 c.721+4C>T/N ? Não descrita
8 e 9 COQ2 1 c.320 G>C/N S107T Não descrita
10 e 16 COQ2 4 c.693‐136A>G/N ? Não descrita
6 ADCK3/CABC1 15 c.1660‐81C>G/N ? Não descrita

Resultados e Discussão
58
a variante c.693‐136A>G foi encontrada também em heterozigotia, na região
flanqueadora do exão 4 (Tabela 22). Os resultados das previsões efectuadas nas bases
informáticas vão ser apresentados neste capítulo.
No COQ9 foram encontradas apenas três variantes descritas em alguns doentes, uma
delas está localizada na região codificante do exão 2 (c.102G> A) (Tabela 18).
No ADCK3 foram detectadas 20 alterações já descritas na literatura e uma nova. Das
alterações descritas, 13 foram encontradas nas regiões intrónicas e 7 nas regiões
codificantes, em que numa há troca de aminoácido, a c.255T>G, presente em
heterozigotia (Tabela 21). A variante não descrita, a c.1660‐81C>G, foi detectada nos
casos 10 e 16 presente em heterozigotia e está localizada na região flanqueadora do exão
15 (Tabela22). Estas alterações vão ser analisadas e apresentadas mais à frente.
4.1. Variantes missense
Para avaliar o impacto destas variantes missense para além de ter em consideração as
características de cada aminoácido foram utilizados os seguintes métodos
bioinformáticos: alinhamento de sequências homólogas efectuado através do programa
Clustal W2 e previsão da patogenicidade das mutações pelo programa Polyphen e
confirmada no Molecular Modeling e Bioinformatics (mmb).
Além das ferramentas bioinformáticas foi efectuada uma análise às características dos
aminoácidos uma vez que para o funcionamento normal de uma proteína a conformação
tridimensional é a principal determinante da sua função biológica, e alterações na
sequência aminoacídica podem mudar a conformação e logo a sua função (configuração
alterada compromete a estabilidade da proteína). Essas alterações são devidas às
características específicas de cada aminoácido, mais concretamente por interacções não
covalentes como ligações de hidrogénio, interacções electrostáticas e hidrofóbicas por
efeitos geométricos impostos pelo aminoácido que uma vez alterados podem
comprometer a estrutura da proteína.

4.1.1
Varia
A sub
básic
do ge
Figur
c.255
O am
em á
uma
dado
conte
da gl
Figurassin
1. Variante
ante c.255T
bstituição c
co, por uma
ene ADCK3
C
ra 12‐ Repr
5T>G, compa
minoácido h
água, e no r
cadeia la
or/receptor
endo grupo
utamina re
ra 13‐ Estrualados. (Nels
es missense
T>G
c.255T>G qu
a glutamina
em heteroz
Caso 21
esentação p
arada com um
histidina tem
radical R ap
ateral ioniz
de electrõ
os funcionai
sulta dos se
utura linear son and Cox, 2
e descritas n
ue origina a
a, aminoácid
zigotia, nos
parcial da s
m controlo n
m carga po
presenta um
zável, facili
ões. A gluta
is que form
eus grupos
(A) dos amino
2004)
59
na literatura
a troca de u
do polar ne
casos 21 (F
sequência do
normal.
sitiva e car
m grupo imi
itando det
amina poss
am ligações
amida (Figu
ácidos gluta
a
um resíduo
eutro (p.H85
Figura 12) e
o caso 21,
acterísticas
idazol. É o ú
erminadas
ui também
s de hidrog
ura 13).
(B)amina (A) e
Re
de histidina
5Q ) foi ide
25.
Controlo
onde se ev
s hidrofílica
único amino
reacções
um radica
énio com a
e histidina (
esultados e D
a, aminoáci
ntificada no
vidência a a
s, ou seja é
oácido que
por actua
al solúvel e
água. A po
(B), com os
Discussão
do polar
o exão 3
alteração
é solúvel
contém
ar como
m água,
olaridade
s radicais

Resultad
Foi efec
bioinfor
diversas
nunca e
encontr
para a e
Figura 1específica
De aco
alteraçã
de 0.00
função
Figura 1program
dos e Discuss
ctuado o a
rmático Clu
s espécies.
está presen
rada na mai
estrutura e/
14‐ Alinhamea o local da oc
ordo com a
ão apresent
0, classificad
ou estrutur
5‐ Previsão dma Polyphen.
são
alinhamento
ustal W2 p
Como pod
nte nas pro
ioria das es
/ou função d
ento entre ecorrência da s
a previsão
ta uma dife
da como u
ra proteica s
da patogenic
o entre vá
para verific
demos veri
teínas hom
pécies. Isto
da enzima.
espécies de substituição)
efectuada
rença de sc
ma alteraç
sejam afect
cidade da alt
60
rias proteín
ar a conse
ificar na Fi
mólogas ma
o sugere que
parte da seq
pelo prog
cores dos am
ção benigna
tadas (Figur
eração c.255
nas homólo
ervação de
gura 14 o
s a sua sub
e esta zona
quência pro
grama bioi
minoácidos
a sendo mu
ra 15).
5T>G no gen
ogas atravé
ste resíduo
resíduo no
bstituição p
parece não
teica do gen
informático
s envolvidos
uito pouco
e ADCK3 atr
és do prog
o, histidina
ormal, hist
pela glutam
o ser impor
ne ADCK3. (
o Polyphen
s na substit
provável q
avés do
grama
, nas
idina,
mina é
rtante
A seta
esta
uição
que a

Para
Hapm
difer
gené
varia
onde
popu
Com
frequ
casos
de C
é um
histó
Cons
nas d
fenó
Figur
pesquisar a
map é um
renças gené
éticas de d
ações genét
e ocorrem
ulação e ent
o é possív
uente na p
s. O caso 21
II‐III de 1,8
m doente q
ória clínica m
siderando a
diferentes
tipo bioquím
ra 16‐ Frequê
a frequênci
projecto i
éticas em s
diferentes
ticas são com
no nosso
tre as popu
vel verificar
opulação e
1 trata‐se d
nmol/min/
que foi sel
muito suges
a análise da
espécies po
mico observ
ência alélica
ia deste po
nternacion
eres human
indivíduos
mpartilhada
DNA e com
lações em d
r este poli
em geral (F
uma crianç
/mg PNC/CS
eccionado
stiva de def
a estrutura
ode‐se con
vado nos do
do polimorf
61
limorfismo
al com o
nos, em qu
para ident
as. Este pro
mo estão d
diferentes p
morfismo
Figura 16).
ça com artro
S e uma bió
não pelos
iciência de
dos amino
siderar que
ois casos.
ismo rs2297
foi utilizado
intuito de
ue o object
tificar regiõ
ojecto descr
distribuídas
partes do m
referido no
Neste estu
ogripose co
ópsia muscu
parâmetro
CoQ10.
oácidos e da
e esta alter
411. (http://w
Re
o a base de
identificar
ivo é comp
ões cromo
reve o que e
entre as p
mundo.
o NCBI (rs2
do aparece
ngénita que
ular sem alt
os bioquím
a conservaç
ração não é
www.hapmap
esultados e D
e dados Hap
as semelh
parar as seq
ossómicas o
essas varian
pessoas da
2297411) é
eu apenas
e apresenta
terações. O
icos mas p
ção do ami
é responsá
p.org/)
Discussão
pmap. O
hanças e
quências
onde as
ntes são,
mesma
é pouco
em dois
a valores
caso 25
por uma
inoácido
vel pelo
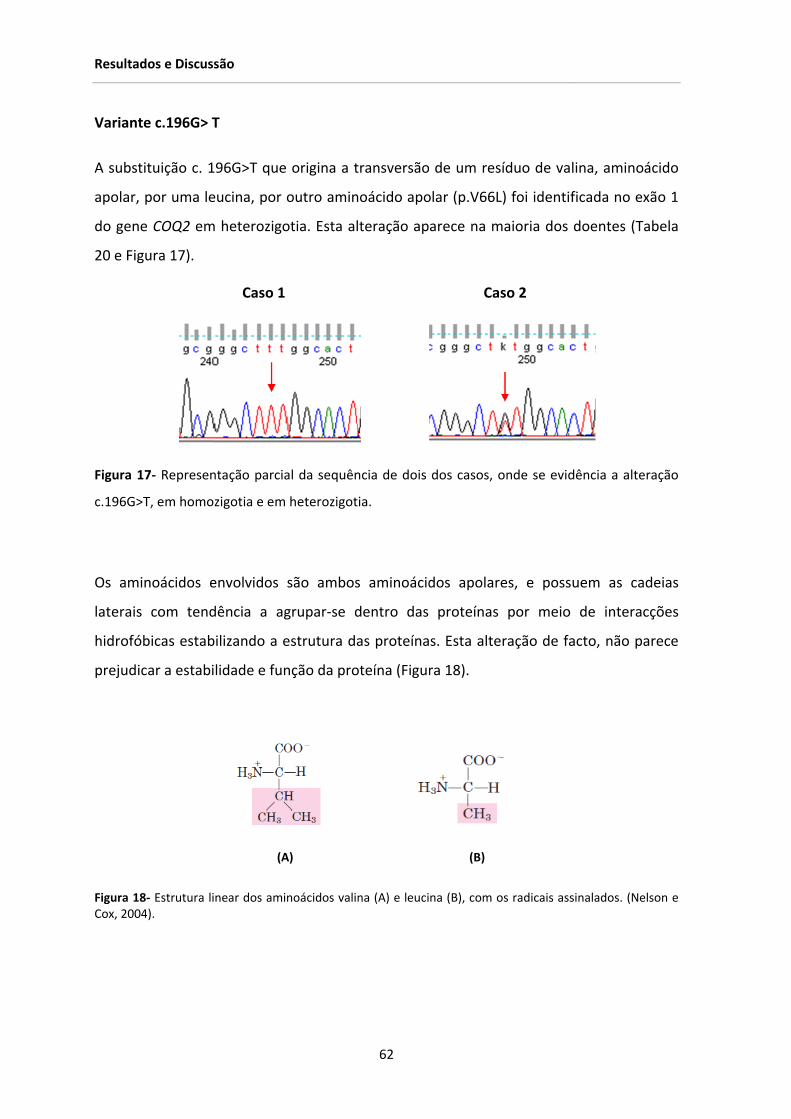
Resultad
Variant
A subst
apolar,
do gene
20 e Fig
Figura 1
c.196G>
Os ami
laterais
hidrofó
prejudic
Figura 18Cox, 2004
dos e Discuss
e c.196G> T
ituição c. 1
por uma le
e COQ2 em
gura 17).
17‐ Represen
T, em homo
noácidos e
com tend
bicas estab
car a estabi
8‐ Estrutura li4).
são
T
96G>T que
eucina, por
m heterozigo
Caso 1
ntação parcia
zigotia e em
envolvidos
dência a ag
ilizando a e
lidade e fun
(A
near dos ami
origina a t
outro amin
otia. Esta al
al da sequên
heterozigot
são ambos
grupar‐se d
estrutura da
nção da pro
A)
noácidos vali
62
transversão
noácido apo
lteração ap
ncia de dois
ia.
s aminoácid
dentro das
as proteína
oteína (Figur
na (A) e leuci
o de um res
olar (p.V66L
arece na m
Ca
dos casos, o
dos apolar
proteínas
s. Esta alte
ra 18).
(B)
na (B), com o
síduo de va
L) foi identif
maioria dos
so 2
onde se evid
es, e poss
por meio
eração de fa
os radicais ass
lina, amino
ficada no ex
doentes (T
dência a alte
uem as ca
de interac
acto, não pa
sinalados. (Ne
ácido
xão 1
abela
eração
adeias
cções
arece
elson e

As s
outra
o res
(valin
algum
Figurespec
A pre
dado
regiã
tradu
músc
pb q
Por e
Foi p
e Bio
enco
equências
as sequênci
síduo em c
na) como a
mas espécie
ra 19‐ Alinhacífica o local d
evisão no P
os utiliza te
ão. Este gen
ução só é
culo esquel
ue não estã
esta razão, a
possível faze
oinformatics
ontrada, c.1
de resíduo
ias proteica
ausa é pou
alteração (
es, como ou
amento entra ocorrência d
Polyphen nã
em apenas
ne tem vário
iniciada n
ético demo
ão presente
alterações n
er a previsã
s (mmb) (ht
96G>T, na p
os que abra
as de outras
uco conserv
(leucina) es
utros amino
re espécies da substituiçã
o foi possív
371 amino
os codões d
o terceiro
onstraram q
es no mRNA
nesta região
o do impac
ttp://mmb.
proteína é n
63
angiam o c
s espécies,
vado. O que
stão presen
oácidos de d
de parte daão)
vel porque a
oácidos (ht
de iniciação
codão ATG
que o exão
A (Forsgren 2
o não altera
cto desta alt
.pcb.ub.es/)
nulo (Figura
codão alter
apresentad
e se verifica
ntes em algu
diferentes c
a sequência
a referência
tp://www.u
(ATG) e alg
G. Estudos
1 possui um
2004).
am a função
teração no
), verificand
a 20).
Re
rado foram
do na Figura
a que tanto
umas prote
característic
proteica do
a da proteín
uniprot.org
guns autore
de expres
ma sequênc
o e a integri
programa M
do‐se que o
esultados e D
m comparad
a 19, revela
o o resíduo
eínas homó
cas.
o gene COQ2
na que esta
) e não inc
s considera
ssão realiza
cia adiciona
idade da pr
Molecular M
o efeito da
Discussão
das com
ndo que
o normal
logas de
2. (A seta
base de
clui esta
am que a
ados no
l de 111
oteína.
Modeling
variante

Resultad
Figura 2
mmb.
O facto
forte ev
4.1.2. V
Foi det
resumid
Tabela 2
Caso
8 e 9
Variant
A subst
neutro,
identific
dos e Discuss
0‐ Previsão d
de esta va
vidência de
Variante m
ectada um
da na Tabela
23 – Descriçã
Ge
CO
e c.320G> C
ituição c.32
por uma
cada no exã
são
da patogenic
ariante esta
se tratar de
issense não
ma variante
a 23.
ão da variant
ene
OQ2
C
20G>C que
treonina, q
ão 1 do gene
cidade da alt
r presente
e uma varia
o descrita n
missense,
te missense e
Exão
1
origina a t
que també
e COQ2 em
64
teração c.19
em todos o
nte comum
na literatura
não descr
encontrada n
Alteração c
c.320 G>C
roca de um
ém é um a
heterozigo
6G>T no gen
os doentes
m e sem imp
a
rita na liter
neste estudo
cDNA Ep
C/N
m resíduo d
aminoácido
otia nos caso
ne COQ2 atra
estudados
pacto ao nív
ratura, e q
o.
Efeito na proteína
S107T
de serina, a
polar neu
os 8 (Figura
avés do prog
é também
vel da prote
que se enc
Referên
Não desc
minoácido
utro, (S107T
a 21) e 9.
grama
m uma
ína.
ontra
ncia
crita
polar
T) foi

Figurcomp
Os a
hidro
solve
as ca
não p
Figur(Nelso
Para
sequ
W2.
cons
(Figu
ra 21‐ Represparada com u
minoácidos
oxilo que t
ente, caract
aracterística
parece alte
ra 22‐ Estruton e Cox, 2004
avaliar a
uência prote
O alinham
ervada, apa
ura 23).
Caso 8
sentação parum controlo
s serina e t
ende a for
terística que
as destes a
rar a estrut
ura linear do4)
conservaçã
eica com seq
mento das s
arecendo e
rcial da sequnormal.
reonina são
rmar ponte
e torna este
aminoácido
ura e/ou fu
(A)
os aminoácid
ão do amin
quências de
sequências
em algumas
65
uência do cas
o do mesm
s de hidrog
es aminoác
s são muit
unção da en
dos serina (A
noácido ser
e outros org
homóloga
s espécies o
so 8, onde se
o grupo e p
génio com
idos hidrofí
o semelhan
zima.
(B)
A) e treonina
rina, foi efe
ganismos re
s mostra q
outros amin
Re
Controlo
e evidência a
possuem no
a cadeia p
ílicos (Figur
ntes, a sub
a (B), com os
ectuada um
ecorrendo a
que esta re
noácidos de
esultados e D
a alteração c
o radical um
principal ou
a 22). Uma
bstituição c
s radicais ass
ma compar
ao programa
egião não
e grupos dif
Discussão
.320G>C,
m grupo
u com o
vez que
.320G>C
sinalados.
ação da
a Clustal
é muito
ferentes

Resultad
Figura 2específica
De aco
alteraçã
de 0.244
estrutur
Figura 2Polyphen
A previs
mmb qu
dos e Discuss
23‐ Alinhamea o local da oc
ordo com a
ão apresent
4, classifica
ra proteica
4‐ Previsão dn.
são do efe
ue prevê um
são
ento entre ecorrência da s
a previsão
ta uma dife
da como um
sejam afect
da patogenic
ito da alter
m efeito neu
espécies de substituição)
efectuada
rença de sc
ma alteraçã
tadas (Figur
cidade da alt
ração enco
utral sobre
66
parte da se
pelo prog
cores dos am
ão benigna s
ra 24).
teração c.32
ntrada foi
a proteína (
equência pro
grama bioi
minoácidos
sendo pouc
0G>C no gen
verificada e
(Figura 25).
oteica do ge
informático
s envolvidos
co provável
ne COQ2 atra
e confirmad
ene COQ2. (
o Polyphen
s na substit
que a funçã
avés do prog
da no prog
A seta
esta
uição
ão ou
grama
grama

Figur
mmb
Os d
cons
alter
susp
miop
suge
fenó
4.2.
Varia
Foi d
Tabe
Tabe
Ca
20
ra 25‐ Previsã
b.
doentes a
ideravelme
rações histo
eita de do
páticas ines
stivas de d
tipo observ
Variant
ante c.64A>
detectada 1
ela 24.
la 24‐ Descri
aso
e 24
ão da patoge
presentam
ente baixos
ológicas na b
oença mitoc
specíficas. A
défice de Co
vado nos 2 c
te nonsen
>T
1 variante n
ição da varia
Gene
COQ2
enecidade da
valores d
s. O caso
biopsia mus
condrial e
Apesar de
oQ10 a alte
casos.
se
nonsense de
ante nonsens
Exão
1
67
a alteração c
de CII‐II d
8 apresent
scular comp
o estudo
os valores
ração enco
escrita na li
se encontrad
Alteraçã
c.64
c.320G>C no
de 1,6 nm
ta hipotoni
patíveis com
da biopsia
s de CII‐III
ontrada não
teratura, e
da neste estu
ão cDNA
4A>T
Re
gene COQ2
mol/min/mg
ia e fraque
m atrofia es
muscular
e as histó
o parece se
que se enc
udo.
Efeito na proteína
R22X
esultados e D
através do p
gPNC/CS q
eza muscu
spinal. No c
revelou alt
rias clínica
er responsá
contra resu
Refe
Não d
Discussão
programa
que são
lar com
aso 9 há
terações
s serem
vel pelo
mida na
erência
descrita

Resultad
Esta alte
e origin
(Figura
aparent
função
codifica
contribu
Figura 2compara
4.3. V
Foram
ADCK3 n
Tabela 2
Caso
16
12
10 e 16
6
dos e Discuss
eração foi e
na a alteraç
26). Esta alt
temente, c
da enzima
ante. Como
ui para a fo
C
6‐ Represenada com um
Variantes
detectadas
não descrita
25‐ Descrição
Ge
PDS
PDS
6 CO
ADCK3/
são
encontrada
ção de um
teração est
ausar uma
a, surge na
referido an
rmação da
aso 20
tação parciacontrolo no
s intrónica
quatro alt
as na literat
o das variant
ene
SS1
SS1
OQ2
/CABC1
em heteroz
a arginina
á considera
paragem
a região do
nteriorment
proteína.
al da sequêncrmal.
as
terações na
tura, e que
tes intrónias
Exão
1
7
4
15
68
zigotia nos
por um co
ada na litera
na traduçã
o exão 1,
te o exão 1
cia do caso 2
as regiões
se encontra
encontradas
Alteração c
c.1‐29C>T
c.721+4C>T
c.693‐136A>
c.1660‐81C>
casos 20 e 2
odão de te
atura como
ão, o que
que segun
possui uma
Cont
20, onde se e
intrónicas
am resumid
s neste estud
cDNA Ep
T/N
T/N
>G/N
>G/N
24 no gene
rminação p
benigna po
resultaria
ndo alguns
a região de
trolo
evidência a a
dos genes
das na Tabe
do.
Efeito na proteína
?
?
?
?
COQ2 no e
prematuro
orque apesa
numa perd
autores, n
111 pb que
alteração c.6
PDSS1, CO
ela 25.
Referên
Não desc
Não desc
Não desc
Não desc
xão 1
(PTC)
ar, de
da de
não é
e não
64A>T,
OQ2 e
ncia
crita
crita
crita
crita

Dura
Exist
que
maio
guan
aden
exão
local
Para
as b
(http
c.1‐2
No c
5`UT
alter
Figur29C>
Pela
prese
ante o proc
em “motivo
actuam co
oria dos c
nina/timina
nina/guanin
o‐intrão exi
izados nos
avaliar o im
bases info
p://www.fru
29C>T
caso 16 foi
TR do gene
ração nos re
ra 27 – ReprT, comparad
análise no
ença desta
cesso de sp
os” conserv
mo sinais p
casos, o lo
(GT); guan
a (AG) (Des
istem outro
exões ou in
mpacto dest
ormáticas:
uitfly.org/) e
detectada
PDSS1 pre
estantes exõ
Caso 16
resentação pda com um co
programa N
variante nã
plicing os i
vados junto
para ocorre
ocal conse
nina/uracilo
sviat L.R. et
os elemen
ntrões que p
tas variante
o Splice
e o Netgene
por seque
esente em
ões ou nas r
6
parcial da seontrolo norm
Netgene 2 e
ão cria nem
69
ntrões são
o das junçõ
er a correc
nso de sp
o (GU) no m
t al., 2007).
tos regulad
permitem o
es intrónica
Site Pred
e 2 (http://w
enciação ge
heterozigot
regiões de j
equência do mal.
e do Splice S
m elimina n
removidos
ões exão‐int
ta identific
plicing em
mRNA, em
Para além
dores (splic
processo n
s no proces
diction by
www.cbs.dt
nómica a a
tia e não f
junção intrã
caso 16, on
Site Predict
enhuma re
Re
s com eleva
trão (local
cação e jun
5’ (dado
3’ (aceitad
destes “mo
cing enhan
normal de sp
sso de splici
Neural N
tu.dk/servic
alteração c.
oi encontra
ão‐exão (Fig
Controlo
nde se evidê
tion foi poss
gião conse
esultados e D
ada especif
de splicing
nção dos ex
or) tem o
dor) tem o
otivos” nas
ncers ou s
plicing.
ing foram u
Network at
ces/NetGen
.1‐29C>T na
ada mais n
gura 27).
ência a altera
sível verific
nso, e por
Discussão
ficidade.
5’ e 3’)
xões. Na
motivo
o motivo
junções
ilencers)
utilizadas
t BDGP
ne2/).
a região
enhuma
ação c.1‐
ar que a
isso não

Resultad
parece
mRNA (
Neste g
com a
multissi
da CRM
sugerind
Pelas pr
encontr
Mas um
mais est
c.721+4
No cas
heteroz
do gene
28).
Figura 2c.721+4C
A anális
score as
o que p
dos e Discuss
haver inter
anexo 8.3.1
gene apenas
tingimento
istémica, co
M verificou‐
do um défic
revisões na
rada e o fen
ma vez que
tudos nome
4C>T
o 12 foi d
zigotia e em
e a partir d
Ca
28‐ RepreseC>T, compar
se bioinform
ssociado ao
poderá resu
são
rferência no
1.).
s foi descrit
cardíaco.
om referênc
‐se uma di
ce primário
as bases de
nótipo apres
se trata de
eadamente
detectada a
mbora tenha
o DNA gen
aso 12
entação parcrada com um
mática atra
o local da al
ltar na perd
o processo
a uma muta
O doent
cia a enfart
minuição n
de CoQ10.
dados, não
sentado.
e uma alter
de cDNA p
a alteração
am sido seq
ómico, não
cial da sequm controlo no
avés do Spl
teração ind
da do local
70
de splicing
ação patogé
te estudad
te cerebral
no valor de
o parece ha
ração locali
ara verifica
o c.721+4C
quenciados
o foi identifi
uência do cormal.
lice Site Pre
dicou uma d
dador (5`)
g, não afect
énica numa
do tem u
e episódio
e CII‐III de
aver uma co
zada na reg
r a ocorrên
>T no intr
todos os e
icada mais
Cont
caso 12, on
ediction rea
diminuição
de splicing
tando o pro
a forma de d
uma apres
s de stroke
1,9 nmol/m
orrelação e
gião 5`UTR
cia de trans
rão 7 do g
exões e junç
nenhuma a
trolo
de se evidê
alizada par
do score de
. A previsão
ocessament
doença neo
sentação c
e‐like. No es
min/mg PN
entre a alte
são necess
scrição.
gene PDSS1
ções intrão‐
alteração (F
ência a alte
ra a previsã
e 0,85 para
o foi verifica
to do
onatal
clínica
studo
C/CS,
ração
sários
1 em
‐exão
Figura
eração
ão do
0,61,
ada e

confi
dado
8.3.2
Esta
regu
pode
por r
Nest
prote
Este
activ
reve
PNC/
c.693
A alt
nos c
terem
Figur136A
irmada nou
or de splicin
2.).
alteração
ladoras, fun
e resultar no
reconhecim
e caso é im
eína.
caso apre
vidades das
lando um a
/CS). O resu
3‐136A>G
teração c.69
casos 10 (F
m sido sequ
ra 29‐ RepreA>G, compara
utro program
ng. Aparen
está locali
ndamentais
o skipping d
mento de ou
mportante o
esentava do
s enzimas d
a proliferaçã
ultado do es
93‐136A>G
Figura 29) e
uenciados to
Casos 10
sentação paada com um
ma, o Netge
temente, o
zada muito
s para a oco
do exão 7 o
tro local crí
estudo do
oença mito
da CRM ve
ão mitocon
studo histol
foi identif
e 16. Não f
odos os exõ
arcial da seq controlo no
71
ene, em que
o local acei
o próximo
orrência de
ou ocorrer u
íptico de sp
mRNA para
ocondrial co
erificou‐se
drial, e um
ógico da bi
icada no in
foi encontra
ões e respec
uência do caormal.
e também s
tador (3’) m
de um lo
splicing. A
uma retenç
licing .
a verificar q
om hiperla
um aumen
a diminuiçã
ópsia musc
ntrão 4 do g
ada mais n
ctivas regiõe
Co
aso 10, onde
Re
se verifica a
manteve‐se
cal de liga
perda de ut
ção total ou
qual o efeito
actacidemia
nto signific
ão do CII‐III
ular não foi
gene COQ2
nenhuma al
es flanquea
ntrolo
e se evidênc
esultados e D
a perda de u
e inalterado
ação das p
tilização de
parcial do
o desta mut
e no estu
ativo da C
(1,7 nmol/
i disponibili
2 em hetero
teração, ap
adoras.
cia a alteraçã
Discussão
um local
o (anexo
roteínas
ste local
intrão 7
tação na
udo das
CS (265),
/min/mg
izado.
ozigotia,
pesar de
ão c.693‐

Resultad
Pela an
encontr
parece
O caso
activida
biopsia
No caso
referido
Este gen
descrita
(Diomed
Apesar
estudar
c.1660‐8
No caso
heteroz
intrão e
Figura 381C>G, c
dos e Discuss
álise dos do
rada nenhu
indicar que
10 apresen
ade do CII‐I
muscular n
o 16 foi ta
o anteriorm
ne tem sido
a uma muta
di‐Camasse
de não ha
r o cDNA de
81C>G
o 6 foi detec
zigotia (Figu
e não foi det
30‐ Represencomparada c
são
ois program
ma variaçã
não há com
ntava doenç
III (1.9 nmo
não foi dispo
mbém dete
mente, em q
o associado
ação em do
ei et al., 200
aver variaç
e forma a es
ctada a alte
ura 30). For
tectada ma
Casos 6
ntação parciacom um cont
mas utilizado
o no score
mprometim
ça mitocond
ol/min/mg
onibilizado.
ectada uma
ue a patoge
ao fenótipo
ois irmãos q
07).
ão de scor
sclarecer o e
eração c.166
ram sequen
is nenhuma
al da sequêntrolo normal
72
os na avalia
que indiqu
mento na pro
drial com h
PNC/CS). O
a alteração
enicidade nã
o de aprese
ue apresen
re nas prev
efeito desta
60‐81C>G n
nciados todo
a alteração.
ncia do casol.
ação das alt
e alteraçõe
odução do m
iperlactacid
O resultado
nova nout
ão está tota
entação nef
ntavam doe
visões que
a alteração
o intrão 15
os os exões
Contro
6, onde se
terações in
es de locais
mRNA (anex
demia e um
o da histolo
tro gene da
almente esc
rótica, cont
nça multiss
se fizeram
a nível da p
do gene AD
s e regiões
olo
evidência a
trónicas, nã
de splicing
xo 8.3.3.).
ma diminuiçã
ogia refere
a biossíntes
clarecida.
tudo foi tam
sistémica in
m, é impor
proteína.
DCK3/CABC
de junção
alteração c.
ão foi
g. Isto
ão da
nte à
se, já
mbém
nfantil
rtante
C1 em
exão‐
1660‐

Resultados e Discussão
73
A análise bioinformática de ambos os programas utilizados neste estudo, não revelou
variação no score, o que parece indicar que e a alteração encontrada não provoca a
criação ou eliminação de locais de consenso de splicing (anexo 8.3.4.). Uma vez que o
processo de splicing é complexo e que intervêm várias proteínas, é importante estudar o
cDNA para verificar se esta alteração não interfere com a maquinaria envolvida no
processo de produção do mRNA.
O caso 6 é um doente já com uma idade muito avançada que foi incluído no estudo pelo
facto de revelar uma deficiência marcada na actividade do CII‐III (0,9 nmol/min/mg
PNC/CS). Apresentava sinais de demência, parkinsonismo e na RMN apresentava
alterações sugestivas de doença mitocondrial.


Conclusão e Perspectivas Futuras


Conclusão e Perspectivas Futuras
77
5. Conclusão
O estudo apresentado nesta dissertação pretende estabelecer um diagnóstico etiológico
em doentes com deficiência primária de coenzima Q10 detectados por disfunção
mitocondrial.
De modo a estabelecer este diagnóstico foi utilizada uma amostra de 24 doentes com
défice do CII‐III e um por suspeita clínica, na qual foram analisados os genes envolvidos na
biossíntese em que até à data, foram identificadas mutações patogénicas. Os genes
estudados foram: PDSS1, PDSS2, COQ2, COQ9 e ADCK3.
A análise dos resultados obtidos permite constatar que foram encontradas 48 variantes, 5
das quais ainda não se encontram descritas na literatura. Através da análise da estrutura
dos aminoácidos e da conservação do aminoácido nas diferentes espécies foi possível
comprovar que a variante missense encontrada parece não interferir na integridade e
função da proteína. Nas variantes intrónicas será necessário a realização de mais estudos
ao nível do cDNA que permitam verificar o impacto destas alterações na proteína.
As alterações novas foram detectadas em sete dos doentes incluídos neste estudo em
heterozigotia, não tendo sido identificada a alteração no segundo alelo, apesar de terem
sido sequenciados todos os exões e junções exões‐intrões de todos os genes estudados.
Isto pode indicar a presença de uma grande delecção ou inserção ou se tratar de uma
alteração no promotor do gene.
A deficiência de CoQ10 é a única doença mitocondrial que actualmente tem um
tratamento eficaz, que é feito com suplementação oral de CoQ10. Após 6 meses de
terapia os doentes demonstram melhorias significativas das funções cognitivas e físicas.
No entanto os danos cerebrais e os danos ao nível do rim quando instalados, são
praticamente irreversíveis apenas verificando‐se apenas uma melhoria dos sintomas. Se o
diagnóstico de deficiência primária de CoQ10 é feito antes do aparecimento dos sintomas,
e o tratamento iniciado nesse momento, o paciente tem um desenvolvimento normal e

Conclusão e Perspectivas Futuras
78
sem qualquer danos ao nível cerebral e ou renais. Daí o diagnóstico precoce ser de
extrema importância e necessidade.
Uma vez que o diagnóstico bioquímico exige uma biópsia muscular e ou pele, a
caracterização molecular revela‐se de muita importância para uma confirmação de
diagnóstico.

Conclusão e Perspectivas Futuras
79
6. Perspectivas Futuras
Este trabalho revela‐se apenas o primeiro passo para a caracterização molecular das
deficiências da biossíntese da CoQ10.
O diagnóstico bioquímico não foi implementado por questões financeiras que surgiram
no decorrer deste trabalho e que nos foi alheio. Mas será de extrema importância pois
permite comprovar e esclarecer a patogénese da doença destes casos.
Uma vez que existem mais genes envolvidos na biossíntese da CoQ10 seria importante
alargar o estudo a esses genes de forma a caracterizar estes doentes.
A utilização duma abordagem integrada de diagnóstico permitirá uma melhor
identificação e caracterização destes e de novos casos.
De forma a completar este estudo seria interessante estudar os mRNAs dos casos em que
se encontraram alterações nas regiões intrónicas dos genes estudados, para verificar se
estas variantes interferem com a transcrição e consequente produção de mRNA.
Seria importante realizar estudos de quantificação dos níveis de mRNA destes pacientes
por PCR em tempo real e avaliar o impacto das mutações encontradas ao nível da
proteína através de técnicas de imunoblot, de forma a contribuir para um possível
estabelecimento correlação genótipo/fenótipo.
Uma vez que se trata de uma doença extremamente rara, todos os doentes identificados
serão relevantes para a investigação desta patologia, e o conhecimento a nível
bioquímico e molecular resultante será importante para uma melhor compreensão da
fisiopatologia da molécula da CoQ10.


Referências Bibliográficas


Referências Bibliográficas
83
7. Referências Bibliográficas
Anderson, S., A. T. Bankier, B. G. Barrell, M. H. de Bruijn, A. R. Coulson, J. Drouin, I. C. Eperon, D. P.
Nierlich, B. A. Roe, F. Sanger, P. H. Schreier, A. J. Smith, R. Staden and I. G. Young (1981).
"Sequence and organization of the human mitochondrial genome." Nature 290(5806): 457‐465.
Artuch, R., G. Brea‐Calvo, P. Briones, A. Aracil, M. Galvan, C. Espinos, J. Corral, V. Volpini, A. Ribes,
A. L. Andreu, F. Palau, J. A. Sanchez‐Alcazar, P. Navas and M. Pineda (2006). "Cerebellar ataxia
with coenzyme Q10 deficiency: diagnosis and follow‐up after coenzyme Q10 supplementation." J
Neurol Sci 246(1‐2): 153‐158.
Aure, K., J. F. Benoist, H. Ogier de Baulny, N. B. Romero, O. Rigal and A. Lombes (2004).
"Progression despite replacement of a myopathic form of coenzyme Q10 defect." Neurology 63(4):
727‐729.
Beg, S., S. Javed and K. Kohli (2010). "Bioavailability enhancement of coenzyme Q10: an extensive
review of patents." Recent Pat Drug Deliv Formul 4(3): 245‐255.
Bentinger, M., K. Brismar and G. Dallner (2007). "The antioxidant role of coenzyme Q."
Mitochondrion 7 Suppl: S41‐50.
Bhagavan, H. N. and R. K. Chopra (2006). "Coenzyme Q10: Absorption, tissue uptake, metabolism
and pharmacokinetics." Free Radical Research 40(5): 445‐453.
Boitier, E., F. Degoul, I. Desguerre, C. Charpentier, D. Francois, G. Ponsot, M. Diry, P. Rustin and C.
Marsac (1998). "A case of mitochondrial encephalomyopathy associated with a muscle coenzyme
Q10 deficiency." J Neurol Sci 156(1): 41‐46.
Crane, F. L. (2008). "The evolution of coenzyme Q." Biofactors 32(1‐4): 5‐11.
Crane, F. L., Y. Hatefi, R. L. Lester and C. Widmer (1957). "Isolation of a quinone from beef heart
mitochondria." Biochim Biophys Acta 25(1): 220‐221.
den Dunnen, J. T. and S. E. Antonarakis (2001). "Nomenclature for the description of human
sequence variations." Hum Genet 109(1): 121‐124.

Referências Bibliográficas
84
Desmet, F. O., D. Hamroun, M. Lalande, G. Collod‐Beroud, M. Claustres and C. Beroud (2009).
"Human Splicing Finder: an online bioinformatics tool to predict splicing signals." Nucleic Acids
Res 37(9): e67.
Desviat L.R., Pérez B. and Ugarte M. (2007). "Functional analysis of splicing mutations causing
organic acidemias." Current Research on Organic Acidemias(6): 17‐19.
Di Giovanni, S., M. Mirabella, A. Spinazzola, P. Crociani, G. Silvestri, A. Broccolini, P. Tonali, S. Di
Mauro and S. Servidei (2001). "Coenzyme Q10 reverses pathological phenotype and reduces
apoptosis in familial CoQ10 deficiency." Neurology 57(3): 515‐518.
DiMauro, S. (2011). "A history of mitochondrial diseases." J Inherit Metab Dis 34(2): 261‐276.
DiMauro, S., E. Bonilla, M. Zeviani, M. Nakagawa and D. C. DeVivo (1985). "Mitochondrial
myopathies." Ann Neurol 17(6): 521‐538.
DiMauro, S. and M. Mancuso (2007). "Mitochondrial diseases: therapeutic approaches." Biosci
Rep 27(1‐3): 125‐137.
DiMauro, S., C. M. Quinzii and M. Hirano (2007). "Mutations in coenzyme Q10 biosynthetic
genes." J Clin Invest 117(3): 587‐589.
Diomedi‐Camassei, F., S. Di Giandomenico, F. M. Santorelli, G. Caridi, F. Piemonte, G. Montini, G.
M. Ghiggeri, L. Murer, L. Barisoni, A. Pastore, A. O. Muda, M. L. Valente, E. Bertini and F. Emma
(2007). "COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal
involvement." J Am Soc Nephrol 18(10): 2773‐2780.
Duncan, A. J., M. Bitner‐Glindzicz, B. Meunier, H. Costello, I. P. Hargreaves, L. C. Lopez, M. Hirano,
C. M. Quinzii, M. I. Sadowski, J. Hardy, A. Singleton, P. T. Clayton and S. Rahman (2009). "A
nonsense mutation in COQ9 causes autosomal‐recessive neonatal‐onset primary coenzyme Q10
deficiency: a potentially treatable form of mitochondrial disease." Am J Hum Genet 84(5): 558‐566.
Duncan, A. J., S. J. Heales, K. Mills, S. Eaton, J. M. Land and I. P. Hargreaves (2005). "Determination
of coenzyme Q10 status in blood mononuclear cells, skeletal muscle, and plasma by HPLC with di‐
propoxy‐coenzyme Q10 as an internal standard." Clin Chem 51(12): 2380‐2382.

Referências Bibliográficas
85
Engel, W. K. and G. G. Cunningham (1963). "Rapid Examination of Muscle Tissue. An Improved
Trichrome Method for Fresh‐Frozen Biopsy Sections." Neurology 13: 919‐923.
Ernster, L. and G. Dallner (1995). "Biochemical, physiological and medical aspects of ubiquinone
function." Biochim Biophys Acta 1271(1): 195‐204.
Ernster, L., D. Ikkos and R. Luft (1959). "Enzymic activities of human skeletal muscle mitochondria:
a tool in clinical metabolic research." Nature 184: 1851‐1854.
Forsgren, M., A. Attersand, S. Lake, J. Grunler, E. Swiezewska, G. Dallner and I. Climent (2004).
"Isolation and functional expression of human COQ2, a gene encoding a polyprenyl transferase
involved in the synthesis of CoQ." Biochem J 382(Pt 2): 519‐526.
Gempel, K., H. Topaloglu, B. Talim, P. Schneiderat, B. G. Schoser, V. H. Hans, B. Palmafy, G. Kale, A.
Tokatli, C. Quinzii, M. Hirano, A. Naini, S. DiMauro, H. Prokisch, H. Lochmuller and R. Horvath
(2007). "The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron‐
transferring‐flavoprotein dehydrogenase (ETFDH) gene." Brain 130(Pt 8): 2037‐2044.
Groneberg, D. A., B. Kindermann, M. Althammer, M. Klapper, J. Vormann, G. P. Littarru and F.
Doring (2005). "Coenzyme Q10 affects expression of genes involved in cell signalling, metabolism
and transport in human CaCo‐2 cells." Int J Biochem Cell Biol 37(6): 1208‐1218.
Grunler, J., J. Ericsson and G. Dallner (1994). "Branch‐point reactions in the biosynthesis of
cholesterol, dolichol, ubiquinone and prenylated proteins." Biochim Biophys Acta 1212(3): 259‐
277.
Haginoya, K., S. Miyabayashi, M. Kikuchi, A. Kojima, K. Yamamoto, K. Omura, M. Uematsu, N.
Hino‐Fukuyo, S. Tanaka and S. Tsuchiya (2009). "Efficacy of idebenone for respiratory failure in a
patient with Leigh syndrome: a long‐term follow‐up study." J Neurol Sci 278(1‐2): 112‐114.
Heeringa, S. F., G. Chernin, M. Chaki, W. Zhou, A. J. Sloan, Z. Ji, L. X. Xie, L. Salviati, T. W. Hurd, V.
Vega‐Warner, P. D. Killen, Y. Raphael, S. Ashraf, B. Ovunc, D. S. Schoeb, H. M. McLaughlin, R. Airik,
C. N. Vlangos, R. Gbadegesin, B. Hinkes, P. Saisawat, E. Trevisson, M. Doimo, A. Casarin, V.
Pertegato, G. Giorgi, H. Prokisch, A. Rotig, G. Nurnberg, C. Becker, S. Wang, F. Ozaltin, R. Topaloglu,
A. Bakkaloglu, S. A. Bakkaloglu, D. Muller, A. Beissert, S. Mir, A. Berdeli, S. Varpizen, M. Zenker, V.
Matejas, C. Santos‐Ocana, P. Navas, T. Kusakabe, A. Kispert, S. Akman, N. A. Soliman, S. Krick, P.

Referências Bibliográficas
86
Mundel, J. Reiser, P. Nurnberg, C. F. Clarke, R. C. Wiggins, C. Faul and F. Hildebrandt (2011).
"COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness." J
Clin Invest 121(5): 2013‐2024.
Hirano, M., C. M. Quinzii and S. Dimauro (2006). "Restoring balance to ataxia with coenzyme Q10
deficiency." J Neurol Sci 246(1‐2): 11‐12.
Holt, I. J., J. M. Cooper, J. A. Morgan‐Hughes and A. E. Harding (1988). "Deletions of muscle
mitochondrial DNA." Lancet 1(8600): 1462.
Holzinger, A., W. Roschinger, F. Lagler, P. U. Mayerhofer, P. Lichtner, T. Kattenfeld, L. P. Thuy, W. L.
Nyhan, H. G. Koch, A. C. Muntau and A. A. Roscher (2001). "Cloning of the human MCCA and
MCCB genes and mutations therein reveal the molecular cause of 3‐methylcrotonyl‐CoA:
carboxylase deficiency." Hum Mol Genet 10(12): 1299‐1306.
Horvath, R., P. Schneiderat, B. G. Schoser, K. Gempel, E. Neuen‐Jacob, H. Ploger, J. Muller‐Hocker,
D. E. Pongratz, A. Naini, S. DiMauro and H. Lochmuller (2006). "Coenzyme Q10 deficiency and
isolated myopathy." Neurology 66(2): 253‐255.
Hsieh, E. J., P. Gin, M. Gulmezian, U. C. Tran, R. Saiki, B. N. Marbois and C. F. Clarke (2007).
"Saccharomyces cerevisiae Coq9 polypeptide is a subunit of the mitochondrial coenzyme Q
biosynthetic complex." Arch Biochem Biophys 463(1): 19‐26.
Iiizumi, M., H. Arakawa, T. Mori, A. Ando and Y. Nakamura (2002). "Isolation of a novel gene,
CABC1, encoding a mitochondrial protein that is highly homologous to yeast activity of bc1
complex." Cancer Res 62(5): 1246‐1250.
Jeya, M., H. J. Moon, J. L. Lee, I. W. Kim and J. K. Lee (2010). "Current state of coenzyme Q(10)
production and its applications." Appl Microbiol Biotechnol 85(6): 1653‐1663.
Johns, D. R. (1995). "Seminars in medicine of the Beth Israel Hospital, Boston. Mitochondrial DNA
and disease." N Engl J Med 333(10): 638‐644.
Kawamukai, M. (2009). "Biosynthesis and bioproduction of coenzyme Q10 by yeasts and other
organisms." Biotechnol Appl Biochem 53(Pt 4): 217‐226.

Referências Bibliográficas
87
Kwong, L. K., S. Kamzalov, I. Rebrin, A. C. Bayne, C. K. Jana, P. Morris, M. J. Forster and R. S. Sohal
(2002). "Effects of coenzyme Q(10) administration on its tissue concentrations, mitochondrial
oxidant generation, and oxidative stress in the rat." Free Radic Biol Med 33(5): 627‐638.
Lagier‐Tourenne, C., M. Tazir, L. C. Lopez, C. M. Quinzii, M. Assoum, N. Drouot, C. Busso, S. Makri,
L. Ali‐Pacha, T. Benhassine, M. Anheim, D. R. Lynch, C. Thibault, F. Plewniak, L. Bianchetti, C.
Tranchant, O. Poch, S. DiMauro, J. L. Mandel, M. H. Barros, M. Hirano and M. Koenig (2008).
"ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme
Q10 deficiency." Am J Hum Genet 82(3): 661‐672.
Lalani, S. R., G. D. Vladutiu, K. Plunkett, T. E. Lotze, A. M. Adesina and F. Scaglia (2005). "Isolated
mitochondrial myopathy associated with muscle coenzyme Q10 deficiency." Arch Neurol 62(2):
317‐320.
Lamperti, C., A. Naini, M. Hirano, D. C. De Vivo, E. Bertini, S. Servidei, M. Valeriani, D. Lynch, B.
Banwell, M. Berg, T. Dubrovsky, C. Chiriboga, C. Angelini, E. Pegoraro and S. DiMauro (2003).
"Cerebellar ataxia and coenzyme Q10 deficiency." Neurology 60(7): 1206‐1208.
Land, J. M., S. J. Heales, A. J. Duncan and I. P. Hargreaves (2007). "Some observations upon
biochemical causes of ataxia and a new disease entity ubiquinone, CoQ10 deficiency." Neurochem
Res 32(4‐5): 837‐843.
Liang, W. C., A. Ohkuma, Y. K. Hayashi, L. C. Lopez, M. Hirano, I. Nonaka, S. Noguchi, L. H. Chen, Y.
J. Jong and I. Nishino (2009). "ETFDH mutations, CoQ10 levels, and respiratory chain activities in
patients with riboflavin‐responsive multiple acyl‐CoA dehydrogenase deficiency." Neuromuscul
Disord 19(3): 212‐216.
Littarru, G. P., L. Ho and K. Folkers (1972). "Deficiency of coenzyme Q 10 in human heart disease.
II." Int J Vitam Nutr Res 42(3): 413‐434.
Littarru, G. P. and P. Lambrechts (2011). "Coenzyme Q10: multiple benefits in one ingredient."
OCL 18(2): 76‐82.
Lopez, L. C., M. Schuelke, C. M. Quinzii, T. Kanki, R. J. Rodenburg, A. Naini, S. Dimauro and M.
Hirano (2006). "Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl
diphosphate synthase subunit 2 (PDSS2) mutations." Am J Hum Genet 79(6): 1125‐1129.

Referências Bibliográficas
88
Lowry, O. H., N. J. Rosebrough, A. L. Farr and R. J. Randall (1951). "Protein measurement with the
Folin phenol reagent." J Biol Chem 193(1): 265‐275.
Luft, R., D. Ikkos, G. Palmieri, L. Ernster and B. Afzelius (1962). "A case of severe hypermetabolism
of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control: a
correlated clinical, biochemical, and morphological study." J Clin Invest 41: 1776‐1804.
Mariotti, C., A. Solari, D. Torta, L. Marano, C. Fiorentini and S. Di Donato (2003). "Idebenone
treatment in Friedreich patients: one‐year‐long randomized placebo‐controlled trial." Neurology
60(10): 1676‐1679.
Matthews, R. T., L. Yang, S. Browne, M. Baik and M. F. Beal (1998). "Coenzyme Q10 administration
increases brain mitochondrial concentrations and exerts neuroprotective effects." Proc Natl Acad
Sci U S A 95(15): 8892‐8897.
Mellors, A. and A. L. Tappel (1966). "The inhibition of mitochondrial peroxidation by ubiquinone
and ubiquinol." J Biol Chem 241(19): 4353‐4356.
Mollet, J., A. Delahodde, V. Serre, D. Chretien, D. Schlemmer, A. Lombes, N. Boddaert, I.
Desguerre, P. de Lonlay, H. O. de Baulny, A. Munnich and A. Rotig (2008). "CABC1 gene mutations
cause ubiquinone deficiency with cerebellar ataxia and seizures." Am J Hum Genet 82(3): 623‐630.
Mollet, J., I. Giurgea, D. Schlemmer, G. Dallner, D. Chretien, A. Delahodde, D. Bacq, P. de Lonlay, A.
Munnich and A. Rotig (2007). "Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH‐benzoate
polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation
disorders." J Clin Invest 117(3): 765‐772.
Montero, R., R. Artuch, P. Briones, A. Nascimento, A. Garcia‐Cazorla, M. A. Vilaseca, J. A. Sanchez‐
Alcazar, P. Navas, J. Montoya and M. Pineda (2005). "Muscle coenzyme Q10 concentrations in
patients with probable and definite diagnosis of respiratory chain disorders." Biofactors 25(1‐4):
109‐115.
Montero, R., M. Pineda, A. Aracil, M. A. Vilaseca, P. Briones, J. A. Sanchez‐Alcazar, P. Navas and R.
Artuch (2007). "Clinical, biochemical and molecular aspects of cerebellar ataxia and Coenzyme
Q10 deficiency." Cerebellum 6(2): 118‐122.

Referências Bibliográficas
89
Montini, G., C. Malaventura and L. Salviati (2008). "Early coenzyme Q10 supplementation in
primary coenzyme Q10 deficiency." N Engl J Med 358(26): 2849‐2850.
Morton, R. A. (1958). "Ubiquinone." Nature 182(4652): 1764‐1767.
Musumeci, O., A. Naini, A. E. Slonim, N. Skavin, G. L. Hadjigeorgiou, N. Krawiecki, B. M. Weissman,
C. Y. Tsao, J. R. Mendell, S. Shanske, D. C. De Vivo, M. Hirano and S. DiMauro (2001). "Familial
cerebellar ataxia with muscle coenzyme Q10 deficiency." Neurology 56(7): 849‐855.
Naini, A., V. J. Lewis, M. Hirano and S. DiMauro (2003). "Primary coenzyme Q10 deficiency and the
brain." Biofactors 18(1‐4): 145‐152.
Nass, M. M. and S. Nass (1963). "Intramitochondrial Fibers with DNA Characteristics. I. Fixation
and Electron Staining Reactions." J Cell Biol 19: 593‐611.
Nelson, D. L. and M. M. Cox (2004). Amino Acids, Peptides, and Proteins. Lehninger Principles of
Biochemistry, W. H. Freeman: 75‐111.
Ogasahara, S., A. G. Engel, D. Frens and D. Mack (1989). "Muscle coenzyme Q deficiency in familial
mitochondrial encephalomyopathy." Proc Natl Acad Sci U S A 86(7): 2379‐2382.
Ohkuma, A., S. Noguchi, H. Sugie, M. C. Malicdan, T. Fukuda, K. Shimazu, L. C. Lopez, M. Hirano, Y.
K. Hayashi, I. Nonaka and I. Nishino (2009). "Clinical and genetic analysis of lipid storage
myopathies." Muscle Nerve 39(3): 333‐342.
Ott, W. H., A. M. Dickinson, A. Van Inderstine, A. W. Bazemore, A. C. Page, Jr. and K. Folkers (1958).
"Studies related to vitamin B13." J Nutr 64(4): 525‐531.
Pineda, M., R. Montero, A. Aracil, M. M. O'Callaghan, A. Mas, C. Espinos, D. Martinez‐Rubio, F.
Palau, P. Navas, P. Briones and R. Artuch (2010). "Coenzyme Q(10)‐responsive ataxia: 2‐year‐
treatment follow‐up." Mov Disord 25(9): 1262‐1268.
Quinzii, C., A. Naini, L. Salviati, E. Trevisson, P. Navas, S. Dimauro and M. Hirano (2006). "A
mutation in para‐hydroxybenzoate‐polyprenyl transferase (COQ2) causes primary coenzyme Q10
deficiency." Am J Hum Genet 78(2): 345‐349.
Quinzii, C. M., S. DiMauro and M. Hirano (2007). "Human coenzyme Q10 deficiency." Neurochem
Res 32(4‐5): 723‐727.

Referências Bibliográficas
90
Quinzii, C. M. and M. Hirano (2011). "Primary and secondary CoQ(10) deficiencies in humans."
Biofactors.
Quinzii, C. M., A. G. Kattah, A. Naini, H. O. Akman, V. K. Mootha, S. DiMauro and M. Hirano (2005).
"Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation." Neurology
64(3): 539‐541.
Rahman, S. (2001). "Neonatal presentation of coenzyme Q10 deficiency." The Journal of Pediatrics
139(3): 456‐458.
Rötig, A., E.‐L. Appelkvist, V. Geromel, D. Chretien, N. Kadhom, P. Edery, M. Lebideau, G. Dallner,
A. Munnich, L. Ernster and P. Rustin (2000). "Quinone‐responsive multiple respiratory‐chain
dysfunction due to widespread coenzyme Q10 deficiency." The Lancet 356(9227): 391‐395.
Rotig, A., J. Mollet, M. Rio and A. Munnich (2007). "Infantile and pediatric quinone deficiency
diseases." Mitochondrion 7 Suppl: S112‐121.
Rustin, P., J. Lebidois, D. Chretien, T. Bourgeron, J. F. Piechaud, A. Rotig, A. Munnich and D. Sidi
(1994). "Endomyocardial biopsies for early detection of mitochondrial disorders in hypertrophic
cardiomyopathies." J Pediatr 124(2): 224‐228.
Saiki, R., A. Nagata, T. Kainou, H. Matsuda and M. Kawamukai (2005). "Characterization of
solanesyl and decaprenyl diphosphate synthases in mice and humans." FEBS J 272(21): 5606‐5622.
Sanger, F., S. Nicklen and A. R. Coulson (1977). "DNA sequencing with chain‐terminating
inhibitors." Proc Natl Acad Sci U S A 74(12): 5463‐5467.
Schaefer, A. M., R. W. Taylor, D. M. Turnbull and P. F. Chinnery (2004). "The epidemiology of
mitochondrial disorders‐‐past, present and future." Biochim Biophys Acta 1659(2‐3): 115‐120.
Shapira, Y., S. Harel and A. Russell (1977). "Mitochondrial encephalomyopathies: a group of
neuromuscular disorders with defects in oxidative metabolism." Isr J Med Sci 13(2): 161‐164.
Shy, G. M. and N. K. Gonatas (1964). "Human Myopathy with Giant Abnormal Mitochondria."
Science 145: 493‐496.

Referências Bibliográficas
91
Shy, G. M., N. K. Gonatas and M. Perez (1966). "Two childhood myopathies with abnormal
mitochondria. I. Megaconial myopathy. II. Pleoconial myopathy." Brain 89(1): 133‐158.
Sobreira, C., M. Hirano, S. Shanske, R. K. Keller, R. G. Haller, E. Davidson, F. M. Santorelli, A. F.
Miranda, E. Bonilla, D. S. Mojon, A. A. Barreira, M. P. King and S. DiMauro (1997). "Mitochondrial
encephalomyopathy with coenzyme Q10 deficiency." Neurology 48(5): 1238‐1243.
Tauche, A., U. Krause‐Buchholz and G. Rodel (2008). "Ubiquinone biosynthesis in Saccharomyces
cerevisiae: the molecular organization of O‐methylase Coq3p depends on Abc1p/Coq8p." FEMS
Yeast Res 8(8): 1263‐1275.
Turunen, M., J. Olsson and G. Dallner (2004). "Metabolism and function of coenzyme Q."
Biochimica et Biophysica Acta (BBA) ‐ Biomembranes 1660(1‐2): 171‐199.
Turunen, M., J. Olsson and G. Dallner (2004). "Metabolism and function of coenzyme Q." Biochim
Biophys Acta 1660(1‐2): 171‐199.
Villalba, J. M., C. Parrado, M. Santos‐Gonzalez and F. J. Alcain (2010). "Therapeutic use of
coenzyme Q10 and coenzyme Q10‐related compounds and formulations." Expert Opin Investig
Drugs 19(4): 535‐554.
Wallace, D. C., G. Singh, M. T. Lott, J. A. Hodge, T. G. Schurr, A. M. Lezza, L. J. Elsas, 2nd and E. K.
Nikoskelainen (1988). "Mitochondrial DNA mutation associated with Leber's hereditary optic
neuropathy." Science 242(4884): 1427‐1430.
Wallace, K. B., J. T. Eells, V. M. Madeira, G. Cortopassi and D. P. Jones (1997). "Mitochondria‐
mediated cell injury. Symposium overview." Fundam Appl Toxicol 38(1): 23‐37.
Wong, L. J. and R. G. Boles (2005). "Mitochondrial DNA analysis in clinical laboratory diagnostics."
Clin Chim Acta 354(1‐2): 1‐20.
Zeviani, M., E. Bonilla, D. C. DeVivo and S. DiMauro (1989). "Mitochondrial diseases." Neurol Clin
7(1): 123‐156.
Zeviani, M. and S. Di Donato (2004). "Mitochondrial disorders." Brain 127(Pt 10): 2153‐2172.


Anexos


Anexos
95
8. Anexos
8.1. Resumo dos resultados das actividades enzimáticas da CRM obtidos neste estudo
Caso CS
(nmol/min/mg PNC)
CII‐III (nmol/min/mg
PNC/CS)
Restantes complexos da
CRM
1 171 1.1 normais
2 115 1.7 Normais
3 57 2.0 Normais
4 242 1.7 normais
5 179 1.8 normais
6 149 0.9 normais
7 86 0.9 normais
8 149 1.6 normais
9 188 1.6 normais
10 110 1.9 normais
11 140 1.8 normais
12 265 1.7 CIV (48%)
13 253 2.0 CIV (41%)
14 185 1.7 CIV (45%)
15 184 1.8 normais
16 125 1.9 normais
17 167 1.6 normais
18 137 1.7 CIV (40%)
19 234 1.4 normais
20 172 1.6 normais
21 163 1.8 CIV (43%)
22 178 1.7 normais
23 149 1.6 normais
24 272 1.8 normais
25 137 5.6 normais
Valores de referência: CS‐85.0‐180.0 nmol/min/mg PNC; CII‐III‐2.6‐12.0 nmol/min/mg PNC/CS
PNC‐ proteínas não colagénicas

Anexos
96
8.2. Determinação das Actividades da CS e do CII‐III da CRM no músculo
‐ Determinação da Actividade da Citrato Sintetase (CS)
1. Interesse Clínico
A citrato sintetase é uma enzima do ciclo de Krebs localizada na matriz mitocondrial que reflecte o potencial redox da célula. Uma alteração na sua actividade pode sugerir uma citopatia mitocondrial.
2. Produto
Músculo deltóide / fígado / outro
O músculo é o tecido preferencialmente investigado, devido ao seu alto metabolismo energético e ao facto de se encontrar quase invariavelmente afectado nas encefalopatias mitocondriais.
3. Amostra
Homogeneizado do fragmento muscular/hepático.
*quantidade ideal – 100 mg
*critérios de rejeição de amostras – má conservação, má colheita, a adição de qualquer tipo de conservante.
3.1. Interferências
Presença na amostra de tecido adiposo, parafina, outros.
3.2. Colheita/transporte/conservação das amostras
3.2.1. Colheita – Biópsia muscular/hepática de um fragmento de cerca de 100 mg para tubo seco. Esta colheita é efectuada no serviços de neuropatologia, anatomia patológica ou cirurgia dos hospitais.
3.2.2. Transporte – Neve carbónica/azoto líquido.
3.2.3. Conservação das amostras – Armazenar em arca a ≈ –70 °C, evitando descongelações.
Anexos
97
4. Reagentes
Os reagentes assinalados poderão ser substituídos por outros equivalentes, desde que devidamente testados e que fique registado internamente, junto com os ensaios a data da alteração (se a mesma for temporária).
5. Preparação da Amostra
A amostra deve ser mantida em gelo durante a sua preparação e manipulação (inclusivamente no transporte da arca até à balança).
• Pesar cerca de 70 mg do fragmento congelado.
• Adicionar ao fragmento o tampão fosfato a 10mmol/l pH 7.4 na proporção 1/15.
• Triturar a amostra no homogeneizador.
• Transferir o homogeneizado para um tubo de 1.5 ml.
• Centrifugar a 6000 rpm durante 10 min a 4 °C.
• Transferir o sobrenadante para outro tubo (para proceder ao ensaio) e congelar o precipitado (para extracção de DNA se necessário).
6. Controlo de qualidade interno (CQI)
6.1. Material
Fragmento músculo/fígado com actividades normais.
6.2. Preparação e armazenamento
Ver preparação e conservação da amostra.
Referência Marca Acondicionamento
Tris 1.08382.0500 Merck TA
HCL 1.00317.1000 Merck TA
DTNB D‐8130 Sigma TA
Acetil CÔA 10101907001 Roche 4ºC
Triton X 92H0250 Sigma TA
Oxaloacetato O‐4126 Sigma ˜ ‐20ºC
KH2PO4 2389213 Merck TA
K2HPO4 1.05099.0250 Merck TA
Água Bidestilada 2652998 Labesfal TA

Anexos
98
CS
6.3. Procedimento
O controlo é processado em paralelo com a série de amostras segundo o mesmo protocolo.
6.4. Critérios de aceitação
A actividade da CS tem de estar dentro dos valores de referência.
7. Método
7.1. Descrição do método
A determinação da CS baseia‐se na cinética desta enzima. A CS catalisa a redução de Acetil‐CoA sintetizando citrato. A Acetil‐CoA reduzida (CÔA‐SH) reage com o reagente de cor (DTNB) sendo a variação da Absorvância devida à formação do produto formado medida espectrofotometricamente.
Acetil‐CoA + oxaloacetato → citrato + CoA‐SH
DTNB + 2 CoA‐SH → 2TNB+CoASSCoA
7.2. Procedimento
• Pipetar para as cuvetes segundo o quadro seguinte:
Reagentes Volume (µl) Concentração final
Tampão Tris HCL 750mM pH8 100 75 mM
DTNB 1mM 100 0.1 mM
H2O 760 ‐‐
Acetil‐CoA 8.5 mM 50 0.42
Amostra / CQI 5 ‐‐
Triton X 100 1% 100 1‰
• Deixar estabilizar a absorvância a 30 °C a 412 nm. Depois adicionar:
Oxaloacetato 10 mM 100 0.5 mM
• Ler a 412nm a 30 °C no programa VisionPro ou PC160PLS (a descrição dos programas está disponível em anexo).

Anexos
99
8. Cálculo de Resultados
As actividades enzimáticas são expressas em nanomoles de substrato transformado por minuto e por mg de proteína, nas condições de reacção escolhidas, de forma a obter uma cinética que é linear em função do tempo.
A actividade específica exprime‐se por proteínas não colagénicas (NCP) presentes no músculo/fígado.
AC = ∆A x Vt x 1 x 1 AC – actividade enzimática
∆t Va ac x d NCP
∆A/∆t ‐ variação da absorvância por minuto
Vt ‐ volume do meio de reacção; Va ‐ volume da amostra; d ‐ trajecto óptico em metros
aC ‐ absorvância = m2 .mole‐1
concentração x d
Unidades/factores de conversão:
ac = 1 = Abosvância
mole.l‐1 x mm concentração x d
ac = litro (1dcm3) = 0,1 m x 0,1 m x 0,1 m
mole x mm (0,001 m) mole x 0,001 m
ac = m2 = m2. mole‐1
mole
NCP : mg.l‐1
Se os factores da fórmula forem expressos nas unidades internacionais o valor de U será respectivamente:
U = (A). min‐1 x 1 x 1
l.mole‐1 . mm‐1 . m mg.l‐1
U = mole. min‐1 x l‐1 x mm x m‐1 x mg‐1 x l
U = 109 nmol x min‐1 x 10‐3m x m‐1 x mg‐1
U = 106 nmole x min‐1 x mg‐1
9. Valores de referência
85.0 – 180.0 nmol/min/mg PNC/CS (em músculo esquelético deltoide)
10. Critérios de Confirmação de Resultados
As amostras são processadas em duplicado no mesmo dia por dois operadores.

Anexos
100
O resultado inicial é confirmado sempre que se encontre fora dos valores de referência. A confirmação é efectuada em nova amostra do mesmo fragmento. Se este for insuficiente solicita‐se mais material biológico ao serviço que efectuou a biópsia e caso se justifique nova biópsia.
11. Interpretação de resultados
Uma actividade diminuída de citrato sintetase está associada a má conversação/transporte /manipulação da amostra. Uma actividade aumentada é muitas vezes indicadora de proliferação mitocondrial e pode sugerir uma patologia mitocondrial.
‐ Determinação da actividade da succinato citocromo c redutase (Complexo II‐III)
1. Interesse clínico
A determinação da actividade de complexos conjugados tem como finalidade esclarecer situações em que as actividades dos complexos isolados, neste caso os CII e III, estão no limite inferior dos valores de referência. Um défice do CII+III isolado poderá igualmente indicar um défice de ubiquinona, a sua determinação é bastante relevante já que o tratamento nestes casos, a administração de coenzima Q10, é eficaz.
2. Produto
Igual à determinação da CS.
3. Amostra
Igual à determinação da CS.
4. Reagentes
Referência Marca Acondicionamento
Citocromo c C‐3131 Sigma ‐ 20 °C
NaCN S‐3296 Sigma TA
KH2PO4 2389213 Merck TA
K2HPO4 1.05099.0250 Merck TA
Succinato de sódio 8.20151.01000 Merck TA
Água Bidestilada 2652998 Labesfal TA, Ultra Pura
Anexos
101
5. Controlo de qualidade interno (CQI)
Igual à determinação da CS.
6. Método
6.1. Descrição do método
A determinação da actividade do CII+III resulta da cinética da enzima succinato citocromo c redutase. A primeira reacção, apresentada no esquema, representa a redução da CoQ pelos equivalentes reduzidos do succinato, transferidos pelo CII. A redução do citocromo c pelos electrões da CoQ, transferidos pelo CIII, é medida e proporcional à actividade do CII+III.
Succinato → CoQ H2 → citocromo c
(2H+, 2e‐) CII CIII
6.2. Procedimento
• Pipetar para as cuvetes segundo o quadro seguinte:
Volume (µL) Concentração
Tampão fosfato 100 mM pH 7,4 550 55 mM
Citocromo c 1 mM 100 0,1 mM
NaCN 30 mM 30 0,9 mM
Amostra/CQI 20 ‐
H2O 200 ‐
• Estabilizar a absorvância a 30 °C a 550 nm
Succinato de sódio 30 mM 100 3 mM
• Ler a 550 nm a 30 °C no programa VisionPro ou PC160PLS (a descrição dos programas está disponível em anexo).
7. Cálculo de resultados
O cálculo da actividade do CII+III é idêntico ao da CS. A actividade do complexo deve ser expressa em relação a da CS.
Exemplo: CII‐III: 10 U CS: 180 U (100%)
Actividade/CS= 10/180 x 100 = 5.6

Anexos
102
8. Valores de referência
2.6‐12.0 nmol/min/mg PNC/CS (em músculo esquelético deltóide).
9. Interpretação dos resultados
Um défice isolado do complexo conjugado II +III está associado a um défice de ubiquinona. Os défices do CII e CIII poderão/deverão ser acompanhados de uma diminuição da actividade complexo II+III.
8.3. Extracção de DNA de biópsia e/ou homogeneizado muscular (Puregene® Tissue Kit ‐
GENTRA)
Preparação da amostra
Triturar aproximadamente 20 mg de músculo fresco ou congelado a ‐70 °C. Também podem ser utilizados os pellets dos homogeneizados usados para o estudo enzimático da CRM. Durante a manipulação manter o músculo em gelo.
Lise Celular
1. Colocar o músculo previamente triturado ou o homogeneizado num eppendorf de 1,5 mL. 2. Adicionar 500 µL de Cell Lysis Solution e homogeneizar, utilizando uma vareta de vidro e
vortex. 3. Adicionar 3 µL de Proteinase K Solution (20 mg/mL)(1). Misturar, invertendo o tubo várias
vezes. 4. Incubar a 55 °C durante a noite, até a solução se tornar homogénea. Se possível inverter o
tubo periodicamente e agitar no vortex, durante a incubação.
Tratamento com RNAse
1. Juntar 3 µL de RNAse Solution (4 mg/mL) (2) à amostra lisada. 2. Misturar a amostra invertendo o tubo várias vezes e incubar a 37 °C durante 15 a 60
minutos.
Precipitação de proteínas
1. Arrefecer a amostra à temperatura ambiente. 2. Adicionar 200 µL de Protein Precipitation Solution à amostra tratada. 3. Agitar no vortex durante 20 segundos e de seguida colocar no gelo 5 minutos. 4. Centrifugar a 14000 rpm durante 3 minutos, a 4 °C. O precipitado de proteínas forma um
fino pellet. Caso não se observe esse precipitado repetir os passos 3 e 4.

Anexos
103
Precipitação de DNA 1. Transferir o sobrenadante, que contém o DNA, para um eppendorf de 1,5 ml contendo
600 µL de Isopropanol a 100% (3). 2. Inverter o tubo lentamente várias vezes. 3. Centrifugar a 14000 rpm, durante 5 minutos, a 4 °C. 4. Rejeitar o sobrenadante e secar o pellet com papel absorvente limpo. 5. Adicionar 600 µl de Etanol a 70% e inverter o tubo várias vezes para ressuspender o DNA. 6. Centrifugar a 14000 rpm, durante 1 minuto, a 4 °C. 7. Decantar o etanol cuidadosamente, invertendo o tubo, de forma a não perder o pellet. 8. Secar com papel e deixar à temperatura ambiente o tempo suficiente para secar.
Hidratação do DNA
1. Adicionar 20 a 100 µL(4) de DNA Hydration Solution, de acordo com o rendimento obtido. 2. Incubar a 65 °C durante 1 hora ou durante a noite à temperatura ambiente, para
completar a rehidratação. Agitar o tubo periodicamente para dispersar o DNA.
Armazenar o DNA a 2‐8 °C ou de ‐20 °C a ‐80 °C.
Observações e Reagentes:
(1) Proteinase K Solution (20 mg/mL), Promega
(2) RNAse Solution (4 mg/mL), Quiagen
(3) Isopropanol a 100%, Sigma
(4) se utilizar pellet de biopsia muscular para extrair DNA, adicionar 20 a 50 µL de DNA Hydration Solution e 50 a 100 µL da solução de hidratação quando partir de tecido muscular.

Anexos
8.3. Pre
PDSS1,
Predicti
8.3.1. c.
Figura 3alteração
evisão dos
COQ2 e AD
ion.
.1‐29C>T
1‐ Previsão o c.1‐29C>T
locais de s
DCK3, atrav
dos locais de(B) através d
splicing pa
vés dos pro
e splicing pado programa
104
ra as nova
ogramas bio
A
B
ara a região 5a NetGene 2.
as alteraçõe
oinformátic
5`UTR do ge
es identific
cos NetGen
ene PDSS1 no
cadas nos g
e 2 e Splice
ormal (A) e c
genes
e Site
com a

Figuraltera
8.3.2
ra 32‐ Previsação c.1‐29C
2. c.721+4C>
ão dos locaiC>T (B) atravé
>T
s de splicingés do progra
105
A
B
g para a regiãama Splice S
A
ão 5`UTR doSite Predictio
o gene PDSS1on.
1 normal (A)
Anexos
) e com a

Anexos
Figura 3e com a
Figura 3e com a
3‐ Previsão dalteração c.7
4‐ Previsão dteração c.72
dos locais de721+4C>T (B
dos locais de21+4C>T (B) a
e splicing paB) através do
e splicing paatravés do p
106
B
ra a região e programa N
A
B
ra a região eprograma Spl
exão7 – intrãNetGene 2.
exão7 – intrãlice Site Pred
ão7 do gene
ão7 do gene diction.
PDSS1 norm
PDSS1 norm
mal (A)
mal (A)

8.3.3
Figurcom a
3. c.693‐136
ra 35‐ Previsãa alteração c
6A>G
ão dos locaisc.693‐136A>
s de splicingG (B) através
107
A
B
para a regiãos do program
o exão4 – intma NetGene
trão4 do gen2.
ne COQ2 nor
Anexos
rmal (A) e

Anexos
Figura 3com a al
6‐ Previsão dteração c.69
dos locais de93‐136A>G (B
e splicing parB) através do
108
A
B
ra a região exo programa S
xão4 – intrãoSplice Site Pr
o4 do gene Crediction.
COQ2 normal (A) e

8.3.4
Figurnorm
. c.1660‐81
ra 37‐ Previsãmal (A) e com
C>G
ão dos locaim a alteração
s de splicingc.1660‐81C>
109
A
B
g para a regiã>G (B) atravé
ão exão15 –és do progra
– intrão15 doma NetGene
o gene ADCKe 2.
Anexos
K3/CABC1

Anexos
Figura 38e com a a
8‐ Previsão doalteração c.16
os locais de sp660‐81C>G (B)
plicing para a r) através do p
110
A
B
região exão15rograma Splic
5 – intrão15 dce Site Predicti
o gene ADCK3ion.
3/CABC1 norm
mal (A)