Embed Size (px)
Citation preview

UNIVERSIDADE FEDERAL FLUMINENSE
INSTITUTO DE QUÍMICA GRADUAÇÃO DE BACHAREL EM QUÍMICA INDUSTRIAL
DALISSA GRANJA VILLA NOVA
DETERMINAÇÃO DE Cd (II) EM ÁGUAS PRODUZIDAS ORIUNDAS DA EXPLORAÇÃO DE PETRÓLEO EMPREGANDO SISTEMA PARA SEPARAÇÃO DA MATRIZ UTILIZANDO MEMBRANAS SEMIPERMEÁVEIS DE POLIETILENO
DE BAIXA DENSIDADE.
NITERÓI 2016

DALISSA GRANJA VILLA NOVA
DETERMINAÇÃO DE Cd (II) EM ÁGUAS PRODUZIDASORIUNDAS DA EXPLORAÇÃO DE PETRÓLEO EMPREGANDO SISTEMA PARA
SEPARAÇÃODA MATRIZ UTILIZANDO MEMBRANAS SEMIPERMEÁVEIS DE POLIETILENO DE BAIXA DENSIDADE.
Monografia de final de curso apresentada ao Curso de Graduação em Química Industrial da Universidade Federal Fluminense como requisito parcial para obtenção do Grau de Bacharel em Química Industrial.
Orientador: Prof. Dr. Ricardo Jorgensen Cassella
Coorientador: Drª. Nicolle Figueira Robaina
Niterói, RJ 2016

N 935 Nova, Dalissa Granja Villa
Determinação de Cd (II) em águas produzidas oriunda da
exploração de petróleo empregando sistema para separação da
matriz utilizando membranas semipermeáveis de polietileno de
baixa densidade./ Dalissa Granja Villa Nova.--Niterói : [s.n.],
2016.
53.
Trabalho de Conclusão de Curso--(Bacharelado em
Química Industrial)--Universidade Federal Fluminense, 2016.
1. Petróleo. 2. Salinidade. 3. Impacto ambiental. 4. Especto-
metria. 5. Otimização. 6. Niterói,RJ. 7. Polietileno. I. Título.
CDD. : 547.83


Dedico essa monografia ao meu avô Sebastião, que tenho certeza, que esteve comigo em toda minha jornada. E sempre estará.
IV

AGRADECIMENTOS
Aos meus pais, Luiz Alberto e Domarcia, e minha irmã Larissa, pelo
carinho, confiança e dedicação.
Ao meu orientador, Ricardo Cassella, pela oportunidade de trabalhar e
aprender, durante esses três anos, com um dos melhores professores que já
tive.
A minha maravilhosa coorientadora, Nicolle Robaina, por todo carinho,
paciência e conhecimento transmitido. Sem você teria sido muito mais difícil!
A todos que passaram pelo laboratório LESPA.
A antiga e nova geração do 901, Ana Clara Reis, Camila Novo, Pedro
Rodrigues, Renata Dantas e Marcele Quevene, que suportaram todo o
estresse e felicidade durante a faculdade e que tornaram tudo mais fácil! Vocês
foram a família que escolhi. Amo vocês.
Aos meus amigos, Caio Pequeno, Caio Guedes, Mayara Müller, Willian
Guedes, que estavam comigo antes mesmo dessa jornada começar e me
ajudaram muito a estar aqui!
Aos amigos maravilhosos que eu fiz aqui e que tornaram essa fase
melhor e mais leve! Vocês estarão para sempre comigo. Aos que me ajudaram
me mandando estudar: Obrigada! E também aos que me atrapalharam me
chamando pra sair: Obrigada, e mesmo assim eu consegui! Amo vocês, Ana
Cecília, Carinne Borges, Carolina Bispo, Claudia Moreira, Claudio Gabriel,
Fabiana Melo, Gian Giordano, Isabella Alves, Isabela Oliveira, Israel Rocha,
Jaqueline Freitas, Jorge Roque, Karina Medeiros, Karla Saboya, Leandro
Machado, Leonardo Gomes, Leonardo Megliorini, Luiz Felipe Santoro, Maria
Carolina, Natália Couto, Pedro Diniz, Rafael Lamarca, Raul Xavier, Ricardo
Rito, Rodrigo Sarcinelli, Samir Costa, Thiago Reitor, Victor Santos, Vitor Keller,
Wesley Costa.
V

SUMÁRIO
Resumo ............................................................................................................................................. VIII
Abstract ................................................................................................................................................ IX
Lista de figuras....................................................................................................................................X
Lista de tabelas ............................................................................................................................... XII
1. Introdução ......................................................................................................................................13
2. Objetivos .........................................................................................................................................16
2.2. Objetivo geral .............................................................................................................16
2.3. Objetivo específico ..................................................................................................16
3. Fundamentação teórica ..........................................................................................................18
3.1. Petróleo.........................................................................................................................18
3.2. Extração do petróleo ..............................................................................................19
3.3. Água produzida .........................................................................................................20
3.4. Contaminação das águas .................................................................................... 20
3.5. Cádmio ..........................................................................................................................22
3.6. Dispositivo de membrana semipermeável .................................................. 23
3.7. Espectometria de absorção atômica em forno de grafite
25
4. Parte experimental..................................................................................................................... 27
4.1. Materiais e reagentes ............................................................................................ 27
4.1.1. Instrumentação ........................................................................................27
4.1.2. Reagentes ................................................................................................ 27
4.1.3. Materiais .....................................................................................................28
VI

4.2. Metodologia ................................................................................................................ 29
4.2.1. Preparo de soluções ............................................................................. 29
4.2.2. Otimização do método de extração............................................... 29
4.2.3. Aplicação do método de extração otimizado em amostras
reais de águas salinas ...................................................................................... 31
5. Resultado e discussão ............................................................................................................. 32
5.1. Otimização do método de extração ................................................................ 32
5.1.1. Solvente extrator .................................................................................... 33
5.1.2. Concentração do complexante ........................................................ 34
5.1.3. Tempo de extração ............................................................................... 36
5.1.4. Influencia do pH ...................................................................................... 38
5.1.5. Volume do solvente extrator .............................................................40
5.1.6. Influencia da força iônica .................................................................... 41
5.2. Aplicação do método otimizado em amostras reais .......................... 43
6. Conclusão ......................................................................................................................................48
7. Bibliografia .....................................................................................................................................49
VII

RESUMO
As águas produzidas oriundas da exploração de petróleo são amostras
de água com elevada salinidade e que muitas vezes são despejadas no
oceano sem o tratamento adequado, podendo acarretar na sua contaminação
por diversas substâncias, como por exemplo, o cádmio, um metal tóxico e
cumulativo. As águas salinas são amostras complexas, particularmente difíceis
de analisar, sendo importante o desenvolvimento de métodos de preparo de
amostra que permitam a separação do analito da matriz e que agreguem
vantagens como baixo custo, simplicidade e limites de quantificação
adequados. O presente trabalho propõe uma metodologia analítica para
determinação de Cd (II) em amostras de águas salinas empregando um
sistema de separação da matriz utilizando membranas semipermeáveis de
polietileno de baixa densidade (DMSP) e determinação espectrométrica através
de absorção atômica em forno de grafite (GF AAS). O processo de extração de
Cd (II) das amostras é baseado na sua conversão em uma espécie de baixa
polaridade, através da sua complexação com dietilditiocarbamato de sódio
(DDTC) e posterior migração para o interior na membrana preenchida com um
solvente extrator apolar. Foram otimizadas as condições de extração dos
analitos: pH = 9,0, volume de 3 mL de clorofórmio, DDTC 3,7 x 10-4 mol L-1 e
extração realizada após um tempo de 2 h. Os limites de detecção e
quantificação para Cd foram de 0,15 μg L-1 e 0,52 μg L-1 e testes de
recuperação com percentuais entre 88 e 133% demonstraram a exatidão do
método proposto. A metodologia foi aplicada em uma amostra de água do mar
proveniente do município de Niterói-RJ, uma amostra de água produzida
sintética preparada em laboratório e duas amostras de água produzida
fornecidas pelo CENPES/Petrobras.
Palavras-chave: Águas salinas, extração, cádmio, membrana semipermeável,
petróleo.
VIII

ABSTRACT
The water produced from petroleum exploration present high salinity and
are often discarded in the ocean without a suitable treatment, which can result
in the ocean contamination by various substances, such as cadmium, a toxic
and cumulative metal. Saline waters are particularly difficult to analyze and, for
this reason, the development of sample preparation methods for the matrix
separation is required. These methods must present low-cost, simplicity and
adequate limits of quantification. This work proposes a novel analytical
methodology for Cd(II) determination in saline water samples using a low
density polyethylene semi-permeable membrane device (SPMD) for analyte
separation. Its determination was performed by graphite furnace atomic
absorption spectronmetry. The extraction of Cd(II) ions was based on their
conversion into a low-polarity complex with sodium diethyldithiocarbamate
(DDTC) and subsequent migration into the membrane filled with a nonpolar
solvent. The extraction conditions were optimized and best results were
observed at pH = 9.0, using a 3 mL of chloroform to fill the membrane, DDTC of
3.7 x 10-4 mol L-1 and an extraction time of 2 h. The limits of detection and
quantification for Cd(II) were 0.15 µg g-1 and 0.52 µg g-1, respectively.
Recovery tests were performed and recovery percentages between 88 and
133% were obtained, which demonstrated the accuracy of the proposed
method. The developed methodology was applied in the determination of Cd(II)
in three saline water samples (one seawater sample and two production water
samples, supplied by CENPES/Petrobras).
Keywords: Saline water, extraction, cadmium, semipermeable membrane, oil.
IX

LISTA DE FIGURAS
Figura 1. Sistema de amostragem com membrana semipermeável preenchida
com solvente (Robaina, 2015). .................................................................................................23
Figura 2. Gráfico comparativo da eficiência de extração entre hexano e
clorofórmio como solvente extrator influente no processo de extração de Cd(II).
32
Figura 3. Estrutura química do complexo Cd(DDTC)2. ................................................33
Figura 4. Gráfico do efeito das concentrações de DDTC na eficiência da
extração de Cd(II). Foi usado 1 μg L-1 de padrão de Cd e 1 hora de agitação na
mesa agitadora. ................................................................................................................................34
Figura 5. Gráfico do tempo de extração de Cd (II) sem agitação. Foi usado 1
μg L-1 de padrão de Cd e DDTC 3,7x10-4 mol L-1. ....................................................... 35
Figura 6. Gráfico do tempo de extração, em minutos, de Cd (II) com agitação
em mesa agitadora horizontal. Foi usado 1 μg L-1 de padrão de Cd e DDTC
3,7x10-4 mol L-1. ............................................................................................................................. 36
Figura 7. Equilíbrio do ácido dietilditiocarbamato com seu respectivo valor de
pKa à 19°C de acordo SOTO et al., 2015. .......................................................................... 37
Figura 8. Gráfico da influência do pH no meio de extração de Cd (II). Foi usado
1 μg L-1 de padrão de Cd, DDTC 3,7x10-4 mol L-1, 2h de agitação em mesa
agitadora horizontal e solução tampão Britton-Robinson em pH proposto. ........ 38
Figura 9. Reações de decomposição do DDTC com formação de (A) dissulfeto
de carbono e de (B) sulfeto de hidrogênio a partir do DDTC..................................... 39
Figura 10. Gráfico da influência do volume do solvente extrator. Foi usado 1 μg
L-1
de padrão de Cd, DDTC 3,7x10-4
mol L-1
, 2h de agitação em mesa agitadora
horizontal e solução tampão Britton-Robinson em pH 9,0. ......................................... 40
Figura 11. Gráfico da influência da força iônica do meio na influência da
extração. Foi usado 1 μg L-1 de padrão de Cd, DDTC 3,7x10-4 mol L-1, 2h de
X

agitação em mesa agitadora horizontal e solução tampão Britton-Robinson em
pH 9,0. ................................................................................................................................................. 41
Figura 12. Curvas analítica e de adição padrão da amostra de água produzida
em laboratório 96,097‰. ............................................................................................................ 44
Figura 13. Curvas analítica e de adição padrão da amostra de água produzida
270‰. ................................................................................................................................................... 44
XI

LISTA DE TABELAS
Tabela 1. Programa de temperatura da absorção atômica em forno de grafite
para determinação de cádmio (Robaina, 2015). .............................................................. 32
Tabela 2. Concentrações de DDTC usadas para estudo da otimização do
método. .................................................................................................................................................35
Tabela 3. Condições ótimas de extração e pré-concentração de Cd (II) de
águas salinas através de membrana semipermeável. .................................................. 43
Tabela 4. Parâmetro de mérito para determinação de Cd (II) pelo método
proposto. ..............................................................................................................................................44
Tabela 5. Resultados de recuperação de Cd (II) através do método
padronizado. ......................................................................................................................................47
XII

1. INTRODUÇÃO
Desde sua descoberta em território nacional, o petróleo transformou
profundamente a economia, a sociedade e o espaço do Brasil, principalmente nas
últimas quatro décadas, fornecendo energia e matérias-primas para o processo de
industrialização (Monié, 2003), gerando além de crescimento econômico, muitos
problemas ambientais, como a poluição de águas e de solos.
O petróleo é o recurso natural mais importante na matriz energética atual,
sendo de interesse das nações investirem em pesquisa e exploração deste, mesmo
que isso signifique gerar impactos ambientais. Esse desenvolvimento do setor
petrolífero tem causado danos irreparáveis ao meio ambiente, ou que exigiriam um
alto investimento para reparar a agressão ambiental, como é o caso da Baía de
Guanabara (Silva, 2008).
Na exploração de petróleo, seja em jazidas em terra (onshore) ou em alto mar
(offshore), existe geração de um efluente aquoso, denominado água produzida. A
chamada água produzida é extraída junto com o petróleo e é oriunda principalmente
da injeção de água para aumentar a produção de petróleo na etapa de recuperação
secundária, onde essa injeção mantém a pressão necessária no reservatório para
que o óleo seja extraído do poço. O processo de recuperação secundária exige um
volume muito grande de água, que geralmente é a própria água do mar coletada
próxima à região da exploração, e após a extração do petróleo é descartada em alto
mar, sendo considerado o maior resíduo, em termos de volume, gerado na produção
de petróleo. A quantidade de rejeito de água produzida gerada em uma atividade
offshore no Brasil está estimada em aproximadamente 3,8 milhões de barris por dia
(Nunes, 2010). Valor este que depende das dimensões do reservatório, da área
explorada e da capacidade da unidade exploradora.

A produção excessiva de água durante a exploração de petróleo é um
problema principalmente nos campos de petróleo denominados maduros, isto é,
aqueles que têm permanecido em operação por longo período de tempo. A eficiência
do separador água/óleo é afetada com a perda da pressão dentro dos poços, tendo
a necessidade de injetar cada vez mais água nos reservatórios (Lima, 2008).
As águas produzidas apresentam, em geral, altos teores de contaminantes
potencialmente tóxicos (metais, tais como Cd, Cr, Cu, Pb, Hg, Ag, Ni, Zn; produtos
químicos adicionados nos reservatórios, tais como inibidores de corrosão, inibidores
de incrustação, desemulsificantes, metanol, glicol, polieletrólitos), além de uma
complexa mistura de compostos orgânicos e inorgânicos, cuja composição varia com
a vida do campo petrolífero (Lima, 2008).
Na atividade offshore o descarte é feito em grandes ambientes receptores,
como o próprio mar, o que poderia ser uma argumentação para o não tratamento da
água produzida. Mas fatores tais como, correntes marítimas, ventos, temperatura da
água, mudança de clima, podem transportar ou mesmo concentrar alguns de seus
constituintes. O descarte de tais volumes de resíduos vem causando preocupações
sobre a poluição ambiental não controlada e irreversível no ambiente marinho (Lima,
2008).
O impacto ambiental é avaliado pela toxicidade dos constituintes e pela
quantidade de compostos presentes. Alguns destes constituintes permanecerão
dissolvidos, enquanto outros são convertidos, seja por decomposição, evaporação,
transformação em outro composto não tóxico, deposição no fundo do mar, etc. Os
efeitos mais nocivos ao meio ambiente são aqueles associados aos compostos que
permanecem solúveis após o descarte, por interagirem diretamente com a vida
presente neste meio (Lima, 2008).
Os metais presentes na água produzida são elementos naturais que se
encontram nos ecossistemas, porém, as atividades humanas têm elevado suas
concentrações naturais, levando a contaminação dos ecossistemas (Almeida, 2009).
Os metais merecem atenção por serem não degradáveis, permanecendo por longos
14

períodos no ambiente, além da elevada toxicidade de alguns metais, o que cria
problemas em zonas costeiras, lagos e rios (Boada et al., 2007).
Com as exigências ambientais, é necessário o monitoramento das
concentrações dos diversos poluentes que a produção de petróleo possa trazer ao
meio ambiente que o cerca. A partir disso, cresce, no mundo, pesquisas que
atendam a essa necessidade, principalmente na área da química analítica, utilizando
diversas técnicas instrumentais, por exemplo, Espectrometria de Absorção Atômica
com Forno de Grafite (GF AAS, Graphite Furnace Atomic Absorption Spectrometry),
uma técnica altamente sensível e seletiva para detectar pequenas concentrações de
metais, além de usar pequenas quantidades de amostras.
Nesse contexto, destaca-se a relevância do presente trabalho, que tem como
objetivo o desenvolvimento de um método analítico para determinação de Cd em
amostras de água salina utilizando membranas semipermeáveis para separação da
matriz complexa. No método são utilizadas membranas de polietileno de baixa
densidade preenchidas com solvente apolar e a extração do Cd é realizada pela sua
conversão em uma espécie de baixa polaridade, através da reação de complexação
do Cd com o complexante DDTC.
15

2 OBJETIVOS
2.1 OBJETIVO GERAL
Desenvolvimento de um método de extração para determinação de Cd (II) em
águas salinas, águas do mar e águas produzidas oriundas da exploração offshore,
através de membranas semipermeáveis de polietileno de baixa densidade
preenchidas com solvente orgânico e detecção por espectrometria de absorção
atômica com forno de grafite (GF AAS).
2.2 OBJETIVOS ESPECÍFICOS
i. Otimizar os parâmetros para melhor eficiência da extração de Cd (II):
a. Tipo de solvente extrator;
b. Concentração do complexante (DDTC);
c. Tempo de exposição das membranas;
d. Agitação do sistema;
e. pH do meio;
f. Volume do solvente extrator;

g. Força iônica; ii. Aplicar a metodologia de extração estudada em amostras salinas reais,
utilizando espectrometria de absorção atômica em forno de grafite.
17

3 FUNDAMENTAÇÃO TEÓRICA
3.1 PETRÓLEO
O interesse pelo petróleo, do ponto de vista econômico, começou no século
XIX, quando ele passou a ser utilizado para produção de energia no lugar do carvão
mineral. Quando Thomas Edison desenvolveu os estudos acerca da energia elétrica,
o petróleo perdeu sua função, porém, no século XX, com a invenção dos motores a
gasolina e diesel, o petróleo voltou ao cenário mundial (Debeir, 1993).
O país pioneiro nas pesquisas sobre petróleo foram os EUA, mas anos mais
tarde o Brasil iria descobrir que a maior parte dos seus reservatórios de petróleo se
encontrava no fundo do mar e não em terra, como nos demais países (Freeman e
Soete, 1997). Sendo assim, os EUA são pioneiros em técnicas onshore, e o Brasil,
através da Petrobrás por intermédio de seu Programa de Capacitação Tecnológica
em Águas Profundas - PROCAP, criado em 1986, desenvolveu as técnicas offshore,
o que a tornou referência mundial em tecnologia de exploração de petróleo em
águas profundas (Neto e Costa, 2007).
O petróleo é constituído de hidrocarbonetos (compostos ricos em carbono e
hidrogênio) saturados, aromáticos, compostos policíclicos, além de elementos
inorgânicos (Souza et al., 2015). A principal teoria de formação do petróleo é
baseada na deposição de um volume alto de produtos orgânicos em uma rocha
específica, denominada geradora, que possui pressão e temperatura adequada para
a formação do óleo, e estar livre de uma possível oxidação, que destrói as ligações
C-H com a ação do oxigênio (Milani, 2000). Essa formação requer um alto resquício
geológico ocorrendo em uma rocha sedimentar e um alto tempo geológico nessa
formação.

3.2 EXTRAÇÃO DO PETRÓLEO
Durante a perfuração do poço e extração do petróleo, a pressão existente no
poço decai bruscamente, fazendo com que nem todo petróleo seja removido, assim,
surgiram maneiras de aproveitar uma maior parte do produto que fica retido nos
poros da rocha. Esse primeiro momento da extração se chama recuperação
primária, que geralmente tem um fator baixo de recuperação, em torno de 15%, mas
um baixo custo de produção porque utiliza energia natural para levar o petróleo até a
superfície (Teixeira, 2007).
Foram desenvolvidos métodos de recuperação para minimizar as perdas de
petróleo durante a exploração, um desses é a recuperação secundária que tem
como objetivo fornecer ao poço a pressão que havia sido perdida. Na recuperação
secundária é realizada a injeção de um fluido nos poços, que nas plataformas
offshore pode ser a própria água do mar onde se encontra o poço, sendo a mais
usada atualmente, por ser mais viável economicamente e de fácil acesso (Teixeira,
2007), fornecendo a pressão necessária para que o óleo que ficou retido seja
liberado para o poço de produção. A água salina injetada nos poços ocupa o espaço
do óleo nos poros da rocha reservatório, sendo que certa quantidade de água fica
retida no reservatório e outra parte é produzida juntamente com o petróleo (Borges,
2009).
A recuperação secundária somada com a primária ainda não é suficiente para
a extração total, somando juntas cerca de 30% da produção (Carrero 2007), mas
ainda assim são as recuperações mais usadas e considerados métodos
convencionais de recuperação. O método de recuperação terciário é considerado um
método de recuperação avançada (Rosa, 2006), para poços mais maduros
recuperar reservatórios que apresentam óleos com alta viscosidade e elevadas
tensões interfaciais (Borges, 2009).
19

3.3 ÁGUA PRODUZIDA
A água produzida é um mistura de água de diferentes origens, como a água
de injeção para recuperação secundária do petróleo e a água presente naturalmente
no reservatório. A sua composição ainda é pouco conhecida quimicamente.
De acordo com um Boletim Técnico da PETROBRAS (Oliveira & Oliveira,
2000), no qual são apresentadas as faixas de concentração e a concentração típica
de diversas espécies presentes em amostras de águas produzidas do mar do norte
(um mar do oceano Atlântico), pode-se destacar as concentrações de Cd: faixa de 0-
100 µg L-1 e concentração típica de 50 µg L-1.
A utilização da técnica secundária de recuperação gera um grande volume de
água produzida onde parte é descartada no mar e parte é reaproveitada para re-
injeção (Thomas, 2004). A água do mar, que é reutilizada como água de injeção,
possui alto teor de salinidade, superior a 30‰ (CONAMA, 2005) e é rica em sulfato e
carbonato. A água de formação, gerada dentro dos reservatórios, de mais elevada
salinidade é rica em cátions precipitantes como bário, estrôncio e cálcio, fazendo
com que favoreça a formação de precipitados e podendo formar incrustações nas
tubulações ocasionando danos econômicos e temporais no processo (Teixeira,
2007; Ribeiro, 2013).
3.4 CONTAMINAÇÃO DAS ÁGUAS SALINAS
A poluição marinha decorrente do descarte de água produzida no mar pelas
indústrias do petróleo vem aumentando gradativamente e, com ela, a preocupação
com o ambiente próximo dos poços de perfuração. Isso, porque essa água possui
diversos componentes nocivos à natureza, podendo conter, por exemplo, minerais
dissolvidos presentes no petróleo, óleo, compostos químicos, sólidos residuais da
produção, gases dissolvidos e microrganismos, e assim, tornando-as complexas se
forem contaminadas por tais substâncias.
20

A água produzida, geralmente, contém (Oliveira & Oliveira, 2000):
- Sais de Na, Cl, Ca, Sr, Mg, K;
- Metais e ametais: Ba, Cd, Cr, Cu, Fe, Pb, Mn, Hg, Mo, Ni, V, Zn;
- Substâncias orgânicas: alifáticos, aromáticos, polares e ácidos graxos;
- Radioisótopos;
- Produtos químicos adicionados nos poços de petróleo: biocidas, inibidores de
corrosão, sequestrantes de oxigênio, anticoagulantes.
Todos esses componentes presentes na água de produção oferecem riscos
ao meio ambiente, em especial os metais pesados, substâncias não degradáveis
que apresentam risco de bioacumulação em seres vivos que entram em contato com
eles. Por eles serem tóxicos, o controle das concentrações desses metais tem sido
cada vez mais pertinente nas águas, devido à introdução na cadeia alimentar que,
posteriormente, podem vir a ser ingeridos por seres humanos (Cruz, 2015).
As águas salinas são classificadas em quatro classes, conforme a resolução
CONAMA nº 357 de 2005:
- Classe especial: águas destinadas à preservação dos ambientes aquáticos em
unidades de conservação de proteção integral e à preservação do equilíbrio natural
das comunidades aquáticas.
- Classe 1: águas que podem ser destinadas à recreação de contato primário,
conforme resolução CONAMA nº 274 de 2000, à proteção das comunidades
aquáticas e à aquicultura e atividade de pesca.
- Classe 2: águas que podem ser destinadas à pesca amadora e à recreação de
contato secundário.
- Classe 3: águas que podem ser destinadas à navegação e à harmonia
paisagística.
A resolução CONAMA nº 357 estipula a concentração máxima permitida de
metais tóxicos para águas salinas, onde o Cd apresenta um valor máximo de 5 μg L-1
para a classe 1 e um valor máximo de 40 μg L-1
para classe 2. A classe especial se
21

deve manter em condições naturais e a classe 3 não possui valores máximos
estabelecidos.
3.5 CÁDMIO
Geralmente o cádmio é encontrado na natureza associado a outros
elementos, como o zinco, sendo um dos subprodutos na mineração de zinco, cobre
e chumbo. Em outras fontes é encontrado como sulfeto, sulfatos, óxidos, nitratos,
cloretos e acetatos. Por sua concentração na crosta terrestre ser de 1,1x10-4 Kg g-1,
o cádmio é tido como um poluente não natural. Sendo encontrado, geralmente, em
concentrações inferiores a 1 mg Kg-1 nos solos e inferiores a 1 µg L-1 em águas,
exceto em áreas de alta atividade industrial envolvendo este metal (Azevedo &
Chasin, 2003).
O cádmio possui ampla aplicação industrial, como revestimento anticorrosivo
de aço e ferro (por eletrodeposição), em pigmentos para plásticos e vidros, na
produção de baterias recarregáveis (níquel-cádmio) e como aditivo estabilizante
térmico do PVC. Além disso, é usado ainda como aditivo na indústria têxtil, em ligas,
em fios de eletricidade, fungicidas, pirotecnia, entre outros. O revestimento de
cádmio sobre aço-carbono por processos eletrolíticos tem como benefícios boa
resistência à corrosão, bom contato elétrico e baixo coeficiente de atrito, acarretando
numa vasta aplicabilidade, por exemplo, parafusos cadmiados são usados em
portas, fechaduras e até em plataformas de petróleo offshore (Oliveira et al., 2011;
Siqueira, 2005; Manahan, 1989).
A principal fonte natural de cádmio no meio ambiente é o vulcanismo, mesmo
em períodos de baixa atividade ou sem erupções. Também podendo ser encontrado
em altas concentrações em rochas sedimentares e fosfáticas. As ocorrências
antropogênicas incluem processos em altas temperaturas, como nas indústrias de
ferro e aço, nestes processos a emissão de cádmio ocorre agregada à partículas
com menos de 10 µm, passíveis de inalação. O cádmio nessa forma agregada tem
potencial de ser transportado a longas distâncias, uma vez que seu tempo de
residência na atmosfera vai de 1 a 10 dias até que seja depositado via úmida ou
22

seca, normalmente sob a forma de óxido ou cloreto de cádmio (formas estáveis à
incineração) (Siqueira, 2005).
Nos últimos anos os estudos sobre o metabolismo e a distribuição do cádmio
no organismo têm se intensificado e sendo estimulado principalmente por ser um
metal tóxico e cumulativo, com um tempo de meia vida biológico de 30 anos. Ao ser
absorvido, se acumula, principalmente, no fígado, pâncreas, pulmões e rins.
Manifestando sua toxicidade através de diversas síndromes e efeitos, como por
exemplo, lesões hepáticas, disfunção renal, hipertensão e, quando inalado, danos
aos pulmões (Vieira, 2004).
3.6 DISPOSITIVO DE MEMBRANA SEMIPERMEÁVEL (DMSP)
Huckins desenvolveu, em 1990, um amostrador com membranas
semipermeáveis, que consiste em uma membrana de polietileno de baixa densidade,
fabricadas sem aditivos, não-porosa, apresentando cavidades transientes formadas
termicamente por movimentos randômicos das cadeias poliméricas de
aproximadamente 10 Å. Essas cavidades livres permitem a dissolução e difusão das
moléculas orgânicas na membrana. Esse processo simula a difusão de
contaminantes através das membranas de organismos vivos (Huckins et al., 1990;
Petty et al., 1998).
Inicialmente, a técnica era utilizada com trioleína como fase aceptora de
analitos no interior da membrana. A tiroleína(1,2,3-tri(cis-9-octadecenoil)glicerol) é
um lipídio apolar de elevado peso molecular (≥ 600 Da), e foi escolhido para simular
o tecido lipídico de organismos aquáticos (Petty et al., 1998).
As substâncias hidrofóbicas interagem com a membrana de polietileno e/ou
com a fase aceptora em seu interior, que vão sendo extraídas e acumuladas no
interior das mesmas, como pode ser visto na Figura 1.
23

Figura 1. Sistema de amostragem com membrana semipermeável preenchida com
solvente (Robaina, 2015).
A capacidade de acúmulo de compostos hidrofóbicos por DMSP está
relacionada com o coeficiente de partição octanol/água (Kow) dos compostos. Kow
consiste na razão das concentrações da substância, no equilíbrio, entre os solventes
imiscíveis octanol e água. Aqueles com valores de Kow mais altos têm maior
possibilidade de serem concentrados significativamente nos dispositivos com níveis
acima do ambiente aquoso, como compostos hidrofóbicos com log Kow≥3. Apesar
dos que possuem valores de log menores de 3 poderem ser concentrados, seu uso
não possui vantagem em relação aos outros procedimentos de amostragem (Petty et
al., 2000). Devido aos altos fatores de concentração alcançados pelos DMSP torna-
se possível analisar níveis de contaminantes em ultra-traços (Lebo et al., 1995).
De acordo com Petty et al. (2000), o uso dos DMSP pode ser vantajoso para
as seguintes aplicações:
- determinar fontes de poluição;
- estimar a concentração média ao longo do tempo (CMLT);
- simular a concentração de compostos químicos biodisponíveis para testes
com bioindicador ou imunoensaios;
24

- seqüestrar contaminantes para os procedimentos de avaliação e
identificação de toxicidade (AIT);
- estimar a exposição de organismos e a bioconcentração potencial.
3.7 ESPECTROMETRIA DE ABSORÇÃO ATÔMICA EM FORNO DE GRAFITE
A Espectrometria de Absorção Atômica em Forno de Grafite (GF AAS) é uma
técnica que possui alta sensibilidade, conferindo baixos limites de detecção (da
ordem de μg L-1), além de possuir a vantagem da utilização de pequenos volumes
de amostra (entre 1 e 50 μL).
O princípio básico do funcionamento do GF AAS consiste na passagem de
uma radiação eletromagnética, de um determinado comprimento de onda por um
vapor atômico contendo átomos livres no estado fundamental, no qual uma parte da
radiação é absorvida e os átomos passam para um estado mais excitado. A
concentração do analito na amostra é determinada por essa energia que foi
absorvida, pois é proporcional ao numero de átomos livres no estado fundamental
que foram excitados (Holler et al., 2009). Assim, é feita analogia com a lei de
Lambert-Beer, em que a absorvância (A) e o coeficiente de absorção (Kⱱ) são
proporcionais a concentração do átomo. A partir dessa correlação, é possível
deduzir o quanto foi absorvido do analito em solução, e assim, determinar sua
concentração através de uma relação linear. (Ebdon et al., 1998).
Equação da lei de Lambert-Beer: A = log (I0/I) = Kⱱ L log e
Onde,
A é a absorvância;
I0 é a intensidade de luz incidente;
I intensidade de luz transmita;
Kⱱ é o coeficiente de absorção;
L é o comprimento do caminho.
25

Após a introdução da amostra no interior de um pequeno tubo de grafite,
localizado no caminho ótico do AAS, são programados estágios de aquecimentos no
programa de temperatura composto por várias etapas para a separação do analito
da matriz,sendo assim minimizadas as possíveis interferências (Freschi et al., 2000).
O programa de temperatura é composto por 4 etapas: Secagem, pirólise,
atomização e limpeza.
A secagem é a etapa em que se eliminam os componentes voláteis e os
solventes presentes na solução. Após a secagem, ocorre a pirólise, onde é
eliminada toda a matriz da amostra ao se atingir uma temperatura muito alta, porém
com cautela para que não ocorra perda de analito. Na atomização, a temperatura é
elevada suficientemente para vaporizar os átomos livres do analito, mas de modo
que não seja alta demais para danificar o tubo de grafite. A etapa final é a limpeza
do tubo, com uma temperatura um pouco acima da de atomização para eliminar
qualquer resíduo de amostra presente nas paredes do tubo comprometendo a
próxima utilização.
É necessária uma programação de temperatura para cada analito em cada
matriz a ser estudada, para que não haja interferência da matriz e nem perda do
analito por elevadas temperaturas. Existem casos em que alguns metais, por
apresentarem baixa estabilidade térmica, não suportam as determinadas
temperaturas, porém suas matrizes as necessitam para que possam ser
completamente eliminadas, então, neste caso, é necessário o uso de modificadores
químicos, onde seu emprego pode acarretar em um aumento da sensibilidade nas
medições e também na diminuição de possíveis interferências espectrais. Um
exemplo é o Pd, que estabiliza termicamente o analito, onde o modificador reduz a
velocidade do analito dentro do forno permitindo que sejam usadas temperaturas
elevadas propícias para a eliminação completa da matriz sem a perda do analito em
questão. Esse metal de transição forma compostos intermetálicos termicamente
estáveis com o metal que deseja ser determinado (Ebdon et al., 1998; Freschi et al.,
2000).
26

4 PARTE EXPERIMENTAL
4.1 MATERIAIS E REAGENTES
4.1.1 INSTRUMENTAÇÃO
i. Agitador horizontal MRII Roler Mixer;
ii. Balança Analítica Shimadzu, modelo AY220;
iii. Espectrômetro de absorção atômica Varian, modelo AA240Z, equipado com
amostrador automático Varian PSD 120, forno de grafite GTA 120, corretor de
fundo por efeito Zeeman e tubos de grafite (eletrolítico) com plataforma de
L´vov;
iv. pHmetro Digimed, modelo DM-22;
v. Seladora Cristofoli;
4.1.2 REAGENTES
i. Acetato de Sódio Trihidratado - Vetec
ii. Ácido Bórico - Caledon
iii. Ácido Clorídrico - Tedia
iv. Ácido Nítrico - Tedia

v. Cloreto de sódio - Vetec
vi. Clorofórmio - Tedia
vii. DDTC – Sigma Aldrich
viii. Etanol - Tedia
ix. Fosfato Monobásico de Sódio - Vetec
x. Hexano - Tedia
xi. Hidróxido de sódio - Vetec
xii. Padrão de Cádmio - Vetec
xiii. Padrão de Paládio - Fluka
4.1.3 MATERIAIS
Todas as vidrarias e plásticos, tubos de polietilenos de 15 e 50 mL, ponteiras
de micropipetas e recipientes do amostrador, foram lavados com água corrente e
detergente comum. Em seguida foram enxaguados com água deionizada e imersos
em solução de HNO3 10% v/v por, pelo menos, 24 h. Antes do uso, esses materiais
foram cuidadosamente lavados com água ultrapura (sistema Milli-Q: Millipore,
Milford, MA, USA) e secos em estufa a 40°C (exceto material volumétrico), evitando
qualquer contato com materiais metálicos e poeira. Rigoroso controle dos brancos foi
realizado para evitar problemas de contaminação.
Foram usadas membranas de polietileno de baixa densidade com 80 μm de
espessura e 2,5 cm de largura. Para o uso no procedimento, foram cortadas com
8 cm de comprimento, lavadas em hexano e secadas a temperatura ambiente.
Os reagentes usados foram de grau analítico e utilizados sem purificação
adicional. Todas as soluções foram preparadas com água deionizada de alta pureza
(resistividade 18,2 MΩ cm-1) obtida em um sistema Direct Q-3 (Millipore, Milford, MA,
USA).
28

4.2 METODOLOGIA
4.2.1 PREPARO DE SOLUÇÕES
As soluções padrões aquosas de Cd utilizadas nos experimentos para
construção de curvas analíticas foram preparadas diariamente pela diluição
adequada da solução estoque de 1000 mg L-1 com água ultrapura e armazenadas
em frascos de polietileno.
Foram preparadas soluções intermediárias de DDTC na concentração 3,7 x
10-3 mol L-1. Para isso foram pesados 84,48 mg do complexante e dissolvido em um
balão volumétrico de 100 mL com água purificada.
Foi utilizada, também, uma solução tampão Britton Robinson na concentração
de 0,01 mol L-1, para manter a faixa de pH em 9,0, preparada através da mistura de
soluções formada pelo ácido bórico, acetato de amônio e fosfato monobásico de
sódio. Para o preparo de 80 mL de solução foram pesados 0,8709 g de acetato,
0,3957 g borato e 0,7677 g de fosfato. O pH foi ajustado pela adição de uma solução
de hidróxido de sódio e/ou ácido clorídrico 2,0 mol L-1. Após a otimização do pH,
optou-se por utilizar uma solução tampão de acetato de sódio 0,1 mol L-1.
As soluções tampão borato (pH = 9,0) na concentração 0,1 mol L-1 foram
preparadas pela pesagem de 1,1808 g de acetato de amônio e avolumados até
100mL em balões volumétricos com água purificada. O pH foi ajustado com a
solução de HCl e/ou NaOH 2,0 mol L-1 previamente preparadas.
4.2.2 OTIMIZAÇÃO DO MÉTODO DE EXTRAÇÃO
Primeiramente, foi realizada uma otimização do método para que fossem
encontradas as melhores condições de pré-concentração e extração Cd através da
membrana semipermeável.
Esta otimização consiste em estudar variáveis como, solvente extrator,
concentração do complexante DDTC, tempo de extração, influência da agitação,
29

influência do pH no meio, volume de solvente extrator e influência da força iônica,
para, assim, ser feita a análise da amostra.
A primeira etapa do método consiste em preparar as membranas
semipermeáveis, as quais foram cortadas com 8 cm de comprimento, imersas em
hexano durante 24h e secadas a temperatura ambiente. Para serem armazenadas,
foram embrulhadas individualmente em papel alumínio e mantidas em refrigerador a-
4ºC. Para seu uso, a membrana foi selada e preenchida com volume de solvente
extrator adequado.
Na etapa de otimização do método foi utilizada uma solução aquosa de 40 mL
contendo o analito e o agente complexante em meio tamponado. As soluções
continham 1 μg L-1 de cádmio, 4 mL de solução aquosa de DDTC 3,7 x 10-3 mol L-1
e 4 mL de solução tampão Britton Robinson 0,01 mol L-1.
Para o estudo da influência da força iônica, foram adicionados solução
estoque de NaCl ao meio contendo DDTC, tampão e padrão do analito, preparando
assim meios com salinidade de 12%, 3,6% e 0,12% m/v.
A solução aquosa foi transferida para um tubo falcon de 50 mL e a membrana,
previamente selada com o solvente extrator, foi introduzida no tubo que é levado
para agitação horizontal por um tempo previamente determinado a velocidade de
cerca de 110 rpm.
Após a agitação, a membrana foi retirada do tubo, e o solvente que estava em
seu interior é diluído adequadamente em etanol para que seja realizada a medição
do analito por absorção atômica em forno de grafite. O volume de injeção da
amostra no equipamento foi de 20 μL e foi utilizado paládio como modificador
químico com injeção de 20 μg deste. As temperaturas de pirólise e atomização,
utilizadas no programa de temperatura, foram 800ºC e 1800ºC, respectivamente, de
acordo com Robaina, 2015.
30

4.2.3 APLICAÇÃO DO MÉTODO DE EXTRAÇÃO OTIMIZADO EM
AMOSTRAS REAIS DE ÁGUAS SALINAS
As amostras foram analisadas nas condições otimizadas: membranas
cortadas com 8 cm de comprimento e seladas com 3 mL de clorofórmio atuando
como solvente extrator, solução de DDTC 3,76 x 10-4 mol L-1, solução tampão de
0,1 mol L-1 de acetato de sódio com pH 9 e agitação horizontal durante 2h.
Para a determinação de cádmio na água do mar, foram adicionados 4 mL de
amostra de água salina, 4 mL de DDTC 3,76 x 10-3 mol L-1, 4 mL de solução tampão
e 28 mL de água deionizada a um tubo falcon de 50 mL. Foi introduzida na solução a
membrana contendo o solvente extrator e levada para agitação horizontal.
Passado o tempo determinado, foi retirada a membrana do tubo, e, após o
estudo de volume do solvente, optou-se por avolumar todo o extrato para o volume
de 5 mL com etanol para que a amostra fosse levada a absorção atômica em forno
de grafite.
Para avaliar o problema de interferência de matriz, na etapa de análise das
amostras foram preparadas curvas analíticas e de adição padrão de Cd para cada
amostra analisada.
As curvas analíticas foram preparadas nas mesmas condições do método
otimizado, ou seja, soluções contendo diferentes concentrações de Cd (II) foram
submetidas ao processo de extração com a DMSP.
31

5 RESULTADO E DISCUSSÃO
5.1 OTIMIZAÇÃO DO MÉTODO DE EXTRAÇÃO
A análise das soluções de Cd (II) na etapa de otimização do método de
extração foi realizada em triplicata real e duplicata instrumental, utilizando o método
GF AAS, desenvolvido por Robaina, 2015. O programa de temperatura utilizado no
GF AAS pode ser visto na Tabela 1.
Tabela 1. Programa de temperatura da absorção atômica em forno de grafite para
determinação de cádmio (Robaina, 2015).
Etapa Temperatura (ºC) Rampa (s) Patamar (s) Vazão de Ar
(mL min-1)
Secagem I 85 5 300
Secagem II 95 40 0 300
Secagem III 120 10 0 300
Pirólise 800 5 4 300
Atomização 1400 1 3 0
Limpeza 2200 2 0 300
Foram realizados testes para otimização do método analítico por GF AAS de
modo a obter a máxima eficiência de extração do Cd (II). Na etapa de otimização
foram avaliados parâmetros que pudessem influenciar na etapa de extração e os
resultados podem ser vistos a seguir.

5.1.1 SOLVENTE EXTRATOR
Foram avaliados dois solventes apolares, hexano e clorofórmio, para que
ajam como extrator e pré-concentrador do complexo apolar formado entre o
complexante (DDTC) e o metal.
Os dois solventes apresentam uma volatilidade muito alta, podendo haver
perda de analito, e acarretando em erro e/ou maior variabilidade de medição no GF
AAS. Para minimizar esse problema foi adicionado, ao final da extração, etanol para
que fosse realizada a medição no GF AAS, pois, além de apresentar um ponto de
ebulição maior, possui baixa toxicidade e disponibilidade no laboratório.
Como pode ser observada na Figura 2, a extração de Cd (II), utilizando
clorofórmio como solvente extrator, foi bem melhor do que com hexano. Essa
afinidade fez com que ocorresse uma maior migração do complexo que estava em
solução para o solvente localizado dentro da membrana.
Sin
al R
ela
tivo
0.40
0.35
0.30
0.25
0.20
0.15
0.10
0.05
0.00 Hexano Cloroformio
Solvente Extrator
Figura 2. Gráfico comparativo da eficiência de extração entre hexano e clorofórmio
como solvente extrator influente no processo de extração de Cd(II).
33

5.1.2 CONCENTRAÇÃO DO COMPLEXANTE
Essa é uma etapa muito importante na otimização do método, pois é o
complexante que vai converter o analito em uma espécie apolar, migrando assim
para o interior da membrana que contém o solvente apolar. O complexante é
essencial para a extração do metal da solução aquosa, já que é um composto
hidrofóbico e faz com que sua transferência para dentro da DMSP seja favorecida.
A membrana possui uma seletividade baseada no diâmetro do composto e
seletividade da fase extratora. Ela possui um diâmetro de passagem do analito de 10
Â, portanto, a escolha do complexante, também foi baseada nessa informação.
Sabe-se que o dietiltiocarbamato (DDTC) possui uma boa capacidade de
complexar metais, como o cádmio, por possuir dois átomos de enxofre, que são
doadores de elétrons, em sua composição, fazendo com que duas moléculas de
DDTC se complexem em um átomo de Cd, como na Figura 3 (Yousefi & Shemirani,
2010; Soto et al., 2015).
Figura 3. Estrutura química do complexo Cd(DDTC)2.
O complexante deve estar em concentração suficiente para que reaja com o
metal e migre para o interior da membrana de forma eficiente. As concentrações de
DDTC avaliadas no estudo podem ser vistas na Tabela 2.
34
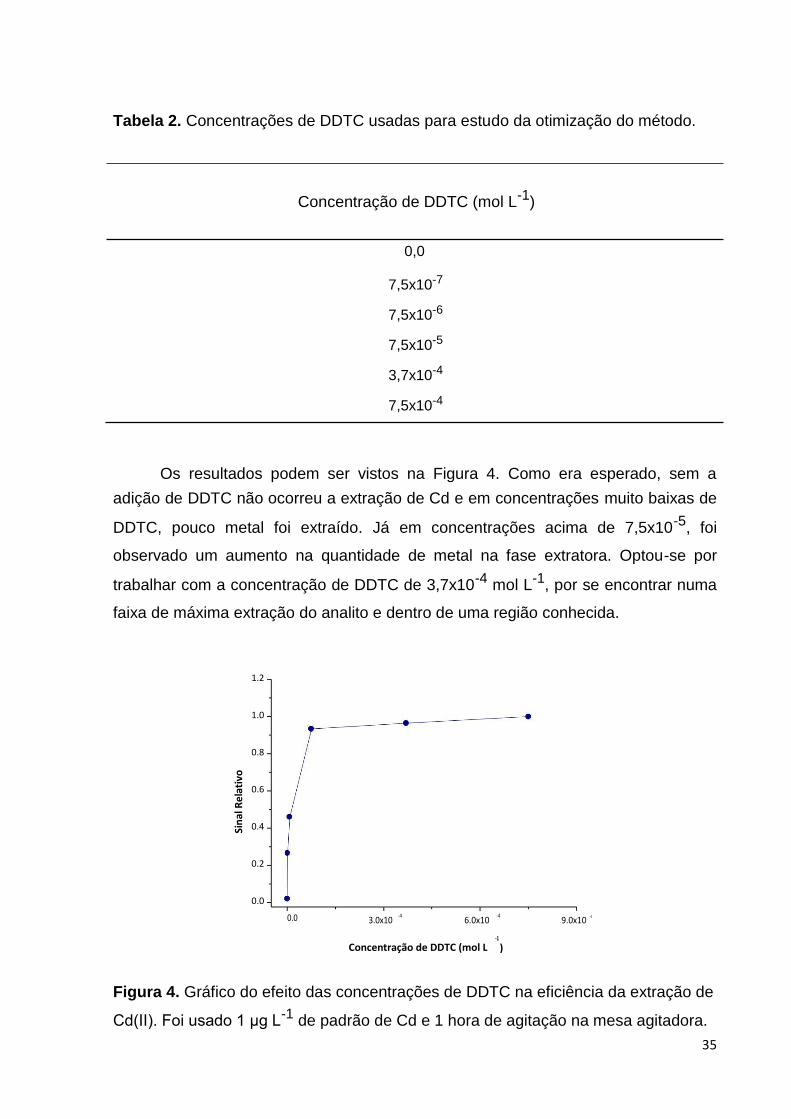
Tabela 2. Concentrações de DDTC usadas para estudo da otimização do método.
Concentração de DDTC (mol L-1)
0,0
7,5x10-7
7,5x10-6
7,5x10-5
3,7x10-4
7,5x10-4
Os resultados podem ser vistos na Figura 4. Como era esperado, sem a
adição de DDTC não ocorreu a extração de Cd e em concentrações muito baixas de
DDTC, pouco metal foi extraído. Já em concentrações acima de 7,5x10-5, foi
observado um aumento na quantidade de metal na fase extratora. Optou-se por
trabalhar com a concentração de DDTC de 3,7x10-4 mol L-1, por se encontrar numa
faixa de máxima extração do analito e dentro de uma região conhecida.
Sin
al R
ela
tivo
1.2
1.0
0.8
0.6
0.4
0.2
0.0
0.0 3.0x10 -4
6.0x10 -4
9.0x10 -4
Concentração de DDTC (mol L
-1 )
Figura 4. Gráfico do efeito das concentrações de DDTC na eficiência da extração de
Cd(II). Foi usado 1 μg L-1 de padrão de Cd e 1 hora de agitação na mesa agitadora.
35
5.1.3 TEMPO DE EXTRAÇÃO
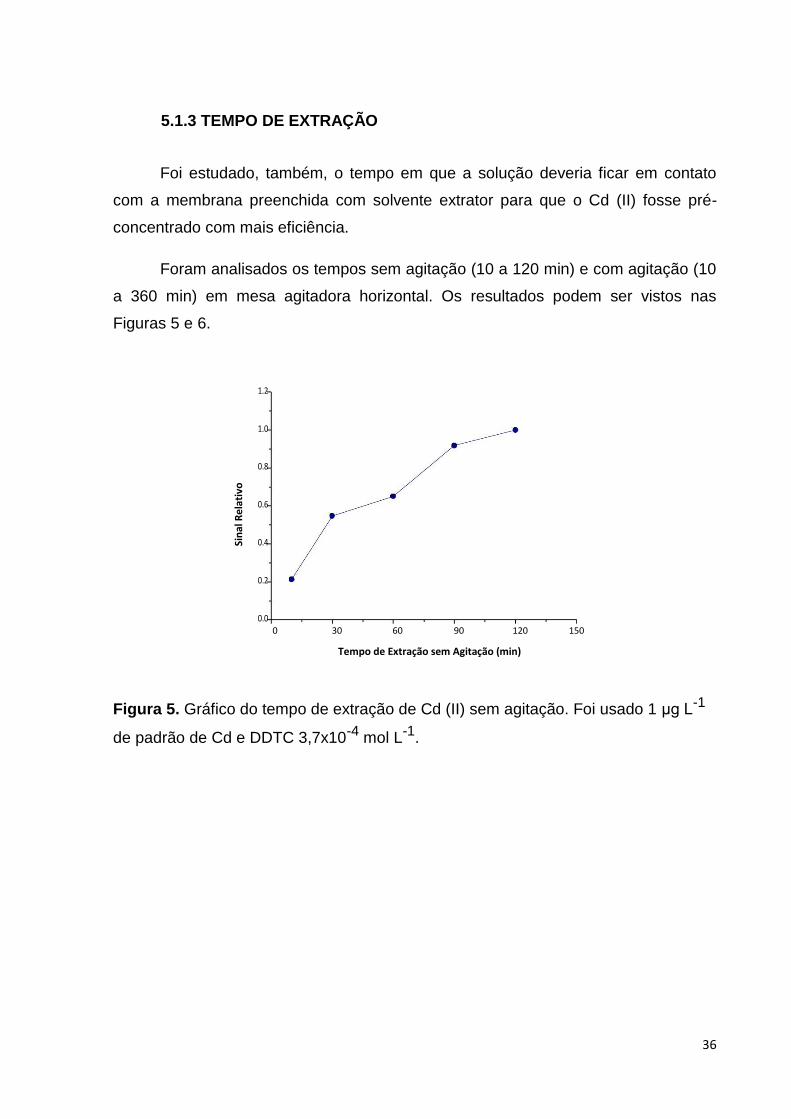
Foi estudado, também, o tempo em que a solução deveria ficar em contato
com a membrana preenchida com solvente extrator para que o Cd (II) fosse pré-
concentrado com mais eficiência.
Foram analisados os tempos sem agitação (10 a 120 min) e com agitação (10
a 360 min) em mesa agitadora horizontal. Os resultados podem ser vistos nas
Figuras 5 e 6.
Sin
al R
ela
tivo
1.2
1.0
0.8
0.6
0.4
0.2
0.0 0 30 60 90 120 150
Tempo de Extração sem Agitação (min)
Figura 5. Gráfico do tempo de extração de Cd (II) sem agitação. Foi usado 1 μg L-1
de padrão de Cd e DDTC 3,7x10-4 mol L-1.
36

Si
nal
Re
lati
vo
1.2
1.0
0.8
0.6
0.4
0.2
0.0
0 100 200 300 400
Tempo de Extração com Agitação (min)
Figura 6. Gráfico do tempo de extração, em minutos, de Cd (II) com agitação em
mesa agitadora horizontal. Foi usado 1 μg L-1 de padrão de Cd e DDTC 3,7x10-4
mol L-1.
Apesar do gráfico da extração sem agitação apresentar um crescimento ao
longo do tempo, observou-se que no mesmo tempo com a agitação, a extração de
Cd foi muito maior, visto que nesse sistema tem-se os fenômenos de difusão e
convecção influenciando na migração do analito para o DMSP. Então, optou-se pelo
procedimento usando a mesa agitadora horizontal.
Como previsto no teste sem agitação, também era esperado o crescimento
conforme o tempo na agitação do sistema, por isso foi realizado um estudo com
tempos mais elevados, até 360 min.
Apesar de se observar um pequeno incremento de extração com tempos
superiores a 240 min, optou-se por trabalhar com o tempo de extração de 120 min,
buscando uma maior velocidade analítica.
37

5.1.4 INFLUÊNCIA DO pH
O estudo do pH é de extrema importância no desenvolvimento do método de
extração proposto, visto que a disponibilidade do complexante e do analito é
dependente desta variável, devido as características ácido-base do molécula de
DDTC, Figura 7, e da formação de hidroxicomplexos de Cd.
Figura 7. Equilíbrio do ácido dietilditiocarbamato com seu respectivo valor de pKa à
19°C de acordo SOTO et al., 2015.
Além disso, de acordo com SOTO et al., 2015, o DDTC pode degradar-se em
meio ácido e formar compostos quimicamente estáveis, fazendo com que sua
disponibilidade em solução para complexar com o metal desejado seja menor,
afetando assim a eficiência da extração.
Foi avaliada uma faixa de pH entre 3,0 e 11,0 utilizando uma solução de
tampão Britton-Robinson, que é preparada com uma mistura de três sistemas
tamponantes: acetato, fosfato e borato.
38
Si
nal
Re
lati
vo
1.2
1.0
0.8
0.6
0.4
0.2
0.0
3.0 6.0 9.0 12.0
pH
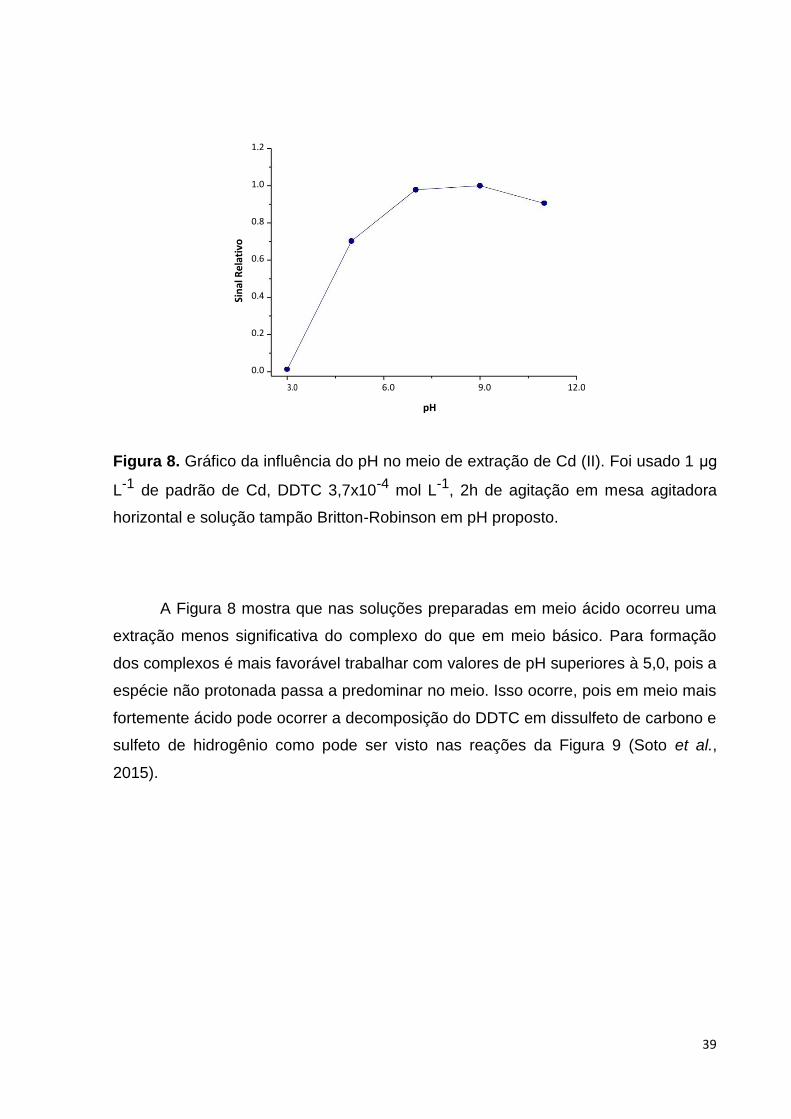
Figura 8. Gráfico da influência do pH no meio de extração de Cd (II). Foi usado 1 μg
L-1 de padrão de Cd, DDTC 3,7x10-4 mol L-1, 2h de agitação em mesa agitadora
horizontal e solução tampão Britton-Robinson em pH proposto.
A Figura 8 mostra que nas soluções preparadas em meio ácido ocorreu uma
extração menos significativa do complexo do que em meio básico. Para formação
dos complexos é mais favorável trabalhar com valores de pH superiores à 5,0, pois a
espécie não protonada passa a predominar no meio. Isso ocorre, pois em meio mais
fortemente ácido pode ocorrer a decomposição do DDTC em dissulfeto de carbono e
sulfeto de hidrogênio como pode ser visto nas reações da Figura 9 (Soto et al.,
2015).
39

Figura 9. Reações de decomposição do DDTC com formação de (A) dissulfeto de
carbono e de (B) sulfeto de hidrogênio a partir do DDTC.
Por isso, foi escolhido trabalhar com pH em torno de 9,0 por se encontrar em
uma região de máxima eficiência. A partir dessa etapa, o ajuste do pH foi realizado
com tampão borato por ser um sistema tamponante mais simples.
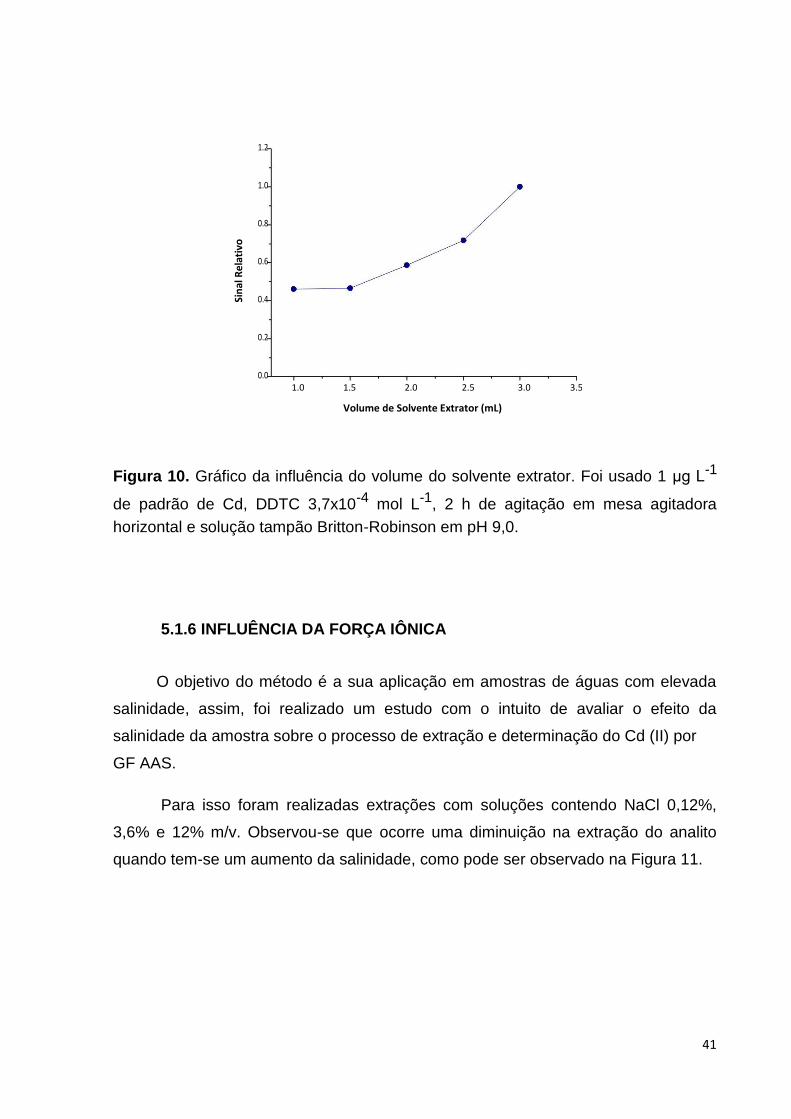
5.1.5 VOLUME DE SOLVENTE EXTRATOR
O volume do solvente extrator está diretamente ligado ao fator de pré-
concentração; volumes menores do solvente resultam em fatores de pré-
concentração maiores, sendo importante a otimização dessa variável. Nesse estudo
foram avaliados os volumes de 1,0 a 3,0 mL de clorofórmio.
Durante a realização do estudo observou-se uma perda de clorofórmio após a
etapa de extração, que foi mais evidente com o volume de 1,0 mL. Para contornar o
problema, optou-se por avolumar todo o solvente extrator em etanol para 5 mL.
Com base nos dados, os volumes menores ocasionaram sinais mais baixos,
indicando que não foram suficientes para captar a quantidade de analito que estava
em solução. Assim, optou-se por utilizar o volume de 3 mL de solvente,pois, além de
possuir o maior sinal, caracterizando maior concentração do analito, diminuem-se as
possíveis perdas após a agitação.
40
Si
nal
Re
lati
vo
1.2
1.0
0.8
0.6
0.4
0.2
0.0 1.0 1.5 2.0 2.5 3.0 3.5
Volume de Solvente Extrator (mL)
Figura 10. Gráfico da influência do volume do solvente extrator. Foi usado 1 μg L-1
de padrão de Cd, DDTC 3,7x10-4 mol L-1, 2 h de agitação em mesa agitadora
horizontal e solução tampão Britton-Robinson em pH 9,0.
5.1.6 INFLUÊNCIA DA FORÇA IÔNICA
O objetivo do método é a sua aplicação em amostras de águas com elevada
salinidade, assim, foi realizado um estudo com o intuito de avaliar o efeito da
salinidade da amostra sobre o processo de extração e determinação do Cd (II) por GF AAS.
Para isso foram realizadas extrações com soluções contendo NaCl 0,12%,
3,6% e 12% m/v. Observou-se que ocorre uma diminuição na extração do analito
quando tem-se um aumento da salinidade, como pode ser observado na Figura 11.
41

Si
nal
Re
lati
vo
1.2
1.0
0.8
0.6
0.4
0.2
0.0 0 2 4 6 8 10 12 14
Salinidade (%)
Figura 11. Gráfico da influência da força iônica do meio na influência da extração.
Foi usado 1 μg L-1 de padrão de Cd, DDTC 3,7x10-4 mol L-1, 2 h de agitação em
mesa agitadora horizontal e solução tampão Britton-Robinson em pH 9,0.
O efeito da diminuição da extração quando a solução possui maior salinidade
é um resultado contrário ao do esperado pelo efeito salting-out, que é quando
moléculas de água estão atraídas pelos sais em solução, fazendo com que a
solubilidade de moléculas apolares diminua devido a queda de disponibilidade de
água para que as mesmas sejam solvatadas. Com o efeito contrario, o aumento da
concentração salina pode dificultar o processo de difusão dos analitos da solução
aquosa para a fase aceptora, uma vez que a presença de outras espécies no meio
pode alterar algumas propriedades da solução, como densidade e viscosidade.
Observou-se que a salinidade afeta a eficiência de extração do Cd, o que
pode ser um indicativo de que, durante a aplicação em amostras reais, pode-se
observar o efeito de interferência de matriz.
42

5.2 APLICAÇÃO DO MÉTODO OTIMIZADO EM AMOSTRAS REAIS
As condições otimizadas do método de extração e pré-concentração de Cd (II)
em águas salinas foram obtidas e serão utilizadas para aplicação em amostras reais.
Essas condições podem ser vistas na Tabela 3.
Tabela 3. Condições ótimas de extração e pré-concentração de Cd (II) de águas
salinas através de membrana semipermeável.
Condições Otimização
Solvente Extrator Clorofórmio
Concentração de DDTC 3,7x10-4 mol L-1
Tempo de Agitação 2 horas
pH 9,0
Volume do Solvente 3 mL
Os limites de detecção (LD) e quantificação (LQ) do método proposto foram
calculados segundo as expressões mostradas a seguir (Brito et al.,2003).
= 3σ
= 10σ
Onde:
σ = desvio padrão do primeiro ponto;
s = sensibilidade da curva analítica.
O LD é a menor concentração de um analito presente em uma amostra que
pode ser detectada. Entretanto, não necessariamente essa porção detectável
43

poderá ser quantificada nas condições experimentais que foram estabelecidas. Já o
LQ é a menor quantidade do analito presente em uma amostra que pode ser
quantificada com precisão e exatidão aceitáveis sob as condições experimentais
estabelecidas (Brito et al., 2003). A Tabela 4 mostra os parâmetros de mérito para o
método desenvolvido.
Tabela 4. Parâmetro de mérito para determinação de Cd (II) pelo método proposto.
Faixa de trabalho LD LQ
0,25 – 2,0 μg L-1 0,15 μg L-1
0,52 μg L-1
O método foi aplicado nas seguintes amostras de água salina:
- Amostra 1: Água produzida sintética preparada no laboratório (96,097‰);
- Amostra 2: Água do mar amostrada próxima ao terminal rodoviário de Niterói-RJ;
- Amostras 3 e 4: Águas produzidas A (234‰) e B (270‰).
Como no estudo da salinidade foi observada influência dessa variável na
extração do analito, esperava-se observar interferência de matriz na análise das
amostras, por isso na análise de todas as amostras foram preparadas curvas
analíticas e de adição padrão, para sua posterior comparação.
As curvas analíticas foram preparadas nas mesmas condições do método
otimizados, ou seja, soluções contendo diferentes concentrações de Cd (II) foram
submetidas ao processo de extração com a membrana. As análises das amostras
reais foram realizadas em duplicata real e duplicata instrumental.
Comparando-se as curvas analíticas com as curvas de adição padrão pode-se
observar que não há indicação de interferência de matriz, visto que os coeficientes
angulares das curvas são compatíveis. Portanto, a quantificação de Cd em águas
salinas pelo método desenvolvido pode ser realizada por meio de método da curva
44
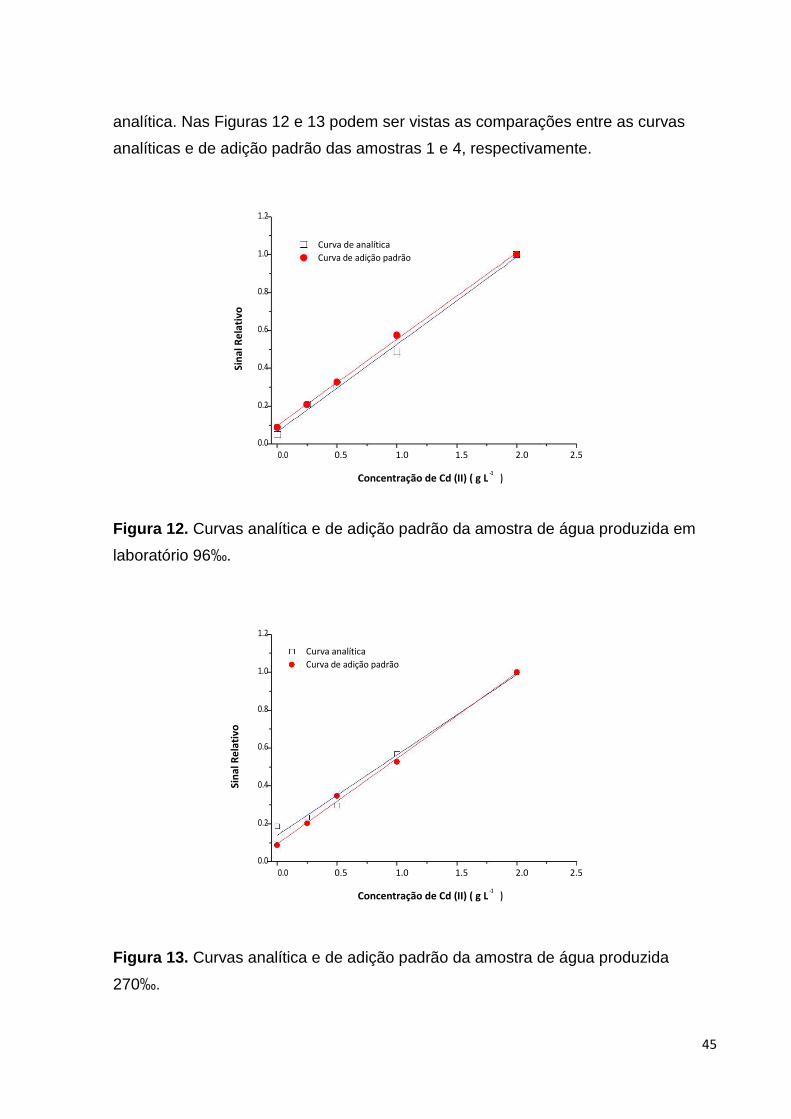
analítica. Nas Figuras 12 e 13 podem ser vistas as comparações entre as curvas
analíticas e de adição padrão das amostras 1 e 4, respectivamente.
Sin
al R
ela
tivo
1.2
1.0 Curva de analítica
Curva de adição padrão
0.8
0.6
0.4
0.2
0.0 0.0 0.5 1.0 1.5 2.0 2.5
Concentração de Cd (II) ( g L
-1 )
Figura 12. Curvas analítica e de adição padrão da amostra de água produzida em
laboratório 96‰.
Sin
al R
ela
tivo
1.2
Curva analítica
1.0 Curva de adição padrão
0.8
0.6
0.4
0.2
0.0 0.0 0.5 1.0 1.5 2.0 2.5
Concentração de Cd (II) ( g L
-1 )
Figura 13. Curvas analítica e de adição padrão da amostra de água produzida
270‰.
45

As amostras analisadas apresentaram concentração de Cd abaixo do limite de
quantificação do método, ou seja, abaixo de 0,52 μg L-1.
A exatidão do método desenvolvido por avaliado por meio de testes de
recuperação, em que faixas de 80 a 120% são consideradas aceitáveis para
exatidão do método proposto, o que representa uma diferença de 20% em relação
ao valor de concentração adicionado.
Na realização do teste de recuperação foram adicionadas quatro diferentes
concentrações (0,25; 0,50; 1,0 e 2,0 μg L-1) de Cd (II) às amostras utilizando um
padrão aquoso. As recuperações foram calculadas de acordo com o descrito na
equação a seguir e podem ser vistas na Tabela 5 (Brito et al., 2003):
Onde,
C1: concentração do analito na amostra com adição do analito padrão;
C2: concentração do analito na amostra sem adição do analito padrão;
C3: concentração do analito adicionado a amostra (com adição de analito).
46

Tabela 5. Resultados de recuperação de Cd (II) através do método padronizado.
Amostras Concentração de Cd(II) Concentração de Cd(II) Recuperação
adicionada (μg L-1) encontrada (μg L-1) (%)
0 < LQ -
0,25 0,26 ± 0,003 104
1 0,50 0,51 ± 0,005 103
1,0 1,05 ± 0,013 105
2,0 1,97 ± 0,049 98
0 < LQ -
0,25 0,29 ± 0,010 116
2 0,50 0,59 ± 0,002 118
1,0 1,00 ± 0,009 100
2,0 1,91 ± 0,046 96
0 < LQ -
0,25 0,33 ± 0,014 133
3 0,50 0,55 ± 0,008 110
1,0 0,88 88
2,0 2,03 101
0 < LQ -
0,25 0,24 ± 0,001 95
4 0,50 0,53 ± 0,002 107
1,0 0,90 ± 0,001 91
2,0 1,88 ± 0,013 94
Os valores de recuperação encontrados foram na faixa de 88-133%, mostra-se
uma boa taxa de recuperação do analito para o método proposto, demonstrando a
exatidão do método proposto.
47

6 CONCLUSÃO
No presente trabalho foi desenvolvida uma metodologia para a determinação
de Cd (II) em amostras de água produzidas oriundas da exploração de petróleo.
Diferentes variáveis relacionadas ao método foram otimizadas: tipo e volume de
solvente extrator, concentração de complexante (DDTC), tempo de extração,
agitação do sistema, força iônica e pH. As condições otimizadas foram: 3 mL de
clorofórmio como solvente extrator, concentração de DDTC de 3,7x10-4 mol L-1,
tempo de extração 2 horas com agitação horizontal e pH 9,0.
O método otimizado apresentou limite de detecção de 0,15 μg L-1, limite de
quantificação de 0,52 μg L-1 e faixa de trabalho de 0,25 – 2,0 μg L-1.
A metodologia desenvolvida foi aplicada em 1 amostra de água do mar obtidas
em uma praia do município de Niterói, no estado do Rio de Janeiro, e 1 amostra de
água produzida em laboratório, e 2 amostras de água produzida doadas pelo
CENPES/Petrobras. Sendo que as concentrações de Cd nas amostras estavam
abaixo do limite de quantificação do método.
Não foi observada interferência de matriz na determinação de Cd nas
amostras pelo método proposto, indicando que se pode empregar o método da curva
analítica, neste caso. Por ter sido observado interferência de matriz nos estudos
preliminares a aplicação nas amostras, a perspectiva é que seja aplicado em mais
amostras com características distintas para que seja verificada a interferência de
matriz.
A sensibilidade do método foi avaliada através de testes de recuperação, que
apresentaram recuperações na faixa de 88-133%, demonstrando uma boa exatidão
para o método proposto.

7 BIBLIOGRAFIA
ALMEIDA, S. I. Avaliação das concentrações de metais pesados em tecido muscular
de siri, Callinectesbocourti (A. Milne Edwards, 1879), da Laguna de
Manguaba (AL). 2009. Trabalho de conclusão de curso (Ciências Biológicas
com ênfase em Ciências Ambientais) - Universidade Estadual do Norte
Fluminense, Campos dos Goytacazes – RJ – 2009.
AZEVEDO, F. A.; CHASIN, A. A. M. Metais: gerenciamento da toxicidade. Atheneu;
São Paulo; 2003.
BOADA, M.; MORENO, M. A.; GIL, H.; MARCANO, J.; MAZA, J. Metales pesados
(Cu+2, Cd+2, Pb+2, Zn+2) em músculo y cefalotórax de camarones silvestres
Litopenaeusschmitti, Farfantepenaeussubtilis, F. notialis y F. brasiliensis de
laregión oriental de Venezuela. FCV-LUZ / Vol. XVII, Nº 2: 186 – 192. 2007.
BORGES, S. M. S. Recuperação Avançada de Petróleo (EOR) com a utilização da
Glicerina Bruta (GB) co-produto da produção de Biodiesel. 2009. Dissertação
de Mestrado – Instituto de Química, Universidade Federal da Bahia, Salvador.
2009.
BRITO, N. M; et al. Validação de métodos analíticos: estratégia e Discussão. R.
Ecotoxicol. e Meio Ambiente, Curitiba, v. 13, 2003.
CARRERO, E.; QUEIPO, N. V.; PINTOS, S., ZERPA, L., E. Global sensitivity
analysis of Alkali–Surfactant–Polymerenhancedoilrecovery processes. Journal
of Petroleum Scence & Engineering, 2006, 58, 30.

CONAMA. Resolução nº 357, Brasil, 17 de março de 2005. Disponível em
<http://www.mma.gov.br/port/conama/res/res05/res35705.pdf> Acesso em 24
de novembro de 2015.
CRUZ, G. F. B. Microextração de cobre e níquel em água produzida utilizando
líquido iônico hidrofóbico como fase extratora. 2015. Tese de Mestrado –
Instituto de Química, Universidade Federal Fluminense, Niterói, 2015.
FEEMA Poluição hídrica da Baía de Guanabara por metais pesados cromo e zinco,
Rio de Janeiro, Brasil, 1992.
FELLENBERG, G. Introdução aos Problemas da Poluição Ambiental, Ed.
Pedagógica e Universitária Ltda: São Paulo, 1980.
FRESCHI, G. P.et al. Espectrometria de absorção atômica multielementar
simultânea com atomização eletrotérmica em forno de grafite – uma revisão
da técnica e aplicações. Eclética Química, vol. 25, 2000.
HOLLER, F. J.; SKOOG, D. A.; CROUCH, S. R. Princípios de Análise Instrumental.
6ª ed. Porto Alegre, BR: Bookman, 2009.
HUCKINS, J.N.; TUBERGEN, M. W.; MANUWEERA, G.K. Semipermeable
membrane devices containing model lipid: anew approach to monitoring the
biovalilability of lipophilic contaminants and estimating their bioconcentration
potencial. Chemosphere,v. 20, n. 5, p. 533-552, 1990.
LEBO, J. A.; GALE, R. W.; PETTY, J. D.; TILLITT, D. E.; HUCKINS, J. N.;
MEADOWS, J. C.; ORAZIO, C. E.; ECHOLS, K.R.; SCHRORDER, D. J. Use
of semipermeable membrane devices as an in situ sampler of waterborne
bioavailable PCDD and PCDF residue at sub-parts-per-quadrillion
concentrations.Environ. Sci. Technol., v. 29, p. 2886-2892,1995.
50

LIMA, R. M. G.et al. Remoção do íon amônio de águas produzidas na exploração de
petróleo em áreas offshore por adsorção em clinoptilolita. Quím.Nova, 2008,
vol.31, no.5, p.1237-1242.
MANAHAN, S. E. Toxicological chemistry a guide to toxic substances in chemistry.
Lewis, Chelsea, Ml; 1989.317 p.
MILANI, E. J. et al. Petróleo na margem continental brasileira: geologia, exploração,
resultados e perspectivas. Rev. Bras. Geof., São Paulo, v. 18, n. 3, p. 352-
396, 2000.
MONIÉ, F. Petróleo, industrialização e organização do espaço regional. In: Piquet,
R. (Org). Petróleo, royalties e região. Rio de Janeiro: Garamond. p257-286.
2003.
NETO, J. B. O.; COSTA, A. J. D. A Petrobrás e a exploração de petróleo offshore no
Brasil: um approach evolucionário. Rev. Bras. Econ., Rio de Janeiro, v. 61, n.
1, p. 95-109, Mar 2007.
NUNES, G.C. Water treatment in brown fields. International seminar on oilfield water
management. Rio de Janeiro. 2010.
OLIVEIRA, M. A. M.; ITABIRANO, J. A. P.; MAINIER, F. B.; MORANI, B. S. L;
ALMEIDA, L. C. M. Estudo da estabilidade térmica dos revestimentos de
cádmio com detecção através de voltametria por pulso diferencial. VIII
Simpósio de excelência em gestão e tecnologia (SEGeT), 2011.
OLIVEIRA, R. C. G.; OLIVEIRA, M. C. K. Remoção de contaminantes tóxicos dos
efluentes líquidos oriundos da atividade de produção do petróleo no mar.
Boletim técnico PETROBRAS, Rio de Janeiro, vol.43, p.129-136, 2000.
PETTY, J. D.; POULTON, B. C.; CHARBONNEAU, C. S.; HUCKINS, J. N.; JONES, S.
B.; CAMERON, J. T.; PREST, H. F. Determination of bioavailable
51

contaminants in the lower Missouri River following the flood of
1993.Environ. Sci.Technol., v. 32, p. 837-842, 1998.
PETTY, J. D.; ORAZIO, C. E.; HUCKINS, J. N.; GALE, R. W.; LEBO, J. A.;
MEADOWS, J. C.; ECHOLS, K. R.; CRANOR,W. L. Considerations involved
with the use of semipermeable membrane devices for monitoring
environmental contaminants. Journal of Chromatography A., v. 879, p. 83-
95, 2000.
RIBEIRO, F. A. L. et al. PCA: uma ferramenta para identificação de traçadores
químicos para água de formação e água de injeção associadas à produção de
petróleo. Quím. Nova, São Paulo, v. 36, n. 9, p. 1281-1287, 2013.
ROBAINA, N. F. Desenvolvimento de novas técnicas de extração para a
determinação de poluentes orgânicos e inorgânicos em águas salinas e
petróleo. 2015. Tese de Doutorado – Instituto de Química, Universidade
Federal Fluminense, Niterói, 2015.
ROSA, A. J.; CARVALHO, R. S.; XAVIER, J.A.D. Engenharia de Reservatórios de
Petróleo, Editora Interciência, 2006.
RUPP, M. T. C. Tese de Doutorado, Universidade Federal do Rio de Janeiro, Brasil,
1996.
SILVA, J. M. C. Impactos Ambientais da Exploração e Produção de Petróleo na
Bacia de Campos, RJ. IV Encontro Nacional de Anppas. Brasília. Jun 2008.
Disponível em <http://www.anppas.org.br/encontro4/cd/ARQUIVOS/GT4-809-
870-20080518190501.pdf> Acesso em 13 de novembro de 2015.
SIQUEIRA, L.C.G. Avaliação do impacto das emissões de metais geradas no
coprocessamento de resíduos em fábricas de cimento. Dissertação
[Mestrado]. Faculdade de Saúde Pública, Universidade de São Paulo. São
Paulo; 2005.
52

SOTO, C. A. T.; COSTA JR, A. C.; VERSIANE, O.; LEMMA, T.; MACHADO, N. C.
F.; MONDRAGÓN, M. A.; MARTIN, A. A.; Surface enhanced Raman
scattering, natural bond orbitals and Mulliken atomic charge distribution in the
normal modes of diethyldithiocarbamate cadmium (II) complex, [Cd(DDTC)2].
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, vol.
146, pp. 192–203, 2015.
SOUZA, M. O. et al. Análise exploratória das concentrações dos metais Na, Ca, Mg,
Sr e Fe em extrato aquoso de petróleo, determinados por ICP OES, após
otimização empregando planejamento de experimentos. Quím. Nova, São
Paulo, v. 38, n. 7, p. 980-986, Ago 2015.
TEIXEIRA, H. M. F. Desenvolvimento e aplicação de metodologias para
caracterização multielementar de água conata em amostras de petróleo.2007.
Tese de Doutorado – Departamento de Química, Pontífica Universidade
Católica do Rio de Janeiro, Rio de Janeiro, 2007.
THOMAS, J. E. Fundamentos de Engenharia de Petróleo, Editora Interciência: Rio
de Janeiro, 2ª ed.; 2004.
VIEIRA, R. K. A determinação voltamétrica multisequencial de Pb, Cd, Zn e Cu em
amostras complexas usando um sistema ternário homogêneo de solventes.
Tese [doutorado]. Instituto de Química, Universidade Estadual de Campinas.
Campinas; 2004.
YOUSEFI, S. R.; SHEMIRANI, F.; Development of a robust ionic liquid-based
dispersive liquid–liquid microextraction against high concentration of salt for
preconcentration of trace metals in saline aqueous samples: Application to the
determination of Pb and Cd. Analytica Chimica Acta, vol. 669, pp. 25–31,
2010.
53





























