
J. J. R. FRAÚSTO DA SILVA
ESTUDOS C O M C O M P L E X O N A S
A ESTABILIDADE DOS COMPLEXOS DOS IÕES MONOVALENTES
L I S B O A 1 9 6 4
V3JWQ1V VI9M3W3 30 OlfUliSNI

A MEUS PAIS
A MINHA MULHER
A MEUS FILHOS

PREFÁCIO 9
SIMBOLOGIA E ABREVIATURAS 10
I. INTRODUÇÃO:
1 — Aspecto geral do problema 11
2 — Âmbito da presente investigação 12
3 — Definições. Bases teóricas da técnica experimental 13
II. RESULTADOS E DISCUSSÃO I
1 — Estudos com o ácido uramildiacético e seus ho
mólogos 15
2 — Estudos com o ácido o-hidroxifeniliminodia-
cético e outras complexonas 24
3 — Relações entre a estabilidade dos complexos
e a basicidade das complexonas 33
4 — Considerações finais 42
III. SECÇÃO EXPERIMENTAL:
A. PREPARAÇÕES:
1 — Síntese das complexonas 44
2 — Soluções 50
B. APARELHAGEM E TÉCNICA:
1 — O titulador com eléctrodo de vidro 52
2 — Técnica experimental 53
C. CONSIDERAÇÕES SCBRE ÀS CONDIÇÕES EXPERI
MENTAIS E RFSULTADOS OBTIDOS ;
1 — Uso de concentrações em vez de activi
dades 55
2 — Electrólito de suporte 56
3 — Determinação das funções termodinâmicas 57
4 — Precisão dos valores obtidos 58
IV. MÉTODOS DE CÁLCULO:
1 — Constantes de dissociação de ácidos polibásicos 59
2 — Constantes de estabilidade de complexos 62
3 — Formação de complexos com o anião do elec
trólito de suporte 65
APÊNDICE: Tabelas de resultados experimentais 67
SUMÁRIO 81
ABSTRACT 83
BIBLIOGRAFIA ,. -...« . a . •.• i-, w :•. 85

Prefácio
A presente investigação iniciou-se no Laboratório de
Química Inorgânica da Universidade de Oxford (Ingla
terra) em Setembro de 1960 e a maior parte do trabalho
experimental foi realizada nos dois anos que se seguiram,
sob a orientação do Prof. H . M . N . H . IRVING. Após
o regresso a Portugal continuámos os estudos no Labo
ratório de Química Analítica do Instituto Superior Téc
nico até à conclusão da Tese na sua forma actual.
Na redacção desta seguiram-se as normas de nomen
clatura adoptadas pela União Internacional de Química
Pura e Aplicada (I. U. P. A. C); deparámos, no entanto,
com inúmeras dificuldades, porque parte da terminologia
ainda não tem equivalente em português. Fomos, pois,
obrigados a escolher traduções que nos pareceram apro
priadas, e até mesmo a introduzir neologismos científicos
cuja legitimidade será, possivelmente, discutível. Sacri
ficámos também, por vezes, a propriedade de certas
designações a uma maior simplicidade de exposição e
à correspondência com o que é hábito noutros idiomas.
Em desacordo com as normas internacionais, utilizámos
porém o termo «complexona» em vez de «complexana»,
que não encontrou aceitação universal.
Ao longo dos anos dedicados a este trabalho criámos
dívidas de gratidão para com diversas pessoas e enti
dades. Entre elas, seja-nos permitido destacar o Instituto
de Alta Cultura, pela concessão de uma bolsa de estudo,
a Universidade de Oxford, pelas facilidades com que nos
honrou, os Doutores M . STACEY, P . D . WILSON e A . O .
MACZEK, por numerosas e estimulantes discussões em
que nasceram muitas das ideias adiante discutidas, o
Eng. J . G . CALADO, que amavelmente leu e criticou o
original, o Eng. C . M . PULIDO, pelo inestimável auxílio
prestado durante a impressão, a Ex.'""' Senhora Dona
ILDA PROENÇA, que pacientemente dactilografou todo o
difícil manuscrito, e, finalmente, em particular, o Prof.
Eng. A . HERCULANO DE CARVALHO, presente em toda
a Tese, pelos seus ensinamentos e conselhos, e o Prof
H . M . N . H . IRVING, D . PHIL., D . S c , F . R . I . C , que
propôs o tema da investigação e em grande parte a orientou.
A todos agradece muito sinceramente
O AUTOR

Simbologia e abreviaturas
M
L
N
¡A
[X = 0,1
[A - > 0
m
fx K
1K
P»
T P »
/c
T / c
X
ião do metal
ligante
raio iónico
carga do ião
carga nuclear efectiva
número de coordenação do complexo
grau de formação do sistema
temperatura
força iónica
M (X) força iónica acertada a 0,1 M por adição
do sal X
força iónica extrapolada a diluição infinita
concentração livre de X
actividade de X
factor de actividade de X
constante de formação (estabilidade) parcial
estequiométrica
constante de formação (estabilidade) parcial
termodinâmica
constante de formação (estabilidade) global
estequiométrica
constante de formação (estabilidade) global
termodinâmica
constante de dissociação parcial estequiomé
trica
constante de dissociação parcial termodinâ-
constante de dissociação global estequiomé
trica
constante de dissociação global termodinâ
mica
G°(X) ou Gí^ energia livre padrão de X, em solução, se
gundo GIBBS
H°(X) ou í?^- entalpia padrão de X, em solução
5 o (X) ou S'^, entropia padrão de X, em solução
A G°
A í f °
A S°
Ca c
CL
CM
CB
pX
R
« .Y
M. M.
P-f-dec.
cale.
anal.
In log
I M D A
MIM DA
H E X D A
NITA
EDTA
PRODTA
variação da energia livre padrão
variação da entalpia padrão (entalpia liga-
cional)
variação da entropia padrão (entropia liga-
cional)
concentração total do ligante
concentração total do metal
concentração da solução da complexona
concentração da solução do sal do metal
concentração do titulante
volume total inicial da solução
grau de neutralização
— log [X]
a C„ + [ í M ] - [ O f f - ] / Ca
i + i P Í [xy ¿•=0
- / ?H ixy
Massa molecular
Ponto de fusão
decomposição
análise elementar calculada
análise elementar experimental
logaritmo natural
logaritmo decimal
ácido iminodiacético
ácido metiliminodiacético
ácido ciclohexiliminodiacético
ácido nitrilotriacético
ácido etilenodiaminotetracético
ácido 1,2-diaminopropanotetracético

ESTUDOS C O M C O M P L E X O N A S
I. Introdução
1 — ASPECTO GERAL DO PROBLEMA
O estudo dos compostos de coordenação, que tanto
interesse têm despertado nos últimos anos, deu origem
a uma renovação total da Química Inorgânica, com
importantes repercussões em vários outros domínios da
química moderna. No campo particular da Química
Analítica, a possibilidade de utilização de comple-
xantes, em especial de ácidos aminopolicarboxílicos,
como agentes sequestrantes e reagentes para deter
minações volumétricas abriu horizontes novos e, em
curto espaço de tempo, assistiu-se a uma expansão
de técnicas e métodos que dificilmente terá sido igua
lada anteriormente ou poderá talvez vir a ser igualada
no futuro.
Os primeiros trabalhos com complexonas devem-se a
BRINTZINGER (1) , PFEIFFER (2) e aos seus colabora
dores, que prepararam, no estado sólido, complexos
dos ácidos etilenodiaminotetracético ( E D T A ) e nitrilo-
triacético ( N I T A ) . Mais tarde, SCHWARZENBACH (3) e
col. retomaram o problema noutra base, analisando a
formação desses complexos em solução aquosa e de
terminando os valores das respectivas constantes de
estabilidade. O seu grupo de investigadores sintetizou nu
merosos ácidos aminopolicarboxílicos («complexonas»)
e estudou-os exaustivamente sob este ponto de vista.
Entretanto, nos E. U . A . , BERSWORTH, MARTELL (4) e
col. desenvolveram um método industrial de síntese
de complexonas, o que possibilitou uma generalização
dos estudos básicos e grande incremento nas aplicações
práticas. Hoje em dia não há praticamente qualquer
indústria que em muitas e variadas instâncias não tenha
feito apelo às extraordinárias propriedades daqueles
compostos.
Como consequência dos estudos teóricos, foram-se de
senvolvendo paralelamente diversos métodos analíticos,
constituindo-se uma nova técnica ora designada por
«complexometria» ou «quelatometria». Como seus pre
cursores devem apontar-se principalmente SCHWARZEN
BACH, IRVING e col., nos aspectos teóricos fundamentais,
e PRTBIL, propriamente nas aplicações.
Mais recentemente, a teoria das «titulações complexo-
métricas» foi substancialmente desenvolvida, em especial
por RINGBOM (5) e WANNINEN (6) na Finlândia, CHA-
BEREK, MARTELL (7) e REILLEY (8) nos E. U. A.
A grandeza fundamental sobre a qual se apoiam os
estudos teóricos, os métodos analíticos e as aplicações
práticas é o valor das constantes de estabilidade termo
dinâmicas dos complexos formados, que permitem prever
a possibilidade de formação dos complexos em diversas
condições, e todos os autores são unânimes em re
conhecer que esses valores dependem não só de diversas
características dos ligantes como da natureza dos iões dos
metais em solução. Entre as características importantes
dos ligantes devem mencionar-se o número de átomos
coordenantes, o seu tipo e a sua basicidade, a possi
bilidade de formação de anéis de quelação, o número
destes anéis e o grau de tensão neles criado, a exis
tência de grupos provocando impedimentos espaciais,
etc. A influência destas características ainda não está
totalmente compreendida.
No que respeita aos metais cujos iões funcionam como
elementos centrais nos complexos, os efeitos dominantes
sobre a estabilidade têm origem na estrutura electrónica
específica de cada um, tendo sido propostas nesta base
diversas classificações. As mais conhecidas são as de
AHRLAND e CHATT (9) e a de SCHWARZENBACH (10),
bastante diferentes na definição e na essência.
Os primeiros autores distinguem duas classes de metais
que designam por classe (à) e classe (b); à primeira
1 1

pertencem os metais que formam complexos mais es
táveis com os elementos mais leves de cada grupo de
doadores no Quadro Periódico, isto é, com o azoto,
o oxigénio e o flúor; à segunda pertencem os metais
que formam complexos mais estáveis com qualquer dos
elementos mais pesados desses grupos de doadores,
isto é, o fósforo, o enxofre, o cloro, etc.
Dentro desta classificação, cada estado de oxidação do
metal deve ser olhado como um receptor distinto, e
assim, por exemplo, o ião Cu+ pertence à classe (b),
mas o ião C M 2 + pertence à classe (a). Os elementos
típicos da classe (b) ocupam uma zona triangular no
Quadro Periódico (fig. 1 ) e incluem, principalmente,
o ródio, o paládio, a prata, o irídio, a platina, o ouro
e o mercúrio; diversos outros elementos, tais como
o cobre, o ruténio, o cádmio, o ósmio, o tálio, e tc ,
apresentam um carácter intermédio.
Cr Mn Fe Co Ni J Cu I Zn Ga Ge As
Mo Tc [ Ru Rh Pd Ag Cd j In Sn Sb
W I Re Os Ir Pt Au Hg Tl Pb Bi |
Fig. 1 — Melais da classe (b).
SCHWARZENBACH propõe uma divisão baseada num cri
tério mais geral: a configuração electrónica apresentada
pelo ião do elemento que se considera. Distingue assim
três grupos, em que as características principais apon
tadas são as seguintes:
1.° grupo A — Catiões que possuem a estrutura de
um gás inerte: 1 s2 ou m2n/>6
É constituído pelos iões dos metais alcalinos e dos metais
alcalino-terrosos, pelos iões Alz+, Scs+, etc. A estabi
lidade dos complexos destes iões é principalmente de
terminada por atracções electrostáticas. Os seus aquo-
complexos são mais estáveis que os aminocomplexos
por ter a água um momento dipolar permanente mais
elevado que o amoníaco.
2.° grupo B — Catiões com subníveis d completamente
preenchidos: (n — 1) d10
Fazem parte deste grupo iões como Cu+, Ag+, Au+,
Zn2+, Cd2+, Hg2+, Ga ( I I I ) , In ( I I I ) , Tl ( I I I ) , etc.
A estabilidade dos complexos destes iões é principal
mente determinada por diferenças de electronegatividade
entre os ligantes e o metal, sendo tanto mais estáveis
quanto mais nobre for o metal e menos electronegativo
for o átomo doador do ligante. Devido ao elevado
poder deformante dos iões, os seus aminocomplexos
são mais estáveis que os aquocomplexos, pois o mo
mento dipolar total na molécula de amoníaco (mo
mento permanente + momento induzido) é superior ao
da água.
3.° grupo C — Catiões com subníveis incompletamente
preenchidos: (n — 1) Í / 1 - 9 e (n — 2) y 1 - 1 3
É o grupo dos metais de transição e transição interna
para os quais se observam propriedades dos dois grupos
anteriores. O predomínio de um ou outro tipo de
características depende fundamentalmente da carga, raio
iónico e potencial de ionização do ião em causa.
O próprio estabelecimento destas classes ou grupos
evidencia a possibilidade de boas correlações entre
valores das constantes de estabilidade dos diversos
complexos que se possam formar dentro de cada tipo
de iões e certas propriedades do elemento central, tais
como valores do raio iónico, potenciais de ionização
e electronegatividades, entre as mais frequentemente
utilizadas.
A comparação de valores para iões de grupos dife
rentes é, no entanto, difícil, pois o número de factores
determinantes da estabilidade varia de caso para caso.
Para fundamentar esta afirmação bastará relembrar que
para os iões dos metais de transição as diferenças de
energia de estabilização do campo de ligantes ( 1 1 , 12)
e a possibilidade de ocorrência de ligações TC ligante-
-metal ou metal-ligante (13 ) podem sobrepor-se a outros
efeitos e invalidar correlações que seriam de esperar
na base de um comportamento «normal» desses iões.
Apesar dos numerosos e excelentes estudos existentes
sobre a influência do tipo de elemento central na esta
bilidade dos complexos com ele formados, o problema
continua longe de estar completamente esclarecido, prin
cipalmente porque alguns grupos de metais foram apenas
superficialmente examinados e só muito recentemente
se tomou consciência da importância de alguns factores.
Encontra-se no primeiro caso a maioria dos iões mono-
positivos e no segundo a mencionada possibilidade de
ocorrência de ligações TT dativas metal-ligante.
2 — ÂMBITO DA PRESENTE INVESTIGAÇÃO
Os nossos trabalhos têm visado sobretudo o esclareci
mento da influência da estrutura dos ligantes e do tipo
de elemento central na estabilidade dos complexos. Esta
Tese é uma contribuição nesse sentido, sendo apresen-
1 2

lados e discutidos os resultados de estudos realizados
com um grupo de iões monopositivos dos grupos la,
\b e LUò do Quadro Periódico: Li+, Na+ e K+ (com
estrutura de gás inerte), Tl+ (com estrutura do tipo
(n — 1) d10 lis2, não considerada até aqui) e Ag+ (com
estrutura do tipo (n—1) d 1 0 ) . Foram também consi
derados em certo pormenor os complexos dos metais
alcalino-terrosos e do zinco.
Os ligantes escolhidos para este trabalho foram, pelo
seu interesse analítico e industrial, as «complexonas»,
e destas, especialmente, as derivadas do ácido imino-
diacético.
Determinaram-se constantes de estabilidade para os
complexos formados pelos iões seleccionados com vá
rios produtos comerciais (ácidos iminodiacético, meti-
liminodiacético, nitrilotriacético, etilenodiaminotetracé-
tico, 1,2-diaminopropanotetracético e 2-hidroxiciclohexi-
liminodiacético) e com uma série de novas complexonas
que sintetizámos para o efeito (ácidos uramildiacético,
1-metiluramildiacético, 1,3-dimetiluramildiacético, o-hi-
droxifeniliminodiacético, o-metoxifeniliminodiacético,
o-mercaptofeniliminodiacético, ciclohexiliminodiacético
e c-carboxifeniliminodiacético).
A análise da influência da estrutura dos novos ligantes
na estabilidade dos complexos por eles formados e a
discussão das relações observadas nos complexos dos
metais alcalinos e alcalino-terrosos constituem os pontos
centrais da Tese.
3 — DEFINIÇÕES. BASES TEÓRICAS
DA TÉCNICA EXPERIMENTAL
Pode definir-se «complexo» como uma entidade formada
pela associação de espécies mais simples capazes de
existência independente.
Segundo esta definição, poderão formar-se «complexos»
pela associação de partículas neutras ou carregadas,
positivas ou negativas. Esta associação pode caracte
rizar-se termodinâmicamente por uma «constante de
estabilidade», que é a constante de equilíbrio da reacção.
B + iiA Zl BAn ( 1 )
Será, pois, por definição,
X = (BAJ I {Af . (B) (2)
onde os valores entre parêntesis representam «activi
dades»; J ( 3 H designa-se neste caso por «constante de
estabilidade termodinâmica global» do complexo BAn.
Na verdade, está já perfeitamente estabelecido que a
formação de um complexo do tipo BAn não é instan
tânea; tem lugar através de uma série de fases e, con
soante as actividades dos reagentes e as variações de
energia livre de GIBBS, pode acontecer que para deter
minadas condições coexistam em equilíbrio vários dos
sucessivos complexos BA, BA2, BAn. A formação
destas espécies pode ser também convenientemente
caracterizada termodinâmicamente por «constantes de
estabilidade parciais», TKn, definidas para a reacção
BAn„x + AZlBAn (3)
Será então
TKn = {BA,) I ÍBAn^) (A) (4)
A constante global é, evidentemente, o produto das
constantes parciais:
R ( 3 ( Í = TKÍ .TK2
rKn = I I TKn (5)
De acordo com as definições anteriores, os ácidos poli
básicos são também «complexos» e podem determi
nar-se constantes de estabilidade (formação) das várias
espécies protonadas. O hábito consagrou, porém, o uso
de «constantes de dissociação», que são constantes de
equilíbrio para reacções do tipo de
HpLim-ph Zl Hp_xÜ
m^+V)~ + H+ (*) (6)
ou seja
Tk?m-P+1) = iH^L) (H) I (HpL) (7)
Definem-se também neste caso constantes de dissociação
parciais e globais, facilmente calculáveis a partir de
valores obtidos na titulação dos ácidos respectivos com
uma solução-padrão de base forte. Da curva de titu
lação obtém-se a concentração molar do ácido, a fracção
neutralizada e a correspondente concentração hidroge-
niónica da solução; será necessário conhecer ainda os
«coeficientes de actividade» das diversas espécies para
determinar as constantes «termodinâmicas», mas, como
mais tarde se verá com maior pormenor, é preferível
trabalhar num meio contendo grande excesso de um
electrólito neutro que actua como «suporte». Definem-
(*) Por uma questão de simplicidade utiMzou-se neste trabalho
o símbolo H + para a espécie i J 3 0 + .
1 3

-se então constantes «estequiométricas» que dependem
apenas da concentração das espécies reaccionáis, sendo
válidas para o meio em que foram determinadas. Para
os complexos formados com iões metálicos o processo
poderá ser análogo, pois os ligantes estudados no pre
sente trabalho são bases conjugadas de ácidos no
sentido de BR0NSTED.
Na presença do ião metálico estabelece-se o seguinte
equilíbrio:
Mq+ + HpLim~p}- Zl ML{m-l]- + pH + (8)
Este tipo de reacção pode ser rigorosamente estudado
por potenciometria, utilizando eléctrodos de hidrogénio,
quinidrona ou vidro, para medir a concentração hidro-
geniónica «livre». Este valor é uma medida da estabi
lidade relativa dos complexos formados com o metal
e com o hidrogenião e, se forem conhecidos os valores
das constantes de dissociação do ligante como ácido,
fácilmente se calculam as constantes de estabilidade
dos complexos com o ião metálico.
No caso das complexonas não se formam em geral
especies de complexidade superior a M £ 2 ; definindo
o «grau de formação» n como o número médio de
moléculas do ligante coordenadas a cada ião metálico,
será (14):
_ concentração total de ligante coordenado n = =
concentração total do metal
= ÁM±±±l±KhL^ = [ ¿ ] ( * i + 2 KiK* M}
[M] + [ML] + [ML2] l + mfa + K&ÍL]}
ou
n + (7i — 1) K} [L] + (n — 2) KXK2 [ L ] 2 = 0 (9)
onde Kx e K2 são constantes de estabilidade estequio
métricas parciais das especies ML e ML 2 e os valores
entre colchetes representam concentrações molares.
Analisando esta equação, é evidente que se poderão
determinar os valores das constantes K± e K2 se se
conhecerem pelo menos dois pares de valores para
n e [ i ] . Em casos mais complicados poderão formar-se
complexos protonados ou hidroxocomplexos e será neces
sário um tratamento matemático mais elaborado, mas,
sob o ponto de vista experimental, o problema é idên
tico. O cálculo de valores de n e [ i ] pode fazer-se conhe
cendo as constantes de dissociação ácida do ligante
e a concentração hidrogeniónica das soluções em várias
fases da neutralização, na presença de uma concen
tração total conhecida de um dado metal. Neste tra
balho utilizou-se a técnica de CALVIN e WILSON (15) ,
onde as várias fases de neutralização são conseguidas
por titulação com uma solução-padrão de base forte.
Este método fornece grande número de pontos expe
rimentais, o que permite a obtenção de valores óptimos
pára as constantes e uma estimativa rigorosa dos erros.
Os resultados são apresentados no capítulo respectivo
à medida que o desenvolvimento da discussão os exige.
Seguem-se secções dedicadas à síntese de complexonas,
à técnica utilizada e aos métodos de cálculo. Os va
lores experimentais são dados no texto na forma de
curvas de titulação e reúnem-se em Apêndice sob a
forma tabular.
14

II — Resultados e discussão
1 — ESTUDOS COM O ÁCIDO URAMILDIACÉ-
TICO E SEUS HOMÓLOGOS
É lícito afirmar que foram os metais alcalino-terrosos
os responsáveis directos pelo enorme interesse suscitado
pela introdução das complexonas na rotina analítica.
Com efeito, foi com vista à determinação da dureza
das águas que SCHWARZENBACH sugeriu pela primeira
vez o emprego daquelas substâncias.
Estava-se então na Primavera de 1945, mas foram ainda
necessários alguns anos para que esta sugestão pro
duzisse os devidos efeitos. Nos numerosos estudos que
se seguiram, os metais alcalino-terrosos continuaram
com lugar de primazia, acompanhados de perto pelos
metais de transição da primeira série e por alguns
outros elementos isolados, devido a interesses espe
cíficos de diversa ordem. Outros elementos foram,
porém, quase completamente esquecidos, entre eles os
metais alcalinos e, de uma maneira geral, os que existem
em solução no estado de catiões monopositivos, como
o tálio, a prata e outros. Este desinteresse foi, natural
mente, motivado pela reduzida estabilidade dos com
plexos formados por aqueles iões, mas o escasso número
de valores existentes originou uma lacuna que tem de
ser preenchida antes de procurar generalizações e for
mular leis.
Com este fim iniciámos uma investigação sistemática
sobre a estabilidade das espécies formadas pelas com
plexonas com diversos tipos de iões, em especial
com os monopositivos. Como base deste estudo foi
escolhido o ácido 5-aminobarbitúrico-7,7-diacético (ura-
mildiacético)-I, uma das primeiras complexonas pro
postas por SCHWARZENBACH (16), que verificou possuir
esta substância uma afinidade invulgar para os iões
Na+ e Li+, formando com eles complexos de esta
bilidade apreciável.
Por não ser um produto comercial, teve de ser encarada
a respectiva síntese e, para permitir comparações e
esclarecer a influência da estrutura dos ligantes na
estabilidade dos complexos, foram igualmente sinteti
zados dois análogos daquela complexona: os ácidos
l-metil-5-aminobarbitúrico-7,7-diacéttco (1-metiluramil-
diacético)-II e l,3-dimetil-5-aminobarbitúrico-7,7-dia-
cético (1,3 dirnetiluramildiacéíico)-!!!.
0 = C 2 5 CH—N,
R,, R2 = H R ^ C H j , R2 = H R r R 2 = C H ,
/ // IH
A primeira amostra de ácido uramildiacético, preparado
segundo as instruções de SCHWARZENBACH (16), apre
sentou um comportamento estranho, dando curvas de
titulação anómalas, quer quando só, quer na presença
de iões metálicos. Estas curvas eram reprodutíveis após
sucessivas recristalizações do produto com água desio-
nizada.
Por ignição obteve-se, porém, um resíduo alcalino, e,
finalmente, uma análise elementar mostrou que a subs
tância preparada era realmente um sal ácido, corres
pondendo à fórmula HsUr.NaH2Ur. H20 (onde Ur =
= CaH707N3); trata-se pois de um caso análogo ao
do tetroxalato de potássio H%Ox. KHOx (Ox=C2Oi).
Este resultado verificou-se estar de acordo com as
curvas de titulação obtidas, consistentes com as cal-
1 5

culadas para uma mistura equimolecular dum ácido
tribásico HzUr com um ácido dibásico hidratado
NaH2Ui\H20.
A tendência para formar sais ácidos de sódio pouco
solúveis e de composição definida foi também notada
com os outros dois homólogos sintetizados, e é de
crer que esta seja a razão de algumas dificuldades
experimentadas por outros autores na síntese destas
substâncias (16, 17). No entanto, foi por nós verificado
que os ácidos livres podiam ser obtidos com facilidade
por recristalizações sucessivas dos sais ácidos com
ácido perclórico diluído, e, no caso do ácido uramil-
diacético, este podia mesmo ser obtido directamente
pelo método de SCHWARZENBACH, usando um grande
excesso de ácido mineral na precipitação da complexona
(vd. secção preparativa).
Os resultados da análise elementar do produto pre
parado por SCHWARZENBACH correspondem, segundo
este autor, a um mono-hidrato — HzlIr.H20 — que
seria extremamente estável, uma vez que não se notou
perda de peso após aquecimento durante 8 horas numa
estufa de vácuo a 100°C e à pressão de 0,005 mm de
mercúrio. Para a nossa amostra, porém, os resultados
da análise elementar correspondem ao produto anidro
HzUr. Também o ácido 1-metiluramildiacético foi obtido
na forma anidra e só o ácido 1,3-dimetiluramildiacético,
preparado pela primeira vez, se obteve no estado de
mono-hidrato, perdendo, no entanto, toda a água após
10 horas de aquecimento a 100°, à pressão de 25 mm
de mercúrio. Tem ainda interesse mencionar que algumas
tentativas infrutíferas para preparar o ácido tiouramil-
diacético conduziram à formação de ácido uramildia-
cético, sempre na forma anidra.
As discordâncias apontadas parecem indicar a possibi
lidade da existência de duas formas, uma hidratada
e outra anidra, para as substâncias preparadas, o que
não é anormal; no entanto, deve notar-se que os resul
tados da análise elementar para o produto obtido por
SCHWARZENBACH correspondem melhor aos calculados
para o sal ácido HzUr.NaH2Ut.H20 do que aos cal
culados para a espécie H3Ur.H20 postulada (tabela II.1).
Tabela II. 1
C H JV
Presente trabalho 3 7 , 1 3 , 6 1 6 , 0
Produto preparado por SCHWARZENBACH 3 4 , 3 5 3 , 6 9 1 5 , 0 2
Calculado para H^Ur 3 7 , 1 3 , 5 1 6 , 2
Calculado para H3Ur.H20 3 4 , 6 4 , 0 1 5 , 2
Calculado para H3Ur.NaHaUr.H20 3 4 , 4 3 , 4 1 5 , 0
Sem se desejar forçar uma conclusão, parece provável
que algumas disparidades nos resultados obtidos em
pontos comuns do presente trabalho e no de outros
autores (16) possam ser explicadas nesta base.
Os valores das constantes de dissociação dos três li
gantes foram determinados a partir das respectivas
curvas de titulação potenciométrica, tal como se des
creve na secção dedicada aos métodos de computação.
Para a determinação da primeira e segunda constantes
utilizou-se como titulante uma solução-padrão de hi
dróxido de potássio isenta de carbonato; como elec
trólito de suporte utilizou-se nitrato de potássio, man
tendo a força iónica constante a 0,100 M. Sendo de
esperar a formação de complexos com o ião potássio,
houve que utilizar para a determinação de pkz outro
titulante e outro electrólito de suporte, respectivamente
hidróxido de tetrametilamónio e nitrato de tetrametila-
mónio. Os valores obtidos apresentam-se na tabela I I . 2 .
Estes resultados estão de acordo com as ideias de
SCHWARZENBACH acerca da localização dos protões na
molécula do ácido uramildiacético; na verdade, os
valores de p/q e pk2 correspondem à ionização de
grupos carboxílicos, enquanto que pkz corresponde à
ionização de um radical amónio. A disparidade entre
os valores obtidos neste trabalho, calculados para uma
força iónica nula usando a equação de DAVIES, e os
valores obtidos anteriormente (16), era já de esperar
na base das considerações feitas acerca dos produtos
utilizados. Para pk3, no entanto, a concordância é
satisfatória.
O aumento progressivo nos valores de p/cx e pkz veri
ficado na série I, II, III estudada deve-se certamente
ao efeito indutivo dos grupos metilo substituintes; o
valor de pk2 mantém-se constante e deve pois corres
ponder à ionização de um dos carboxilos menos afec
tados pelos grupos metilo. O primeiro protão, porém,
não pode ser concretamente localizado na molécula das
complexonas, como se verá recordando o caso dos
barbituratos.
NOLLER (18) afirma que o ácido barbitúrico (IVa) pode
ser estabilizado na forma enólica por ressonância no
//° / N H - S
0 = C CH,
NH—C
IVa
OH /
N - C , / / \ \
HO-C CH W
\ OH
m
o II
/ N H - C R,
o=c c NH—C V
1 6

ciclo pirimidinico (IVb) e que as propriedades elec-
trófilas dos dois átomos de azoto aumentam o carácter
ácido do grupo ^> C-OH situado entre eles, pelo que
este ácido (pk = 4 ,0 ) é ainda mais forte que o ácido
acético (pk = 4,7) . Em favor da sua teoria, NOLLER
aponta que a inserção de dois alquilos na posição 5
impede a ressonância na molécula destes derivados do
ácido barbitúrico (V), e, uma vez que os grupos imino
simples nas posições 1, 3 e 5 , ou por substituição simul
tânea nas posições 1 e 3 . Pelo contrário, a substituição
dupla na posição 5 impede completamente a resso
nância. Sendo assim, o primeiro protão ionizável nos
5,5-barbituratos (V) não pode ser o mesmo dos res
tantes casos; é mesmo surpreendente que, nestas con
dições, o ácido barbitúrico não seja tribásico, uma
vez que os barbituratos são dibásicos (19) , mas é natural
Tabela 11.2
CONSTANTES D E DISSOCIAÇÃO D O ÁCIDO URAMILDIACÉTICO E SEUS HOMÓLOGOS
(T = = 20,0°C)
Composto P*i Ref.:
Ácido uramildiacético (I) 1,7 ± 0,3
1,9
2,86
2,67 ± 0,02
3,1
3,76
9,63 ± 0,03
10,33
10,44
Presente trabalho (\x = 0,1)
Presente trabalho
([X = 0, calculado)
Valores de SCHWARZENBACH ( * )
Ácido 1-metiluramildiacético (II) 1,85 ± 0,05 2,67 ± 0,02 9,81 ± 0,01 Presente trabalho (jx = 0,1)
Ácido 1,3-dimetiluramildiacético (III) 2,05 ± 0,05 2,67 ± 0,02 10,12 ± 0,01 Presente trabalho ([x = 0,1)
(*) Ret . (16). Valores obt idos a pa r t i r de titulações com hidróxido de te t ramet i lamónio , sem electrólito de supor te , e calculados p a r a uma
força iónica p. = 0.
situados entre dois carbonilos são bastante menos
ácidos, serão de esperar valores de pk mais elevados.
É nossa opinião, porém, que nem a teoria exposta
nem a sua confirmação são satisfatórias, o que se
torna evidente examinando os valores das primeiras
constantes de dissociação dos ácidos 1-metilbarbitúrico
e 1,3-dimetilbarbitúrico, para os quais a ressonância no
anel pirimidinico é também impossível (tabela II.3).
Parece-nos, portanto, mais provável que o carácter
ácido pronunciado de algumas das espécies indicadas
esteja antes ligado à possibilidade de ressonância na
base conjugada da 4,6-dicetona, que constitui parte
das respectivas moléculas (VI e VII).
Na verdade, a estabilização adquirida por ressonância
deste anião não será influenciada por substituições
que a carga formal negativa no híbrido VII obrigue
a que a terceira ionização tenha lugar a um pH ina
cessível na prática.
Estas conclusões podem aplicar-se aos casos do ácido
uramildiacético e seus homólogos, que são, no entanto,
mais complexos, porque a introdução do grupo imino-
diacetato facilita muito a ionização do protão residual
na posição 5. Então, a estrutura da molécula não
Tabela II.3
CONSTANTES D E DISSOCIAÇÃO D O ÁCIDO
BARBITÚRICO E A L G U N S DOS SEUS DERIVADOS (*)
( r = 25°C, ¡X = 0)
Composto
—C
CH,
—c; +i-r
— C
.CH
Ácido barbitúrico
Ácido 1-metilbarbitúrico
Ácido 1,3-dimetilbarbitúrico
Ácido 5-isopropilbarbitúrico
Ácido 5,5-dietilbarbitúrico
Ácido 5-metil 5-fenilbarbitúrico
Ácido 5-etil 5-fenilbarbitúrico
4,035
4,348
4,679
4,94
7,97
7,73
7,45
VI VII (*) Ref. (69)
1 7

ionizada deve ser uma das abaixo representadas (A,
B ou C) ou um correspondente equilíbrio tautomérico.
Por ionização do primeiro protão obtém-se em qualquer
dos casos a estrutura D. A linha ponteada representa
uma insaturação parcial das ligações, correspondendo
pois a um híbrido de ressonância.
juntamente com outros valores já conhecidos para estas
substâncias.
Os resultados mostram que a ordem de estabilidades
dos complexos normais ML é K+ < Na+ < Li+ <
< Tl+ para os catiões univalentes, e Ba2+ < St2+ <
< Ca2+, Mg2+ < Bei+ para os catiões bivalentes.
N R , — C M C H , - C O O
0 = C C H — N H +
y / V - c { X C H 2 - C O O H V H O
/ N R R _ C \ / C H 2 - C O O H U _ B _ / m ^ \ e / C H ^ C 0 °
0 = C C H —N 0 = C e ) C - N H
YlR2— C ^ N C H 2 - C O O H p YlR—c/ X C H 2 - C O O H
0 ^ NR,—ó{ C H , - C O O H 0
- A - ° = \ Q/f-"\
NR—C C H 2 - C O O H " O
- C -
As fases de dissociação que se seguem são, evidente
mente, a ionização do grupo carboxilo e a do radical
amónio.
Embora este mecanismo pareça razoável, alguns pontos
ficam por explicar; em especial, torna-se difícil com
preender o elevado valor de pks para qualquer das
complexonas, quando o valor correspondente de pk2
para o ácido feniliminodiacético é apenas 4,96 em
condições idênticas; possivelmente, resultará do efeito
da carga formal negativa na zona adjacente do anel
dos primeiros compostos.
A formação de complexos com as novas substâncias
é evidenciada pelas respectivas curvas de titulação po-
tenciométrica, das quais se dá um exemplo (ííg. 2).
Pode verificar-se que se formam na verdade complexos
de estabilidade apreciável, mesmo com os iões K+ e
2?e 2 + , o que não tinha sido notado até ao presente.
Na tabela II.4 apresentam-se os valores das constantes
de estabilidade calculadas para os complexos das três
substâncias estudadas. Em alguns casos formam-se com
plexos ácidos MHL e noutros foi possível determinar
os valores para os complexos MLV Incluem-se nesta
tabela, para comparação, resultados também por nós
obtidos com duas complexonas comerciais, os ácidos nitri-
lotriacético (NITA) e etilenodiaminotetracético (EDTA),
- 1 0
1 i
pH
- 9
- 8 1 / h
- 7 / w / 1
1 y?^ / Á - 6
1 rf^ / 1 1
/ yn 1
- 5 / / \ ' A \
- 4 /
«a» ? 1 2 3
~ 2 1 1 1
Fig. 2 — Curvas de titulação do ácido uramildiacético na pre
sença de diversos iões.
1. NMe+ (100:1) 2. K+ (100:1) 3. Na+ (10:1)
4. Li+ (1:1) 5. 77+ (1:1) 6. Ba2 + (1:1)
7. Sr*+ (1:1) 8. Cc?+, Mg2+ (1:1) 9. Be2 + (1:1)
1 8

Tabela ¡1.4
Valores de log K para as espécies ML, MHL e ML2, complexos de metais com os ácidos uramildiacético (I),
1-metiluramildiacético (II), 1,3-dimetiluramildiacético (III), NITA e EDTA. Resultados válidos para T = 20,0°C e ¡x = 0,1 Aí
t ão Espécie I I I I I I N I T A E D T A
Li + ML 4,90 ± 0,02 4,86 ± 0,01 4,91 ± 0,01 2,51 ± 0,01 2,79 (b)
Na+ ML 2,72 ± 0,01 2,67 ± 0,01 2,53 ± 0,01 1,32 ± 0,02 1,66 (6) K+ ML 1,23 ± 0,03 1,11 ± 0,02 0,94 ± 0,01 — — T1+ ML 5,99 ± 0,02 5,79 ± 0,01 5,73 ± 0,01 4,74 ± 0,01 6,55 ± 0,01
MHL — — — — 2,06 ± 0,02 Be2 + ML 10,36 ± 0,02 10,42 ± 0,02 10,54 ± 0,02 7,11 (a) 9,27 (a)
MHL 3.44 3,32 3,54
Mg2 + ML 8,19 I 0,02 8,23 ± 0,01 8,29 ± 0,01 5,41 (è) 8,69 (b) MLZ 3,62 ± 0,05 3,72 ± 0,06 3,78 ± 0,02
Ca* + ML 8,31 ± 0,01 8,22 ± 0,01 8,13 ± 0,01 6,41 (6) 10,70 (6) ML<¡ 5,27 ± 0,02 5,38 ± 0,01 5,40 ± 0,02
S>2 + ML 6,93 + 0,02 6,83 ± 0,02 6,82 ± 0,02 4,98 (è) 8,63 (6) ML2 4,06 I 0,10 4,19 ± 0,02 4,27 ± 0,10
Bir + ML 6,13 ± 0,02 6,06 ± 0,01 6,00 ± 0 , 0 1 4,82 (&) 7,76 (b) 3,68 ± 0,10 3,85 ± 0,05 3,88 ± 0,10
Todos os valores foram obtidos no presente t raba lho , excepto (a) ref." 2 0 e (b) ref.» 21 . Valores p a r a Z.Í + , Na+ e K+ obt idos n u m meio com
(X = 0,1 M ({CH3)Í N.N03); todos os res tan tes n u m meio com [X = 0 ,1 M (KN0.ò).
A substituição sucessiva nas posições 1- e 1,3- não
tem influência na estabilidade dos complexos do lítio
mas produz um ligeiro abaixamento de estabilidade nos
complexos dos outros catiões univalentes, do cálcio,
do estrôncio e do bário. Com o magnésio e com o
berílio observa-se o efeito contrário e a ordem de
estabilidades Mgi+ < Cài+, verificada para os com
plexos do ácido uramildiacético, é invertida para o seu
homólogo dissubstituído. Quanto à ordem de estabili
dades dos complexos ML2 é sempre Mg2+ < Ba2+ <
< Sr2+ < Ca2+ e os valores das constantes aumentam
na série I < II < III, de acordo com o aumento de
basicidade do átomo de azoto respectivo; na formação
destes complexos devem pois estar envolvidos apenas os
grupos iminodiacetato de cada ligante.
A comparação entre as estabilidades dos complexos dos
ligantes sintetizados e dos ligantes comerciais NITA e
EDTA pode apreciar-se melhor sob uma forma gráfica.
É o que se faz na fig. 3, escolhendo para representante
do primeiro grupo o ácido uramildiacético.
Nesta figura vê-se que, exceptuando o caso do berílio,
os complexos do ácido uramildiacético com os metais
bivalentes são menos estáveis do que os complexos
do EDTA. Isto não constitui surpresa, uma vez que
este último ligante é potencialmente hexadentado, en-
6
4
2
111/ P——Ó
R I I \ 1 1 1 1 1 K* Na- L r TL' B a " S r " C a " MG" B e "
Fig. 3 — Comparação das constantes de estabilidade dos com
plexos dos ácidos etilenodiaminotetracético (1), ura
mildiacético (2) e nitrilotriacético (3).
1 9

quanto que o primeiro poderá, quando muito, ser
pentadentado, se os átomos de oxigénio nas posições
4- e 6- puderem coordenar-se simultaneamente aos
metais. O estudo de modelos moleculares mostra, porém,
que o plano do anel pirimidínico acomoda-se em relação
ao grupo iminodiacetato — N(CH2COO~)2 de um modo
tal que apenas um dos átomos de oxigénio, na posição
4- ou 6-, pode participar na coordenação. Isto é,
o ácido uramildiacético só pode actuar como tetraden-
tado, tal como acontece com o ácido nitrilotriacético.
É, pois, tanto mais digno de nota quanto inesperado
o facto de serem os complexos do ácido uramildiacético
bastante mais estáveis que os do ácido nitrilotriacético
e, com os metais alcalinos e berílio, mais estáveis do
que com qualquer dos outros ligantes.
Desde logo, a sugestão de SCHWARZENBACH (16) de que
o ácido uramildiacético deveria actuar como tridentado
e que o efeito indutivo do anel pirimidínico seria res
ponsável pelas invulgares estabilidades observadas é to
talmente inaceitável; um tal efeito influenciaria também
as constantes de dissociação do ligante na forma ácida
e os valores obtidos não deveriam exceder, por exemplo,
os correspondentes ao ácido metiliminodiacético, cujo
p/c2 é da ordem de grandeza de pk3 para a primeira
complexona. A explicação para o comportamento desta
família de substâncias terá, pois, de apoiar-se noutro
ou noutros factores.
O exame das constantes apresentadas na tabela 11.4
mostra que ao longo da série ácido uramildiacético (I),
ácido 1 -metiluramildiacético (JI), ácido 1,3-dimetilura-
mildiacético ( I I I ) há um aumento de estabilidade para
os complexos dos iões de menores dimensões, Mg2+
e Be2+, que contrasta com a diminuição de estabilidade
verificada nos complexos dos iões de maiores dimensões,
como Tl+ e K+; as variações são pequenas, sugerindo
um efeito indirecto de obstrução por parte dos grupos
metilo substituintes em II e III. Estes resultados são
coerentes, por um lado, com o papel pouco relevante
da possibilidade de ressonância completa do ciclo
pirimidínico na estabilidade dos complexos formados,
e por outro, com uma provável estrutura em «túlipa» *
resultante da orientação do grupo iminodiacetato em
relação aos átomos de oxigénio 4- ou 6-, provocada
pela coplanaridade dos átomos de carbono C-4, C-5
e C-6. A fig. 4 — fotografia do modelo molecular do
ácido 1,3-dimetiluramildiacético — ilustra este ponto.
Poderá acontecer que a estrutura indicada seja. espe-
* D o inglês cage.
cialmente favorável à complexação de iões pequenos
e, em especial, certamente, daqueles que têm tendência
a uma coordenação tetraédrica; daqui resultariam as
tendências observadas nos valores das constantes de
estabilidade determinadas. Compreender-se-ia também
a razão pela qual o berílio forma com o ácido uramil
diacético e os seus homólogos complexos mais estáveis do
que com o EDTA: devido às suas reduzidas dimensões,
o ião Be2+ «ajusta-se» bem à estrutura favorável dos
primeiros compostos, enquanto que a molécula do
EDTA tem dificuldade em tomar uma configuração
propícia à sua «acomodação». Isto é, as repulsões entre
os grupos carboxilato carregados negativamente e a
perda de entropia configuracional, resultante da dis
torção necessária para permitir uma interacção efectiva
entre os ligantes e o ião Be2+, diminuem considera
velmente a estabilidade do complexo que se possa
formar com o EDTA.
Fig. 4 — Fotografia do modelo molecular do ácido l.S-dime-
tiluramUdiacèúco.
Quanto aos complexos dos iões dos metais alcalinos
e do tálio, terá de admitir-se outra explicação, sendo
provável que as anomalias resultem não de qualquer
peculiaridade dos derivados do uramil, mas apenas de
uma impotência do EDTA para exercer todas as suas
possibilidades coordenativas em relação a estes iões;
de facto, é conhecida a reduzida afinidade dos ligantes
azotados pelos metais com estrutura de gás inerte (10),
e é natural que no EDTA só o átomo de azoto mais
2 0

fortemente básico (cf. valores de p/c para este ácido) possa efectivamente ligar-se com iões dos metais alcalinos. Assim, este ligante seria bastante menos poderoso que o que se poderia esperar em face do seu comportamento habitual, talvez mesmo menos poderoso que o ácido uramildiacético, que possui um só átomo de azoto mas com força básica relativamente elevada e, como se viu, uma estrutura propícia à complexação. Esta hipótese é, porém, bastante discutível, e, de qualquer modo, permanece inexplicado o aspecto mais enigmático do problema: a razão por que o ácido uramildiacético e os seus homólogos, que só podem actuar como ligantes tetradentados, dão complexos mais estáveis do que as outras complexonas tetradentadas típicas, como se evidencia na fig. 2 com a mais poderosa destas — o ácido nitrilotriacético. Para tentar esclarecer esta anomalia poderemos encarar o problema sob dois ângulos: ou as elevadas estabilidades verificadas são devidas a uma favorável variação de entalpia na reacção de complexação (entalpia liga-cional) — e portanto originadas por particularidades estruturais do ligante influindo na energia de ligação—, ou são devidas a favoráveis variações de entropia (entropia ligacional), frequentemente originadas pela «libertação» de moléculas de água das camadas de hidratação do catião ou do próprio ligante, com o concomitante aumento da energia translacional do sistema.
Para avaliar a influência deste último efeito poderá , recordar-se o exemplo clássico das reacções do M 2 +
com a amónia e com a metilamina, estudadas por BJERRUM (22)
M 2 + (aq.) + 6 NH3 (aq.) ZZ Ni(NH.fa+ (aq.)
A H° = — 19 .fixai A5» = - 2 2 u.e.
M 2 + ( a q . ) + 6 CH3NH2 (aq.) ZZ Ni(CH3MHz)l + (aq.)
A H° = + 9,7 Atai A 5 o = + 73 u.e.
Os complexos formados têm uma estabilidade da mesma ordem de grandeza, mas devida, como se vê, a causas diferentes. No primeiro caso, a influência preponderante é a energia de ligação; no segundo, é a favorável variação de entropia, que resulta, certamente, da libertação de moléculas áz água da camada de hidratação do ligante.
Nos complexos do ácido uramildiacético e homólogos
poderia admitir-se uma situação semelhante e decidimos portanto determinar as variações de entalpia e entropia em algumas reacções de formação de complexos daquelas substâncias. Como exemplos escolheram-se as reacções entre o ácido uramildiacético e os iões monovalentes Li+, Na+, K+ e Tl+. Não foi possível estudar a reacção com o ião Ag+ devido a efeitos secundários não esclarecidos (decomposição do ligante). As variações de entalpia e entropia — AH0 e AS0 — podem obter-se a partir de valores de constantes de estabilidade dos complexos a várias temperaturas. Este método não é tão preciso como o de calorimetria directa, mas é suficiente para o fim em vista. Na tabela I I . 5 apresentam-se os valores obtidos.
Tabela 11.5
Constantes de dissociação e de estabilidade (p/c e log K) para
os complexos do ácido uramildiacético com protões e outros
iões monovalentes, ¡x = 0,1 M ( (Cí7 3 ) 4 N. N03)
Temperatura (°C) 20° 27» 34° 39"
Hidrogénio, pk1 1,7 1,82 1,90 2,21
P><2 2,67 2,83 2,88 2,90
P'<3 9,63 9,47 9,38 9.31
Lítio 4,90 4,70 4,57 4,60
Sódio 2,72 2,54 2,42 —
Potássio 1,23 1,00 0,81 0,70
Tálio 5,99 5,76 5,41 5,33
A variação de entalpia nas reacções calculou-se a partir do coeficiente angular da recta obtida ao representar os
valores de log KML em função de °K~X; a variação
de entropia calculou-se pela relação A G3 = AH0 — TAS°,
verificando-se ser constante na zona de temperaturas considerada. Os resultados obtidos apresentam-se na tabela II.6. juntamente com valores da variação de energia livre A G° = — R.T lnfi, valores da entropia padrão dos iões em solução S° (M) e valores de soma AS° + S" (M).
Uma vez que a formação dos complexos é favorecida por variações negativas de entalpia e por variaçõss positivas de entropia, a primeira conclusão a tirar em face dos valores da tabela anterior é que os complexos dos iões estudados são principalmente estabilizados pela entalpia ligacional. No entanto, é curioso notar que os valores de A H° para os complexos dos metais
2 1

alcalinos se tornam progressivamente mais negativos
na ordem Li+, Na+, K+, enquanto que as variações
de energia livre seguem a ordem inversa, que é a de
estabilidades crescentes. Deve pois concluir-se que em
bora os complexos sejam estabilizados pela variação
de entalpia na reacção de complexação, é a variação
de entropia que efectivamente controla a ordem de
estabilidades.
Tabela II.6
VALORES D A S F U N Ç Õ E S T E R M O D I N Â M I C A S PARA
AS REACÇÕES D E F O R M A Ç Ã O D E COMPLEXOS D O
ÁCIDO URAMILDIACÉTICO
Ião — AG°(20°C) AH" As° S» (M)» AS°+
+ S»(M)
Hidrogénio (HL~ ") 12,89 — 6,9 + 1 9 ± 1 0 1 9 ± 1
Lítio 6,55 — 7,0 — 1 ± 5 + 3,4 2 ± 5
Sódio 3,64 — 8,7 —18 ± 1 + 14,4 — 4 ± 1
Potássio 1,65 —11,8 —35rnl + 24,5 —10 ± 1
Tálio 8,02 —15,4 —25 ± 4 + 30,4 5 ± 4
Valores de A G° e A H° em cal / ião-grama; valores de A 5 o e 5°(M)
em cal / ião-grama X grau (u. e. — u n i d a d e s de entropia) .
* RD.» (23).
Os valores de A 5 O são especialmente desfavoráveis para
os catiões de maiores d imensões—K + e Tl+—; este
resultado é compatível com o menor grau de hidra
tação destas espécies. Realmente, as variações de en
tropia parecem estar estreitamente relacionadas com as
energias de hidratação dos iões metálicos e é possível
obter boas relações lineares com os valores apresen
tados por LATIMER (23 ,24) . Mais directamente, porém,
poderemos relacionar A S° com uma função do raio
iónico dos catiões; na fig. 5 apresenta-se uma corre-
1 lação deste tipo, sendo a função f(f) — — •
r
Incluem-se na figura alguns valores para os complexos
de metais bivalentes com o EDTA, obtidos por STAVELEY
e RANDALL (25) . A semelhança de comportamento dos
diversos grupos de metais é evidente, mostrando que
em todos os casos a entropia ligacional depende essen
cialmente dos mesmos factores.
Estes resultados demonstram que as elevadas estabi
lidades encontradas para os complexos do ácido uramil-
diacético e seus homólogos não derivam de variações
de entropia favoráveis, presumivelmente atribuíveis a
um estado de hidratação pouco usual daqueles ligantes.
Por exclusão, será a energia de ligação a responsável
por tais estabilidades, e é nas particularidades estru
turais daqueles compostos que deverá procurar-se uma
explicação.
0.6 0.8 1.0 1.2 1,4 1.6 1.8
Fig. 5 — Valores da variação de entropia padrão nas reacções
de formação de complexos em função do inverso do
raio iónico dos metais.
Iões monovalentes — ácido uramildiacético
Iões bivalentes — ácido etilenodiaminotetracético.
Se a nossa hipótese de estruturas em «túlipa» for válida,
é de esperar que os complexos formados tenham apro
ximadamente a mesma forma e difiram pouco no seu
volume. Uma vez que em tais condições a sua acção
sobre as moléculas de água na vizinhança será análoga,
a entropia dos complexos em solução deverá ser quase
igual ou variará gradualmente com o raio do ião me
tálico no complexo. Ora a soma A S° + S°(M) é exac
tamente uma medida da entropia padrão dos complexos
em solução S°(ML), diferindo desta apenas pelo valor,
constante em cada caso, da entropia em solução do
ião L. Na verdade
A S° (reacção) = S°(ML) — S°(M) — S°(L)
Então, segundo a nossa teoria, será de esperar uma
relação entre os valores de AS0 + S° (M) e os valores
dos raios iónicos dos catiões estudados. Na fig. 6 poderá
verificar-se que existe na verdade uma relação para os
2 2

iões dos metais alcalinos e para o ião H (cuja posição
não pode ser exactamente fixada), mas o comporta
mento do 7 7 + é anómalo, embora este ião tenha apro
ximadamente as dimensões do K+. Um caso semelhante
foi encontrado por CARE e STAVELEY (26) nos com
plexos de Pb2+ e Sr2+ com o EDTA.
- 2 0 _
- 1 5 \
- 1 0 \ Tl*
" 5 \ Li* O
- 0 \ . Na*
- - 5
10
- - 1 5 , i \ . r¡ -Â
- - 1 5 , i 1 , 1 , 1 , 1 0 0,4 0,8 1,2 1,6
Fig. 6 — Valores de A S" + S° (M) em função do raio iónico
dos metais.
É claro que não se trata realmente de uma anomalia,
pois as propriedades termodinâmicas dos comple
xos não são determinadas exclusivamente por fac
tores de forma; devem também depender da natureza
da interacção entre o ião metálico e os átomos ligantes.
No nosso caso, esta interacção é com certeza diferente
com o Tl+, que é um ião do tipo d10s2, e que, não
obstante a presença de par inerte 6s 2, deverá com
portar-se como se possuísse uma carga positiva mais
elevada, uma vez que os electrões d têm um poder de
«blindagem» relativamente baixo. Então, não só os
valores de A H° serão desproporcionalmente mais ne
gativos (o que de facto se verifica), mas também a
interacção dos complexos do Tl+ com as moléculas
de água na sua vizinhança será diferente da dos com
plexos dos metais alcalinos e não se poderão esta
belecer correlações gerais tão simples como a da figura
anterior.
O poder «neutralizante» ou de «blindagem» das várias
classes de electrões é, porém, tomado em linha de
conta na escala de electronegatividades «absolutas» de
ALLRED e ROCHOW (27) . A electronegatividade é defi
nida por estes autores como a razão Z^/r 2, onde
é a carga nuclear efectiva e r0 é o raio covalente do
átomo no estado de valência considerado; representa
portanto a intensidade do campo eléctrico a uma dis
tância igual ao raio covalente. A fig. 7 mostra que os
valores dos logaritmos das constantes de estabilidade
(e portanto a «energia livre ligacional») dos complexos
formados pelo ácido uramildiacético com os iões mo
novalentes considerados é uma função linear da elec
tronegatividade absoluta destes, calculada por NYHOLM
para os metais alcalinos (28 ) e por nós, segundo o
seu método, para o Tl+.
- 9
0,2 0,3 0 , 4 0,5 0.6
Fig. 7 — Valores dos logaritmos das constantes de estabilidade
dos complexos do ácido uramildiacético em função
da electronegatividade absoluta dos metais.
A relação é bastante satisfatória e mostra que a esta
bilidade dos complexos formados pelo ácido uramil
diacético e seus homólogos depende, de um modo
regular, dos mesmos factores.
Nestas condições não há qualquer anormalidade no com
portamento destas substâncias, como sugeria a fig. 3, e
a nossa hipótese de um menor número de coordenação
operativo nos complexos do EDTA com o Tl+ e com
os metais alcalinos recebe um apoio considerável. Nos
complexos deste ligante há, portanto, a possibilidade de
números de coordenação variáveis, o que, com toda
a probabilidade, não acontece com o ácido uramil-
2 3

diacético e seus homólogos. Ora como òs Complexos
formados por esta família de ligantes são uniformemente
mais estáveis que os complexos formados por outros
ligantes tetradentados típicos, a conclusão a tirar é que
nos primeiros, por qualquer razão ainda não identifi
cada, o número de coordenação efectivo é superior a
quatro.
Para esclarecer este ponto, impõe-se uma comparação
entre o comportamento destas complexonas e o de
outras com estrutura análoga. Convirá pois estudar
substâncias do tipo de viu,
À comparação entre a estabilidade dos complexos for
mados por IX e X e por XIII e XIV deverá mostrar
até que ponto o grupo hidroxilo em IX e XIV estará
envolvido na coordenação; a comparação de IX e XI
permitirá verificar o efeito da substituição do átomo
de oxigénio por enxofre.
O H
IX
O C H ,
X
S H
XI
C H , C O O H
VIII
em que X é um doador potencial. O número reduzido
destas torna necessário sintetizar novas complexonas,
o que oferece oportunidade para estudos mais por
menorizados sobre esta classe de ligantes.
\ R
C O O H
XII XIII
O H
XIV
CH, — COOH
2 — ESTUDOS COM O ÁCIDO o-HIDROXIFENILI-
MINODIACÉTICO E OUTRAS COMPLEXONAS
Dos compostos com a estrutura indicada, o mais con
veniente para comparação directa será, segundo as
nossas hipóteses, o ácido o-hidroxifeniliminodiacético
(IX) — X — OH— ; esta substância foi por nós sin
tetizada e, para profundar tanto quanto possível o
estudo deste tipo de reagentes, preparámos ainda os
análogos: ácido o-metoxifeniliminodiacético (X) — X =
= OCH3 — , ácido o-mercaptofeniliminodiacético (XI)
— X = SH — , ácido o-carboxifeniliminodiacético (XII)
—X=COOH—e ácido ciclohexiliminodiacético (XIII)
— X.= H, saturado—. Adquirimos também uma amos
tra de ácido 2-hidroxiciclohexiliminodiacético (XIV)
— X = OH, saturado—único explorado comercialmente
(Geigy).
As complexonas XII e XIV foram já estudadas ante
riormente (29, 30), mas apenas se calculou um reduzido
número de constantes de estabilidade para os respec
tivos complexos. Também a complexona XIII foi exa
minada antes (31), mas nós verificámos que o produto
utilizado não era suficientemente puro, pelo que os
resultados publicados não são válidos, devendo ser
substituídos pelos obtidos no presente trabalho. Na
secção experimental são dados os pormenores de uma
preparação melhorada desta substância.
A síntese do ácido o-metoxifeniliminodiacético foi con
seguida por condensação da o-anisidina com o ácido
cloroacético; a reacção dá-se facilmente até à formação
de ácido o-metoxifeniliminomonoacético, mas a intro
dução do segundo equivalente de ácido requer con
dições drásticas, evidenciando o estereo-impedimento
exercido pelo volumoso grupo metoxilo. Este impedi
mento faz-se também sentir posteriormente, provocando
uma descarboxilação rápida do composto; todas as
medidas foram, portanto, efectuadas em soluções re
centes.
Outro aspecto da interacção fácil entre os grupos na
posição «orto» e os substituintes no átomo de azoto
24

foi verificado na preparação das complexonas IX e
XI. Na verdade, estes compostos não puderam pre
parar-se no estado sólido, sendo obtidas em seu lugar
as correspondentes 8-lactonas formadas por eliminação
intramolecular de água: ácidos 3,4-dihidro-2-oxo-l,4-
-benzomorfolina-4-acético (IXa) e 3,4-dihidro-2-oxo-
-1,4-benzotiazina-4-acético (Xla).
CH,-COOH
CH2
0 - C .
\\ //
CH2-COOH
.CH,
s-c.
IXa Xla
Istas lactonas comportam-se como simples ácidos mo-
lobásicos, mas o anel lactónico abre com relativa
acuidade e, após a adição de três equivalentes de
•ase, a titulação por retorno corresponde ao sal dos
icidos tribásicos IX e XI. Deste modo, a formação
las lactonas não impede o estudo das complexonas
desejadas.
A preparação destas substâncias é descrita na secção
respectiva e na presente limitar-nos-emos a uma breve
menção de alguns pormenores curiosos.
A formação de um anel lactónico em IXa foi confir
mada pela frequência de absorção no infravermelho
a 1760 c m - 1 (suspensão em Nujol); a identificação de
uma frequência de absorção a 1715 cm - 1 , correspon
dente a um grupo carboxílico, prova que IXa não
tem, no estado sólido, a estrutura de betaína.
s—c, / C H 2
Ós outros dois produtos foram obtidos acidificando a
mistura reaccional, e diversos ensaios permitiram esta
belecer que se tratava da mesma substância — Xla —
em duas formas polimórficas com pontos de fusão
69-71°C e 143-5°C.
A banda de absorção no infravermelho correspondente
ao anel lactónico aparece localizada a 1660 cm - 1 , en
quanto que a banda do grupo carboxílico livre se en
contra a 1703 c m - 1 nos dois casos. A forma de ponto
de fusão mais elevado é a mais estável e a de ponto
de fusão mais baixo transforma-se lentamente nesta,
mesmo no estado sólido, o que requer cerca de dois
meses. A substância XV resulta, evidentemente, da
descarboxilação destas últimas. Todas estas substâncias
estão em estudo no que se refere a possíveis aplicações
terapêuticas, pois conhecem-se produtos com estrutura
semelhante que apresentam propriedades sedativas e
analgésicas.
A formação de complexos de IXa e Xla foi estudada
após a abertura dos respectivos anéis lactónicos por
fervura com a quantidade equivalente de álcali.
Titulações com ácido na presença e ausência de iões
metálicos permitiram obter os valores necessários. Para
os restantes ligantes seguiu-se o processo normal de
titulação directa com uma solução de base forte. As
constantes de dissociação das várias complexonas apre
sentam-se na tabela II.7, juntamente com os valores
correspondentes de outros ligantes análogos: ácidos
feniliminodiacético (XVI), 2-hidroxietiliminodiacético
(XVII), 2-metoxietiLiminodiacético (XVIII) e 2-carbo-
xietiliminodiacético (XIX).
XVI
,R
H O - C H 2 - C H 2 - N '
XVII
/ I
XV
A condensação de o-aminotiofenol com ácido cloroacé-
tico em meio alcalino deu origem a três produtos: o
produto principal cristaliza por simples arrefecimento
e foi identificado como 3,4-dihidro-4-metil-l,4-benzo-
tiazina-2-ona (XV); a ausência de grupos carboxílicos
livres foi evidenciada pelo espectro no infravermelho.
XVIII
R R
-J HOOC - C H , - C H , - N \ \ X R X R
XIX
R = -CHj-COOH
2 5

Os valores achados para pkx correspondem à ionização
do protão de um grupo carboxílico, sendo mais baixos
no caso das complexonas alifáticas do que no caso
das aromáticas, devido ao efeito indutivo negativo dos
grupos — OH, — OCH3 e —COOH nas primeiras. Os
c3=°< — e O * <
valores de pk2 correspondem à ionização dos protões
dos radicais amónio, excepto nos casos dos ácidos
uramildiacético (I), o-carboxifeniliminodiacético (XII) e
2-carboxietiliminodiacético (XIX), para os quais corres
ponde à ionização do segundo grupo carboxílico. Os
- 1 1 ! pH
- 1 0 (3) -
- 9 /A /' ' i - 8 [ey /
1 1
- 7
- 6
~ 5 ^
- 3 1 1
2 1
«a» 3 1
Fig. 8 — Curvos de titulação do ácido o-hidroxifeniliminodia-
cético na presença de diversos iões.
(1) K+ (100:1) (2) Li+ (1:1) (3) Ba2+ (1:1)
(4) Sr2+ (1:1) (5) 77+ (1:1) (6) Ça?+ (1:1)
(7) M g 2 + (1:1)
casos de IX, X e XI devem comparar-se com o do
ácido feniliminodiacético (XVI), em que a participação
do átomo de azoto no sistema ressonante do ciclo
benzénico origina uma diminuição da respectiva basi-
cidade. Este facto compreende-se facilmente atendendo
o=s< e
às formas canónicas mesómeras em que o átomo de
azoto tem uma carga formal positiva (XX).
Os valores de pk3 obtidos para as complexonas I, XII
e XIX correspondem também à remoção do protão
do respectivo radical amónio, mas o mesmo não sucede
-11
pH
- 3 «a»
1 2 I I I I
Fig. 9 — Curvas de titulação do ácido o-metoxifeniliminodia-
cético na presença de diversos iões.
(1) K+ (100:1) (2) Sr*+ e Ba2+ (1:1)
(3) C a 2 + e Ag+ (1:1) (4) 27+ (10:1)
2 6

Tabela II.7
CONSTANTES D E DISSOCIAÇÃO D E A L G U N S ÁCIDOS AMINOPOLICARBOXÍLICOS
T = 20,0°C e (x = 0,1 Aí (KN03 ou KCl)
gigan tes P ' ; i Rcf.»
Ácido uramildiacético (I) 1,7 ± 0 , 3 2,67 ± 0,02 9,63 ± 0,02 (a) Ácido o-hidroxifeniliminodiacético (IX) 2,98 ± 0,01 5,43 ± 0,01 11,08 ± 0,03 (a) Ácido o-metoxifeniliminodiacético (X) 2,69 ± 0,01 5,58 ± 0,01 — (a)
Ácido o-mercaptofeniliminodiacético (XI) 2,85 ± 0,01 6,30 ± 0,01 9,54 ± 0,02 {a)
Ácido o-carboxifeniliminodiacéüco (XII) 2,33 ± 0,02 2,98 ± 0,01 7,75 + 0,01 (a)
Ácido ciclohexiliminodiacético (XIII) 2,15 ± 0,02 10,81 ± 0,01 — (a) Ácido 2-hidroxiciclohexüiminodiacético (XIV) 2,32 ± 0,01 9,57 ± 0,01 — («) Ácido feniliminodiacético (XVI) 2,40 4,96 (*) Ácido 2-hidroxietiliminodiacético (XVII) 2,2 8,73 (P) Ácido 2-metoxietiliminodiacético (XVIII) 2,2 8,96 <P) Ácido 2-carboxietiliminodiacético (XIX) 2,06 3,69 9,66 (b)
{a) Presente t rabalho. Valores válidos pa ra [X = 0,1 (KN03).
(b) Ref.» 21 . Valores válidos pa ra [X = 0,1 (KCl).
Tabela II.8
CONSTANTES D E ESTABILIDADE (log K) D O S COMPLEXOS D E DIVERSOS IÕES
T = 20,0°C e [x = 0,1 Aí (KNO 3 OU KCl)
IÁgante Espécie Ag + Tl + Li + Na + M g 2 + C « a + Sr* + BaÍJr Réf.»
Ácido uramildiacético (I) ML red. 5,99 4,90 2,72 8,19 8,31 6,93 6,13 (.a) ± 0 , 0 2 ± 0 , 0 2 ±0 ,01 ± 0 , 0 2 ±0 ,01 ± 0 , 0 2 ± 0 , 0 2
Ácido o-hidroxifeniliminodiacético (IX) MHL red. 2,34 2,67 3,21 2,67 2,50
± 0 , 0 2 ± 0 , 0 2 ±0 ,01 ± 0 , 0 2 ± 0 , 0 4 (a) ML red. 4,79 2,20 1,0 6,86 6,27 4,65 4,27
± 0 , 0 3 ± 0 , 0 2 ± 0 , 1 ±0 ,01 ± 0 , 0 3 ±0 ,01 ±0 ,01
Ácido o-metoxifeniliminodiacético (X) ML 2,75 2,46 — — — 2,75 2,13 2,08
± 0 , 0 2 ±0 ,01 ±0 ,01 ± 0 , 0 2 ± 0 , 0 2 («) Ácido o-mercaptofeniliminodiacético (XI) ML pp. — — — 1,84 2,79 2,6 pp. (a)
± 0 , 0 3 ± 0 , 0 4
Ácido o-carboxifeniliminodiacético (XII) ML 3,54 2,93 2,05 0,89 3,91 5,06 3,91 3,57 («). ,b) ±0,01 ±0 ,01 ±0 ,01 ±0 ,01
Ácido ciclohexiliminodiacético (XIII) ML 4,94 3,40 1,74 0,90 3,46 3,34 2,55 2,37 («) ± 0 , 0 2 ±0 ,01 ± 0 , 0 2 ± 0 , 0 3 ± 0 , 0 2 ± 0 , 0 2 ±0 ,01 ± 0 , 0 2
Ácido 2-hidroxiciclohexiliminodiacético (XIV) ML 3,83 3,07 2,19 0,76 4,27 5,19 3,81 3,26 ±0 ,01 ±0 ,01 ±0 ,01 ±0 ,01 ±0 ,01 ±0 ,01 (a), (b)
Ácido feniliminodiacético (XVI) ML ~ 1 — — 1,15 1,5 ~ 1 ~ 1 (61 Ácido 2-hidroxietiliminodiacético (XVII) ML — — — — 3,44 4,63 3,77 3,42 (b) Ácido 2-metoxietiliminodiacético (XVIII) ML — — — — 3,31 4,53 3,84 3,56 (b) Ácido 2-carboxietiliminodiacético (XIX) ML — — — 5,28 5,04 3,87 3,40 (b)
[a) Presente t rabalho. Valores válidos pa ra [X = 0,1 (KN03).
(b) Réf.» 21 . Valores válidos pa ra (X = 0,1 {KCl).
2 7

corn as complexonas IX e XI, ácidos o-hidroxifenili-
minodiacético e o-mercaptofeniliminodiacético, para as
quais pks corresponde à ionização do grupo funcional
substituinte, evidenciando o aumento de carácter ácido
dos fenóis e tiofenóis em relação aos álcoois e tióis,
devido à interacção dos átomos de oxigénio e enxofre
com o ciclo aromático.
A estabilidade dos complexos formados pelos novos
ligantes foi estudada por potenciometria, como habi
tualmente, e nas fig. 8, 9 e 10 apresentam-se algumas
curvas de titulação.
- 3
«a»
Fig. 10 — Curvas de titulação do ácido o-mercaptofenilimino
diacético na presença de diversos iões
(1) K+ (100:1) (2) C a 2 + e S r 2 + ( 1 : 1 )
(3) Mg2+ (10:1)
As constantes de estabilidade determinadas a partir
dos dados destas titulações encontram-se agrupadas na
tabela II.8 com os valores correspondentes para ligantes
análogos, de modo a permitir a comparação directa.
Os valores apresentados permitem algumas conclusões
interessantes: vê-se desde já que o grupo hidroxílo na
complexona IX intervém certamente na coordenação
dos iões metálicos, pois os complexos desta substância
são mais estáveis que os da complexona X, embora
os átomos de azoto de ambas tenham aproximadamente
a mesma basicidade (p/c2=5,43 e 5,58, respectivamente).
A diferença de estabilidades aumenta à medida que
os raios iónicos diminuem, dentro da mesma série, o
que sugere que a estrutura de IX é geometricamente
favorável para a complexação de iões pequenos, ana
logamente ao que acontecia com o ácido uramildiacé-
tico e seus homólogos.
No caso das complexonas XIII e XIV, a introdução
de um grupo hidroxilo na posição 2- do ácido ciclo-
hexiliminodiacético baixa o valor de pA2 de 10,81 para
9,57; não obstante, os complexos formados pelo ácido
2-hidroxiciclohexiliminodiacético com os metais alca-
lino-terrosos são mais estáveis que os formados pelo
primeiro ligante. Sendo XIII uma complexona tri-
dentada típica, XIV deverá actuar como tetradentada,
embora em certos casos, como possivelmente o dos
iões 7 7 + , Ag+ e Na+, talvez se formem menos anéis
de quelação.
Os valores publicados para o ácido feniliininodiacético,
ligante obviamente tridentado, confirmam que, em prin
cípio, IX e X deverão actuar também como tetraden-
tados. De qualquer modo, é evidente pelos valores
da tabela II.8 que os complexos formados pelo
ácido uramildiacético (I) são bastante mais estáveis
que os formados pelo ácido o-hidroxifeniliminodiacé-
tico (IX) (cerca de 2-3 Kcal). À primeira vista este
resultado é surpreendente, pois as duas complexonas
são geometricamente equivalentes e a segunda é, apa
rentemente, bastante mais básica que a primeira. Numa
segunda análise, porém, verifica-se que a comparação di
recta não é inteiramente válida, pois os valores de pk de
terminados referem-se a grupos diferentes: o ião amónio
no caso de I e o grupo hidroxilo no caso de IX. O átomo
de azoto desta última complexona é mesmo bastante
pouco básico, pelas razões já comentadas anteriormente,
e este facto poderia, em princípio, explicar a menor esta
bilidade dos complexos formados por esta substância.
Acontece, porém, que em relação a outras complexonas
com átomos de azoto igualmente bastante básicos (ácido
nitrilotriacético, por exemplo) continuam a verificar-se
diferenças de estabilidade da mesma ordem de gran
deza, isto é, 2-3 Kcal, correspondentes aproximada
mente à variação de energia livre por formação de
mais um anel de quelação nos complexos do ácido
uramildiacético. Ora como os átomos de oxigénio nas
posições 4- e 6- desta complexona não podem estar
simultaneamente envolvidos na coordenação, o que é
2 8

contrário à teoria e à evidência fornecida pelos mo
delos moleculares, somos obrigados a ir mais longe
e a concluir que a estabilidade anormal dos complexos
do ácido uramildiacético se deve a uma afinidade
especial dos metais por esta substância, conferindo-lhe
um carácter de complexona potencialmente pentaden-
tada.
A única explicação que poderemos propor para este
facto é que a carga formal negativa distribuída no anel
pirimidínico em toda a zona adjacente ao metal desem
penha um papel cuja importância tem sido subestimada
até aqui (vd. XXI).
/ \ r 0 = C e';-C—N —
X NH—C I •A
0
XXI
A existência desta carga, que resulta da possibilidade
de ionização do grupo metilénico — 5 no ácido ura
mildiacético e seus homólogos e que constitui a única
característica verdadeiramente distinta destas comple-
xonas em relação a todas as outras estudadas, poderá
contribuir substancialmente para a variação de entalpia
na formação dos complexos, aumentando a energia de
atracção electrostática entre o metal e o ligante e,
em virtude da conformação estrutura] propícia deste,
levando à saturação da constante dieléctrica da água
dentro da esfera de coordenação. Esta constante poderá
pois ter um vaJor bastante menor que o habitual valor
macroscópico, e as atracções de natureza electrostática
serão correspondentemente maiores, conforme se poderá
prever pela expressão da lei de COULOMB.
Esta hipótese não é susceptível de demonstração ine
quívoca; é, no entanto, admissível, e parece fornecer
a única explicação para o comportamento anormal do
ácido uramildiacético e seus homólogos. Como alter
nativa só seria possível admitir que os dois átomos
de oxigénio nas posições 4- e 6- intervêm simultanea
mente na coordenação, mas, como foi já referido, tal
ocorrência é contrária à teoria e, a verificar-se, poria
em cheque as ideias actuais sobre a estrutura mole
cular.
A solução definitiva deste problema terá pois de
aguardar uma análise estrutural pormenorizada dos
complexos destes ligantes, possivelmente por difracção
dos raios X. Tal análise é, no entanto, extremamente
difícil no estado actual de desenvolvimento desta
técnica.
Em relação aos outros ligantes sintetizados há ainda
algumas observações a fazer.
A complexona XI, ácido o-mercaptofeniliminodiacético,
cuja síntese foi realizada para evidenciar diferenças
entre os poderes coordenativos dos átomos de oxigénio
e enxofre, forma complexos de reduzida estabilidade
com os metais alcalino-terrosos e mostra uma ten
dência pronunciada para dar com eles precipitados
pouco solúveis que não foram examinados em por
menor. Os valores determinados, em especial para o
complexo de estrôncio, não são pois de inteira con
fiança, embora permitam conclusões qualitativas rela
tivamente seguras. Estes valores não correspondem ao
que seria de esperar de uma complexona potencial
mente tetradentada, mesmo tomando em linha de conta
a menor afinidade dos metais alcalino-terrosos por
ligantes sulfurados em relação aos oxigenados. Possi
velmente formam-se menos anéis de quelação, talvez
devido às dimensões do átomo de enxofre que podem
forçar excessivamente o anel de cinco membros em
que participaria aquele átomo.
Os resultados obtidos com as outras complexonas con
firmam esta hipótese; a comparação dos valores das
constantes de estabilidade dos complexos dos ácidos
2-hidroxiciclohexiliminodiacético e 2-hidroxietilimino-
diacético sugere que a complexação dos iões de me
nor raio é favorecida pela posição do grupo hidroxilo
no ciclohexano, e podem fazer-se comparações análo
gas para os valores obtidos com os pares de ácidos o-me-
toxifeniliminodiacético e 2-metoxietiliminodiacético e
ácidos o-hidroxifeniliminodiacético e 2-hidroxietili-
minodiacético.
Deve pois concluir-se que a habitual asserção de
especial estabilidade dos anéis de cinco membros
não se pode aceitar sem outras considerações, pois
o raio iónico do metal complexado é um factor crítico.
Os valores das constantes de estabilidade dos com
plexos do ácido o-carboxifeniliminodiacético são apre
ciavelmente maiores que os que seriam de esperar teo
ricamente, sobretudo tendo em linha de conta a reduzida
basicidade deste ligante; o mesmo acontece com o ácido
o-sulfonofeniliminodiacético (21). Este facto foi já des
crito por outros investigadores (32), que sugeriram
que a estrutura parcialmente plana destas complexonas
2 9

poderia favorecer a interacção dos átomos doadores
com o elemento central (o que é um argumento essen
cialmente entálpico) e que a diminuição da liberdade
de rotação dos ligantes poderia também contribuir para
uma sobrestabilização dos complexos (o que é, cla
ramente, um argumento entrópico). No entanto, pare
ce-nos que nenhuma destas explicações é satisfatória,
pois não se aplicam ao ácido nitri lotriacético (p. 19)
nem a outros casos evidenciados no presente trabalho.
Cremos que dois outros efeitos diferentes poderão for
necer uma justificação mais aceitável para as estabili
dades observadas: um, a elevada polarizabilidade dos
grupos carboxilato e sulfonato (33); outro, a presumível
elevada hidratação destes mesmos grupos, uma vez que
se sabe existir uma forte interacção do ião acetato
com as moléculas da água (34). No entanto, os valores
da variação de entalpia na formação de pares iónicos
com o ião acetato são, em geral, baixos e, às vezes,
positivos, ao passo que os valores da variação de
entropia são elevados e favoráveis, isto é, positivos (26).
iões dos metais, dando origem a variações de entropia
favoráveis nas reacções de complexação com os res
pectivos ligantes (22).
Estes efeitos têm um carácter mais geral que os ante
riormente propostos e poderão explicar, pelo menos
parcialmente, o comportamento anormal dos ligantes
considerados. A existência de desvios em relação ao
comportamento «normal» será adiante demonstrada es
tatisticamente.
Resta-nos considerar o ácido ciclohexiliminodiacético
( X I I I ) . Este Iigante foi, como dissemos, estudado ante
riormente (31); porém, no processo de preparação uti
lizado, o isolamento do produto foi conseguido por
intermédio do respectivo complexonato de bário, e,
em virtude de a remoção deste metal não ter sido
eficiente, a substância obtida não era suficientemente
pura.
Um método em que a precipitação da complexona foi
induzida por sementeira permitiu-nos obter um pro
duto em boas condições, com o qual se conduziram
Tabela II.9
CONSTANTES D E DISSOCIAÇÃO D E DIVERSAS COMPLEXONAS (p/c)
T = 20,0°C [JL = 0,1 M (.KN03 e KCl)
g igan te p&i p / í 2 pk3 p/í-4 Ref.»
Ácido iminodiacético 2,65 9,38 — — (6)
Ácido metiliminodiacético 2,12 9,65 — — (ô)
Ácido ciclohexiliminodiacético 2,15 ± 0,02 10,81 ± 0,01 — — («)
Ácido nitrilotriacético 1,89 2,49 9,73 — (/>)
Ácido etilenodiaminotetracético 1,99 2,67 6,16 10,26 (b)
Ácido 1,2-diaminopropanotetracético 1,83 2,79 6,25 10,86 ± 0,02 (a), (c)
(a) Presente t raba lho .
(b) Ref.» 2 1 . (c) Ref.» 36.
MARTELL (35) observou mesmo, para um grupo de
complexos de ácidos aminopolicarboxílicos, que os va
lores de AS 0 aumentavam com o número de grupos
acetato que se poderiam coordenar, tendo obtido uma
boa relação linear entre aqueles valores e Z/rf, em
que Z representa o número de grupos acetato e r{ o
raio iónico dos metais. Todos estes resultados parecem
apoiar a hipótese de que o «iceberg» de hidratação
dos grupos carboxilato e sulfonato é desagregado pelos
novos estudos. Os resultados obtidos neste trabalho
substituem, portanto, os obtidos anteriormente.
Nas tabelas II.9 e 11.10 apresentam-se novos valores de
constantes de dissociação e de estabilidade dos com
plexos deste Iigante e também, para aumentar o nú
mero de constantes conhecidas e permitir comparações,
valores determinados por nós e por outros autores
para os complexos formados por diversas complexonas
comerciais: ácidos iminodiacético ( I M D A ) , metilimino-
3 0

diacético (MIMDA), nitrilotriacético (NITA), etileno-
diaminotetracético (EDTA) e 1,2-diaminopropanote-
tracético (PRODTA).
HM / •CH2-COOH
vCH 2-COOH
IMDA
CH-N' / V
CH,-COOH
xCH 2-COOH
MIMDA
Esta sugestão, apoiada no estudo de espectros no infra
vermelho e de ressonância magnética nuclear, continua,
porém, a manter dificilmente explicável a diferença de
valores entre pk3 e p/<4 para o EDTA e, uma vez que
a estrutura proposta para o ião HLT do ácido metili-
minodiacético é semelhante a A, é incompreensível
a disparidade dos valores de p/c2 para esta complexona
e p/c3 para aquela.
/ CK-COOH
H O O C - C H T - N ,
NCH2-COOH
NITA
°\ /° C-CH / C H r C
H N - C H - C H — N . H.
o; / C - C H , C H - C :
V
H O O C - C H 2 . ^ C H 2 - C O O H
< N — C H — C H — N
H O O C - C H / V H , - C O O H
A : H,Y~
EDTA
HOOC - CH, f H 3 ^ C H 2 - C O O H
, N — C H — C H — N .
H O O C - C H 2 \ H 2 - C O O H
PRODTA
0
I I I CO CO
...N (CH 2COO) 2
CH, CH
B : HY-
XXII XXIII
Os dois primeiros produtos são homólogos de XIII
e constituem exemplos de ligantes potencialmente tri-
dentados; NITA pode actuar como tetradentado em
virtude de possuir mais um grupo carboxilo; os dois
últimos ligantes poderão, em princípio, actuar como
hexadentados, embora, como já foi sugerido (p. 20 ) ,
nem sempre assim aconteça.
As constantes determinadas anteriormente foram dis
cutidas por diversos autores, mas estes, em especial
no caso dos valores de pA:3 para o EDTA e PRODTA,
não estão inteiramente de acordo entre si ( 36 , 37 , 38 ,
39, 40) .
CHAPMAN sugeriu recentemente (40) que as estruturas
dos ácidos aminopolicarboxílicos em solução envolvem
a formação de pontes de hidrogénio, e propôs as
estruturas A e B (XXIII) para os iões H2Y~2 e HY"3
do ácido etilenodiaminotetracético.
Os novos valores não necessitam discussão; p/cx e p/<2
do ácido ciclohexiliminodiacético correspondem à ioni
zação de um dos carboxilos e do radical amónio resi
dual, evidenciando o efeito indutivo positivo do grupo
ciclohexilo, superior ao do grupo metilo e ao do hi
drogénio. O valor de p/c4 para o ácido 1,2-diamino-
propanotetracético foi por nós redeterminado, pois,
devido a uma troca de sinal, foi calculado incorrecta
mente pelo investigador anterior ( 3 6 ) ; o novo valor
— 10 ,86 — pode comparar-se directamente com o valor
de p/c2 do ácido ciclohexiliminodiacético—10,81—e
a concordância não surpreende, uma vez que em ambos
os casos há que considerar principalmente a influência
de um grupo isopropilo sobre o radical amónio que
se ioniza (vd. XXIV).
3 1

Os valores de constantes de estabilidade obtidos reque- entanto, o aumento da estabilidade dos complexos do
rem apenas um breve comentário: no caso do ácido metiliminodiacético, em relação aos complexos
e XHj- j—CH 2 CH2-COO
/ ¡ \ œ / CH2 C H - N H
\ / \ C H H — CH, CH,-COO
OOC-CHx
OOC-CH,"
CH, ,CH,-COO
N-7-CH7-CH —NH
(HEXDA) PRODTA
XXIV
PRODTA são uniformemente mais elevados que no caso do ácido iminodiacético, é bastante superior ao que
do EDTA, devido ao aumento da basicidade no primeiro seria de esperar de uma variação de apenas 0,27 uni-
ligante (pki = 10,86, comparado com pkt = 10,26 para dade nos valores de p/<2; deverá, pois, existir nos com-
Tabela 11,10
CONSTANTES D E ESTABILIDADE (log K) DOS COMPLEXOS D E DIVERSAS COMPLEXONAS
T = 20,0°C
I ão Bspécie I M D A MIMDA H E X D A N I T A E D T A P R O D T A
Li+ ML 0,96 ± 0,02 1,20 ± 0,01 1,74 ± 0,02 2,51 ± 0,01 2,79 3,43 ± 0,01
Na+ ML 0,36 ± 0,06 0,61 ± 0,02 0,90 ± 0,03 1,32 ± 0,01 1,66 2,24 ± 0,01
77+ MHL — — — — 2,06 ± 0,02 2,47 ± 0,01
ML 1,78 ± 0,01 3,05 ± 0,01 3,40 ± 0,01 4,74 ± 0,01 6,55 ± 0,01 7,02 ± 0,01
Ag+ MHL — — — — 3,07 3,10 ± 0,01
ML 3,38 ± 0,02 4,06 ± 0,01 4,94 + 0,02 5,03 ;fc 0,01 7,32 8,05 ± 0,01
Mgz + MHL — — — — 2,28 —
ML 2,94 3,44 3,46 ± 0,02 5,41 8,69 10,02 ± 0,01
C« 2 + MHL — — — — 3,51 —
ML 2,59 3,75 3,34 ± 0,02 6,41 10,70 11,40 ± 0,01
ó>'+ MHL — — — — 2,30
ML 1,81 ± 0,01 2,85 2,55 ;t 0,01 4,98 8,63 9,67 ± 0,01
Ba2 + MHL — — — — 2,07 — ML 1,67 2,59 8,37 ± 0,02 4,82 7,76 8,57 ± 0,01
Todos os valores apresentados com desvio-padrão pertencem ao presente trabalho e são válidos para [a = 0 , 1 M (KN03);
os restantes foram coligidos na referência (21) e são válidos para [x = 0,1 M (KCl).
o EDTA). Os complexos do ácido ciclohexiliminodiacé-
tico são, porém, menos estáveis que os do ácido me
tiliminodiacético, não obstante o primeiro ligante ser
mais básico que o segundo (pÂ:2 = 10,81 e 9,65 res
pectivamente). À primeira vista poder-se-ia pensar que
este facto seria devido a um estereo-impedimento exer
cido pelo grupo ciclohexilo de XIII, o que resultaria
num efeito contrário ao do aumento de basicidade. No
plexos daquele ligante uma favorável contribuição en-
trópica, analogamente ao que acontece nos complexos
da metilamina (p. 21).
Nestas condições, o ácido metiliminodiacético não
pode ser tomado como referência, sendo natural,
como veremos adiante, que o efeito do grupo
ciclohexilo na complexona XIII seja puramente in
dutivo.
3 2

3 — RELAÇÕES ENTRE A ESTABILIDADE DOS
COMPLEXOS E A BASICIDADE DAS COM-
PLEXONAS
Temo-nos referido até aqui a um «comportamento
normal» das complexonas como base de previsão da
estabilidade dos complexos formados, e comparámos
frequentemente valores de constantes de estabilidade
experimentais com os valores «teoricamente» expectá
veis a partir da basicidade ( *) das respectivas comple
xonas. Este modo de proceder é, naturalmente, limi
tado, e terá de se justificar para que as conclusões
anteriores possam ser aceites.
Interessa pois demonstrar a existência de uma depen
dência funcional entre constantes de estabilidade e
constantes de dissociação, em especial do tipo
l o g KML = a pkHL + b ( 1 )
sendo
KML = [ML] I [M] [L] e KHL = [HL] / [H] [L]
Uma vez que
AG 0 = — R T ln K virá:
ln Kn •ln KflL —
RT (G°M G°ML ~ G°H + GHL> (4)
Visto que = 0, por convenção, esta expressão pode
simplificar-se; além disto pode efectuar-se uma sepa
ração conveniente, resultando
ln K„ ]nKH
1 1 R T <
G H L — GML) + G M (5)
isto é, uma relação linear entre os logaritmos das cons
tantes de estabilidade dos complexos de um mesmo
metal e os logaritmos das constantes de dissociação
das diversas complexonas (41).
As entidades envolvidas no problema são M, ião do
metal considerado, H, o hidrogenião e L, anião de
uma complexona que, para simplificar e sem perda de
generalidade, consideraremos monobásica. As cargas
das diferentes espécies não serão representadas para
não sobrecarregar excessivamente a impressão.
As reacções que vão ter lugar podem representar-se
de um modo geral por
M+ HL ML + H (2)
Ignoram-se, de momento, as interacções com o sol
vente — neste caso, a água — e considera-se a força
iónica do meio controlada por adição de um electrólito
de suporte inerte.
A constante de equilíbrio para a reacção (2) é
K [ML] . [H] Kk
[M] . [HL] KH
(3)
(*) Como medida de «basicidade» utilizamos, em geral, o valor
de —log kn = p/c„, sendo kn a última constante de dissociação
da complexona considerada. Assim, a elevados valores de pkn
corresponde uma elevada «basicidade» da complexona,
O último termo é independente da natureza do ligante
e é constante para todos os complexos de um dado
metal, desde que o solvente e a força iónica da solução
sejam os mesmos.
Se o termo 1
RT (GHL — G M I ) f ° r também constante,
é evidente que haverá uma relação linear, com coefi
ciente angular unitário, entre os valores de In KUL
e ln KHL ou, como é óbvio, entre log KML e log KHL.
1 Se (GHL—GMI)
n ã o for constante, poderão obter-se RT
relações lineares com coeficientes angulares diferen
tes da unidade se este termo for uma função linear
de ln KHL, ou não haverá qualquer espécie de relação
se a diferença (G°HL — G M L ) variar de um modo irre
gular.
Ora a diferença (G°HL — G°Ml) depende fundamental
mente da natureza do ligante e da interacção metal-
-ligante, podendo-se, para auxiliar a discussão, separar
as respectivas contribuições entálpicas e entrópicas:
G°HL — GML = (HHL — HMl) — T (SHL ~ SMl) (6)
A diferença de entropias-padrão em solução {SHL—S^i)
é, a menos de um valor constante, igual à variação
de entropia A 5 na reacção de complexação; ora os
33

valores que têm sido obtidos até ao presente parecem
indicar que:
a) AS é uma função linear de 5 ^ , com coeficiente
angular unitário (42).
b) Os valores de A S dependem do número de grupos
acetato coordenados no complexo (26, 35).
Poderemos pois inferir que nas reacções de formação
de complexos de um dado metal com complexonas
de igual potencialidade coordenativa as variações de
entropia dependerão sobretudo das variações de hidra
tação das espécies, e serão aproximadamente idênticas
desde que a variação no número de hidratação dos
complexos e dos ligantes seja a mesma, hipótese não
muito restritiva para uma família de complexantes tal
como a que consideramos aqui.
Parece portanto que a linearidade da relação entre
log KML e log KHL depende fundamentalmente da di
ferença HffL — HliL e portanto da energia das ligações
H-L e M-L.
isto é, o ganho de energia obtido pelo desdobramento
dos orbitais d do elemento central no campo dos li
gantes, com novo arranjo dos electrões de modo a
ocuparem os níveis de energia mais baixos (E).
Deste modo, o efeito energético total pode exprimir-se
pela soma das três contribuições especificadas, e como,
em princípio, é de esperar uma proporcionalidade
entre AH e ln KHL quando a natureza da interacção
metal-ligante for semelhante à da interacção protão-
-ligante, é evidente que terá de ser E nulo ou desprezável
e inexistentes as ligações n.
Nestas condições, o valor de C é função, até certo
ponto, das cargas e raios dos elementos centrais, tal
como W, e para uma série de ligantes intimamente
relacionados obtêm-se, com toda a probabilidade, re
lações lineares entre log KML e log KHL (ou pkHL) do
tipo (1):
•og KML = a pkHL + b
Tabela 11.11
CONSTANTES D E ESTABILIDADE (log K) DOS COMPLEXOS D E A L G U M A S COMPLEXONAS TETRADENTADAS
7" = 20,0°C
Complexona H Mg Ca 'Sr Ba Zn Ref.»
Ácido o-metoxifeniliminodiacético (1) 5,58 < o 2,75 2,13 2,08 — 0) Ácido sulfoetiliminodiacético (2) 8,16 3,48 4,15 3,26 3,01 7,05 (b) Ácido hidroxietiliminodiacético (3) 8,73 3,44 4,63 3,77 3,42 8,33 (b) Ácido metoxietiliminodiacético (4) 8,96 3,31 4,53 3,84 3,56 8,43 (*) Ácido tetrahidropirano-2-metiliminodiacético (5) 9,04 3,70 4,86 3,97 3,61 9,06 (a)
Ácido 2-hidroxiciclohexiliminodiacético (6) 9,57 4,27 5,19 3,81 3,26 9,19 («.) (P) Ácido carboxietiliminodiacético (7) 9,66 5,28 5,04 3,87 3,40 10,07 (b) Ácido fosfonoetiliminodiacético (8) 10,46 6,33 5,44 4,10 3,64 11,24 (b) Ácido o-hidroxifeniliminodiacético (9) 11,08 6,86 6,27 4,65 4,27 — (a) Ácido o-hidroxibenziliminodiacético (10) 11,79 7,28 6,74 4,99 4,40 — (b)
a) P resen te t raba lho . [X = 0,1 M (KN03).
b) Ref.» 21 . ¡X = 0,1 M (KCl).
Ora as energias de ligação metal-ligante podem divi
dir-se esquematicamente em três partes (43):
1) Energia de interacção electrostática do tipo ião-ião
ou ião-dipolo, dependendo das cargas das partículas,
dos seus raios e dos seus momentos dipolares (W).
2) Energia de formação de ligações covalentes a ou TZ,
dependendo das diferenças de electronegatividade dos
elementos centrais e dos ligantes e da disponibilidade
de orbitais em cada um para aceitar pares electrónicos
dos outros (C).
3) Energia de estabilização do campo dos ligantes,
Teoricamente, é pois de esperar uma relação deste tipo
se determinadas condições forem obedecidas. Em alguns
casos observam-se desvios que são principalmente devi-
vidos a efeitos da substituição causando estereo-impe-
dimentos; se esse não for o caso, deverão ter-se in
fringido as condições de comparação.
Os elementos centrais com estrutura de gás inerte são
os mais favoráveis para exemplificar a relação a que
chegámos. Com efeito, nos seus complexos não se
formam ligações n e a energia de estabilização do
campo de ligantes é nula; por outro lado, a interacção
3 4
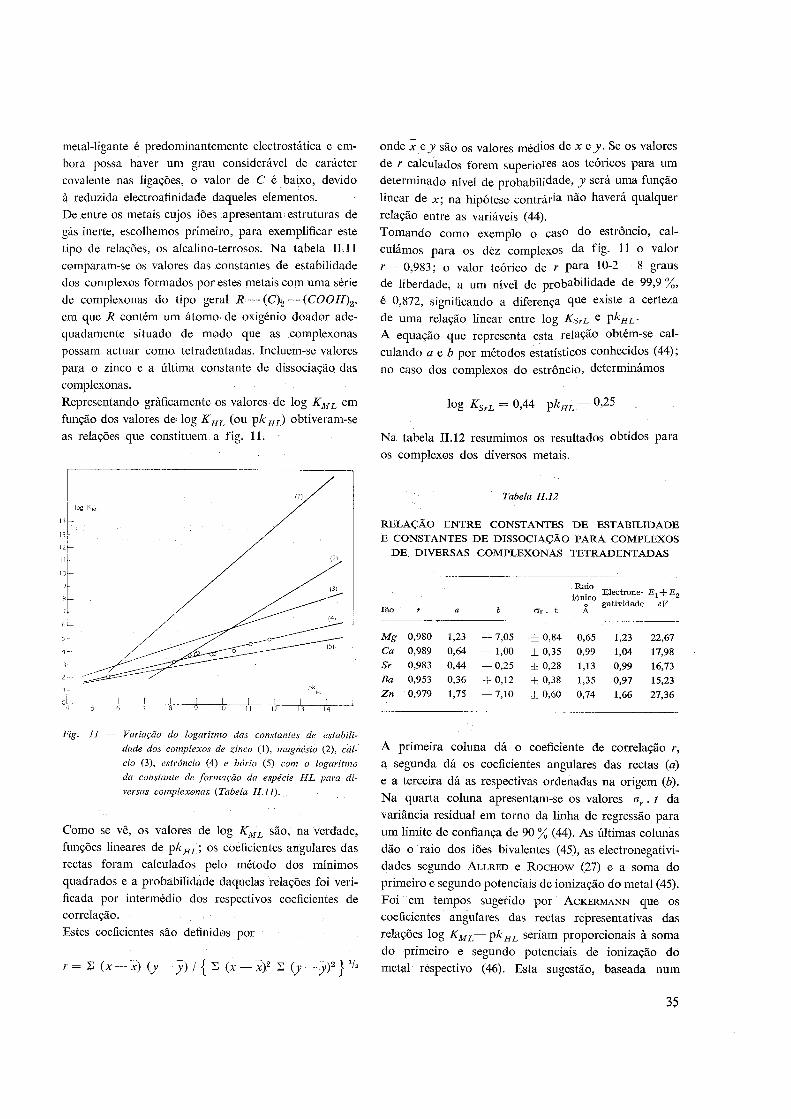
metal-ligante é predominantemente electrostática e em
bora possa haver um grau considerável de carácter
covalente nas ligações, o valor de C é baixo, devido
à reduzida electroafinidade daqueles elementos.
De entre os metais cujos iões apresentam estruturas de
gás inerte, escolhemos primeiro, para exemplificar este
tipo de relações, os alcalino-terrosos. Na tabela I I . 11
comparam-se os valores das constantes de estabilidade
dos complexos formados por estes metais com uma série
de complexonas do tipo geral R — ( Q 2 — (COOH)2,
em que R contém um átomo de oxigénio doador ade
quadamente situado de modo que as complexonas
possam actuar como tetradentadas. Incluem-se valores
para o zinco e a última constante de dissociação das
complexonas.
Representando graficamente os valores de log KML em
função dos valores de log KHL (ou ~çkHL) obtiveram-se
as relações que constituem a fig. 11.
o
lo
-(2)
-
mi
-• 5 T • 5 T
1 1 1 i i i i i i i 4 5 6 7 8 9 10 1 I 12 13 14
Fig. 11 — Variação do logaritmo das constantes de estabili
dade dos complexos de zinco (1), magnésio (2), cál
cio (3), estrôncio (4) e bário (5) com o logaritmo
da constante de formação da espécie HL para di
versas complexonas {Tabela 11.11).
Como se vê, os valores de log KML são, na verdade,
funções lineares de pkHL; os coeficientes angulares das
rectas foram calculados pelo método dos mínimos
quadrados e a probabilidade daquelas relações foi veri
ficada por intermédio dos respectivos coeficientes de
correlação.
Estes coeficientes são definidos por
r = S (x - x) {y—y) I { S (x - xf S (y -~y? }
onde x e>y são os valores m é d ¡ o s de x e_y. Se os valores
de r calculados forem superiores aos teóricos para um
determinado nível de probabilidade, y será uma função
linear de x; na hipótese contrária não haverá qualquer
relação entre as variáveis (44).
Tomando como exemplo o caso do estroncio, cal
culámos para os dez complexos da fig. 11 o valor
r = 0,983; o valor teórico de r para 10-2 = 8 graus
de liberdade, a um nível de probabilidade de 99,9 %,
é 0,872, significando a diferença que existe a certeza
de uma relação linear entre log KsrL e V^HL-
A equação que representa esta relação obtém-se cal
culando a e b por métodos estatísticos conhecidos (44);
no caso dos complexos do estroncio, determinámos
log KSrL = Q,AA ? k H L - 0 , 2 5 .
Na tabela 11.12 resumimos os resultados obtidos para
os complexos dos diversos metais.
Tabela 11.12
RELAÇÃO ENTRE CONSTANTES D E ESTABILIDADE
E CONSTANTES D E DISSOCIAÇÃO P A R A COMPLEXOS
D E DIVERSAS COMPLEXONAS T E T R A D E N T A D A S
i ã o r a b cr . t
. Ra io
iónico
A
Elec t rone-
ga t iv idade £ i + £ 2
eV
Mg 0,980 1,23 — 7,05 ± 0,84 0,65 1,23 22,67 Ca 0,989 0,64 — 1,00 ± 0,35 0,99 1,04 17,98 Sr 0,983 0,44 — 0,25 ± 0,28 1,13 0,99 16,73 Ba 0,953 0,36 + 0,12 ± 0,38 1,35 0,97 15,23 Zn 0,979 1,75 — 7,10 ± 0,60 0,74 1,66 27,36
A primeira coluna dá o Coeficiente de correlação r,
a segunda dá os coeficientes angulares das rectas (a)
e a terceira dá as respectivas ordenadas na origem (b).
Na quarta coluna apresentam-se os valores <sr. t da
variância residual em torno da linha de regressão para
um limite de confiança de 90 % (44). As últimas colunas
dão o raio dos iões bivalentes (45), as electronegativi-
dades segundo ALLRED e ROCHOW (27) e a soma do
primeiro e segundo potenciais de ionização do metal (45).
Foi em tempos sugerido por ACKERMANN que os
coeficientes angulares das rectas representativas das
relações log KML—pkHL seriam proporcionais à soma
do primeiro e segundo potenciais de ionização do
metal respectivo (46). Esta sugestão, baseada num
35

grupo de valores muito escasso (apenas três), pode
agora ser confirmada. Na fig. 12 verifica-se que existe,
na verdade, uma relação linear entre os valores dos
coeficientes angulares a e a soma dos potenciais de
ionização Ej_ + E2.
A 1 1 1 1 [ 1 1 1
2,2
2,0
1.8 Zn
/m
Zn p
1.6
1.4
1.2 : Mg Mg
1.0 -
0.8
0.6
0.4
Ca
Sr
Ba y Elecrronegar v idade
Ca •
Ba —
0,2 - 0,5
1 0,7
1 0.9
1 1.1
1 1.3 L
1.5 1,7 ± 1
1.9 t
2.1
0 15 17 19 21 23 25 27 29 31
Fig. 12 — Coeficiente angular das rectas da Figura 11 em
função da soma dos potenciais de ionização dos
metais (1) e da electronegatividade destes (2).
Devemos porém salientar que não tem significado
a utilização da soma Ex + E2 ou de qualquer dos
valores individuais E± ou E2, pois nós verificámos que
estes valores, para o grupo de metais considerado,
estão relacionados linearmente entre si (fig. 13) e todas
as correlações serão equivalentes.
E, eV
- 1 0
- 9
- 8
^ ^ B e "
- 7
- 6
- 5
- 4
S r "
Ba"
C a "
- 3
- 2
- 1 10 1 1 1 i i
15 • 1 , i i i
E ; cV
20 1 , ,
Fig, 13 — Relação entre o primeiro e o segundo potenciais
de ionização dos metais do grupo Ha e do zinco.
Para os metais alcalino-terrosos pode também veri
ficar-se uma excelente relação entre a e as electrone-
gatividades dos elementos considerados (fig. 12,) mas
esta relação não é extensiva ao zinco, que é um metal
de um subgrupo B no Quadro Periódico.
A fig. 11 permite ainda algumas conclusões interes
santes: a ordem de estabilidade dos complexos dos
metais estudados é Mg > Ca > Sr > Ba, ou seja a
ordem dos potenciais iónicos dos metais, para com-
plexonas em que pkHL > 10; porém, à medida que
o valor de pkHL desce, dão-se inversões, especialmente
com os complexos do magnésio. Parece evidente que
esta inversão da ordem de estabilidades é uma conse
quência dos diferentes coeficientes angulares das rectas
(e, portanto, do potencial de ionização dos metais),
sendo de esperar que se dê uma inversão total para
valores de pkHL suficientemente baixos, isto é, para
substâncias de força ácida relativamente elevada. Este
efeito foi, na verdade, verificado nos complexos de
ligantes monodentados, todos eles bases conjugadas de
ácidos bastante fortes (tabela 11.13).
Tabela 11.13
CONSTANTES D E ESTABILIDADE (log K) D E A L G U N S
COMPLEXOS DOS METAIS ALCALINO-TERROSOS
T = 25°C [x -> 0
Ligante H + Afg2 + Caï + Sf2 + Baí +
lodato 0,77 0,72 0,89 0,96 1,05
Sulfato 1,92 2,29 2,31
Tiossulfato 1,72 1,84 1,92 2,04 2,33
Nitrato (*) — 1,3 cpx? 0,28 0,82 0,92
Ferricianeto 1 2,79 2,83 2,85 2,88
(*) T = 18°C.
Valores obt idos na referência (47).
Para estes ligantes a ordem de estabilidades é Ba >
> Sr > Ca > Mg, e W i l l i a m s ( 48 ) interpretou este
resultado admitindo a possível formação de pares
iónicos segundo reacções do tipo de
M (H20\ + LZZM (H20\ L
Se os catiões nos pares iónicos fossem igualmente hi
dratados, a estabilidade destes seguiria a ordem Mg >
> Ca > Sr > Ba, porque o diâmetro dos catiões hi
dratados diferiria do diâmetro dos catiões isolados
3 6

apenas por uma constante. No entanto, ê possível que
as estabilidades determinadas se refiram a diferentes
graus de hidratação dos catiões, o que origina uma
ordem de estabilidades contrária à ordem dos potenciais
iónicos dos metais visto que o ião bário isolado Ba2+
é menor que o ião magnésio hidratado Mg(H20)\+.
Em resumo, os valores experimentais das constantes
de estabilidade diriam respeito a verdadeiros com
plexos no caso dos catiões de maiores dimensões e a
pares-iónicos no caso dos de menores dimensões.
Parece-nos natural que, à medida que a força básica
dos ligantes aumenta, se torna energéticamente mais
favorável a competição destes com as moléculas de
água do catião hidratado, que assim serão deslocadas
segundo a equação
M (H20)nL Zl ML (H20)m + (n-m) H20
Este processo resulta, por um lado, na diminuição do
calor de formação dos complexos (porque a interacção
M — L será maior) e, por outro, numa variação de
Iyigantes
Ácido uramildiacético exp.
pfc, = 9,63 cale.
A Ácido o-carboxifeniliminodiacético exp.
pks — 7,75 cale.
A Ácido o-sulfonofeniliminodiacético exp.
pfe3 = 6,29 cale.
A Ácido etilenodiamina-NN-diacético exp.
pk2 = 11,05 cale.
A Ácido metiltioetiliminodiacético exp.
pfc¡ = 8,91 cale.
A Ácido mercaptoetiliminodiacético exp.
p/c 3 = 10,79 cale.
A
entropia, crescente em valor positivo, à medida que
aumenta o número de partículas na solução. Se assim
for, e dentro de certos limites, obter-se-á uma propor
cionalidade entre as variações de entalpia e as variações
de entropia na formação das espécies ML(H20)m,
até que a ocorrência de ligações covalentes e outros
factores alterem o esquema simples proposto.
Esta hipótese poderá constituir uma explicação acei
tável para a distinção, um tanto quanto arbitrária, de
DUNCAN entre «pares-iónicos» e «complexos» (49). Tal
distinção é baseada na proporcionalidade das variações
de entalpia e entropia, que parece verificar-se nos
«pares-iónicos» e não nos «complexos»; no esquema
proposto, as formações de pares-iónicos e de com
plexos aparecem como extremos numa interacção gra
dualmente crescente entre o catião hidratado e os
ligantes.
As observações anteriores ilustram com clareza o in
teresse das relações log KML — pkHL na interpretação
de alguns factos experimentais. Fica também justificada
a possibilidade da previsão teórica de constantes de
estabilidade, desde que se obedeçam às condições de
comparação exigidas. Assim, na elaboração da fig. 11
alguns ligantes tetradentados não foram tomados em
8,19 8,31 6,93 6,73
4,80 5,16 3,99 3,59 9,75
+ 3,39 + 3,15 + 2,94 + 3,14 — 3,91 5,06 3,91 3,57 — 2,48 3,96 3,16 2,91 6,46
+ 1,43 + 1,10 + 0,75 + 0,66 — 2,68 4,57 3,50 3,48 — 0,68 3,03 2,52 2,38 3,91
+ 2,00 + 1,54 + 0,98 + 1,10 —
4,53 4,63 3,55 3,19 11,93
6,54 6,07 4,61 4,04 12,24
— 2,01 — 1,44 — 1,06 — 0,85 — 0,31
3,02 3,34 2,71 2,62 8,28
3,91 4,70 3,67 3,33 8,49
— 0,89 — 1,36 — 0,96 — 0,71 — 0,21
4,32 4,88 3,62 3,55 15,92
6,22 5,91 4,50 4,00 11,78
— 1,90 — 1,03 — 0,88 — 0,45 + 4,14
consideração, nomeadamente os ácidos uramildiacético,
o-carboxifeniliminodiacético e o-sulfonofeniliminodiacé
tico, cujo comportamento anormal já comentámos (p. 30);
Tabela 11.14
DIFERENÇAS ENTRE VALORES D E KUL EXPERIMENTAIS E TEÓRICOS
Log K M J Mg Ca Sr Ba Zn
3 7

também não foram incluídos os ácidos nitrííotriacético
e fosfonoiminodiacético, que não são geometricamente
comparáveis com os outros, e os ácidos em que o átomo
doador no grupo R não é o oxigénio. Resta-nos de
monstrar estatisticamente que estas omissões são jus
tificáveis.
Para uma complexona tetradentada «normal», isto é,
que se comporte como qualquer das dez mencionadas
na tabela 11.11, haverá 9 0 % de probabilidade de que
a diferença entre os valores de KML experimentais e
teóricos, calculados por log KML = a pkHL + b, caiam
no intervalo [— ar.t, + csr. t] (tabela 11.12). Qualquer
desvio substancial deste intervalo indicará um com
portamento anómalo.
Na tabela 11.14 resumimos os resultados de compa
rações desta natureza para os Complexos de algumas
das complexonas mencionadas.
Verifica-se que todos os complexos do ácido ura-
mildiacético são, na verdade, mais estáveis que o que
seria de prever na base de um comportamento «normal»
daquela complexona. Também os dois ligantes aromá
ticos que se seguem na tabela dão complexos de esta
bilidade excepcional, sendo de interesse notar que os
acréscimos A diminuem à medida que o raio iónico
dos metais aumenta (comparar com p. 28 ) .
metais alcalino-terrósós são rherios estáveis. Com—NB^
e — S~~ como doadores a diminuição de estabilidade
acentua-se à medida que o raio iónico do metal de
cresce, atingindo o máximo para o magnésio. Para
o zinco, porém, os valores de A nos complexos dos
ácidos etilenodiamina-NN-diacético e metiltioetllimino-
diacético estão bem dentro da zona a, . t = ± 0 ,60 ,
indicando que a força das ligações Zn-N e Zn-S é com
parável à da ligação Zn-O. Já com o ácido mercapto-
etiliminodiacético o desvio positivo é notável, eviden
ciando a tendência do ião S~ para formar uma ligação
excepcionalmente forte com o zinco, provavelmente
com um certo carácter TC.
Deve salientar-se que em tudo o que se disse está im
plícita a hipótese de que os valores de log KHL utili
zados na fig. 11 se referem a uma combinação entre
o protão e o anião das complexonas com uma estrutura
análoga à dos complexos dos metais. Aliás, um recente
trabalho de CHAPMAN (40) , já mencionado, dá um
apoio considerável a esta hipótese (v. p. 31).
Em relação a outros catíões com estrutura de gás
inerte não tem sido até aqui possível discutir relações
do tipo das exemplificadas por escassez dos valores
necessários.
O número substancial de constantes determinadas no
Tabela 11.15
CONSTANTES D E ESTABILIDADE (log K) D E COMPLEXOS DOS METAIS ALCALINOS COM DIVERSAS
COMPLEXONAS. T = 20,0°C
Ligante H Li Na K Ref.»
Ácido iminodiacético 9,38 0,96 0,36 (a)
Ácido metiliminodiacético 9,65 1,20 0,61 — (a) Ácido ciclohexiliminodiacético 10,81 L74 0,90 — (a)
Ácido 2-hidroxiciclohexiliminodiacético 9,57 2,19 0,76 — («) Ácido o-mdroxifeniliminodiacético 11,08 2,20 1,0 (a)
Ácido 0-carboxifeniliininodiacético 7,75 2,05 0,89 (a)
Ácido nitrilotriacético 9,74 2,51 1,32 — («) Ácido etilenodiaminotetracético 10,26 2,79 1,66 — (b) Ácido 1,2-diaminopropanotetracético 10,86 3,43 2,24 — (a)
Ácido uramildiacético 9,63 4,90 2,72 1,23
(a) Presente t raba lho . (X = 0,1 M Í.KN03).
[b) Ref," (21). [X =- 0,1 M (KCl). . .
Quando o átomo doador no grupo R das complexonas
de fórmula geral R — (C)2—JV (CH2COOH)2 é o azoto
oú o enxofre em vez do oxigénio, os complexos dos
presente trabalho permite já encarar a discussão quan
titativa do comportamento dos metais alcalinos, em
especial do sódio e do lítio, já que para o potássio
3 8

só foi possível obter valores mensuráveis com a série
de derivados do ácido uramildiacético.
Reunimos na tabela 11.15 os valores das constantes
de estabilidade dos complexos formados por estes
metais e pelo hidrogénio com diversas complexonas
de todos os tipos.
Na fig. 14 representam-se graficamente os valores de
log KLiL em função dos correspondentes valores de
PkHL-
log K
Fig. 14 — Valores de KML para complexos do titio em função
dos valores correspondentes de pkjjj^ (Tabela 11.15).
Complexonas representadas: (1) ácido iminoãiacético;
(2) ácido metiliminodiacético; (3) ácido ciclohexil-
iminodiacético; (4) ácido o-hidroxifeniliminodiacético;
(5) ácido 2-hidroxiciclohexiliminodiacètico; (6) ácido
nitrilotriacético; (7) ácido etilenodiaminotetracético;
(8) ácido \,1-diaminopropanotetracético; (9) ácido
o-carboxifeniliminodiacético; (10) ácido uramildiacé
tico.
É evidente que se podem definir duas linhas rectas:
uma onde se acham os pontos dos complexos dos
ácidos iminodiacético, metiliminodiacético e ciclohexi-
liminodiacético, que actuam, no máximo, como ligantes
tridentados; outra com os pontos dos complexos dos
ácidos 2-hidroxiciclohexiliminodiacético e nitrilotriacé-
cético, que actuam, no máximo, como tetradentados.
Para os complexos do sódio obtém-se um resultado
idêntico, e as duas linhas correspondem, provavelmente,
a complexos nos quais o número de coordenação é
3 e 4.
Notam-se algumas excepções, nomeadamente com os
ácidos o-carboxifeniliminodiacético e uramildiacético,
já anteriormente discutidas.
Resultados surpreendentes são, porém, os que se obtêm
com os ácidos o-hidroxifeniliminodiacético, etilenodia
minotetracético e 1,2-diaminopropanotetracético, pois
todos estes ligantes se comportam como se possuíssem
menores possibilidades coordenativas.
Já anteriormente fizemos algumas observações a este
respeito (p. 20) e cremos que as anomalias resultam
da reduzida tendência que os ligantes azotados pos
suem para se combinarem com os iões dos metais
alcalinos. Serão de esperar ligações fracas em todos
os casos, e o número de ligações «efectivas», respon
sáveis pela estabilidade dos complexos, será menor
que o número de ligações possíveis.
Ora já foi mencionado, a propósito do ácido uramil
diacético, que as variações de entalpia e entropia na
formação dos complexos daquele ácido com os iões
dos metais alcalinos eram função dos respectivos raios
iónicos; o mesmo deverá acontecer com as variações
de energia livre de GIBBS e, portanto, com os valores
de log KML. DuNCAN (50) demonstrou que, na zona
dos raios iónicos existentes, diversos tipos de função
eram equivalentes; no caso presente a relação
- 6
Fig. 15 — Logaritmo das constantes de estabilidade dos com
plexos formados pelos metais alcalinos com diversas
complexonas em função da raiz quadrada do inverso
dos respectivos raios iónicos.
Complexonas representadas: (1) ácido iminodiacético;
(2) ácido metiliminodiacético; (3) ácido ciclohexilimi-
nodiacético; (4) ácido o-carboxifeniliminodiacético;
(5) ácido o-hidroxifeniliminodiacético; (6) ácido ni
trilotriacético; (7) ácido etilenodiaminotetracético;
(8) ácido 1,2-diaminopropanotetracético; (9) ácido
uramildiacético.
3 9

é a mais satisfatória. Esta relação está representada
na fig. 15 para os complexos do lítio e do sódio; no
caso do ácido uramildiacético incluiu-se também o valor
para o potássio. Excluiu-se o ácido 2-hidroxiciclohe-
xiliminodiacético, que parece dar um valor anómalo
para o complexo do sódio.
Se a nossa hipótese for correcta, ao representar log KML
I T em função de (27V— 3,35) i / — deverá obter-se uma
V r i
família de rectas paralelas. A fig. 16 mostra que, na
verdade, assim acontece.
Por outro lado, B deve depender apenas de caracte
rísticas do ligante, e, em casos simples, apenas do
valor de pkn. Representando graficamente o valor das
ordenadas na origem das rectas da fig. 16 em função
- 6
Fig. 16 — Logaritmo das constantes de estabilidade dos com
plexos formados pelos metais alcalinos com diver
sas complexonas em função do número de coorde
nação e da raiz quadrada do inverso dos raios
iónicos dos metais.
Complexonas representadas: (1) ácido iminodiacético;
(2) ácido metiliminodiacético; (3) ácido ciclohexilimi-
nodiacético; (4) ácido o-carboxifeniliminodiacético;
(5) ácido o-hidroxifeniliminodiacético; (6) ácido ni-
trilotriacético; (7) ácido etilenodiaminotetracético;
(8) ácido 1,2-diaminopropanotetracético; (9) ácido
uramildiacético.
A maior parte dos ligantes divide-se em dois grupos,
correspondendo a complexos nos quais o número de
coordenação TV é o mesmo.
Admitindo que o coeficiente angular das rectas da
fig. 15 depende linearmente de TV, facilmente se de
termina.
A = 2 TV—3,35 (8)
Combinando as equações (7) e (8) obtém-se
log KML = (2N- 3,35) yj±- + B (9)
0 2 4 6 8 10 12 14 16
Fig. 17 — Comparação entre os valores calculados e os va
lores experimentais das constantes de estabilidade
dos complexos formados pelos iões com estrutura de
gás-inerte com diversas complexonas (Tabelas 11.16
e 17).
dos valores de pkn das complexonas respectivas, obtém-
-se para B a seguinte expressão, em termos de TV e pkn:
B = 0,45 pkn — (TV + 3,5) (10)
E, combinando (9) e (10)
log KML = (2 TV-3,35) \J~ + °> 4 5 VK -
- ( A - + 3 , 5 ) (11)
40

Esta equação pode, a despeito de algumas inconsis
tências, reproduzir com bastante fidelidade a maior
parte dos resultados obtidos para os complexos dos
metais alcalinos com os ácidos aminopolicarboxílicos;
probabilidade de uma relação linear com coeficiente
angular unitário entre os valores de log KML experi
mentais e os calculados pela equação empírica que
propomos.
Tabela 11.16
COMPARAÇÃO ENTRE VALORES D E log KML CALCULADOS E EXPERIMENTAIS
Li + Na+ C«2 + Sf2 + Ba2 +
legante AT cale. exp. cale. exp. cale. exp. cale. exp. calc. exp.
Ácido iminodiacético 3 1,15 0,96 0,45 0,36 2,98 2,59 2,71 1,81 2,28 1,67
Ácido metiliminodiacético 3 1,27 1,20 0,58 0,61 3,13 3,75 2,84 2,85 2,41 2,59 Ácido ciclohexiliminodiacético 3 1,80 1,74 1,10 0,90 3,64 3,34 3,36 2,55 2,93 2,37
Ácido 2-hidroxiciclohexiliminod ¡acético 3 e 4 1,90(*) 2,20 1,17(* ) 1,00 6,85 6,27 5,54 4,65 4,80 4,27 Ácido o-hidroxifenilirainodiacético 3 e 4 2,80 2,19 1,23(* )0 ,76 6,08 5,19 3,49(*) 3,81 2,94( *) 3,26 Ácido o-carboxifeniliminodiacético 4 2,03 2,05 0,77 0,89 5,26 5,06 4,73 3,91 3,75 3,57
Ácido nitritotriacético 4 2,88 2,51 1,67 1,32 6,12 6,41 5,72 4,98 4,75 4,82
5 9,40 8,62 7,44
Ácido etilenodiaminotetracético 4 e 3,10 2,79 1,91 1,66 10,70 8,63 7,76 6 12,38 11,39 9,86 5 9,68 8,89 7,72
Ácido 1,2-diaminopropanotetracético 4 e 3,41 3,43 2,19 2,24 11,70 9,67 8,57
6 12,68 11,67 10,74
(*) Menor valor de N.
o mais notável, porém, é que pode tornar-se aplicável
a todos os metais cujos iões possuam a estrutura de
gás inerte, tomando apenas em consideração a res
pectiva carga iónica Z{.
A equação geral será
log KML = (27V— 3,35) Z,. 0,45 p V
•(N+ 3,5) (12)
As tabelas 11.16 e 17 e a fig. 17 demonstram a exce
lente concordância verificada entre os valores calculados
e os valores experimentais das constantes de estabilidade
dos complexos do lítio, sódio, cálcio, estrôncio, bário,
e mesmo os lantanidos, com diversas complexonas de
todos os tipos. Excluiu-se o magnésio porque as redu
zidas dimensões do ião Mg2+ forçam os ângulos de
valência na estrutura dos ligantes e provocam frequen
temente valores anormais das constantes de estabili
dade dos respectivos complexos.
Os pontos representados na fig. 17 (e outros não repre
sentados para não a sobrecarregar demasiado) podem
ser considerados estatisticamente, evidenciando a forte
Esta é pois uma boa aproximação, com inegável uti
lidade na previsão teórica de constantes da estabilidade
dos complexos dos metais cujos iões apresentam a
estrutura de gás inerte. Os maiores desvios verificam-se
para complexos nos quais o número de coordenação
«efectivo» é menor que o suposto TV, o que acontece
Tabela 11.17
COMPARAÇÃO ENTRE VALORES D E log KML CAL
CULADOS E EXPERIMENTAIS (TERRAS R A R A S E ÍTRIO)
N I T A (¡V = 4) R D T A (N - 5)
Metais cale. exp . cale. exp.
La 10,40 10,37 15,46 15,40
Ce 10,56 10,71 15,67 15,80
Pr 10,70 10,89 15,87 16,16
Nd 10,81 11,09 16,03 16,47
Sm 10,95 11,39 16,43 16,90
Gd 11,29 11,43 16,72 17,20
Dy 11,49 11,62 17,12 17,75
Yb 11,91 12,09 17,67 18,70
Y 11,31 11,41 16,80 17,8
4 1

quando o ião do metal tem uma afinidade reduzida
para um ou mais dos átomos ligantes na complexona.
É o caso dos ligantes com azoto alifático, oxigénio-éter,
álcoois, etc. Se fosse possível fazer uma estimativa
realista do número de coordenação «efectivo», é pro
vável que uma equação com a mesma forma de (12)
pudesse reproduzir todos os valores experimentais com
elevado grau de fidelidade.
A generalidade da equação obtida dá maior apoio
à ideia de que nos complexos dos metais alcalinos com
o EDTA o número de coordenação não excede 4, embo
ra nos complexos dos metais alcalino-terrosos e lantani-
dos seja 5 < N < 6, o que parece implicar que os
seus seis átomos ligantes potenciais estão de facto
coordenados.
Este resultado não serve, porém, como prova conclu
dente, uma vez que a equação geral foi estabelecida
por raciocínio indutivo; por outro lado, no caso dos
lantanidos, há que contar ainda com a estabilização
originada pelo campo dos ligantes, originada pelo
preenchimento preferencial de orbitais / .
4 — CONSIDERAÇÕES FINAIS
Até ao presente não tinha ainda sido possível discutir
a estabilidade dos complexos dos iões monovalentes
por escassez de valores de constantes. Com os valores
agora disponíveis podem fazer-se alguns comentários
a este respeito.
O comportamento dos metais alcalinos foi largamente
comentado nas alíneas anteriores, embora a discussão
fosse limitada ao lítio e ao sódio por não ter sido
possível, com a técnica potenciométrica utilizada, de
terminar mais valores para os complexos do potássio.
Verificou-se, no entanto, que a ordem de estabilidades
normal é Li+ > Na+ > K ' e, presumivelmente, será
Li+ > Na+ > K+ > Rb+ > Cs+. Com bases conju
gadas de ácidos fortes como ligantes poderão dar-se
inversões, o que se verifica com o N03, S203 , HSO^ ,
C107¿ e 101.
A natureza das ligações nos complexos dos metais
alcalinos é, como se sabe, predominantemente electros
tática; pode, porém, admitir-se um certo grau de cova-
lência, em especial nos complexos do lítio (pois o
ião possui um poder deformante elevado devido às
suas reduzidas dimensões); alguns complexos deste ele
mento serão pois um pouco beneficiados em relação
aos complexos dos outros metais alcalinos e até mesmo
aos do tálio, que possui um raio iónico demasiado
grande.
No entanto, os complexos deste metal e os da prata
são, em geral, mais estáveis que os dos metais alca
linos, e com as complexonas obtivemos em todos os
casos a ordem Ag+ > Tl+ > Li+ > Na+ > K+.
Esta constatação experimental está de acordo com
o que seria de esperar teoricamente na base da força
atractiva exercida pelos iões, dada por / = Z e ///-^(*)
(28, 51). Para idênticos raios covalentes, esta força au
menta com o aumento da carga nuclear efectiva Ze¡,
e, uma vez que esta depende da capacidade de blin
dagem dos electrões presentes nos átomos (que diminui
na ordem Í > p> d> / ) , infere-se imediatamente que
a estabilidade dos complexos de Ag+ deve ser, em
princípio, superior à dos complexos dos outros iões.
Ainda na mesma base devem colocar-se em segundo
lugar os complexos do Tl+, se bem que o baixo poder
neutralizante das subcamadas 4 / 1 4 e 5a"10 seja par
cialmente compensado pela blindagem eficiente da suh-
camada 6 s2. Na verdade, é por esta razão que o com
portamento do ião 77 + se assemelha frequentemente
ao dos iões dos metais alcalinos, mas não se pode
compensar totalmente o efeito dos electrões d e / e a
força atractiva exercida por aquele ião terá de ser
superior à exercida por estes. Finalmente, os valores
de r°M determinam a ordem de estabilidades teórica, que
será paralela à da força atractiva, isto é, será Ag+ >
> 77+ > Li+ > Na+ > K+.
Quando a interacção metal-ligante é predominante
mente electrostática, a estabilidade dos complexos for
mados por estes iões variará de um modo regular com
Zeflr°M e não serão de esperar grandes diferenças. Este
é o caso das complexonas estudadas neste trabalho;
a ordem de estabilidades observada é a teórica e os
valores das constantes não são muito afastados entre si.
É claro que se conhecem exemplos em que a estabi
lidade dos complexos dos iões 77+ e Ag+ é muito
semelhante e até mesmo a ordem poderá ser invertida;
acontece isto com ligantes que são bases conjugadas de
ácidos fortes, como o NOs~ e o ffi?04
_ (tabela 11.18),
e trata-se mais uma vez do efeito de diferenças de
hidratação (p. 37).
* Utilizou-se o raio covalente, que é da ordem das distâncias
metal-ligante verificadas nos complexos (28).
4 2

Quando se torna possível a formação de ligações cova
lentes, a ordem de grandeza das estabilidades pode ser
consideravelmente alterada; se o ião do metal actua
como receptor de electrões, a função Zej\r°M continua
a ser uma medida da intensidade de tais ligações, pois
representa o poder polarizante do ião à distância r°M.
No entanto, a formação de ligações covalentes depende
também da polarizabilidade dos ligantes e da disponi
bilidade de orbitais no metal, e neste particular os
iões 77 + e Ag+ são realmente muito diferentes. No 77 +
os dois electrões do subnível 6s constituem um «par
inerte» (52) e os orbitais utilizáveis para ligações cova
lentes são os 6p. A energia necessária para desacoplar
o par inerte e a necessária para provocar a promoção
s -> p não são conhecidas mas são certamente muito
elevadas (28); não é pois surpreendente que o tálio
forme um número tão reduzido de compostos cova
lentes. Por outro lado, o ião Ag+ tem uma vasta gama
de possibilidades, e a formação de compostos deste
tipo depende unicamente dos ligantes. Quando a for
mação de ligações covalentes for possível, devem
portanto esperar-se diferenças de estabilidade conside
ráveis nos complexos do tálio e da prata. Gomo exem
plos típicos apresentam-se os casos da amónia e da
etilenodiamina (tabela 11.18).
Diferenças- ainda mais consideráveis resultarão quando
o lígante possua orbitais p ou d disponíveis e possa
aceitar pares de electrões d dos metais para formar
ligações dn—p^ ou dK — dn. Como o ião Tl"1" não
pode formar tais ligações (pois os seus electrões d
estão «isolados» pelos electrões exteriores da subcamada
6 s) e, por outro lado, o ião Ag+ é um dos que maior
tendência têm a formá-las, a estabilidade dos complexos
destes catiões com ligantes que possam actuar como
receptores iz deve diferir notavelmente. Este efeito é
ilustrado de modo espectacular com os valores das
Constantes de estabilidade dos iodo e ciano-complexos
na tabela 11.18.
Tabela 11.18
CONSTANTES D E ESTABILIDADE (log K) D E A L G U N S
COMPLEXOS DOS IÕES Ag+ E 77+
I . igautes Ag + Tl + A log K
NO¡¡~ — 0,29 0,33 — 0,62
HSO,{- 1,3 1,33 0,0
2,3 0,82 1,48
cr 3,04 0,68 2,36
NH3 3,32 — 0,92 4,24
Br" 4,38 1,05 3,33
SCN~ 4,75 0,80 3,95 (H2N—CH2—)2 5,0 0,40 4,60
I- 8,13 0,72 7,41
CN~ 21,4 (log [i2) cpx.? 21,4
Valores a temperatura e força iónica variáveis. Ret> (21).
Resumindo: quando as ligações entre o metal e o
ligante têm um carácter covalente acentuado, a órderh
de estabilidades dos complexos formados deverá sèr
Ag+> 77+, Li+ > Na+ > K+.
O tálio assemelha-se, neste caso, aos metais alcalinos,
e a prata aos restantes elementos do seu subgrupo,
isto é, ao cobre e ao ouro. Outros iões pseudomo-
novalentes por nós estudados, tais como o catião
C2H&Hg+ (53), assemelham-se também aos iões mono
valentes dos elementos deste subgrupo.
O possível interesse prático de uma complexona pos
suindo um receptor potencial de electrões incorporado
levou-nos já a sintetizar o ácido piridil-2,6-metilimi-
nodiacético (54); os resultados obtidos com este ligante
não provam sem ambiguidade a formação de liga
ções ir, contrariamente ao que se esperava. Este é,
todavia, um campo que aguarda futuras investigações.
43

III. Secção experimental
A) PREPARAÇÕES
1 — SÍNTESE DAS COMPLEXONAS
Introdução
A síntese de complexonas muito puras é, na maioria
dos casos, um problema bastante delicado. Algumas,
tal como o EDTA, são muito pouco solúveis em água
e precipitam em elevado estado de pureza quando
soluções alcalinas dos seus sais de sódio são acidi
ficadas com ácidos minerais. Outras, porém, são extre
mamente solúveis e não podem ser precipitadas nem
recuperadas por outros processos usuais, como a ex
tracção com solventes, visto que nenhum dos ácidos
aminopolicarboxílicos é apreciavelmente solúvel em tais
substâncias. Por outro lado, as complexonas, quando
razoavelmente puras, podem ser recristalizadas com
água sem grandes dificuldades; a presença de impu
rezas, em especial outros aminoácidos, ácido glicólico
ou sais inorgânicos, cria problemas dificilmente reso
lúveis.
Foram já descritas diversas sínteses de complexonas (55),
embora nem todas aplicáveis à preparação de substân
cias adequadas para estudos rigorosos. O método mais
geral consiste na condensação de aminas com cloroace-
tato de sódio em meio alcalino, normalmente a um pH
mantido entre 8 e 10, de modo a impedir a formação
de ácido glicólico por reacção com a água, o que levaria
a uma mistura viscosa difícil de cristalizar. Este método
é especialmente útil se a complexona é pouco solúvel
em água e precipita por acidificação da solução alcalina
do seu sal de sódio. O processo mais frequentemente
usado para fins industriais consiste numa espécie de sínte
se de STRECKER, conduzindo à formação de nitritos que
em seguida se hidrolisam aos ácidos correspondentes.
Neste trabalho utilizámos a primeira técnica em todos
os casos, variando porém as condições experimentais
e as particularidades de separação e purificação dos
produtos. Por esta razão todas as preparações são
descritas com certo pormenor.
Das complexonas cuja síntese se relata, três eram já
conhecidas: os ácidos uramildiacético, o-carboxifenil-
iminodiacético e ciclohexilimínodiacético. Contudo, e
tal como se disse anteriormente, estes produtos não
foram antes obtidos em estado de pureza suficiente, pelo
que foi necessário elaborar métodos mais perfeitos que
ora se descrevem.
a. Síntese do ácido 5-aminobarbitúrico-l ,1-diacético (ura
mildiacético)
/ / ° / N H - C X
0 = C C H - N H , \ /
N H — C
N H -/
0 = C
C / C H 2 C O O H
C H - N
\ | H c / V H , C O O H
Uramil (M. M. = 143) Ácido uramildiacético (M. Af .=259)
Suspenderam-se 10 g de uramil (0,07 moles) em 14 ml
de NaOH 5 M (0,07 moles); adicionaram-se 19 g de
ácido cloroacético ( — 0,2 moles), previamente neutra
lizado com NaOH, agitando constantemente. Após a
adição, a solução foi aquecida à ebulição e adiciona
ram-se 28 ml de NaOH 5 M (0,14 moles) a uma velo
cidade tal que o pH da solução se manteve sempre
4 4

entre 8 e 10. Esta adição levou cerca de 30 minutos,
após o que se obteve uma solução rósea que se ferveu
durante mais dez minutos sem que se notasse variação
de p/J.
Arrefeceu-se em seguida num banho de gelo moído
e adicionaram-se cerca de 40 ml de HCl concentrado,
obtendo-se uma suspensão facilmente flltrável. Deixou-
-se esta repousar durante cerca de 1 hora, filtrou-se,
e o sólido obtido recristalizou-se sucessivamente
com HClOi 2M e água.
O produto secou-se a pressão reduzida sobre P2Oh,
obtendo-se na forma de pequenas lamelas brancas
fundindo com decomposição a 240°C.
C 8/J 97V 30 7 (M.M. = 259,2)
Cale. C 37,1 %, H 3,5 %, TV 16,2%
Anal. C 37,1 %, H 3,6%, TV 16,0%,
M.M. (titulação) = 259
Seguindo o método descrito por outros autores (16)
obteve-se um sal ácido de sódio (p. 15) cuja compo
sição foi estabelecida por titulação e análise.
C8H807N3Na . CsH907N3 . H20
Cale. C 34,4%, H 3,4 %, TV 15,05 %, Na 4,1 %
Anal. C 34,4 %, H 3,55 %, TV 14,9 %, Na 4,1 %
Obteve-se também ácido uramildiacético puro a partir
deste sal ácido, por recristalizações sucessivas com
HClOl 2 M e por eluição através de uma coluna de
resina catiónica na forma ácida, seguida de concen
tração do eluído até à cristalização do produto. O pri
meiro método deu, porém, melhores resultados.
b. Síntese do ácido l-metil-5-aminobarbitúrico-7,7-diacé-
tico (1-metiluramildiacético)
A síntese desta complexona foi realizada em três fases:
1. Síntese do ácido 1-metilbarbitúrico
C H ,
KW C 2 H 5 O C ^
0 = C ^ + C H 2
N H 2 C , H 5 O C ^
Meti/ureia Malonato de dietilo
(Aí. Aí. = 74) (Aí. Aí. = 160)
• 0 = C CH, \ /
N H C
Acido 1-metilbarbitúrico
(Aí. M. - 142)
Adicionaram-se 3 g de sódio metálico bem limpo
( - 0 , 1 5 átomos-grama) a 75 ml de álcool absoluto,
previamente destilado sobre aparas de cálcio, num
balão de três tubuladuras munido com agitador e
condensador de refluxo. Depois da dissolução adicio
naram-se, de uma só vez, 20 g de malonato de dietilo
(0,125 moles) e em seguida, gota a gota e com forte
agitação, uma solução de 9,3 g de metilureia (0,125
moles) em 50 ml de álcool absoluto. Após a adição
manteve-se a mistura em refluxo durante 5 horas, pre
cipitando, entretanto, o metilbarbiturato de sódio. Arre
feceu-se então a suspensão, filtrou-se, lavou-se com
etanol e secou-se. Obtiveram-se 12 g do produto (ren
dimento 60%).
Dissolveram-se 8,2 g deste (0,05 moles) em 200 ml de
água e adicionaram-se 2,6 ml de £f2SOi concentrado
(0,05 moles). O ácido 1-metilbarbitúrico não precipitou
imediatamente e a solução foi logo usada para a se
gunda fase de preparação.
2. Síntese do ácido l-metil-5-aminobarbitúrico (1-me-tiluramil)
C H , C H ,
0 = C ,CH, • o=c ^NH—C
Ãcido 1 - metilbarbitúrico
(Aí. Aí. = 142)
N H - < ; C H - N H 2
O
Ácido 1 - metil
5 - aminobarbitúrico
(Aí. Aí. = 157)
A solução obtida na fase anterior foi tratada com
3,8 g de nitrito de sódio, (0,055 moles) desenvolvendo-se
imediatamente a cor característica do derivado nitroso.
Levou-se a solução à ebulição e adicionaram-se de
uma só vez 30 g de ditionito de sódio (Na^O^ 2 H20)
dissolvidos em 200 ml de amónia 1:3. A solução, que
descorou em poucos segundos, foi fervida durante cerca
de 1 hora até começo de precipitação. Por arrefecimento
separou-se o 1-metiluramil, que foi filtrado, lavado com
água e seco na estufa.
Obtiveram-se 4 g (rendimento 50 %) de um pó branco
com ponto de fusão 260-l°C dec. (literatura: 253-6°).
3. Síntese do ácido l-metil-5-aminobarbitúrico-7,7-dia-cético.
Condensaram-se 3,1 g de ácido l-metil-5-aminobarbi-
túrico (0,02 moles) com 5,5 g de ácido cloroacético
45

( ~ 0,06 moles) usando a técnica já descrita na prepa
ração do ácido uramildiacético. Depois de acidificar
a mistura reaccional com 6 ml de HCl concentrado,
a solução foi deixada repousar de um dia para o outro,
C H , ri
/ 0 = C
N H -
C H ,
I N —
! C H — N H 2
Acida l-metil-5-aminqbarbitúrico
(Aí. Aí. = 157)
o=c N H -
/ / 0
C H - N
- C C H
, C H , - C O O H
- C O O H
Acido l-metil-5-aminobar-
bilúrico-7,7-diacâtico
(Aí. Aí. = 273)
formando-se um precipitado branco que se filtrou.
Devido à elevada solubilidade em água deste produto,
o método de recristalização com ácido perclórico não
era aconselhável. Obtiveram-se bons resultados passando
uma solução aquosa da substância por uma coluna ca-
tiónica na forma ácida e concentrando o eluído até
se verificar a cristalização (para pormenores ver p. 50).
O ácido l-metil-5-aminobarbitúrico-7,7-diacético foi fi
nalmente obtido sob a forma de um pó branco, p. f. =
= 205-6°C dec.
Rendimento: 2,7 g (50%).
C9Hn07Ns (M. M. = 273,2)
Cale. C 39,6%, H 4,0%, TV 15,4%
Anal. C 39,7 %, H 4,3 %, TV 15,3 %,
M. M. (titulação) = 273
c. Síntese do ácido 1,3-dimetil-5-aminobarbitúrico-7,7-
diacético (l,-3dimetiluramildiacético)
A síntese desta complexona foi também realizada em
três fases:
1. Síntese do ácido 1,3-dimetilbarbitúrico
Ç H 3
N
0 = C +
/ / C 2 H 5 O C ^
PH2
C , H 5 O C
C H , ^0
T - ff
H C / \
0 = C C H , \ /
N C 1 \ \
C H , O
Dimetilureia Malonato de dieíilo Acido 1,3-dimetilbarbitúrico
(Aí. Aí. = 88) (Aí. Aí. = 160) (Aí. Aí. = 156)
Seguindo a técnica descrita para a preparação do ácido
1-metilbarbitúrico, condensaram-se 40 g de malonato
de dietilo (0,25 moles) com 22 g de dimetilureia (0,25
moles).
As quantidades de álcool absoluto e sódio foram du
plicadas e o período de refluxo aumentado para 7 horas.
O precipitado de 1,3-dimetilbarbiturato de sódio foi
filtrado, lavado com etanol e seco na estufa. Dissolve
ram-se 8,9 g (0,05 moles) em 200 ml de água e adicio
naram-se 2,6 ml de H2SOi concentrado ( ~ 0,05, moles).
O ácido 1,3-dimetilbarbitúrico precipitou imediatamente
em agulhas incolores que se filtraram e secaram a
pressão reduzida. O rendimento foi quantitativo e o
produto apresentou um ponto de fusão nítido a 124°C
(literatura: 123°C).
2. Síntese do ácido l,3-dimetil-5-aminobarbitúrico (1,3-
-dimetiluramii)
C H ,
N -/ /
C H , /
--C
\— / C H 3 \ )
Acido 1,3-dime
tilbarbitúrico
(Aí. Aí. = 156)
C H ,
/
N —
! C H ,
< O
C H — N H ,
Acido l,3-dimetil-5-
-aminobarbitúrico
(Aí. Aí. = 171)
7,9 g de ácido 1,3-dimetilbarbitúrico (0,05 moles) foram
dissolvidos em 350 ml de água e a solução tratada
com 3,9 g de nitrito de sódio (0,055 moles). A solução
tomou a cor púrpura devida à formação do derivado
nitroso. Após a adição de ditionito de sódio, fervura
e aquecimento, tal como foi descrito anteriormente,
obteve-se o produto desejado em agregados cristalinos
muito leves. Rendimento: 7 g (82%). p.f.~ 200°C
dec. (literatura ~ 200°C dec).
3. Síntese do ácido l,3-dimetil-5-aminobarbitúrico-7,7-
diacético
C H
I N -
o-c/ \ ,
/ / Ç H 3
, N — // ,0
C H - N H •
N ' —
I C H ,
Acido 1,3-dimetil-5-ami-
noburbitúrico
(Aí. Aí. = 171)
^ C / C H 2 - C O O H
- 0 = C ^ C H - N
N C V H , - C O O H 1 W
C H , b
Acido l,3-dimetil-5-ami-
nobarbitúrico-7,7-diacético
(Aí. Aí. = 305)
4 6

4,3 g de 1,3-dimetiluramil (0,025 moles) foram condensa
dos com ácido cloroacético (6,5 g). seguindo a técnica
habitual. Depois de acidificar a mistura com 7,5 ml de
HCl concentrado a solução foi deixada repousar de
um dia para o outro, obtendo-se uma substância que
se recristalizou duas vezes com HClOl 2 Me, finalmente,
com água. O produto cristalino obtido funde a 198°C
dec. Rendimento: 4 g (53%).
O peso molecular calculado por titulação correspondia
ao da complexona desejada com uma molécula de
água; após secagem a 100°C, sob pressão reduzida,
durante cerca de 10 horas, o produto perdeu 5,85 %
do seu peso, o que confirmou a presença de 1 mole
de água de cristalização por mole da complexona (per
centagem calculada: 5,90%). O produto anidro foi
analisado.
água, obtendo-se um produto cristalino em palhetas
brancas,'/»./. = 212-4°C (literatura: 212°C).
Rendimento: 10 g (40%)
CnHu06N (M. M. = 253,2)
Cale. C 52,2 %, H 4,4 %, TV 5,5 %
Anal. C 52,1 %, H 4,5 %, N 5,4%
e. Síntese do ácido o-metoxifeniliminodiacético
NH, /=Z\ / C H j - C O O H
OCH,
o-Anisidina
(Aí. Aí. = 123)
\ X CH 2 -COOH \ > C H ,
A eido o-metoxifeniliminodiacético (Aí. Aí. = 239)
C10H13O7N3 (M. M. = 305,2)
Cale. C 41,8 %, H 4,5 %, N 14,5 %
Anal. C 41,6%, H 4,7%, TV 14,5%,
M. M. (titulação) = 305
d. Síntese do ácido o-carboxifeniliminodiacético (antra-
nildiacético )
VoOH
/ = \ / C H 2 - C O O H
x —Y N C H 2 - C O O H VOOH
Acido antranilico Ácido o-carboxifeniliminodiacético (Aí. Aí. = 137) (Aí. Aí. = 253)
Dissolveram-se 13,7 g de ácido antranilico (0,1 moles)
em 20 ml de NaOH 5 M (0,1 moles) e juntaram-se
30 g de ácido cloroacético ( ~ 0 , 3 moles), previamente
neutralizado com soda cáustica. A solução foi aquecida
à ebulição e adicionaram-se 40 ml de NaOH 5 M
(0,2 moles) gota a gota, de modo que o p / í da solução
se mantivesse sempre entre 8 e 10. Esta operação foi
facilmente controlada, pois a p/f 9 a mistura toma
uma cor verde-escura.
Após a adição, que levou cerca de 30 minutos, a so
lução foi fervida durante 10 minutos sem que se notasse
variação de pH; em seguida arrefeceu-se com gelo
e acidificou-se com 30 ml de HCl concentrado. Obteve-
-se um precipitado que se recristalizou com 200 ml de
12,3 g de o-anisidina recentemente destilada (0,1 moles)
foram emulsionadas com 25 ml de água e adicionaram-se
40 g de ácido cloroacético ( ~ 0,4 moles), previamente
neutralizado com soda cáustica. A técnica habitual de
adição de NaOH 5 M gota a gota, a uma temperatura
próxima do ponto de ebulição, deu resultados satis
fatórios apenas até à condensação com o primeiro
equivalente de cloroacetato, levando à formação de
ácido o-metoxifeniliminomonoacético; a condensação
com o segundo equivalente exigiu adição com a mis
tura em refluxo e levou cerca de 6 horas. Por acidi
ficação com 30 ml de HCl concentrado precipitou um
óleo solúvel em excesso de ácido que solidificou rapi
damente, sendo filtrado, lavado e recristalizado com
água e gotas de etanol. Obteve-se finalmente um pó
branco, p.f.= 104-5°C.
Rendimento: 2,4 g (10%)
CnH13OòN (M. M. = 239,2)
Cale. C 55,5 %, H 5,3 %, TV 6,0 %
Anal. C 55,3 %, H 5,4 %, N 5,9 %,
M. M. (titulação) = 238
O baixo rendimento da preparação e as condições enér
gicas exigidas evidenciam a obstrução exercida pelo
grupo metoxilo à introdução do segundo radical ace
tato. Com menor tempo de reacção ou menor excesso
de ácido cloroacético obtém-se uma mistura de mono-
e di- derivados. O primeiro é facilmente sintetizado
pelo método usual, sendo obtido em agulhas incolores,
p.f. = 143-4°C (de etanol a 50 %).
4 7

f. Síntese do ácido o-hidroxifeniliminodiacético
N H ,
*0H
o - Aminofenol
(Aí. Aí. = 109)
^ C H - , - C O O H
Ácido 3,4-dihidro - 2-oxo
1,4-benzomorfolina - 4-acético
(Aí. Aí. = 207)
o - Hidroxifeniliminodiacetato
(Aí. Aí. do ácido = 225)
Suspenderam-se 10,9 g de o-aminofenol recristalizado
(0,1 moles) em 25 ml de água e adicionaram-se 30 g
de ácido cloroacético (~ 0,3 moles) previamente neu
tralizado com soda cáustica.
A suspensão foi levada à ebulição e manteve-se 4 horas
em refluxo, durante as quais se adicionaram 60 ml de
NaOH 5 M (0,3 moles), conservando o pH da mistura
entre 8 e 10, tanto quanto possível. O sólido dissol
veu-se totalmente e o aquecimento foi interrompido
quando deixou de se observar variação no pH.
Por arrefecimento e acidificação com 30 ml de HCl
concentrado precipitou uma substância que se filtrou
e recristalizou duas vezes com etanol a 50 %. Obteve-se
um produto cristalino em lamelas incolores, p.f. =
= 178-9°C.
A titulação deste mostrou tratar-se de um ácido mo
nobásico com peso molecular igual a cerca de 207;
no entanto, o produto dava reacção positiva com o
reagente cálcio-eriocromo T (ver p. 50) e, ao adicionar
três equivalentes de base, titulando em seguida com
um padrão ácido, obteve-se uma curva com três pa
tamares de neutralização, correspondendo a um ácido
tribásico com peso molecular igual a cerca de 225.
Estes factos sugerem a possível formação de uma 8-lac-
tona derivada do ácido o-hidroxifeniliminodiacético
por eliminação intramolecular de água.
A presença do anel lactónico foi confirmada pelo es
pectro infravermelho, que apresenta uma banda de
absorção com \ m a x = 1760 c m - 1 (emulsão em Nujol).
A existência de outra banda a 1715 cm - 1 , correspon
dente a um grupo carboxilo normal, mostra ainda que
a lactona não é dipolar no estado sólido.
/ C H 2 - C O O H _ Q I H _
\ \\ // C H , — C O O H
OH
• C H , - C O O H
0 - C '
) C H 2
A análise elementar apoia também a hipótese sugerida.
C 1 0/f 9O 4/V (M. M. = 207,2)
Cale. C 58,0 % H 4,3 %, N 6,8 %
Anal. C 58,0 %, H 4,4 % N 6,6 %,
M. M. (titulação) = 207
A preparação eventual do ácido 3,4-dihidro-2-oxo-l,4-
-benzomorfolina-4-acético, />•/•= 180-2°C, foi descrita
anteriormente (56). As nossas conclusões estão de
acordo com os resultados dos investigadores anteriores,
que apontam uma frequência de absorção máxima a
1 769 cm""1; os dados analíticos apresentados por estes
estão, no entanto, errados, pois correspondem a uma
substância de fórmula C' 1 6 i í 1 4 0 4 A' 2 , que precede a re
ferida lactona no texto do artigo (loc. cit., p. 1297).
g. Síntese do ácido o-mercaptofeniliminodiacético
< W ^ - N H 2
\ H
o - Aminotiofenol
(Aí. Aí. = 125)
\ \ / /
C H , - C O O H
C H , / 2
S — C
Acido 3,4-dihidro - 2-oxo
1,4-benzotiazina - 4-acético
(Aí. Aí. = 223)
= \ / C H , - C O O
X C H , - C O O
o - Mercaptofenilimifiodiacetato
(Aí. Aí. do ácido = 241)
12,5 g de o-aminotiofenol (0,1 moles) foram emulsio-
nados com 25 ml de água e adicionaram-se 30 g de
48

ácido cloroacético (~ 0,3 moles) previamente neutra
lizado com soda cáustica.
A mistura foi aquecida a 50-60° e adicionaram-se 60 ml
de NaOH 5 M (0,3 moles) mantendo o pH da solução
entre 8 e 10, o que levou cerca de 24 horas. Arrefeceu-se
em seguida com gelo e o precipitado obtido foi filtrado
(produto C). Acidificou-se o filtrado com 30 ml de
HCl concentrado e deixou-se repousar de um dia para
o outro; obteve-se um precipitado branco que se filtrou
e recristalizou com etanol a 50%; separou-se primeiro
um óleo que solidificou rápidamente e foi filtrado
(produto A) e das águas-mães obtiveram-se ainda cris
tais incolores após repouso prolongado (produto B). A titulação dos produtos A e B mostrou tratarem-se
de ácidos monobásicos, ambos com peso molecular
igual a cerca de 223; após adição de três equivalentes
de base e fervura prolongada, a titulação com ácido
deu uma curva idêntica com três patamares de neu
tralização. Por analogia com o caso anterior pensou-se
que se teria formado uma S-tiolactona derivada do
ácido pretendido por eliminação intramolecular de
O produto C formou-se em quantidades variáveis, au
mentando com a temperatura da reacção, e sendo
mesmo o produto principal quando se seguiu a técnica
habitual. É uma substância extremamente estável,
que pode ser recristalizada sem decomposição com
ácido acético glacial ou ácido clorídrico concentrado,
formando longas agulhas incolores comp.f. = 174-5°C.
O seu comportamento e modo de formação levaram
a supor que poderia tratar-se de um produto da des-
carboxilação de A e B, correspondendo à estrutura
JZH,
A análise elementar confirmou esta hipótese
C9H9ONS
Cale. C 60,3 %, H 5,0 %, N 7,3 %
Anal. C 60,3 %, H 5,1 %, N 7,3 %
C K - H i °
SH
N C H 2 - C O O H xn /ri CHj-COOH
C H ,
A análise e os espectros no infravermelho confirma
ram esta hipótese.
C10H9O3NS
Cale. C 53,8 %, H 4,0 %, TV 6,3 %
A : C 53,7%, H 4,1 %, N 6,0%
Anal.
B : C 53,8 %, H 4,0 %, N 6,5 %
No espectro infravermelho, por outro lado, verifica-se
o desaparecimento da banda correspondente ao grupo
carboxilo livre que se notava nos espectros dos pro
dutos A e B.
Produto Frequência de absorção
máxima: c m - 1
A e B 1660 Anel lactónico
(ácido 3,4-dihidro-2- 1703 Grupo carboxilo livre
-oxo-l,4-benzotia-
zina-4-acético)
(4-metil-3,4-dihidro
-2-oxo-1,4-benzotiazina)
1640 Anel lactónico
Acontece, porém, que os pontos de fusão dos pro
dutos A e B são muito diferentes (p.f.A = 69-71°C e
P-f-v. = 143-5°C), o que sugere que o produto A de
veria estar contaminado com qualquer impureza. No
entanto, notou-se que a hidrólise alcalina de A à tempe
ratura de ebulição era completa em 30 minutos,
enquanto que B necessitava pelo menos de 90 minutos;
por outro lado, o ponto de fusão de A variou com
o tempo e, após cerca de dois meses, era praticamente
o mesmo de B. Estes factos parecem compatíveis com
a existência da 8-tiolactona em duas formas polimór-
ficas, mas o problema não foi profundado por estar
fora do âmbito do presente trabalho.
Estas substâncias não parece terem sido sintetizadas
anteriormente e encontram-se em estudo sob o ponto
de vista das suas propriedades farmacológicas.
h. Síntese do ácido ciclohexiliminodiacético
C H , - C O O H
- N H ,
Ciclohexilamina
(Aí. Aí. = 99)
\ — / Y H J - C O O H
Ácido ciclohexiliminodiacético
(Aí. Aí. = 215)
5 g de ciclohexilamina (0,05 moles) foram condensadas
com ácido cloroacético da maneira habitual. A com-
49

plexona não precipitou por acidificação, mesmo após
repouso prolongado e tratamentos usuais. Tomou-se,
portanto, uma parte alíquota da mistura reaccional e
tratou-se com excesso de carbonato de bário; preci
pitou o complexonato de bário, que se filtrou e lavou,
sendo em seguida tratado com um pequeno excesso
de ácido sulfúrico diluído. O sulfato de bário formado
aglomerou-se por fervura e, depois de repouso prolon
gado, filtrou-se por papel de filtro quantitativo. O fil
trado foi levado à secura em banho-maria, obtendo-se
a complexona impura em cristais incolores. Alguns
destes foram então usados para induzir a precipitação
no restante da mistura reaccional; a técnica deu resul
tado e o ácido ciclohexiliminodiacético precipitou len
tamente em agulhas incolores. Após recristalização com
etanol a 50%, o produto foi seco a pressão reduzida
e a 100°C até peso constante. Os cristais perderam a
água de cristalização, transformando-se num pó branco,
p.f. = 194-6°C.
Rendimento: 6 g (56 %)
CwH11NOí (A/. M. = 215,2)
Cale. C 55,8 %, H 8,0 %, N 6,5 %
Anal. C 55,9 %, H 8,2 %, N 6,8 %,
M. M. (titulação) = 215
i. Purificação das complexonas com resinas permuta
doras de catiões
Construiu-se uma coluna com um condensador vertical
de LIEBIG, em cujo tubo interno se colocou a resina;
deste modo foi possível, por circulação da água de
um termostato, manter a temperatura da coluna a
60-70°C, de modo a evitar a precipitação das comple
xonas no seu interior.
A resina utilizada foi a Zeo-Carb 225, que se verificou
ser a que possuía menos tendência para adsorver os
ligantes a purificar. Antes de encher a coluna, a resina
foi tratada com HCl 2 N para assegurar a remoção
completa de todos os metais e, em seguida, lavada
com água desionizada para remover o excesso de
ácido, etanol para dissolver as impurezas orgânicas e,
finalmente, água desionizada outra vez para remover
todo o álcool. Estas operações foram sempre repeti
das antes de cada purificação.
As complexonas a purificar dissolveram-se no mínimo
volume de água a 60-70°C e as soluções foram pas
sadas na coluna com o débito regulado a cerca de
uma gota por segundo. Em seguida passoú-se água
desionizada até o eluído não dar reacção positiva com
o reagente cálcio-eriocromo 7"; em geral foi necessário
cerca de 1 1 de água por cada grama de complexona
a tratar.
As complexonas recuperaram-se por evaporação do
eluído até começo de cristalização ou até à secura,
quando necessário.
j . Ensaios com o reagente cálcio-eriocromo T
A reactividade das complexonas, que tem de se verificar
em diversas circunstâncias (preparação, eluição de
colunas de resinas, etc), foi determinada por uma
adaptação do método de SCHWARZENBACH e IRVING (57).
Utiliza-se uma solução alcoólica de eriocromo T,
tamponizada a píf 10 com amónia e cloreto de amónio,
que apresenta uma cor azul brilhante; adiciona-se uma
gota de uma solução diluída de um sal de cálcio (água
corrente é uma solução adequada), com o que o reagente
toma a cor vermelha do complexo de cálcio. Se se adi
cionarem em seguida algumas gotas de solução de
uma complexona, a cor do reagente reverte imediata
mente ao azul inicial (ou passa a púrpura, se a com
plexona der complexos de cálcio pouco estáveis —
2 — SOLUÇÕES
a) Material
Utilizou-se exclusivamente material de Pyrex, previa
mente desengordurado com mistura cromo-sulfúrica,
lavado com uma solução concentrada e quente de
EDTA para eliminar traços de metais e, finalmente,
passado com água desionizada em abundância.
As soluções foram conservadas em frascos de polie
tileno, não mostrando qualquer sinal de alteração
ou variações de concentração durante um período de
vários meses.
b) Água
Toda a água utilizada neste trabalho é referida como
«desionizada» e foi obtida por passagem de água des
tilada normal através de uma resina mista «Elgastat».
O efluente sai com uma resistência superior a 4 me-
gohms cm" 1 e possíveis impurezas orgânicas são inde-
tectáveis.
c) Complexonas
A pureza das complexonas foi cuidadosamente veri-
5 0

ficada em todos os casos; o carvão animal e o vegetal
usados para a purificação e descoloração foram previamente tratados com uma solução concentrada de
EDTA, lavados com água desionizada e secos na estufa
ao abrigo de contaminações.
As soluções foram feitas com uma concentração da
ordem dos IO - 3 M e aferidas por titulação com hidró
xido de potássio-padrão, sendo os pontos de equiva
lência calculados pelo método de GRAN (58).
d) Catiões
As soluções dos catiões foram preparadas a partir dos
respectivos nitratos, utilizando-se produtos pró-análise,
quando possível; nos outros casos, os produtos foram
purificados por recristalizações sucessivas.
As soluções foram feitas com uma concentração da
ordem dos IO - 2 M e, em alguns casos, 10 _ 1 M e 1,2 M.
Os títulos foram verificados periodicamente, em geral
por titulações complexométricas.
e) Electrólitos de suporte
Utilizaram-se como electrólitos de suporte os nitratos
de potássio e de tetrametilamónio; este último foi
obtido por neutralização do hidróxido de tetrametila
mónio comercial com ácido nítrico, evaporação da solu
ção à secura e cristalizações repetidas do produto com
etanol a 80 %. A análise correspondeu à fórmula
{CH^N.NOs.
Prepararam-se soluções 1,200 M destas substâncias para
ajustar a força iónica das tomas a titular e soluções
0,100 M para enchimento da ponte salina na mon
tagem experimental.
/) Ácidos para calibrações
Para a calibração do aparelho de pH utilizaram-se
soluções I O - 3 M de ácidos clorídrico e acético prepa
radas por diluição de produtos concentrados pró-aná
lise. Preferiu-se o ácido clorídrico ao ácido perclórico
porque com este não se poderia utilizar um electrólito
de suporte contendo potássio. No entanto usou-se este
ácido em titulações por retorno, dado que a concen
tração final de KCIO^ não excede a solubilidade deste
sal.
g) Tampões
Para a aferição do aparelho de p/l utilizaram-se tam
pões de ftalato de potássio 0,05 M e tetraborato de
sódio 0,01 M, preparados com água previamente desio
nizada e fervida. As soluções foram renovadas men
salmente e tomaram-se precauções especiais para im
pedir o acesso do CO a atmosférico ao tampão de bórax.
li) Hidróxidos de tetrametilamónio e de potássio isentos
de carbonato
Para a preparação das soluções dos titulantes seguiu-se
um método análogo ao recomendado por SCHWARZEN
BACH e BIEDERMAN (59). Utilizou-se o aparelho esque
matizado na fig. 18, que é constituído essencialmente
-— Azoto
— - Trompa de água
Fig. 18 — Esquema do aparelho utilizado para a preparação
dos hidróxidos de potássio e de tetrametilamónio
isentos de carbonato.
por dois balões de 1 1 com duas tubuladuras ligados
entre si por um tubo com cerca de 10 cm de
comprimento, no interior do qual se soldou uma placa
de vidro sinterizado. Um dos balões (A) é provido
de um tubo com enchimento de cal-sodada; o ou
tro (B) possui um sistema adequado para se poder
passar azoto purificado através do conjunto e aplicar
sucção, de modo a conseguir-se a filtração através da
placa de vidro sinterizado. Descreve-se a seguir a
técnica utilizada.
INSTITUTO 0£ Eíáí.R©lA ATÔMICA
5 1

Dissolveram-se 16,987 g (0,100 moles) de nitrato de
prata pró-análise em 100-200 ml de água desionizada
e precipitou-se o Ag20 no balão A com uma solução
concentrada de soda cáustica, tanto quanto possível
isenta de carbonato.
O sólido lavou-se diversas vezes, por decantação, com
água fervente, até não se verificar alteração no pH das
águas de lavagem. Na última lavagem ferveu-se a sus
pensão durante alguns minutos para converter em óxido
todo o carbonato que pudesse existir. Decantou-se a
última porção de água e adicionaram-se 14,910 g de
cloreto de potássio (ou 20,106 g de iodeto de tetra-
metilamónio) dissolvidos em 500 ml de água desioni
zada e fervida recentemente. Fez-se borbulhar azoto
purificado no balão durante uma hora, após o que
se tapou hermeticamente, colocando-se num agitador
durante 3 horas.
Cerca do fim da reacção fez-se passar azoto pelo balão B e tubo de ligação durante alguns minutos e, com o
feriu por pressão de azoto para o reservatório do
aparelho de titulação.
Diluiu-se em seguida a solução a cerca de 1 1, aferi u-
-se e ajustou-se a força iónica a 0,100 M por conveniente
adição de nitratos de potássio ou tetrametilamónio,
tendo em conta a neutralização dos iões hidroxilo que
tem lugar nas reacções com as complexonas.
Com os cuidados referidos conseguiram-se soluções de
titulante absolutamente isentas de carbonato.
B. APARELHAGEM E TÉCNICA EXPERIMENTAL
l — O TITULADOR COM ELÉCTRODO DE VIDRO
O titulador (fig. 19), que foi por nós projectado e mon
tado, é constituído por duas células de vidro com
parede dupla e capacidades aproximadas de 250 e 150 ml,
sendo a primeira a célula de titulação e a segunda a
célula de referência.
A célula de titulação é vedada por um tampão de
: í : i.;
jjk. II i
Fig. 19 — Fotografia do titulador utilizado nas determinações
aparelho na posição invertida (isto é, com B em cima),
adaptou-se rapidamente o balão A, interrompendo a
passagem de azoto. Invertendo lentamente o conjunto,
aplicou-se uma leve sucção em B. Obteve-se assim uma
solução límpida de KOH ou (CH^NOH, que se trans-
polietileno provido de entradas para o eléctrodo de
vidro, terminal da bureta, ponte salina, termómetro,
entrada e saída de azoto e, se necessário, outro eléc
trodo. Para evitar efeitos capacitivos, envolveu-se a
célula com uma rede metálica ligada à terra,
52

A célula de referência, onde se introduz Um eléctrodo de
calomelanos saturado, é simples, protegendo-se apenas
o conteúdo contra as poeiras por meio de uma tampa
de plástico.
A ponte salina, necessária devido à natureza dos iões
a estudar (prata, tálio), é do tipo aconselhado por
LAITINEN (60) e constituída por um tubo em U com
cerca de 8 mm de diâmetro, sendo as duas extremi
dades fechadas com placas de vidro sinterizado com
1,5 mm de espessura. O ramo horizontal possui um
cone esmerilado 519 onde se adapta um funil de de
cantação que actua como reservatório; este funil permite
também obter a pequena pressão necessária para esgotar
um pouco de líquido pela extremidade mergulhada na
célula de titulação, de modo a renovar a superfície de
contacto em cada operação.
Para impedir que a ponte actue como sifão e que o
mesmo aconteça na outra extremidade, vedou-se esta
com uma camada de agar-agar com cerca de 3 mm
de espessura.
Este tipo de ponte é excelente e tem diversas vantagens
sobre os tipos mais comuns, como, por exemplo, as
pontes de gel de agar-agar, nas quais o equilíbrio se
estabelece com maior lentidão e a possível presença de
impurezas pode dar origem a erros. Possui além disso
uma baixa resistência eléctrica, permite um contacto
directo líquido-líquido e oferece a possibilidade de
redução ou mesmo eliminação completa das variações
dos potenciais de junção líquida no decurso das titu
lações. No caso presente, conseguiu-se este resultado
utilizando na ponte soluções com a mesma força iónica
das tituladas.
A representação simbólica do conjunto é, pois, a se
guinte :
Solução a titular
[¿=0,1 M(KN03)
KCl KNOz KNOz
sat. N/10 N/10
eléctrodo de vidro
Nas titulações utilizámos uma bureta E-MIL Green
Line (classe A), graduada em 0,01 ml, e, para maior
rigor nas leituras, adaptou-se-lhe uma lente móvel. Os
erros na leitura podem, com este dispositivo, consi
derar-se inferiores a 0,002 ml.
As soluções aferidas dos titulantes conservaram-se em
frascos de Pyrex, sendo todos os contactos com a
atmosfera feitos através de tubos com cal-sodada.
O enchimento da bureta é conseguido por pressão de
ar, também através de um tubo com cal-sodada.
Como eléctrodo de referência utilizámos um eléctrodo de
calomelanos saturado Radiometer tipo K401; como
eléctrodos de medida, os eléctrodos de vidro Radiometer
tipo G202B e Electronic Instruments Ltd. tipo GHS 23.
O instrumento de medida, que se vê na figura, é um
aparelho de pH «Radiometer» tipo pHM4, que tem
um poder discriminador de 0,003 unidades de loga
rítmicas. Este aparelho exige um conjunto de medida
tal que a diferença de potencial se anule a pH 6,3 ±
1,5; esta exigência limita a escolha de eléctrodos de vi
dro utilizáveis, mas a dificuldade foi torneada interca
lando no circuito um acessório com uma fonte de
potencial variável, em série com o eléctrodo de calo
melanos *.
Na fig. 20 apresenta-se o esquema deste acessório;
a pilha, potenciómetro e resistências estão encerradas
numa caixa de alumínio ligada à terra e o circuito é
provido de um interruptor, para evitar a descarga
contínua.
Todas as partes metálicas do aparelho estão também
ligadas à terra e bem assim a própria água do ter
mostato. Deste modo evita-se o uso de óleo como
fluído termostático, sem que se notem efeitos capaci
tivos sobre o eléctrodo de vidro. Também não se no
taram quaisquer efeitos devidos à humidade atmos
férica.
•TÉCNICA EXPERIMENTAL
d) Calibração do aparelho de medida
Desejando-se calcular constantes «estequiométricas», é
necessário calibrar o aparelho de pH para que as lei
turas correspondam ao pcH, isto é, a -log [H+]. Na
verdade, os valores obtidos normalmente são os de pH
«operacional», definido em relação a um tampão ou
sistema de tampões escolhidos.
Para calibrar o aparelho, este foi primeiramente aferido
como se indica na alínea seguinte, de modo a repro
duzir exactamente um conjunto de condições; em se
guida titulou-se uma solução de ácido clorídrico I O - 3 M
com força iónica 0,1 M ajustada com nitrato de po-
(*) Este acessório foi construído nas oficinas do Centro de Es
tudos de Electrónica da C. E. E. N. , a cujo director, Sr. Prof.
A B R E U F A R O , manifestamos a nossa gratidão.
53

tássio, e uma de ácido acético nas mesmas condições.
No primeiro caso pode calcular-se exactamente a con
centração hidrogeniónica correspondente a cada fase
de neutralização, pois trata-se de um ácido forte; no
segundo caso o cálculo é igualmente possível, pois a
constante de dissociação do ácido acético é rigorosa
mente conhecida nos mais diversos meios e numa larga
gama de temperaturas (61). As diferenças entre os valores
calculados e os valores lidos (que devem ser constantes
a ± 0,01 unidades) dão a correcção a introduzir na
zona de pH < 7.
termostato, tendo ambos os instrumentos sido previa
mente calibrados. Introduz-se o primeiro tampão (ftalato
ácido de potássio 0,050 M) na célula de titulação e
deixa-se atingir a temperatura; equilibra-se então o
aparelho com os devidos comandos. Os eléctrodos são
novamente lavados e repete-se a operação com um
segundo tampão (tetraborato de sódio 0,010 M, em
geral) sem alterar a posição desses comandos. Lê-se
o novo valor de pH e, se este concordar com o valor
nominal do tampão, com desvios não excedendo ± 0,02
unidades, considera-se o aparelho em condições de
Radiometer
pHM4
Fig. 20 — Esquema do acessório compensador do potencial do
conjunto de medida.
As correcções na zona alcalina foram também deter
minadas por titulações de soda cáustica e amónia com
uma solução aferida de ácido clorídrico.
Finalmente, a calibração foi verificada redeterminando
as constantes de dissociação bem conhecidas das com-
plexonas ácidos iminodiacético, nitrilotriacético, o-car-
boxifeniliminodiacético e etilenodiaminotetracético (21).
A concordância foi perfeita, com desvios máximos de
± 0,02 nos valores de p/c.
b) Aferição
Antes de cada série de operações, hora a hora e no
final da série, o aparelho de pH foi aferido utilizando
a seguinte técnica: após uma hora de aquecimento para
estabilização, os eléctrodos e células lavam-se cuidado
samente, secando-se com papel absorvente macio. O con
trolador de temperatura fixa-se a 20,0°C, tal como o
operação satisfatórias. Caso contrário, todas as ope
rações são repetidas até se conseguir a concordância.
No presente trabalho, após os ajustes iniciais, o apa
relho mostrou excelente reprodutibilidade em diversas
condições de trabalho.
c) Determinação das curvas de titulação das comple-
xo/ias — Técnica
Medem-se para a célula de titulação 100,0 ml de uma
solução aproximadamente IO - 3 M da complexona utili
zando uma pipeta calibrada. Adicionam-se 10,0 ml de
água desionizada e 10,0 ml de uma solução de nitrato
de potássio (ou de tetrametilamónio) 1,200 M.
Quando a solução atinge a temperatura de 20,0 ± 0,1°C
inicia-se a titulação com uma solução aferida de hi
dróxido de potássio ou de tetrametilamónio isenta de
carbonato, com uma concentração aproximadamente
54.

0,1 M. As adições sucessivas de 0 ,100 ou 0 , 0 5 0 ml
são efectuadas com intervalos adequados, para que o
equilíbrio na reacção e com o eléctrodo seja atingido,
o que aliás acontece em poucos segundos.
As curvas de titulação obtêm-se representando os va
lores de p / / corrigidos em função do volume de titu-
lante adicionado (*); o ponto de equivalência calcula-se
pelo método de GRAN (58) e a pureza da complexona
é determinada em relação ao título da solução-padrão
da base.
d) Determinação da curva de titulação das complexonas
na presença de iões dos metais—Técnica
O método usado é exactamente o indicado na alínea
anterior, adicionando-se na célula de titulação 10,0 ml
de soluções 0 , 0 1 0 M de sais dos metais, em vez de
10,0 ml de água desionizada. Obtém-se deste modo uma
relação metal-ligante de 1 :1 .
Para a determinação das constantes de estabilidade
dos complexos ML2 convém utilizar a relação 3:2,
adicionando-se na célula 5,0 ml das soluções 0 , 0 1 0 M
dos sais dos metais e 5,0 ml de água.
Em alguns casos, quando os complexos formados são
pouco estáveis, usaram-se relações 10:1 e mesmo 1 0 0 : 1 ;
no primeiro caso, a força iónica da solução a titular
foi ajustada por adição da quantidade calculada de
nitratos de potássio ou tetrametilamónio; no segundo
caso, a força iónica da solução é já 0 , 1 0 0 M como se
pretende.
Claro que, em qualquer das circunstâncias, o aparelho
de \oH tem de ser calibrado de novo e as aferições
repetidas.
C. CONSIDERAÇÕES SOBRE AS CONDIÇÕES
EXPERIMENTAIS E RESULTADOS OBTIDOS
actividades e são independentes da força iónica do
meio; são portanto constantes «termodinâmicas» na
verdadeira acepção.
Acontece, porém, que a determinação destas constantes
é, em geral, impraticável, por não ser possível medir
as actividades de todas as espécies consideradas; isto
é especialmente problemático no caso dos sistemas
em que coexistem diversos complexos em equilíbrio.
Por outro lado, o cálculo dos coeficientes de actividade
necessários para a conveniente conversão das concen
trações das várias espécies (cuja medida é, em geral,
acessível) não é directo nem fácil, excepto em soluções
muito diluídas; mesmo neste caso, é difícil seleccionar
valores razoáveis para os coeficientes de actividade dos
complicados aniões orgânicos das complexonas, que
possuem frequentemente vários grupos iónicos simples
e dipolos na mesma molécula.
Por estas razões, prefere-se actualmente exprimir as
constantes de equilíbrio como razões de concentrações,
isto é, como «constantes estequiométricas», válidas para
um meio com força iónica e composição determinadas.
A relação entre as constantes «termodinâmicas» e as
«estequiométricas» deduz-se facilmente; para o equi
líbrio
M + L ML
a constante «termodinâmica» é
T K _ JML1_ M L (Aí) . (L)
onde os parênteses representam «actividades»; a cons
tante «estequiométrica» é
KM
[ML]
[M].[L]
onde os colchetes representam concentrações molares.
J — USO DE CONCENTRAÇÕES EM VEZ DE
ACTIVIDADES
As constantes de equilíbrio para as reacções de for
mação de complexos em solução a uma determinada
temperatura definem-se rigorosamente como razões de
(*) É, no entanto, preferível usar valores de a = moles de titu
lante adicionado/mole de complexona em vez de v = volume
de titulante.
Visto que ( x ) = fx . [ x ] , é evidente que
J ML ' K ML — KML
fM * fL
onde os ff são os coeficientes de actividade.
É claro que as constantes estequiométricas só serão
verdadeiras constantes se os coeficientes de actividade
das diversas espécies se mantiverem constantes e não
55.

variarem durante as titulações ou de titulação para
titulação. Se assim for
e, sob a forma logarítmica habitual,
log TKML = log KML + log C
Isto é, as constantes «estequiométricas» diferem das
«termodinâmicas» apenas de um valor constante.
Consegue-se este resultado efectuando todas as medi
ções num meio contendo um grande excesso de um
electrólito de suporte inerte, isto é, que não forma
complexos com as espécies em estudo. Nestas condi
ções, supõe-se que os coeficientes de actividade de
pendem apenas da concentração do electrólito inerte,
uma vez que este determina praticamente a força iónica
do meio, e diz-se que se trabalha «a força iónica ¡x constante». No entanto, isto só é verdade para soluções
muito diluídas, quando [x < 0,05 (62); rigorosamente,
tanto a concentração como a natureza do electrólito
de suporte influem no valor dos coeficientes de acti
vidade. Além disso, os iões do electrólito (X e Y)
formam geralmente complexos, ainda que pouco está
veis, com as espécies em estudo; segue-se que as cons
tantes de formação do complexo MLn determinadas
por via experimental são realmente uma medida da
estabilidade das espécies 2 2 2 MLn Xx Y (H20)z em x y z
relação à estabilidade das espécies 2 2 2 LXxYy (H20)z
x y z
e 2 2 2 M XxY(H20\ (63). x y z
Em princípio, portanto, os resultados publicados por
investigadores diferentes só serão comparáveis quando
obtidos exactamente nas mesmas condições; no en
tanto, para forças iónicas da ordem de 0,1 M, pode
considerar-se, sem grande erro, que os coeficientes de
actividade das espécies em jogo são independentes da
natureza do electrólito de suporte. Os resultados de
origem diferente serão pois comparáveis quando obtidos
à mesma temperatura e em meios de igual força iónica.
Por esta razão se escolheu a temperatura de 20°C e a
força iónica 0,100 M para o presente trabalho, con
dições em que foi determinada a maioria dos valores
existentes na literatura (21).
2 — ELECTRÓLITO DE SUPORTE
A escolha do nitrato como anião do electrólito de
suporte foi imposta pela necessidade de evitar a
presença de cloretos nas medições com os iões Ag+
e Tl+ e de percloratos nas medições com o ião K+;
por outro lado tiveram de se excluir os sais de sódio
e lítio devido à possibilidade da formação de complexos
relativamente estáveis entre estes metais e alguns dos
ligantes a estudar, o que deixou a escolha limitada aos
sais de potássio entre os meios mais vulgarmente utili
zados. No caso dos derivados do ácido uramildiacétiço,
que formam complexos com o ião K+, utilizou-se, em
vez do nitrato de potássio, o nitrato de tetrametila-
mónio.
Os valores das constantes determinadas são válidos,
portanto, para um meio de nitrato 0,1 M, e não foram
corrigidos para o efeito de formação de pares-iónicos
entre este anião e os metais estudados. Não quer isto
dizer que a correcção seja desprezável, como se poderá
avaliar examinando os valores das constantes de for
mação destas associações (tabela III. 1).
Tabela 111.1
CONSTANTES D E ASSOCIAÇÃO (log K) D E PARES-
-lÓNICOS FORMADOS PELO IÃO NO~3 T= 18°C. fx = 0
(a) (a) (a)
Ião Ag+ Tl+ Li+ Na + K+ A í g 2 + Caz+ Sr2+ Ba2+
log KMN0¡¡ —0,27 0,41 —1,0 - 0 , 4 —0,22 0,0 0,28 0,82 0,92
(a) T = 25°C
R e i ' (21)
É claro que se poderia fazer uma correcção, obtendo
valores válidos para um meio ideal não complexante;
bastaria, para tal, adicionar aos valores de log KML
determinados o valor log (1 + KMNOs [NO~]) = log (1 +
+ 0,1 KMNOa), como se verá na secção seguinte.
Nos casos estudados no presente trabalho esta correcção
variaria de 0,01 a 0,25 unidades, sendo mínima para
os complexos do Li+ e máxima para os complexos
do Baz+.
Para estudar alguns complexos pouco estáveis, utili
zaram-se, como se disse, relações metal-ligante de 10:1
5 6
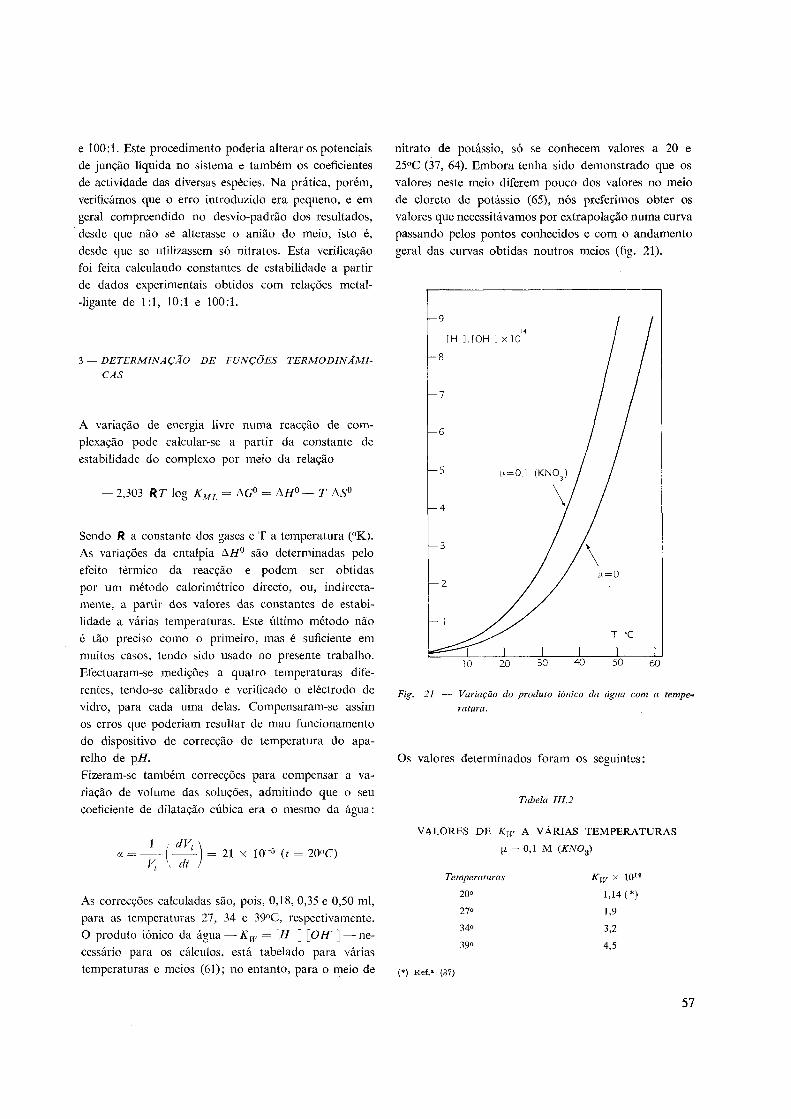
e 100:1. Este procedimento poderia alterar os potenciais
de junção líquida no sistema e também os coeficientes
de actividade das diversas espécies. Na prática, porém,
verificámos que o erro introduzido era pequeno, e em
geral compreendido no desvio-padrão dos resultados,
desde que não se alterasse o anião do meio, isto é,
desde que se utilizassem só nitratos. Esta verificação
foi feita calculando constantes de estabilidade a partir
de dados experimentais obtidos com relações metal-
-ligante de 1:1, 10:1 e 100:1.
3 — DETERMINAÇÃO DE FUNÇÕES TERMODINÂMI
CAS
A variação de energia livre numa reacção de com-
plexaçâo pode calcular-se a partir da constante de
estabilidade do complexo por meio da relação
— 2,303 RT log KML = AG0 = AH° — T AS0
Sendo R a constante dos gases e T a temperatura (°K).
As variações da entalpia AH0 são determinadas pelo
efeito térmico da reacção e podem ser obtidas
por um método calorimétrico directo, ou, indirecta
mente, a partir dos valores das constantes de estabi
lidade a várias temperaturas. Este último método não
é tão preciso como o primeiro, mas é suficiente em
muitos casos, tendo sido usado no presente trabalho.
Efectuaram-se medições a quatro temperaturas dife
rentes, tendo-se calibrado e verificado o eléctrodo de
vidro, para cada uma delas. Compensaram-se assim
os erros que poderiam resultar de mau funcionamento
do dispositivo de correcção de temperatura do apa
relho de pH.
Fizeram-se também correcções para compensar a va
riação de volume das soluções, admitindo que o seu
coeficiente de dilatação cúbica era o mesmo da água:
1 l d v i \
a = = 21 x 10~6 (t = 20°C) Vt \ dt )
As correcções calculadas são, pois, 0,18, 0,35 e 0,50 ml,
para as temperaturas 27, 34 e 39°C, respectivamente.
O produto iónico da água — Kw = [H+] [OH~] — ne
cessário para os cálculos, está tabelado para várias
temperaturas e meios (61); no entanto, para o meio de
nitrato de potássio, só se conhecem valores a 20 e
25°C (37, 64). Embora tenha sido demonstrado que os
valores neste meio diferem pouco dos valores no meio
de cloreto de potássio (65), nós preferimos obter os
valores que necessitávamos por extrapolação numa curva
passando pelos pontos conhecidos e com o andamento
geral das curvas obtidas noutros meios (fig. 21).
10 20 30 40 50 60
Fig. 21 — Variação do produto iónico da água com a tempe
ratura.
Os valores determinados foram os seguintes:
Tabela III.2
VALORES D E Kw A VÁRIAS TEMPERATURAS
[X = 0,1 M (KN03)
Temperaturas R~yy x IO 1 4
20° 1,14 (*)
27° 1,9
34° 3,2
39° 4,5
(*) Ref.» (37)
57

4 —PRECISÃO DOS VALORES OBTIDOS
Os valores obtidos neste trabalho são, em geral, apre
sentados na forma S ± c, sendo cr o desvio-padrão
dos valores calculados a partir de uma série de N
pontos experimentais em cada titulação:
r 2 S 2 — ( 2 SfjN ~V2
Deve notar-se que cr liga-se mais à precisão das me
didas que ao rigor dos resultados; pequenos valores de
cr são consequência do uso de reagentes muito puros,
calibrações rigorosas, perfeita linearidade dos eléctrodos,
potenciais de junção líquida constantes durante as titu
lações e hipóteses correctas quanto às espécies for
madas.
Há, porém, erros intrínsecos do método que podem
causar desvios não previsíveis, e se relacionam sobretudo
com a natureza dos eléctrodos de vidro. Este problema
foi recentemente discutido por ANDEREGG (66) e, de
acordo com este autor, nós verificámos que a concor
dância entre constantes obtidas a partir de curvas de
titulação diferentes era da ordem de ± 0,03 unidades
logarítmicas.
Nestas condições, os desvios em relação aos valores
reais serão, possivelmente, da ordem de ± 0,05 e, em
alguns casos, ± 0,1.
5 8

IV. Métodos de cálculo (*}
1 — CONSTANTES DE DISSOCIAÇÃO DE ÁCIDOS [K+] + [H+] = [0H~] + [_Hn_1L~~\ + 2 [#„_ 2L2~]
POLIBÁSICOS p[H^L*-]+ + n II-
Os ácidos polibásicos dissociam-se em fases sucessivas,
cada uma delas correspondendo à formação do protão
e da base conjugada da espécie que se dissocia
= // + II. , L
Conhecendo a concentração do ácido, a fracção neu
tralizada e a concentração hidrogeniónica da solução,
podem calcular-se as constantes de dissociação de
todas as espécies.
Para um ácido polibásico de fórmula geral HnL esta
belecem-se os seguintes equilíbrios:
HL H - A í ' \H+]I[H,,L\
Hn_xL-=H^% L*-+H + ... k2=[Hllr_2L2~][H+}l[Hn_1Lr
(D
Se definirmos o parâmetro a, grau de neutralização,
como
número de equivalentes de base adicionados a —
número de moles do ácido
sera
[K+] = a Ca
a Ca + [H+] - [O /T] = 2 p [H,,^] (3) P=i
As concentrações das diferentes espécies podem obter-se
a partir das equações (1):
[HnL] = [L«-] [H+r I k» .k» k%
[H^IT] = [L"~] [H + ] « / * ? . A f (4)
HL [HL-n+1] = [£-] [H+] I k*
onde os kf são, por definição, as «constantes de dis
sociação estequiométricas parciais».
Em qualquer momento, a concentração total de ácido
será:
Ca = [H„L] + [HH_LL-] + + \Hn_pL"-} +
+ + [L"-] (2)
Pela condição de electroneutralidade da solução (su
pondo ser KOH o titulante):
(*) Os métodos de cálculo utilizados baseiam-se no sistema
de BJERRUM ( 1 4 ) , adaptado por IRVING e STACEY (67) e estru
turado pelo autor (68).
Substituindo em (2):
Ctt = { [H+Y I k?.kH C + [H+r1 i
I kg.14
Usando as constantes de formação das espécies pro-
tonadas, que estão relacionadas com as constantes de
dissociação pelas expressões
Af = l / / v f , A f = l , < = l / / c ?
e introduzindo as constantes de formação globais
ftf = Kf , ftf = . Kf , ,tf = K?.K? K%
5 9

a expressão anterior toma o seguinte aspecto:
Ca = \in { tf [h+t + 'A: i r '•1 + ....
+ p f [H+] + 1 }
O U
.2 Pf [H+Y + i }
Em geral designa-se 2 p¿ [H+]1 + 1 por a f í . Logo
(5a)
Para eliminar [LM ] multiplica-se a equação (2) por n
e subtrai-se a equação (3) do produto, resultando:
(«-«) Ca-[H + ] + [OH~] = n[HnL] +
+ ( « - 1 ) [Hn_t L-] + +
Introduzindo as constantes de formação obtém-se
(« - fl) C 8 - [H+ ] + [Off-] = [ L - ] { « p f [Tí + ] " +
+ o, - d \ c [H+r1 + + p f [H+]}
ou
n (n-a) C -[H + ] + [OH-] = [Ln~] 2 i p? [ # + ]* (6)
i = l
n Designando 2 / p f [H + ]% por p H virá
i — l
(n - a) C B - [JÏ + ] + [O/ / " ] = [L*-] . p „ (6a)
Dividindo (6a) por (5a)
(n — d)Ca — [H + ] + [OH-] p w
1 + 2 p f
(7)
O valor deste quociente corresponde à «função de for
mação n», definida por B j e r r u m para complexos de
metais (14); representa neste caso o número médio
de protões ligados a L.
Ordenando a equação (7) obtém-se
+ D p f [H + ] + (nH-2) p f [H+f +
+ + ( / 7 h - b ) p? [ # + ] " = 0 (8)
(5) sendo
2 (nH~i) p f [H + J = 0 ¿ = 0
pf = k" = 1, por definição.
(9)
A resolução desta equação é fácil quando n não é
muito grande; felizmente, na prática, n varia normal
mente de 1 a 4, e em muitos casos os valores das cons
tantes diferem suficientemente (A p/c > 2,5) para que
algumas simplificações se tornem possíveis.
Dado, no entanto, que é mais corrente exprimir os
equilíbrios com ácidos e bases em termos de constantes
de dissociação, convém modificar a expressão (9) de
modo a tornar possível o cálculo directo de tais cons
tantes. Consegue-se este resultado substituindo as
«constantes de formação» por «constantes de disso
ciação» e os valores de nH por (n — R), sendo 7?
definido por
a Ca
R = [H + ]-[OH-]
(10)
Assim, de (8):
R [H+]n + (R — 1) [H + f1 /cf +
+ (R-2)[H+Y ^ . , 2
(R-n + D [H + ] * f . * f kl,
+ (R — n)k?.k% /cf = 0 (11)
Representando por X; as «constantes de dissociação
globais», será
2 ( 7 ? - / ) \ f [// + ]* = 0 » = o
(12)
onde xf = k" 1, por definição.
Também esta equação é susceptível de simplificações
nos casos práticos; passemos pois a examinar cada
caso em particular.
I. Ácidos monobásicos (n = 1)
Das equações (10) e (12):
6 0

* [ g + ]
a - i ? )
Quando [ # + ] » [ O f f - ] :
a C a + [ff + ] i ? =
C.
(13) Quando [/f + ] — [OH ] a expressão anterior simplifica-
-se ainda mais:
( a - 1 ) [/-/ + ] (19)
( 2 - a )
No ponto a = 1,5 será /cf = [ f f + ] , ou seja p/cf = pff.
Logo [ff+] { a C a + [ff+] }
Ca~{a Ca + [H+]}
II. Ácidos dibásicos (n = 2)
Da equação (12) obtém-se
(14)
R [H+f + (R - 1) [H+] + (R- 2) /cf . fc£ 0
E, dividindo por (R — 2) /cf e f? [ f f+] 2 /cf . k% , vem
respectivamente:
(R—\)[H+] R[H+f 1
( f ? - 2 ) C R - 2 )
(2 —*k — D i
/c, f? [ í f+] 2 j e [ £ r + ]
w i k2 (15)
T T T T (16) * 1 • K-2
I I I . Ácidos tribásicos
Nos casos práticos /cf é, em geral, da ordem de /cf,
e /cf é bastante menor. Estes casos poderão portanto
ser tratados como a mistura de um ácido dibásico
e um monobásico: /cf e /cf calculam-se por meio das
equações (15) e (16) e /cf por uma expressão análoga
a (14), substituindo a por (a—2) para tomar em conta
a neutralização dos dois primeiros hidrogénios ácidos.
Será, portanto,
/cf = — - H - (20) C a - { ( a - 2 ) Ca-[OH-} }
[Tf +] { ( a - 2 ) C a - [ O f T ] }
quando [Ofí ] > [ f f + ] , ou, o que é o mesmo,
„ (R-2)[H+]
( 3 - f ? ) (21)
IV. Ácidos tetrabásicos
Ambas as expressões são equações de rectas do tipo
y — ax + b; representando graficamente os valores de
y em função dos valores de x, os coeficientes angu
lares das rectas dão Ijkf e l//cf e as ordenadas na
origem dão /cf e l//cf . /cf.
Quando /cf e /cf são bastante diferentes (A p/c > 2,5)
podem obter-se, a partir da equação geral, expressões
mais simples:
R [H + ] [H + ]{aCa [H + ]}
c a
quando [H + ]>[OH~]
[H + l )
(17)
, / ; H _ ( f ? - l ) [ f f ] _ [ # + ] { ( a - 1 ) C a - [ O f f - ] } 2 ( 2 - i ? ) c a - { ( a - l ) C B - [ 0 # - ] }
quando [ O f f - ] > [ f f + ] (18)
Nos casos práticos comportam-se como misturas de
dois ácidos dibásicos ou de um dibásico com dois
monobásicos. Normalmente, portanto, será /cf - /cf
e /cf - /cf ou Acf > / c H
•3 - ""4 u u ^ 3 •
As duas primeiras constantes podem calcular-se pelas
expressões (15) e (16); quando as duas últimas são
muito diferentes, /cf pode calcular-se pela expressão (20)
e /cf por uma expressão semelhante:
[ g + ] { ( a - 3 ) C.-IOH-] }
Ca-{ ( a - 3 ) Ca-[OH~] } (22)
quando [OH ] > [ f f + ] , ou, tal como anteriormente,
tf - 3 ) [ff-
( 4 - Ä ) (23)
Se /c 3 ~ /c 4 , a equação geral (12) resolve-se em ordem
a kf, obtendo-se
61

- * f = R [H+f+(R-1)[H + ] 3 / c f + t f - 2 ) [ H + fkf kg-I-
+ tf-3)[#+]/cf A £ * f / tf-4) * f Af
E ordenando:
tf-3) [/?+] tf-2) [H+] •M2
( 4 - i í ) ( 4 - 1 ? ) fcf
tf-1) [H+f 1 tf [ i ¥+] 4 1 + (4-R) k" . /cf + ~ Õ T ^ 1 j 7 ' kfkfkf ( 2 4 )
Quando /cf , /<f ¡8* /cf , /cf, o que acontece nos casos
correntes, o terceiro e quarto termos na equação (24)
podem desprezar-se, resultando
tf-3)[17+] ( * - 2 ) [//+]* 1 • —g-- + /c 4 (25)
( 4 - 1 ? ) tf-4)
Esta é, mais uma vez, a equação de uma linha recta
de coeficiente angular l/kz, em que a ordenada na
origem é /cf.
Vê-se também pela equação (24) que quando kf > kf
esta última constante é dada pela expressão (22) sem
erro sensível.
2 — CONSTANTES DE ESTABILIDADE DE COM
PLEXOS
Suporemos no que se segue que os complexos pre
sentes na solução são dos tipos MHL, ML, ML2,
ML3, , MLn. Não se consideram, portanto, com
plexos polinucleares nem hidroxo-complexos.
A formação das espécies acima indicadas, corresponde
aos equilíbrios
M+L^ML Kx = [ML ] / [M ] [L] (26)
ML + L = ML2 K2= [ML2] / [ML] [L]
MLn__x + L = MLn Kn= [ML,Ü / [ML^] [l]
Os K{ são constantes de estabilidade estequiométricas
parciais, tal como na alínea anterior, e do mesmo
modo se definem as constantes de estabilidade glo
bais:
Pi = K i , h = Kx.K2, , p„ = K1.Ki ...... Kn
Numa solução contendo o complexo MHL, todos os
complexos ML,- e as diversas espécies H^L, a con
centração total do ácido (*) será
Ca = [H.L] + [HSL] + [H2L] + [HL] + [L] +
+ [MHL] + [ML] + 2 [ML2] + + n [MLn] (27)
E, por outro lado, a concentração total de metal pre"
sente será
C„ = [M] + [MHL] + [ML] + [ML2] +
+ [ML,,] (28)
Pela condição da electroneutralidade, considerando o
hidróxido de potássio como titulante e o nitrato
como anião do sal adicionado, obtém-se:
[K+] + [H+] + 2 [ M 2 + ] = [HzLr] + 2[H2L2-] +
+ Z[HL3~] + ¿L^] + 2[ML2-] + [ MHLr] +
+ 6 [ML| - ] + + (4b - 2 ) [ML^~] +
+ [OH~] + [NOz~] (29)
Mas
[*+] = a Ca
[NO~z] = 2C,„ = 2[M] + 2[MHL] + 2[ML] -|-
+ 2[ML2] + + 2 [ M L J (30)
Substituindo na expressão anterior e ordenando, obtém-se
aCa + [H + ]~ [OH-] = [HZL] + 2[H2L] +
+ 3[HL] + 4 [ L ] + S[MHL] + 4 [ML] +
¿ML2] + + 4n[ML„] (31)
Multiplicando a equação (27) por 4 e subtraindo a
equação (31), obtém-se
(4 -a)Ca-[H+] + [OH-] = 4 [HtL] +
+ 3 [HZL] + 2 [H2L] + [HL] + [MHL] (32)
ou, em termos de constantes de formação globais das
espécies H{L,
(*) Para maior clareza e sem perda de generalidade considera-se
o ácido tetrabásico e o metal bivalente ; omitem-se as cargas,
excepto quando necessárias à compreensão das deduções.
6 2

(A-a) Ca - [ff+] + [OÍT] = [L] { 4 p f [ff+]* +
+ 3 p f [ff+]» + 2 p f [H+f + p f [ff+] +
+ KMHL[M] [ff] p f } = [ £ ] { P H + * W [ M ] [ / f ] p f } (33)
sendo KMHL = [MHL] /[M] [HL] e p f í = 2 / p f [ í í + r
Desta expressão obtém-se o valor de [L]:
P / Í + ^ W - P f [Aí] [ f f ]
De um modo geral será
(i i-a) Ca-[H+] + [OH~]
[Aí] { KMHL [H] [L] Pf + I / P ¿ }
[Aí] { 1 + À - M H i . [ff] [L] p f + 2 p ¿ [L] ¿ }
[ i * - ] = 1H + KMHL. p f [Aí] [L]
(35)
Para aplicar esta expressão terá de conhecer-se o valor
de [Aí]; subtraindo (27) de (28), obtém-se
Cm - Ca = [Aí] - [Aí í J - 2 [AÍZ,3] - -
- (n - 1) [AÍLj - { [ff4L] + [ff3L] + [ff 2L] + (36)
+ [HL] + [L] }
ou
Cm ~Ca+ »H [L] = [Aí] { 1 - ( P 2 [ i ] 2 +
2 P 3 M3 + + ( « - D P.M")}
[Aí] C M - C a + « H . [ L ]
i - { p 2 [ í ] H 2 p 3 [ t:] 3 + + ( « - DP„M"} (37)
Por outro lado, o «grau de formação» dos complexos
— n — que é igual ao quociente entre a concentração
do ligante combinado com o metal e a concentração
total do metal, é dado por
_ C a - { [ f f 4 L ] + [ff sL] + [ff2L] + [HL] + [L]} n = — =
C Ca — °=H • [L\
(38)
E, também pela definição:
[MHL] + [ML] + 2 [ML2] + + « [AÍZ,J
[Aí] + [AíffL] + [AÍL] + [AÍL2] + + [AíXJ
(39)
Definindo, por analogia com o caso da alínea an
terior, os valores
n
a L = l + 2 ML]1
P L = 2 i p, [L] ' vem
KMHL [ g ] M Pf + P¿
1 + fff] M Pf + « L
Í39a>
E, ordenando:
« + ( « — ! ) Pi [L] + (n — 2) p 2 [Lf + +
+ Çn - ri) PM [Lf + (n - 1) í í : m h l . p f [ff] [ i ] = 0
Isto é:
2 ( J i - O^Lj + in-l) KMHL.tf .[H][L] = 0 (40) t = 0
Este é um sistema' de equações que permite calcular as
constantes desejadas. Os valores de n dependem dos valo
res de [L], mas estes, por sua vez, exigem o conhecimento
de KUHL. Como esta é uma das incógnitas a deter
minar, é evidente que só poderemos conseguir a so
lução final por aproximações sucessivas.
Na primeira aproximação considera-se KMHL = 0; isto
permite calcular os primeiros valores de [L] pela equação
(34) e, a partir destes, os correspondentes valores de n.
Pode então resolver-se o sistema de equações (40) e
determinar os primeiros resultados aproximados para
KMHL e para os pt- . Usando o valor de KMHL assim
determinado calculam-se novos valores para [L] e para
os correspondentes n . Resolve-se de novo o sistema de
equações (40) e determinam-se soluções mais aproxi
madas para KMHL e para os p á . O processo é repetido
até não se obterem modificações sensíveis nas solu
ções determinadas.
E claro que este método se torna excessivamente moroso
se não se dispuser de um computador electrónico; nos
casos práticos, porém, são possíveis diversas simpli
ficações e o cálculo torna-se bastante mais rápido.
Isto é especialmente verdade no caso das complexonas,
6 3

com as quais, normalmente, só se formam os com
plexos MHL, ML e ML2 e, às vezes, apenas ML.
Nestas condições aplicam-se as expressões simplificadas
que se deduzem a seguir.
I. Caso em que se forma apenas o complexo ML
É o caso mais geral com as complexonas; as equações
(35), (37) e (40) transformam-se em:
(n — a) C — [H + ] + [OH~] [L] = * - - (41)
ÍM] = Cm-Ca + zH.[L] (42)
n+(n—l) K1[L] = 0 (43)
o valor de n é dado por (38), e, no caso presente,
_ Ca-aH.[L] [ML]
C c
Substituindo os valores de [ML], [M] e [L] na defi
nição de Kx = KML, obtém-se
Ca — u.H. [L]
[L]{Cm-Ca+*H.[L]}
I I . Caso em que não existe o complexo MHL
b + (n— 1) Pi [L] + ( « - 2 ) p 2 [Lf = 0
Dividindo por (n — 2) [Lf obtém-se
(»—1)
+ • (n~2)[Lf (n-2)[L]
0 — n )
KL + Ã-j K, = 0
(n — 2)[Lf (n — 2)[L] . ^ - ^ K , (45)
Representando gráficamente os valores de (« - 2) [Lf
( l — n ) em função de — , obtém-se uma recta cujo
(« - 2) [L] 3
coeficiente angular dá o valor de Kx e cuja ordenada
na origem é KXK%, permitindo calcular as duas cons
tantes desejadas.
Quando estas constantes são bastante diferentes (A log K
> 2,5), Kx pode calcular-se a partir da expressão (43);
para calcular K2 considera-se o seguinte equilíbrio
ML + L = ML„
(44) Por definição
[ML] + 2 [ML2]
[m] + [mlJT[ml2]
anterior:
Neste caso [L] é dado pela mesma expressão do caso [m] é, em geral, muito pequeno em relação a [ML]
e [ML2], logo
[ ML] + 2 [ML2] = [M]{K1[L] + 2K1K2[Lf}
[ML] + [ML2] [M] { A, [L] + A, K.2 [Lf}
E, simplificando,
[L] = (n -d) Ca-[H + ] + [OH~]
A equação (40) toma a seguinte forma
n + (n-l) p x [L] + (n~2) p 2 [ L ] 2
+ (n-n) PM [ L f = 0
que é uma equação linear do tipo a Pj + b p 2 + c p 3 +
+ + p„ + 1 = 0, facilmente resolúvel por de
terminantes.
No caso das complexonas só se formam os complexos
ML e ML2 e a equação (40) é, nestas condições. Kn
1 + 2 K2 [L]
1 . A2 [L]
( ã - 1 )
(2 — n) [L] (46)
64

I I I . Caso em que existem apenas os complexos MHL e ML
Como [Aí] é, normalmente, da ordem dos 10 4 Aí, os limites de aplicação serão aproximadamente
Se a estabilidade dos dois complexos é da mesma ordem de grandeza, tem de se aplicar o método de aproximações sucessivas indicado na dedução geral. Se KMHL e KML são bastante diferentes, pode ignorar-se KMHL no cálculo de KML obtendo-se uma primeira aproximação deste valor.
incale. A ML
[ML]
[M] [L]
Como
[AÍL] c a l c ' = [ M L ] r e a l + [MHL]*
K [AÍL] r e a l [MHLJ
ML [M] [L] [M] [L] — K ML +
[H] K
H MHL
(47)
P # < P * .
l 0 g KMHL < 4
Na prática estes limites devem considerar-se menores, e, tanto quanto possível, o cálculo deve ser feito por aproximações sucessivas.
3 — FORMAÇÃO DE COMPLEXOS COM O ANIAO
DO ELECTRÓLITO DE SUPORTE
Quando se formam apenas os complexos MHL e ML,
a concentração do metal é, segundo a equação (37), dada por
[M] = Cm-Ca+*H.[L] (48)
Se M forma complexos com o anião do meio, a con
centração total do metal passará a ser
Representando graficamente os valores calculados de
[H] KML em função de —-g, obtém-se uma linha recta
cujo coeficiente angular é o verdadeiro valor de KMHL
e cuja ordenada na origem é o valor real de KML. Este método só pode ser utilizado quando o produto KMHL. p f [H] [M] for desprezável em relação a p H , pois os valores de [L] são calculados pela expressão (35) considerando KMHL = 0. As condições de aplicação são dadas, portanto, por:
KMHL-^I [H] [ M ] < p H < p f [H]
sendo p f [H] o termo dominante em p ^ , o que acontece nas soluções alcalinas. Segue-se que
[H+]
Terá pois de ser
I 1 KMHL• [aí] } ;
[H + ]>kH
n
* W < 1 / [ A Í ]
Cm = [Aí] + [ML] + [MHL] + [MX]+[MX2]
+ + [MXJ (49)
em que X é o anião do meio. A expressão (49) substitui portanto a expressão (28). Subtraindo agora (27) de (49), obtém-se
c » - c a + «H. [l] = [m] + [m] 2 p ; [ x r =
= [ a í ] { i + I p; [xj}
E tirando o valor de [Aí]:
Cm-Ca + *H [L] [Aí]
i + i p; [*Y
(50)
Comparando (50) com (48), verifica-se que, pelo facto de se formarem complexos com o anião do meio, a concentração real do metal, que podemos designar por [A í ] ' , é igual a [Aí] multiplicada por um factor
/ = 1 / 1 + I p; [XJ , isto é: [Aí]' = [ A í ] . /
A constante de estabilidade calculada ignorando a for
mação destes complexos — KML — estará relacionada
65

com a constante ideal para um meio nâo complexante
— K'ML — do seguinte modo
_ [ML] [ML] . / _
[ M ] [ L ] " [ M ] : [ L ] " ^ ' ^
Logo
ou
log KML = log K'L + log /
log K'L = log R~ML + log d + .2 ß- [*]') (51) í = I
Como a concentração do electrólito de suporte se
pode considerar constante por este estar presente em
grande excesso, virá:
log K'ML = log K M L + log (1 (52)
em que Cx é a concentração do electrólito de suporte.
Nos casos estudados no presente trabalho é Cx — n ,
força iónica da solução, e em geral forma-se apenas
um complexo (aliás, um par iónico) com o anião do
meio. Nestas condições, a expressão que relaciona as
constantes «experimentais» com as constantes «ideais»
é, evidentemente,
log K'ML = log KML + log (1 + KMX . ( i ) (53)
sendo KMX a constante de formação do referido par
iónico.
66

A p ê n d i c e
T A B E L A S D E V A L O R E S E X P E R I M E N T A I S
Nas tabelas que se seguem apresentam-se os valores
obtidos nas titulações potenciométricas efectuadas, cuja
técnica foi descrita na secção anterior.
Para maior facilidade de consulta aponta-se de novo
o significado da simbologia usada nesta secção:
CL concentração da solução da complexona
CM concentração da solução do sal do metal
Cb concentração da base titulante
Vt volume total no início da titulação (a = 0)
H força iónica da solução
T temperatura ( °C)
a grau de neutralização: equivalentes de base adi
cionada por mole da complexona
pH — log [ f f + ] (valores corrigidos)
67

I. ÁCIDO URAMILDlACÉTICO Tabela 3
A) T = 20,0°C
Tabela 1
DETERMINAÇÃO D A S CONSTANTES D E DISSOCIAÇÃO
pkx e p/c 2 : ¡j. = 0,1 M (KNOs)
pks : ¡x = 0,1 M «CH^N.NOg)
CL = 9,768 X I O - 4 M Cj = 0 , 1 1 1 M
a pH a pH a pH
0,000 2,787 1,019 3,112 2,045 8,290
0,114 2,817 1,136 3,170 2,159 8,885
0,227 2,844 1,250 3,230 2,272 9,175
0,341 2,871 1,363 3,310 2,386 9,378
0,454 2,903 1,477 3,400 2,499 9,545
0,568 2,940 1,591 3,509 2,613 9,690
0,682 2,979 1,704 3,653 2,727 9,820
0,795 3,020 1,818 3,880 2,840 9,944
0,909 3,064 1,931 4,344 2,954 10,057
Tabela 2
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
= 9,768 X 10~ 4 M CM = 0,010 M (77+)
cb = 0,111 e 1,200 M ( r e s t a n t e s )
pH
K + Na + Li + 27 + (*)
2,045 7,574 6,540 4,629 5,490
2,159 8,458 7,230 5,038 6,080
2,272 8,780 7,560 5,321 6,432
2,386 9,005 7,800 5,555 6,741
2,499 9,187 8,003 5,762 7,015
2,613 9,352 8,204 5,975 7,317
2,727 9,525 8,418 6,210 7,676
2,840 9,688 8,676 6,531 8,153
2,954 9,850 9,010 7,254 8,810
3,067 10,005 9,345 9,135 9,460
(*) [X = 0,1 M ( (C / í 3 ) 4 N.N03)
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
(A = 0,1 M ( (C7Í 3 ) 4 N.N03)
CL = 9,768 X 10" 4 M (Li+) e 9,670 X 10" 4 M (Na+)
CM= 0,0108 M (Lí+) e 0,100 M (Na+)
Cb = 0 , 1 1 2 M (Li+) e 9,524 X 10~ 2 M ( A / a + )
pH pH
Li + Na +
2,000 5,250 1,970 5,050
2,113 6,815 2,068 7,650
2,227 7,320 2,167 8,160
2,340 7,650 2,265 8,450
2,454 7,940 2,364 8,626
2,568 8,201 2,462 8,840
2,681 8,427 2,561 9,000
2,795 8,750 2,659 9,157
2,908 9,040 2,758 9,310
3,022 9,330 2,856 9,470
2,955 9,640
Tabela 4
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
V- = 0,1 M (KN03) cL = = 1,0 X 10" 3 M
CM = 0,005 M cb -= 0,100 M
pB
a Afg2 + C«2 + 5f2 + B<& +
2,500 7,591 6,448 7,774 8,106
2,550 — — — —
2,600 8,661 7,210 8,402 8,671
2,650 8,875 — — 8,865
2,700 9,035 7,695 8,788 9,020
2,750 9,165 — — 9,175
2,800 9,302 8,190 9,090 9,295
2,850 9,410 — — 9,415
2,900 9,540 8,775 9,380 9,540
2,950 — — — —
3,000 9,750 9,395 9,658 9,770
68

Tabela S
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
= 0,1 Aí (KN03) 9,838 X IO"4 M
= 0,0108 (Mg2+)
e 0,010 M ( r e s t a n t e s )
cb = 9,524 X IO"2 M
a
pH
a Be2 + Ca¿ T- S r 2 +
0 2,730
0,484 2,815
0,968 2,920
1,065 2,943
1,162 2,969
1,259 2,993
1,356 3,022
1,452 3,051
1,549 3,085
1,646 3,120
1,743 3,160
1,840 3,200 3,729 3,711
1,936 3,241 3,842 3,829 4,310 4,510
2,033 3,291 3,979 3,959 4,742 5,370
2,130 3,349 4,127 4,100 5,111 5,871
2,227 3,408 4,285 4,261 5,400 6,187
2,324 3,479 4,458 4,440 5,647 6,441
2,420 3,563 4,645 4,630 5,880 6,680
2,517 3,662 4,852 4,850 6,122 6,919
2,614 3,791 5,098 5,099 6,373 7,170
2,711 3,970 5,381 5,401 6,670 7,457
2,808 4,234 5,750 5,801 7,041 7,810
2,905 4,744 6,289 6,400 7,589 8,280
3,002 6,146 7,800 7,800 8,600 8,940
B) T = 27,0°C
Tabela 6
DETERMINAÇÃO DAS CONSTANTES D E DISSOCIAÇÃO
pfcj e p / í 2 : [j. = 0,1 Aí (KNOJ
= 0.1 M ((CH^.N.NO
tabela 7
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
CL = 1,096 X IO"3 M (K+) e 9,500 X 10~~4 M (Tl+, Li+ e Na+)
CM = 0,010 M (77+), 0,0108 M ( i í + ) e 1,200 M (Na+ e K+)
Cb = 0,112 M
a
pH
a
pH
a
Ti+(») Li + C) Na +
a
K +
1,989 4,870 5,069 5,120 2,090 8,030
2,106 5,940 6,850 6,900 2,985 8,489
2,223 6,384 7,358 7,350 2,280 8,742
2,340 6,688 7,685 7,639 2,375 8,935
2,457 6,957 7,952 7,860 2,470 9,095
2,574 7,218 8,195 8,054 2,565 9,240
2,691 7,510 8,436 8,252 2,660 9,374
2,808 7,830 8,681 8,460 2,755 9,509
2,925 8,240 8,935 . 8,701 2,850 9,639
3,042 8,720 9,200 8,995 2,945 9,769
(*) [L = 0 , 1 M ( ( C J / 3 ) 4 JV. N03)
O T = 34,0°C
Tabela 8
DETERMINAÇÃO D A S CONSTANTES D E DISSOCIAÇÃO
pkí e p í : 2 : ¡j, = 0,1 M (KN03)
pfeg : ¡X = 0,1 M «CH^N.NOJ r * " á CL = 1,016 X 1 0 - 3 M Cb = 0 , 1 1 2 M CL = 9,500 X IO"4 M Cb = 0,112 M
0,000 2,911 1,560 3,649 2,339 9,145 0,000 2,921 1,543 3,516 2,535 9,300
0,223 2,970 1,671 3,800 2,451 9,316 0,220 2,979 1,763 3,931 2,645 9,425
0,446 3,031 1,782 4,031 2,562 9,458 0,441 3,040 1,984 4,950 2,755 9,532
0,668 3,108 1,894 4,540 2,674 9,578 0,661 3,121 2,094 8,270 2,865 9,631
0,891 3,195 2,005 7,710 2,785 9,690 0,882 3,149 2,204 8,743 2,975 9,728
1,114 3,300 2,117 8,584 2,896 9,795 1,102 3,301 2,314 8,981 3,086 9,810
1,337 3,442 2,228 8,928 3,008 9,895 1,322 3,428 2,424 9,160
6 9
p H a pH a pH a pH a pH a pH
INSTITUTO DE ENERGIA ATÔMICA

Tabela 9 Tabela 11
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
CL — 1,016 X I O - 3 M CL = l , 0 1 6 x l 0 " 3 M'Li+ e Afa+) e 9,941 X 10-* M (77+ e K+)
CM = 0,010 M (77+), 0,0108 (Xi'+) e 1,200 M (Na+ e K+) CM = 0,010 M (77+), 0,0108 M (Li+) e 1,200 M (Na+ e Í T + )
Cb — 0,112 M C 6 = 0 , 1 1 2 M
pi f p i f p i f p i f
(*) [X = 0,1 M {(CH3)i N. NOs)
D) T = 39,0°C I I . ÁCIDO 1 -METIL UR A MI LDI A CÉTICO
7 7 + (•) Li+ (•) Na + K + a
Li + Na+ a
77 + a
K +
2,094 6,130 6,923 6,939 8,128 2,094 6,741 6,870 2,039 5,767 2,095 8,184
2,204 6,582 7,352 7,345 8,540 2,204 7,245 7,294 2,152 6,391 2,208 8,652
2,314 6,893 7,670 7,610 8,785 2,314 7,569 7,565 2,264 6,750 2,310 8,900
2,424 7,179 7,921 7,816 8,975 2,424 7,837 7,770 2,376 7,035 2,432 9,085
2,535 7,460 8,172 8,000 9,130 2,535 8,081 7,960 2,489 7,306 2,545 9,225
2,645 7,750 8,421 8,191 9,268 2,645 8,321 8,151 2,601 7,594 2,657 9,354
2,755 8,125 8,665 8,400 9,400 2,755 8,561 8,350 2,713 7,909 2,769 9,460
2,865 8,495 8,919 8,636 9,525 2,865 8,787 8,580 2,826 8,286 2,882 9,559
2,975 8,920 9,144 8,912 9,646 2,975 8,908 8,841 2,938 8,736 2,994 9,649
3,086 9,275 9,346 9,219 9,753 3,086 9,201 9,129 3,050 9,125 3,106 9,726
Tabela 10
DETERMINAÇÃO D A S CONSTANTES D E DISSOCIAÇÃO
p/<j e pk2 : (X = 0,1 M (KNO^
p/f3 : [± = 0,1 M (CHZ)I N.NO?)
9,941 x 10~ 4 M cb = 0,112 M
a pH a p H a pH
0,000 2,940 1,573 3,700 2,545 9,225
0,242 3,001 1,798 4,064 2,657 9,354
0,466 3,066 1,910 4,531 2,769 9,460
0,691 3,141 2,095 8,184 2,882 9,559
0,918 3,232 2,208 8,652 2,994 9,649
1,124 3,331 2,310 8,900 3,108 9,226
1,348 3,460 2,432 9,085
7 0
Tabela 12
DETERMINAÇÃO D A S CONSTANTES DE DISSOCIAÇÃO
T = 20,0°C
pk1 e pk2 : ¡X = 0,1 M (KNOs)
pk3 : (X = 0,1 M ( (C/J 3 ) 4 N.NOs)
CL = 9,619 X 10-4 M = 9,524 X 10" 2 M
a p H a pH a pH
0,000 2,871 1,287 3,348 2,376 9,511
0,198 2,920 1,485 3,495 2,475 9,651
0,396 2,974 1,683 3,719 2,574 9,775
0,594 3,033 1,881 4,180 2,673 9,881
0,792 3,105 2,079 8,590 2,772 9,980
0,990 3,188 2,178 9,080 2,871 10,071
1,089 3,287 2,277 9,334 2,970 10,153

j
J Tabela 13
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
T = 20,0°C
CL = 9,619 X 10~ 4 M CM = 0,010 M (77+ e Li+),
Cb = 9,524 X 10~ 2 M 0,100 M (Na+) e 1,200 M (K+)
pH
a Tl+ (*) Li+ (*) A 7 « + (*) 7Í +
2,079 6,032 6,909 7,870 8,040
2,178 6,508 7,430 8,430 8,696
2,277 6,820 7,741 8,709 8,990
2,376 7,081 8,000 8,909 9,196
2,475 7,330 8,230 9,075 9,360
2,574 7,572 8,465 9,226 9,502
2,673 7,840 8,701 9,364 9,634
2,772 8,142 8,950 9,501 9,760
2,871 8,511 9,202 9,634 9,880
2,970 8,975 9,456 9,769 9,991
3,069 9,450 9,689 9,900 10,050
(*) [A = 0,1 AT ( (CHs)iN. N03)
Tabela 14
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
T = 20°,0°C u. = 0,1 M (KNOs)
CL = 1,000 X IO"3 M CM = 0,005 M
Cb = 9,524 X IO- 2 M
pH
a ———— —— •— — ——
C«2 - f - 5 / 2 + BaZ-Y
2.475 6,291 7,358 7,930
2,524 6,768 7,775 8,211
2,571 7,145 8,210 8,501
2,619 7,440 8,515 8,755
2,646 7,686 8,740 8,960
2,714 7,920 8,924 9,130
2,761 8,150 9,085 9,267
2,809 8,387 9,230 9,391
2,856 8,625 9,361 9,506
2,904 8,881 9,486 9,610
2,952 9,131 9,605 9,707
3,000 9,370 9,717 9,800
Tabela 15
T = 20,0°C y. = 0,1 M (.KNOJ
CL = 9,619 X 10" "4 M CM = 0,0108 M (Mg2+)
ch = 9,524 X 10" " 2 M e 0,010 M (restantes)
pH
BE2 + M G 2 + C « 2 + SF2 + B « 2 +
0 2,750
0,495 2,837
0,990 2,949
1,089 2,970
1,188 2,997
1,287 3,024
1,386 3,057
1,485 3,090
1,584 3,121
1,683 3,160
1,782 3,201 3,710 3,710
1,881 3,244 3,830 3,834 4,101
1,980 3,296 3,967 3,981 4,560 4,765
2,079 3,351 4,121 4,150 5,130 5,799
2,178 3,410 4,292 4,330 5,510 6,251
2,277 3,481 4,480 4,521 5,789 6,557
2,376 3,569 4,676 4,721 6,035 6,811
2,475 3,670 4,888 4,941 6,270 7,050
2,574 3,800 5,114 5,180 6,514 7,295
2,673 3,972 5,380 5,460 6,780 7,557
2,772 4,220 5,695 5,799 7,100 7,859
2,871 4,732 6,122 6,250 7,522 8,239
2,970 5,602 6,897 7,065 8,205 8,741
3,069 — 9,225 9,220 9,275 9,324
71
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE

III . ÁCIDO 1,3-DIMETIL VRAMILDIACÉTICO
Tabela 16
DETERMINAÇÃO D A S CONSTANTES D E DISSOCIAÇÃO
T = 20,0 °C
pkx e pfc2 : ¡x = 0,1 M (KN03)
pk3 : (JL = 0,1 M ( (C /J 3 ) 4 N.N03)
cL = 1,000 X 10" "3 M cb = 9,524 X 10' ~2 M
a pH a pH a pH
0,000 2,860 1,332 3,370 2,380 9,760
0,190 2,905 1,523 3,530 2,476 9,882
0,380 2,952 1,713 3,779 2,571 9,988
0,571 3,010 1,904 4,375 2,666 10,080
0,761 3,079 2,095 9,023 2,761 10,160
0,952 3,156 2,190 9,391 2,856 10,233
1,142 3,255 2,285 9,601 2,952 10,300
Tabela 17
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
T = 20,0 °C CM = 0,010 M (T7+ e Li+),
C L = 1,000 x 10~ 4 M 0,100 M (Na+)
Cb = 9,524 X IO"2 M e 1,200 M (K+)
pH
a
77 f (*) Li+ (») Na+ K +
2,000 5,720 5,815 6,072 6,630
2,095 6,548 7,330 8,401 8,740
2,190 6,937 7,739 8,805 9,140
2,285 7,220 8,029 9,050 9,371 2,380 7,470 8,270 9,231 9,550
2,476 2,711 8,500 9,386 9,690
2,571 7,955 8,725 9,520 9,810
2,666 8,228 8,954 9,642 9,924
2,761 8,538 9,185 9,750 10,025
2,856 8,903 9,411 9,870 10,120
2,952 9,303 9,633 9,979 10,210
3,047 9,655 9,830 10,080 10,290
(*) [L = 0,1 M ((CH3)i iV .A 'Og)
Tabela 18
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
T = 20,0"C ¡i = 0,1 M (KN03)
CL = 1,000 X IO"3 M CM = 0,0108 M ( M g a + )
e 0,010 M (restantes)
Cb = 9,524 X 10--2 M
pH
a Be2 + Mg1 + C«2 + S/-2 +
0 2,750
0,476 2,835
0,952 2,939
1,047 2,962
1,142 2,990
1,237 3,015
1,332 3,044
1,428 3,074 3,420 3,430
1,523 3,109 3,497 3,503
1,618 3,141 3,581 3,598
1,713 3,181 3,685 3,710
1,808 3,220 3,805 3,848 3,830 3,839
1,904 3,265 3,950 4,019 4,150 4,189
2,000 3,314 4,118 4,220 4,876 5,420
2,095 3,370 4,300 4,433 5,515 6,291
2,190 3,430 4,495 4,660 5,869 6,671
2,285 3,501 4,692 4,880 6,140 6,952
2,380 3,588 4,900 5,100 6,386 7,202
2,476 3,689 5,121 5,336 6,620 7,440
2,571 3,811 5,360 5,586 6,865 7,685
2,666 3,980 5,635 5,885 7,142 7,956
2,761 4,220 5,984 6,250 7,485 8,274
2,856 4,650 6,452 6,790 7,935 8,647
2,952 5,350 7,370 7,780 8,618 9,088
3,000 — 9,340 9,348 9,340 9,492
7 2

Tabela 19
pH
a Mg2 + Cá¿ + S»2 + 7Ja2 +
2,524 6,566 6,921 8,038 8,560
2,571 7,800 7,390 8,455 8,825
2,619 8,750 7,725 8,725 9,050
2,666 9,091 7,989 8,969 9,221
2,714 9,300 8,220 9,145 9,357
2,761 9,459 8,431 9,291 9,481
2,809 9,585 8,641 9,420 9,592
2,856 9,693 8,845 9,540 9,691
2,904 9,789 9,049 9,646 9,780
2,952 9,870 9,240 9,743 9,862
3,000 9,949 9,420 9,833 9,938
IV. ÁCIDO o-CARBOXIFENILIMINODIACÉTICO
Tabela 20
DETERMINAÇÃO D A S CONSTANTES DE DISSOCIAÇÃO
T = 20,0°C \L = 0,1 M (KN03)
c L = 1,260 X IO"3
M c» = 0,111 M
a PH a pH a pH
0,000 2,888 1,499 3,559 2,381 7,546
0,176 2,933 1,588 3,650 2,470 7,704
0,353 2,985 1,676 3,763 2,558 7,854
0,529 3,040 1,764 3,910 2,646 8,011
0,706 3,105 1,852 4,140 2,734 8,183
0,882 3,175 1,940 4,602 2,822 8,385
1,058 3,259 2,029 6,265 2,911 8,655
1,235 3,358 2,117 6,870 3,000 9,053
1,323 3,423 2,205 7,165 3,087 9,530
1,411 3,482 2,293 7,380 3,175 9,861
Tabela 21
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
T = 20,0°C
CL = 1,260 X 10~ 3 M CM = 0,010 M (Tl+ e Ag+)
Cb = 0,111 M e 1,200 M (Zi+ e Na+)
pH pH
a a
Tl + {*) Li+ Na+ Ag + (*)
2,205 6,949 6,070 6,912 2,134 6,405
2,293 7,162 6,279 7,121 2,223 6,700
2,381 7,359 6,455 7,298 2,311 6,925
2,470 7,509 6,615 7,450 2,400 7,121
2,558 7,665 6,773 7,602 2,489 7,302
2,646 7,824 6,939 7,760 2,578 7,477
2,734 8,008 7,119 7,932 2,667 7,660
2,822 8,219 7,345 8,134 2,756 7,853
2,911 8,505 7,679 8,406 2,845 8,086
3,000 8,941 8,421 8,822 2,934 8,391
(*) [X = 0 , 1 Aí (KN03)
V. ÁCIDO o-METOXIFENILIMINODIACÉTICO
Tabela 22
DETERMINAÇÃO D A S CONSTANTES D E DISSOCIAÇÃO
T = 20,0°C ¡i = 0,1 M (KN03)
CL = 1,173 X IO"3
M Cj, = 0,111 M
a pH a pH a pH
0,000 3,135 0,758 3,787 1,515 5,607
0,095 3,189 0,852 3,989 1,610 5,775
0,189 3,240 0,947 4,263 1,705 5,961
0,284 3,298 1,042 4,600 1,799 6,190
0,379 3,361 1,136 4,880 1,894 6,531
0,474 3,440 1,231 5,095 1,989 7,620
0,568 3,531 1,326 5,277 2,083 9,519
0,663 3,642 1,421 5,446 2,178 9,917
73
T = 20,0°C \x = 0,1 M (KN03)
CL = 1,000 X IO"3 M CM = 0,005 M
Cb = 9,524 X IO- 2 M
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE

Tabela 23
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
T = 20,0°C jx = 0,1 Aí (KN03)
cL = 9,375 X 10-* M CM = 0,100 M (77+)
c b = 0,100 M e 0,010 M (Ag+ e S V 2 + )
pH
d Ag+ Tl+ ÍÍ-2 +
0,959 4,324 4,130 4,400
1,066 4,595 4,328 4,710
1,173 4,838 4,521 4,960
1,280 5,047 4,710 5,170
1,387 5,241 4,882 5,351
1,493 5,430 5,060 5,530
1,600 5.630 5,250 5,720
1,707 5,868 5,470 5,931
1,813 6,183 5,758 6,219
1,920 6,797 6,270 6,740
2,027 9,170 8,850 9,070
Tabela 24
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
T = 20,0°C [X = 0,1 Aí (KNO.ò)
CL = 1,173 X 10~ 3 Aí CM = 0,010 Aí
Cb = 0 , 1 1 1 M
pH
a —• —
Ca9--\- Ba% +
1,042 4,477 4,600
1,136 4,739 4,850
1,231 4,955 5,045
1,326 5,137 5,235
1,421 5,307 5,409
1,515 5,470 5,570
1,610 5,639 5,742
1,705 5,824 5,930
1,799 6,054 6,165
1,894 6,397 6,530
1,989 7,361 7,640
VI. ÁCIDO O-HIDROXIFENILIMINODIACÉTICO
Às soluções preparadas do modo habitual adicionou-se
a quantidade equivalente de KOH 0,105 M, titulando-se
em seguida por retorno com HCIOi 0,100 M. Nestas
condições Vt = 125,85, decrescendo 0,1 ml por cada
aumento de 0,105 unidades de a.
Tabela 25
DETERMINAÇÃO D A S CONSTANTES D E DISSOCIAÇÃO
T = 20,0°C ¡X = 0,1 Aí (KN03)
pfex e pk3 — titulação directa com KOH 0,105 Aí (Vt= 120,0 ml)
p £ 2 —titulação por retorno com HCIO^ 0,100 Aí
CL = 1,000 X IO- 3 Aí Cb = 0,105 Aí
a pH a pH a pH
0,000 3,258 1,050 4,642 2,105 9,550
0,105 3,310 1,110 4,785 2,210 9,941
0,210 3,376 1,215 4,973 2,315 10,152
0,315 3,444 1,320 5,147 2,420 10,300
0,420 3,527 1,425 5,310 2,525 10,411
0,525 3,621 1,530 5,481 2,630 10,501
0,630 3,740 1,635 5,670 2,735 10,580
0,735 3,886 1,740 5,901 2,840 10,641
0,840 4,078 1,845 6,225 2,945 10,700
0,945 4,337 1,950 6,915 3,000 10,727
74

Tabela 26
DETERMINAÇÃO DE CONSTANTES D E ESTABILIDADE
T = 20,0 °C (x = 0,1 Aí (KN03)
CL = 1,000 x IO"3 Aí CM = 1,200 Aí e 7Va+)
e 0,010 Aí (restantes)
p/í
Afg2 + Cít2 + Sf2 + Ha2 + 77.+ Li + Na +
1,000 4,433 4,271 4,335 4,381 4,446
1,100 4,653 4,457 4,544 4,610 4,660
1,200 4,851 4,640 4,759 4,810 4,858
1,300 5,025 4,810 4,955 5,000 5,030
1,400 5,190 4,976 5,120 5,109 5,201
1,500 5,350 5,147 5,291 5,346 5,370
1,600 5,510 5,330 5,489 5,530 5,551
1,700 5,688 5,540 5,677 5,740 5,750
1,800 5,887 5,800 5,925 5,995 6.011
1,900 6,116 6,146 6,222 6,325 6,401
2,000 6,370 6,606 6,781 7,050 7,450 7,960 7,770
2,100 6,631 7,130 8,411 8,780 8,497 8,812 9,398
2,200 6,900 7,471 8,961 9,248 8,880 9,179 9,781
2,300 7,140 7,725 9,239 9,518 9,128 9,410 10,011
2,400 7,379 7,950 9,456 9,709 9,336 9,585 10,171
2,500 7,625 8,170 9,634 9,870 9,515 9,735 10,291
2,600 7,887 8,391 9,790 10,009 9,681 9,856 10,380
2,700 8,185 8,641 9,931 10,121 9,825 9,970 10,465
2,800 8,550 8,935 10,058 10,231 9,971 10,069 10,533
2,900 9,000 9,270 10,178 10,326 10,094 10,152 10,594
3,000 9,435 9,602 10,290 10,405 10,210 10,230 10,649
VTI. ÁCIDO o-MERCAPTOFENILIMINODIA CÉ-
TICO
As respectivas soluções foram obtidas a partir da
Iactona correspondente por fervura durante duas horas
com a quantidade equivalente de KOH. Utilizou-se
HClOi 9,400 x IO"2 M nas titulações por retorno; nes
tas condições Vt = 123,2 ml, decrescendo 0,1 ml por
cada aumento de 0,094 unidades de a.
Tabela 27
DETERMINAÇÃO D A S CONSTANTES D E DISSOCIAÇÃO
T = 20,0 °C [X = 0,1 Aí (KN03)
CL =•- 9,964 X IO"4 Aí
a pH a pH a pH
0,005 3,241 1,042 5,019 2,080 8,468
0,099 3,290 1,137 5,510 2,174 8,862
0,193 3,340 1,231 5,790 2,269 9,067
0,288 3,400 1,325 6,000 2,363 9,251
0,382 3,466 1,420 6,150 2,457 9,399
0,478 3,540 1,514 6,285 2,552 9,516
0,571 3,630 1,608 6,470 2,646 9,627
0,665 3,740 1,703 6,669 2,740 9,736
0,759 3,881 1,797 6,880 2,835 9,824
0,854 4,081 1,891 7,142 2,929 9,917
0,948 4,420 1,986 7,647 3,023 10,011
Tabela 28
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
T = 20,0 »C [X = 0,1 Aí (KN03)
CL = 9,964 X 10~ 4 Aí CM = 0,100 Aí ( A í g 2 + )
e 0,010 Aí (Ca2+ e Sr2+ )
pH
a
MgZ+ Ca'ï+ S/-2 +
2,174 8,350 8,621 8,650
2,269 8,685 8,911 8,940
2,363 8,927 9,110 9,151
2,457 9,109 9,270 9,306
2,552 9,254 9,400 9,440
2,646 9,380 9,510 9,561
2,740 9,499 9,620 9,666
2,835 9,610 9,720 9.769
2,929 9,710 9,814 9,861
3,023 9,810 9,905 9,951
75

VIII. ÁCIDO 2-HIDROXIClCLOHEXILIMINODIÁ-
CÉTICO
Tabela 29
DETERMINAÇÃO D A S CONSTANTES D E DISSOCIAÇÃO
T = 20,0 °C v- -= 0,1 Aí (JsT/VOg)
CL = 1,000 X IO""3 Aí cb •• = 0,100 M
a pH a pH a pH
0,000 3,140 0,700 3,687 1,400 9,340
0,100 3,182 0,800 3,885 1,500 9,498
0,200 3,232 1,900 4,239 1,600 9,641
0,300 3,292 1,000 5,805 1,700 9,770
0,400 3,362 1,100 8,455 1,800 9,892
0,500 3,448 1,200 8,898 1,900 10,010
0,600 3,550 1,300 9,151 2,000 10,125
Tabela 30
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
T = 20,0 "C [X = 0,1 Aí (KNOa)
cL = 1,000 X 10 - 3 M CM = 1,200 Aí (Li + )
c„ = 0,100 Aí e 0,010 Aí ( res tantes)
pH
a Ag + Tl + S>2 + BaZ + Li +
1,000 5,649 5,698 5,761 5,631 5,560
1,100 7,730 8,185 7,775 8,090 7,210
1,200 8,167 8,630 8,200 8,550 7,636
1,300 8,455 8,892 8,485 8,824 7,902
1,400 8,691 9,098 8,716 9,032 8,110
1,500 8,898 9,269 8,921 9,210 8,291
1,600 9,091 9,424 y , m 9,374 8,468
1,700 9,284 9,581 9,306 9,527 8,648
1,800 9,471 9,725 9,496 9,675 8,843
1,900 9,658 9,870 9,685 9,822 9,070
2,000 9,832 10,010 9,875 9,964 - 9,340
Tabela 31
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
T = 20,0°C
CL = 9,300 x 10~ 4 Aí (L;+) e 9,810 X IO"4 Aí (Na+)
CM= 0,100 Aí (Li+) e 1,200 Aí (Na+)
Cb = 0,100 Aí (Lí+) e 9,524 X IO"2 Aí (Na+)
pH pH
a
Li+ Na+
1,075 7,942 1,068 8,070
1,182 8,518 1,165 8,620
1,290 8,800 1,262 8,896
1,397 9,011 1,359 9,091
1,505 9,190 1,456 9,253
1,612 9,359 1,553 9,399
1,720 9,514 1,650 9,530
1,827 9,671 1,747 9,649
1,935 9,830 1,844 9,761
2,042 9,985 1,941 9,876
IX. ÁCIDO IMINODIACÉTICO
Tabela 32
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
T = 20,0°C CL = 1,000 X I O - 3 Aí
CM = 1,200 Aí Cb = 0,112 Aí
pH
Li+ Na +
1,111 8,193 8,408
1,222 8,583 8,780
1,333 8,828 9,015
1,444 9,025 9,200
1,555 9,185 9,364
1,666 9,340 9,520
1,777 9,488 9,670
1,888 9,631 9,815
1,999 9,773 9,959
2,111 9,910 10,069
7 6

Tabela 33 X I . ÁCIDO C1CLOHEXILIM1NODIACÉTICO
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
pH
Ag+ Tl+ Sr2 +
1,037 7,400 6,619 7,650
1,140 8,160 8,335 8,392
1,244 8,470 8,694 8,695
1,347 8,698 8,921 8,912
1,451 8,896 9,114 9,098
1,554 9,071 9,280 9,261
1,658 9,240 9,435 9,420
1,702 9,423 9,585 9,570
1,865 9,595 9,740 9,730
1,969 9,746 9,895 9,890
Tabela 35
DETERMINAÇÃO D A S CONSTANTES D E DISSOCIAÇÃO
T = 20,0°C (x = 0,1 M (KNOz)
CL = 1,000 X 10~ 3 Aí Cb = 0,100 Aí
a pH a pH a pH
0,000 3,121 0,700 3,656 1,400 10,176
0,100 3,170 0,800 3,850 1,500 10,290
0,200 3,220 0,900 4,197 1,600 10,382
0,300 3,280 1,000 6,629 1,700 10,460
0,400 3,345 1,100 9,480 1,800 10,530
0,500 3,425 1,200 9,830 1,900 10,598
0,600 3,528 1,300 10,032 2,000 10,645
X. ÁCIDO METILIMINODIACÉTICO
Tabela 34
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
T = 20,0"C CL = 1,000 X IO- 3 M
Cu= 0,010 M (Ag+), 0,100 M e 1,200 M (Li+ e Na+)
Cb = 0,105 M
pH
Ag+ Tl+ Li+ Na +
1,036 7,107 6,325 7,720 8,000
1,140 7,900 7,790 8,425 8,710
1,244 8,243 8,140 8,740 8,995
1,347 8,498 8,385 8,952 9,201
1,451 8,732 8,571 9,130 9,370
1,554 8,943 8,752 9,290 9,519
1,658 9,150 8,930 9,430 9,660
1,762 9,359 9,110 9,571 9,795
1,865 9,576 9,316 9,707 9,919
1,969 9,775 9,549 9,840 10,036
Tabela 36
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
= 20,0°C ¡X = 0,1 M (KNOz) — Ag+ e T
• = 1,000 X IO"3 M CM = 0,010 M (Ag+ e 77+)
= 0,100 M e 1,200 M (Li+ e Na+)
p/ í ]
Ag+ Tl + Li + Na +
1,000 6,045 6,150 6,665 6,150
1,100 8,041 9,160 8,950 9,350
1,200 8,460 9,560 9,320 9,709
1,300 8,750 9,787 9,540 9,918
1,400 8,990 9,957 9,701 10,064
1,500 9,205 10,089 9,831 10,181
1,600 9,413 10,198 9,940 10,275
1,700 9,611 10,290 10,036 10,351
1,800 9,791 10,374 10,120 10,421
1,900 9,961 10,450 10,159 10,481
2,000 10,110 10,515 10,540 10,645
7 7
T = 20,00c [X = 0,1 M (KN03)
CL = 1,000 X IO"3 M CM = 0,010 Aí (Ag+)
Cb = 0,105 Aí e 0,100 M {Tl+ e Sr*+)

Tabela 37 XIII. ÁCIDO ETILENO DI AM IN O TETRA CÉTICO
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
pH
a — ——
M g 2 + c « 2 + Sr2+ B«2 +
1,000 6,238 6,695 6,270 6,000
1,100 8,460 9,229 9,140 9,180
1,200 8,830 9,600 9,499 9,570
1,300 9,067 9,816 9,709 9,791
1,400 9,248 9,980 9,870 9,950
1,500 9,400 10,109 9,992 10,079
1,600 9,540 10,218 10,100 10,182
1,700 9,670 10,310 10,194 10,270
1,800 9,796 10,390 10,280 10,350
1,900 9,914 10,461 10,352 10,420
2,000 10,022 10,522 10,421 10,485
X I I . ÁCIDO NITRILO TRIACÉTICO
Tabela 38
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
T = 20,0 °C u, = 0,1 Aí (KN03)
CL = 1,000 X IO"3 Aí CM = 0,100 Aí (Ll+ e Na+)
e 0,010 Aí (Ag+ e 77+)
Cb = 0,100 M (Li+) , 0,105 Aí (Na+ e Ag+)
e 0,111 Aí (77+)
pH
a
pH PH
a Li +
a
Na+ Ag + a
Tl+
2,000 6,950 2,072 8,500 6,762 2,111 7,251
2,100 8,232 2,175 8,965 7,200 2,222 7,643
2,200 8,571 2,279 9,215 7,510 2,333 7,949
2,300 8,800 2,382 9,406 7,779 2,444 8,215
2,400 8,982 2,486 9,561 8,039 2,555 8,480
2,500 9,146 2,589 9,709 8,316 2,666 8,757
2,600 9,300 2,692 9,840 8,621 2,777 9,051
2,700 9,450 2,796 9,961 8,970 2,888 9,367
2,800 9,603 2,899 10,080 9,360 2,999 9,660
2,900 9,760 3,003 10,190 9,715 3,111 9,920
78
Tabela 39
DETERMINAÇÃO D E CONSTANTES
D E ESTABILIDADE (27+)
T = 20,0 °C [X = 0,1 Aí (KNOs)
CL = L000 X 10" 3 Aí CM = 0,010 Aí
Cb = 0,112 Aí
a pH a pH a pH
1,962 4,451 2,725 6,256 3,488 7,220
2,071 5,050 2,834 6,370 3,597 7,442
2,180 5,433 2,943 6,489 3,706 7,741
2,289 5,674 3,052 6,607 3,815 8,097
2,398 5,850 3,161 6,741 3,924 8,576
2,507 6,002 3,270 6,886 4,033 9,169
2,618 6,132 3,379 7,041 4,142 9,610
XIV. ÁCIDO 1,2-DlAMINOPROPANOTETRACÉ-
TICO
Tabela 40
DETERMINAÇÃO D A S CONSTANTES D E DISSOCIAÇÃO
7' = 20,0 °C V-= 0,1 Aí (KN03)
1,015 X IO"3 Aí cb = 0,105 Aí
a pH a pH a pH
0,000 2,943 1,862 4,205 3,205 9,862
0,207 3,000 2,062 5,236 3,309 10,065
0,414 3,058 2,275 5,840 3,412 10,213
0,621 3,125 2,482 6,219 3,516 10,321
0,828 3,201 2,689 6,595 3,619 10,415
1,034 3,293 2,792 6,826 3,723 10,491
1,241 3,410 2,896 7,181 3,826 10,560
1,448 3,562 2,999 8,318 3,930 10,619
1,655 3,785 3,102 9,515 4,034 10,670
T = 20,0°C [i = 0,1 M (KNOz)
CL = 1,000 X IO"3 Aí CM = 0,010 Aí ( C a 2 + )
Cb = 0,100 Aí e 0,100 Aí (restantes)

Tabela 41 Tabela 42
DETERMINAÇÃO D E CONSTANTES D E ESTABILIDADE
T = 20,0°C \x = 0 , 1 M (KNO.A)
CL = 1,015 X 10~ 3 M CM= 0,010 M (Li + )
Cb = 0,105 M e 0,100 M (Na+)
pH
a ' •—"~ ~
3,102 9,284 9,351
3,205 9,625 9,680
3,309 9,845 9,888
3,412 9,982 10,039
3,516 10,122 10,151
3,619 10,240 10,257
3,723 10,331 10,331
3,826 10,422 10,422
3,930 10,495 10,495
4,034 10,565 10,565
4,134 10,620 10,620
DETERMINAÇÃO DE CONSTANTES D E ESTABILIDADE
T = 20,0»C y, = 0,1 M (KNOs)
CL = 1,000 X IO"3
M c M = 0 j 0 1 0 M
Cb = 0,101 M
pH
a Ag + 77 + Afg2 + C«2 + S»-2 + B«2 +
2,538 5,770 6,100 4,941 4,180 5,110 5,604
2,640 5,875 6,240 5,010 4,237 5,180 5,688
2,741 5,971 6,360 5,076 4,291 5,245 5,761
2,843 6,065 6,480 5,141 4,349 5,310 5,831
2,944 6,159 6,600 5,203 4,408 5,376 5,911
3,048 6,250 6,721 5,271 4,470 5,447 5,982
3,147 6,345 6,855 5,340 4,531 5,511 6,060
3,249 6,449 6,999 5,412 4,600 5,585 6,140
3,350 6,559 7,151 5,492 4,677 5,662 6,231
3,452 6,681 7,321 5,581 4,761 5,751 6,330
3,553 6,827 7,526 5,683 4,856 5,851 6,440
3,655 7,010 7,761 5,803 4,972 5,972 6,590
3,756 7,225 8,052 5,961 5,120 6,125 6,771
3,858 7,631 8,437 6,190 5,330 6,350 7,058
3,959 8,350 8,955 6,621 5,695 6,771 7,638
79

umário
Contribuindo para o esclarecimento da influência da
estrutura dos ligantes e do tipo de elemento central
na estabilidade dos respectivos complexos, apresentam-
-se neste trabalho os resultados do estudo das reacções
duma série de iões mono- e bivalentes com diversos
ácidos aminopolicarboxílicos («complexonas»).
Utilizaram-se como ligantes alguns produtos comerciais
correntes (ácidos iminodiacético, metiliminodiacético,
2-hidroxiciclohexiliminodiacético, nitrilotriacético, etile-
nodiaminotetracético e 1,2-diaminopropanotetracético),
bem como outros produtos por nós sintetizados,
a maioria pela primeira vez (ácidos uramildiacético,
1-metiluramildiacético, 1,3-dimetiluramildiacético, o-hi-
droxifeniliminodiacético, o - metoxifeniliminodiacético,
o-mercaptofeniliminodiacético, o - carboxifeniliminodia-
cético e ciclohexiliminodiacético). Deste segundo grupo,
só o primeiro e os dois últimos compostos tinham sido
estudados anteriormente, mas crê-se que não se encon
travam no estado de pureza requerido, com excepção
do ácido o - carboxifeniliminodiacético.
No aspecto preparativo, a síntese de derivados orto-
substituídos do ácido feniliminodiacético apresentou al
guns aspectos curiosos, nomeadamente o isolamento das
S-lactonas correspondentes aos ácidos c-hidroxifenilimi-
nodiacético e o-mercaptofeniliminodiacético, formadas
por eliminação intramolecular de uma molécula de água.
As reacções de formação de complexos dos novos
ligantes foram estudadas por meio de uma técnica
potenciométrica adequada, utilizando-se como titulantes
os hidróxidos de potássio e tetrametilamónio e como
suporte iónico os nitratos dos mesmo catiões. Em
todos os casos a força iónica foi ajustada a 0,100 M.
Calcularam-se assim as constantes de estabilidade dos
complexos de diversos iões, em especial dos metais
alcalinos e alcalino-terrosos, merecendo referência espe
cial os valores obtidos para os complexos do potássio
e do berílio com a série de homólogos do ácido uramil
diacético; até ao presente não se conheciam valores de
constantes de estabilidade para complexos de qualquer
destes metais com as complexonas.
Dada a tendência notável para a complexação dos iões
dos metais alcalinos evidenciada por esta mesma série
de homólogos, pareceu de interesse o estudo termodi
nâmico das respectivas reacções. Os resultados mostra
ram que os complexos são estabilizados por uma variação
de entalpia favorável; a variação de entropia é negativa
e bastante desfavorável nos casos dos iões de maior
raio, o que se relaciona com a baixa hidratação destas
espécies.
Uma vez que o ácido uramildiacético e seus homólogos
só podem actuar como tetradentados, como se demonstra
teoricamente e se confirma por meio de modelos mole
culares, é difícil propor uma explicação satisfatória para
as elevadas estabilidades dos complexos que formam;
no entanto, a comparação com os resultados obtidos
para a série de derivados oz-to-substituídos do ácido
feniliminodiacético sugere que devem resultar da elevada
basicidade do átomo de azoto no grupo iminodiacetato
daqueles compostos, aliada a uma estrutura envolvente
favorável para os grupos coordenantes.
Os valores obtidos no presente trabalho permitiram
aumentar consideravelmente o número de constantes de
estabilidade conhecidas para os complexos dos iões
8 1

dos metais alcalinos, alcalino-terrosos, prata e tálio.
Com os novos valores foi possível examinar mais
detalhadamente o problema da influência da basicidade
dos ligantes na estabilidade dos complexos, demons
trando-se que em casos simples existe uma relação
linear entre as energias livres de formação dos com
plexos com os iões dos metais e com os protões.
No caso dos metais alcalino-terrosos verificou-se que
os coeficientes angulares das rectas representativas
destas relações são proporcionais ao primeiro e segundo
potenciais de ionização dos metais, não havendo razões
para discriminar entre estes ou a sua soma uma vez que
todos estão também relacionados linearmente en
tre si.
As variações na ordem de estabilidades dos complexos
destes metais são interpretadas com base nas relações
estabelecidas.
Para os metais alcalinos obtêm-se igualmente relações
lineares e, por generalização, estabeleceu-se uma equação
empírica válida para estes metais, para os metais alcalino-
-terrosos e para as terras raras. Esta equação permite
calcular um valor aproximado para a constante de
estabilidade do complexo de um ião com estrutura
exterior de gás-inerte, sendo dado o raio iónico do
metal, a basicidade da complexona e o número de
coordenação efectivo nesse complexo.
Em conclusão discute-se a ordem de estabilidades nos
complexos dos iões monovalentes, verificando-se ser
Ag+ > Tl+ > Li+ > Na+ > K+, quando a interacção
com os ligantes é predominantemente electrostática, e
Ag+ > T7 + , Li+ > Na+ > K+, quando a interacção é
predominantemente covalente. A diferença de compor
tamento entre o tálio e a prata é especialmente notável
com ligantes possuindo orbitais desocupados que possam
aceitar pares electrónicos d do ião Ag+ formando-se
assim ligações n .
82

A b s t r a c t
The present work is concerned with the stabilities of the complexes formed by several, mono- and divalent ions with the aminopolycarboxylic acids («complexo-nes»). It is a contribution to the study of the influence of the structure of the ligands and the type of the central element on the stabilities of their complexes. Some of the ligands used were commercial products (iminodiacetic acid, methyliminodiacetic acid, 2-hydroxycy-clohexyliminodiacetic acid, nitrilotriacetic acid, ethylene-diaminetetracetic acid and 1,2-diaminepropanetetra-cetic acid); others had to be synthesized, most of them for the first time (uramildiacetic acid, 1-methyluramil-diacetic acid, 1,3-dimethyluramildiacetic acid, o-hydro-xiphenyliminodiacetic acid, o-methoxyphenyliminodia cetic acid, o-mercaptophenyliminodiacetic acid, o-car-boxyphenyliminodiacetic acid and cyclohexyliminodia-cetic acid). Among these, the first and the two last products had already been studied before, but they were not in the state of required purity, except o-car-boxyphenyliminodiacetic acid.
On the preparative side some interesting results were obtained in the attempted synthesis of o-hydroxyphenyl-iminodiacetic acid and o-mercaptophenyliminodiacetic acid, e.g. the isolation of their respective S-Iactones, formed by intramolecular elimination of water. The reactions of complexation of the new ligands were studied by a potentiometric technique, using potassium and tetramethylammonium hydroxides as titrants and the respective nitrates as the ionic background. In every case the ionic strength was adjusted to 0.100 M. Stability constants of several complexes were calculated, mainly for alkali and alkaline-earth metals, silver and
thallium; the values for the complexes of potassium and beryllium with uramildiacetic acid and its homologues deserve special mention since they are the first ones reported up to now. These same ligands showed an unusual affinitiy for the alkali metals and a thermodynamic study of the respective reactions seemed of interest. From the results it appears that the complexes are stabilised by the enthalpy change in the reactions; the entropy change is negative and especially unfavourable for the largest ions, a result which can be related to the low hydration of these species. Since molecular models and theory show that these ligands can only act as tetradentate, a satisfactory explanation for the unusual stabilities observed cannot be proposed; nevertheless, the comparison with the behaviour of a series of ligands derived from phenyli-minodiacetic acid suggests that it may be the result of the high basicity of the nitrogen atom in the diacetate group; the tendency to form complexes with the smallest ions suggests, on the other hand, that an especially favourable coordinating «cage» structure may be operative. The range of available values of stability constants of the complexes of alkali and alkaline-earth metals, which has been considerably extended in this work, allowed a more detailed examination of the problem of the influence of the basicity of the ligands on the stability of the complexes, and it is shown that in simple cases there exists a linear relationship between the free energies of formation of metal and proton complexes.
8 3

In the ease of the alkaline-earth metals it was found that the slopes of the straight lines representative of these relationships are proportional to the first and second ionization potentials of the metals, and that the plots against any of these values or its sum are all equivalent, since they are also linearly related. The order of stabilities for the complexes of these metals is discussed on the basis of the relationships established. For the alkali metals similar relationships can be obtained, and it was possible to deduce, by generalization, an empirical equation valid for all ions with an external inert-gas structure, which allows approximate values
of stability constants of their complexes to be calculated if the ionic radius of the metal, the basicity of the complexone and the coordination number of the complex are known. In conclusion, the order of stabilities for the complexes of monovalent ions is examined and found to be Ag+ > > 7 7 + > Li+ > Na+ > K+, when the interaction with the ligands is predominantly electrostatic; Ag+ > 7 7 + , Li+ > Na+ > K+ holds when the interaction is predominantly covalent. The difference of behaviour between silver and thallium is especially remarkable when the ligand has vacant orbitals which can accept d electrons from Ag+ to form K bonds.
8 4

Bibl iograf ia
1. Brintzinger, H. e Hesse, G., Z. Anorg. Allgem. Chem., 249, 133 (1942). Brintzinger, H. e Hesse, G., Z. Anorg. Allgem. Chem., 249, 299 (1942).
2. Pfeiffer, P. e Offermann, P., Ber., 75B, 1 (1942). 3. Schwarzenbach, G., Kampitsch, E. e Steiner, R., Helv.
Chim. Acta, 28, 828 (1945). 4. Martell, A . e Bersworth, F., Tech. Bull., n.° 2 (1950),
Bersworth Chemical Co., Framingham, Mass. 5. Ringbom, A., «Complexation Reactions» in Kolthoff e
Elving, «Treatise on Analytical Chemistry», Interscience, New York, 1959, p. 543.
6. Wanninen, E., «Thesis», Abo Akademi, Finlândia, 1960. 7. Chaberek, S. e Martell, A., «Organic Sequestering Agenis»,
J. Wiley & Sons, New York, 1959. 8. Reilley, G., Schmid, R. e Sadek, I., J. Chem. Educ, 36,
555 (1959). 9. Ahrland, S., Chatt, J. e Davies, N . R., Quart. Rev.
(London), 12, 265 (1958). 10. Schwarzenbach, G., Experientia Suppl., n." 5, 162 (1956). 11. Orgel, L., «Transition Metal Chemistry», Methuen & Co.,
London, 1961. 12. Ballhausen, C. J., introduct ion to Ligand Field Tbeory»,
J. Wiley & Sons, New York, 1963. 13. Coulson, C. A., «Valence», 2 . a ed., Oxford Press, 1961,
p. 292. 14. Bjerrum, J., «Metal-ammine Formation in Aqueous Solu
tion», P. Haase & Son, Copenhagen, 1957, p. 21. 15. Calvin, M. e Wilson, K., Am. Chem. Soc, 67, 2003
(1945). 16. Schwarzenbach, G., Kampitsch, E. e Steiner, R., Helv.
Chim. Acta, 29, 364 (1946). 17. Stein, A., Gregor, H. e Spoerri, P., J. Am. Chem. Soc,
78, 6185 (1956). 18. Noller, C. R., «Chemistry of Organic Compounds», Saun
ders & Co., Philadelphia, 1955, p. 603. 19. Näsänen, R. e Heikkilä, Suomen Kemistilehti, 32B, 163
(1959). 20. Stàry, J., Anal. Chim. Acta, 28, 132 (1963).
21. Bjerrum, J., Schwarzenbach, G. e Sillen, L., «Stability Constants*, The Chemical Society Special Publication n.° 6, London, 1958.
22. cf. Frank, H. e Evans, M., J. Chem. Phys., 13, 507 (1945).
23. Powell, R. e Latimer, W., / . Chem. Phys., 19, 1139 (1951).
24. Latimer, W., «Oxidation Potentials», Prentice Hall, New York, 1952.
25. Staveley, L. e Randall, T., Discussions Faraday Soc, n.° 26, 157 (1958).
26. Care, R. e Staveley, L., / . Chem. Soc, 4571 (1956). 27. Altred, A. e Rochow, E., / . Inorg. Nucl. Chem., 5, 264
(1958). 28. Nyholm, R. S., Proc. Chem. Soc, 273 (1961). 29. Schwarzenbach, G., Willi, A. e Bach, R. O., Helv. Chim.
Acta, 30, 1303 (1947). 30. Anderegg, G. e Schwarzenbach, G., Helv. Chim. Acta,
38, 1940 (1955). 31. Irving, H. e Pettit, L., «Advances in Co-ordination Che
mistry», McGraw-Hill, New York, 1961, p. 412. 32. Martell, A. e Calvin, M., «Chemistry of the Metal Chelate
Compounds», Prentice Hall, New York, 1959, p. 278. 33. Syrkin, Y. e Dyatkyna, M., «Structure of Molecules»,
Butterworth's Scientific Publications, London, 1950, p. 200.
34. Robinson, R. e Stokes, R., «Electrolyte Solutions», Butterworth's Scientific Publications, London, 1959, p. 302.
35. Martell, A. E., Ree. Trav. Chim., 75, 781 (1956). 36. Gillard, R., «Part II Thesis», Oxford, 1960. 37. Schwarzenbach, G. e Ackermann, A. , Helv. Chim. Acta,
30, 1798 (1947). 38. Tillotson, M., «B. Sc. Thesis», Oxford, 1957. 39. Chapman, D . , J. Chem. Soc, 1766 (1955). 40. Chapman, D . , Lloyd, D . R. e Prince, R. H., J. Chem.
Soc, 3645 (1963). 41. Irving, H. e Rossotti, H., Acta Chem. Scand., 10, 72
(1956). 42. Charles, R. G., J. Am. Chem. Soc, 76, 5854 (1954).
8 5

43. Yatsimirskii, K. B., «Advances in the Chemistry of the Coordination Compounds», MacMillan Co., New York, 1961, p. 96.
44. Deining, W. E., «Statistical Adjustment of Data», J. Wiley & Sons, London, 1948.
45. Durrant, P. J. e Durrant, B., «Advanced Inorganic Che-mistry», Longman's, London, 1962, p. 328.
46. Schwarzenbach, G., Ackermann, H. e Ruckstuhl, P., Helv. Chim. Acta, 32, 1175 (1949).
47. Monk, C. B., «Electrolytic Dissociation)), Academic Press, New York, 1961, p. 277.
48. Williams, R. J. P., J. Chem. Soc, 3770 (1952). 49. Duncan, J. e Keepert, D . , «The Structure of Electrolyte
Solutions», J. Wiley & Sons, London, 1959, pp. 26-27. 50. Duncan, J., Australian J. Chem., 12, 356 (1959). 51. Basolo, F. e Pearson, R., «Mechanisms of Inorganic Reac
tions*, J. Wiley & Sons, London, 1958, p. 72. 52. Cotton, F. A. e Wilkinson, J., «Advanced Inorganic Che-
mistry», Interscience, London, 1962, p. 333.
53. Irving, H. e Silva, J. J. R. F. da, «Proc. 7 ICCC», Stockholm, 1962, p. 326.
54. Irving, H. e Silva, J. J. R. F. da, / . Chem. Soc, 945 (1963).
,55. Aitken, J. K., Chem. Ind. (London), 45, 1334 (1956). 56. Freedman, H. e Frost, A „ / . Org. Chem., 23, 1292 (1958). 57. Schwarzenbach, G., «Complexometric Titrations», Me-
thuen & Co., London, 1957. (Trad, e rev. por H. Irving). 58. Gran, R., Analyst, 77, 661 (1952). 59. Schwarzenbach, G. e Biederman, W., Helv. Chim. Acta,
31, 331 (1948). 60. Laitinen, H. A., Ind. Eng. Chem. (Anal. Edition), 13,
393 (1941). 61. Harned, H. e Owen, B., «The Physical-Chemistry of Elec
trolytic Solutions», 3 . a ed., Reinhold, London, 1960, p. 752. 62. Lewis, G. e Randall, M., / . Am. Chem. Soc, 43, 1140
(1921). 63. Rossotti, F. e Rossotti, H., «Determination of Stability
Constants)), McGraw-Hill, New York, 1961, p. 24. 64. Thompson, L. C , Inorg. Chem., I, 491 (1962). 65. Moeller, T. e Ferrius, R., / . Inorg. Nucl. Chem., 20, 261
(1961). 66. Anderegg, G., Helv. Chim. Acta, 44, 1673 (1961). 67. Irving, H. e Stacey, M„ J. Chem. Soc, 2019 (1961). 68. Silva, J. J. R. F. da, «D. Phil. Thesis», Oxford, 1962. 69. Kortüm, G., Vogel, W. e Andresson, K., «Dissociation
Constants of Organic Acids in Aqueous Solutions», Butter worth's Scientific Publications, London, 1961.
86
Recommended




![EX-FQA715-EE-2014-CC - IAVEiave.pt/images/arquivo_de_provas/fqa_atual/EX_FQA715_EE...dá origem a iões OH-em solução. B) O aumento da concentração de [iões] OH-(aq) conduz a](https://img.document.onl/doc/110x75/5e5c2bc630dcad3ee1662ff7/ex-fqa715-ee-2014-cc-d-origem-a-ies-oh-em-soluo-b-o-aumento-da-concentrao.jpg)












