
UNIVERSIDADE DE LISBOA
FACULDADE DE CIÊNCIAS
DEPARTAMENTO DE BIOLOGIA VEGETAL
Inibição da desaminase celular APOBEC3G
pela proteína Vif do HIV-1: O papel da
localização celular
Dissertação orientada pelo Prof. Doutor João Gonçalves (Faculdade de
Farmácia da Universidade de Lisboa) e pela Prof. Doutora Margarida
Meireles (Faculdade de Ciências da Universidade de Lisboa)
Diana Isabel Máximo Rodrigues
Mestrado em Biologia Molecular Humana
2009

UNIVERSIDADE DE LISBOA
FACULDADE DE CIÊNCIAS
DEPARTAMENTO DE BIOLOGIA VEGETAL
Inibição da desaminase celular APOBEC3G
pela proteína Vif do HIV-1: O papel da
localização celular
Dissertação orientada pelo Prof. Doutor João Gonçalves (Faculdade de
Farmácia da Universidade de Lisboa) e pela Prof. Doutora Margarida
Meireles (Faculdade de Ciências da Universidade de Lisboa)
Diana Isabel Máximo Rodrigues
Mestrado em Biologia Molecular Humana
2009

iii
Agradecimentos
Em primeiro lugar, ao Professor Doutor João Gonçalves, pela oportunidade de realizar o
meu projecto de mestrado no seu laboratório, pela orientação em todos os aspectos e pela
correcção da tese.
Em segundo, aos colegas de laboratório, em especial ao Nuno, à Patrícia e à Inês, por me
ensinarem tudo o que sei e por me terem ajudado em tudo, no mais básico possível e nas
dúvidas “existenciais”. Aos restantes colegas, Andreia, Catarina, Soraia, Lídia, Luís, Carina,
Sylvie, Sara e Paula, pela ajuda diária constante. Aos colegas e amigos Joana, Ana
Catarina e André por todos os favores que jamais vos poderei recompensar, pelo espírito de
entre-ajuda, por me aturarem, por estarem lá nas horas difíceis, pelo apoio incondicional,
pelos vossos ombros amigos.
Agradeço ainda à Professora Doutora Maria Margarida Meireles, pela disponibilidade e
prontidão em todo o apoio que me prestou e pela correcção da tese.
Em último mas não menos importante, à minha família e aos meus amigos. Obrigada Mãe e
Pai por tudo aquilo que as palavras não chegam para descrever. Pela educação, pelos
sermões, pela compreensão, pela amizade, pela ajuda, pelo amor. A vocês e à minha
restante família porque serem o meu chão, as minhas raízes, o meu lar. Aos amigos mais
chegados e aos velhos amigos, pela paciência, pela cumplicidade e pelo carinho, por me
ajudarem a ser a pessoa que sou hoje.
A todos vocês, muito obrigada.

iv
Índice Geral
Agradecimentos ................................................................................................................. iii
Índice Geral ......................................................................................................................... iv
Índice de Figuras ............................................................................................................... vii
Índice de Tabelas ............................................................................................................. viii
Símbolos e Abreviaturas ................................................................................................... ix
Resumo .............................................................................................................................. xii
Abstract ............................................................................................................................. xiii
1. Introdução ........................................................................................................................ 1
1.1. O HIV e a SIDA .......................................................................................................... 1
1.1.1 Etiologia e Patogenia ........................................................................................... 1
1.1.2. O impacto demográfico ...................................................................................... 1
1.2. Cataracterização do HIV-1 ......................................................................................... 2
1.2.1. Taxonomia ......................................................................................................... 2
1.2.2. Genoma e Estrutura ........................................................................................... 2
1.2.3. Ciclo de replicação ............................................................................................. 3
1.3. A proteína viral Vif e o seu alvo celular ...................................................................... 4
1.3.1. A importância da Vif ........................................................................................... 4
1.3.2. A interacção Vif-APOBEC3G .............................................................................. 6
1.3.3. Especificidade à espécie .................................................................................... 7
1.4. O factor anti-viral APOBEC3G ................................................................................... 8
1.4.1. A família APOBEC .............................................................................................. 8
1.4.2. Regulação intracelular da desaminase ............................................................... 9
1.5. A necessidade de novas terapias:
o potencial terapêutico da interacção Vif-APOCEC .................................................. 10
1.6. Hipótese de trabalho e Objectivos ............................................................................ 10

v
2. Materiais e Métodos ...................................................................................................... 11
2.1. Clonagem ................................................................................................................ 11
2.1.1. Extracção de DNA plasmídico em média escala por kit (midiprep) ................... 11
2.1.2. Reacção de polimerização em cadeia (PCR, Polymerase Chain Reaction) ...... 12
2.1.3. Hidrólise enzimática de DNA ............................................................................ 12
2.1.4. Reacção de ligação inserto-vector .................................................................... 13
2.1.5. Preparação de bactérias electrocompetentes ................................................... 13
2.1.6. Transformação de bactérias electrocompetentes ............................................. 14
2.1.7. Selecção e confirmação de colónias positivas para a clonagem ....................... 14
I) Selecção de colónias e extracção de DNA plasmídico em pequena escala
sem kit (lise alcalina) .......................................................................................... 14
II) Confirmação de clones positivos por hidrólise enzimática de DNA
e electroforese em gel ....................................................................................... 15
III) Extracção de DNA plasmídico em pequena escala por kit (miniprep)
e sequenciação automática de DNA ................................................................. 15
2.2. Expressão proteica .................................................................................................. 16
2.2.1. Linha celular – HEK 293T ................................................................................. 16
2.2.2. Transfecção pelo método de fosfato de cálcio .................................................. 16
2.2.3. Extracção e quantificação de proteínas ............................................................ 17
2.2.4. Separação de proteínas por electroforese em gel de poliacrilamida
com dodecil sulfato de sódio (SDS-PAGE) ....................................................... 17
2.2.5. Western blot ..................................................................................................... 17
2.2.6. Extracção diferencial de proteínas do citosol e do núcleo ................................. 18
2.2.7. Transcrição e Tradução in vitro e Imuno-precipitação (IP) ................................ 19
2.3. Análise da função proteica em células eucariotas .................................................... 20
2.3.1. Ensaio de degradação dos clones NLS-A3G .................................................... 20
2.3.2. Ensaio de infecciosidade do HIV-1 na presença de NLS-A3G .......................... 20
I) Extracção de DNA plasmídico em média escala por kit (midiprep) .................... 20
II) Co-transfecção, produção e concentração das partículas virais ........................ 21
III) Quantificação de partículas virais
pelo ensaio imunosorvente ligado a enzima (ELISA) ........................................ 21
IV) Linha celular – TZM-bl, infecção
e quantificação da inibição da infecciosidade viral por CPRG .......................... 22

vi
3. Resultados ..................................................................................................................... 23
3.1. Produção de clones pcDNA3.1/Zeo(+)-NLS-A3G-HA .............................................. 23
3.2. A expressão dos clones pcDNA3.1/Zeo(+)-NLS-A3G-HA
resulta em proteínas menores que o esperado ........................................................ 23
3.3. A proteína NLS-A3G-HA é mantida no citoplasma da célula .................................... 24
3.4. A localização celular não influencia o tamanho da NLS-A3G-HA ............................. 25
3.5. A Vif do HIV-1 consegue induzir a degradação da NLS-A3G-HA ............................. 25
3.6. A proteína NLS-A3G-HA é encapsidada nos novos viriões ...................................... 26
3.7. A infecção viral é inibida pela NLS-A3G ................................................................... 27
4. Discussão dos resultados e Conclusões .................................................................... 28
5. Referências Bibliográficas ............................................................................................ 30
6. Anexos ........................................................................................................................... xiv

vii
Índice de Figuras
Figura 1 | Organização do genoma do HIV-1 ......................................................................... 2
Figura 2 | Estrutura do virião do HIV-1 .................................................................................. 3
Figura 3 | Ciclo replicativo do HIV-1 ...................................................................................... 3
Figura 4 | A APOBEC3G causa a hipermutação do cDNA viral ............................................. 5
Figura 5 | Motivos-chave na proteína Vif ............................................................................... 6
Figura 6 | Degradação da APOBEC3G induzida pela Vif ....................................................... 7
Figura 7 | Os dois domínios catalíticos da APOBEC3G ......................................................... 8
Figura 8 | Obtenção de quatro clones positivos ................................................................... 23
Figura 9 | Expressão de NLS-A3G-HA em células HEK 293T ............................................. 24
Figura 10 | Localização citoplasmática de NLS-A3G-HA ..................................................... 24
Figura 11 | Expressão de NLS-A3G-HA in vitro ................................................................... 25
Figura 12 | Degradação de NLS-A3G-HA induzida pela proteína viral Vif ............................ 26
Figura 13 | Encapsidação de NLS-A3G-HA nos viriões produzidos ..................................... 26
Figura 14 | Inibição da infecção por NLS-A3G-HA ............................................................... 27
Figura 15 | Mapas genómicos de plasmídios utilizados ................................................. xv, xvi
Figura 16 | Representação esquemática da construção efectuada
para produzir os clones pcDNA3.1/Zeo(+)-NLS-A3G-HA ................................... xvi

viii
Índice de Tabelas
Tabela 1 | Composição da mistura e condições da reacção de PCR .................................. xiv
Tabela 2 | Representação esquemática das sequências de reconhecimento
das enzimas NotI e XhoI .................................................................................... xiv
Tabela 3 | Anticorpos utilizados na detecção de proteínas por Western blot ...................... xiv
Tabela 4 | Constituição das soluções utilizadas ..................................................................xvii

ix
Símbolos e Abreviaturas
Unidades e Símbolos
pg Picograma(s)
µg Micrograma(s)
mg Miligrama(s)
µL Microlitro(s)
mL Mililitro(s)
mm Milímetro(s)
M Molar
mM Milimolar
U Unidade enzimática
pb Pares de bases
h Hora(s)
min Minuto(s)
seg Segundo(s)
kDa Kilodalton(s)
mA Miliamper(es)
mm Milímetro(s)
V Volt(s)
Ω Ohm(s)
ºC Graus Celsius
t.a. Temperatura ambiente
rpm Rotações por minuto
x g Unidade de força centrífuga relativa
Reagentes e Abreviaturas
APOBEC3G (A3G) Apolipoprotein B editing catalitic polypeptide 3G
Abs Absorvância
DMEM Dulbecco’s Modified Eagle’s Medium
PBS Phosphate Buffered Saline
TBS-T Tris-Buffered Saline-Tween 20
EDTA Ácido etilenodiaminotetra-acético
SOB Super Optimal Broth
LB Luria-Bertani Broth

x
HRP Horseradish peroxidase, Peroxidase de rábano selvagem
TE Tris-EDTA
BSA Bovine serum albumin, albumina de soro bovino
HBS HEPES Buffered Saline
HEPES Ácido 4-(2-hidroxietil)-1-Piperazinaetanosulfónico
m.o.i. Multiplicidade de infecção
SDS-PAGE Electroforese em gel de poliacrilamida com dodecil sulfato de sódio
PMSF PhenylMethaneSulphonylFluoride
CA Cápside
CCR5 Chemokine (C-C motif) Receptor 5
CD4 Cluster of Differentiation 4
CXCR4 Chemokine (C-X-C motif) Receptor 4
DNA Deoxyribonucleic acid, ácido desoxirribonucleico
RNA Ribonucleic acid, ácido ribonucleico
cDNA DNA complementar
E. coli Escherichia coli
Env Poliproteina do envelope
Gag Group-specific antigen polyprotein
ART Anti-retroviral therapy
HAART Highly active anti-retroviral therapy
HIV-1 Human Immunodeficiency Virus type 1
HA Hemaglutinina
HIV-2 Human Immunodeficiency Virus type 2
IP Imunoprecipitação
IN Integrase
LTR Long terminal repeat, repetição terminal longa
MA Matriz
mRNA RNA mensageiro
NC Nucleocápside
Nef Negative factor, factor negativo
ORF Open reading frame, grelha de leitura aberta
Pol Polimerase
PR Protease
Rev Reguladora da expressão do virião
SIDA Síndrome da Imunodeficiência Adquirida
SU Glicoproteína de superfície
Tat Trans-activator of transcription

xi
TAE Tampão de Tris-acetato EDTA
TM Transmembranar
Vif Viral infectivity factor, factor de infecciosidade viral
Vpr Viral protein R
Vpu Viral protein U
ELISA Enzyme-linked immunosorbent assay
dNTP Trifosfato de desoxiribonucleótido
PCR Polymerase chain reaction, reacção de polimerização em cadeia
VSV-G Glicoproteína do Vírus da Estomatite Vesicular (Vesicular Stomatitis
Virus)
GAPDH Glyceraldehyde 3-Phosphate Dehydrogenase
PARP Poly [ADP-ribose] polymerase 1
CPRG Chorophenol red-β-D-galactopyranoside

xii
Resumo
A desaminase de citidinas APOBEC3G (A3G) (apolipoprotein B-editing catalitic
polypeptide 3G), confere uma importante estratégia anti-viral às células humanas, a qual é
neutralizada pelo factor de infecciosidade viral (Vif, viral infectivity factor) do Vírus da
Imunodeficiência Humana (HIV, Human Immunodeficiency Virus). Esta enzima é o
componente central desta defesa intrínseca de células do sistema imunitário, como os
linfócitos T e os macrófagos, ao promover a hipermutação do genoma do vírus mutado em
vif, impedindo uma infecção produtiva destas células-alvo. Em contra-partida, a selecção
deste gene durante a evolução do HIV-1 permitiu que uma proteína viral conseguisse induzir
de forma eficiente a degradação da A3G, através da via-dependente de proteossomas
celulares.
Neste trabalho, a interacção que ocorre entre a proteína viral e a enzima celular foi
investigada, nomeadamente através de uma estratégia que inibe esta ligação e que
pretende devolver a capacidade inibidora da infecção à desaminase. Através da adição de
um sinal de localização nuclear (NLS, nuclear localization signal) à extremidade N-terminal
da A3G, foi tentada a separação física destas duas proteínas em diferentes sub-localizações
celulares. A proteína Vif foi aferida quanto à capacidade de induzir a degradação da proteína
construída, e foram testadas as capacidades da NLS-A3G de incorporação nas partículas
virais produzidas nas células infectadas e de inibição da replicação viral nas células-alvo. As
diferenças detectadas na sensibilidade à acção da Vif e a total inibição da infecção que
ocorre na ausência da proteína viral, poderão ser bastante promissoras na descoberta de
novas regiões na estrutura de ambas proteínas, ou até mesmo na revelação de novos
mecanismos de evasão da desaminase celular à acção da proteína viral. Poderá, assim,
atingir-se uma maior compreensão da adaptação evolutiva deste vírus ao sistema imunitário
do Homem, uma adaptação muito eficiente que culmina no desenvolvimento da Síndrome
da Imunodeficiência Adquirida (SIDA).
Palavras-chave: HIV-1, Vif, APOBEC3G, NLS, domínio catalítico.

xiii
Abstract
Cytidine deaminase APOBEC3G (A3G) (apolipoprotein B-editing catalitic polypeptide 3G),
provides an important anti-viral strategy to human cells, which is neutralized by the viral
infectivity factor (Vif) from the Human Immunodeficiency Virus (HIV). This enzyme is the
main component of this intrinsic defense of immune system cells, as T lymphocytes and
macrofages, by promoting the hypermutation of the viral genome with mutation on vif,
preventing a productive infection of these target cells. Nevertheless, the selection of this
gene during HIV-1 evolution allowed that a viral protein could efficiently induce A3G
degradation, through the cellular proteasomes-dependent pathway.
In this work, the interaction that occurs between the viral protein and the cellular enzyme
was investigated, namely by a strategy that inhibits this connection and that acts on returning
the infection inhibitory ability to the deaminase. Through the addition of a nuclear localization
signal (NLS) to the N-terminal end of A3G, the physical separation of this two proteins in
different cellular sub-locations was attempted. Vif protein was evaluated as far as its ability to
induce de degradation of the designed protein, and the A3G capabilities of incorporation in
the viral particles produced in the infected cells and viral replication inhibition in the target
cells were tested. The detected differences in sensitivity to Vif’s action and the complete
inhibition of infection that occur in the absence of the viral protein, may be very promising in
the discovery of new regions in the structure of both proteins, or even in the unveiling of new
mechanisms of cellular deaminase evasion to the viral protein action. In this way, a better
comprehension of the evolutional adaptation of this virus to the Human immune system may
be achieved, a very efficient adaptation that ends up in the development of the Acquired
Immunodeficiency Syndrome (AIDS).
Key-words: HIV-1, Vif, APOBEC3G, NLS, catalytic domain.

1
1. Introdução
1.1. O HIV e a SIDA
1.1.1 Etiologia e Patogenia
Os Vírus da Imunodeficiência Humana tipo-1 e tipo-2 (HIV-1 e HIV-2) são os agentes
etiológicos actualmente conhecidos de SIDA. O HIV-1, principal responsável pela epidemia
mundial, foi isolado em 1983(1), enquanto o HIV-2 apenas foi descoberto três anos mais
tarde(2). Ambos derivaram de Vírus da Imunodeficiência Símia (SIV, Simian
Immunodeficiency Virus) que infectam chimpazés e macacos fuliginosos, respectivamente.
Apesar das semelhanças morfológicas e biológicas, estes diferem significativamente em
alguns componentes antigénicos e moleculares, o que faz com que o HIV-2 apresente
períodos de latência consideravelmente mais longos e seja menos virulento, tornando-o
responsável por epidemias mais localizadas, principalmente em certas regiões da África
Ocidental(2).
Estes vírus infectam primariamente linfócitos T CD4+ e macrófagos por estes
apresentarem na sua superfície um receptor específico (CD4) e os correceptores CCR5 e
CXCR4(3); contudo, outras células do sistema imunitário como os linfócitos B e as células
dentríticas também podem expressar estes receptores, tornando-se susceptíveis à infecção
pelo HIV. Como consequência, o sistema imunitário é destruído progressivamente, levando
ao desenvolvimento da síndrome que surge associada a múltiplas infecções oportunistas,
complicações neurológicas e neoplasias(4).
1.1.2. O impacto demográfico
De acordo com os últimos dados do Programa das Nações Unidas para o
HIV/SIDA/Organização Mundial de Saúde referentes ao ano de 2007, existem actualmente
33,2 milhões de pessoas infectadas com HIV no mundo, das quais mais de 30 milhões
vivem em países de desenvolvimento. Apesar da terapia antiretroviral (ART, antiretroviral
therapy) actual ser capaz de atrasar o aparecimento dos sintomas da SIDA para mais de 15
anos após a infecção, apenas 3 milhões de pessoas em 2007 estavam a receber
tratamento, correspondendo a 31% do total de infectados com necessidade imediata desta
terapia. A somar registaram-se só nesse ano 2,5 milhões de novos casos e 2,1 milhões de
mortes, estando estas incluídas num total de 25 milhões de pessoas que já morreram devido
a esta doença contabilizados deste 1981.
Particularmente em países em desenvolvimento, o HIV/SIDA continua a ser um dos
maiores problemas mundiais em saúde pública. Esta crescente pandemia mantém a

Intr
odução
2
exigência do estudo aprofundado deste vírus, dos seus mecanismos e interacções com os
componentes das células, para alcançar terapêuticas mais eficazes a nível global.
1.2. Cataracterização do HIV-1
1.2.1. Taxonomia
O HIV-1 pertence à família Retroviridae, a qual engloba vírus cujo material genético é
ácido ribonucleico (RNA, ribonucleic acid) de cadeia simples positiva não segmentada. Tal
como o nome indica, os vírus desta família são caracterizados por realizarem a transcrição
reversa do seu RNA genómico em ácido desoxirribonucleico (DNA, deoxyribonucleic acid)
de cadeia dupla, processo desempenhado por uma enzima característica do seu proteoma,
a transcriptase reversa(5).
Este vírus está incluído ainda no género Lentivirus, que significa vírus “lentos”, uma vez
que estes vírus são caracterizados por longos períodos de latência. Estes distinguem-se dos
restantes retrovírus porque possuem cápside em forma de bala(6) e genomas complexos
com um grande número de proteínas acessórias que aumentam fortemente a eficiência da
infecção(5). Para além disso, possuem um invólucro formado por uma bicamada lipídica que
é derivada da membrana da célula hospedeira (Figura 1).
1.2.2. Genoma e Estrutura
O genoma do HIV-1 é formado por duas moléculas de RNA, cada uma com cerca de 9
kb. Estas são flanqueadas por repetições terminais longas (LTR, long terminal repeats),
sequências vitais para a integração e transcrição do genoma viral, e possuem 9 grelhas de
leitura abertas (ORF, open reading frame) que originam 15 proteínas distintas(7).
Três ORFs codificam as poliproteínas precursoras Gag, Pol e Env (características de
todos os retrovírus), as quais são posteriormente proteolizadas originando 9 proteínas. As
poliproteínas estruturais Gag e Env constituem, respectivamente, o núcleo e o invólucro
externo do virião, sendo quatro as proteínas Gag: matriz (MA, p17), cápside (CA, p24),
nucleocápside (NC, p7) e p6; e duas as proteínas Env: superfície (SU, gp120) e
transmembranar (TM, gp41). O virião de forma esférica (com cerca de 100nm de diâmetro) é
revestido por um invólucro lipídico proveniente da membrana da célula produtora, no qual
estão inseridos inúmeros complexos formados por trímeros de gp120 e gp41. A poliproteína
Figura 1 | Organização do genoma do HIV-1. As poliproteínas Gag, Pol e Env, são precursoras das proteínas do núcleo, do
invólucro e das enzimas virais. Adicionalmente, 6 ORF originam as proteínas acessórias que regulam a expressão dos genes
virais e que participam no processo de encapsidação, aumentando fortemente a eficiência da infecção.

Intr
odução
3
Pol origina três enzimas: protease (PR), transcriptase
reversa (RT) e integrase (IN), as quais fornecem as
funções enzimáticas essenciais e são também
encapsidadas na partícula viral (Figuras 1 e 2)(3,6,7).
As seis restantes ORFs originam pequenos transcritos
que sofreram splicing. Estes codificam duas proteínas
reguladoras de genes Tat (trans-activadora) e Rev
(reguladora da expressão do virião) que não constituem a
partícula, e quatro proteínas acessórias: Nef (factor
negativo), Vif (factor de infecciosidade viral) e Vpr
(proteína viral R), que são encapsidadas, e Vpu (proteína
viral U) que assiste na formação do virião(3,5).
1.2.3. Ciclo de replicação
A fase precoce do ciclo de replicação inicia-se quando uma partícula infecciosa encontra
uma célula-alvo que apresenta na sua membrana o receptor CD4, e este se liga à
glicoproteína de superfície do vírus, gp120 (SU). Esta sofre alterações conformacionais que
permitem o reconhecimento dos co-receptores CCR5 e CXCR4, possibilitando a fusão do
invólucro viral com a membrana celular e consequentemente a entrada de todas as
moléculas encapsidadas na partícula no citoplasma da célula(3). Aí, o RNA genómico viral
em cadeia simples é convertido pela transcriptase reversa (com o auxílio da proteína da
nucleocápside) num intermediário de DNA de cadeia dupla, o provírus, que é então
transportado para o núcleo e integrado no genoma da célula, por acção da integrase (Figura
3)(5,6). Na fase tardia do ciclo, estes genes de DNA são transcritos e traduzidos pela
maquinaria da célula hospedeira em
proteínas virais. As proteínas
estruturais são transportadas para a
membrana plasmática onde se
acumulam; a poliproteína Gag liga-se
ao RNA genómico do vírus e então
interactua com a membrana plasmática,
a qual é forçada a se projectar e
eventualmente a se separar, formando-
se uma partícula (cujo invólucro lipídico
é proveniente da membrana da célula)
que é libertada para o exterior da célula
Figura 2 | Estrutura do virião do HIV-1.
Na partícula viral são encapsidadas duas
cópias do genoma que não sofreram
slicing, que se associam a milhares de
proteínas NC para formar um complexo
ribonucleoproteico estável. São ainda
encapsidadas as proteínas estruturais
Gag e Env, as enzimas virais (Pol) e as
três proteínas acessórias (não
representadas) Nef, Vif e Vpr.
Figura 3 | Ciclo replicativo do HIV-1. Após fusão do virião com a
membrana da célula, o RNA viral é transcrito em cDNA de cadeia
dupla, o qual se integra no genoma da célula por acção da
integrase, completando-se a fase precoce do ciclo. Na fase tardia as
proteínas virais, sintetizadas pela maquinaria celular, acumulam-se
próximo da membrana, juntamente com o RNA viral, e o novo virião
forma uma gémula, acabando por sair da célula.
MA
RT
PR
SU
Invólucro
lipídico
CA
NC
RNA
IN
TM
Núcleo Célula produtora
Fusão
Gemulação
Integração

Intr
odução
4
infectada (Figura 3)(6,8). A formação do virião infeccioso é completada já fora da célula, ao
ocorrer a maturação da partícula que consiste no processamento proteolítico da Gag e da
Gag-Pol pela transcriptase reversa, levando à formação do núcleo e activação das enzimas
virais(3,5).
1.3. A proteína viral Vif e o seu alvo celular
1.3.1. A importância da Vif
A proteína Vif, codificada pelo genoma do HIV-1 e expressa na fase tardia do ciclo de
replicação por um mecanismo dependente de Rev, é uma pequena (23 kDa) proteína
acessória básica formada por 192 aminoácidos, que é incorporada no virião (7-100
moléculas por cada partícula) através da interacção como RNA genómico viral(7,9). Na célula
localiza-se predominantemente no citoplasma, mas também é encontrada no núcleo(10) e
associada a membranas celulares. O gene vif, inicialmente designado sor, foi descoberto
1987 ao ser demonstrado que o vírus mutado HIV-1(Δvif) (com o gene vif delectado),
apresentava uma perda da infecciosidade viral de 100 a 1000 vezes, em relação ao vírus
selvagem, embora sem diminuição da produção viral(11). Esta proteína é essencial para a
replicação do vírus em células humanas ditas não-permissivas (como linfócitos T CD4+ e
macrófagos, que são as principais células-alvo do HIV, e várias linhas celulares de células T
leucémicas), ou seja, células que quando infectadas pelo vírus mutado HIV-1(Δvif) (com o
gene vif delectado), produzem novas partículas virais não infecciosas(8,12,13). De facto, a Vif é
vital para neutralizar uma potente via anti-viral que existe neste tipo de células, uma vez a
replicação das partículas virais produzidas na ausência de Vif é inactivada eficientemente
por esta via inata. Em outro tipo de células, designadas de permissivas (como as linhas
celulares HeLa e 293T), mesmo o vírus mutado é capaz de replicar de forma eficaz(12,14,15).
Em 2002, Sheehy e os seus colaboradores descobriram que uma proteína celular é o
alvo da Vif(16), a subunidade 3G da desaminase do mRNA da apolipoproteína B -
APOBEC3G (A3G) (Apolipoprotein B-editing catalitic polypeptide 3G) inicialmente conhecida
como CEM-15(9,15,17). Esta enzima é um membro da família de desaminases de citidinas
(enzimas que modificam/editam ácidos nucleicos), e o seu nome foi-lhe atribuído pela
homologia considerável com a primeira enzima desta família (APOBEC1)(12). Ao se
expressar A3G em células permissivas, estas adquirem um fenótipo não-permissivo, o que
revelou/desvendou que esta desaminase é o factor anti-viral cuja expressão natural está
restrita às células não-permissivas, e que permite inactivar o HIV-1(Δvif). Portanto, a
proteína viral Vif é crucial para proteger o vírus da actividade anti-viral da A3G, permitindo
assim que o vírus replique em células que expressam este factor de restrição(12,13).

Intr
odução
5
Na ausência da Vif, a A3G presente
no citoplasma da célula produtora é
encapsidada (3 a 11 moléculas) nos
viriões de HIV-1(Δvif) em formação ao
ligar-se à porção N-terminal da região
da nucleocápside da poliproteína Gag,
bem como ao RNA viral, no local da
membrana celular onde ocorre a
gemulação das partículas virais(15,18).
Como resultado da fusão viral, é
introduzida no citoplasma da próxima
célula alvo (Figura 4), e ao ocorrer a
transcrição reversa de genoma de
RNA do vírus, esta enzima vai catalizar
a excessiva desaminação de
desoxicitidinas (dC) a desoxiuridinas
(dU) na cadeia negativa (-) do DNA
complementar (cDNA) formado,
preferencialmente em sequências 5’-
CC CU(8,14,15,19). A cadeia negativa é
o alvo da desaminação uma vez que a A3G apenas consegue utilizar moldes em cadeia
simples de DNA, ao passo que a cadeia positiva existe quase totalmente como DNA de
cadeia dupla(13).
O DNA viral contendo uridinas (U) pode ser reconhecido e degradado pela acção
combinada de duas enzimas de reparação do DNA, a glicosilase de uracil-DNA (que remove
os uracilos originando locais abásicos) e a endonuclease apurínica-apirimidínica (que cliva
os locais abásicos), bloqueando assim a replicação viral(8,9,15,19,20,21). Porém, um número
reduzido de cadeias negativas pode resistir à degradação e servir de molde para formar a
cadeia positiva (+) do cDNA. Nesta, as mutações ficam registadas como transições de
guanosina (G) para adenosina (A) em mais de 10% de todos os resíduos G(14,15), e este
provírus pode integrar-se no genoma da célula hospedeira, embora as sequências
geneticamente comprometidas não darão origem a uma progenia de viriões infecciosos. A
A3G pode ainda inibir a replicação de uma forma independente da desaminação quando
ligada ao RNA viral, ao competir fisicamente com o movimento da transcriptase reversa na
cadeia molde de RNA, resultando numa diminuição inicial transcrição(22). Contudo, esta
inibição é frequentemente incompleta e ocorre a produção da cadeia negativa do cDNA que
é então alvo da acção da A3G dependente da desaminação.
Núcleo Célula produtora Proteossoma 26S
Ubiquitinação e degradação Incorporação
Gemulação
Fusão
Degradação?
Integração
Hipermutação C/G T/A
Degradação?
Célula alvo
Figura 4 | A APOBEC3G causa a hipermutação do cDNA viral. A
desaminase é encapsidada nos novos viriões, actuando na cadeia
negativa (-) do cDNA durante a transcrição reversa. O DNA viral
hipermutado pode ser degradado por enzimas de reparação ou pode
ser integrado no genoma da célula. A proteína viral Vif liga-se à
APOBEC3G e induz a sua poli-ubiquitinação, de forma a marcá-la
para degradação pelo proteossoma 26S.

Intr
odução
6
Assim explica-se como é possível que células não permissivas possam produzir níveis
normais de viriões, mas que estes não sejam capazes de replicar de forma eficiente nas
próximas células-alvo. Apesar de tudo, a desaminação das citidinas pode ser construtiva em
vez de destrutiva, e pode ajudar as sequências do HIV-1 a evoluir, contribuindo
possivelmente para o surgimento de variantes com vantagens selectivas que lhes permitam
escapar a respostas imunes adaptativas ou a adquirir resistência a fármacos anti-virais(13).
1.3.2. A interacção Vif-A3G
Sabe-se então que, de forma a se protegerem deste mecanismo de defesa das células,
todos os lentivírus (à excepção do vírus da anemia infecciosa equina) desenvolveram os
genes vif, cuja função primária é prevenir a encapsidação da A3G nos viriões produzidos,
impedindo-a de realizar a sua função anti-viral de desaminase(9). Para tal, a Vif liga-se
directamente à A3G e diminui a sua estabilidade ao induzir a sua degradação por uma via
dependente de proteossomas, eliminando-a das células produtoras(8,12,13,15,23). Assim, os
níveis celulares de A3G são reduzidos, prevenindo-se desta forma a incorporação da
desaminase nas novas partículas virais e permitindo a preservação da infecciosidade
viral(8,20,24). A expressão de Vif pode então resultar numa depleção da A3G das células T
infectadas pelo HIV-1 enquanto os níveis de mRNA da A3G permanecem inalterados(13).
Não obstante, a ligação da Vif à A3G não é suficiente para neu tralizar o fe nótipo anti-
viral(12). A porção N-terminal da Vif (aminoácidos 14-17 e 40-44) interactua com a
extremidade N-terminal na A3G (aminoácidos 54-124 e 128)(9,15); contudo, outros dois
domínios da Vif são fundamentais para que ocorra a degradação da desaminase(12). Um
deles é um motivo muito conservado perto da extremidade C-terminal, designado motivo
SLQ (aminoácidos 144-149), cuja sequência SLQ(Y/F)LA (seguido de 4 a.a. hidrofóbicos) é
semelhante a um motivo conservado na caixa BC das proteínas supressoras de sinalização
de citocinas (SOCS) e intervém na ligação da Vif à elongina C, um componente do
complexo da ligase de ubiquitina E3(13,17,25). É de salientar que este motivo SLQ é a
sequência mais conservada nas proteínas Vif do HIV-1 e de outros lentivírus, o que sugere
que fármacos direccionados para este local podem ser resistentes a mutações do vírus(12). O
outro motivo altamente conservado, também designado de motivo HCCH (aminoácidos 108-
139), é a sequência H-X5-C-X17-18C-X3-5-H, localizada a montante da caixa BC, e que
estabelece a interacção com a
culina-5(15,17). Os dois resíduos
de cisteína que fazem parte do
motivo HCCH são componentes
críticos de um domínio de dedos-
Figura 5 | Motivos-chave na proteína Vif. Os motivos SLQ e HCCH, bem
como os aminoácidos 14-17 e 40-44 são sequências vitais para a interacção
com a A3G e com dois constituintes do complexo da ligase de ubiquitina E3,
a culina-5 e a elongina C.
108H-X5-C-X17-18-C-X3-5-H139
Motivo SLQMotivo HCCH
144SLQ(Y/F)LA149N-terminal C-terminal
14RMRD17 40YHHRY44128 124-54

Intr
odução
7
-de-zinco, sendo a ligação do zinco
vital para a função da Vif pois
permite a correcta conformação da
proteína(9,13). Estas interacções são
essenciais para que a Vif consiga
recrutar o complexo da ligase de
ubiquitina E3, formado pela
elongina C, a elongina B, a culina-5
e a ring-box-1 (Rbx1)(14,17,25). Este
complexo pode assim interagir com
a A3G e encaminhá-la para a
proximidade da enzima conjugadora E2-ubiquitina, induzindo a poli-ubiquitinação da
desaminase – uma modificação pós-traducional que marca a proteína para degradação pelo
proteossoma 26S (Figura 6)(14,15). Para além desta função, existem também evidências que
a Vif pode ajudar a suprimir a tradução do mRNA da A3G, sendo que este efeito combinado
com a aceleração da degradação proteossomal resultam na depleção eficiente dos níveis
intracelulares da desaminase contrapondo a sua actividade anti-viral(20,23,24,26). Têm sido
propostas propriedades funcionais adicionais da Vif que previnem a encapsidação da A3G
nos viriões de uma forma independente da degradação, como a exclusão física da A3G dos
locais de formação das particulas virias, ou inibição da encapsidação da desaminase por
competição da ligação com componentes virais como a nucleocápside na poliproteína Gag
ou no RNA viral(15,23).
1.3.3. Especificidade da espécie
Como praticamente todos os lentivírus têm um gene vif, é fácil extrapolar que a A3G ou
proteínas da mesma família (por exemplo, APOBEC3F (A3F)) podem exercer actividade
contra uma variedade de vírus deste género. Contudo, existe uma elevada especificidade ao
nível da espécie na actividade da A3G, a qual está relacionada com determinantes
genéticos muito específicos. Assim, a A3G humana é marcada para degradação pela Vif do
HIV-1, enquanto que, por exemplo, a A3G do macaco verde africano não é afectada pela Vif
do vírus humano. Esta especificidade também se aplica às proteínas Vif do HIV-1 e do SIV
do macaco verde africano. Contudo, a Vif do SIV do macaco rhesus actua de uma forma
mais alargada e já consegue neutralizar a A3G dos humanos e do macaco verde africano,
bem como a da própria espécie(13,25,27). É de salientar de as Vif do SIV dos chimpazés e do
SIV dos macacos fuliginosos conseguem degradar com sucesso a A3G humana, o que
explica como estes vírus representam os percursores dos HIV-1 e 2, respectivamente(15,23).
Um único resíduo de aminoácido na posição 128 na A3G humana é responsável por esta
Figura 6 | Degradação da APOBEC3G induzida pela Vif. Após
interacção com a proteína viral, a desaminase sofre ubiquitinação pelo
complexo E3 ubiquitina ligase que interage com a Vif.
Ubiquitinação
Degradação
proteossomal

Intr
odução
8
especificidade, sendo que a mudança deste resíduo da sequência do humano (aspartato)
para o da sequência do macaco verde africano (lisina), D128K, é suficiente para reverter a
sensibilidade às Vif do HIV-1 e do SIV do macaco(19,27). Igual função apresentam os
aminoácidos mente, os aminoácidos 14-17 da Vif também conferem a restrição à espécie(15).
Assim, demonstra-se que as defesas celulares que restringem a transmissão dos lentivírus
dos macacos para o Homem podem ser ultrapassadas por polimorfismos subtis dentro de
genes como o vif(8).
1.4. O factor anti-viral A3G
1.4.1. A família APOBEC
No Homem, os genes da enzimas APOBEC estão espalhados por vários cromossomas e
resultam na produção de onze enzimas: AID (activation-induced deaminase), APOBEC1,
APOBEC2, sete enzimas APOBEC3 e ainda APOBEC4(9). Os genes da subfamília
APOBEC3 estão codificados em tandem no cromossoma humano 22q.13.1: APOBEC3A-B-
C-DE-F-G-H. Todas as APOBECs têm os mesmos domínios característicos, um pequeno
domínio em hélice-α seguido por um domínio catalítico (CD), um pequeno péptido linker, e
um domínio pseudocatalítico (PCD); nas A3G e A3F (e também nas APOBEC3B e
APOBEC3DE) estas unidades estão duplicadas formando uma estrutura hélice1-CD1-
linker1-PCD1-hélice2-CD2-linker2-PCD2. Cada domínio catalítico contém o motivo
conservado His-X-Glu-(X)23-28-Pro-Cys-(X)2-4-Cys, no qual os resíduos de histidina e cisteína
se ligam a um ião zinco e o resíduo de glutamato está envolvido no transporte de protões
durante a reacção hidrolítica de desaminação. Estes resíduos são fulcrais para que ocorra,
na presença de água, a remoção de um grupo amina na posição C4 da base azotada 2´-
desoxiciditina convertendo-a a 2’-desoxiuridina (Figura 7a)(13,14,15,27).
Figura 7 | Os dois domínios catalíticos da APOBEC3G. a | Tal como a A3F, a A3B e a A3DE, a A3G possui dois domínios
catalíticos no entanto apenas o domínio N-terminal participa na reacção de desaminase, sendo o domínio C-terminal
cataliticamente inactivo, estando envolvido na ligação ao RNA viral e, portanto, na encapsidação da desaminase no virião. b |
O mecanismo de desaminação. Os resíduos de histidina e cisteína ligam-se a um ião zinco, e na presença de água o resíduo
de glutamato promove o transporte de protões que culmina na libertação de um grupo amina na posição C4 da citosina,
convertendo-a uracilo.
a
b CD1 CD2
C-terminalN-terminal

Intr
odução
9
Apesar da A3G apenas realizar a sua actividade de desaminase em cadeias simples de
DNA(15), esta também se consegue ligar a DNA em cadeia dupla, RNA em cadeia simples e
dupla e a híbridos RNA:DNA. Nas APOBEC que contêm dois domínios de desaminase (CD1
e CD2), geralmente apenas um dos domínios está cataliticamente activo, estando o
segundo domínio envolvido na ligação ao ácido nucleico (neste caso do HIV-1, RNA) e
encapsidação no virião. Desta forma, as propriedades de ligação a ácidos nucleicos da A3G
estão associadas com os seus dois domínios de desaminase: o domínio C-terminal fornece
a actividade catalítica, ligando-se ao DNA em cadeia simples, enquanto o domínio N-
terminal é cataliticamente inactivo mas pode ser vital para a ligação ao RNA viral, permitindo
que a enzima seja encapsidada nas partículas virais na forma de oligodímero(13,28).
A A3G foi a primeira enzima desta família que se descobriu inibir o HIV-1(20). Contudo,
actualmente sabe-se que outros membros da família também são potentes inibidores da
infecção pelo HIV, embora a A3G seja a que tem um maior efeito anti-viral. Depois da A3G,
a A3F é o segundo membro desta família mais eficiente a suprimir infecções de HIV-1(Δvif).
Ambas as enzimas são altamente neutralizadas pela Vif, por mecanismos de degradação
dependentes da ubiquitinação, mas a Vif também é capaz de prevenir a encapsidação de
A3G e A3F através de mecanismos independentes da degradação(9,15,27,28).
1.4.2. Regulação intracelular da desaminase
O estudo intensivo da A3G fez despoletar variadas questões sobre a regulação desta
actividade mutagénica potencialmente promíscua e do seu efeito tóxico no genoma celular.
Estudos de localização subcelular indicam que a A3G é uma proteína citoplasmática que, ao
contrário de outros dois membro s da família APOBEC – AID e APOBEC1 – não é
transportada para o núcleo, sendo fortemente retida no citoplasma por acção de uma
sequência de 16 aminoácidos (113 a 128) próxima do CD1 e que se sobrepõe a alguns
resíduos cruciais à interacção com a poliproteína Gag e com a Vif(29,30).
Contudo, a exclusão nuclear não deve ser o único mecanismo existente na célula para
limitar a actividade A3G, uma vez que os componentes citoplasmáticos e nucleares se
misturam aquando da divisão celular após a disrupção da membrana nuclear. De facto, a
A3G expressa no citoplasma ocorre na forma de complexos ribonucleoproteicos (RNP) de
alta massa molecular (HMM – high-molecular-mass) (5-15kDa) nos quais a actividade de
desaminase da A3G é altamente inibida. A A3G na forma de complexos de baixa massa
molecular (LMM – low-molecular-mass) funciona como um potente factor de restrição ao
impedir a replicação do vírus logo após a entrada na célula. De notar que a acção da A3G
LMM não é antagonizada pela Vif uma vez que esta é encapsidada em pouca quantidade
nos viriões e só é produzida Vif na fase tardia do ciclo de replicação por intermédio da
poliproteína Rev; logo, este efeito antiviral que ocorre mal o vírus entra na célula acontece

Intr
odução
10
independentemente da incorporação da A3G nas partículas virais e é igualmente eficiente
tanto contra o vírus selvagem como contra o HIV-1(Δvif). Mais, este bloqueio pós-entrada
não depende exclusivamente da desaminação(9,15).
1.5. A necessidade de novas terapias: o potencial terapêutico da
interacção Vif-APOBEC
Actualmente, a terapia antiretroviral ART (ou HAART, highly active antiretroviral therapy),
consiste na administração de pelo menos 3 diferentes fármacos anti-retrovirais inibidores da
transcriptase reversa e da protease, no sentido de reprimir a replicação do vírus(4). Contudo,
a ART não representa uma cura, é uma terapêutica dispendiosa que apenas atrasa a
progressão da infecção e deve ser administrada durante toda a vida.
A compreensão dos mecanismos de interacção das proteínas Vif viral e A3G celular
potencia a descoberta de novas terapêuticas anti-retrovirais mais eficazes. A caracterização
de novas moléculas capazes de bloquear a degradação da A3G pela proteína Vif, de inibir a
ligação da proteína viral à desaminase ou a componentes do complexo da ligase de
ubiquitina E3 são alvos a considerar para que o tratamento definitivo da infecção HIV/SIDA
seja uma realidade conseguida num futuro não muito distante.
1.6. Objectivos e Hipótese de trabalho
Esta dissertação visa o objectivo global de criação de uma estratégia de inibição da
interacção da proteína Vif do HIV-1 com o factor anti-viral celular A3G. Neste sentido, a
ligação entre as duas proteínas é impedida pela sua separação física em diferentes sub-
localizações celulares, tendo esta divisão sido conseguida pela formação de uma proteína
A3G contendo um sinal de localização nuclear (NLS, nuclear localization signal). Desta
forma, sendo a Vif uma proteína citoplasmática, a A3G retida no núcleo não poderia ser
marcada para degradação pela Vif nem degradada no proteossomas celulares. Contudo,
para poder desempenhar a sua actividade anti-viral, esta A3G modificada teria de revelar
uma capacidade de se translocar para a proximidade da membrana celular para que posse
ser encapsidada nos novos viriões, todavia evitando o reconhecimento por parte da Vif. Esta
estratégia poderia ser dependente da ligação à porção da nucleocápside da poliproteína
Gag do vírus, a qual dificulta a ligação da Vif, ou por desfasamento temporal da localização
citoplasmática das duas proteínas, causado pelos diferentes tempos de semi-vida
característicos de cada uma. Sob esta hipótese, e através de estudos ex vivo, esta proteína
A3G modificada foi sujeita a análise quanto às seguintes propriedades: capacidade de
evasão à marcação para a degradação provocada pela Vif; encapsidação nos viriões
formados nas células produtoras e inibição da replicação viral nas células-alvo.

11
2. Materiais e Métodos
Os anticorpos utilizados para a detecção de proteínas em Western blot, bem como as
diluições usadas, estão sumarizados na Tabela 3 dos Anexos.
Os mapas dos plasmídios citados encontram-se na Figura 16 dos Anexos.
Na Tabela 4 dos Anexos encontra-se listada a composição de todas as soluções
referidas.
2.1. Clonagem
2.1.1. Extracção de DNA plasmídico em média escala por kit (midiprep)
Antes de mais, para começar o processo de clonagem foi necessário realizar a
extracção de DNA plasmídico clonado em bactérias, nomeadamente do vector de expressão
em células eucariotas pcDNA3.1/Zeo(+) (Invitrogen) que será utilizado na construção dos
clones; e do plasmídio contendo o gene humano da APOBEC3G (com o epitopo HA)
clonado em pcDNA3.1/Zeo(+) (abreviadamente: pcDNA3.1/Zeo(+)-A3G-HA) (AIDS
Research and Reference Reagent Program, Division of AIDS, NIAID, NIH), que irá servir de
DNA molde inicial. Ambos os plasmídios estão clonados numa estirpe de E.coli, TOP10F’
(Invitrogen) (genótipo: F'[lacIqTn10(TetR)] mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15
ΔlacX74 recA1 deoR nupG araD139 Δ(ara-leu)7697 galU galK rpsL (StrR) endA1 λ-) e
devido à possibilidade de contaminação todos os procedimentos de manipulação de culturas
bacterianas foram realizados em condições de esterilidade. Para obter uma cultura crescida
destas bactérias transformadas com os plasmídios, foi realizado um inóculo de uma única
colónia em 250mL (média escala) de meio LB (líquido) com 100μg/mL de ampicilina (USB
Corporation) (resistência conferida pelo vector), com posterior incubação a 37ºC com
agitação a 225rpm, durante a noite. No dia seguinte, toda a cultura é sujeita a centrifugação
a 2400 x g durante 30 minutos e o sobrenadante desprezado, recolhendo-se assim o pellet
de células. A extracção é então efectuada recorrendo ao Jetstar 2.0 Plasmid Purification
MIDI Kit (Genomed), seguindo as instruções fornecidas pelo fabricante e todas as
indicações opcionais que permitem obter melhor resultados em termos de quantidade e
pureza do DNA obtido. O DNA foi eluído em água destilada, desionizada e autoclavada. A
quantificação do DNA foi efectuada através do espectrofotómetro NanoDrop 1000 (Thermo
Scientific), sendo posteriormente armazenado a -20ºC.
Para a preparação de um stock destas bactérias, foram necessários 5mL de uma
cultura crescida, a qual foi sujeita a centrifugação a 3500 x g durante 10 minutos, o
sobrenadante foi desprezado e o pellet ressuspenso em 1mL de uma solução de glicerol
10%(v/v). O stock ficou então armazenado a -80ºC.

Ma
teria
is e
Mé
todos
12
2.1.2. Reacção de polimerização em cadeia (PCR, Polymerase Chain Reaction)
O gene da proteína A3G de Homo sapiens clonado no vector pcDNA3.1/Zeo(+) e
contendo a sequência codificante do epitopo HA a jusante do gene, foi amplificado por PCR
utilizando o seguinte par de oligonucleótidos sintéticos: forward NotI-NLS-A3G-F (5’ – ATA
AGA ATG CGG CCG CTA AAC TAT ATG CCC AAG AAG AAG CGC AAG GTA GGA GGC
AAG CCT CAC TTC AGA AAC ACA GTG – 3’) produzido pela InvitrogenTM, e reverse HA-
XhoI-R (5’ – CCG CTC GAG CGG CTA AGC GTA GTC CGG AAC GTC G – 3’) produzido
pela Thermo Fisher Scientific. Estes oligonucleótidos iniciadores, além da amplificação,
permitem ainda a criação de sequências de reconhecimento de enzimas de restrição e a
introdução de uma sequência (que codifica o sinal de localização nuclear) que será
traduzida em fusão com o gene. Nesta reacção foi utilizada a polimerase de DNA Phusion
(Finnzymes) uma vez que esta enzima apresenta uma forte actividade de revisão de provas
e uma alta fidelidade, permitindo uma amplificação mais eficiente, com tempos de extensão
menores e mais produtiva. A mistura e as condições da reacção estão descritas na Tabela 1
dos Anexos.
Posteriormente, recorreu-se a electroforese em géis de agarose SeaKem LE
(Lonza) a 1% em tampão TAE 1X com 0,5μg/mL de brometo de etídio, para a visualização
do DNA obtido separado de acordo com o tamanho molecular (em pb). Para tal, adicionou-
se à reacção de PCR tampão de aplicação e esta foi aplicada no gel, assim como 5μL do
marcador de pesos moleculares (GeneRuler 1Kb DNA Ladder, 0,5µg/µL - Fermentas). A
electroforese da amostra e do marcador ocorreu em paralelo a uma voltagem de 0,5-
5,0V/cm em tampão TAE, até atingir o nível de resolução desejado, seguindo-se a
visualização do DNA através de luz ultravioleta utilizando o equipamento ChemiDoc (Bio-
Rad) que permitiu fotografar a imagem obtida.
Após confirmação que o produto deste PCR tem um tamanho semelhante ao gene da
A3G (~1200pb), este foi purificado através de electroforese em gel de agarose de
purificação SeaKem GTG (Lonza) a 1%, do volume total da reacção. O gel foi visualizado
num transiluminador, a banda de interesse foi extraída e o DNA purificado pelo Jetquick Gel
Extraction Spin Kit (Genomed) ou pelo Invisorb Spin DNA Extration Kit (Invitek).
Este DNA corresponde ao inserto que irá ser utilizado na clonagem pretendida, e para
simplificação, será designado doravante pela abreviatura NLS-A3G-HA. Após quantificação
no NanoDrop o DNA foi mantido a -20ºC.
2.1.3. Hidrólise enzimática de DNA
Para proceder à clonagem do inserto no vector de expressão utilizado
(pcDNA3.1/Zeo(+)) foi necessário realizar a digestão dos dois DNAs com enzimas de

Ma
teria
is e
Mé
todos
13
restrição, nomeadamente NotI e XhoI (Fermentas), as quais originam extremidades
coesivas, 5’P projectadas. Os locais de clivagem no vector e as sequências de
reconhecimento das enzimas estão esquematizados na Figura 16 e na Tabela 2 dos
Anexos, respectivamente. Em condições de digestão dupla (com as duas enzimas em
simultâneo) foi necessário utilizar 5U de NotI e 10U de XhoI por cada μg de DNA, em
tampão O (Fermentas) para que a actividade de cada enzima atingisse uma percentagem
de 100%. Assim, o volume total da reacção foi calculado tendo em conta o volume e a
quantidade de DNA da amostra de inserto purificado, de seguida foi feito o cálculo dos
volumes das enzimas necessários, adicionou-se o volume do tampão (10X) de modo a que
ficasse a 1X no volume final de reacção; para arredondar o valor final completou-se com
água destilada, desionizada e autoclavada. No caso do vector pcDNA3.1/Zeo(+), a digestão
foi realizada usando DNA em excesso (5μg). As reacções ocorreram durante a noite a 37ºC,
após a qual o volume total de cada reacção foi purificado por electroforese em gel de
agarose de purificação (como descrito em 2.1.2.), o DNA purificado por kit, quantificado no
NanoDrop e armazenado a -20ºC.
2.1.4. Reacção de ligação inserto-vector
Para ligar inserto e vector digeridos, utilizou-se a enzima ligase de DNA T4(New
England Biolabs) e o respectivo tampão (10X). A ligação foi efectuada em 3 diferentes
proporções de quantidade de DNA vector:inserto, preparadas em reacções distintas: 1:0, 1:1
e 1:3. A quantidade de vector utilizada foi de 60ng, sendo que a reacção 1:0 foi realizada
como controlo uma vez que não incluía DNA do inserto. A reacção com a proporção 1:1
continha igualmente 60ng de inserto, e a reacção 1:3 levou 3 vezes mais DNA do inserto
que do vector, ou seja, 180ng. As misturas de cada reacção foram preparadas utilizando o
volume necessário de DNA de vector e de inserto que correspondesse à quantidade
desejada; a estes foi adicionado 1μL de ligase (400000U/mL) e 1μL de tampão (de modo a
ficar 1X no volume final), e o volume das 3 reacções independentes foi acertado com água
destilada, desionizada e autoclavada, de forma a perfazer 10μL de volume final.
2.1.5. Preparação de bactérias electrocompetentes
Para clonar as construções efectuadas em bactérias TOP10F’, foi necessário torná-las
competentes por electroporação. Nesse sentido, foi preparado um pré-inóculo com uma
cultura de TOP10F’ de um stock armazenado a -80ºC em 10mL de meio SOB sem
antibiótico e foi incubado a 37ºC em agitação (225rpm) durante a noite. No dia seguinte,
5mL desse pré-inóculo foram inoculados em 500mL de meio SOB sem antibiótico e
incubados sob as mesmas condições até atingirem uma OD600 entre 0,5 e 0,6; nessa altura
a cultura foi colocada imediatamente em gelo para parar o crescimento. De seguida toda a

Ma
teria
is e
Mé
todos
14
cultura foi centrifugada a 2400 x g por 30 minutos a 4ºC, utilizando copos de centrífuga de
250mL frios, e desprezado o sobrenadante, tendo sido necessário repetir novamente este
passo até esgotar o volume total da cultura. O pellet de células foi ressuspenso em 50mL de
10%(v/v) de glicerol frio, através de movimentos circulares sobre gelo, e centrifugado a 2400
x g por 15 minutos a 4ºC, desprezando-se o sobrenadante de seguida. O último passo foi
repetido novamente, sendo que o pellet foi ressuspenso no glicerol residual, aliquotado em
tubos de 1,5mL com 40μL da suspensão e congelado de imediato através de azoto líquido,
ficando as células armazenadas a -80ºC.
2.1.6. Transformação de bactérias electrocompetentes
A competência destas células foi testada pela adição a um dos tubos preparados de
10pg do plasmídio pUC19 (Invitrogen), seguindo-se o protocolo de electroporação descrito a
seguir. Para além deste controlo, também foram transformadas a posteriori bactérias
competentes com o DNA das 3 reacções de ligação, adicionando para tal 2μL de cada
reacção aos tubos preparados com 40μL de bactéria, previamente descongelada em gelo.
As misturas de bactéria e plasmídio/ligação foram mantidas em gelo durante 15 minutos,
sendo que após esse tempo toda a cultura foi transferida para cuvettes e a electroporação
foi realizada a 1,8V e 200Ω num electroporador (Bio-Rad). A cada cuvette foram adicionados
500μL de meio SOC sem antibiótico e o volume total foi transferido para um tubo de
centrífuga de 15mL, que foi colocado sob incubação a 37ºC com agitação a 225rpm durante
1 hora. Este pré-inóculo, além de permitir o início (arranque) do crescimento da cultura mais
rápido, tem como objectivo permitir a expressão pela maquinaria celular do gene de
resistência à ampicilina existente no plasmídio controlo pUC19 e no vector pcDNA3.1/Zeo(+)
usado nas construções. Desta forma, após esta incubação, o volume total do pré-inóculo foi
plaqueado em meio LB agar (sólido) suplementado com 100μg/mL de ampicilina, e
novamente incubado a 37ºC, mas durante a noite (~16 horas). O aparecimento de colónias
na placa inoculada com bactérias transformadas com pUC19 revela um teste positivo à
competência das células.
2.1.7. Selecção e confirmação de colónias positivas para a clonagem
I) Selecção de colónias e extracção de DNA plasmídico em pequena escala sem kit
(lise alcalina)
Das placas correspondentes às proporções de ligação 1:1 e 1:3, seleccionaram-se no
total 7 colónias isoladas que foram inoculadas em 6mL de meio LB com 100μg/mL de
ampicilina, seguindo-se incubação a 37ºC com agitação a 225rpm durante a noite. Da
cultura obtida, 1,5mL foram utilizados para extracção do DNA plasmídico por lise alcalina

Ma
teria
is e
Mé
todos
15
para confirmar que a construção foi bem sucedida. Este protocolo é realizado em pequena
escala sem recurso a kit, e é utilizado nesta situação em que o DNA obtido não necessita
elevada pureza. Após obtenção do pellet de células por centrifugação a 2300g, 5 minutos, a
4ºC e desprezo do sobrenadante, este foi ressuspenso em 150μL de solução I contendo
100μg/mL de RNase, lisado com 200μL de solução II e agitado por inversão. Após 2 minutos
de incubação a temperatura ambiente, o lisado foi neutralizado por inversão com 150μL de
solução III (previamente arrefecida a -20ºC), incubado a -20ºC por 9 minutos, e centrifugado
durante 10 minutos, a 15700 x g. O sobrenadante obtido foi transferido para outro tubo de
1,5mL, centrifugado à mesma velocidade por 5 minutos, e novamente transferiu-se o
sobrenadante para novo tubo; a este adicionou-se 500μL de isopropanol e centrifugou-se a
15700 x g durante 15 minutos, para precipitar o DNA. O pellet obtido foi lavado com
70%(v/v) de etanol e centrifugado a 15700 x g por 5 minutos, com subsequente aspiração
do etanol e incubação a 37ºC durante 10 minutos para eliminação total do etanol. O DNA já
seco foi resssuspendo em água destilada, desionizada e autoclavada, quantificado no
NanoDrop e conservado a -20ºC.
II) Confirmação de clones positivos por hidrólise enzimática e electroforese em gel
O DNA plasmídico obtido foi então sujeito a hidrólise enzimática, utilizando 1μg de
DNA numa reacção com condições semelhantes às descritas em 2.1.3. O volume total da
reacção foi aplicado em gel de agarose a 1% e a electroforese decorreu como descrita em
2.1.2.; após visualização do gel no ChemiDoc a identificação de construções que
apresentavam duas bandas simultaneamente, correspondentes ao vector (~5000pb) e ao
gene da A3G (~1200kb), confirmou a existência de clones positivos.
III) Extracção de DNA plasmídico em pequena escala por kit (miniprep) e
sequenciação automática de DNA
A partir da cultura obtida no ponto I) acima, utilizaram-se os restantes 4,5mL para
extrair DNA dos clones que se confirmaram ser positivos, desta vez através dos kit Invisorb
Spin Plasmid Mini Two (Invitek) e JETQuick Plasmid Miniprep Spin Kit (Genomed), de forma
a recolher DNA com um elevado nível de pureza, tendo sido efectuadas todas as
recomendações fornecidas pelo fabricante nesse sentido. A eluição do DNA foi feita com
água destilada, desionizada e autoclavada, e após quantificação no NanoDrop foi
conservado a -20ºC. Mais tarde, uma alíquota foi sequenciada (Macrogen) para se poder
confirmar em máxima resolução que a clonagem efectuada foi bem sucedida. Os resultados
obtidos foram analisados recorrendo à aplicação Jellyfish v3.3.1.

Ma
teria
is e
Mé
todos
16
2.2. Expressão proteica
Os procedimentos realizados nos seguintes pontos 2.2.1., 2.2.2. e 2.3.1. foram
realizados sob condições de esterilidade numa câmara de fluxo vertical para assegurar o
correcto manuseamento das culturas celulares.
2.2.1. Linha celular – HEK 293T
Uma vez obtidos clones positivos, estes foram aferidos quanto à expressão da
proteína codificada em células eucariotas. Para esse efeito, recorreu-se à linha celular HEK
293T derivada de células embrionárias de rim humano (AIDS Research and Reference
Reagent Program, Division of AIDS, NIAID, NIH), as quais são células epiteliais que
apresentam crescimento em aderência. Estas células foram mantidas em cultura em meio
DMEM completo: DMEM (Dulbecco's Modified Eagle Medium - Lonza) suplementado com
10%(v/v) de soro fetal bovino (Thermo Scientific), 2mM L-glutamina (Lonza), 100U/mL de
penicilina (Lonza), 100µg/mL de estreptomicina (Lonza), 0,25μg/mL de anfotericina B
(fungizona) (Lonza). As células foram mantidas até 90% de confluência em frascos para
cultura celular de 75cm3 (Orange Scientific), a 37ºC numa estufa com atmosfera de 5% de
CO2, e foram utilizadas em ensaios até atingirem um máximo de 15 passagens.
2.2.2. Transfecção pelo método de fosfato de cálcio
Para iniciar o protocolo de transfecção, recorreu-se a uma cultura de células HEK
293T cultivadas num frasco de 75cm3 com uma confluência entre 70% a 90%. Após
contagem, foram semeadas 4 x 105 células por poço, em placas de 6 poços (34,5mm de
diâmetro dos poços – TPP, ou 34,7mm de diâmetro dos poços – Orange Scientific) e
mantidas na estufa. Cada poço de células semeadas foi usado na transfecção com DNA do
clone respectivo, tendo-se reservado mais dois poços que serviram de controlo positivo e
negativo: um que foi transfectado com o DNA do vector pcDNA3.1/Zeo(+) sem inserto, e
outro com DNA do plasmídio pcDNA3.1/Zeo(+)-A3G-HA.
No dia seguinte, após observação das células ao microscópio (confluência entre 50%
e 70%), foram preparados 2 microtubos por cada amostra de DNA a transfectar, referentes à
mistura A e à mistura B. A mistura A continha o volume correspondente a 5μg de DNA, 25μL
de CaCl2 a 2,5M, e tampão TE 0,1X até perfazer 250μL; a qual foi então adicionada, gota a
gota sob agitação no vortéx, à mistura B composta por 250μL de tampão HBS 2X. Esta
solução obtida, após incubação a temperatura ambiente durante 40 minutos, foi rapidamente
misturada no vórtex (10 segundos) e aplicada gota a gota sobre a camada de células
aderentes ao fundo do poço respectivo, de forma a distribuir homogeneamente o precipitado
de pequenas partículas escuras visíveis ao microscópio. Durante os dois dias seguintes as

Ma
teria
is e
Mé
todos
17
células permaneceram sob incubação na estufa, tendo sido substituído o meio de cultura 24
horas após transfecção por novo meio DMEM completo previamente aquecido a 37ºC.
2.2.3. Extracção e quantificação de proteínas
Ao fim de 48 horas após a transfecção, o meio foi retirado e as células foram
recolhidas por ressuspensão em 1mL de tampão PBS 1X frio, transferidas para um
microtubo de 1,5mL e centrifugadas a 200 x g 5 minutos a 4ºC. Depois de desprezado o
sobrenadante, o pellet foi ressuspenso em 200μL de tampão de lise e passados 30 minutos
de incubação em gelo centrifugou-se a 15700 x g durante 5 minutos. O sobrenadante foi
recolhido para novo tubo de 1,5mL e armazenado a -80ºC.
A quantificação de proteínas dos extractos celulares foi efectuada recorrendo ao
método de Bradford utilizando o reagente Bio-Rad protein assay (Bio-Rad), seguido as
indicações do fabricante, num espectrofotómetro Infinite M200 (Tecan). As amostras foram
normalizadas e o volume correspondente a 50μg de proteína total foi aliquotado para novo
tubo, ao qual se adicionou tampão de aplicação 5X (de forma a ficar 1X no volume final),
seguindo-se a desnaturação durante 5 minutos a 95ºC e armazenamento a -20ºC.
2.2.4. Separação de proteínas por electroforese em gel de poliacrilamida com
dodecil sulfato de sódio (SDS-PAGE)
Para separar as proteínas extraídas das células de acordo com o tamanho molecular
(em kDa), aplicaram-se as amostras desnaturadas e 5μL de marcador de pesos moleculares
(PageRuler Prestained Protein Ladder – Fermentas) num gel de poliacrilamida formado por
uma fase de gel de concentração a 4% seguido de uma fase de gel de resolução a 10%
(com espessura de 1,5mm), conforme as indicações do fabricante (National Diagnostics). A
electroforese das amostras e do marcador decorreu em paralelo, através do sistema Mini-
PROTEAN Tetra cell (Bio-Rad) no qual os mini-géis ficaram verticalmente imersos em
tampão de corrida frio, a uma voltagem constante inicial de 100V e de 120V uma vez as
amostras já se encontrassem no gel de resolução, até ao nível de resolução pretendido.
2.2.5. Western blot
Após a electroforese de separação, as proteínas foram transferidas do gel para uma
membrana de nitrocelulose (Hybond ECL - Amersham Biosciences). A transferência
decorreu a uma amperagem constante de 250mA durante 2 horas, e através do sistema
Mini Trans-Blot Cell (Bio-Rad), onde o gel e a membrana ficaram submersos em tampão de
transferência frio juntamente com um bloco de gel congelado. Depois, a membrana foi
incubada rapidamente em reagente de Ponceau S Solution (Sigma Aldrich) pois este marca

Ma
teria
is e
Mé
todos
18
todas as proteínas de uma forma não-específica e dessa forma foi possível confirmar o
sucesso da separação das proteínas e da transferência. Após lavagem da membrana com
tampão TBS-T (Tween 20 – VWR) para eliminar vestígios de Ponceau, esta ficou a 4ºC
durante a noite a bloquear em solução de 5%(p/v) de leite magro em pó em TBS-T.
No dia seguinte, a membrana foi incubada durante 1 hora a temperatura ambiente com
uma diluição do anticorpo α-HA conjugado com a enzima peroxidase de rábano selvagem
(HRP - horseradish peroxidase), lavada durante 10 minutos com TBS-T e repetindo a
lavagem 4 vezes com novo TBS-T. Depois, a membrana foi incubada com um reagente de
detecção da actividade da HRP (ECL Plus – Amersham Biosciences, ou Immobilon Western
– Millipore) de acordo com as indicações do fabricante, permitindo a detecção rápida de
sinal emitido por quimioluminescência por exposição da membrana a uma película
fotográfica (Hyperfilm ECL - Amersham Biosciences) durante cerca de 15 minutos, sendo
seguidamente revelada e fixada.
A proteína de expressão constitutiva GAPDH (37kDa) foi escolhida como controlo
endógeno ara normalizara a quantidade de células presente em cada amostra. Para a sua
detecção, a membrana ficou a incubar a 4ºC durante a noite com uma solução de anticorpo
primário α-GAPDH diluído, no dia seguinte foi lavada durante 5 x 10 minutos com TBS-T, e
incubada com uma diluição do anticorpo secundário α-ratinho, que se liga ao anticorpo α-
GAPDH. Posteriormente a mais uma série de lavagens (5 x 10 minutos), foi novamente
impressionada uma película fotográfica à membrana, como descrito no parágrafo anterior.
Ao longo do decorrer do trabalho prático, devido os resultados obtidos, surgiu a
necessidade de recorrer a mais técnicas para avaliar a expressão proteica:
2.2.6. Extracção diferencial de proteínas do citosol e do núcleo
Foi realizado todo o procedimento descrito em 2.2.2., com as seguintes alterações: o
número de células semeadas foi de 3 x 106, uma vez que se utilizaram caixas de Petri
(100mm de diâmetro – Orange Scientific) em vez de placas de 6 poços; por esse motivo a
quantidade de DNA utilizada foi de 20μg.
Após 48h pós-transfecção, retirou-se o meio de cultura e as células foram lavadas com
1mL de PBS 1X frio duas vezes, na segunda lavagem as células foram ressuspensas,
recolhidas para um microtubo de 1,5mL e centrifugadas a 100 x g durante 5 minutos a 4ºC,
desprezando-se o sobrenadante. O pellet de células foi ressuspenso em 300μL de tampão
de lise de citosol completo e depois de centrifugado a 3000 x g, 4 minutos a 4ºC, o
sobrenadante contendo as proteínas do citosol foi recolhido para novo tubo e armazenado a
-80ºC. O pellet foi lavado novamente com 300μL de tampão de lise de citosol completo,
centrifugado a 3000 x g, 4 minutos a 4ºC, e o sobrenadante desprezado, para eliminar por
completo as proteínas do citosol. Seguiram-se 3 lavagens do pellet com 1mL de PBS 1X e

Ma
teria
is e
Mé
todos
19
centrifugação a igual velocidade, duração e temperatura. Posteriormente adicionaram-se
30μL de tampão de lise de núcleos completo, mantiveram-se as amostras em gelo durante
20 a 25 minutos e agitaram-se os tubos 3 vezes no vórtex durante período de tempo.
Centrifugaram-se os tubos a 10000 x g durante 10 minutos, a 4ºC, e o sobrenadante
contendo as proteínas do núcleo foi recolhido para novo tubo e armazenado a -80ºC. De
seguida os extractos foram quantificados e as proteínas analizadas por SDS-PAGE e
Western blot como descrito nos pontos 2.2.3., 2.2.4. e 2.2.5. Adicionalmente à proteína
GAPDH detectada conforme descrito em 2.2.5., foi detectada ainda a proteína PARP,
utilizada como controlo da completa separação dos extractos do citosol e do núcleo. A
porção nuclear desta proteína (PARP-1) foi detectada com soluções diluídas do anticorpo
primário α-PARP e do anticorpo secundário α-coelho, incubados de forma semelhante à
descrita para os anticorpos α-GAPDH e α-ratinho, respectivamente.
2.2.7. Transcrição e Tradução in vitro e Imuno-precipitação (IP)
A reacção de transcrição e tradução in vitro foi realizado através do kit TnT® T7 Quick
Coupled Transcription/Translation System (Promega), seguindo as instruções do fabricante.
Em complemento desta reacção, as amostras resultantes, correspondentes a cada
clone e ao controlo positivo, foram sujeitas a uma imunoprecipitação das proteínas NLS-
A3G-HA construídas. Ao volume total da reacção (50µL) foram adicionados 800μL de
tampão de IP frio e as amostras foram misturadas por inversão num girador de tubos,
durante 20 minutos. Então os lisados foram centrifugados a 15700 x g durante 5 minutos, a
4ºC, e o sobredanante transferido para um novo tubo. Uma pequena amostra (50μL) foi
recolhida do lisado, para servir como controlo de aplicação. Para realizar uma pré-limpeza
dos detritos celulares, foram adicionados 10μL de beads de agarose conjugadas com o
anticorpo α-proteína A (n Protein A Sepharose 4 Fast Flow, Immunoprecipitation Starter
Pack - GE Healthcare) e misturados pelo mesmo método durante 20 minutos. Após
centrifugação 1 minuto a 15700 x g a 4ºC, os sobrenadantes foram recolhidos para novos
tubos. Para imunoprecipitar a proteína de interesse, foram adicionados 20μL de matriz de
beads de agarose conjugadas com o anticorpo α-HA(HRP) (anti-HA affinity matrix - Roche) e
as amostras foram misturadas por inversão no girador durante a noite. No dia seguinte, após
centrifugação 1 minuto a 15700 x g, o sobrenadante foi recolhido para novos tubos,
representando a amostra que foi imunodelectada. O pellet de beads foi lavado 3 vezes com
800μL de tampão de IP, centrifugado nas condições já descritas e desprezado o
sobrenadante. Finalmente, as beads foram fervidas a 95ºC com tampão de aplicação 5X na
quantidade necessária para ficar a 1X no volume final, e armazenadas a -20ºC para
posteriormente serem analizadas por SDS-PAGE e Western blot, tal como as alíquotas de
controlo de aplicação, como descrito em 2.2.4. e 2.2.5.

Ma
teria
is e
Mé
todos
20
2.3. Análise da função proteica em células eucariotas
2.3.1. Ensaio de degradação dos clones NLS-A3G
O protocolo de co-transfecção pelo método de fostato de cálcio utilizado
corresponde ao protocolo de transfecção já descrito em 2.2.2., apenas com alterações no
que diz respeito ao DNA transfectado. Assim, cada poço plaqueado com células HEK 293T
foi usado na co-transfecção com uma mistura do DNA do respectivo clone e do DNA do
plasmídio com o gene vif clonado em pcDNA3.1/Zeo(+) (abreviadamente: pcDNA3.1/Zeo(+)-
vif). Para além destes poços, reservaram-se mais três poços com as seguintes misturas de
DNA: plasmídios pcDNA3.1/Zeo(+)-A3G-HA e pcDNA3.1/Zeo(+)-vif; plasmídio
pcDNA3.1/Zeo(+)-vif com vector pcDNA3.1/Zeo(+) sem inserto; e plasmídio
pcDNA3.1/Zeo(+)-A3G-HA com vector pcDNA3.1/Zeo(+) sem inserto. A mistura A continha o
volume correspondente a 1μg de DNA dos clones ou do plasmídio pcDNA3.1/Zeo(+)-A3G-
HA, adicionado ao volume correspondente a 4μg de DNA do plasmídio pcDNA3.1/Zeo(+)-vif,
e ainda DNA do vector pcDNA3.1/Zeo(+) nos casos em que foi preciso completar a
quantidade de DNA total para 5μg. As proteínas produzidas foram extraídas e analizadas
por SDS-PAGE e Western blot como descrito em 2.2.3., 2.2.4. e 2.2.5.
2.3.2. Ensaio de infecciosidade do HIV-1 na presença de NLS-A3G
Por questões de segurança, os procedimentos realizados na seguinte alínea II), e os
que envolvem o manuseamento de amostras infectadas até ocorrência de lise descritos
alíneas nas III) e IV) deste ponto, foram realizados num laboratório de contenção biológica
de nível 3 sob condições de esterilidade numa câmara de fluxo vertical, para assegurar
ainda o correcto manuseamento das culturas celulares.
I) Extracção de DNA plasmídico em média escala por kit (midiprep)
Para realizar este ensaio foi necessário primeiro obter o genoma do vírus HIV-1 para
conseguir produzir partículas virais infecciosas. O plasmídio contendo o DNA da estirpe
NL4-3 com o gene vif delectado (pNL4-3Δvif - AIDS Research and Reference Reagent
Program, Division of AIDS, NIAID, NIH) foi utilizado com esse intuito, assim como o
plasmídio pHEF-VSV-G (AIDS Research and Reference Reagent Program, Division of AIDS,
NIAID, NIH) que codifica para a glicoproteína G do vírus da estomatite vesicular, a qual é
incorporada na partícula viral e facilita a fusão do invólucro viral com a célula hospedeira.
Ambos plasmídios encontram-se clonados numa estirpe de E. coli, JM109 (New England
Biolabs) (genótipo: F´traD36 proA+B+ lacIq Δ(lacZ)M15/ Δ(lac-proAB) glnV44 e14– gyrA96
recA1 relA1 endA1 thi hsdR17). A extracção do DNA plasmídico foi realizada em média

Ma
teria
is e
Mé
todos
21
escala por kit como descrito em 2.1.1., assim como a preparação de um stock, com a
excepção que a cultura bacteriana foi incubada a 30ºC e não a 37ºC.
II) Co-transfecção, produção e concentração das partículas virais
Células HEK 293T foram novamente co-transfectadas como descrito em 2.3.1., com
duas diferenças: a cada mistura A foram adicionados os volumes correspondentes a 2μg do
plasmídio pNL4-3Δvif e a 0,4μg do plasmídio pHEF-VSV-G; e as quantidades de DNA
referidas em 2.3.1. foram alteradas para 0,5μg de DNA dos clones/plasmídio
pcDNA3.1/Zeo(+)-A3G-HA, 3μg do DNA do plasmídio pcDNA3.1/Zeo(+)-vif, e a quantidade
de DNA total de 5,9μg foi completada quando necessário com DNA do vector
pcDNA3.1/Zeo(+). Cada co-transfecção foi produzida em dois poços-réplica.
Dois dias após a co-transfecção não foram as células mas sim o meio de cultura que
continha os vírus produzidos que foi recolhido para tubos de centrífuga de 15mL, de forma a
juntar o meio dos poços duplicados. Depois de centrifugado durante 5 minutos a 200 x g o
sobrenadante foi recolhido para novo tubo, de forma a garantir a exclusão de células ou
detritos, e deste foram separados 100μL para um microtubo de 1,5mL que foram
quantificados através de um ensaio imunosorvente ligado a enzima (ELISA, enzyme linked
immuno sorbent assay) (descrito na alínea III) desde ponto), 2mL foram usados para
ultracentrifugar os vírus e o restante foi armazenado durante a noite a 4ºC. Os 2mL
recolhidos para concentrar as partículas virais produzidas foram centrifugados numa
ultracentrífuga Beckman a 50000rpm durante 1 hora a 4ºC, com 500μL de uma solução de
sacarose a 20% como gradiente. Após se ter desprezado o sobrenadante, o pellet de vírus
foi ressuspenso em 30μL de tampão de aplicação 5X, transferido para um microtubo de
1,5mL e analizado por SDS-PAGE e Western blot como descrito em 2.2.4. e 2.2.5.
III) Quantificação de partículas virais por ELISA
As partículas virais presentes na alíquota de sobrenadante foram quantificadas
recorrendo ao HIV-1 p24CA Antigen Capture Assay Kit (AIDS & Cancer Virus Program of the
NAtional Cancer Institute Frederick Cancer Research), através do qual é realizado uma
ELISA contra o antigénio p24. Foram seguidas as instruções do fabricante com as seguintes
excepções: o tampão de lavagem usado foi 0,05%(v/v) de Tween20 em PBS 1X; as
soluções de diluição das amostras e dos anticorpos utilizadas foram 1%(p/v) de albumin de
soro bovino (BSA, bovine serum albumin) em PBS 1X, e 3%(p/v) BSA em tampão PBS 1X,
respectivamente, e não as indicadas; a placa não foi lavada com tampão de lavagem antes
da colocação das amostras como indicado, nem as lavagens posteriores foram realizadas
num aparelho automático, mas sim manualmente com 5 x 200μL de tampão de lavagem e
recorrendo a uma micropipeta multi-canal; as amostras standard não foram aplicadas na

Ma
teria
is e
Mé
todos
22
placa em duplicado, apenas uma vez; as amostras apenas foram lisadas após terem sido
adicionadas à placa, e por esse motivo a incubação subsequente decorreu por um período
de 3 horas. A cada pg de p24 obtida correspondem 10 unidades de transdução (TU,
transduction units).
IV) Linha celular – TZM-bl, infecção e quantificação da infecciosidade viral por CPRG
A linha celular utilizada para testar a infecciosidade das partículas virais produzidas
designa-se TZM-bl, é uma linha de células HeLa derivada de carcinoma cervical (AIDS
Research and Reference Reagent Program, Division of AIDS, NIAID, NIH), que tal como as
HEK 293T são células humanas epiteliais que apresentam crescimento em aderência. Para
além disso, expressam elevados níveis de CD4 e CCR5, o que as torna boas candidatas a
células-alvo para infecção por HIV-1, e permitem uma análise simples e quantitativa do HIV
por expressarem os genes repórteres da luciferase e da β-galactosidase sob o controlo de
uma longa repetição terminal (LTR, long terminal repeat) do vírus. Estas células foram
mantidas em cultura nas mesmas condições que as HEK 293T descritas em 2.2.1.
No dia de recolha dos sobrenadantes (48 horas pós-transfecção) contendo as
partículas virais produzidas, recorreu-se a uma cultura de células TZM-bl cultivadas num
frasco de 75cm3 com uma confluência entre 70% a 90%, e após contagem, semearam-se 1
x 104 células por poço, em placas de 96 poços (6,9mm de diâmetro dos poços – Orange
Scientific) e foram mantidas na estufa. No dia seguinte, como ocorreu duplicação do número
de células em cada poço, estas foram infectadas com igual número (20000) de unidades de
transdução (TU), para que dessa forma o rácio entre partículas virais infecciosas e células,
que se designa por multiplicidade de infecção (m.o.i., multiplicity of infection), fosse igual a 1.
Após a quantificação das partículas virais, o meio de cultura das células plaqueadas
foi substituído pela quantidade calculada de sobrenadante (armazenado a 4ºC) e meio
DMEM completo novo de forma a completar um volume final de 200μL por poço. Durante os
dois dias seguintes as células permaneceram sob incubação na estufa, tendo sido
substituído o meio de cultura 3 horas após infecção por novo meio DMEM completo
previamente aquecido a 37ºC.
Ao fim de 48 horas após a infecção, o meio foi retirado e as células foram lavadas com
200μL de PBS 1X, e de seguida foram adicionados 100μL de tampão de lise do CPRG a
cada poço. A placa foi envolvida em papel de prata e colocada a -80ºC durante 40 minutos,
seguindo-se a incubação a 37ºC até descongelar (cerca de 30 minutos) e repetiu-se a
congelação/descongelação novamente. Então, adicionou-se a cada poço 100μL de uma
diluição de 6mM de CPRG em tampão de lise, e a placa foi novamente incubada a 37ºC
durante 1 hora. A leitura foi feita no espectrofotómetro Infinite M200 a 550nm com 620nm de
referência.

23
3. Resultados
3.1. Produção de clones pcDNA3.1/Zeo(+)-NLS-A3G-HA
A reacção de amplificação por PCR do gene humano de A3G permitiu a adição, a
montante e a jusante deste gene, das sequências codificantes do sinal de localização
nuclear NLS (PKKKRKV) e do epitopo HA (YPYDVPDYA), respectivamente. A sequência
NLS é responsável por encaminhar a proteína traduzida para o compartimento nuclear,
enquanto o epitopo HA, por sua vez, é utilizado para permitir a detecção imunológica da
proteína através de um anticorpo específico. Locais de clivagem para as enzimas de
restrição NotI e XhoI também foram adicionados, respectivamente, a 5’ da sequência de
NLS e a 3’ da sequência de HA (Figura 16 dos Anexos). Após hidrólise enzimática com as
enzimas NotI e XhoI do inserto e do vector de expressão pcDNA3.1/Zeo(+), ligação
vector:inserto e transformação da construção em bactérias electrocompetentes, obtiveram-
se colónias de bactérias TOP10F’ resistentes à marca de selecção (ampicilina) fornecida
pelo vector. Do total obtido, seleccionaram-se sete colónias isoladas e com crescimento
uniforme, extraiu-se o DNA plasmídico por lise alcalina e após hidrólise com as enzimas NotI
e XhoI observou-se o DNA por electroforese em gel de agarose. Este permitiu detectar, em
quatro dos clones escolhidos, a presença de duas bandas distintas, uma com o tamanho
aproximado do vector pcDNA3.1/Zeo(+) (~5000pb) e outra com tamanho correspondente ao
gene humano da A3G (~1200pb). (Figura 8), Estes clones (#2, #4, #5 e #6) foram então
classificados de positivos, por se ter comprovado o sucesso da construção. Mais tarde (após
os ensaios de expressão e de degradação) a análise das
sequências nucleotídicas, obtidas por sequenciação
automática, revelou que o clone #5 apresentava algumas
mutações pontuais em relação ao gene da A3G e que não
se observavam nos outros clones, pelo que este foi
desconsiderado para os ensaios de infecciosidade
realizados posteriormente.
3.2. A expressão dos clones pcDNA3.1/Zeo(+)-NLS-A3G-HA resulta
em proteínas menores que o esperado
O DNA dos quatro clones positivos pcDNA3.1/Zeo(+)-NLS-A3G-HA foi expresso em
células HEK 293T, por transfecção pelo método de fosfato de cálcio, bem como o DNA dos
controlos negativo e positivo (pcDNA3.1/Zeo(+) e pcDNA3.1/Zeo(+)-A3G-HA, respectiva-
mente).Os extractos proteicos totais foram analisados por SDS-PAGE seguida de Western
~ 5000pb
~ 1200pb
Figura 8 | Obtenção de quatro clones
positivos. Visualização por electroforese em
gel de agarose dos clones que apresentam
as bandas correspondentes ao vector
pcDNA3.1/Zeo(+) e ao inserto NLS-A3G-HA.

Resultados
24
blot, e após três réplicas o resultado manteve-se:
as proteínas codificadas pelo clones apresentam
um tamanho menor (menos ~5kDa) que a do
controlo positivo (46kDa) (Figura 9). No entanto,
em alguns dos clones como é o caso do #5, é
possível visualizar uma banda com tamanho
superior muito semelhante ao tamanho da
proteína expressa pelo controlo (ver seta ).
Este resultado poderá demonstrar que as proteínas dos clones sofrem algum tipo de
clivagem, à qual a proteína selvagem não está sujeita; no clone #5 essa clivagem poderá
não ter ocorrido em todas as moléculas de NLS-A3G-HA existentes nas células e por isso é
detectada um pequeno número de moléculas com tamanho semelhante à A3G-HA.
Sabendo que, teoricamente, a diferença entre os clones e o plasmídio pcDNA3.1/Zeo(+)-
A3G-HA, reside apenas na sequência que codifica para a porção NLS, parece claro que
esta sequência seja a responsável pela redução no tamanho das proteínas expressas.
Pensa-se que, por algum motivo não identificado, a sequência-sinal NLS, juntamente com
uma porção da extremidade N-terminal da A3G, são removidas da proteína sintetizada,
possivelmente por mecanismos envolvidos na exclusão do núcleo. Outros ensaios de
expressão foram tentados no sentido de compreender a origem desta diferença de tamanho.
3.3. A proteína NLS-A3G-HA é mantida no citoplasma da célula
Para analisar a capacidade da sequência NLS em exercer a função de encaminhamento
para o núcleo das proteínas construídas, estas foram expressas novamente em células HEK
193T, no entanto, o extracto proteico foi recolhido com separação diferencial das proteínas
do citosol e do núcleo, para se poder analisar qual a sua localização dentro da célula. Nas
duas réplicas deste ensaio, após separação em SDS-PAGE e análise por Western blot, foi
possível verificar que as proteínas produzidas se encontravam exclusivamente no
citoplasma (Figura 10). De notar que neste caso é possível distinguir com clareza em todos
os clones uma forma de NLS-A3G-
HA com um tamanho muito
próximo do da proteína do controlo
positivo, assim como a forma mais
pequena (41kDa) já detectada
anteriormente. A acrescentar, é
detectada uma outra proteína
ainda mais pequena (~36kDa) Figura 10 | Localização citoplasmática de NLS-A3G-HA. Detecção por
Western blot da localização sub-celular das proteínas clonadas em células
HEK 293T, por extracção diferencial de proteínas do núcleo e do citosol.
Figura 9 | Expressão de NLS-A3G-HA em células
HEK 293T. Visualização por Western blot da expressão
das proteínas codificadas pelos quatro clones
pcDNA3.1/Zeo(+)-NLS-A3G-HA.
α-HA
α-GAPDH
46 kDa
37kDa
α-HA 46 kDa
α-PARP
α-GAPDH
116 kDa
37 kDa

Resultados
25
resultante da clivagem de uma porção maior da extremidade N-terminal das proteínas
construídas. Verificou-se assim que a sequência NLS não era capaz de realizar o seu efeito
sinalizador, ou que, após o transporte, a NLS-A3G-HA seria novamente transportada para o
citoplasma por um outro mecanismo, envolvendo possivelmente a clivagem da sequência
NLS e de parte da extremidade N-terminal da A3G.
3.4. A localização celular não influencia o tamanho da NLS-A3G-HA
No sentido de tentar descortinar
o fenómeno que envolve a clivagem
da porção N-terminal das proteínas
construídas, foi realizado um outro
ensaio de expressão. Desta vez, as
construções foram analisadas fora do
contexto celular, através de uma
reacção de transcrição e tradução in
vitro, que utiliza o DNA dos clones
como molde. Após a reacção, o extracto proteico obtido foi purificado através de uma IP
com beads de agarose conjugadas com anticorpo α-HA(HRP). Novamente os extractos
foram analisados por SDS-PAGE e Western blot, sendo que os resultados obtidos não
permitiram tirar novas conclusões: manteve-se a diferença de tamanho entre proteínas
codificadas pelos clones (~41KDa) e a proteína usada como controlo positivo (46KDa)
(Figura 11). Neste ensaio já não foram detectadas bandas superiores com tamanho muito
semelhante ao da A3G-HA, mas verificaram-se novamente as proteínas mais pequenas de
36kDa anteriormente detectadas no ensaio de separação de extractos do núcleo e do
citosol. Surpreendentemente, também foi possível visualizar uma versão truncada da
proteína A3G-HA usada como controlo positivo, com sensivelmente a mesma diferença de
tamanho (~5kDa) verificada nas proteínas NLS-A3G-HA. No entanto, esta proteína de
menor tamanho apenas é visualizada após a IP, o que indica que os seus níveis celulares
são muito baixos. Este resultado, embora corresponda apenas a um ensaio, representa um
forte indício que a clivagem que ocorre nas proteínas construídas não é devida à entrada
e/ou saída do núcleo das proteínas construídas, uma vez que mesmo quando os
compartimentos celulares não estão presentes esta degradação se mantém.
3.5. A Vif do HIV-1 consegue induzir a degradação da NLS-A3G-HÁ
Apesar da redução no tamanho verificado em todas as proteínas codificadas pelos clones
pcDNA3.1/Zeo(+)-NLS-A3G-HA, estas foram testadas no sentido de averiguar se
Figura 11 | Expressão de NLS-A3G-HA in vitro. Visualização por
Western blot das proteínas codificadas pelos clones pcDNA3.1/Zeo(+)-
NLS-A3G-HA, produzidas através de uma reacção de transcrição e
tradução in vitro e purificadas (ou não, no caso dos controlos) por IP.
Imunoprecipitados Controlos
α-HA 46 kDa

Resultados
26
mantinham a capacidade de
interacção com a proteína Vif
do HIV-1, e se consequente-
mente eram degradadas pelos
proteossomas celulares. Para
tal, o DNA dos clones foi co-
transfectado com o DNA do
plasmídio pcDNA3.1/Zeo(+)-vif
em células HEK 293T e os extractos proteicos separados em gel de poliacrilamida. A análise
por Western blot nas duas réplicas deste ensaio permitiu detectar uma diminuição da
quantidade de NLS-A3G-HA na presença da proteína Vif, em comparação com a proteína
expressa na ausência de Vif (Figura 12). Mais uma vez, são visíveis os dois tipos de
proteínas truncadas que sofreram clivagem por um processo ainda não desvendado. É de
salientar que as proteínas com cerca de 41kDa são altamente sensíveis à degradação,
principalmente no caso dos clones #5 e #6, em que a expressão de NLS-A3G-HA é
significativamente diminuída para valores muito baixos ou indetectáveis. As proteínas com
tamanho aproximado de 36kDa, por seu lado, aparentam ser ligeiramente menos sensíveis
à Vif. Estes resultados demonstram que estas proteínas conseguiram interagir de forma
eficiente com a proteína viral, a qual induziu a sua destruição. Curiosamente, neste ensaio a
versão truncada da proteína produzida pelo controlo positivo apenas surge na presença de
Vif, e não quando o controlo positivo é transfectado isoladamente, o que também
permanece por justificar.
3.6. A proteína NLS-A3G-HA é encapsidada nos novos viriões
Após se demonstrar que a proteína NLS-A3G-HA é sensível à indução para degradação
pela proteína Vif, surgiu a necessidade de averiguar se, tal como a desaminase selvagem, a
proteína construída é ou não encapsidada nas novas partículas virais de NL4-3(Δvif)
produzidas em células HEK 293T. A produção dos viriões foi conseguida pela co-
transfecção de pNL4-3(Δvif) (e de pHEF-VSV-G) com o DNA dos clones que codificam as
proteínas que codificam as proteínas
NLS-A3G-HA e do DNA do plasmídio
pcDNA3.1/Zeo(+)-vif, assim como de
vários controlos. Após concentração e
lise dos viriões produzidos, os
extractos totais foram separados por
electroforese em SDS-PAGE, e por
α-HA 46 kDa
Figura 13 | Encapsidação de NLS-A3G-HA nos viriões produzidos.
Demonstração por Western blot da encapsidação das proteínas
codificadas pelos clones nas partículas virais produzidas em células
HEK 293T, tanto na ausência como na presença de Vif.
Figura 12 | Degradação de NLS-A3G-HA induzida pela proteína viral Vif.
Detecção por Western blot da degradação das proteínas clonadas na presença
de Vif, em células HEK 293T.
α-HA 46 kDa
α-GAPDH 37 kDa
pcDNA3.1/Zeo(+)-vif – – – – + + + + + + – –
pNL4-3(Δvif) + + + + + + + + + –
pcDNA3.1/Zeo(+)-vif – – – + + + + + – –

Resultados
27
0
0,5
1
1,5
2
2,5
3
Ab
s (
55
0n
m)
Figura 14 | Inibição da infecção por NLS-A3G-HA. Níveis de
infecciosidade viral em células Tzm-BL por viriões NL4-3(Δvif), produzidos
em células HEK 293T. Valores representados correspondem a três
réplicas simultâneas.
Western blot foi possível visualizar que a NLS-A3G-HA é encapsidada tanto na ausência
como na presença de Vif (Figura 13). Aqui apenas a versão truncada de NLS-A3G-HA com
41kDa é observada nas amostras correspondentes aos clones. Porém, em relação ao clone
#6 é notória uma diminuição da quantidade de desaminase encapsidada quando a Vif está
presente, mas esta diferença pode não ser significativa uma vez que a quantidade de viriões
em cada amostra não foi normalizada e este ensaio apenas foi realizado uma vez. Mesmo
assim, é de notar que este clone foi o que demonstrou uma maior sensibilidade à
degradação induzida pela Vif (Figura 12), embora a sequenciação automática tenha
excluído qualquer diferença na sequência dos três clones #2, #4 e #6, pelo que esta
variação considerável na sensibilidade à inibição pela Vif continua por esclarecer.
3.7. A infecção viral é inibida pela NLS-A3G
Por último, foi aferida a actividade
anti-viral da proteína NLS-A3G-HA.
Após a produção de viriões de NL4-
3(Δvif), estes foram utilizados para
infectar células Tzm-BL e a
infecciosidade viral foi medida
através da degradação do substrato
CPRG pela enzima β-galactosidase,
expressa por esta linha celular na
ocorrência de produção viral. Os
resultados obtidos revelam que os
viriões produzidos na ausência de Vif não são infecciosos, pois a infecção é abolida pela
actividade anti-viral das proteínas NLS-A3G-HA (Figura 14). Quando a Vif está presente na
célula produtora, esta consegue contrabalançar o efeito anti-viral das proteínas construídas,
atingindo-se assim valores de infecção tão elevados quanto os obtidos na ausência da
desaminase, embora continue por justificar a razão pela qual se observam variações tão
grandes na capacidade de inibição dos três clones. Aliás, quando estão presentes as
proteínas dos clones, a inibição na ausência de Vif é ainda mais potente do que a verificada
aquando da produção viral na presença de A3G-HA do controlo positivo. Em contra-partida,
os níveis de infecção com partículas produzidas na presença das proteínas dos clones e de
Vif são ainda maiores do que quando a produção ocorre em células que expressam A3G-HA
e a proteína viral. Estes resultados demonstram que seria necessário repetir várias vezes
este ensaio no sentido de garantir com certeza os resultados e as assumpções que deles
provêm.
pNL4-3(Δvif) + + + + + + + + + + –
pcDNA3.1/Zeo(+)-vif – – – + + + + – + – –

28
4. Discussão dos resultados e Conclusões
Com este trabalho pretendeu-se modificar a proteína A3G para que esta fosse capaz
de manter uma localização nuclear no contexto celular, de forma a evitar a acção da
proteína viral Vif que decorre no citoplasma e que culmina na degradação proteossomal da
desaminase celular. Neste sentido, foram desenvolvidas construções que visavam a
produção de uma proteína A3G, contendo uma sequência-sinal NLS, capaz de ser expressa
numa linha celular humana caracterizada por um fenótipo permissivo – células HEK 293T.
Contudo, os resultados obtidos não corresponderam totalmente ao esperado: esta
proteína expressa é sujeita a modificações pós-traducionais que modificam a sua
sensibilidade à Vif e a sua capacidade de inibir a infecção nas próximas células-alvo, para
além de impedirem a sub-localização nuclear desejada. De facto, os resultados apontam
para a ocorrência de uma clivagem a nível da extremidade N-terminal da proteína, que para
além de remover a sequência NLS também elimina alguns aminoácidos dessa extremidade
(Figuras 9, 10 e 11). Esta clivagem parece ocorrer à priori do transporte para o núcleo, uma
vez que num contexto in vitro esta restrição é conservada (Figura 11), podendo-se
extrapolar que esta degradação parcial é efectuada por um (ou mais) factor(es)
citoplasmático(s). É possível ainda afirmar que esta clivagem terá de ocorrer por um (ou
mais) mecanismo(s) que permite(m) o reconhecimento de duas localizações distintas da
sequência da A3G, uma mais externa e outra mais interna, de forma a originar as duas
versões truncadas detectadas (com 41kDa e com 36kDa, respectivamente).
Esta hipótese de exclusão do núcleo já tinha sido mostrada por outros autores
(Bennett et al., 2006 e Bennett et al., 2008), que no entanto não verificaram qualquer
clivagem ou degradação da proteína, embora os estudos efectuados não visassem a análise
do tamanho das proteínas construídas mas sim a identificação da sub-localização celular
por métodos de fluorescência. Segundo esta publicação, este facto deve-se à presença de
uma sequência com forte poder de retenção citoplasmática (localizada entre os aminoácidos
113 e 128), que mesmo na presença de uma sequência localizadora do núcleo, tem a
capacidade de sobrepor o seu efeito e anular o sinal NLS.
Não obstante, uma outra forma de justificar a manutenção da localização
citoplasmática pode corroborar a hipótese de trabalho proposta. Esta supõe que a proteína
A3G modificada é capaz de se translocar para a proximidade da membrana celular de forma
a ser encapsidada nos novos viriões, conseguindo evitar a ligação à Vif por ligação à
poliproteína Gag ou por desfasamento temporal dos níveis citoplasmáticos das duas
proteínas. No entanto, outros estudos seriam necessários para que esta hipótese fosse
consolidada e aceite como forte representante do que acontece ao nível da célula.

Dis
cussão d
os r
esultados e
Conclu
sões
29
Através da análise da interacção Vif-A3G, foi possível detectar que a versão truncada
da proteína A3G com aproximadamente 41kDa é mais sensível à degradação induzida pela
Vif do que a proteína com cerca de 36kDa, e mesmo até do que a proteína A3G selvagem
(Figura 12). A grande diferença na sensibilidade à acção da proteína viral registada entre os
quatro clones testados pode ser indicadora de uma certa promiscuidade no reconhecimento
dos aminoácidos que representam o local de corte. Deste modo, diferenças num número
reduzido de aminoácidos não provoca uma alteração visível no tamanho da proteína mas
pode ser suficiente para se manifestar em diferenças significativas no fenótipo de
sensibilidade à Vif. Assim sendo, é possível concluir que o local de corte poderá localizar-se
dentro da região identificada como vital para a ligação à extremidade N-terminal da Vif
(aminoácidos 54 a 124 e 128), dificultando o reconhecimento por parte da Vif destas
proteínas truncadas; ou na porção N-terminal adjacente a este local, que embora resulte
numa proteína de menores dimensões que a proteína selvagem é igualmente ou até mais
eficientemente marcada para a degradação. No que diz respeito à proteína com 36kDa, a
diminuição da sensibilidade à acção da Vif demonstra que esta clivagem ocorre
garantidamente dentro da região de ligação à Vif (ou seja, entre os aminoácidos 54 e 128).
A determinação da capacidade de inibição da infecção viral destas proteínas
modificadas pareceu revelar-se denunciadora de uma localização mais restrita dos locais
onde ocorre a clivagem. Visto que todas as proteínas expressas pelos clones testados foram
capazes de abolir totalmente a infecção (algo que não se verificou com a proteína A3G
selvagem) (Figura 14), é possível concluir que os cortes que resultam nas proteínas
truncadas ocorrem dentro da região de interacção com a proteína Vif mas antes do domínio
CD1; ou seja, entre os aminoácidos 54 e 64, uma vez que a encapsidação da desaminase
(Figura 13) e a sua função não são afectadas, pelo contrário, são potenciadas.
Controversamente, a presença de Vif nas células produtoras torna as partículas virais
produzidas na presença das proteínas construídas mais infecciosas do que as produzidas
na presença da A3G selvagem, o que não corrobora as deduções retiradas até agora. Se
por um lado a clivagem origina proteínas com um poder anti-viral mais potente, por outro
lado este efeito é mais fortemente inibido pela proteína do vírus.
Por estas razões aqui sumariadas, demonstra-se que permanece a necessidade de
efectuar mais estudos que permitam desvendar os mecanismos de interacção destas duas
proteínas, de forma a se atingir uma compreensão mais profunda dos efeitos provocados
pelo HIV-1 nas células do Homem, possibilitando a descoberta de novas terapias anti-virais.

30
5. Referências Bibliográficas
1. Barré-Sinoussi, F., Chermann, J.C., Rey, F., Nugeyre, M.T., Chamaret, S., Gruest, J., Dauguet, C., Axler-Blin, C., Vézinet-Brun, F., Rouzioux, C., Rozenbaum, W., and Montagnier, L. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220 (4599), 868-871 (1983).
2. Clavel, F., Guétard, D., Brun-Vézinet, F., Chamaret, S., Rey, M.A., Santos-Ferreira, M.O., Laurent, A.G., Dauguet, C., Katlama, C., Rouzioux, C., et al. Isolation of a new human retrovirus from West African patients with AIDS. Science 233 (4761), 343-346 (1986).
3. Freed, E. O. HIV-1 Replication. Somatic Cell and Molecular Genetics 26, 13-33 (2001).
4. Levy, J. A. HIV Pathogenesis: Knowledge Gained after Two Decades of Research. Advances in Dental Research 19, 10-16 (2006).
5. Cimarelli, A., Darlix, J.-L. Assembling the human immunodeficiency virus type 1. Cellular and Molecular Life Sciences 59, 1166-1184 (2002).
6. Turner, B. G., Summers, M. F. Structural Biology of HIV. Journal of Molecular Biology 285, 1-32 (1999).
7. Frankel, A. D., Young, J. A. T. HIV-1: Fifteen Proteins and an RNA. Annual Review of Biochemistry 67, 1-25
(1998).
8. Stevenson, M. Developments in Basic Science Research. Topics in HIV Medicine 12 (1), 4-7 (2004).
9. Henriet, S., Mercenne, G., Bernacchi, S., Paillart, J.-C., Marquet, R. Tumultuous Relationship between the Human Immunodeficiency Virus Type 1 Viral Infectivity Factor (Vif) and the Human APOBEC-3G and APOBEC-3F Restriction Factors. Microbiology and Molecular Biology Reviews 73 (2), 211-232 (2009).
10. Gonçalves, J., Jallepalli, P., Gabuzda, D. H. Subcellular Localization of the Vif Protein of Human Immunodeficiency Type 1. Journal of Virology 68 (2), 704-712 (1994).
11. Strebel, K., Daugherty, D., Clouse, K., Cohen, D., Folks, T., Martin, M. A. The HIV ‘A’ (sor) gene product is essencial for virus infectivity. Nature 328 (6132), 728-730 (1987).
12. Marin, M., Rose, K. M., Kozak, S. L., Kabat, D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nature Medicine 9 (11), 1398-1403 (2003).
13. Goila-Gaur, R., Strebel, K. HIV-1 Vif, APOBEC, and Intrinsic Immunity. Retrovirology 5, 51 (2008).
14. Harris, R. S., Liddament, M. T. Retroviral Restriction by APOBEC Proteins. Nature Reviews Immunology 4,
868-877 (2004).
15. Chiu, Y.-L., Greene, W. C. The APOBEC3 Cytidine Deaminases: An Innate Defensive Network Opposing Exogenous Retroviruses and Endogenous Retroelements. Annual Review of Immunology 26, 317-353 (2008).
16. Sheehy, A. M., Gaddis, N. C., Choi, J. D., Malim, M. H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418, 646-650 (2002).
17. Rose, K. M., Marin, M., Kozak, S. L., Kabat, D. The viral infectivity factor (Vif) of HIV-1 unveiled. Trends in Immunology 10 (6), 291-297 (2004).
18. Alce, T. M., Popik, W. APOBEC3G Is Incorporated into Virus-like Particles by a Direct Interaction with HIV-1 Gag Nucleocapsid Protein. The Journal of Biological Chemistry 279 (33), 34083-34086 (2004).
19. Gonçalves, J., Santa-Marta, M. HIV-1 Vif and APOBEC3G: Multiple roads to one goal. Retrovirology 1, 28
(2004).
20. Malim, M. H. Natural resistance to HIV infection: The Vif-APOBEC interaction. Comptes Rendus Biologies 329, 871-875 (2006).
21. Harris, R. S., Bishop, K. N., Sheehy, A. M., Craig, H. M., Peterson-Mahrt, S. K., Watt, I. N., Neuberger, M. S., Malim, M. H. DNA Deamination Mediates Innate Immunity to Retroviral Infection. Cell 113, 803-809 (2003).
22. Bishop, K. N., Holmes, R. K., Malim, M. H. Antiviral Potency of APOBEC Proteins Does Not Correlate with Cytidine Deamination. Journal of Virology 80 (17), 8450-8458 (2006).
23. Mariani, R., Chen, D., Schröfelbauer, B., Navarro, F., König, R., Bollman, B., Münk, C., Nymark-McMahon, H., Landau, N. R. Species-Specific Exclusion of APOBEC3G from HIV-1 Virions by Vif. Cell 114, 21-31 (2003).
24. Kao, S., Khan, M. A., Miyagi, E., Plishka, R., Buckler-White, A., Strebel, K. The Human Immunodeficiency Virus Type 1 Vif Protein Reduces Intracellular Expression and Inhibits Packaging of APOBEC3G (CEM15), a Cellular Inhibitor of Virus Infectivity. Journal of Virology 77 (21), 11398-11407 (2003).
25. Navarro, F., Landau, N. R. Recent insights into HIV-1 Vif. Current Opinion in Immunology 16, 477-482 (2004).
26. Stopak, K., Noronha, C., Yonemoto, W., Greene, W. C. HIV-1 Vif Blocks the Antiviral Activity of APOBEC3G by Impairing Both Its Translation and Intracellular Stability. Molecular Cell 12, 591–601 (2003).
27. Holmes, R. K., Malim, M. H., Bishop, K. N. APOBEC-mediated viral restriction: not simply editing? Trends in Biochemical Sciences 32 (3), 118-128 (2007).
28. Haché, G., Liddament, M. T., Harris, R. S. The Retroviral Hypermutation Specificity of APOBEC3F and APOBEC3G Is Governed by the C-terminal DNA Cytosine Deaminase Domain. The Journal of Biological Chemistry 280 (12), 10920-10924 (2005).
29. Bennett, R. P., Diner, E., Sowden, M. P., Lees, J. A., Wedekind, J. E., Smith, H. C. APOBEC-1 and AID are Nucleo-cytoplasmic Trafficking Proteins but APOBEC3G Cannot Traffic. Biochemical and Biophysical Research Communications 350 (1), 214-219 (2006).
30. Bennett, R. P., Presnyak, V., Wedekind, J. E., Smith. H. C. Nuclear Exclusion of the HIV-1 Host Defense Factor APOBEC3G Requires a Novel Cytoplasmic Retention Signal and Is Not Dependent on RNA binding. The Journal of Biological Chemistry 283 (12), 7320-7327 (2008).

xiv
6. Anexos
Tabela 1 | Composição da mistura e condições da reacção de PCR, referida no ponto 2.1.2. dos Materiais e Métodos:
Tabela 2 | Representação esquemática das sequências de reconhecimento das enzimas NotI e XhoI, utilizadas na
clonagem:
Tabela 3 | Anticorpos utilizados na detecção de proteínas por Western blot, e respectivas diluições:
1 Todos os anticorpos foram diluídos em soluções bloqueantes de 3%(p/v) de leite magro em pó em TBS-T.
Mistura de reacção – total: 50 μL Condições de reacção
Componente Concentração final Passo Temperatura Duração
DNA molde 0,02ng/μL Desnaturação
inicial 98ºC 3 min
Oligonucleótidos (NotI-NLS-A3G-F e HA-XhoI-R) 0,6μM cada Desnaturação 98ºC 30 seg
X 350 ciclos
Nucleótidos - dNTP (dATP, dCTP, dGTP, dTTP) 160μM cada Hibridação 61ºC 30 seg
Tampão HF (fornecido com a enzima) 1 X Extensão 72ºC 1 min 30 seg
Polimerase de DNA Phusion
0,04 U/μL Extensão final 72ºC 10 min
Enzima Sequência de reconhecimento
NotI 5’ – G CG G C C G C – 3’ 3’ – C G C C G GC G – 5’
XhoI 5’ – CT C G A G – 3’ 3’ – G A G C TA – 5’
Anticorpo Espécie Diluição1 Referência
α-HA HRP Rato 1:2500 Roche
α-GAPDH Rato 1:5000 Santa Cruz
Biotechnology
α-ratinho IgG HRP Cabra 1:8000 Sigma
α-PARP Coelho 1:3000 Santa Cruz
Biotechnology
α-coelho IgG HRP Ovelha 1:8000 Chemicom

Anexos
xv
a
b
pNL4-3 Δvif
14635bp
HIV-1 5’LTR
HIV-1 sinal encapsidação Ψ
gag
pol
viftat I
rev Ivpu
envtat II
rev II
nef
HIV-1 5’LTR
pUC ori
Ampicilina
pAmpicilina
VSVG
EF1α Enhancer
Ampicilina
SVpA
pHEF-VSV-G
5471bp

Anexos
xvi
APOBEC3GNLS HApcDNA3.1/Zeo(+) pcDNA3.1/Zeo(+)
c
Figura 15 | Mapas genómicos de plasmídios utilizados. a | Plasmídio p-HEF-VSV-G, codificante da glicoproteína G do vírus
da estomatite vesicular, utilizado em co-transfecção com o plasmídio pNL4-3(Δvif) com o intuito de aumentar a eficiência de
infecção. b | Plasmídio pNL4-3(Δvif), codificante do genoma da estirpe NL4-3 do HIV-1; c | Vector pcDNA3.1/Zeo(+) utilizado
nas clonagens efectuadas e na transfecção de células HEK 293T; são referidos os locais de clivagem das enzimas NotI e XhoI
utilizadas para a clonagem efectuada.
Figura 16 | Esquema representativo da construção efectuada para produzir os clones pcDNA3.1/Zeo(+)-NLS-A3G-HA.
NotI XhoI
pcDNA3.1/Zeo(+)
5023bp
pCMV
BGH pA
SV40 pA
pUC ori
Ampicilina
pAmpicilina
pT7
f1 ori
SV40 ori
Zeomicina
HindIII 920
KpnIII 930
BamHI 938
EcoRI 961
PstI 970
NotI 988
XhoI 994
XbaI 1000
ApaI 1010
EcoRV 973
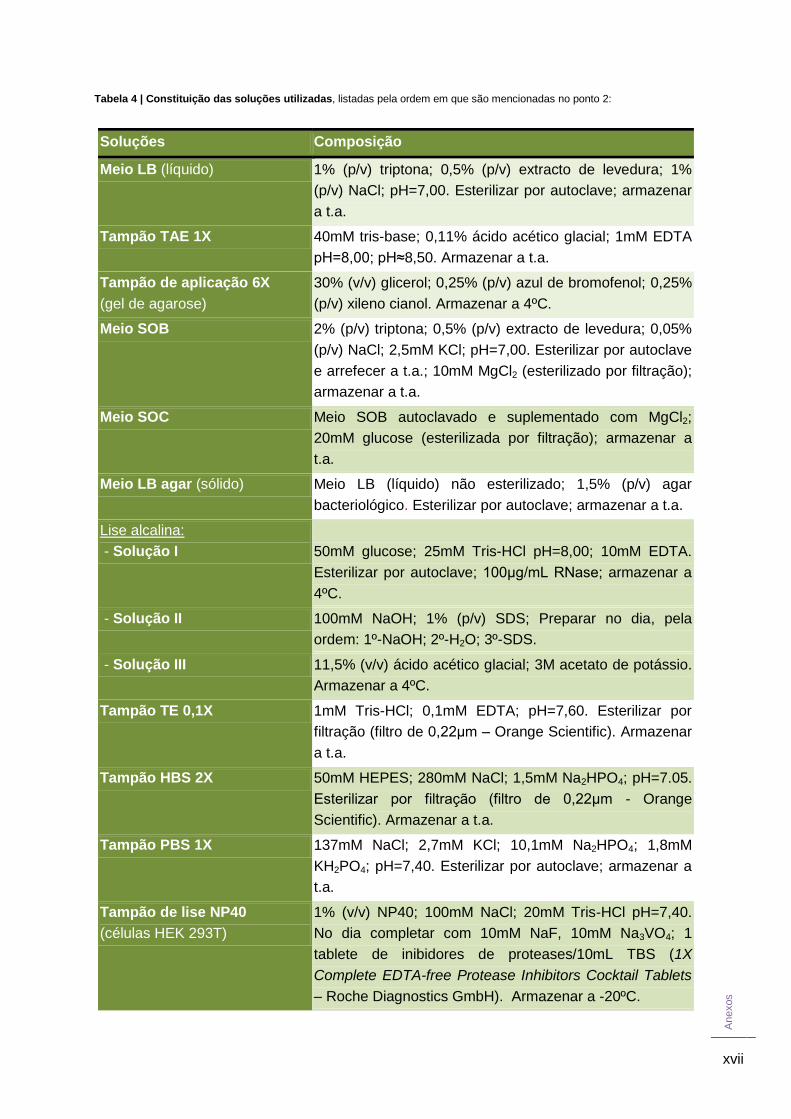
Anexos
xvii
Tabela 4 | Constituição das soluções utilizadas, listadas pela ordem em que são mencionadas no ponto 2:
Soluções Composição
Meio LB (líquido) 1% (p/v) triptona; 0,5% (p/v) extracto de levedura; 1%
(p/v) NaCl; pH=7,00. Esterilizar por autoclave; armazenar
a t.a.
Tampão TAE 1X 40mM tris-base; 0,11% ácido acético glacial; 1mM EDTA
pH=8,00; pH≈8,50. Armazenar a t.a.
Tampão de aplicação 6X
(gel de agarose)
30% (v/v) glicerol; 0,25% (p/v) azul de bromofenol; 0,25%
(p/v) xileno cianol. Armazenar a 4ºC.
Meio SOB 2% (p/v) triptona; 0,5% (p/v) extracto de levedura; 0,05%
(p/v) NaCl; 2,5mM KCl; pH=7,00. Esterilizar por autoclave
e arrefecer a t.a.; 10mM MgCl2 (esterilizado por filtração);
armazenar a t.a.
Meio SOC Meio SOB autoclavado e suplementado com MgCl2;
20mM glucose (esterilizada por filtração); armazenar a
t.a.
Meio LB agar (sólido) Meio LB (líquido) não esterilizado; 1,5% (p/v) agar
bacteriológico. Esterilizar por autoclave; armazenar a t.a.
Lise alcalina:
- Solução I
50mM glucose; 25mM Tris-HCl pH=8,00; 10mM EDTA.
Esterilizar por autoclave; 100μg/mL RNase; armazenar a
4ºC.
- Solução II 100mM NaOH; 1% (p/v) SDS; Preparar no dia, pela
ordem: 1º-NaOH; 2º-H2O; 3º-SDS.
- Solução III 11,5% (v/v) ácido acético glacial; 3M acetato de potássio.
Armazenar a 4ºC.
Tampão TE 0,1X 1mM Tris-HCl; 0,1mM EDTA; pH=7,60. Esterilizar por
filtração (filtro de 0,22μm – Orange Scientific). Armazenar
a t.a.
Tampão HBS 2X 50mM HEPES; 280mM NaCl; 1,5mM Na2HPO4; pH=7.05.
Esterilizar por filtração (filtro de 0,22μm - Orange
Scientific). Armazenar a t.a.
Tampão PBS 1X 137mM NaCl; 2,7mM KCl; 10,1mM Na2HPO4; 1,8mM
KH2PO4; pH=7,40. Esterilizar por autoclave; armazenar a
t.a.
Tampão de lise NP40
(células HEK 293T)
1% (v/v) NP40; 100mM NaCl; 20mM Tris-HCl pH=7,40.
No dia completar com 10mM NaF, 10mM Na3VO4; 1
tablete de inibidores de proteases/10mL TBS (1X
Complete EDTA-free Protease Inhibitors Cocktail Tablets
– Roche Diagnostics GmbH). Armazenar a -20ºC.

Anexos
xviii
Gel de poliacriamida:
- Tampão de aplicação 5X
125mM Tris-HCl pH=6,80; 4% (p/v) SDS; 20% (v/v)
glicerol; 200μM β-mercaptoetanol; 2% (v/v) azul
bromofenol. Armazenar a t.a., no escuro.
- Tampão de corrida 1X
25mM tris; 192mM glicina; 0,1% (p/v) SDS; pH=8,3.
Armazenar a 4ºC.
- Tampão de transferência 1X 25mM tris; 192mM glicina; 20% (v/v) metanol; pH=8,3.
Armazenar a 4ºC.
Tampão TBS-T 50mM tris; 150mM NaCl; pH=7,5. Esterilizar por
autoclave; 0,1% (v/v) Tween-20; armazenar a t.a.
Tampão de lise de citosol 50mM HEPES-KOH pH=7,20; 2mM EDTA; 10mM NaCl;
250mM sacarose. Esterilizar a 95ºC durante 30 minutos;
armazenar a 4ºC. No dia completar com 1 tablete de
inibidores de proteases/10mL TBS (1X Complete EDTA-
free Protease Inhibitors Cocktail Tablets – Roche
Diagnostics GmbH); 10mM NaF; 10mM Na3VO4, 1mM
PMSF; 0,1M DTT; 0,1% (v/v) NP-40.
Tampão de lise de núcleos 50mM HEPES-KOH pH=7,20; 2mM EDTA; 400mM NaCl;
20% (v/v) glicerol. Esterilizar por autoclave; armazenar a
4ºC. No dia completar com 1 tablete de inibidores de
proteases/10mL TBS (1X Complete EDTA-free Protease
Inhibitors Cocktail Tablets – Roche Diagnostics GmbH);
10mM NaF; 10mM Na3VO4, 1mM PMSF; 0,1M DTT.
Tampão de IP 50mM Tris-HCl pH=7,40; 50mM NaCl; 1mM EDTA; 0,2%
(v/v) Triton X-100. Armazenar a 4ºC.
Tampão de lise do CPRG 5mM MgCl2; 0,1% (v/v) NP40; em PBS 1X. Armazenar a
t.a.
Recommended

















