Embed Size (px)
Citation preview

Caso Clínico
19/06/2012
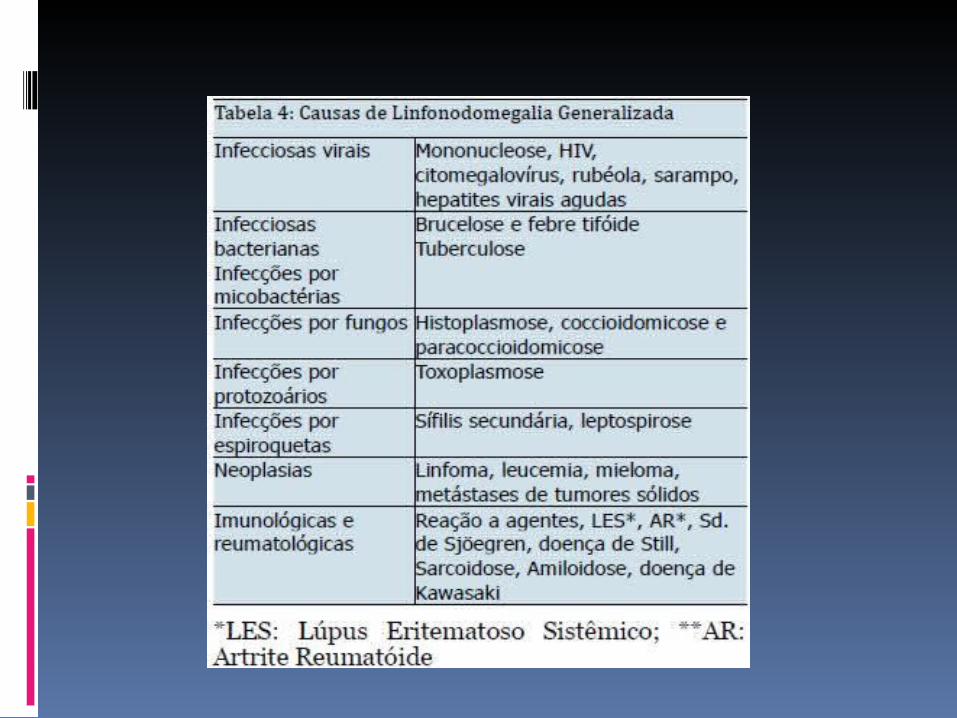
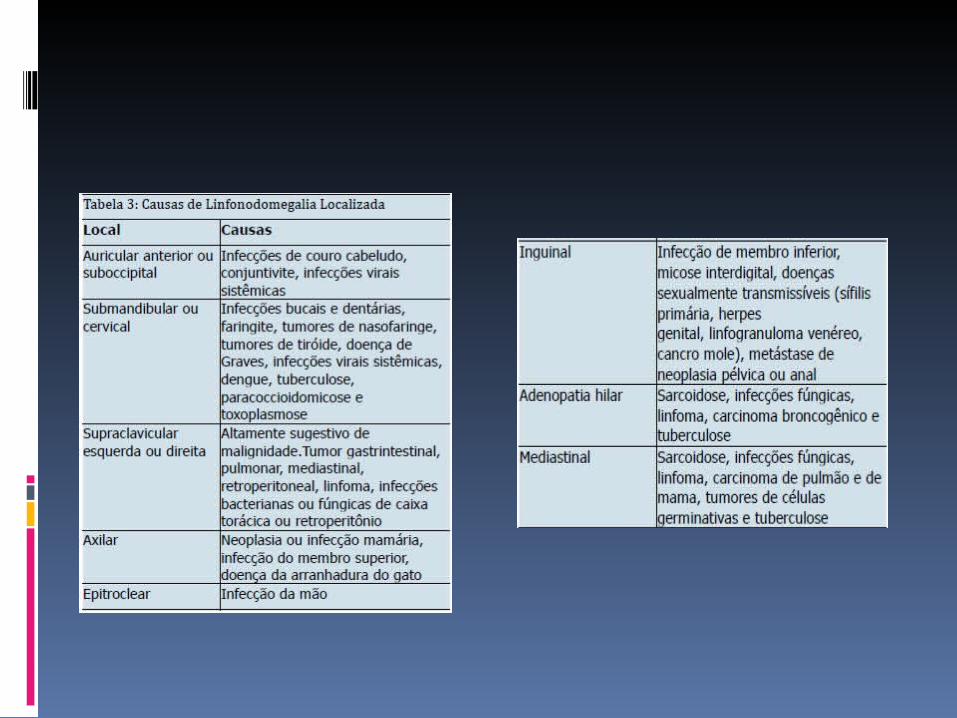
LINFONODOMEGALIA GENERALIZADA
Secretaria de Estado de Saúde do Distrito FederalHospital Regional da Asa Sul
Unidade de Pediatria
José Messias e Élida SantosInternato – 6ª série
ESCSCoordenação: Luciana Sugaiwww.paulomargotto.com.br
Brasília, 23/6/12

CASO CLÍNICO
Identificação: I S B, 14 anos, sexo feminino , , natural de Gama , residente em Valparaíso. Peso: 43.3Kg.Acompanhante: mãe – Silvana..QP: Febre + dores nas juntas + adenites há 10 dias

Caso Clínico

CASO CLÍNICO
Exame físico do PS: BEG, EUPNEICO, HIDRATADO, AFBRIL, ANICTERICO, ACIANOTICOMV PRESENTE,S/RARC 2T SEM SOPROABDOMEM FLACIDO SEM MEGALIASMM -SEM EDEMA,PULSOS CHEIOSOROSCOPIA HIPEREMIADAPELE SEM PETEQUIAS SEM SINAIS MENINGEOS.

EM TEMPO -MENOR APRESENTA EXANTEMA ,ALGUMASMACULOPAPULAR ESPARSAS,NÃO PRURIGINOSASADENOMEGALIA DOLOROSO EM AXILA D.CONDUTA -INTERNAÇÃO -SOLICITAÇÃO DA HEMATOLOGISTA.
CASO CLÍNICOHDA: Mãe refere que adolescente iniciou há 14 dias, da admissão com quadro de aumento de linfonodos de regiões cervical anterior, bilateralmente, além de linfonodo em região axilar a direita e em regiões inguinais, com presença de dor local, mas sem eritema ou calor. Também iniciou em associação com febre de 38-39ºC,diaria, com um pico pela manhã e outro pela tarde, dor em cotovelos e joelhos, sem sinais inflamatórios e dorsalgia de moderada intensidade, prejudicando movimentação e manchas eritematosas, palpáveis, difusas pelo corpo, predominando em tronco, face e MMSS, indolores e não pruriginosas, que iniciam em 1x1cm e evoluem para cerca 2x2cm com manifestação de região central esbranquiçada.
CASO CLÍNICOHá 10 dias permanecia com mesmos sintomas e passou a ter disfagia, devido a compressão da área de linfonodomegalia de região cervical. Procurou serviço do HMIB há cerca de 5 dias e não foi atendida devido a idade (sic), buscando posto de saúde próximo a sua casa recebeu amoxicilina por via oral, por 5 dias, sem apresentar melhora do quadro, interrompendo seu uso.Retornou há um dia ao HMIB com mesmo quadro: febre, artralgia, manchas na pele, adenites e disfagia.Eliminações normais.Alimentação pouco diminuída devido a desconforto para deglutição.

CASO CLÍNICO
Antecedentes pessoais fisiológicos
Nascido de parto normal, a termo, não se lembra de peso ou comprimento e não trouxe cartão da criança. Nega intercorrências na gestação e no parto.Vacinação em dia segundo diz a mãe.Apresentou menarca no dia 27.05.2012.
Antecendentes pessoais patológicos:
Cerca de 5 Internações prévias. 2010: Teve investigação para Síndrome de Evans. Apresentou um episodio de PTI , tratado com corticoterapia. 2011: um episodio semelhante ao atual (febre, adenite, manchas na pele e artralgias) , melhorando após uso de antibioticoterapia. Estava bem até há 2 semanas.Nega trauma, alergias, cirurgias e uso de medicações.

CASO CLÍNICOAntecedentes familiares:
Pai saudável;Mãe saudável; Irmãos: Quatro irmãs por parte de pai – saudáveis (27 anos, 21 anos, 18 anos, 11 anos) e três irmãs por parte de mãe saudáveis (16 anos, duas gêmeas de 11 anos). Família do pai com histórico de câncer (de mama e útero em tias) e também de diabetes melitus.
Hábitos de vida:
Reside em casa de alvenaria, sem mofo e sem forro no teto. Com presença de gato em casa. Saneamento básico completo.Alimentação: equilibrada. Frequenta escola com bom rendimento escolar, no 9ºano.
CASO CLÍNICO
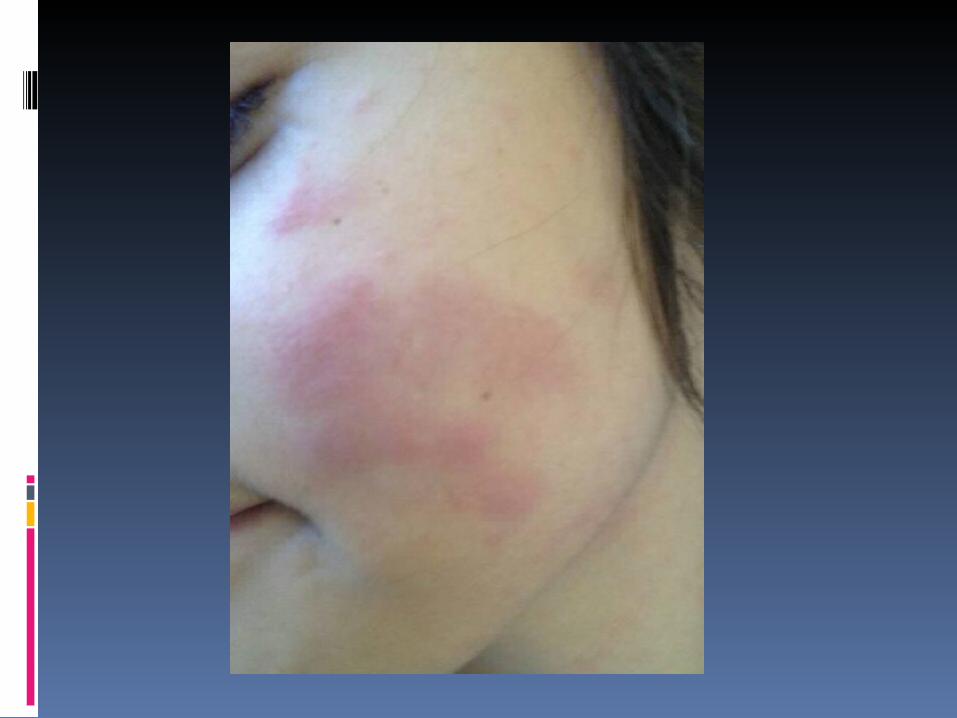
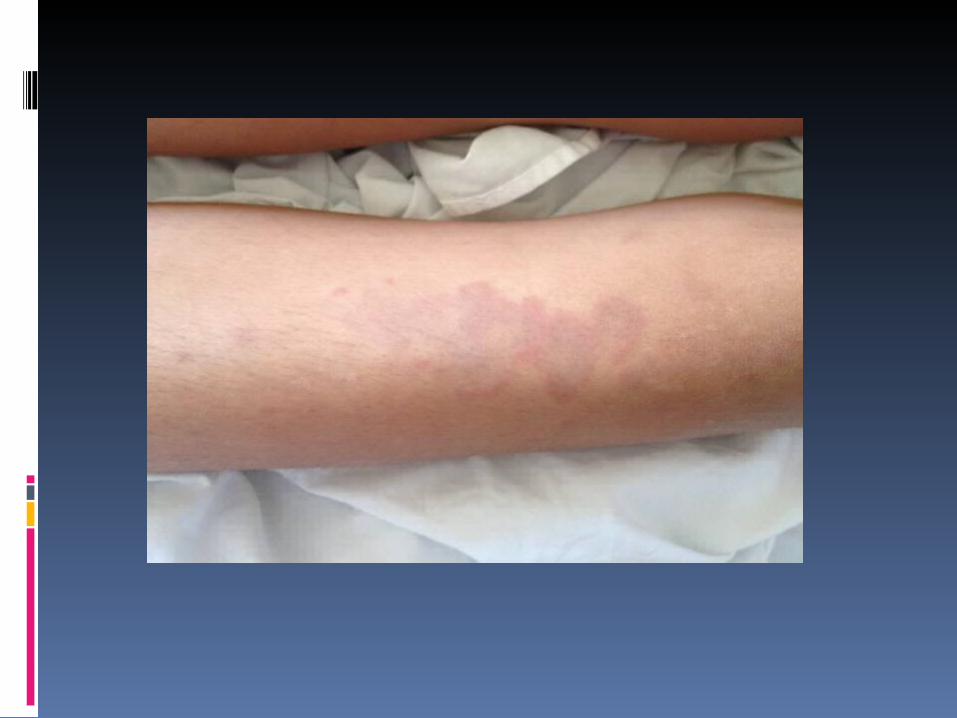
Exame físico em 05/06/2012:BEG, corada, nutrida, hidratada, anictérica, ativa, reativa, acianótica, febril 38.8ºC e eupneica.FC= 99bpm FR=15irpmAR: MVF sem RA.ACV: RR em 2T, BNF sem sopros.ABD: Plano, flácido, indolor, sem VMGs, ausência de dor a descompressão.Pele: presença de lesões palpáveis de cerca de 2x2cm, eritematosas, com região central empalidecida, não dolorosas ou pririginosas, localizadas em tronco, face, MMSS e poucas em MMII. Desaparecem a digitopressão.Ext: bem perfundidas (tempo de enchimento capilar menor que 2 segundos) e sem edema.Orofaringe: sem hiperemia e placas purulentas.Neurológico: Ausência de sinais meningeos.

Evolução
Na data 05/06/2012: O exame permanecia inalterado,
sendo a conduta: Encaminhar a adolescente para a Ala
B Solicitar provas de atividades
inflamatórias e sorologias Inicia Ampicilina+sulbactam
(adenite?)
06/06/2012 Nesta data a paciente já aceitava bem
a dieta com diminuição considerável da adenomegalia cervical, matinha de 2 a 3 picos febris/dia de 38 a 39°C. Com o seguinte exame físico, que não apresentou alterações até a data 19/06/2012:
Adolescente em bom estado geral, ativa, alerta, hidratada, normocorada, acianótica, anicterica, afebril ao toque.

Cont. AP CARD: ritmo cardíaco regular, 2
tempos, bulhas normofonéticas, sem sopros.
AP RESP: murmúrio vesicular fisiológico, sem ruídos adventícios. Sem sinais de esforço respiratório.
ABDOME: Plano, ruídos hidroaéreos presentes, normotenso, sem visceromegalias ou hiperestesias.
Cont. PELE: presença de lesões palpáveis de
cerca de 2X2cm, eritematosas, com região central empalidecida, não dolorosas ou pruriginosas, localizadas em tronco, face, MMSS e poucas em MMII. Que desaparecem a digitopressão.
EXTREMIDADES: sem edemas e bem perfundidas.
LINFONODOS: palpáveis em região cervical anterior D de 1 cm, um de 3 cm doloroso em região axilar D, 1 em região inguinal E de 1 cm.
SN: sem sinais meníngeos.

Na data 07/06/2012 Foi respondido o parecer da reumatologia: Não houve retorno da citopenia. No
momento febril, com adenomegalias generalizadas, alem da artralgia (essencialmente na coluna lombar).
Histórico de anemia hemolítica auto-imune + plaquetopenia (Sd. De Evans).
Raynaud em pés. Adenomegalia difusa cervicais, inguinais , axilares. Placas hiperemiadas em face e membros- Vasculite?
Foi sugerido pedir: FAN, anti-SM, anti-DNS, anti-la, anti-Ro, anti-
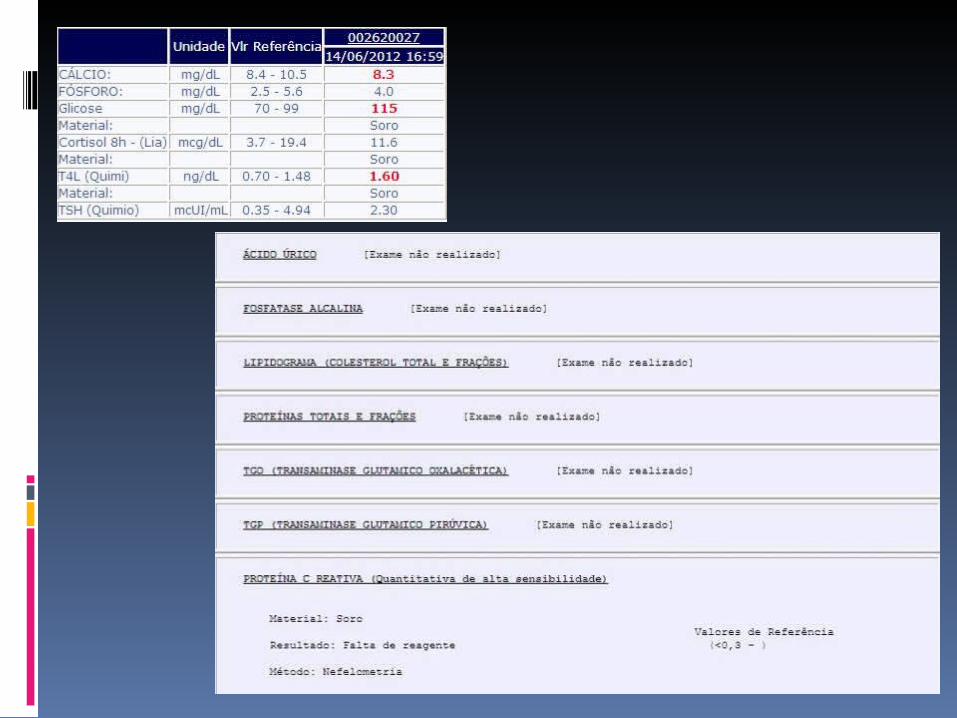
cardiolopina IgM e IgG, anti-coagulante lúpico, anti-RNP, C3, C4, VDRL, Coombs direto, lipidograma, proteinograma, TGO, TGP, FAL, proteinúria de 24h, cistatina C, clearence de creatinina, VHS, PCR, GAC, EAS, LDH, acido úrico, bilirrubinas
TC de tórax de alta resolução (linfonodos mediastinais).
Biopsia de pele (vasculite?) e Biopsia de gânglios Mielograma Ecocardiograma (serosite?)
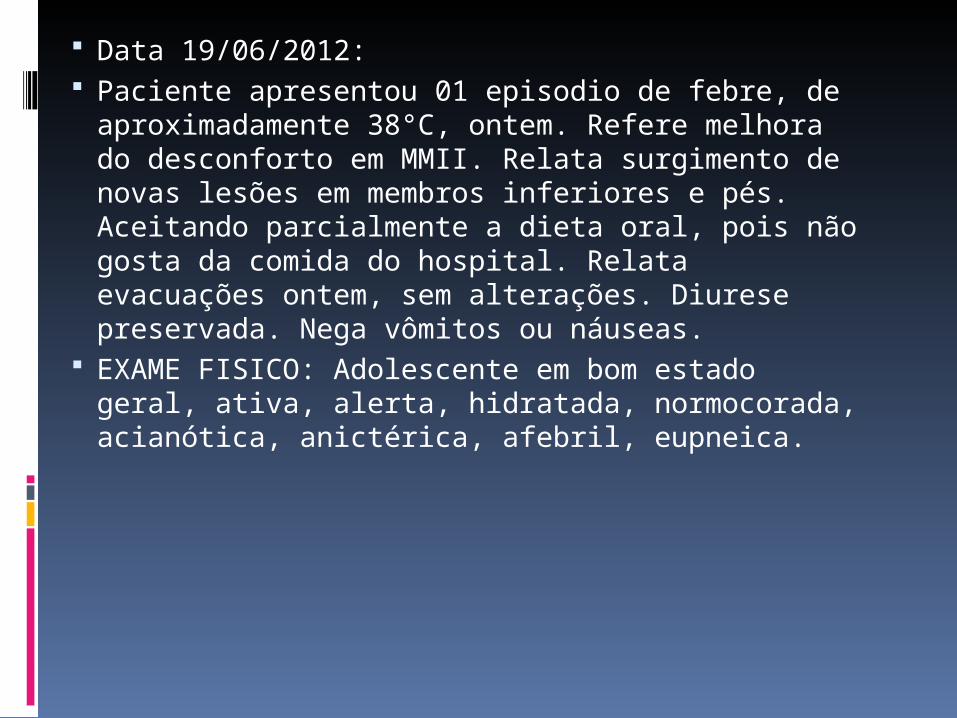
Data 19/06/2012: Paciente apresentou 01 episodio de febre, de
aproximadamente 38°C, ontem. Refere melhora do desconforto em MMII. Relata surgimento de novas lesões em membros inferiores e pés. Aceitando parcialmente a dieta oral, pois não gosta da comida do hospital. Relata evacuações ontem, sem alterações. Diurese preservada. Nega vômitos ou náuseas.
EXAME FISICO: Adolescente em bom estado geral, ativa, alerta, hidratada, normocorada, acianótica, anictérica, afebril, eupneica.

AP CARD: ritmo cardíaco regular, 2 tempos, burlhas normofonética, sem sopros.
AP RESP: murmúrio vesicular fisiológico, sem ruídos adventícios, sem utilização de músculos acessórios.
ABDOME: Plano, ruídos hidroaéreos presentes, normotenso, sem visceromegalias ou massas palpáveis, indolor.
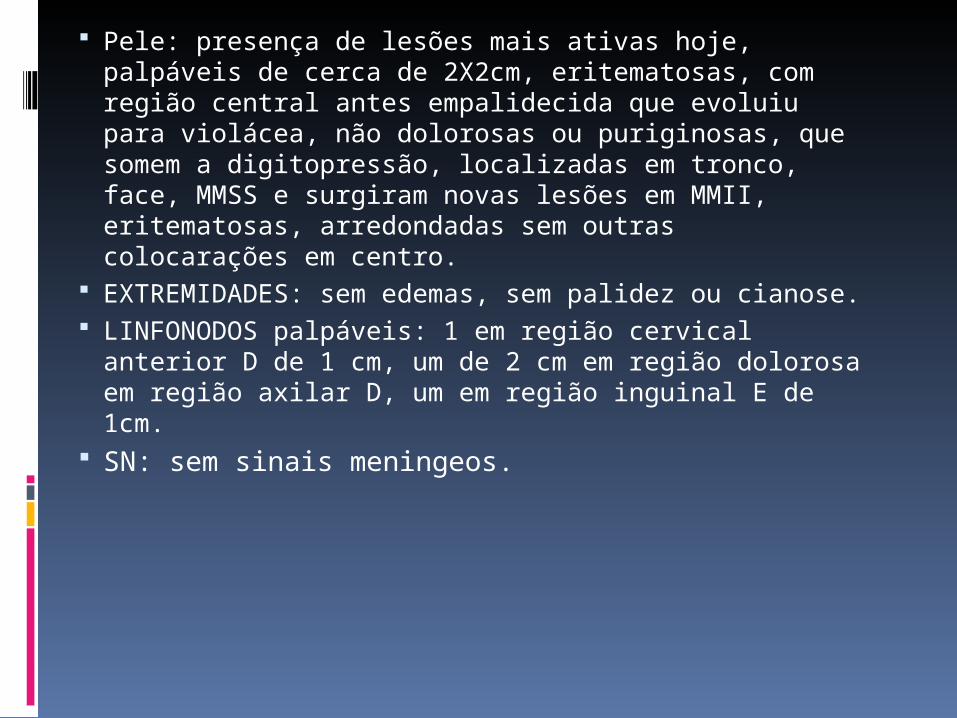
Pele: presença de lesões mais ativas hoje, palpáveis de cerca de 2X2cm, eritematosas, com região central antes empalidecida que evoluiu para violácea, não dolorosas ou puriginosas, que somem a digitopressão, localizadas em tronco, face, MMSS e surgiram novas lesões em MMII, eritematosas, arredondadas sem outras colocarações em centro.
EXTREMIDADES: sem edemas, sem palidez ou cianose.
LINFONODOS palpáveis: 1 em região cervical anterior D de 1 cm, um de 2 cm em região dolorosa em região axilar D, um em região inguinal E de 1cm.
SN: sem sinais meningeos.

A Biopsia de linfonodo axilar e de pele foram realizadas na tarde da data 12/06, aguardamos resultados.
A TC foi realizada na data 15/06, também aguardamos resultado
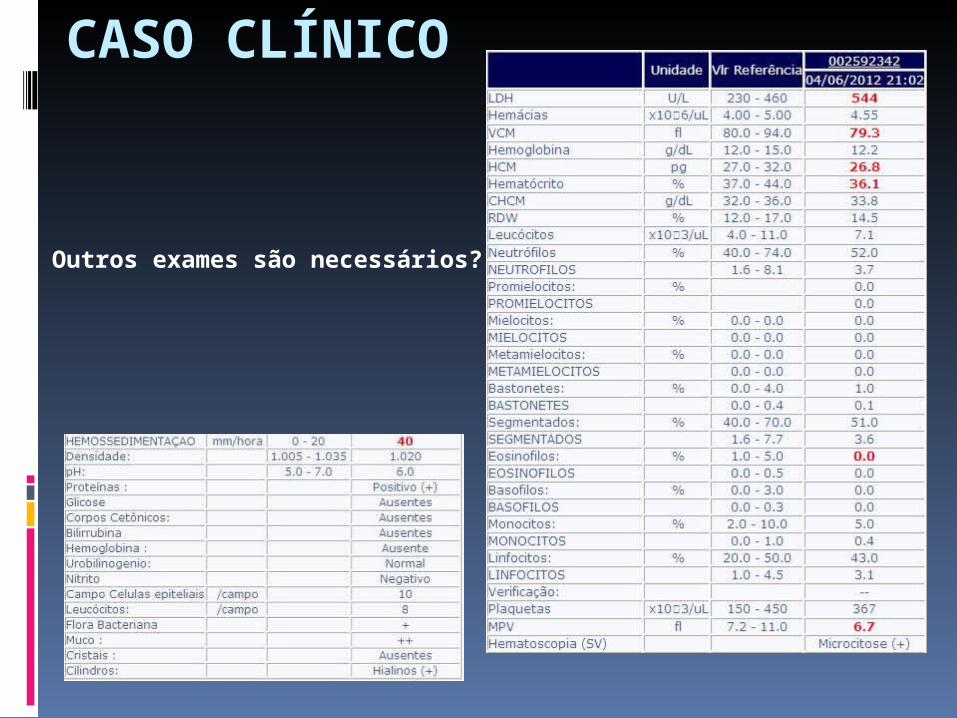
CASO CLÍNICO
Outros exames são necessários? Quais?
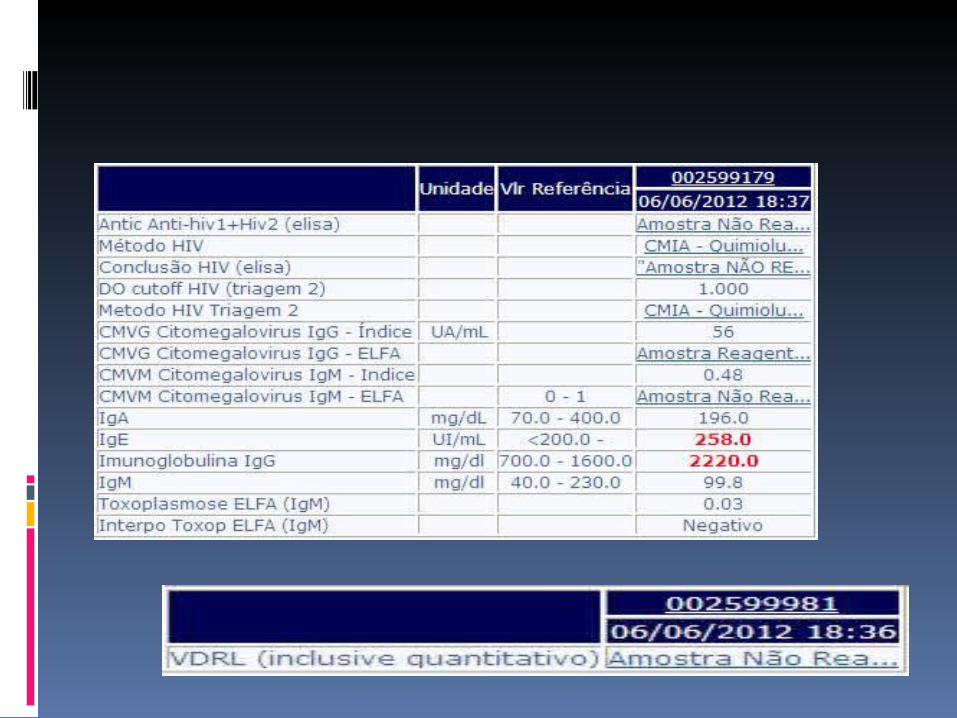

Hipótese diagnóstica? Conduta?


Sindrome de Evans
Evans e colaboradores: 1951 Anemia hemolítica autoimune Coombs
positivo + trombocitopenia imunologicamente mediada.
Desordem de imuno-regulação. Auto anticorpos afetam diferentes
antígenos nos eritrócitos e plaquetas, que seriam os responsáveis por episódios isolados de anemia e trombocitopenia
Diagnóstico de exclusão

O Síndrome linfoproliferativo Autoimune considerado uma doença da
homeostasia linfocitária causada por defeitos na via apoptótica do Fas/CD952,5. A definição restrita de caso de ALPS inclui acumulo de linfócitos, expansão de células DNT e apoptose linfocitária in vitro anormal, frequentemente associado a fenômenos auto-imunes

Epidemiologia
- A doença ocorre em ambos os sexos, com ligeiro predomínio do sexo masculino e sem predomínio de
raças. - Clinicamente apresenta-se na infância, com
idade média de aparecimento de 24 meses, variando desde os primeiros dias de vida até aos 15 anos,
- Adenopatias persistentes e/ou esplenomegália4 e doença autoimune, em particular hematológica.

Classificação
Classificação genotípica do ALPS –
Tipo de ALPS Proteína mutada Ia TNFRSF6 Fas, apoptose no receptor major
linfocitário Ia (mutação somática) TNFRSF6 Igual a Ia Ib TNFSF6 Ligando Fas II CASP10, CASP8 Caspase-10, caspase-8,
protease intracellular na cascada apoptótica III Desconhecida;
*ALPS – Síndrome linfoproliferativo autoimune

Quadro Clínico
A linfoproliferação é a manifestação clínica mais dramática e consistente.
As adenomegalias estão presentes em virtualmente todos os doentes, dimensões variáveis; tipicamente indolores e generalizadas,
Envolve especialmente as regiões cervical e axilar. A esplenomegalia, moderada a maciça, está quase
sempre presente antes dos cinco anos de idade, persistindo com dimensões flutuando ao longo do tempo.
As dimensões dos gânglios linfáticose baço permanecem relativamente estáveis, podendo inclusivamente diminuir com a idade.

Diagnóstico
Cerca de 80% dos doentes apresentam anticorpos detectáveis, mais frequentemente anticorpos anticardiolipina ou antieritrocitários
Cerca de metade destas crianças têm doença autoimune, sobretudo hematológica .
Frequentemente anemia hemolítica autoimune.
O estudo histopatológico dos gânglios linfáticos

Tratamento
O controle das manifestações autoimunes. Perante doença autoimune grave um ciclo de corticóides pode ajudar a controlar a doença (metilprednisolona 5-30 mg/Kg/dia, endovenosa, seguido de prednisolona, oral, 1-2 mg/Kg/dia, durante alguns meses)
A imunoglobulina endovenosa pode ser utilizada, 1-2 g/Kg para tratamento de episódios de hemólise e trombocitopenia autoimunes e o G-CSF (granulocyte colony stimulating factor) para a neutropenia (1-2 μg/Kg, subcutâneo, uma dose diária ou três vezes por semana).

Tratamento
A longo prazo, pode ser utilizado o micofenolato de mofetil, 600 mg/m2/dose, duas tomas diárias, oral4.
Perante uma doença autoimune refratária, podem ser necessários outros fármacos imunossupressores.
A esplenectomia é por vezes necessária para controlar o Hiperesplenismo.

Prognóstico
Embora a linfoproliferação seja inicialmente benigna,cerca de 10% dos doentes com ALPS do tipo Ia desenvolvem linfoma4, quase exclusivamente de células B. O risco de linfoma de Hodgkin e não Hodgkin pode estar aumentado até cerca de 51 vezes.
A evolução de um doente com ALPS é variável, mas frequentemente melhora ao longo dos anos e a esperança média de vida sobrepõe-se à restante população.

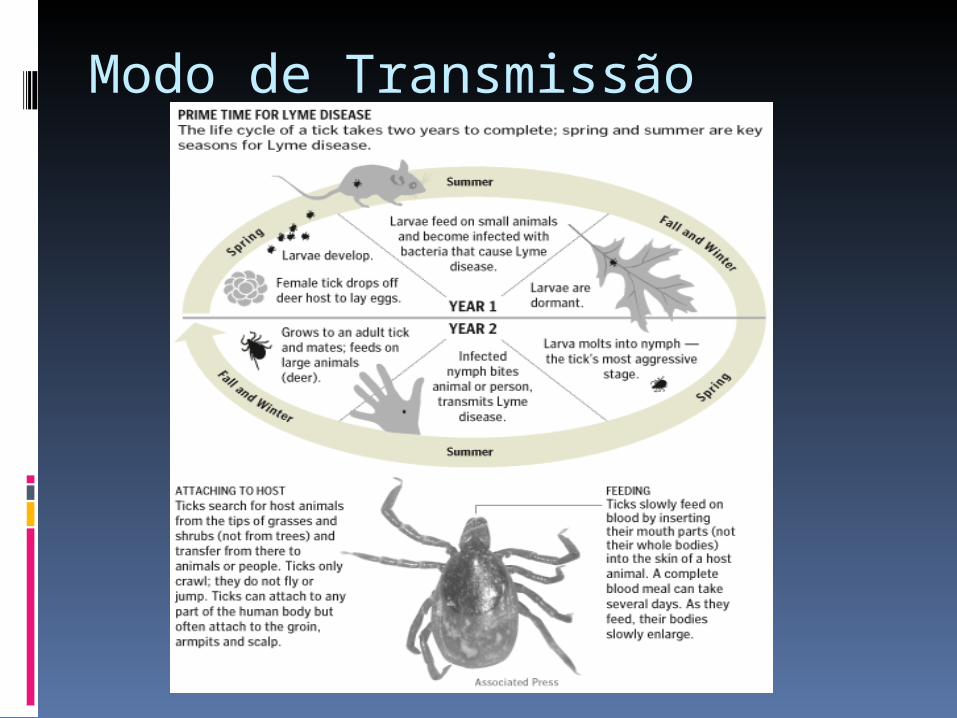
Borreliose de Lyme/ Doença de Lyme As borrelioses constituem de doenças
infecciosas causadas por espiroquetas do gênero Borrelia.
Borreliose de Lyme (BL), é uma doença, infecciosa não contagiosa, causada por espiroquetas pertecentes ao complexo Borrelia burgdoferi.
Eritema Marginado recidivante (EMR), relevante achado.

Classificação
a) febre recurrente epidêmica humana, causada pela B. recurrentis, e febre recurrente endêmica,
(b) borreliose aviária, a qual é determinada por uma única espécie, a B. anserina, (c) borreliose bovina, causada pela B. theileri. (d) aborto enzoótico bovino, (e) borreliose de Lyme (ou doença de Lyme) e borreliose de Lyme simile, as quais são causadas pelo grupo da B. burgdorferi lato sensu.

Epidemiologia
Diagnosticada em todos os continentes Endêmica: na Alemanha, na Áustria, na
Suécia, na Dinamarca, na Noruega, na Eslovênia, na Polônia e frequentemente observada no Reino Unido, na Espanha, em Portugal, na Suíça, na Croácia, na Itália, na República Tcheca . Também nos Estados Unidos;
Relato de casos no Brasil, no México, no Canadá, no Chile, na Costa Rica, na Colômbia e na Venezuela

No Brasil, os primeiros três casos de EM no estado do Amazonas.(1987)
Diante da impossibilidade de identificação do agente etiológico, foram denominados borreliose-símile (Borrelia-like).

Modo de Transmissão

Quadro Clínico
1º estágio ou fase aguda, com lesões predominantemente cutâneas;
2º estágio, no qual podem ocorrer manifestações articulares, neurológicas, cardíacas e oftalmológicas;
3º estágio, com quadros reumatológicos, neurológicos, oftalmológicos e cutâneos crônicos.
Manifestações concomitantes

Máculas ou pápulas eritematosas, que aumentam de tamanho, formando placas isoladas ou múltiplas, com bordas descontínuas e centro claro, cianótico e/ ou descamativo, que se expande centrifugamente, podendo atingir grandes diâmetros. Prurido e ardência pode surgir.

Quadro Clínico

Quadro Clínico
sintomas gerais extracutâneos : febre (55%), cansaço (48%), desconforto
musculoesquelético (47%) e cefaléia (38%). Dores articulares e sintomas neurológicos também foram observados.
Manifestações gerais outras, como mal-estar, rigidez de nuca, fotossensibilidade, conjuntivite, linfoadenopatias, são descritas e podem durar várias semanas ou mais, na ausência de tratamento

DIAGNÓSTICO
Epidemiológicos, clínicos e laboratoriais
Sorologia ELISA (enzime-linked immunosorbent assay)e a imunofluorescência indireta (IFI)
Exames histológico Imunoistoquímico, PCR e cultura

Tratamento
Eritema migratório, linfocitoma cutis e outras manifestações iniciais: a droga de escolha é a doxiciclina, na dose de 100mg, de 12/12 horas, por via oral (VO), durante 14 dias. Em crianças com menos de 12 anos, utiliza-se a amoxicilina, 500mg, VO, de 8/8 horas, ou azitromicina, 20mg/kg/dia, VO, em dose única diária, por 14 dias. Em grávidas, recomenda-se a eritromicina, na dose de 500mg, de 6/6 horas, VO, durante 14 dias;

Tratamento
b) Manifestações neurológicas, cardíacas e oftalmológicas: ceftriaxona, 2g/dia, por via endovenosa (EV), de 21 a 28 dias. Outras opções terapêuticas são a cefotaxima 2g/dia, EV, ou penicilina cristalina, 18-24 milhões de unidades/dia, EV, divididas em seis doses diárias, durante 21 a 28 dias;
c) Manifestações articulares: doxiciclina, 100mg, de 12/12 horas, VO, no mínimo, durante 28 dias. Na impossibilidade de se usar a doxiciclina, emprega-se a amoxicilina ou a eritromicina;
d) Acrodermatite crônica atrofiante (ACA): doxiciclina, 100mg, de 12/12 horas, VO, ou amoxicilina, 500mg, de 8/8 horas, VO, por 21 dias
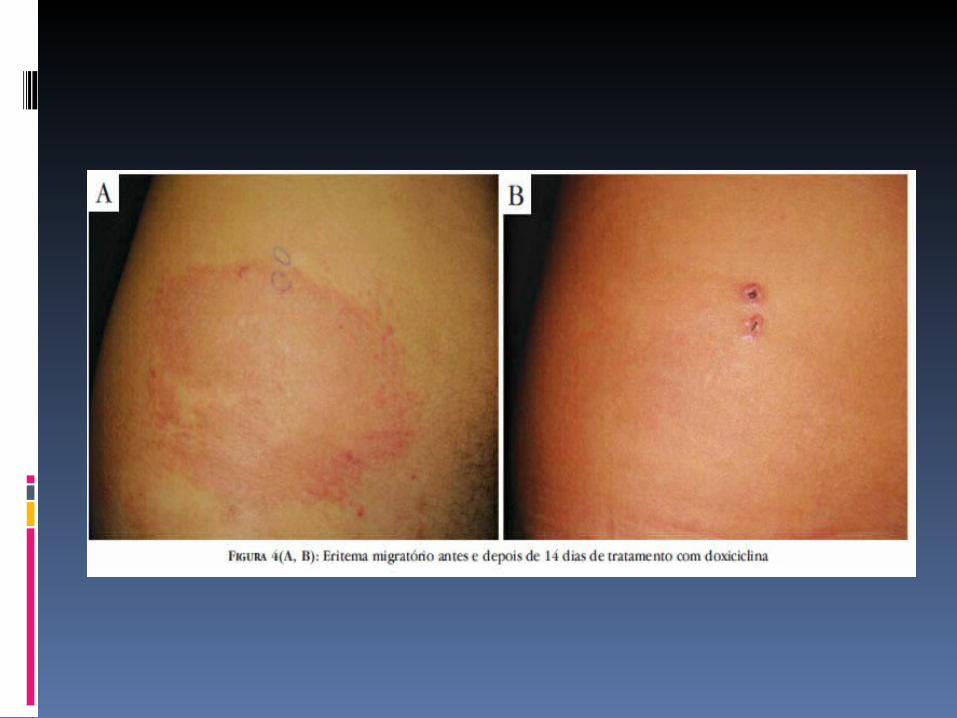

LÚPUS ERITEMATOSO SISTEMICO JUVENIL CONCEITO: é uma doença auto-imune
caracterizada por uma inflamação generalizada crônica e difusa em todo o corpo. A depender do paciente, há possibilidade de acometimento de alguns ou vários órgãos e sistemas do corpo ao mesmo tempo.
Caracteristicamente, o LESJ inicia-se até os 18 anos de idade. Se iniciar após 19 anos será acompanhado pelo reumatologista de adulto e não o pediátrico.

EPIDEMIOLOGIA
Terceira doença mais frequente nos ambulatórios de Reumatologia Pediátrica, seguido pela artrite idiopática juvenil e vasculites
Predomínio do sexo feminino, em todas as idades, sendo que após a adolescência é de 8a 13 vezes mais frequente no sexo feminino.
A real prevalência de LESJ é desconhecida em nosso país.
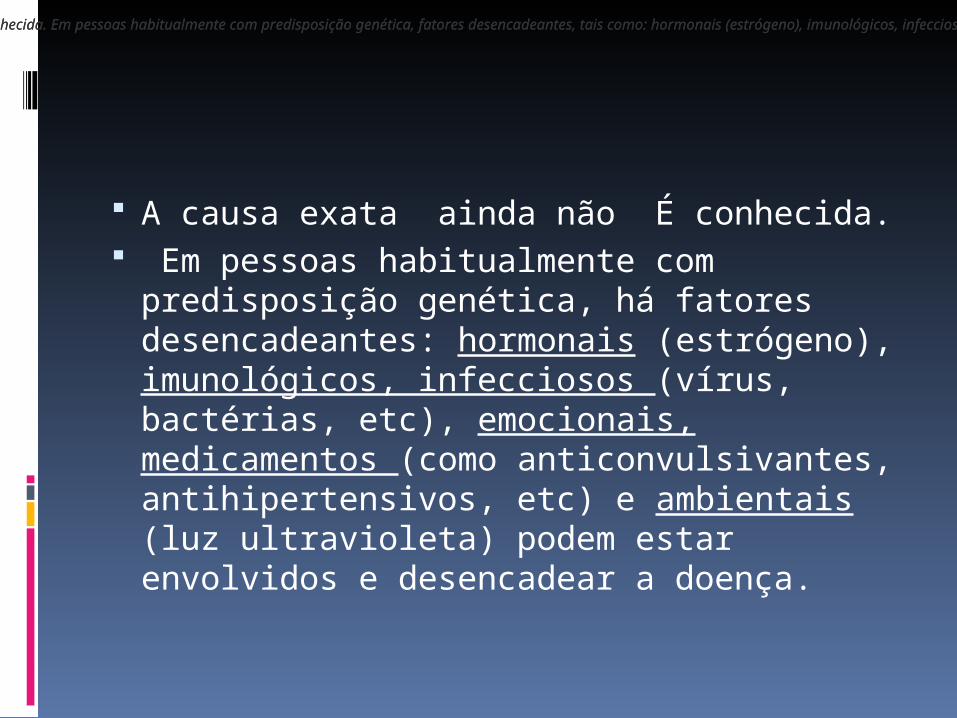
A causa exata ainda não É conhecida. Em pessoas habitualmente com
predisposição genética, há fatores desencadeantes: hormonais (estrógeno), imunológicos, infecciosos (vírus, bactérias, etc), emocionais, medicamentos (como anticonvulsivantes, antihipertensivos, etc) e ambientais (luz ultravioleta) podem estar envolvidos e desencadear a doença.
A causa exata da LESJ ainda não é conhecida. Em pessoas habitualmente com predisposição genética, fatores desencadeantes, tais como: hormonais (estrógeno), imunológicos, infecciosos (vírus, bactérias, etc), emocionais, medicamentos (como anticonvulsivantes, antihipertensivos, etc) e ambientais (luz ultravioleta) podem estar envolvidos e desencadear a doença.A causa exata da LESJ ainda não é conhecida. Em pessoas habitualmente com predisposição genética, fatores desencadeantes, tais como: hormonais (estrógeno), imunológicos, infecciosos (vírus, bactérias, etc), emocionais, medicamentos (como anticonvulsivantes, antihipertensivos, etc) e ambientais (luz ultravioleta) podem estar envolvidos e desencadear a doença.

O indivíduo produz anticorpos direcionados contra seus próprios tecidos (os auto-anticorpos, como anti-DNA, anticorpos anti-fosfolípides, etc). Esses auto-anticorpos vão se depositando em vários órgãos e com o evoluir da doença pode haver progressivo comprometimento de suas funções.
O lúpus não é uma doença infecto-contagiosa

A principal diferença entre a doença do adulto e a juvenil se dá no comprometimento renal, mais frequente e com formas mais graves na forma juvenil.
Os critérios diagnósticos utilizados para o LESJ são os mesmos e o tratamento bastante similar ao adulto, com ênfase nas vacinações e no acompanhamento do crescimento e desenvolvimento da criança e adolescente.

DIAGNÓSTICO
Para o diagnóstico de LESJ é necessária a presença de quatro ou mais dos 11 critérios. Estes critérios podem aparecer todos ao mesmo tempo ou demorar alguns meses para o diagnóstico definitivo da doença (em média de 4 meses).

CRITÉRIOS
1. Eritema malar2. Eritema discóide3. Fotossensibilidade4. Úlcera de mucosa oral ou nasal5. Artrite não erosiva6. Serosites (pleurite e/ou pericardite)7. Alterações renais (proteinúria superior a 500 mg/dia e/ou presença de cilindrúria)

CRITÉRIOS
8. Alterações neurológicas (convulsão e/ou psicose na ausência de distúrbios metabólicos, hipertensão arterial ou infecções)9. Alterações hematológicas [anemia hemolítica com reticulocitose e/ou leucopenia (menos que 4.000/mm3) e/ou linfopenia (menos que 1.500/mm3) e/ou plaquetopenia (menos que 100.000/mm3), em duas ou mais ocasiões]

CRITÉRIOS
10. Alterações imunológicas [presença de anticorpos anti-fosfolípides (anti-cardiolipina IgM ou IgG e/ou anti-coagulante lúpico e/ou reações sorológicas falsamente positivas para sífilis) e/ou anticorpo anti-DNA e/ou anticorpo anti-Sm]
11. FAN positivo

QUADRO CLINICO
É uma doença de evolução imprevisível, possui caráter crônico com períodos de remissão e exacerbação das manifestações clínicas.
As principais manifestações clínicas iniciais do LESJ compreendem febre prolongada, redução do apetite, perda de peso, comprometimentos articular, da pele e dos rins.
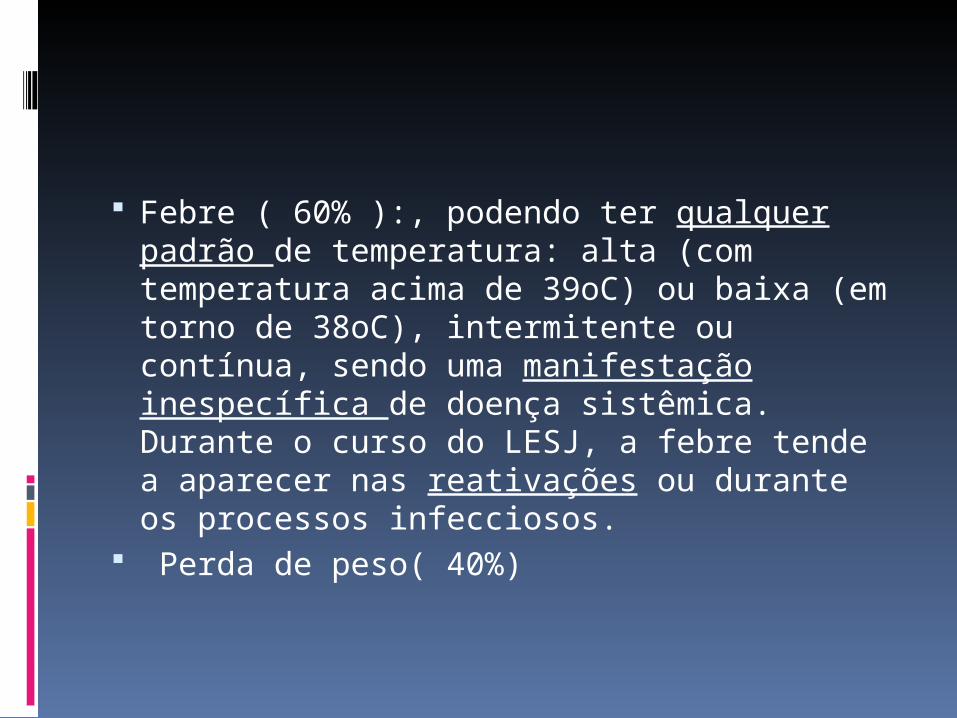
Febre ( 60% ):, podendo ter qualquer padrão de temperatura: alta (com temperatura acima de 39oC) ou baixa (em torno de 38oC), intermitente ou contínua, sendo uma manifestação inespecífica de doença sistêmica. Durante o curso do LESJ, a febre tende a aparecer nas reativações ou durante os processos infecciosos.
Perda de peso( 40%)

Adenomegalia (50%) localizada ou generalizada.
Hepatomegalia e esplenomegalia (30 a 40%): discretas a moderadas.
Icterícia: é um achado raro e associa-se habitualmente com a presença de anemia hemolítica.

Comprometimento articular, músculos e ossos ( 88%): dores articulares ou artrites, podendo envolver articulações dos joelhos, tornozelos, punhos, mãos, pés, entre outras.
Miosite, com fraqueza muscular: pode ocorrer nos períodos de atividade da doença.
Necrose avascular: principalmente nos ossos de sustentação (fêmur e tíbia) secundária à atividade da doença ou como complicação do tratamento prolongado com corticosteróide.

Lesões cutâneas: eritema malar, úlceras de mucosa, púrpura palpável, urticária, eritema polimorfo, eritema palmo-plantar, pequenos infartos digitais, livedo reticular, nódulos, lúpus discóide e bolhas.
O eritema desencadeado ou exacerbado pela exposição solar (fotossensibilidade) é encontrado em cerca de 50% dos pacientes, habitualmente localizado em face, braços pescoço e pernas.

OBS: O eritema malar em asa de borboleta, apesar de altamente sugestivo de LES, não é específico, podendo ocorrer em outras condições como reações a drogas, dermatomiosite juvenil, dermatite seborréica e acne rosácea.

Lúpus discoide (20%): lesões crônicas e iniciam como placas ou pápulas eritematosas cobertas com escamas aderentes. Com a progressão, as escamas se tornam grossas e podem desenvolver atrofias e cicatrizes, com lesões esbranquiçadas ou escuras. Há uma predileção pelo couro cabeludo, face e orelhas.

Nefrite (70-100%): é precoce, geralmente nos primeiros dois anos da doença. As alterações no exame de urina são variadas: hematúria, leucocitúria ou proteinúria.
Os pacientes podem apresentar HAS, edema e insuficiência renal.
A biópsia renal é indicada quando há manifestações clínicas da doença renal e/ou alterações laboratoriais persistentes e/ou graves, para avaliar o tipo da nefrite e propor tratamento específico.

Envolvimentos do sistema nervoso e psiquiátrico (50%): dor de cabeça de forte intensidade, convulsão, distúrbios do comportamento (alucinações, depressão, etc), raramente derrame (acidente vascular cerebral - AVC) e coma.

Comprometimento hematológico: pode-se manifestar com plaquetopenia, anemia hemolítica, leucopenia e linfopenia. O hemograma com plaquetas, dosagem de reticulócitos e a pesquisa do teste de Coombs são necessários para avaliar este comprometimento.
Estas manifestações podem iniciar o LESJ ou se apresentarem durante a evolução da doença.
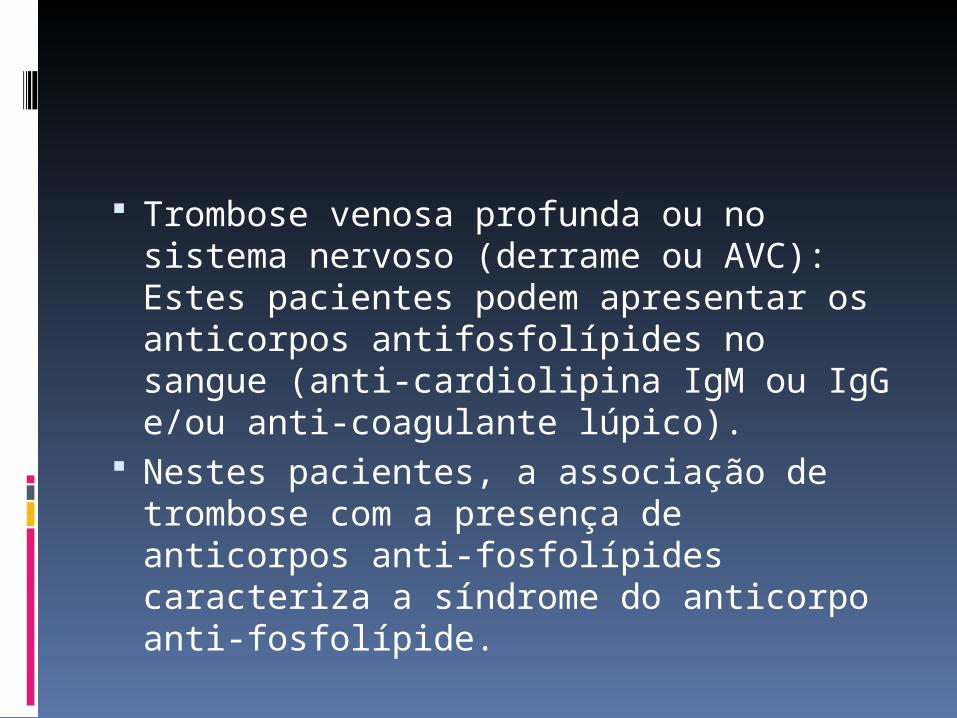
Trombose venosa profunda ou no sistema nervoso (derrame ou AVC): Estes pacientes podem apresentar os anticorpos antifosfolípides no sangue (anti-cardiolipina IgM ou IgG e/ou anti-coagulante lúpico).
Nestes pacientes, a associação de trombose com a presença de anticorpos anti-fosfolípides caracteriza a síndrome do anticorpo anti-fosfolípide.

Comprometimento pulmonar ( 50%): pleurite, e raramente pneumonite intersticial, hipertensão pulmonar e hemorragia pulmonar.
Pleurite e derrame pleural (10-37%). O comprometimento pleural geralmente é
assintomático ou simplesmente um achado radiológico. Ele é usualmente pequeno e evidenciado em um pulmão, devendo ser excluídas causas infecciosas.

Coração ( 50%): pericardite (12 a 40%), que é geralmente assintomática, tendo como manifestações clínicas: dor no peito que piora com a posição e/ou taquipnéia e/ou taquicardia.

TRATAMENTO
Boa relação entre médico, paciente e familiares, uma vez que a sua duração é prolongada.
Nutrição balanceada. Esportes e atividades de lazer são
indicados, nas fases sem atividade importante.
Protetor solar que bloqueia os raios ultravioletas A e B (UVA e UVB).

Os corticosteróides e os anti-maláricos são os medicamentos iniciais de escolha.
Prednisona, dose inicial de 0,5 a 2 mg/Kg/dia. Doses altas (1 a 2 mg/Kg/dia) são utilizadas
no controle da crise aguda inicial, envolvimento do sistema nervoso, anemia hemolítica e nefrite.
Baixas doses de prednisona (≤ 0,5 mg/Kg/dia) indicadas no controle da febre, comprometimento da pele, artrite,pericardite e pleurite.

CUIDADOS NO TRATAMENTO
Evitar o uso excessivo de sal e restringir a ingestão de alimentos calóricos.
Suplementar cálcio e vitamina D. Efeitos colaterais: face em lua cheia,
obesidade, estrias, aumento dos pêlos, infecções, catarata, parada do crescimento, úlcera do estômago, hipertensão arterial e osteoporose.

Pulsoterapia endovenosa com metilprednisolona é indicada em situações graves: nefrite grave, envolvimento grave do SNC ou psiquiátrico, alterações no sangue e hemorragia pulmonar ou para poupar ou reduzir o uso de doses altas de corticosteróide por via oral.
Efeitos colaterais: HAS, risco de infecções e manifestações vasomotoras transitórias (como visão embaçada e intensificação do vermelhão na face).

O tratamento tópico, com corticosteróide está indicado no lúpus discóide e lesões vasculíticas .
Os corticosteróides tópicos não fluorados, como hidrocortisona a 1%, são aplicados 1 a 3 vezes ao dia, diretamente na lesão em média por duas semanas.
Acetonide de triamcinolona em forma de pasta dental emoliente (Orabase) pode ser usado nas feridas na boca, nariz e lábio 2 a 3 vezes ao dia.

Anti-maláricos (difosfato de cloroquina ou sulfato de hidroxicloroquina) devem ser utilizados em todos os casos de LESJ.
Têm mostrado boa resposta no controle da atividade da doença, melhorando a sobrevida, reduzindo os anticorpos anti-fosfolípides, melhorando a fotossensibilidade, além de funcionar como imunomodulador, auxilia na redução do colesterol e das doses dos corticosteróides.

Os antiinflamatórios não hormonais são usados no controle das artrite e miosite, particularmente o naproxeno.
O Ibuprofeno é um antiinflamatório não hormonal que pode causar reação de hipersensibilidade e meningite asséptica e não deve ser utilizado em pacientes com LESJ.

Outros medicamentos podem ser utilizados no controle dos pacientes que não respondem aos corticosteróides e anti-maláricos ciclofosfamida, metotrexate, azatioprina, micofenolato mofetil, ciclosporina, talidomida, dapsona, rituximabe (anti-CD20), entre outros.
Nos pacientes internados e em situações de gravidade clínica pode-se utilizar a gamaglobulina endovenosa e/ou plasmaferese

A ciclofosfamida endovenosa é realizada em pacientes com LESJ, habitualmente com sete infusões mensais e manutenção com 10 infusões trimestral (total de 36 meses), ou ao invés da ciclofosfamida a manutenção é realizada com outro imunossupressor (azatioprina ou micofenolato mofetil).
Antes, no momento e após a infusão da ciclofosfamida recomenda-se a ingestão de líquidos.

É indicado nas nefrites graves (cuja biópsia do rim evidenciou formas proliferativa focal ou difusa), envolvimento do SNC ou psiquiátrico grave, alterações no sangue (plaquetopenia e anemia hemolítica), hemorragia pulmonar, etc.
Efeitos adversos: enjôos, vômitos e hipertensão arterial, cistite hemorrágica, infertilidade e risco de câncer futuro.

O tratamento da síndrome do anticorpo anti-fosfolípide inclui anti-coagulantes (heparina de baixo peso molecular e warfarin) que são utilizados por um período muito prolongado, preferencial a vida inteira.
O uso de aspirina em doses baixas (3 a 5 mg/Kg/dia) é indicado nos casos com presença dos anticorpos anti-fosfolípides persistentemente sem comprometimento clínico.

Deve-se evitar a aplicação de vacinas de vírus vivos em pacientes em uso de corticosteróides e imunossupressores.

Bibliografia da Fonseca , A. H.; Salles ,R. de S.; Salles, S. de A. N. ;
Madureira, R. C.; Yoshinar , N. H. Borreliose de Lyme simile: uma doença emergente e relevante para a dermatologia no Brasil. An Bras Dermatol. 2005;80(2):171-8.
Santos ,M. ; Júnior ,V. H.; Rodrigues ,R. R.; Talhari , S. Borreliose de Lyme ; An. Bras. Dermatol. Nov./Dec. 2010 vol.85 no.6 Rio de Janeiro ; 85(6):930-8.
Vera RODRIGUES et al, Síndrome linfoproliferativo autoimune, Acta Med Port. 2011; 24(5):833-836.
Sacchetti.S.B, Marini.R, Terreri.M.T; Lupus eritematoso sistêmico juvenil. Rev . Bras. Reumatol Maio/2005
Machado, Claudia and Ruperto, Nicolino Consenso em reumatologia pediátrica: parte II - definição de melhora clínica para o lúpus eritematoso sistêmico e dermatomiosite juvenil. Rev. Bras. Reumatol., Fev 2005,

![Zitai - AsianNet24Pcs (42LV~56LV) / 36Pcs (90LV~125LV) Z2 Ø Ú Z2 System ( C-Series close loop feedback control ) Z2 Control System Z2 Ø Ú 3 á ½ ú ê ] Ø Ú Y c E d _ g î …](https://img.document.onl/doc/110x75/60f94c1414bd8406a033fcf3/zitai-asiannet-24pcs-42lv56lv-36pcs-90lv125lv-z2-z2-system-c-series.jpg)




![U q÷í KV sM Ä »ç±Ù Ä w] º...ºp ÝzK Mx Ø C {w !Z t `o '"9y ¢ Ú è£ l¢ q Í ú ïù¢ ½ ´Ã ¬© È è ~ O] l¢ GÝ Ê j ? é ¢ Ú è£ '"9 Ú è£ l Í ú Ó l v](https://img.document.onl/doc/110x75/5ea63cfafca6062015765e76/u-q-kv-sm-w-p-zk-mx-c-w-z-t-o-9y-.jpg)























