Embed Size (px)
Citation preview

Curso de Farmácia - Profª Cristina Maria Pereira dos Santos
Interações intermoleculares - Estado de agregação da matéria
A formação de uma molécula ou de agregados de moléculas é devida,
respectivamente, à existência de interações intramoleculares (relacionadas aos diferentes
tipos de ligações químicas) e intermoleculares ( obtenção de um arranjo energeticamente
estável).
No processo de agregação, as substâncias são submetidas a forças de atração e
repulsão simultâneas que determinarão, junto com a energia cinética, os estados sólido,
líquido e gasoso das substâncias.
gEstado Sólido
As forças de atração e repulsão são significativas, estando de forma equilibrada. As
partículas apresentam-se ordenadas.
gEstado Líquido
As forças de atração e repulsão apresentam papel moderado.
gEstado Gasoso
As forças de atração são pequenas e as forças de repulsão atuam apenas durante as
colisões (que são elásticas, sem perda de energia cinética). As partículas estão em constante
movimento (en. cinética maior do que no líquido).
Sólido Líquido Gasoso
Obs.: Energia potencial º distância entre partículas; energia cinética º movimento
Fluido º termo para líquidos e gases; estado condensado º termo para sólidos e líquidos
GASES
O comportamento físico de um gás pode ser determinado pelas variáveis volume,
pressão e temperatura. O volume do gás corresponde ao volume do recipiente em que ele está
contido. A pressão (definida como força por unidade de área) é função da temperatura e da
altitude (nível do mar ou km acima ou abaixo).
No Sistema Internacional a pressão é dada em Pascal (Pa), o volume em m , a3
temperatura em K e a constante dos gases (R) em Joule (J). (R = 8,314 J/K mol = 0,082 atm L/K mol
= 1,987 cal/K mol; 1 atm = 101325 Pa; 1 dm = 1 L; 1 cal = 4,184 J; 1Latm = 101,3 J; 1J = 24,2 cal).3
*Gases ideais
1

Um gás ideal é um modelo de substância gasosa que obedece a uma equação de estado,
permitindo a determinação aproximada das propriedades do sistema
Equação de estado: pV = nRT
Num gás ideal as interações entre as moléculas são nulas. Como não ocorre atração não faz
psentido falar de energia potencial (E = 0), pois não importa a distância entre as moléculas. O gás
ideal apresenta apenas energia cinética, que é diretamente proporcional à temperatura.
A origem do conceito de idealidade está nos estudos de:
Boyle (séc. XVII): V % 1/P ou PV = cte
A uma temperatura constante, o volume ocupado por uma quantidade fixa de gás é
inversamente proporcional à pressão aplicada.
Ex.: Bomba para encher o pneu da bicicleta
O gás ao ser comprimido pelo êmbolo da bomba (diminuição de V) tem sua
pressão aumentada. Havendo uma conexão com o pneu, o gás escapará para a região onde a
pressão for menor, enchendo, assim, o pneu da bicicleta.
Charles: V % T ou V/T = cte
À pressão constante, o volume de uma dada quantidade de gás varia diretamente com
a temperatura. O ponto onde todas as retas se interceptam corresponde ao zero absoluto de
temperatura (-273,15°C)
Gay-Lussac: P % T ou P/T = cte
Quando um gás é aquecido num recipiente de volume constante sua pressão aumenta.
p V p
1/V -273°C T T
Boyle Charles Gay-Lussac
2

*Gases reais
Quando estudamos a leis de Charles, percebemos que a diminuição de temperatura leva
a uma diminuição do volume, fazendo com que as moléculas do gás fiquem mais próximas e que
comece a ocorrer força de atração entre elas. Chegará um momento em que ocorrerá a
condensação do gás e ele não mais atingirá o zero absoluto previsto pela lei de Charles.
Esse desvio da idealidade percebido a baixas temperaturas e altas pressões levou ao
desenvolvimento de modelos que descrevessem o comportamento de um gás real.
Van der Waals propõe as seguintes correções para a pressão e o volume do gás real:
Como o volume da molécula de um gás real não é desprezível, o volume do gás real deve ser
maior do que o do gás ideal de uma quantidade nb.
real ideal ideal real V = V + nb þ V = V - nb
Como a colisão das moléculas do gás com a parede ocorrem de forma menos energética devido
às forças atrativas entre as moléculas, a pressão do gás real é menor do que do gás ideal de uma
quantidade diretamente proporcional a n /V .2 2
real ideal ideal realp = p - a n /V þ p = p + a n /V2 2 2 2
Assim, substituindo as correções na equação dos gases ideais, obtém-se a equação de estado de
van der Waals
[p + (n a/V )] (V - nb) = nRT,2 2
onde a e b são constantes tabeladas e n o nº de moles. Na correção, a está relacionado às forças
atrativas e b estabelece um volume finito. A tabela a seguir apresenta os valores experimentais
atribuídos às constantes de correção para alguns gases.
3

Termodinâmica Química
O universo encontra-se dividido em sistema e ambiente.
Reagentes e produtos þ sistema
Ambiente þ pode sofrer influência dos componentes do sistema
A termodinâmica estuda as trocas de energia ocorridas durante os processos químicos.
Assim, esses processos químicos podem ser:
a) Adiabático þ não ocorre transferência de calor através da fronteira sistema-ambiente.
Ex.: Nas reações rápidas, o calor gerado não é dissipado havendo aumento de temperatura.
b) Isotérmico þ ocorre transferência de calor através do contato térmico entre o sistema e o
ambiente, mantendo-se a temperatura constante.
Ex.: As reações bioquímicas do nosso corpo ocorrem mantendo a temperatura em ±36 °C.
A discussão das transformações químicas necessita, inicialmente, da definição das
propriedades do sistema através da pressão, temperatura, do número de moles de cada
componente, estado físico, etc, que correspondem às funções de estado. As funções de estado só
dependem do estado inicial e final do sistema e têm suas relações estabelecidas através de uma
equação de estado.
Princípio zero da termodinâmica
Uma transferência de calor ocorrerá de um sistema mais quente para um mais frio.
1° Princípio da Termodinâmica
Os processos ocorrem com conservação de energia. A energia, manifestada sobre a
forma de calor ou trabalho, pode sofrer interconversão, sendo a quantidade total do sistema
mantida a mesma. Considerando U a energia interna do sistema e que o sistema realiza trabalho
sobre o ambiente, o que diminui sua energia interna, teremos
final inicial ÄU = U - U = q - W
A energia interna e ÄU dependem apenas dos estados inicial e final, sendo funções de
estado. A quantidade de calor q e o trabalho W, dependerão dos processos realizados, o que faz
com que eles não sejam funções de estado.
* Trabalho (W)
Na termodinâmica, o trabalho ocorre na maioria das vezes na forma de trabalho de
exp.expansão W Ou trabalho termoelétrico. Existem outras formas de trabalho , que são o trabalho
elástico, elétrico e de expansão de superfície, que são chamadas trabalho útil (W’).
exp. W = W + W’
Quando W’ é considerado nulo,
exp.W = W
4

I- Trabalho relacionado à expansão de um gás ideal contra uma pressão constante
W = Força x distância
C W = Pressão x Área x distância
Pressão = Força/ Área - Força = Pressão x Área W = P x V
*Numa mudança de volume contra uma pressão externa constante, o trabalho de expansão será
exp. exp.W = P ÄV
Considerando um processo isotérmico e espontâneo:
P.Atmosf. P.Atmosf.
÷ ÄV = Äh x área
Gás sob pressão Gás à pressão atmosférica
c pÄU = ÄE + ÄE
p cPara um gás ideal, ÄE = 0 - ÄU = ÄE
c Considerando o processo isotérmico, ÄT = 0 -ÄE = 0,pois a energia cinética apresenta
dependência direta com a temperatura. Assim,
ÄU = q - W = 0 - q = W
*Numa mudança de volume contra um sistema de vácuo, considerando o mesmo processo
isotérmico
P.Atmosf. P.Atmosf.
÷
Gás com vácuo sobre o pistão, num cilindro fechado
O gás irá expandir-se, sem haver resistência alguma - W = 0
Nesse caso, ÄU = 0 - q = W = 0
Obs: Um exemplo de aplicação de um sistema a vácuo são as expansões e compressões de gases
(reais) que circulam isoladamente na tubulação de uma geladeira ou de um ar condicionado.
5

II- Processos Reversíveis e Irreversíveis
Os processos de expansão, como o de compressão, nem sempre ocorrem contra uma
pressão externa constante. Ao aumentarmos o número de etapas de expansão, o valor total de
trabalho, que será a soma dos trabalhos envolvidos em cada etapa, será maior.
Existirá, porém, uma quantidade de trabalho máxima que poderá ser obtida em um
número infinito de etapas, onde a pressão oposta será ligeiramente maior do que a pressão
exercida pelo gás. Como as variações de volume e de trabalho serão muito pequenas teremos
ÄV - dV e W -d W.
Sendo no processo reversível as variações pequenas, a pressão oposta, num processo
ext. gásisotérmico, será praticamente a pressão do gás, o que nos permite dizer que p = p = nRT/V.
Id W = I p dV - Id W = nRT I1/V dV
máx. f iW = nRT ln(V /V )
O trabalho efetuado numa transformação reversível poderá ser determinado calculando-
se a área sob a curva representada num diagrama p x V.
Nota: Uma transformação real espontânea é irreversível e o valor do trabalho será menor
máx.que o valor calculado para W reversível.
*Calor (q)
Forma de energia que pode manifestar-se sob a forma de:
# Calor sensível " quando o calor que flui do sistema ou para o sistema ocasiona
uma variação de temperatura (aquecimento da água), sem que ocorram reações químicas
ou mudanças de fase. Relacionado à variação da energia cinética.
# Calor latente " quando o calor absorvido ou liberado pelo sistema não se
manifesta através de alterações de temperatura (calor envolvido nas mudanças de fase.
Relacionado à variação da energia potencial
6

CAPACIDADE CALORÍFICA (C)
�Encontra-se relacionada ao calor sensível (troca de calor com variação de temperatura)
Idq = IC(T) dT $ Idq = C IdT $ q = C ÄT
�Capacidade calorífica molar (&c = C/n)
Quantidade de calor necessária para elevar de 1°C 1 mol de uma substância.
�Calor específico (c = C/m)
Quantidade de calor necessária para elevar de 1°C 1 g de uma substância.
�Processo de expansão, compressão ou aquecimento, em gases ideais, à pressão ou volume
constante:
p p p pq = &c n ÄT ou q = c m ÄT
v v v vq = &c n ÄT ou q = c m ÄT
�Capacidade calorífica de gases reais são valores próximos aos dos gases ideais.
�Capacidade calorífica de gases ideais:
v pGases monoatômicos: &c = 3/2 R; &c = 5/2 R
v pGases diatômicos: &c = 5/2 R; &c = 7/2 R
v pGases triatômicos: &c = 7/2 R; &c = 9/2 R
Nota: O parâmetro de correlação entre as capacidades caloríficas é o ã
p v 1 2 2 1 ã = c /c o que fornece as relações: T / T = (V /V ) ;ã ! 1
1 1 2 2p V = p V ;ã ã
1 2 1 2T / T = (p /p ) / (ã ! 1) ã
* Entalpia (H)
&Propriedade termodinâmica necessária para caracterizar o calor trocado (absorvido ou
liberado) durante uma transformação química ocorrida à pressão constante.
H = U + pV $ ÄH = ÄU + p ÄV
&Corresponde a uma função de estado:
final inicialÄH = H - H
7

&Processo de expansão, à pressão constante
p exp. pÄU = q - W = q - p ÄV
p pÄH = ÄU + p ÄV = q - p ÄV + p ÄV $ ÄH =q
&Transformações químicas envolvendo fases condensadas (líquidos e sólidos) apresentam
variações de volume muito pequenas $ ÄH � ÄU
&Entalpia das transformações de expansão, compressão ou aquecimento
�Processos adiabáticos (não há troca de calor $ q = 0)
p p v vÄH = q = c m ÄT e ÄU = q = &c n ÄT
. vÄU = q - W = 0 - W $ W = -ÄU $ W = - &c n ÄT
�Processos à pressão constante:
p p v v ÄH = q = c m ÄT e ÄU = q = &c n ÄT
�Processos isotérmicos (ÄT = 0), à pressão ou volume constante:
ÄH = ÄU = 0
ÄU = q - W $ 0 = q - W $ q = W
rev f iPara proc. Reversíveis: q = W = n R T ln (V /V )
�Processos a volume constante:
p ÄH = c m ÄT
v v(ÄU = &c n ÄT ou ÄU = c m ÄT)
vÄU = q - W = q ! p ÄV = q - 0 $ ÄU = q = &c n ÄT
8

Tabela: Valores de trabalho, calor, energia interna e entalpia para
transformações reversíveis envolvendo gases ideais.
Transformação w = p ÄV q Ä U = q - W Ä H
f i f iIsotérmica n R T ln (V /V ) n R T ln (V /V ) Zero Zero
Isobárica p ÄV = n R Ä Tp v p n &c Ä T n &c Ä T n &c Ä T
Isométrica Zerov v p n &c Ä T n &c Ä T n &c Ä T
Adiabáticav -n &c Ä T Zero
v p n &c Ä T n &c Ä T
&Entalpia dos processos físicos
Relacionada apena às mudanças de fase, uma vez que nos processos físicos não ocorrem
mudanças na natureza química das espécies do sistema. (Calor latente)
ÄH negativo - reação exotérmica;
ÄH positivo - reação endotérmica
� Entalpia de vaporização ( líq. ! vap ) e de condensação ou liquefação (vap ! líq.)
líq. ! vap: o calor fornecido corresponde à energia necessária para romper as
forças de atração no líquido e afastar uma molécula da outra.
2 2 vapH O(l) ! H O(g) ÄH° = 44,01 kJ (25°C e 1 atm)+
� Entalpia de fusão (sól. ! líq.) e de solidificação ou congelamento ( líq. ! sól. )
As forças atrativas entre as moléculas são mais fortes no sólido do que no líquido.
2 2 fusH O(s) ! H O(l) ÄH = 6,02 kJ (0°C e 1 atm)+
� Entalpia de sublimação (sól. ! vap.)
subl fus vapSendo a entalpia uma função de estado: ÄH = ÄH + ÄH
Nota: Clausius & Clapeyron verificaram a dependência da pressão de vapor de um líquido
vappuro com a temperatura ( ln p = -ÄH / T + c )te
9

&Entalpia dos processos reacionais - Lei de Hess
(Estabelece que a entalpia de uma reação será a mesma independente de estar ocorrendo
em uma ou em várias etapas.
2 2 combustãoC (graf.) + O (g) ! CO (g) ÄH
ou
2 1C (graf.) + 1/2O (g) ! CO (g) ÄH
2 2 2CO (g) +1/2O (g) ! CO (g) ÄH
1 2ÄH = ÄH + ÄH
(Os calores de formação poderão ser usados, segundo a lei de Hess, para calcular a
entalpia de uma reação.
reação i f,i produtos i f,i reagentesÄH° = (3í ÄH° ) !(3í ÄH° )
2 2 2 2 4Exemplo: Estudo da reação C H (g) + H (g) ! C H (g)
2 2 4 2 4 2C(graf) + 2H (g) ! C H (g) ÄH° form. C H = 12,496 kcal /g mol (Handbook D-82)
2 2 2 2 2 C H (g) ! 2C(graf) + H (g) !ÄH° form. C H = !54,194 kcal /g mol (Handbook D-82)
2 2 2 2 4 C H (g) + H (g) ! C H (g) ÄH° reação = 12,496 x 4,184 kJ/mol !54,194 x 4,184 kJ/mol
ÄH° reação = 52,283 ! 226,75 kJ mol = ! 174,46 kJ mol !1 !1
Ou simplesmente, sem montar a combinação sugerida por Hess:
f,p C2H4 f,i C2H2 f,i H2 ÄH° reação = (1 x ÄH° ) ![( 1 x ÄH° ) + (1 x ÄH° ) ]
ÄH° reação= (1 x 52,283 kJ mol ) ! [(1 x 226,75 kJ mol ) + 0] = ! 174,46 kJ mol !1 !1 !1
10

*Variação da entalpia de reação com a temperatura:
T2 T1 pÄH = ÄH° + ÄC ÄT
p i p,i produtos i p,i reagentesÄC = (3í C ) ! (3í C )
p ionde C é a capacidade calorífica dos reagentes e produtos; í é o coeficiente estequiométrico
(A entalpia de uma reação também poderá ser obtida através das entalpias de combustão
reação i c,i reagentes i c,i produtosÄH° = (3í ÄH° ) ! (3í ÄH° )
2 2 2 2 4Exemplo: Estudo da reação C H (g) + H (g) ! C H (g)
Dados os calores de combustão:
2 2 2 2 2 c C2H2 C H (g) + 5/2O (g) ! CO (g) + H O(l) ÄH° = !1300 kJ mol !1
2 4 2 2 2 c C2H4 C H (g) + 3O (g) ! 2CO (g) + 2H O(l) ÄH° = !1411 kJ mol !1
2 2 2 c H2 H (g) + 1/2O (g) ! H O(l) ÄH° = !285,8 kJ mol !1
Arranjando as equações segundo Hess, teremos:
2 2 2 2 4 reação C H (g) + H (g) ! C H (g) ÄH° = !174,8 kJ mol !1
reaçãoOu, simplesmente, empregando a expressão indicada anteriormente para ÄH° :
reação ÄH° = [(1 x !1300) + (1 x !285,8)] ! [ 1 x !1411] = !1585,8 + 1411 = !174,8 kJ mol !1
3Reagentes 3Produtos
(Calor de solução (entalpia de solução)
O processo de dissolução ou diluição de substâncias em um determinado solvente
apresenta um efeito térmico devido à interação entre as moléculas e pode ser dado como uma
reação química.
2 4 2 2 4 2H SO (l) + 4H O(l) ! H SO .4H O(l) ÄH = -54,06 kJ mol !1
2 4 2 2 4 2H SO .2H O(l) ! H SO (l) + 2H O(l) ÄH = +41,49 kJ mol !1
2 4 2 2 2 4 2H SO .2H O(l) + 2H O(l) !H SO .4H O(l) ÄH = -12,57 kJ mol !1
11

(Determinação da entalpia da reação através das entalpias de ligação
A entalpia de ligação corresponde à variação de entalpia que acompanha a dissociaçãode uma ligação química, estando o composto inicial na fase gasosa.
reação reagentes produtosÄH° = (3Lig. rompidas) - (3Lig. formadas)
2 2 2Dada a reação: 2H (g) + O (g) ! 2H O(g)
*Valores tabelados de entalpia de ligação:
H-H ! ÄH = 436,0 kJ mol ; O=O ! ÄH = 495,4 kJ mol ; O-H ! ÄH = 462,7 kJ mol!1 !1
!1
reaçãoÄH° = [(2x436,0) + 495,4] - [(4x462,7)] = -483,4 kJ mol !1
Exercício: Calcular a entalpia de formação do ácido acético líquido a partir dos seguintes
dados:
Entalpia de ligação: H-H ! ÄH = 436,0 kJ/mol;
O=O ! ÄH = 495,4 kJ/mol;
O-H ! ÄH = 462,7 kJ/mol;
C-H ! ÄH = 412,0 kJ/mol;
C-C ! ÄH = 348,0 kJ/mol;
C=O ! ÄH = 743,0 kJ/mol;
C-O ! ÄH = 360,0 kJ/mol;
Entalpia de vaporização do ácido acético = 61,9 kJ/mol;
Entalpia de atomização do carbono grafite = 716,7 kJ/mol.
12

2° Princípio da termodinâmica
(Estabelece critérios para a espontaneidade e a irreversibilidade observadas nas
transformações naturais.
Um processo ocorre de forma espontânea:
� na busca de um estado mais estável (de energia mais baixa) - liberação de energia;
2 2 2H (g) + 1/2O (g) ! H O(g)
� no sentido de aumentar o estado caótico das partículas
- matéria e energia tende a sofrer o máximo de dispersão possível.
2Exemplo de proc. espontâneo com absorção de energia: KI(s) + H O(l) ! KI(aq)
( ao se dissolver, o sal perde sua estrutura cristalina altamente ordenada
e se difunde pelo líquido.
(Para representar o grau de desordem de um sistema cria-se a propriedade termodinâmica
chamada entropia (S)
Entropia
(Sendo função de estado, a variação de entropia (ÄS) depende dos estados inicial e final.
rev(A adição de calor provoca um aumento na desordem (dispersão da matéria) - ÄS % q
(ÄS é inversamente proporcional à temperatura
- em proc. reversíveis isotérmicos:
rev ÄS = q / T
- em proc. irreversíveis:
irrev ÄS > q / T
(Em sistemas isolados (Universo) - q = 0:
ÄS = 0 (p/ proc. reversíveis)
ÄS > 0 (p/ proc. irreversíveis)
Desigualdade de Clausius:
Universo sistema ambienteÄS = ÄS + ÄS $ 0
(Enunciado da 2ª lei: “Todo processo espontâneo causa aumento da entropia do universo ”.
13

(Processo de aquecimento ou resfriamento
p p*Considerando a pressão constante: ÄS = I(C (T)/T dT
p p p C varia com a temperatura, sendo ÄS a área sob a curva descrita pelo gráfico C X ln
T.
p p p f iP/ ÄT pequenos: ÄS = C I1/T dT = c m ln(T /T )
V V*Considerando o volume constante: ÄS = I(C (T)/T dT
V V V f iP/ ÄT pequenos: ÄS = C I1/T dT = c m ln(T /T )
(Processo de expansão e compressão
Expansão " aumento de entropia
Compressão " diminuição de entropia
Transformação isotérmica para um gás ideal
c ÄT = 0 - ÄE = 0
c p � ÄU = ÄE + ÄE = 0
p Gás ideal - ÄE = 0
ÄU = q ! W - q = W (pois ÄU = 0)
f iQuando p é constante:Idq = IdpV � q = Ip dV - q = nRT ln(V /V )
f i q = !IV dp - q = ! nRT ln(p /p )
p f i V f iÄS = q/T � ÄS = nR ln(V /V ) ; ÄS = ! nR ln(p /p )
(Entropia de reação
reação i i produtos i i reagentesÄS° = (3í S° ) - (3í S° )
*Variação da entropia de reação com a temperatura:
T2 T1 p 2 1ÄS = ÄS° + ÄC ln(T /T )
p i p,i produtos i p,i reagentesÄC = (3í C ) - (3í C )
p ionde C é a capacidade calorífica dos reagentes e produtos; í é o coeficiente estequiométrico
14

(Critério de espontaneidade dos processos
À p e T constantes, o calor fornecido ao sistema é igual ao calor fornecido ao
ambiente, com sinal contrário:
sistema ambiente ambiente sistemaÄH = !q - q = !ÄH
ambiente ambiente sistemaÄS = q /T = !ÄH /T
sistema ambiente sistema sistemaÄS = ÄS + ÄS - ÄS = ÄS + (!ÄH /T)
sistema sistemaTÄS = TÄS ! ÄH
sistemaTÄS = !(ÄH !TÄS)
Para um proc. espontâneo ÄS > 0 e, como T, em Kelvin, também é positivo, o produto T ÄS
sistematambém terá que ser positivo. Isso implica que (ÄH !TÄS) < 0
�Energia Livre de Gibbs (G)
Grandeza que expressa o critério de espontaneidade
G = H - TS - ÄG = ÄH !TÄS
Para um proc. espontâneo ÄG = ÄH !TÄS < 0
Na verdade, a reação só caminha até que ÄG = 0
Variação de entropia na mudança de fase sól.W líq.
No equilíbrio: ÄG = 0 - ÄH = TÄS
fusãoSendo ÄH da água, a 0°C = 1437 cal:
sistema fusão fusão ÄS = ÄH / T = 1437 cal / 273 K = 5,26 cal/K
ambiente sistema ÄS = !ÄS = !5,26 cal/K
Universo ÄS = 0
No caso da fusão estar ocorrendo num ambiente numa temperatura de 5°C (=278K),
teremos:
sistema fusão fusãoÄS = ÄH / T = 1437 cal / 273 K = 5,26 cal K!1
ambiente fusão ambienteÄS = !ÄH / T = !1437 cal / 278 K = !5,17 cal K!1
Universo sistema ambienteÄS = ÄS + ÄS = +0,09 cal K - ÄS > 0 e o processo é espontâneo!1
15

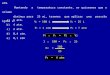
�Variação de G dependente da temperatura
ÄH ÄS ÄG
Negativo Negativo Negativo p/ T baixa; Positivo p/ T alta
Positivo Negativo Positivo (P. Não-espontâneo)
Negativo Positivo Negativo (P. espontâneo)
Positivo Positivo Positivo p/ T baixa; Negativo p/ T alta
Nota: Para processos a volume e temperaturas constantes usa-se a energia livre de
Helmholtz (F)
F = U - TS
3º Princípio da termodinâmica
“A entropia de qualquer substância cristalina pura no zero absoluto é igual a
zero”.
(Permite a determinação da entropia absoluta de uma substância no estado padrão a 25°C
e 1 atm.
2 21º ex.: C(s, grafite) + O (g) ! CO (g)
f 2 fÄH° (CO ) = -94,1 kcal/mol (ÄH° - calor de formação padrão a 25°C e 1 atm.
fLembrar que ÄH° para elemento na forma
natural = 0 )
f CO2 C O2ÄS° = S° - (S° +S° ) = 51,1 - (1,36 + 49,0) = 0,7 cal/mol K
f f fÄG° = ÄH° - TÄS° = -94,1 x 10 - 298. 0,7 3
f- ÄG° = -94,3 kcal/mol
ÄG° corresponde à variação de energia livre para o caso de uma transformação total de
produtos a reagentes.
(Para o avanço da reação:
16

(MG/Mî) = ÄG° + RTlnQ
No caso do equilíbrio: (MG/Mî) = 0 - ÄG° = -RT lnk (onde k é a constante de equilíbrio)
*Variação de k com a temperatura:
Nem sempre temos dados tabelados para todas as temperaturas, por isso é útil a
relação:
ln(k2/k1) =(- ÄH°/R) [(1/T2) - (1/T1)]
*Variação de ÄG° com o pH :
ÄG°’ = ÄG° + RTln(1/[H+]) - ÄG°’ = ÄG° + 2,303RTlog(1/[H+] )
Como pH = - log[H+] = log(1/[H+] ) - ÄG°’ = ÄG° + 2,303RT pH
17

Curso de Farmácia
1ª Lista de Exercícios - Gases / Termodinâmica
1-a)Defina e explique: sistema, ambiente, função de estado, equação de estado e entalpia.
b)De que forma os estudos de Boyle, Charles e Gay-Lussac contribuíram para o estudo dos
gases reais?
2-Como você caracterizaria o volume e a pressão de um gás real em relação ao gás ideal?
3- 10,0 dm de um gás ideal a 1500 kPa, expande-se isotermicamente em duas etapas. Na3
primeira, a pressão externa é mantida em 750 kPa e na segunda em 100 kPa. a) Quais são
as variações globais de energia interna do sistema e do ambiente? b) Quais os valores de q
e w para cada etapa?
4-Em que se baseia o primeiro princípio da termodinâmica? De que forma o primeiro
princípio da termodinâmica é verificado no sistema de resfriamento de um aparelho de ar
condicionado? Por que em determinados pontos do circuito do gás o processo é dito
adiabático?
5- Qual a diferença entre processo reversível e irreversível. Que expressões caracterizam o
trabalho verificado em cada um deles? Na prática, qual dos dois processos (reversível ou
irreversível) ocorre?
vap. 26-Sendo ÄH (H O)=43,9 kJ/mol. Calcule q, w, e ÄU para a vaporização da água.25 EC
7- Se 500 cm de um gás são comprimidos a 250 cm , sob uma pressão externa constante de3 3
300 kPa, absorvendo 12,5 kJ de calor, qual o valor para q, w e ÄU, em kJ. Qual o valor de
ÄU para o ambiente? Respostas no SI.
fus vap8- Se o calor latente de fusão e vaporização da água são iguais a ÄH =6,02 kJ/mol e ÄH
= 40,7 kJ/mol, a 0 EC e 100 EC, respectivamente, calcule o ÄS para cada um desses processos
e explique porque um é maior do que o outro.
f9-A partir dos valores de ÄH E e SE a 25 EC dados a seguir, calcule o ponto de ebulição da
água.
f 2 f 2Valores a 25 EC: ÄH (H O(l)) = !285,830 kJ/mol; ÄH (H O(g)) = !241,826 kJ/mol;o o
2 2S (H O(l) = 69,950 J K mol ; S (H O(g) = 188,834 J K molo -1 -1 o -1 -1
10-Quais os postulados do segundo e terceiro princípios da termodinâmica? Que parâmetros
estão envolvidos em cada um deles?
18