Embed Size (px)
Citation preview

GENÉTICA DO DESENVOLVIMENT
O
Profa. Dra. Ana Elizabete Silva Genética Humana

BIOLOGIA E GENÉTICA DO DESENVOLVIMENTO
Duas hipóteses da embriologia:
Preformacionismo (século XVII): visão de que todos os órgãos adultos estariam pré configurados no esperma ou óvulo.
Hartsoecker (1677): publicou em revista científica a descrição do homúnculo presente na porção cefálica do espermatozóide, fortalecendo a teoria do animalculismo.
“Um organismo
desenvolve de si
próprio”

Epigênese: propunha que um organismo adulto se desenvolveria a partir de uma forma indiferenciada
ZIGOTO
ADULTOTRANSFORMAÇÃO
Como é possível que grupos de células assumam diferentes funções já que elas tem genoma idênticos?
DIFERENCIAÇÃO CELULAR: regulação e diferenciação da atividade de genesExpressão gênica: quando, onde e quanto?
Duas Hipóteses Da Embriologia:
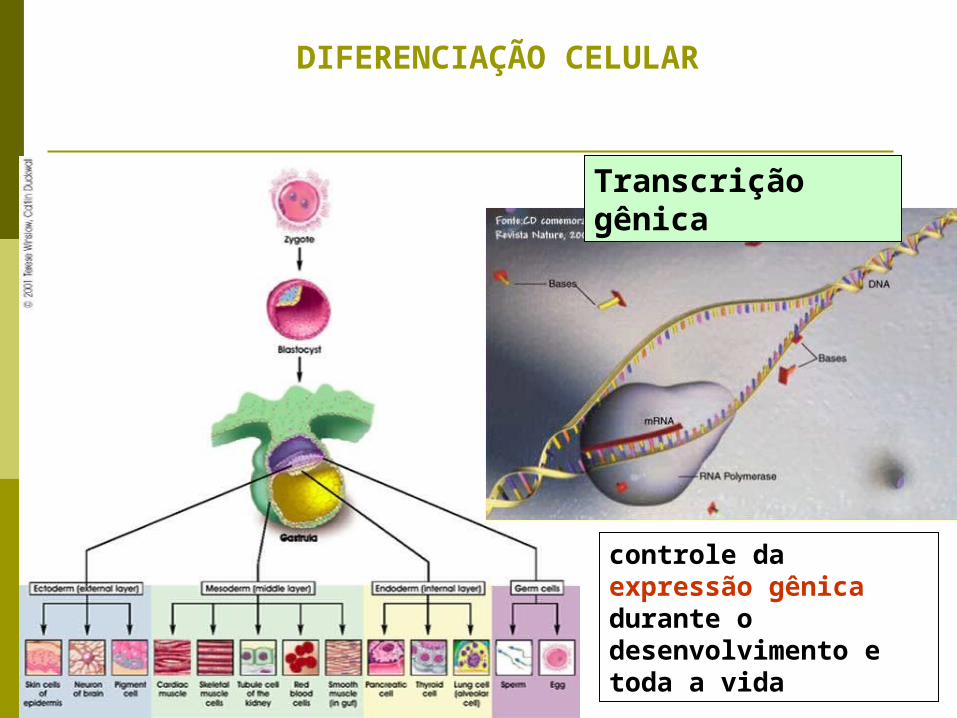
DIFERENCIAÇÃO CELULAR
Transcrição gênica
controle da expressão gênica durante o desenvolvimento e toda a vida
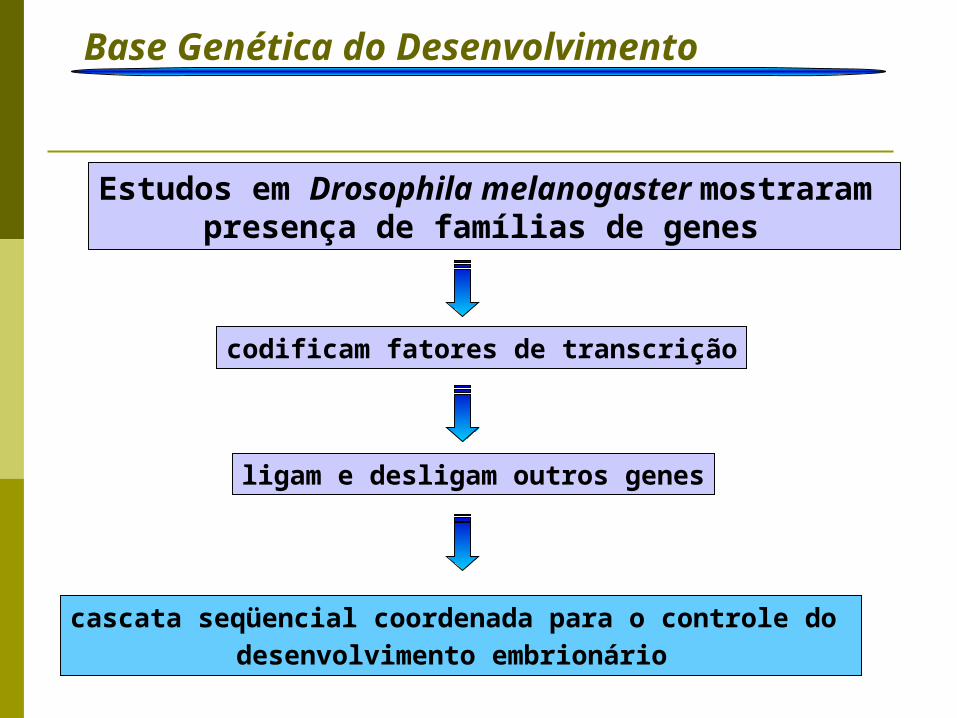
Base Genética do Desenvolvimento Introdução
Estudos em Drosophila melanogaster mostraram presença de famílias de genes
codificam fatores de transcrição
ligam e desligam outros genes
cascata seqüencial coordenada para o controle do
desenvolvimento embrionário
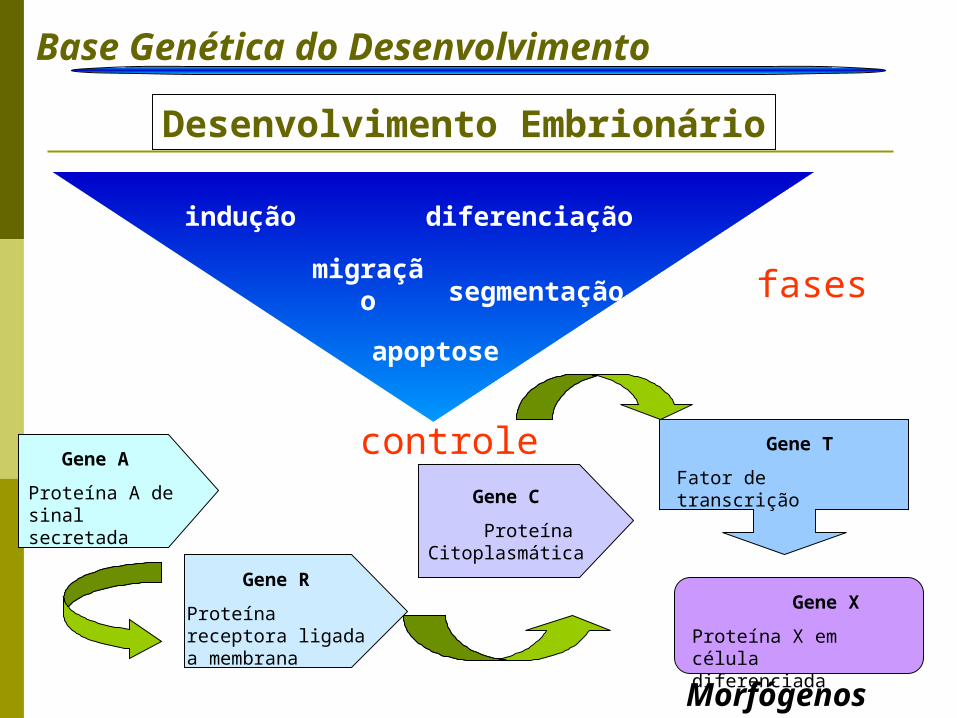
Base Genética do Desenvolvimento Introdução
Desenvolvimento Embrionário
Morfógenos
segmentação
indução
migração
diferenciação
apoptose
controle
fases
Gene A
Proteína A de sinal secretada
Gene R
Proteína receptora ligada a membrana
Gene C
Proteína Citoplasmática
Gene T
Fator de transcrição
Gene X
Proteína X em célula diferenciada
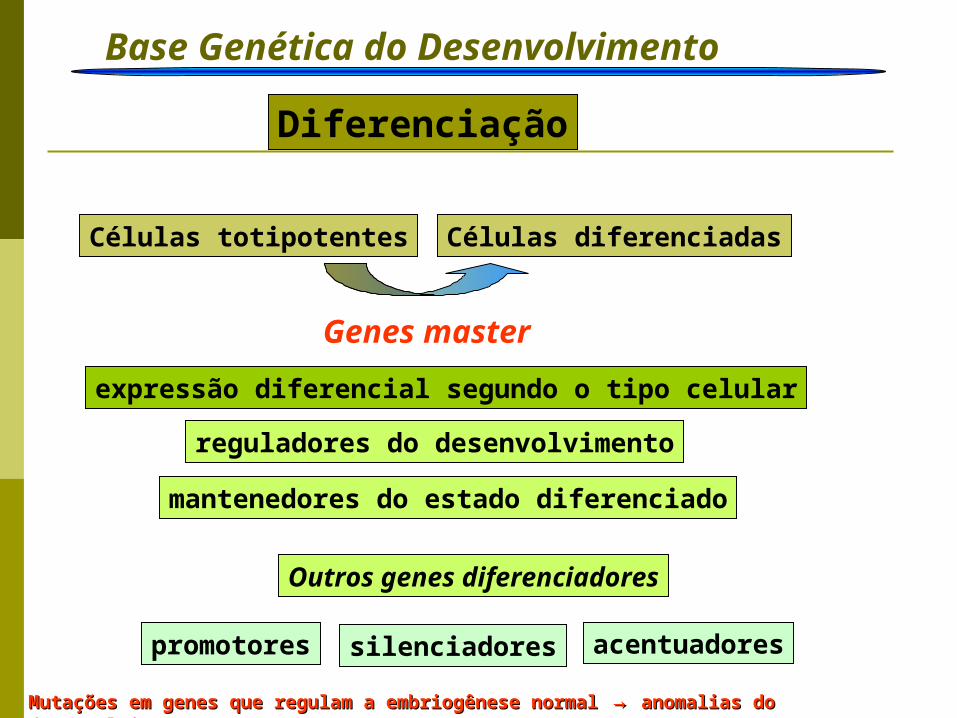
Base Genética do Desenvolvimento Ação gênica
Diferenciação
Células totipotentes Células diferenciadas
Genes master
expressão diferencial segundo o tipo celular
reguladores do desenvolvimento
mantenedores do estado diferenciado
Outros genes diferenciadores
promotores silenciadores acentuadores
Mutações em genes que regulam a embriogênese normalMutações em genes que regulam a embriogênese normal → → anomalias do desenvolvimentoanomalias do desenvolvimento
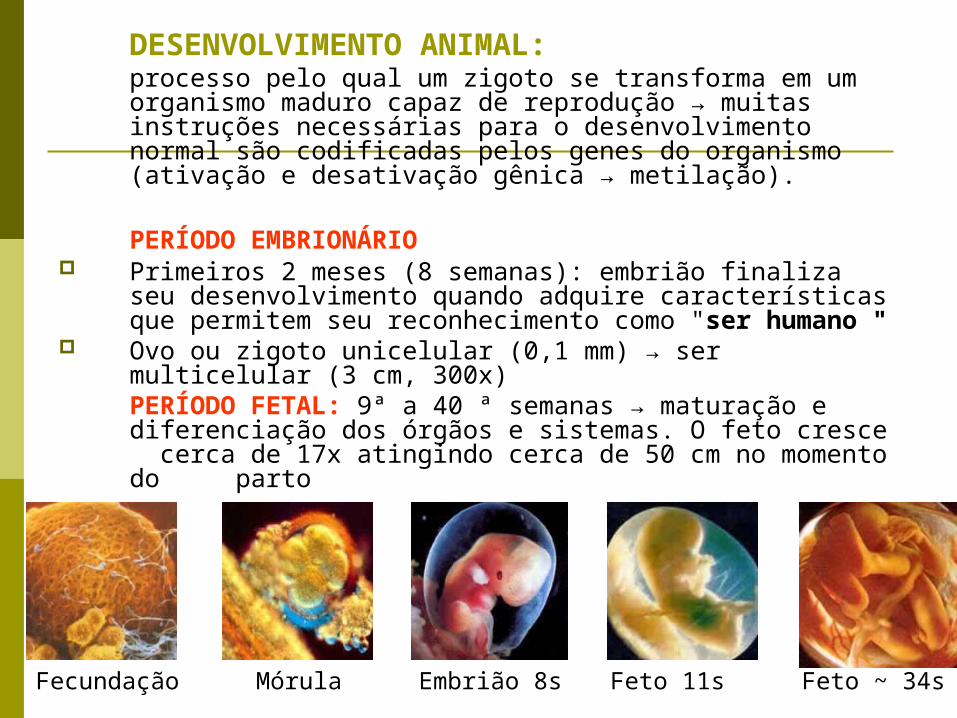
DESENVOLVIMENTO ANIMAL: processo pelo qual um zigoto se transforma em um organismo maduro capaz de reprodução → muitas instruções necessárias para o desenvolvimento normal são codificadas pelos genes do organismo (ativação e desativação gênica → metilação).
PERÍODO EMBRIONÁRIO Primeiros 2 meses (8 semanas): embrião finaliza seu
desenvolvimento quando adquire características que permitem seu reconhecimento como "ser humano "
Ovo ou zigoto unicelular (0,1 mm) → ser multicelular (3 cm, 300x)PERÍODO FETAL: 9ª a 40 ª semanas → maturação e diferenciação dos órgãos e sistemas. O feto cresce cerca de 17x atingindo cerca de 50 cm no momento do parto
Fecundação Mórula Embrião 8s Feto 11s Feto ~ 34s
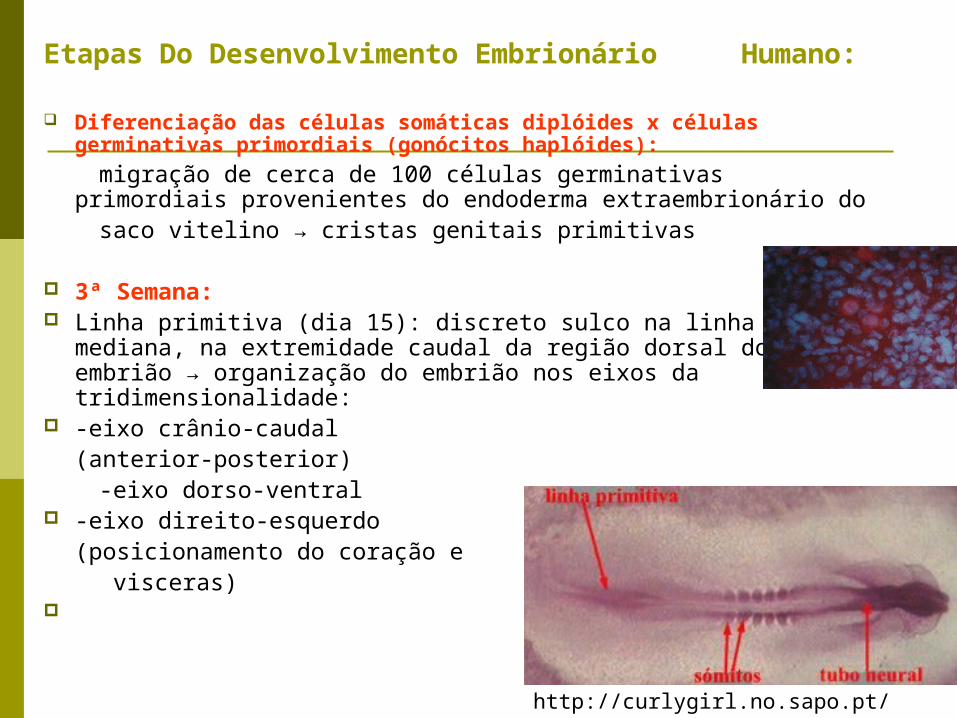
Etapas Do Desenvolvimento Embrionário Humano:
Diferenciação das células somáticas diplóides x células germinativas primordiais (gonócitos haplóides):
migração de cerca de 100 células germinativas primordiais provenientes do endoderma extraembrionário do
saco vitelino → cristas genitais primitivas
3ª Semana: Linha primitiva (dia 15): discreto sulco na linha mediana, na
extremidade caudal da região dorsal do embrião → organização do embrião nos eixos da tridimensionalidade:
-eixo crânio-caudal (anterior-posterior)
-eixo dorso-ventral -eixo direito-esquerdo
(posicionamento do coração e visceras)
http://curlygirl.no.sapo.pt/desan.htm
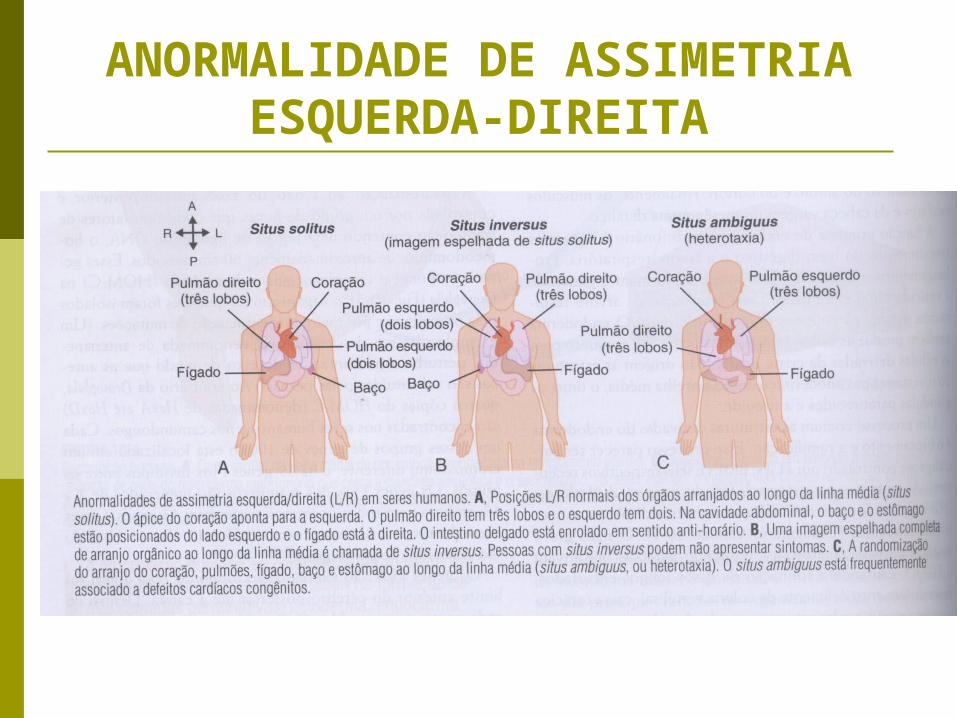
ANORMALIDADE DE ASSIMETRIA ESQUERDA-
DIREITA
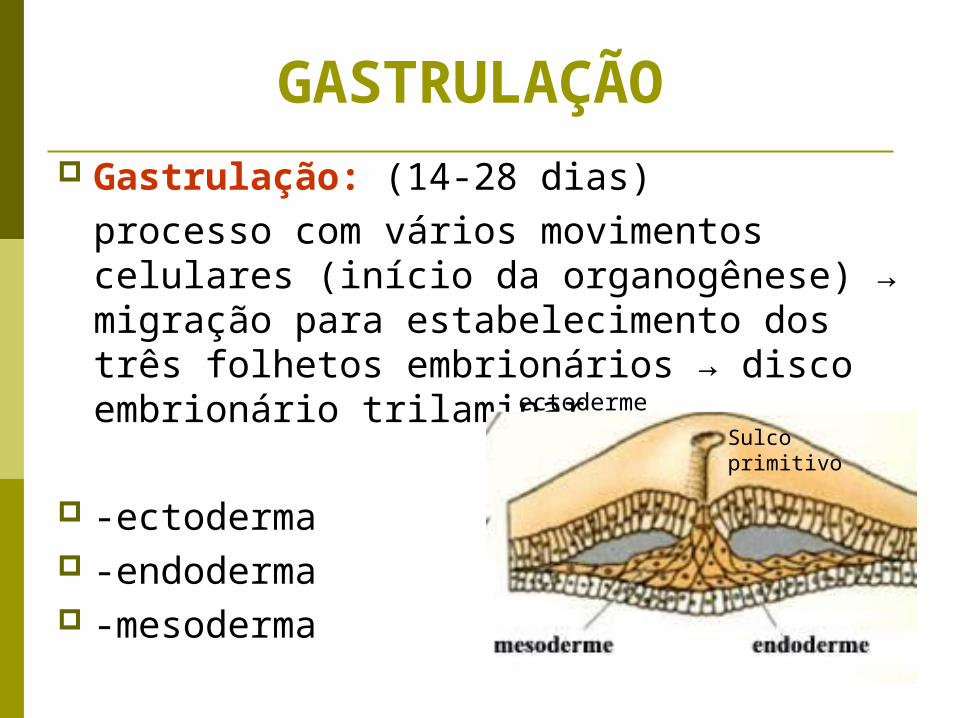
GASTRULAÇÃO Gastrulação: (14-28 dias)
processo com vários movimentos celulares (início da organogênese) → migração para estabelecimento dos três folhetos embrionários → disco embrionário trilaminar
-ectoderma -endoderma -mesoderma
ectodermeSulco primitivo


NEURULAÇÃO Neurulação:
diferenciação do ectoderma superficial e mesoderma dorsal em tubo neural (epitélio neural ou neuroectoderma) → precursor do SNC
extremidade craniana da placa neural → encéfalo: prosencéfalo-1,mesencéfalo-2 e rombencéfalo-3
extremidade caudal da placa neural → medula espinhal-4
http://www.uc.cl/sw_educ/neurociencias/html/035.html

ENDODERMA ENDODERMA: tubo endodérmico (intestino primitivo ou
arquenteron) → origem ao revestimento do trato digestório, árvore respiratória e de outros derivados do tubo intestinal.
Bifurcação da árvore respiratória: pulmões INTESTINO PRIMITIVO: no período da organogênese →
todo aparelho digestório e anexos : pâncreas, fígado, vesícula biliar e pulmão
Intestino anterior: -Faringe -Esôfago -Intestino anterior abdominal (diafragma à
porção média do duodeno
Intestino médio: Segunda metade do duodeno até 2/3 proximais do cólon transverso
Intestino posterior: cólon transverso ao canal anorretal

MESODERMA MESODERMA: dividido em 5 componentes Notocórdio: formação do tubo neural e eixo do corpo
Mesoderma dorsal (nos dois lados do notocórdio): esqueleto axial e músculos esqueléticos e tecido conjuntivo da pele
Mesoderma intermediário: rins e sistema genitourinário
Mesoderma lateral: coração, esqueleto apendicular, tecido conjuntivo das vísceras e das paredes corporais
Mesenquima cefálico: musculos oculares e da cabeça

MESODERMA segmenta em somitômeros (unidade de
pequenos corpos) → formarão os somitos no sentido crânio-caudal- 7 primeiros somitômeros → criarão estruturas da cabeça e pescoço
Somitos (formam-se 42 a 43) → desenvolvem na futura região da base do crânio (dia 20) → 5 a 7 mais caudais se degeneram : total 37 somitos : -4 occipitais-8 cervicais-12 torácicos-5 lombares-5 sacrais-3 coccígeos

ALTERAÇÕES NOS PADRÕES DE
DESENVOLVIMENTO
Gastrulação anormal: Humanos → ciclopia, sirenomelia
Alteração eixo antero-posterior: Drosófila mutação antenapédia e bitórax
Dismorfogênese da notocorda: Humanos → hemivértebras
Patologias da crista neural: Humanos → S. Waardenburg

ANOMALIAS CONGÊNITAS RELACIONADAS À TERCEIRA SEMANA : GASTRULAÇÃO ANORMAL
Ciclopia: (holoprosencefalia) → desenvolvimento anormal da linha média cerebral, RM grave e morte precoce → mutação no gene SHH (sonic Hedgehog)
http://www.conganat.org/7congreso/imagenes_trabajos/396-ciclope1.jpg

ANOMALIAS CONGÊNITAS RELACIONADAS À TERCEIRA SEMANA : GASTRULAÇÃO ANORMAL
Sirenomelia: anormalidade da formação mesodérmica na eminência caudal (membros de sereia) → defeito da linha mediana no estágio 11 → monopodium, simpodium ou rudimentos dos membros inferiores, ausência de tecidos como estruturas sacrococcígeas, períneo, bexiga, intestino posterior, associado à agenesia renal e anomalias vertebrais.
http://galenored.com/bolivia/images/sirenom%201%20gral%20.jpg
http://www.mediresource.com/CPNews/images/x020807A.jpg

ANOMALIAS CONGÊNITAS RELACIONADAS À QUARTRA SEMANA - SEGMENTAÇÃO
plano de segmentação: orientado pelos genes homeóticos → Hox → manifesto na anatomia humana definitiva através das vértebras, costelas, dermátomos e nervos espinhais
GENES HOMEÓTICOS: genes envolvidos na segmentação → a sequência destes genes se expressam em todas as classes de vertebrados →
denominados homeobox (Hox)
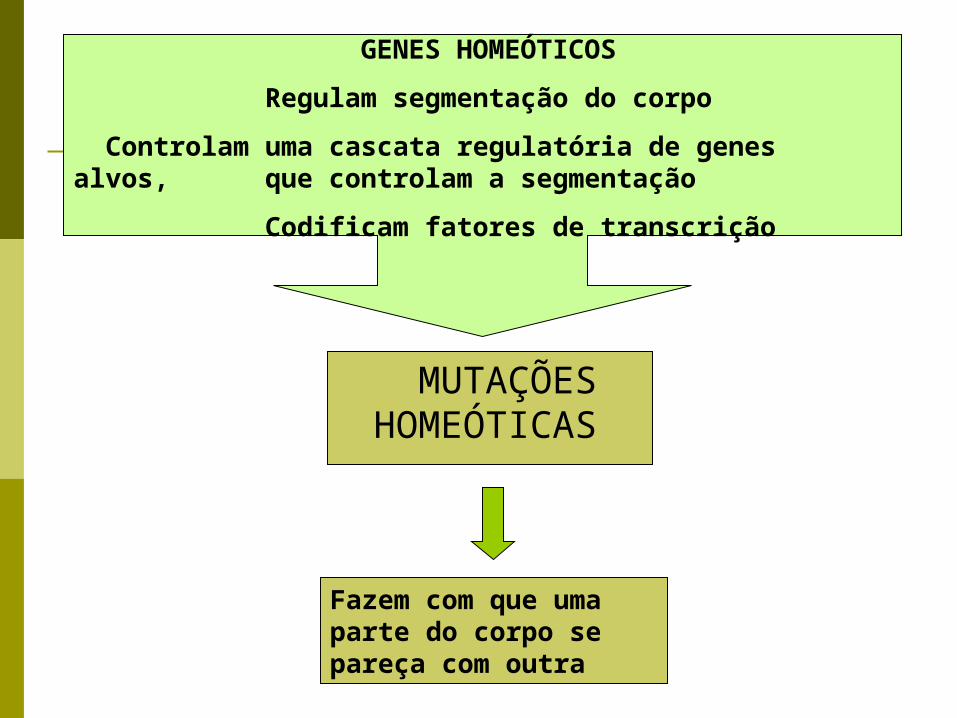
MUTAÇÕES HOMEÓTICAS
GENES HOMEÓTICOS
Regulam segmentação do corpo
Controlam uma cascata regulatória de genes alvos, que controlam a segmentação
Codificam fatores de transcrição
Fazem com que uma parte do corpo se pareça com outra

http://www.ncbi.nlm.nih.gov/books/bv.fcgi?rid=iga.figgrp.3681
Alteração Eixo Anterto-Posterior
Formação do eixo anterior/posterior → extremidade da linha primitiva → estrutura denominada nódulo (gene nodal) → início e manutenção da linha primitiva e formação do eixo direito/esquerdo.
-complexo gênico homeótico (HOM-C): controla o padrão ao longo do eixo anterior/posterior Drosophila (mutação antenapédia) pertuba a padronagem do eixo → antenas são substituídas por pernas
Mutação → causando mudança do local de
desenvolvimentode uma parte do corpo

MUTAÇÃO BITHORAX EM DROSÓFILA
Mutação em gene homeótico de segmentação (bx): afeta dois segmentos torácicos
a) Asas rudimentares no lugar de altéres; b) dois pares de asas

SEGMENTAÇÃO DO CORPO
Muitos invertebrados → corpo formado por unidades adjuntas denominadas segmentos
Exemplo: Drosófila
3 segmentos da cabeça (H) 3 segmentos do tórax (T1-T3) 8 segmentos do abdomen
(A1-A8)
embrião larva adulto

GENES HOMEÓTICOS
Insetos: genes HOM-C Seres humanos e camundongo: 4 cópias de Hox
(HoxA até HoxD) 39 genes Hox: cada grupo gênico de 100 Kb está
situado em um cromossomo diferente genes Hox de mamíferos são numerados de 1 a 13 os genes Hox são expressos ao longo do eixo dorsal
do limite anterior do rombencéfalo até a cauda Os genes Hox são expressos de 3’ a 5’ ao longo do
eixo antero-posterior
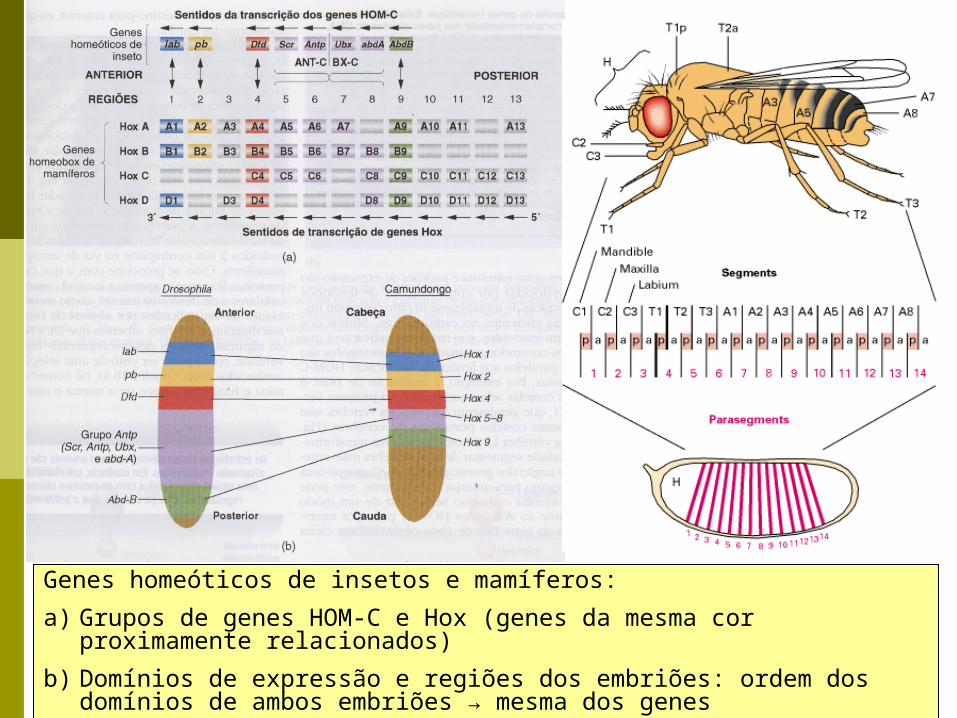
Genes homeóticos de insetos e mamíferos:
a) Grupos de genes HOM-C e Hox (genes da mesma cor proximamente relacionados)
b) Domínios de expressão e regiões dos embriões: ordem dos domínios de ambos embriões → mesma dos genes
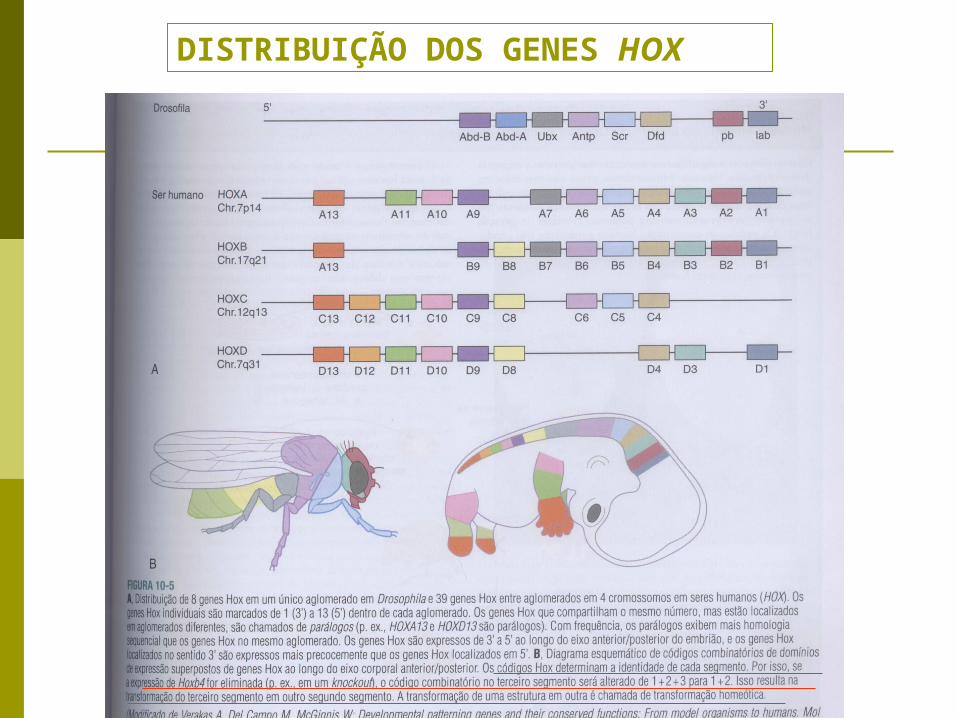
DISTRIBUIÇÃO DOS GENES HOX
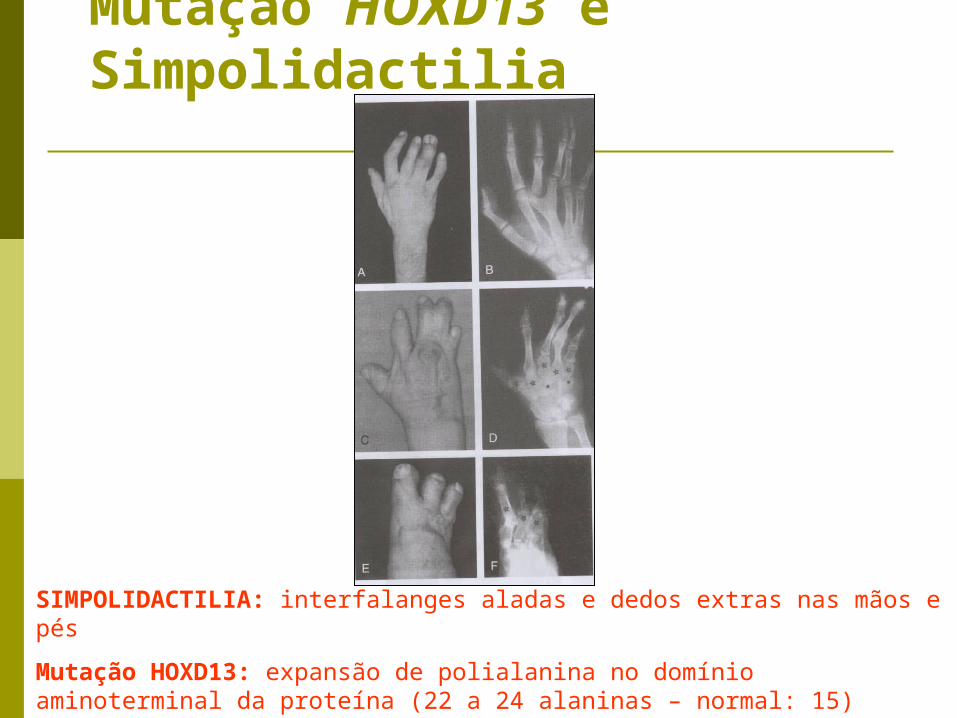
Mutação HOXD13 e Simpolidactilia
SIMPOLIDACTILIA: interfalanges aladas e dedos extras nas mãos e pés
Mutação HOXD13: expansão de polialanina no domínio aminoterminal da proteína (22 a 24 alaninas – normal: 15)

Dismorfogênese da notocorda Dismorfogênese da notocorda: leva a
malformação do corpo vertebral : hemivértebras (escoliose) e falha produzida pelo tubo neural (alteração no arco vertebral) → espinha bífida
http://www.srs.org/patients/review/images/9c.jpg
http://www.emedicine.com/orthoped/images/748748748ort0618-08.jpg
http://paginas.terra.com.br/saude/maninho/sss.html
hemivértebras
Espinha bífida

PATALOGIAS ASSOCIADAS A CRISTA NEURAL
A- Defeito na migração ou morfogênese da crista neural (neuroectoderma): origina melanócitos, medula adrenal, sistema nervoso autônomo (SN entérico), neurônios sensoriais, tecido conjuntivo da cabeça e face
fenda labial ou palatina Síndrome de Waardenburg (PAX3) Associação CHARGE (coloboma,
cardiopatia, SNC, genital e auditivo)
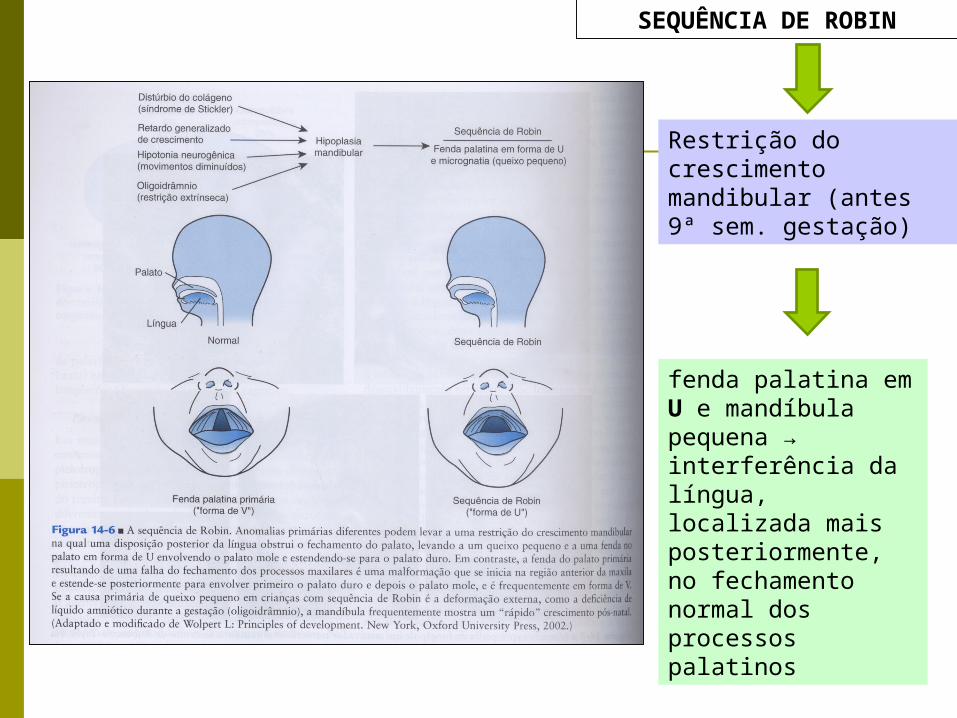
SEQUÊNCIA DE ROBIN
Restrição do crescimento mandibular (antes 9ª sem. gestação)
fenda palatina em U e mandíbula pequena → interferência da língua, localizada mais posteriormente, no fechamento normal dos processos palatinos

Mutações em PAX 3
Síndrome de Waardenburg: redução ou deficiência de melanócitos nos cabelos, olhos e ouvido interno → mecha branca, olhos claros e heterocromia,surdez
PATALOGIAS ASSOCIADAS A CRISTA NEURAL
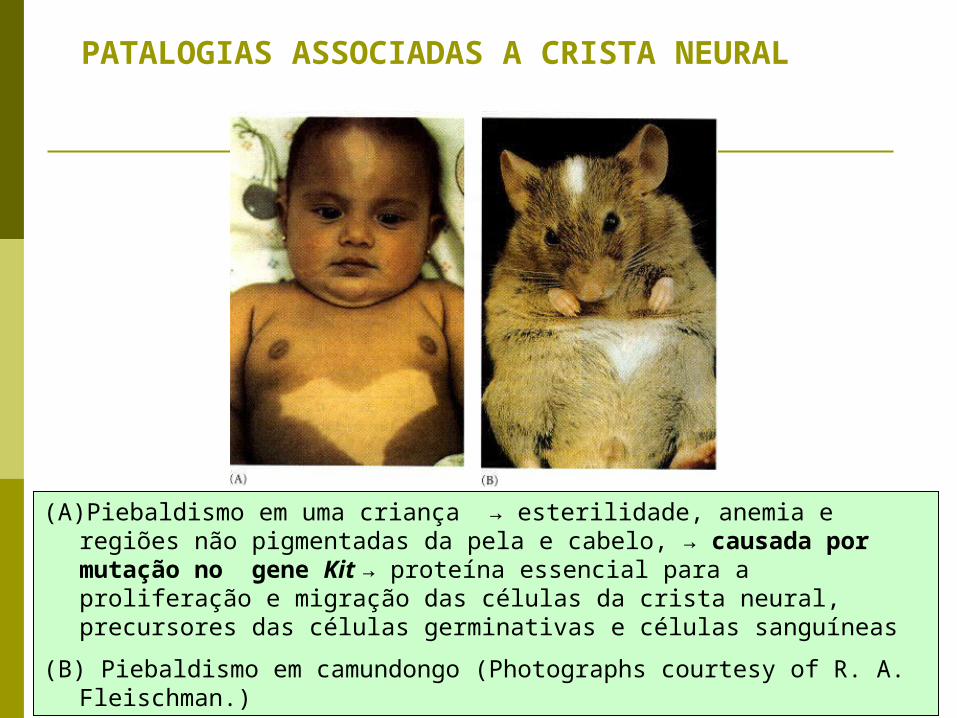
(A)Piebaldismo em uma criança → esterilidade, anemia e regiões não pigmentadas da pela e cabelo, → causada por mutação no gene Kit → proteína essencial para a proliferação e migração das células da crista neural, precursores das células germinativas e células sanguíneas
(B) Piebaldismo em camundongo (Photographs courtesy of R. A. Fleischman.)
PATALOGIAS ASSOCIADAS A CRISTA NEURAL

VIAS DE DESENVOLVIMENTO
Consiste em eventos envolvidos na diferenciação de tecidos e órgãos, com participação de diferentes produtos gênicos → criando um fenótipo
Codificam muitos produtos diferentes:
-moléculas sinalizadoras e seus receptores-transdutores de sinais-fatores de transcrição do DNA-componentes da matriz extracelular-enzimas-sistemas de transporte e outras proteínas
GENES NECESSÁRIOS AO DESENVOLVIMENTO NORMAL

Moléculas de sinalização parácrina
São fatores parácrinos (secretado no espaço circundante das células) atuam nas células vizinhas através de pequenas distâncias
Quatro famílias de moléculas de sinalização parácrina:-família do fator de crescimento de fibroblasto (FGF)-família Hedgehog-família wingless-família do fator de transformação do crescimento (TGF-)
Cada uma das moléculas sinalizadoras se ligam a um ou mais receptores para efetuar uma resposta mutações nestes genes podem levar a uma comunicação anormal entre as células

Doenças dos Receptores do Fator de Crescimento de Fibroblastos – FGFR3
FGFRs: são receptores para ~22 fatores de crescimento de fibroblastos
Estrutura: peptídeo sinal, 3 domínios semelhantes aos da Ig, domínio transmembrana e um domínio intracelular tirosina quinase.
Processos biológicos: migração, crescimento e diferenciação celular.
Expressão: ossos em desenvolvimento
Síndromes: displasias ósseas AD

Doenças dos Receptores do Fator de Crescimento de Fibroblastos – FGFR3
Acondroplasia: mutação substituição de glicina – arginina no domínio transmembrana de FGFR3 – ativação constitutiva do receptor
Superativação de FGFR3 aumenta a inibição do crescimento de condrócitos – defeitos do esqueleto
Hipocondrodisplasia: graus mais baixos de ativação
Displasia tanatofórica (letal): ativação acentuada
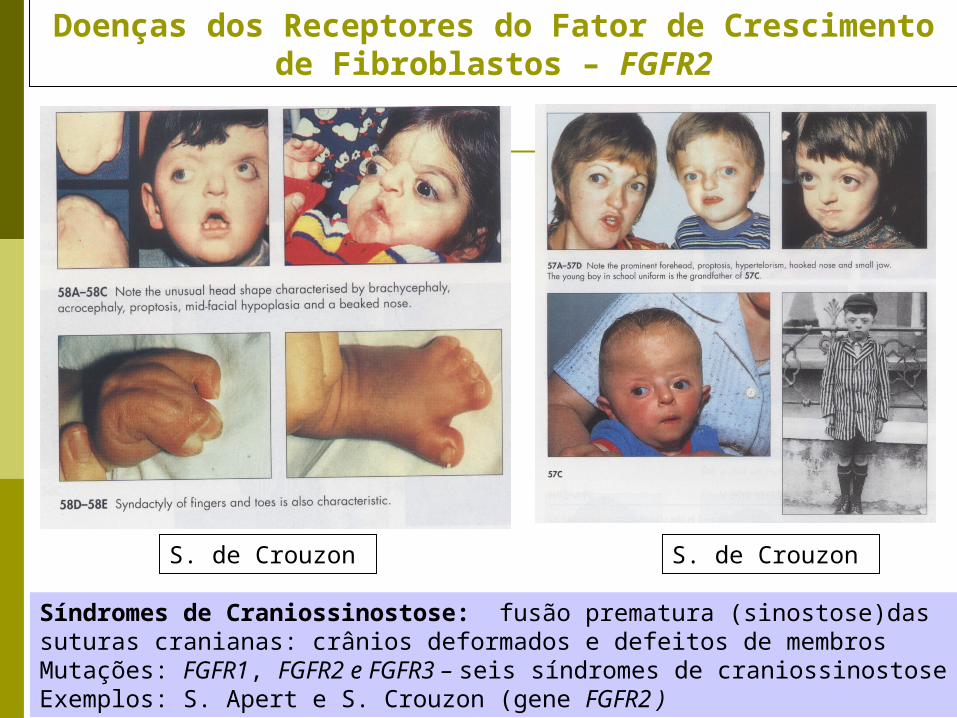
Doenças dos Receptores do Fator de Crescimento de Fibroblastos – FGFR2
Síndromes de Craniossinostose: fusão prematura (sinostose)das suturas cranianas: crânios deformados e defeitos de membrosMutações: FGFR1, FGFR2 e FGFR3 – seis síndromes de craniossinostoseExemplos: S. Apert e S. Crouzon (gene FGFR2 )
S. de CrouzonS. de Crouzon

Gene hedgehog
Descoberto em Drosophila
Gene de segmentação
mutações em hedgehogolhos funcionais em asas e pernas
38% de homologiacom SHH de vertebrados (Sonic
hedgehog)
Genes no desenvolvimento humano e suas homologias

Via de Sinalização Sonic hedgehog Síndromes relacionadas
HoloprosencefaliaS. Smith-Lemli-Opitz S. Gorlin
S. Rubenstein-Taybi
S. Greig
Funções de SHH: especificação de eixo; indução de neurônios motores na placa neural;padronização dos membros.
S. Gorlin: mutação de PTC causa S. Gorlin e nas células somáticas causa câncer pele

Moléculas de sinalização parácrina
2-Família Wnt (gene de Drosophila wingless - sem asas): polaridade durante a formação de membros de Drosophila
-Genes WNT: glicoproteínas responsáveis pela especificação do eixo dorsal/ventral, formação do cérebro, músculos, gônadas e rins.
-Mutação em WNT3 causa tetra-amelia (ausência dos quatro membros) em humanos
http://elacuarista.com/alimentos/drosofilas.htm

Moléculas de sinalização parácrina3-Família do supergene TGF-: pelo
menos 30 genes estruturais
-famílias TGF--família de proteína morfogenética óssea
BMP formação do osso-família de activina-família Vg1

Moléculas de sinalização parácrina MUTAÇÕES NA FAMÍLIA BMP: proteína 1
morfogenética derivada de cartilagem (CDMP1) causa várias anormalidades esqueléticas mutações diferentes podem produzir fenótipos diferentes.1-mutação sem sentido em CDMP1: braquidactilia (AD)
2-duplicação em 22pb em CDMP1: braquidactilia e encurtamento dos ossos longos dos membros (displasia acromesomélica - AR)
3-mutação homozigoto de sentido trocado: condrodisplasia AR de Grebe encurtamento dos ossos longos e dedos

http://www.oftalmo.com/seo/2002/07jul02/09.htm
Braquidactilia
Síndrome de Grebe
MUTAÇÕES NA FAMÍLIA BMP
1.
Displasia acromesomélica

VIA DE TRANSDUÇÃO DE SINAL – RAS-MAPK transdução de sinais extracelulares para célula: regula várias funções celulares (expressão, divisão, diferenciação e morte celular
S. Noonan: mutação de ganho de função do gene PTPN11 proteína que interage com a via RTK-MAPK
S. Costello e S. carciofaciocutânea: mutações em outros componentes da via

Fatores de Transcrição do DNA
São proteínas codificadas por genes que ligam (ativam) ou desligam (inativam) outros genes regulam a transcrição de muitos genes que regulam outros genes efeito cascata mutações nesses genes tem efeitos pleiotrópicos
Exemplos: genes contendo homeobox: família HOX, PAX
(domínios de ligação ao DNA)Pax6 camundongo: olhos pequenos anormais;
PAX6 humano: catarata e aniridia homólogo de Drosophila gene eyeless (fator de transcrição → ativa vários genes)

Mutações em PAX 6
Aniridia
Anoftalmia
http://www.ucm.es/info/genetica/AVG/practicas/Drosophila/Drosophila.htmhttp://www.exploratorium.edu/imaging_station/gallery.php?Category=drosophila
Drosophila eyeless

Mutações em PAX 6
Genes PAX6 humano, Pax6 de camundongo e Eyeless de Drosophila: são homólogos e codificam fatores de transcrição
(os genes e vias do desenvolvimento do olho foram conservados durante a evolução)

Proteínas da Matriz Extracelular (EMP) Macromoléculas secretadas que servem de
arcabouços para todos os tecidos e órgãos colágeno, fibrilinas, proteoglicanas e glicoproteínas (fibronectina, laminina e tenascina) mediadores ativos do desenvolvimento
EMPs: se aderem à superfície das células para permitir a migração (pelos receptores: integrinas e glicosiltransferases)
Integrinas: integram a matriz extracelular ao citoesqueleto
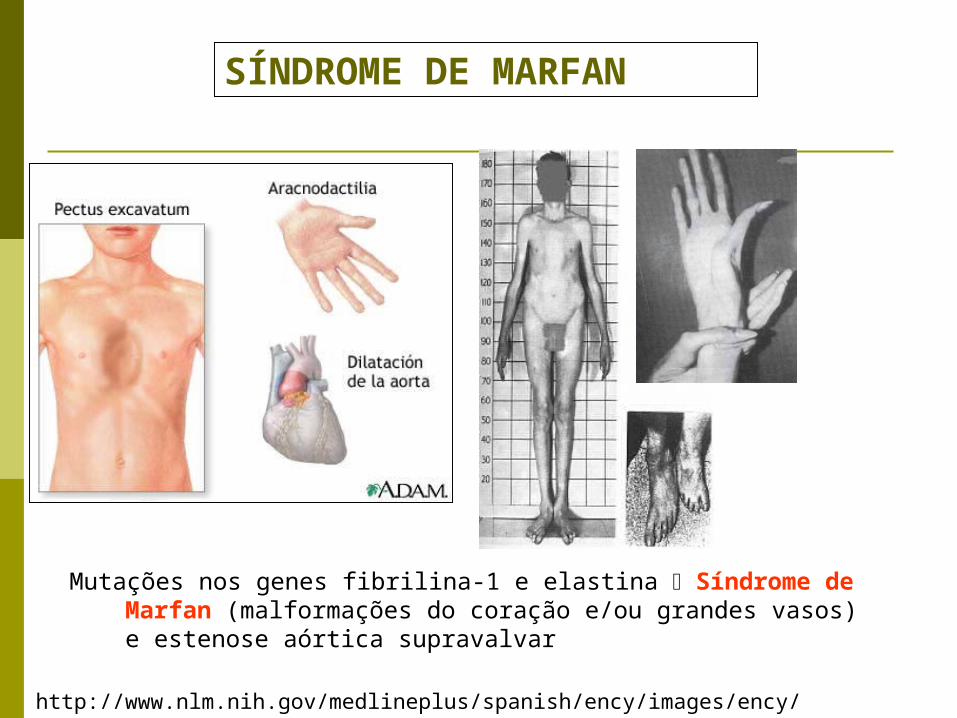
http://www.nlm.nih.gov/medlineplus/spanish/ency/images/ency/fullsize/9611.jpg
SÍNDROME DE MARFAN
Mutações nos genes fibrilina-1 e elastina Síndrome de Marfan (malformações do coração e/ou grandes vasos) e estenose aórtica supravalvar

DESENVOLVIMENTO DOS MEMBROS
Membros dos vertebrados: -elementos derivados da placa mesodérmica lateral: ossos, cartilagem e tendões
-mesoderma somítico: músculos, nervos e vasculatura
Sinal que inicia indução dos membros anteriores e posteriores mesoderma intermediário
Fator 8 de crescimento de fibroblastos (FGF8):
- expresso nos membros anteriores e posteriores induz o programa de brotamento dos membros
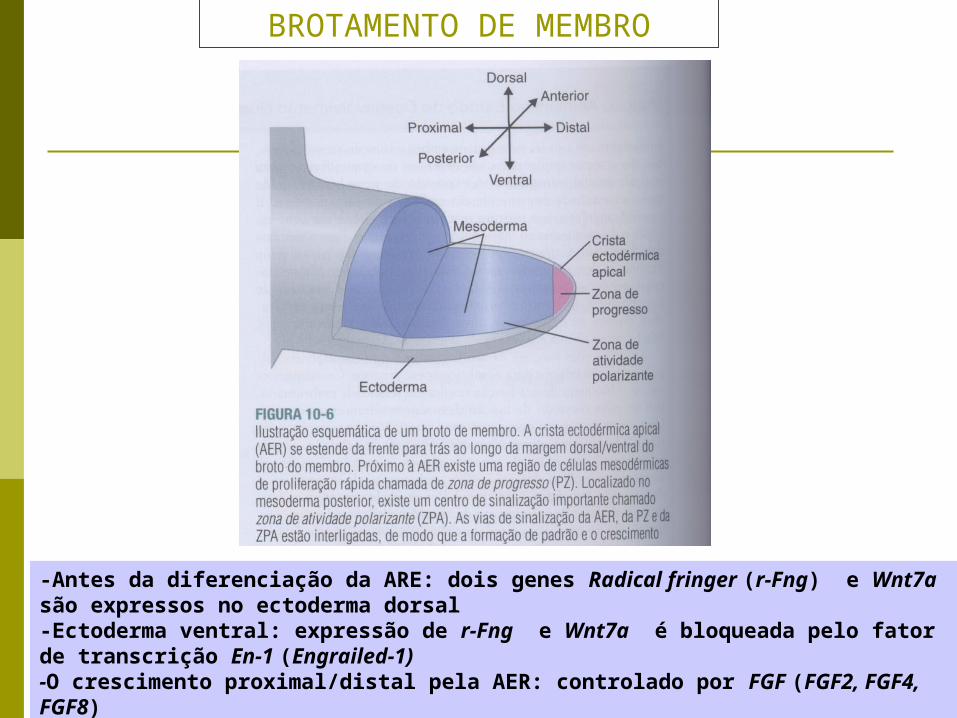
BROTAMENTO DE MEMBRO
-Antes da diferenciação da ARE: dois genes Radical fringer (r-Fng) e Wnt7a são expressos no ectoderma dorsal -Ectoderma ventral: expressão de r-Fng e Wnt7a é bloqueada pelo fator de transcrição En-1 (Engrailed-1)-O crescimento proximal/distal pela AER: controlado por FGF (FGF2, FGF4, FGF8)
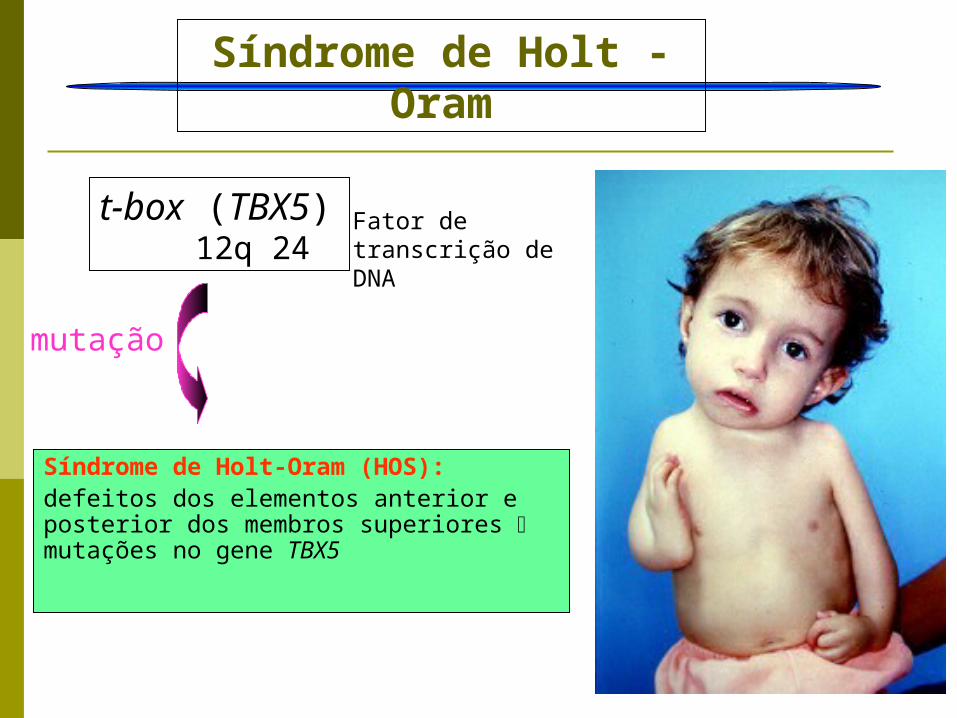
t-box (TBX5) 12q 24
Síndrome de Holt - Oram
mutação
Síndrome de Holt-Oram (HOS): defeitos dos elementos anterior e posterior dos membros superiores mutações no gene TBX5
Fator de transcrição de DNA
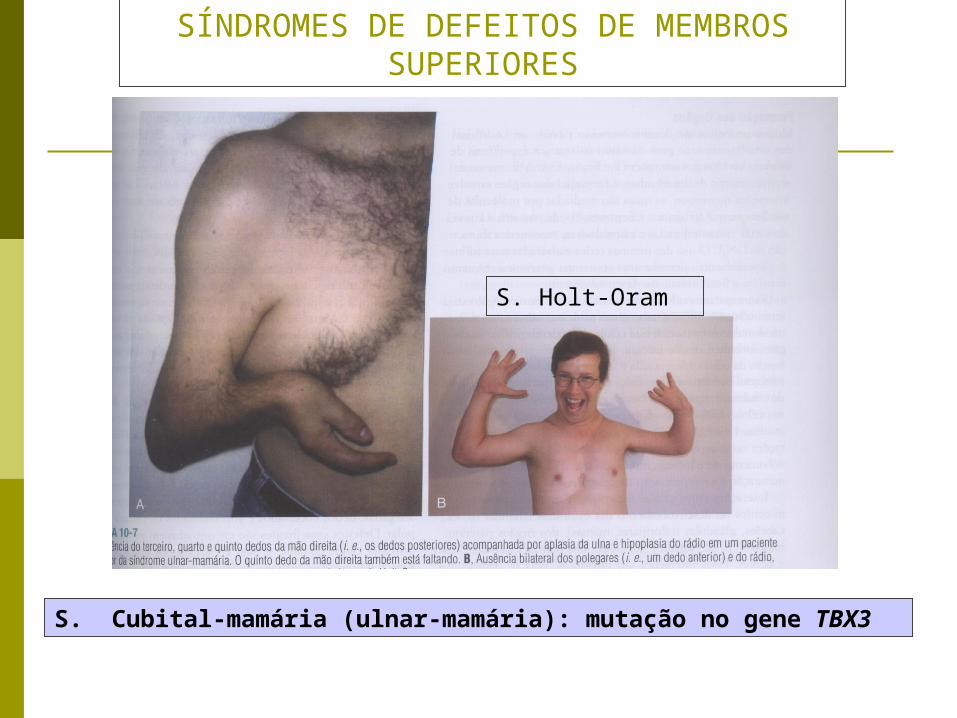
SÍNDROMES DE DEFEITOS DE MEMBROS SUPERIORES
S. Cubital-mamária (ulnar-mamária): mutação no gene TBX3
S. Holt-Oram
GENÉTICA DO DESENVOLVIMENTO
Vias de sinalização do desenvolvimento (conservadas) → usadas p/diferentes programas morfogenéticos no embrião
Mutações
Fenótipos
Homólogos em vertebrados:
sondas de genes de invertebrados → clonar genes homólogos em outros organismos
Identificação de genes relevantes → vias de desenvolvimento
LIM-2 – Genética do Desenvolvimentohttp://www.cienciablogada.com.br/2011/12/biologia-do-desenvolvimento-segundo-as.html - Teorias epigenistashttps://www.youtube.com/watch?v=tbOlV150P8U - Da concepção ao nascimentohttp://www.sogab.com.br/apostiladeembriologiaegenetica.pdf - Apostila de embriologia e genéticahttp://www.portalsaofrancisco.com.br/alfa/desenvolvimento-embrionario-humano/desenvolvimento-embrionario-humano.php Desenvolvimento embrionário humanohttp://www.publico.pt/ciencia/noticia/descobertos-genes-que-ajudam-a-construir-a-cara-que-temos-1610446 - Genes reguladores do desenvolvimento da face e crâniohttp://www.biocel.icb.usp.br/~ireneyan/EMBRIOLOGIAMOLECULAR_arquivos/SemataTEMBio1.pdf - genes homeóticos http://www.drscope.com/pac/pediatria-1/d5/p1d5_p17.htm - Família Hox humanohttp://enfermedadesraras2.blogspot.com.br/2011/10/el-sindrome-de-la-sirenomelia.html - síndrome da sirenomelia http://www.fecolsog.org/userfiles/file/revista/Revista_Vol58No1_Enero_Marzo2007/v58n1a11.pdf - artigo ciclopia http://www.hermespardini.com.br/atual_manual/pdf_genetica_novos_exames/Acondroplasia.pdf - Estudo Genético das várias formas de Nanismo http://geneticanaescola.com.br/wp-home/wp-content/uploads/2012/10/Genetica-na-Escola-12-Artigo-14.pdf - Genética do desenvolvimento e evoluçãohttp://dreyfus.ib.usp.br/bio208/arquivos2011/evodevo_2011.pdf - Genética do desenvolvimento e evolução

Questões:
1. As síndromes de craniossinostose, como S. de Apert e S. de Crouzon, apresentam características clínicas semelhantes:
Consulte o link: http://www.ncbi.nlm.nih.gov/omim
Compare ambas as síndromes e descreva:a-padrão de herança, gene mutado e localização cromossômica
b- características clínicas
c- qual o principal sinal clínico que diferencia estas síndromes
Obs.: procure no google imagens fotos das síndromes

2. Caracterize os genes homeóticos e faça uma comparação entre os genes homeóticos de insetos (HOM) e mamíferos (Hox).
3. Alterações no processo de gastrulação provoca anomalias como ciclopia e sirenomelia. Descreva o processo que provoca essas alterações, as características clinicas, causas prováveis e genes envolvidos.
4. Compare e descreva a base genética das várias formas de Nanismo devido mutações no gene do Receptor 3 do Fator de Crescimento de Fibroblastos (FGFR3): Acondroplasia, Hipocondroplasia e Displasia
Tanatofórica.





























