Embed Size (px)
Citation preview

LUCIANA BOFFONI GENTILE
PAPEL DE FOSFOLIPASES A2 (PLA2s) NA ATIVAÇÃO
POR ESTRESSE MICROAMBIENTAL IN VITRO DE
LINHAGEM CELULAR DE CÂNCER DE CÓLON
UNIVERSIDADE FEDERAL DO RIO DE JANEIRO
CENTRO DE CIÊNCIAS DA SAÚDE
INSTITUTO DE CIÊNCIAS BIOMÉDICAS
2007

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

UNIVERSIDADE FEDERAL DO RIO DE JANEIRO
CENTRO DE CIÊNCIAS DA SAÚDE
INSTITUTO DE CIÊNCIAS BIOMÉDICAS
LUCIANA BOFFONI GENTILE
PAPEL DE FOSFOLIPASES A2 (PLA2s) NA ATIVAÇÃO POR
ESTRESSE MICROAMBIENTAL IN VITRO DE LINHAGEM
CELULAR DE CÂNCER DE CÓLON
Tese de Doutorado submetida ao Programa de Pós-graduação em Ciências Morfológicas, Instituto de Ciências Biomédicas, Centro de Ciências da Saúde, Universidade Federal do Rio de Janeiro (UFRJ), como parte dos requisitos necessários à obtenção do título de Doutor em Ciências Morfológicas
Orientador: Dr. Bruno Lourenço Diaz
Rio de Janeiro
Janeiro de 2007

Gentile, Luciana Boffoni
Papel de fosfolipases A2 (PLA2s) na ativação por estresse microambiental in vitro de linhagem celular de câncer de cólon / Luciana Boffoni Gentile. Rio de Janeiro: UFRJ,CCS, 2007.
xiv, 158 f. : il. ; 31 cm. Orientador: Bruno Lourenço Diaz
Tese de Doutorado – UFRJ, CCS, ICB, Pós-graduação em
Ciências Morfológicas, 2007 Referências bibliográficas: f. 108-138
1. Fosfolipases A. 2. Neoplasias do colo. 3. Prostaglandinas E.4. Fatores de Crescimento do Endotélio Vascular. 5. CiênciasMorfológicas - Tese. I. Diaz, Bruno Lourenço. II. UniversidadeFederal do Rio de Janeiro, Centro de Ciências da Saúde, Instituto deCiências Biomédicas. III. Papel de fosfolipases A2 (PLA2s) naativação por estresse microambiental in vitro de linhagem celular decâncer de cólon.

FOLHA DE APROVAÇÃO
Papel de Fosfolipases A2 (PLA2s) na ativação por estresse microambiental in vitro de linhagem celular de câncer de cólon
Luciana Boffoni Gentile
Tese de Doutorado submetida ao Programa de Pós-graduação em Ciências Morfológicas, Instituto de Ciências Biomédicas, Centro de Ciências da Saúde, Universidade Federal do Rio de Janeiro (UFRJ), como parte dos requisitos necessários à obtenção do título de Doutor em Ciências Morfológicas. Rio de Janeiro, 17 de janeiro de 2007.
Aprovado por:
Dr. Bruno Lourenço Diaz, Professor Adjunto, UFRJ (Orientador) Dra. Cláudia dos Santos Mermelstein, Professora Adjunta, UFRJ (Revisora) Dra. Iolanda Margherita Fierro, Professora Adjunta, UERJ (Membro da banca) Dr. José A. Morgado Díaz, Pesquisador Associado, INCA (Membro da banca) Dra. Tatiana Lobo Coelho de Sampaio, Professora Adjunta, UFRJ (Membro da banca) Dra. Clarissa Menezes Maya Monteiro, Assistente de Pesquisa, FIOCRUZ (Suplente)

Este trabalho foi desenvolvido na Divisão de Biologia Celular da Coordenação de Pesquisa do Instituto Nacional de Câncer (INCA) sob a orientação do Dr. Bruno Lourenço Diaz e com o auxílio financeiro do Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) e da Fundação Ary Frauzino (FAF/INCA).

À Clarinda, Elvira e Annita.

AGRADECIMENTOS
À minha madrinha Clarinda (in memoriam) por todo apoio que me deu durante meu caminho.
À minha querida mãe Annita pela dedicação e carinho.
À minha avó Elvira (in memoriam) pela sua presença maravilhosa.
Ao Dr. Bruno Lourenço Diaz pela oportunidade de aguçar meu senso crítico, minha organização, pelas discussões, sempre proveitosas, e por ter me permitido enxergar que quase tudo depende de sua própria vontade e determinação. Ao Programa de Pós-graduação em Ciências Morfológicas pelo apoio financeiro no último ano de Doutoramento, crucial para definir meus rumos em 2007. Ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) e à Fundação Ary Frauzino (FAF/INCA) pelo financiamento deste trabalho. Ao Dr. João Viola e ao Dr. Marcello André Barcinski pela oportunidade de trabalho em um laboratório com boa infra-estrutura. À Dra. Cláudia Mermelstein pelo acompanhamento e revisão da tese. À minha grande amiga Priscila pelas risadas, que tanto me acalmaram e ajudaram a esquecer os problemas científicos. Aos amigos que conheci no laboratório, Júlia, Fausto, João Paulo, João Luiz, Elaine, Bruno Robbs, Sheila, Giuliana e Ivana, pelas conversas animadas, conturbadas, loucas e divertidíssimas. Aos meus amigos da UERJ, Bebeeeé, Cabeção, Lalica, Evita e Naninha que deram mais Arte ao meu mundo. Às nossas noites boêmias na Lapa!!! À Lurdes, que, com respeito e responsabilidade, me ajudou a entender quais caminhos desejo trilhar. Ao pessoal das Divisões de Biologia Celular e Medicina Experimental. Ao pessoal do apoio técnico da Divisão de Medicina Experimental pelo trabalho eficiente, me permitindo dedicar maior tempo aos experimentos. Às secretárias Sueli (Susu) e Marilene pela super competência em resolver questões burocráticas.

RESUMO
A Prostaglandina (PG)E2, produto da metabolização do ácido araquidônico (AA) pela enzima Ciclooxigenase (COX )-2, controla cada aspecto do desenvolvimento do câncer de cólon, com função desde a proliferação celular e resistência à apoptose até a angiogênese, invasão e metástase. A primeira etapa na geração dos eicosanóides, tais como PGE2, é a liberação de ácido araquidônico (AA) esterificado em fosfolipídeos, que é realizada por uma ou mais fosfolipases (PL)A2. A PLA2 citosólica-α (cPLA2-α), ou grupo IVA, é considerada como a PLA2 mais importante para a liberação do AA e síntese de eicosanóides, porém outras PLA2s, de baixo peso molecular, as PLA2 secretórias (sPLA2), também podem participar na liberação do AA para a síntese de PGE2. O câncer de cólon e reto é a terceira neoplasia que mais mata entre homens, e a quarta entre mulheres. Há vários fatores de risco para esta neoplasia e um deles é a dieta pobre em fibras e rica em produtos processados. Esta dieta pode gerar um microambiente favorável para o desenvolvimento tumoral, incluindo o estresse provocado por aumento da osmolaridade no cólon. A associação entre a produção de PGE2 e o desenvolvimento de câncer colo-retal tem sido bem definida, porém os estímulos e mecanismos induzindo esta produção em células de câncer de cólon ainda não estão elucidados. O objetivo principal deste trabalho é o de avaliar o papel das PLA2s na ativação de células de câncer de cólon por estresse microambiental. Também foram investigadas as vias de sinalização que conduzem à geração de PGE2 e de Fator de Crescimento do Endotélio Vascular (VEGF). A linhagem de câncer de cólon humano Caco-2 foi ativada com adição de NaCl (100 mM) ao meio de cultura para mimetizar o ambiente intestinal hipertônico. A liberação do AA, a geração de PGE2 e de VEGF foram analisadas no sobrenadante da cultura celular, enquanto que, a expressão de COX-2, e a fosforilação das MAP Kinases foram analisadas nos lisados celulares por Western Blot. Os inibidores farmacológicos das PLA2s, de COX-2 e das MAPKs foram adicionados de 15 a 30 minutos antes da ativação de Caco-2. Os resultados indicaram que o estresse hipertônico e o CoCl2 induzem a expressão de COX-2 e a produção de PGE2 e de VEGF em Caco-2. A produção de PGE2 por Caco-2 estimuladas depende de COX-2 e cPLA2. A cPLA2 fornece AA como substrato para a enzima COX-2 resultando na produção de PGE2, que, por sua vez, modula a produção de VEGF, via receptor EP2, no estresse hipertônico. Já em condições de hipóxia, a PGE2 não possui papel regulatório sobre a produção do fator angiogênico por Caco-2. A cPLA2 é ativada pelas MAPKs, ERK1/2, p38 e JNK, nos dois tipos de estresse microambiental. ERK1/2 induz a expressão de COX-2 em Caco-2 ativadas por estresse hipertônico. A fosforilação de p38 possui função na indução da produção de VEGF por Caco-2 no estresse hipertônico. Nossos resultados sugerem que as células de câncer de cólon respondem ao estresse microambiental com ativação de vias de sinalização de MPAKs e produção de PGE2 e VEGF, provavelmente como uma forma de adaptação às condições externas. O papel autócrino da PGE2 na produção de VEGF parece depender do tipo de estresse ambiental e sugere vias de sinalização diferentes com potencial impacto no desenvolvimento tumoral.

ABSTRACT
Prostaglandin (PG)E2, product of arachidonic acid metabolized by Cyclooxygenase (COX)-2 enzyme, controls every aspect of colon cancer development, from cell proliferation and resistance to apoptosis to angiogenesis, invasion, and metastasis. The first step in generation of eicosanoids, such as PGE2, is the release of arachidonic acid (AA) sterified to phospholipids, which is performed by one or more phospholipases (PL)A2. Cytosolic (c)PLA2-α or group IVA, is considered to be the most important PLA2 for AA release and eicosanoid synthesis, however other PLA2s, in particular low molecular weight secretory (s)PLA2s, can also participate in AA release for eicosanoid synthesis. Colon cancer ranks third in deaths among men and fourth among women. There are several risk factors to this neoplasia and one of them is diet poor in fibers and rich in manufactured products. Such diet may create a microenvironment suitable for tumoral development that includes increased colonic osmolarity-evoked stress. While association between PGE2 production and development of colorectal cancer has been well defined, the stimuli and mechanisms inducing this production in colorectal cells are not clear. The main objective of this study is to evaluate the role of the PLA2s in the activation of colon cancer cells by environmental stress. We also investigated the signaling pathways involved in PGE2 and VEGF generation. Human colon cancer cell line Caco-2 was activated in complete culture medium with addition of NaCl (100 mM) to emulate the colon hyperosmotic environment. AA release, PGE2 and VEGF generation were analyzed in cell culture supernatants, while, COX-2 expression and MAPKs phosphorylation were analyzed in cell lysates by western blotting. Pharmacological inhibitors of PLA2s, COX-2 and MAPKs were added 15 to 30 minutes before Caco-2 activation. Hypertonic stress and CoCl2 induced expression of COX-2 and production of PGE2 and VEGF in Caco-2. PGE2 production by stimulated Caco-2 depends on COX-2 and PLA2. The cPLA2 supplies AA as substrate for COX-2 resulting in PGE2 production, which, in turn, modulates VEGF production, through receptor EP2, in hypertonic stress. However, the PGE2 does not regulate the VEGF production by Caco-2 in hypoxic conditions. The PLA2 is activated by MAPKs, ERK1/2, p38 and JNK, in both environment stress. ERK1/2 induces the COX-2 expression in Caco-2 activated by hypertonic stress. The phosphorylation of p38 has a role in the induction of the VEGF production for Caco-2 in hypertonic stress. Our results suggest that colon cancer cells respond to environmental stress with activation of MAPK signaling pathways, PGE2 and VEGF production to adapt to the external conditions. The autocrine role of PGE2 on VEGF production seems to depend on environmental stress type and suggests different signaling with potential impact on tumoral development.

LISTA DE ABREVIATURAS E SIGLAS
γGT Gama glutamil transpeptidase 3H Trítio 5-LO 5-Lipoxigenase AA Ácido araquidônico AACOCF3/AATFMK/ATK Araquidonil trifluorometil cetona ACF Focos de cripta aberrante APC Gene da polipose adenomatosa colônica BAEC Célula endotelial de aorta bovina BEL Bromoenol lactona CamKII Quinase calmodulina II cAMP Adenosina monofosfato cíclico CBL Alça de ligação para cálcio CoCl2 Cloreto de cobalto COX-2 Ciclooxigenase-2 cPLA2 Fosfolipase A2 citosólica CREB Proteína ligante do elemento de resposta ao AMP cíclico DFX Desferrioxamina DMEM Meio de Eagle modificado por Dulbecco DMSO Dimetilsulfóxido DNA Ácido desoxirribonucléico DTT Ditiotreitol ELISA Ensaio de imunoadsorção enzimática ERK Quiinase regulada por sinal extracelular FACL4 Ligase 4 de ácido graxo-CoA FAP Polipose adenomatosa familiar FLAP Proteína ativadora de 5-lipoxigenase GAPDH Gliceraldeído 3-fosfato desidrogenase HDL Lipoproteína de alta densidade HEPES Ácido hidroxietil piperazina etanosulfônico HETE Ácido hidroxieicosatetraenóico HGF Fator de crescimento para hepatócito HIF-1α Fator induzível por hipóxia-1alfa HPETE Ácido hidroperoxieicosatetraenóico HRP Peroxidase de Armoracia rusticana HUVEC Célula endotelial de veia umbilical humana IL Interleucina iPLA2 Fosfolipase A2 independente de cálcio JNK Quinase de região terminal NH2 de c-Jun LDL Lipoproteína de baixa densidade LPS Lipopolissacarídio bacteriano LTA4 Leucotrieno A4 MAFP Metil araquidonil fluorofosfonato

MAPK Proteína quinase ativada por mitógeno MNK1 Quinase que interage com MAPK NaCl Cloreto de sódio NFAT Fator nuclear de célula T ativada PAF Fator ativador plaquetário PBS Tampão fosfato salina PC Fosfatidilcolina PDGF Fator de crescimento derivado de plaqueta PE Fosfatidiletanolamina PGE2 Prostaglandina E2 PGF Fator de crescimento placentário PKA Proteína quinase dependente de AMP cíclico PKC Proteína quinase C PS Fosfatidilserina Ras Oncogene do vírus do sarcoma murino de Harvey e Kirsten RNAm Ácido ribonucléico mensageiro SDS Duodecil sulfato de sódio SFB Soro fetal bovino sPLA2 Fosfolipase A2 secretória TBS Tampão Tris salina Tcf Fator de célulaT TNFα Fator de necrose tumoral alfa TXA2 Tromboxana A2 VEGF Fator de crescimento endotelial vascular

LISTA DE ILUSTRAÇÕES
Figura 1: Metabolismo do AA pelas vias de 5-LO e ciclooxigenase (PGHS). 16
Tabela 1: Comparação das enzimas PLA2 secretórias. 18
Tabela 2: Funções da cPLA2α definidas pelo fenótipo do camundongo knockout para cPLA2α.
29
Figura 2: Representação espacial das taxas brutas de incidência por 100.000 homens e 100.000 mulheres, estimadas para o ano 2006, segundo a Unidade da Federação (neoplasia maligna do cólon e reto).
36
Figura 3: O estresse hipertônico induz a liberação de AA, a expressão de COX-2 e a produção de PGE2 em Caco-2.
63
Figura 4: A produção de PGE2 é dependente da atividade de cPLA2 e de COX-2 em Caco-2 estimulada por estresse hipertônico.
65
Figura 5: cPLA2 regula o fornecimento de AA em Caco-2 estimulada por estresse hipertônico.
66
Figura 6: As quinases ERK1/2, JNK e p38 estão envolvidas na produção de PGE2 em Caco-2 estimulada por estresse.
68
Figura 7: O estresse hipertônico modula a fosforilação das três MAP quinases em Caco-2.
70
Figura 8: MAPKs modulam a expressão de COX-2 em Caco-2 estimulada por estresse hipertônico.
71
Figura 9: As três MAP quinases participam da liberação de AA por cPLA2 em Caco-2 estimuladas por estresse hipertônico.
72
Figura 10: O estresse hipertônico induz a produção de VEGF em Caco-2 estimulada por estresse hipertônico.
74
Figura 11: PGE2 modula a produção de VEGF em Caco-2 estimulada por estresse hipertônico.
75
Figura 12: Papel da cascata do AA sobre a produção de VEGF em HCT116 estimulada por estresse hipertônico.
76
Figura 13: A adição de PGE2 ao meio reverte o efeito de sua inibição sobre a produção de VEGF em Caco-2 estimulada por estresse hipertônico.
77
Figura 14: O receptor EP2 está envolvido na regulação da produção de VEGF por PGE2 em Caco-2 estimulada com estresse hipertônico.
79
Figura 15: A adição de CoCl2 ao meio induz a produção de PGE2 e a expressão de COX-2 em Caco-2.
81
Figura 16: A produção de PGE2 é dependente de COX-2 e cPLA2 em Caco-2 estimulada por CoCl2.
82
Figura 17: A adição de CoCl2 ao meio induz a produção de VEGF em Caco-2 e PGE2 não modula a produção do fator angiogênico neste tipo de estímulo.
83
Figura 18: ERK1/2, JNK e p38 estão envolvidas no aumento da produção de VEGF em Caco-2 estimulada por CoCl2.
85

Figura 19: p38 e ERK1/2 são as quinases envolvidas na produção de VEGF em Caco-2 estimulada por estresse hipertônico.
86
Figura 20: Modelos de regulação da expressão de COX-2 e produção de PGE2 e VEGF por cPLA2 em células de câncer de cólon.
106

SUMÁRIO
1. INTRODUÇÃO 15
1.1. Fosfolipases A2 (PLA2s) 15
1.2. Fosfolipase A2 citosólica (cPLA2) 24
1.3. Inibição Farmacológica de Fosfolipases A2 30
1.4. Câncer de Cólon 34
1.5. Fosfolipase A2 e Câncer 42
1.6. Angiogênese e Enzimas do Metabolismo do Ácido Araquidônico 48
2.OBJETIVOS 53
3. MATERIAIS E MÉTODOS 54
3.1. Linhagens Celulares 54
3.2. Reagentes 55
3.3. Tratamentos com PGE2 e inibidores farmacológicos 56
3.4. Enzyme Linked Immuno Sorbent Assay (ELISA) e Enzyme Immuno Assay (EIA) 57
3.5. Liberação de Ácido Araquidônico 57
3.6. Western Blot 58
3.7. Análise Estatística 60
4. RESULTADOS 61
4.1. O estresse hipertônico induz a liberação de AA, o aumento da expressão de COX-2 e a produção de PGE2 em Caco-2
62
4.2. A produção de PGE2 é dependente da atividade de cPLA2 e de COX-2 em Caco-2 estimulada com estresse hipertônico
64
4.3. cPLA2 regula o fornecimento de AA em Caco-2 estimulada com estresse hipertônico
64

4.4. As quinases ERK 1/2, JNK 1/2/3 e p38 estão envolvidas na produção de PGE2 por Caco-2 estimuladas com estresse hipertônico
67
4.5. O estresse hipertônico modula a fosforilação das três MAPKs em Caco-2 69
4.6. MAPKs modulam a expressão de COX-2 e a liberação de AA por cPLA2 em Caco-2 estimulada por estresse hipertônico
69
4.7. O estresse hipertônico induz a produção de VEGF em Caco-2 e PGE2 modula a produção do fator angiogênico neste tipo de estresse microambiental
73
4.8. O receptor EP2 está envolvido na regulação da produção de VEGF por PGE2 em Caco-2 estimulada com estresse hipertônico
78
4.9. A adição de CoCl2 ao meio induz a produção de PGE2 e a expressão de COX-2 por Caco-2
80
4.10. A produção de PGE2 é dependente de COX-2 e cPLA2 em Caco-2 estimulada por CoCl2
80
4.11. ERK1/2, JNK e p38 estão envolvidas na produção de VEGF por Caco-2 estimulada por CoCl2
84
4.12. p38 e ERK1/2 estão envolvidas na produção de VEGF por Caco-2 estimulada por estresse hipertônico
84
5. DISCUSSÃO 87
6. CONCLUSÕES 107
7. REFERÊNCIAS BIBLIOGRÁFICAS 108
8. ANEXO 139

15
1. INTRODUÇÃO
1.1. Fosfolipases A2 (PLA2s)
As PLA2s representam o primeiro passo metabólico da via do ácido araquidônico (AA)
que leva à produção de eicosanóides, uma classe de mediadores químicos que participa de
processos tão diversos quanto a manutenção da integridade da mucosa gástrica, coagulação e
inflamação (Balsinde J et al., 1999a; Yedgar S et al., 2000). Até o momento, existem
aproximadamente 20 genes codificantes para enzimas PLA2s em mamíferos. A superfamília das
PLA2s é composta por um amplo espectro de enzimas que catalizam a hidrólise da ligação éster
na posição 2 (sn-2) do esqueleto de glicerol de fosfolipídeos (Six DA e Dennis EA, 2000; Yedgar
S et al., 2000). Os produtos desta reação de hidrólise são um ácido graxo livre e um
lisofosfolipídeo. O ácido graxo liberado pela atividade fosfolipásica pode funcionar, como um
segundo mensageiro ou como precursor de eicosanóides, neste último caso quando o ácido graxo
liberado for o AA (Six DA et al., 2000). Outro composto, liberado pela ação destas enzimas sobre
a fosfatidilcolina, é o 1-éter 2-lisofosfolipídeo (ou liso-PAF), precursor do Fator Ativador
Plaquetário (PAF), outro potente mediador inflamatório (Balsinde J et al., 1999a). O AA
liberado dos fosfolipídeos das membranas celulares pela ação das PLA2s, é metabolizado por
uma das vias enzimáticas para a produção dos eicosanóides. Podendo ser metabolizado pela
enzima 5-lipoxigenase (5-LO) até leucotrieno A4 (LTA4), um substrato para enzimas terminais da
via do leucotrieno, ou por uma das isoformas da enzima ciclooxigenase (COX ou sintase
endoperóxido prostaglandina) até PGH2, um substrato para enzimas terminais da biosíntese de
prostanóides (Figura 1) (Smith WL, 1992; Griffiths RJ, 1999). Leucotrienos e prostaglandinas
possuem função em vários processos fisiopatológicos, incluindo asma, inflamação,

16
carcinogênese, dor, febre, hemostasia, manutenção da função renal e parto (Lewis RA et al.,
1990; DuBois RN et al., 1998). Outros produtos do AA incluem ácidos hidroxieicosatetraenóicos
(HETEs), as lipoxinas, os ácidos epoxieicosatriênicos e isoprostanos, que também podem
modular as respostas inflamatórias (Serhan CN e Oliw E, 2001). Além dos metabólitos do AA, o
próprio AA também pode ter ações biológicas independente de modificações enzimáticas (Brash
AR, 2001).
Figura 1: Metabolismo do AA pelas vias de 5-LO e ciclooxigenase (PGHS). PLA2, fosfolipase A2; FLAP, proteína ativadora de 5-LO; LT, leucotrieno; HETE, ácido hidroxieicosatetraenóico; HPETE, ácido hidroperoxieicosatetraenóico; GSH, glutationa; γGT, gama glutamil transpeptidase; PG, prostaglandina (Diaz BL e Arm JP, 2003).
Glicerofosfolipídeos
Ácido Araquidônico
5-LO + FLAP
Fosfolipase A2cPLA2sPLA2
PGG2
PGHS-1PGHS-2
PGH2
PGD2, PGE2, PGI2, PGF2α, TXA2
Sintases específicas
LTA4
LTB4
LTA4 Hidrolase 6-t-LTB4
GSH
LTC4 Sintase
LTC4 LTD4 LTE4
DipeptidaseγGT
5-HPETE
5-HETE
Glicerofosfolipídeos
Ácido Araquidônico
5-LO + FLAP
Fosfolipase A2cPLA2sPLA2
PGG2
PGHS-1PGHS-2
PGH2
PGD2, PGE2, PGI2, PGF2α, TXA2
Sintases específicas
LTA4
LTB4
LTA4 Hidrolase 6-t-LTB4
GSH
LTC4 Sintase
LTC4 LTD4 LTE4
DipeptidaseγGT
5-HPETE
5-HETE
Glicerofosfolipídeos
Ácido Araquidônico
5-LO + FLAP
Fosfolipase A2cPLA2sPLA2
PGG2
PGHS-1PGHS-2
PGH2
PGD2, PGE2, PGI2, PGF2α, TXA2
Sintases específicas
LTA4
LTB4
LTA4 Hidrolase 6-t-LTB4
GSH
LTC4 Sintase
LTC4 LTD4 LTE4
DipeptidaseγGT
LTC4 LTD4 LTE4
DipeptidaseγGT
5-HPETE
5-HETE

17
A superfamília das PLA2s está dividida em quatro sub-famílias, a das PLA2 secretórias
(sPLA2), que abrange os grupos I, II, V, X e XII, a da PLA2 citosólica (cPLA2), constituída pelo
grupo IV, as Acetilhidrolases de PAF, que compreende o grupo VII. e a sub-família da PLA2
independente de cálcio (iPLA2), que abrange o grupo VI (Six DA et al., 2000; Yedgar S et al.,
2000). Estas sub-famílias de PLA2 diferem em alguns pontos de sua estrutura, em relação ao
requerimento de cálcio para ativação, pH ótimo, especificidade de substrato e suscetibilidade aos
diferentes inibidores farmacológicos (Tischfield JA, 1997; Dennis EA, 1997; Balsinde J et al.,
1999a).
Entre as sub-famílias de PLA2, a cPLA2 foi distinguida das enzimas secretórias existentes
pelo seu maior tamanho de 85 kDa, por ausência de pontes di-sulfeto, pelo resíduo de serina em
seu sítio catalítico, por sua seletividade em clivar o AA dos fosfolipídeos e por sua dependência
de cálcio, na faixa de micromolar, para ser ativada e translocada do citosol para o envelope
nuclear (Smith WL, 1992; McNish RW e Peters-Golden M, 1993; Griffiths RJ, 1999). Já a sub-
família das sPLA2s caracteriza-se pelo sítio catalítico com uma histidina que é conservada em
todos os membros do grupo; tamanho relativamente pequeno (em torno de 14 kDa) e por sua
estrutura terciária com muitas pontes di-sulfeto, o que as torna sensíveis à inibição por ditiotreitol
(DTT). Elas requerem concentrações de cálcio na faixa de milimolar para a atividade catalítica,
não possuem preferência pelo AA na posição sn-2 dos fosfolipídeos, pois o funil catalítico é curto
e não se liga ao AA, e se distinguem umas das outras por certos aspectos de sua estrutura e
distribuição tecidual, sugerindo funções diversas para cada grupo (Tabela 1) (Six DA et al.,
2000). A sub-família das Acetilhidrolases de PAF (PLA2 VII) possuem uma serina em seu sítio
catalítico, tamanho entre 40 e 45 kDa, possuem atividade hidrolase, especialmente sobre PAF,
mas podem também hidrolisar ácidos graxos oxidados de cadeia curta e não dependem de cálcio
para sua ativação. A sub-família da iPLA2, assim como a cPLA2, possui uma serina em seu sítio

18
catalítico e tamanho de 85 kDa, mas não depende de cálcio para sua ativação (Six DA et al.,
2000).
PLA2 Cr Mr (kDa)
Pares de Cisteínas
pI Principal local de expressão dos transcritos de RNA
Outras características
IB 5 14.1 7 6.7 Pâncreas, pulmão, fígado, intestino delgado
Pró-peptídeo; par de
Cys11...Cys77 na alça
elapídica IIA 4 13.9 7 9.2 Intestino delgado IIC 4 14.6 8 8.3 Testículos, pâncreas IID 4 14.2 7 8.7 Pâncreas, baço, timo, pulmões IIE 4 14.4 7 8.1 Útero, tireóide, testículos IIF 4 16.8 7 5.9 Embrião,testículos
Extensão carboxi-
terminal; par de
Cys50...Cys137IIIa n.c. 55.3 5 9.1 Coração, rins, fígado, músculo
esquelético Extensões terminais
longas NH2...e COOH...
V 4 13.8 6 8.1 Olhos, coração, pâncreas X 16 13.9 8 5.9 Testículos, estômago Pró-peptídeo
XII 3 18.7 7 6.3 Células Th2 Estrutura nova Tabela 1: Comparação das enzimas PLA2 secretórias. Detalhes são fornecidos sobre a localização de cada gene nos cromossomos (Cr) de camundongo, massa molecular relativa (Mr) (em kDa), o número de pares de cisteínas (Cys) ligadas à pontes di-sulfeto, ponto isoelétrico (pI), principal local de expressão dos transcritos em tecidos de camundongo e outros aspectos relevantes. aMolécula humana (não reportada em camundongo); n.c., não conhecido (Diaz BL e Arm JP, 2003).
As PLA2s possuem diferenças em relação ao seu modo de ação, podendo ter atividade
interfacial, afinidade por PC/PS ou por proteoglicanos, características funcionais específicas,
como no caso da sPLA2 IIF, ou se ligar à receptores de membrana. A cPLA2 possui preferência
por substratos apresentados na interface da membrana à substratos na forma monomérica
(Nalefski EA et al., 1994). Este comportamento, chamado de ativação interfacial, é devido à
existência de uma espécie de tampa anfipática (resíduos 413-457), que evita que o ácido graxo

19
dos fosfolipídeos entre no funil catalítico (Dessen A et al., 1999). Em condições fisiológicas, a
cPLA2 interage com membranas intracelulares contendo lipídeos aniônicos, e esta interação abre
a tampa do sítio ativo através de repulsão eletrostática, permitindo a entrada do ácido graxo,
preferencialmente o ácido araquidônico, no funil catalítico (Das S e Cho W, 2002). A sPLA2
deve primeiro ancorar à membrana para ter acesso ao seu substrato. As sPLA2s dos grupos X e V
possuem forte ligação interfacial com PC e usam um resíduo de triptofano aromático para se
acoplarem ao fosfolipídeo na membrana plasmática (Bezzine S et al., 2000). Ao contrário da
enzima do grupo IIA, cuja carga de sua superfície permite sua interação com fosfolipídeos
aniônicos. Logo, a enzima do grupo IIA pode se ligar à fosfatidilserina (PS) sobre a superfície de
células apoptóticas ou sobre a superfície de células ativadas, após a ação da escramblase (Atsumi
G et al., 1997). Já as sPLA2s IIA, IID e V possuem afinidade por proteoglicanos. Quando estas
enzimas que se ligam à heparina foram transfectadas em células HEK293, que eram estimuladas
com ionóforo de cálcio ou com SFB e IL-1β, elas eram liberadas da célula e se ligavam ao
glipican na cavéola na membrana plasmática, sendo, então, internalizadas e acopladas à COX-2
para a produção de PGE2 (Murakami M et al., 2001). Porém, o fato da enzima ser internalizada e
dirigida para uma via de produção de eicosanóides não depende somente de suas propriedades
bioquímicas, mas também do tipo celular envolvido. Em certas células hematopoiéticas, tais
como mastócitos (Enomoto A et al., 2000) e neutrófilos (Kim KP et al., 2001), a ligação aos
proteoglicanos pode direcionar as sPLA2s internalizadas para uma via de degradação. Além de
sua afinidade com proteoglicanos, estudos revelaram que PLA2IID recombinantes de murinos e
humanos, secretadas de células COS e CHO, eram ativas contra vesículas de fosfatidilglicerol,
fosfatidilcolina (PC) e fosfatidiletanolamina (PE), com afinidade por sítio ativo similar em todos
os três substratos (Six DA et al., 2000). Uma enzima que é única em sua habilidade de liberação

20
de AA independente de qualquer afinidade significante pela heparina ou PC, é a sPLA2IIF, que
por causa de sua extensão carboxi-terminal de 30 aminoácidos, pode se ligar diretamente aos
fosfolipídeos da membrana (Murakami M et al., 2002a). Além dos mecanismos de ligação das
enzimas às cabeças polares dos fosfolipídeos de membrana ou com proteoglicanos, já foram
descritos receptores de membrana tipo manose, específicos para sPLA2 dos grupos IB e X, o que
sugere que estas enzimas podem ter ações independentes de sua capacidade enzimática (Valentin
E e Lambeau G, 2000; Yokota Y et al., 2000).
A sPLA2 do grupo IB foi a primeira fosfolipase não oriunda de veneno de cobra
identificada. Ela foi identificada primeiramente em suco pancreático de suínos e, posteriormente,
em humanos. Esta enzima é secretada como um zimogênio sendo clivada pela tripsina. Por causa
de sua alta expressão em sucos pancreáticos foi atribuída à sPLA2IB função na digestão de
fosfolipídeos da dieta (Six DA et al., 2000). Porém, com a produção do camundongo knockout
para sPLA2 IB, verificou-se que não havia quaisquer anormalidades na digestão de lipídeos
quando estes animais eram alimentados com dieta normal (Richmond BL et al., 2001). Contudo,
a sPLA2IB parece ter função no uptake de lipídeos, pois quando os camundongos knockout foram
alimentados com dieta rica em gordura os animais eram protegidos da obesidade e resistência à
insulina. Portanto, tal enzima pode ter importante papel no desenvolvimento do diabetes tipo II,
sendo potencial alvo terapêutico nesta doença (Huggins KW et al., 2002).
As sPLA2s do grupo II são expressas em tecidos como fígado, baço, plaquetas e fluido
sinovial e são secretadas por várias células do sistema imune em resposta a estímulos apropriados
(Ono T et al., 1988; Seilhamer JJ et al., 1989; Kramer RM et al., 1989; Komada M et al., 1989;
Aarsman AJ et al., 1989; Kusunoki C et al., 1990). A sPLA2 do grupo IIA está envolvida em
diversos processos patológicos como artrite reumatóide (Seilhamer JJ et al., 1989), choque
séptico (Vadas P et al., 1992), câncer de cólon (Cormier RT et al., 1997; Kucherlapati R et al.,

21
2001), queimaduras (Yamada Y et al., 1998), bem como em sinalização celular para regular a
formação de mediadores lipídicos derivados de AA (Seilhamer JJ et al., 1989; Murakami M et al.,
1995; Fujishima H et al., 1999). Até o descobrimento de outros membros desta sub-família era
conhecida como a PLA2 inflamatória. Certas cepas de camundongos, como C57Bl/6; 129 e A/J,
possuem uma disrupção natural do gene desta enzima (Kennedy BP et al., 1995; MacPhee M et
al., 1995). Entretanto, células provenientes de camundongos C57Bl/6 não apresentam defeito em
relação à liberação de AA ou à produção de eicosanóides apesar da ausência desta enzima
(Bingham CO et al., 1996). A sPLA2IIA humana também é conhecida por se ligar a interfaces
contendo fosfolipídeos aniônicos como membranas bacterianas, sendo implicada em resposta de
defesa do hospedeiro com sua atividade anti-bacteriana (Buckland AG e Wilton DC, 2000).
Não há presença de transcritos de PLA2 IIC em tecidos humanos, pois é um pseudogene
em humanos, e uma mutação frequente nonsense no exonII elimina a possibilidade de um
produto gênico funcional em homozigotos. Esta enzima é expressa em testículos de roedores e
não possui função conhecida (Tischfield JA et al., 1996). As enzimas do grupo IID, IIE e IIF,
assim como as do grupo XII foram identificadas recentemente e, apesar de serem capazes de
liberar AA e favorecer a produção de eicosanóides em células transfectadas (Murakami M et al.,
1998; Murakami M e Kudo I, 2001), pouco se sabe sobre suas funções biológicas.
O RNAm para sPLA2V é encontrado de forma abundante em coração humano e murino
(Chen J et al., 1994; Bingham COIII et al., 1999). A participação desta enzima na produção de
eicosanóides, mediada por COX-2, em conjunto com a cPLA2, foi demonstrada em uma
linhagem macrofágica murina P388D1 (Balboa MA et al., 1996; Balsinde J et al., 1999a).
Quando estas células foram marcadas com [3H] AA por 5 horas, primadas com lipopolissacarídeo
(LPS) por 1 hora, e então, ativadas com PAF, havia liberação bifásica de AA marcado. Na
primeira fase, que terminava aos 2 minutos, o AA era liberado, porém retido dentro da célula,

22
enquanto que, na segunda fase a liberação extracelular de AA se dava aos 10 minutos (Balsinde J
et al., 1994). Estudos com inibidores farmacológicos e com oligonucleotídeos anti-senso
demonstraram que a liberação intracelular inicial de AA foi dependente de cPLA2α, ao passo que
a liberação extracelular subseqüente de AA era dependente da sPLA2V (Balboa MA et al., 1996).
Ainda, a ação inicial de cPLA2α parece ser necessária para a ação da sPLA2V, sugerindo um
crosstalk entre cPLA2 e sPLA2V (Balsinde J e Dennis EA, 1996). Além disso, o papel da enzima
sPLA2V em promover a indução de COX-2 e produzir uma fase tardia de produção de PGE2
também foi demonstrado nesta mesma linhagem celular (Balsinde J et al., 1999b).
A expressão de transcritos de PLA2X em humanos foi detectada em baço, timo e
leucócitos, enquanto que a proteína tem sido detectada em células endoteliais pulmonares. Já em
camundongos os transcritos da enzima são identificados em testículos e estômago (Six DA et al.,
2000). A sPLA2 X apresenta reduzida capacidade de liberar AA quando transfectada em diversas
linhagens celulares (Six DA et al., 2000), entretanto, devido às suas características físico-
químicas que permitem sua ligação às membranas ricas em PC, é capaz de liberar AA e PGE2 de
células monocíticas humanas THP-1 quando adicionada exogenamente (Hanasaki K et al., 1999).
A subfamília das acetilhidrolases de PAF compreende os grupos VIIA e VIIB. A PLA2
VIIA é a enzima mais bem conhecida por sua atividade acetilhidrolase de PAF, podendo também
hidrolizar ácidos graxos oxidados de cadeia curta com mais de nove carbonos na posição sn-2 de
PC ou fosfatidiletanolamina (PE) independente da natureza ou comprimento da cadeia sn-1. Em
humanos, tal enzima está associada com a apolipoproteína B100 (apo B100) da lipoproteína de
baixa densidade (LDL) e lipoproteína de alta densidade (HDL). A presença de fosfolipídeos
oxidados na LDL é associada com condições patológicas como a aterosclerose e, portanto, a ação
da PLA2VIIA clivando os lipídeos oxidados pode funcionar protegendo contra a doença. A

23
PLA2VIIA não é uma enzima interfacial, o que significa que ela não acessa fosfolipídeos como
substratos agregados à membrana, mas somente acessa PAF em solução como um substrato
monomérico. A PLA2VIIB é uma enzima intracelular primeiramente purificada e caracterizada
de cérebro bovino, tendo grande expressão em fígado e rins. Apesar de sua presença no citosol,
está também localizada no retículo endoplasmático próximo ao envelope nuclear. Células
superexpressando a PLA2VIIB foram resistentes à apoptose induzida por estresse oxidativo,
sugerindo que enquanto a PLA2VIIA possui função protetora contra oxidação no plasma, a
PLA2VIIB intracelular protege células hepáticas e renais de danos oxidativos (Six DA et al.,
2000).
A primeira PLA2 clonada e caracterizada sem nenhuma dependência de cálcio foi a PLA2
cálcio-independente (iPLA2), sendo também classificada como PLA2VIA. Além de inibidores
farmacológicos específicos para esta enzima, como o bromoenol lactona (BEL), outros inibidores
farmacológicos da PLA2 citosólica também inibem tal enzima, confirmando o papel da serina
como o nucleófilo. A PLA2VIA tem como funções o remodelamento de fosfolipídeos da
membrana, regulando o pool de AA disponível para liberação por outras PLA2s (Six DA et al.,
2000); a liberação de AA e geração de eicosanóides iniciada pela agregação de receptores para
imunoglobulina G (FcγRs) em células monocíticas humanas (Tay HK e Melendez AJ, 2004).
Ainda, em situações onde o nível intracelular de cálcio está diminuído, tal enzima pode regular o
influxo do íon. A ativação do Fator de Influxo de Cálcio (CIF) pela depleção de cálcio, faz com
que CIF desloque a calmodulina inibitória da iPLA2 resultando na ativação da enzima e produção
de lisofosfolipídeos, que ativam canais store-operated (SOCs) estimulando o influxo de cálcio
(Smani T et al., 2004).

24
Ao agir no passo requerido para a biossíntese de eicosanóides, as PLA2s possuem
importante papel em vários processos fisiopatológicos, abrangendo desde inflamação até
neoplasias. Logo, novas descobertas que esclareçam cada vez mais as funções de cada grupo de
PLA2 são necessárias, tornando possíveis futuras abordagens terapêuticas com a inibição destas
enzimas, bloqueando a síntese de mediadores inflamatórios, como os eicosanóides e o PAF.
1.2. Fosfolipase A2 citosólica (cPLA2)
A cPLA2α, também conhecida como PLA2IVA, foi a primeira PLA2 caracterizada de sítio
catalítico com uma serina, ao invés de uma histidina, tendo peso molecular de 85 kDa. A
disrupção do gene da cPLA2-α por recombinação homóloga revelou o seu papel essencial na
liberação de AA para geração de eicosanóides, potentes mediadores da inflamação e de vias de
transdução de sinal (Bonventre JV et al., 1997; Fujishima H et al., 1999).
A família da PLA2 do grupo IV contém seis membros, grupo IVA, grupo IVB, grupo
IVC, grupo IVD, grupo IVE e grupo IVF. Estas enzimas são também conhecidas como cPLA2α,
β, γ, δ, ε e ζ, respectivamente (Ghosh M et al., 2006). A cPLA2α tem sido muito estudada por
conta de sua especificidade pelo AA na posição 2 do esqueleto de glicerol de fosfolipídeos (sn-2)
e por seu papel na produção de eicosanóides (Leslie CC, 2004a; Leslie CC, 2004b). Tal enzima é
conservada evolutivamente com mais de 95% de identidade de aminoácidos entre as enzimas
homólogas de humanos e de camundongos (Clark JD et al., 1991). A principal característica da
família do Grupo IV é um sítio díade ativo conservado composto de uma serina nucleofílica
(Ser228 na cPLA2α) e um ácido aspártico (Asp549 na cPLA2α), sendo também necessária para a

25
atividade enzimática uma arginina conservada (Arg200 na cPLA2α) (Sharp JD et al., 1994;
Pickard RT et al., 1996; Huang Z et al., 1996; Dessen A, 2000). Em mamíferos os parálogos do
grupo IV, cPLA2 β, γ, δ, ε e ζ possuem aproximadamente 30 a 37% de identidade de aminoácidos
com a cPLA2α (Ohto T et al., 2005). Todos os membros do Grupo IV, com exceção da cPLA2γ,
contêm um domínio C2. Um grande número de proteínas em mamíferos possuem este domínio,
que promove a interação das proteínas com membranas (Nalefski EA e Falke JJ, 1996; Cho W,
2001; Jimenez JL et al., 2003). Eles são compostos de aproximadamente 120 aminoácidos e
possuem alças de ligação para cálcio (CBL). Quando a proteína não está ligada à membrana, os
resíduos ácidos do domínio C2 conferem eletronegatividade à face da proteína que se ligaria à
membrana, não favorecendo a interação com a mesma. A ligação com o cálcio neutraliza os
resíduos aniônicos e muda o potencial para eletropositivo, caso resíduos básicos estejam
presentes nas alças de ligação à membrana, permitindo, assim, a ligação da enzima com a
membrana (Murray D e Honig B, 2002).
O gene codificante para a cPLA2α humana está no cromossoma 1q25 próximo ao gene
codificante para COX-2 (Tay A et al., 1995). Ao contrário de COX-2, a cPLA2α está
constitutivamente expressa na maioria das células e tecidos (Clark JD et al., 1995). A expressão
desta fosfolipase é aumentada por algumas citocinas pro-inflamatórias e fatores de crescimento
em vários tipos celulares. Este aumento na expressão muitas vezes ocorre juntamente com o
aumento na expressão de COX-2 e contribui para a fase tardia da produção de eicosanóides. A
regulação da expressão de cPLA2α pelas citocinas pode envolver efeitos pós-transcricionais, com
a estabilidade do RNA mensageiro, e mecanismos transcricionais (Dolan-O'Keefe M et al.,
2000). Recentemente demonstrou-se que a expressão de cPLA2α é induzida nas células de
músculo liso pelo Fator de Crescimento Derivado de Plaqueta (PDGF) e pela trombina e é

26
dependente do fator de transcrição STAT-3 (Neeli I et al., 2004; Dronadula N et al., 2005).
Ainda, foram encontrados vários elementos de resposta a interferon-γ, uma seqüência ativada por
interferon-γ e dois elementos responsivos a glicocorticóide no promotor da cPLA2α, sugerindo
regulação transcricional da enzima (Wu T et al., 1994; Miyashita A et al., 1995).
Há muitas evidências apontando para o papel da PGE2 na promoção da tumorigênese.
Como a cPLA2α fornece AA para a produção do prostanóide, a expressão de cPLA2α em células
de câncer tem sido avaliada. A expressão aumentada de cPLA2α foi observada em vários tipos de
neoplasias (Laye JP e Gill JH, 2003), como é o caso de linhagens celulares de câncer pulmonar
humano (NSCLC). As células NSCLC possuem expressão constitutiva de cPLA2α e COX-2, o
que propicia a produção de PGE2. O aumento da transcrição de cPLA2α em células NSCLC é
dependente do oncogene Ras e possui papel importante na promoção da transformação celular
(Heasley LE et al., 1997). Ras tem papel na ativação das proteínas quinases ativadas por
mitógeno (MAPKs), entre elas a quinase de região terminal NH2 de c-Jun (JNK) e a quinase
regulada por sinal extracelular (ERK), que são necessárias para a indução de cPLA2α. O RNA
mensageiro de cPLA2α é também super-regulado em adenocarcinomas coloretais, contudo, tal
regulação não está relacionada com mutações no gene Ras, sugerindo que os mecanismos de
indução de cPLA2α diferem dependendo do tipo de câncer (Osterstrom A et al., 2002).
Um importante passo na regulação da cPLA2α envolve a sua translocação do citosol para
as membranas para acessar o substrato. A translocação é induzida em resposta ao aumento na
concentração de cálcio intracelular, que se liga ao domínio C2 e aumenta a afinidade da enzima
por membranas (Leslie CC, 1997). Em muitas células expostas a agonistas que mobilizam cálcio,
a cPLA2α transloca principalmente para a região perinuclear, especificamente para o envelope
nuclear, retículo endoplasmático e Golgi. A preferência na translocação para uma ou outra

27
membrana varia de acordo com os níveis de cálcio e com a duração do estímulo, em estímulos de
curta duração e de baixa concentração a translocação se dá, preferencialmente, para o Golgi. O
domínio C2 é suficiente para direcionar a localização subcelular da cPLA2α em resposta a
estímulos que liberam cálcio, pois transloca para as mesmas membranas intracelulares da mesma
forma que a enzima selvagem (Evans JH et al., 2001). As CBLs 1 e 3, que possuem resíduos
hidrofóbicos necessários para a ligação da proteína com a membrana, são essenciais na ligação ao
Golgi (Evans JH et al., 2004). Outro domínio importante para a ligação da cPLA2α à membrana é
o domínio catalítico, que estabiliza tal ligação por mecanismos independentes de cálcio. Em
células, a cPLA2α transloca mais vagarosamente para o Golgi e exibe associação mais
prolongada com a membrana depois da diminuição dos níveis de cálcio intracelular, em
comparação com o domínio C2 isolado (Evans JH e Leslie CC, 2004). Esta associação
prolongada independente de cálcio é atribuída a um resíduo de triptofano (Trp464) localizado no
domínio catalítico e que interage principalmente com PC (Han SK et al., 1999; Feng J et al.,
2002).
A fosforilação da cPLA2α é importante para a regulação da liberação de AA nas células.
O domínio catalítico da enzima contém vários sítios de fosforilação, como Ser505, Ser727 e
Ser515, que são fosforilados por MAPKs, quinase interagindo com MAPK (MNK1), ou quinase
calmodulina II (CamKII), respectivamente (Lin LL et al., 1993; Kramer RM et al., 1996; Borsch-
Haubold AG et al., 1998; Hefner Y et al., 2000; Muthalif MM et al., 2001). Estudos em sistemas
celulares têm demonstrado que a fosforilação de cPLA2α sobre a Ser505 ou Ser727 possui
importante papel na regulação da liberação de AA sob condições de aumento transitório no cálcio
intracelular, porém, é menos importante em situações onde há aumento de cálcio sustentado
(Hefner Y et al., 2000). Contudo, poucos estudos têm investigado o papel da fosforilação na

28
regulação da translocação de cPLA2α em células. Dois estudos demonstraram que cPLA2α
mutada na Ser505 possui propriedades de translocação similares às da enzima selvagem em
células ativadas com aumento transitório de cálcio (Schievella AR et al., 1995; Evans JH et al.,
2002). Entretanto, a translocação de cPLA2α em cardiomiócitos estimulados pelos receptores β-
adrenérgicos, ocorre sem aumento de cálcio intracelular, e requer a ativação de MAPKs,
sugerindo um papel para a fosforilação de cPLA2α na Ser505 (Magne S et al., 2001). Desde que
a fosforilação de cPLA2α sobre a Ser505 não é suficiente para promover a ligação na membrana,
outros mecanismos devem agir, juntamente com a fosforilação, para a regulação da ligação da
enzima com a membrana quando a translocação ocorre na ausência de aumento de cálcio (Gijon
MA et al., 1999).
A disrupção do gene para cPLA2α por recombinação homóloga em camundongos não
revelou somente sua função essencial na liberação de AA para a produção de eicosanóides
(Bonventre JV et al., 1997; Uozumi N et al., 1997; Fujishima H et al., 1999), mas também
evidenciou que a cPLA2α contribui para a patogênese de uma variedade de doenças
especialmente aquelas nas quais a inflamação possui função primária como nas reações alérgicas,
injúrias pulmonares agudas, fibrose pulmonar, injúria cerebral, artrite e em alguns tipos de
neoplasias (Tabela 2) (Diaz BL e Arm JP, 2003).

29
Fisiologia ou modelo de doença
Principais achados em camundongos knockout para cPLA2α
Fertilidade Implantação do blastocisto e/ou decidualização defeituosas Fecundidade Ovulação e/ou fertilização defeituosas Parto Retardo no trabalho de parto; morte perinatal Excreção urinária Aumento da perda de água e osmolaridade plasmática devido
a insuficiência na concentração da urina Isquemia cerebral Redução no volume de cérebro infartado e déficit neurológico
após isquemia/reperfusão Doença de Parkinson Reduzida neurotoxicidade induzida pela 1-metil-4-fenil-
1,2,3,6-tetrahidropiridina (MTPT) Asma Reduzida hiperreatividade das vias aéreas Ulceração gastrointestinal Lesões ulcerativas espontâneas no intestino delgado Injúria pulmonar aguda Edema pulmonar reduzido e seqüestro de
polimorfonucleares; melhora na troca gasosa Fibrose pulmonar Fibrose pulmonar induzida por bleomicina e inflamação
reduzidas Câncer coloretal Número e tamanho dos pólipos reduzidos
Tabela 2: Funções da cPLA2α definidas pelo fenótipo do camundongo knockout para cPLA2α (Diaz BL e Arm JP, 2003).
A ablação genética de cPLA2α suprime a tumorigênese no intestino delgado em
camundongos Apc (Takaku K et al., 2000; Hong KH et al., 2001). De maneira semelhante,
camundongos knockout para COX-2 são também protegidos da tumorigênese no intestino
delgado sugerindo um papel para as prostaglandinas na progressão do câncer (Oshima M et al.,
1996). Contudo, um estudo recente demonstrou que a cPLA2α protege contra o câncer de cólon
(Ilsley JNM et al., 2005). Os camundongos knockout para a enzima desenvolvem mais tumores
no intestino grosso em resposta a um carcinógeno organotrópico de cólon. A susceptibilidade
destes camundongos ao câncer de cólon se correlaciona com a menor freqüência de células
apoptóticas e níveis reduzidos de ceramida no epitélio colônico, já que o AA, liberado pela
cPLA2α, possui ação pró-apoptótica dependente de ceramida (Cao Y et al., 2000; Taketo MM e
Sonoshita M, 2002; Levine LAWR, 2003). Talvez o papel pró-tumorigênico dos mediadores

30
liberados pela ação de cPLA2α e COX-2 seja menos importante do que a ação pró-apoptótica do
AA no balanço das forças que ditam o desenvolvimento tumoral (Ghosh M et al., 2006).
1.3. Inibição Farmacológica de Fosfolipases A2
A abordagem mais utilizada pela maioria dos estudos para avaliar a implicação de uma
PLA2 específica em determinado processo patofisiológico é a inibição da atividade enzimática
pelo uso de inibidores químicos.
As primeiras PLA2s a serem identificadas, isoladas e caracterizadas foram as secretórias e
uma das ferramentas metodológicas usadas para elucidar suas funções foram os inibidores
farmacológicos, que, até meados da década de 90, ainda eram citados em diversos artigos como
sendo específicos para sPLA2s. Tal grupo de inibidores incluía drogas antimalária, como a
mepacrina, aminoglicosídeos, álcoois e poliaminas. Porém, geralmente estas moléculas não
inibem as PLA2s per se, mas agem por bloquear a interação da enzima com seus substratos ou
com o cálcio (Chang J et al., 1987; Mukherjee AB et al., 1994). Logo, atualmente sabe-se que a
falta de especificidade de tais compostos indica que eles não devem ser usados como inibidores
destas enzimas.
Da mesma forma, agentes inespecíficos que se ligam covalentemente às PLA2s, como a
manoalida (Lombardo D e Dennis EA, 1985; Reynolds LJ et al., 1991; Bianco ID et al., 1995) ou
o brometo de p-bromofenacil (Roberts MF et al., 1977; Mayer RJ e Marshall LA, 1993), já foram
considerados inibidores destas enzimas. Na verdade, estes compostos inibem sPLA2 in vitro, mas
não cPLA2 ou iPLA2, tal inibição se deve ao bloqueio covalente por estes compostos dos resíduos
expostos de lisina ou histidina. Então, em sistemas celulares, estes compostos provavelmente irão

31
interagir com várias proteínas, tornando impossível conclusões sobre seus efeitos. Outros
compostos utilizados, são os fosfolipídeos análogos de substrato, que funcionam como inibidores
reversíveis de PLA2s e podem ter preferência por sPLA2, como é o caso dos fosfolipídeos amida
tioéter (Plesniak LA et al., 1993; Plesniak LA et al., 1995) e dos análogos de estado de transição
de fosfonato (Yuan W et al., 1990; Yu L e Dennis EA, 1993; Gelb MH et al., 1994; Gelb MH et
al., 1995). Infelizmente, nenhuma destas drogas parecem ser potentes in vitro ou in vivo e, além
disso, tais compostos são anfipáticos e frequentemente se agregam ou se dividem dentro de
micelas ou membranas. Devido a estas propriedades de competitividade e reversibilidade, o uso
de tais inibidores requer avaliação cinética especial (Carman GM et al., 1995). Apesar de tal
inibição não ser potente o suficiente para intervenção farmacológica, estes compostos podem ser
úteis nos estudos mecanísticos.
Em 1995, um grupo de inibidores mais específico para sPLA2, em especial para sPLA2
humana não-pancreática, foram descritos. Entre eles estava o ácido sulfônico propano 3-(3-
acetamida-1-benzil-2-etilindolil-5-oxi), também denominado LY311727, que apresentou inibição
de 1500 vezes quando colocado juntamente com a sPLA2 pancreática de porco (Schevitz RW et
al., 1995). O composto LY311727 é, provavelmente, o inibidor melhor caracterizado e mais
eficiente em estudos sobre o papel da enzima secretória na mobilização de AA em sistemas
celulares (Lio YC e Dennis EA, 1998). Este inibidor é um derivado do indol, cuja estrutura
química foi refinada por cristalografia de raio-X, usando o sítio ativo da sPLA2 IIA como molde
(Schevitz RW et al., 1995). O LY311727 se liga à enzima em faixa de concentração de
nanomolar e inibe as sPLA2s dos grupos IIA, IB e V, sendo mais seletivo para a sPLA2 do Grupo
IIA. Contudo, ele não necessariamente distingue entre outros grupos de sPLA2s (Chen Y e
Dennis EA, 1998). Além do LY311727, no ano de 2006, foi sintetizado outro grupo de inibidores
com estrutura derivada do indol. Por meio de estudos cristalográficos da interação entre um

32
derivado do indol, o Me-Indoxam, e a sPLA2 X humana, foi modificada a estrutura química deste
inibidor em diversos pontos gerando um novo grupo de compostos. Estas modificações tinham
como objetivo aumentar a afinidade do inibidor pela enzima e alterar a estrutura físico-química
do indol para otimizar seu uso em futuros estudos em animais. Entre as modificações estava a
substituição do grupamento 2-metil pelo 2-etil no local onde o composto interagia com o bolso
hidrofóbico do sítio ativo da enzima, verificando-se maior interação entre o inibidor e a enzima.
Entre estes inibidores gerados por estas modificações estruturais, o mais potente possuía um valor
de IC50 de 75 nM sobre a sPLA2 X humana e seu ortólogo em camundongo. Tal composto é o
mais potente inibidor de sPLA2 X humana reportado até o presente e também inibe um subgrupo
de outras sPLA2s humanas e de camundongos, como as dos grupos IIA, IB e V (Smart BP et al.,
2006).
Em relação à cPLA2, dois inibidores têm sido amplamente usados, o araquidonil
trifluorometil cetona (AATFMK), também conhecido como ATK ou AACOCF3 (Street IP et al.,
1993; Riendeau D et al., 1994; Bartoli F et al., 1994) e o metil araquidonil fluorofosfonato
(MAFP) (Balsinde J et al., 1996; Huang Z et al., 1996). Os dois compostos compartilham uma
estrutura química comum, que é uma cauda araquidonil acoplada a um grupo Serina reativo,
sendo inibidores potentes in vitro. O ATK é um inibidor reversível e de ligação lenta, isto é, ele
leva um tempo maior para que sua atividade inibitória seja plena, além de também inibir as
ciclooxigenases (Riendeau D et al., 1994; Reddy ST e Herschman HR, 1997). Outro problema
que poderá surgir em decorrência do uso, tanto de ATK, quanto de MAFP, é a inibição
concomitante da iPLA2 do Grupo VI (Ackermann EJ et al., 1995; Lio YC et al., 1996). Isto
acontece devido ao fato de ambas enzimas, cPLA2 e iPLA2 usarem uma Serina central para
catálise e possuírem mecanismos catalíticos semelhantes. Porém, a maneira de se distinguir se as
consequências do efeito inibitório destes compostos (ATK ou MAFP) se devem à ação da cPLA2

33
ou da iPLA2 é o uso paralelo do bromoenol lactona (BEL), um inibidor seletivo para iPLA2
(Hazen SL et al., 1991; Balsinde J et al., 1996). Portanto, se determinada resposta é inibida por
ATK e MAFP mas não por BEL, isto indicaria o envolvimento de cPLA2 (Balsinde J et al., 1996;
Balsinde J et al., 1999a).
Como revisado anteriormente, a família da cPLA2 engloba seis grupos. Foram utilizados,
em 2000, inibidores derivados de pirrolidina, que bloqueavam a atividade da isoforma α (ou
grupo IVA) in vitro além da liberação de AA em células THP-1 estimuladas com ionóforo de
cálcio (Seno K et al., 2000). O composto de maior potência deste grupo de inibidores era a
pirrolidina-1, que, no ano seguinte, foi testada por Ghomashchi e colaboradores em diferentes
tipos celulares. Os estudos de inibição in vitro demonstraram que tal composto era um potente
inibidor para a isoforma α da cPLA2, inibindo em 50% a atividade enzimática quando utilizado
em IC50 de 1,8 µM, sendo menos potente sobre a isoforma γ da cPLA2, sobre a iPLA2 IV e sobre
as secretórias dos grupos IIA, X e V. A pirrolidina também bloqueava toda a liberação de AA em
células CHO estimuladas por ionóforo de cálcio estavelmente transfectadas com cPLA2α, em
macrófagos peritoneais de camundongo estimulados com zimozam e ácido ocadaico e em células
MDCK estimuladas com ionóforo de cálcio e ATP (Ghomashchi F et al., 2001).
Em síntese, uma revisão de Lucas e Dennis, publicada, em 2005, na Prostaglandins &
other Lipid Mediators, aborda de maneira bem clara o uso dos inibidores farmacológicos para
distinguir a ação das PLA2s por meio de ensaios grupo-específicos, onde é possível diferenciar
entre as várias PLA2s encontradas nas amostras biológicas. A especificidade de cada ensaio foi
testada pela observação do efeito da adição dos inibidores de PLA2 sobre a atividade. Os
inibidores e compostos testados para a atividade de PLA2 neste estudo foram: o MAFP, que é um
inibidor enzimático de cPLA2 IVA, IVB e IVC e iPLA2 VIA; o BEL, que é um inibidor

34
enzimático de iPLA2 VIA e VIB; o LY311727 e o Indoxam, que são inibidores enzimáticos de
todas as sPLA2s; o EDTA, um quelante de íons divalentes e que, conseqüentemente, inibe todas
as sPLA2s e a cPLA2 IVA e IVB e o DTT (ditiotreitol), que reduz as pontes dissulfeto e desnatura
a estrutura protéica. Para cada ensaio, foram utilizados como amostra biológica, 50 µL de
homogeneizado de medula espinhal de rato, pois este tipo de tecido expressa, pelo menos, uma
enzima de cada uma das sub-famílias (sPLA2, cPLA2 e iPLA2). O homogeneizado foi incubado
com substrato por 1 hora na presença ou ausência de 0,8 mol% de inibidor de PLA2, 10 mM de
DTT ou 5 mM de EDTA. Para cada ensaio, a especificidade foi verificada não só pela inibição da
atividade de interesse, mas também pela inibição de outras PLA2s que pudessem estar
interferindo no sistema. Como por exemplo, o uso de MAFP, BEL e DTT, para verificar as ações
de cPLA2, iPLA2 e sPLA2, respectivamente (Lucas KK e Dennis EA, 2005). Neste estudo, os
pesquisadores descobriram algo peculiar a respeito do inibidor MAFP. Tal inibidor de fonte
comercial, comumente utilizado na pesquisa como um inibidor de cPLA2 IVA e de iPLA2 VIA,
inibiu, nestes ensaios com medula espinhal de rato, as sPLA2 IIA e V, mas não a cPLA2 IVA,
provavelmente devido à oxidação do composto durante o processamento, resultando em uma
molécula diferente com especificidade alterada (Lucas KK et al., 2005).
1.4. Câncer de Cólon
O Brasil é um país que se caracteriza pelo seu desenvolvimento sócio-econômico
desigual, que é a causa das discrepâncias no quadro sanitário, onde se observam tanto doenças
ligadas à pobreza como doenças crônico-degenerativas, típicas de países mais desenvolvidos.
Entre as doenças crônico-degenerativas está o câncer, que embora tenha diferentes posições na

35
análise das taxas de mortalidade por cada região brasileira, sempre figura entre as primeiras
causas de morte, ao lado de doenças do aparelho respiratório, afecções do período peri-natal e
doenças infecciosas e parasitárias.
Atualmente, o câncer se constitui na segunda causa de morte por doença no Brasil.
Somente na região Nordeste, as neoplasias representam a terceira causa de morte por doença,
consistindo de 6,34% dos óbitos atestados. Nas demais regiões, as neoplasias seguem-se às
doenças cardio-vasculares e sua proporcionalidade aumenta à medida que se desloca para o sul
do país, com 7,83% dos óbitos na região Norte, 9,89% na região Centro-Oeste, 11,93% na região
Sudeste e 15,19% na região Sul. No Brasil, as estimativas para o ano de 2006 apontam que
ocorrerão 472.050 casos novos de câncer, sendo um dos tumores mais incidentes, tanto para o
sexo masculino (11 mil) quanto para o feminino (14 mil), o câncer de cólon e reto (Instituto
Nacional de Câncer - MS, 2006).
Mundialmente, o câncer de cólon e reto é a terceira neoplasia que mais mata entre
homens, e a quarta entre mulheres (Pisani P et al., 1999). No Brasil, o número de casos novos
esperados para o câncer de cólon e reto no ano de 2006 é de 11.390 casos em homens e 13.970
em mulheres. Estes valores correspondem a um risco estimado de 12 casos novos a cada 100 mil
homens e 15 para cada 100 mil mulheres. Desconsiderando os tumores de pele não melanoma, o
câncer de cólon e reto em homens é o quarto mais freqüente nas regiões Sul (22/100.000),
Sudeste (17/100.000) e Centro-Oeste (10/100.000). Nas regiões Nordeste (4/100.000) e Norte
(3/100.000), ocupa a quinta e sexta posição, respectivamente. Para as mulheres é o segundo mais
freqüente (21/100.000) na região Sudeste, o terceiro nas regiões Sul (22/100.000), Centro-Oeste
(10/100.000) e Nordeste (5/100.000), enquanto na região Norte (4/100.000) ocupa a quinta
posição (Figura 2) (Instituto Nacional de Câncer - MS, 2006).

36
Figura 2: Representação espacial das taxas brutas de incidência por 100.000 homens e 100.000 mulheres, estimadas para o ano 2006, segundo a Unidade da Federação (neoplasia maligna do cólon e reto) (Instituto Nacional de Câncer - MS, 2006).
Embora o câncer de cólon e reto tenha fatores de risco de natureza hereditária, como por
exemplo, na Polipose Adenomatosa Familiar (FAP), que explicam aproximadamente um quarto
da ocorrência do tumor, fatores ambientais são também relevantes na sua gênese. Fatores de
risco, que poderiam servir de base para programas de prevenção primária, incluem um baixo
consumo de frutas, de vegetais frescos e de alimentos ricos em fibras; alto consumo de carnes
vermelhas e processadas e de bebidas alcoólicas; bem como a falta de exercícios físicos. Um
estudo com células de carcinoma de cólon humano transfectadas com vetor plasmidial contendo o
promotor de COX-2, importante enzima na carcinogênese de cólon, como discutido mais adiante
nesta seção, mostrou que componentes da dieta, como lipídeos, e do lúmen colônico, como os
ácidos butírico e deoxicólico, são capazes de induzir a atividade do promotor da enzima.
Também foi verificado o efeito destes componentes do ambinete colônico sobre a indução da
expressão de COX-2 em outra linhagem de célula, que expressava, constitutivamente, a enzima, e

37
se verificou aumento na expressão da proteína. Estes resultados comprovam a influência do
conteúdo luminal sobre a transcrição de COX-2, podendo influenciar o desenvolvimento do
câncer de cólon (Glinghammar B e Rafter J, 2001). Ainda dentro da influência do microambiente
sobre o desenvolvimento tumoral está a alta concentração de sais, presente no cólon intestinal.
Tal condição recebe o nome de hiperosmolaridade e há vários trabalhos na literatura que
evidenciam que o aumento na osmolaridade é um fator estimulador para a produção de citocinas,
fato que pode propiciar o desenvolvimento neoplásico. A hiperosmolaridade é fator estimulador
para IL-1β em células endoteliais de aorta humana (Asakawa H et al., 1997), para IL-6 em
macrófagos peritoneais de rato (Yassad A et al., 1997), para IL-8 em células mononucleraes de
sangue periférico humano (Shapiro L e Dinarello CA, 1995) e em células epiteliais de intestino
(Nemeth ZH et al., 2002) e brônquio (Hashimoto SHU et al., 1999) humanos e para IL-8 em
linhagem de câncer de cólon humano (Caco-2) (Hubert A et al., 2004). Portanto, estes resultados
sugerem que a hiperosmolaridade pode agir como um sinal pro-inflamatório em diversos tipos
celulares. Outro órgão, além do cólon intestinal, onde a hiperosmolaridade é fator determinante
na patofisiologia é o rim, especificamente na medula renal. Uma série de experimentos vêm
demonstrando que o acúmulo de osmólitos compatíveis tais como sorbitol, inositol, taurina,
glicerofosforilcolina e betaína é essencial para a sobrevivência de células medulares sob
condições hipertônicas. Recentes estudos sugerem que a resposta das células à osmolaridade do
meio é um processo complexo envolvendo a participação de várias MAPKs. Este processo foi
demonstrado primeiramente em leveduras, que possuem uma proteína homóloga da quinase p38,
que é ativada por hipertonicidade e é responsável pela indução da dehidrogenase glicerol-3-
fosfato, a enzima essencial para a produção de glicerol, o principal osmólito orgânico em
leveduras (Albertyn J et al., 1994; Ruis H e Schuller C, 1995). A ativação das vias de sinalização

38
envolvendo as MAPKs resulta na indução da expressão de COX-2 em células de medula renal
(IMCD), que teria função na sobrevivência destas células sob condições hipertônicas (Yang T et
al., 2000). Outro trabalho, neste mesmo tipo celular, demonstrou que a produção de Espécies
Oxigênio-Reativas (ROS) de origem mitocondrial, mediavam a fosforilação de MAPK induzida
pela hipertonicidade e a expressão de COX-2 (Yang T et al., 2005). A associação entre a
expressão de COX-2 e o desenvolvimento de câncer colo-retal tem sido bem definida. Porém, os
estímulos e mecanismos induzindo esta expressão em células de câncer de cólon têm sido
paulatinamente elucidados, e um deles é que, em condições de hiperosmolaridade, a proteína
quinase p38 é ativada e induz a expressão de COX-2, contribuindo para o fenótipo neoplásico
(Arbabi S et al., 2001).
A aceitação da premissa de que pólipos adenomatosos representam uma lesão precursora
da maioria de tumores malignos de cólon e reto faz com que a remoção de pólipos possa ser uma
estratégia importante na prevenção deste câncer (Instituto Nacional de Câncer - MS, 2002;
Kligerman J, 2002). O prognóstico deste tipo de câncer pode ser considerado de moderado a
bom, sendo a sobrevida média mundial estimada de 44%. A detecção dos pólipos adenomatosos
coloretais e de tumores localizados é possível e é feita através de pesquisa de sangue oculto nas
fezes e métodos endoscópicos, porém mesmo em países com recursos abundantes como os EUA,
têm se encontrado dificuldades na realização de avaliação diagnóstica com exames endoscópicos
em pacientes com presença de sangue oculto nas fezes, impossibilitando a implantação de
rastreamento populacional (Instituto Nacional de Câncer - MS, 2006).
As neoplasias são um grupo particular de doença que se caracteriza pela expansão
desregulada e invasão de células nos tecidos vizinhos. Todas as neoplasias possuem um aspecto
em comum, resultam de mutações genéticas. Contudo, as neoplasias diferem de doenças
genéticas de duas maneiras. A primeira é que diversas mudanças genéticas devem ocorrer para

39
um câncer se desenvolver por completo, contrastando com doenças genéticas, como anemia
falciforme e fibrose cística, que se desenvolvem a partir de um único defeito genético. A segunda
é que, enquanto algumas das alterações genéticas podem ser inerentes a linhagem germinativa, a
vasta maioria destas mutações em câncer são somáticas (Kinzler KW e Vogelstein B, 1995). Este
processo de vários passos sequenciais é bem ilustrado pelo câncer de cólon, que se desenvolve,
tipicamente, durante décadas, e passa por uma série de alterações genéticas para seu completo
desenvolvimento.
Em relação à morfologia, o câncer de cólon progride por uma série de estágios
histopatológicos que vão desde pequenos tumores benignos (pólipos adenomatosos), que se
originam do epitélio glandular e têm como característica a morfologia displásica e diferenciação
anormal das células epiteliais, até neoplasias malignas (carcinomas), com infiltração de células
neoplásicas e inflamatórias no estroma intestinal e acentuada angiogênese. Esta progressão é o
resultado de mudanças genéticas seriadas que envolvem ativação de oncogenes e inativação de
genes supressores tumorais. Certas síndromes hereditárias predispõe à formação de pólipos e,
consequentemente, aumentam o risco do surgimento de lesões neoplásicas. Entre as síndromes
estão a Polipose Adenomatosa Familiar (FAP) e Câncer Coloretal Não-polipose Hereditário
(HNCC). A melhor caracterizada destas é a FAP (Kinzler KW et al., 1995). As mutações no gene
da polipose adenomatosa do cólon (APC) são as responsáveis por esta forma de câncer de cólon.
Indivíduos com FAP desenvolvem centenas a milhares de pólipos no intestino grosso, dos quais
um subgrupo invariavelmente progride até neoplasias malignas se não forem removidos
cirurgicamente. Pacientes com mutações de APC na linhagem germinativa não desenvolvem,
necessariamente, câncer de cólon, porém possuem um risco aumentado em desenvolver a doença
em relação ao resto da população (Kinzler KW e Vogelstein B, 1996). Estudos em pacientes com
FAP, bem como em camundongos com mutações análogas do homólogo de APC (camundongos

40
Min), sugerem que o passo limitante na iniciação do tumor é uma mutação somática do alelo
selvagem do APC da linhagem parental não afetada, fornecendo suporte para a hipótese de two-
hit (Kinzler KW et al., 1996).
As manifestações clínicas nos pacientes com FAP variam consideravelmente, deixando
uma lacuna entre o genótipo e o fenótipo nesta síndrome. Em alguns casos, esta variação é devida
ao tipo de mutação, como, por exemplo, nas mutações entre os códons 1403 e 1578, associadas
com manifestações extra-colônicas, tais como tumores desmóides e lesões mandibulares. Um
exemplo bem conhecido de distinção entre fenótipo e genótipo APC é observado em
camundongos Min. Estes camundongos desenvolvem múltiplos adenomas intestinais e possuem
uma mutação no gene APC em posição similar àquela encontrada em muitos pacientes com FAP.
E dependendo da localização desta mutação nas diferentes linhagens murinas o número de
pólipos varia significantemente (Kinzler KW et al., 1996).
Os estudos elucidando a função de APC vêm de sua interação com a β-catenina. As
cateninas são proteínas citoplasmáticas que se ligam às caderinas, uma família de moléculas de
adesão. A proteína APC interage com a β-catenina para regular os níveis desta molécula no
citosol. Com o truncamento da APC, a proteína não se liga à β-catenina e o acúmulo e/ou
redistribuição da β-catenina poderia contribuir para o crescimento celular anormal ou
desregulação da sinalização transmitida ao núcleo pelo complexo β-catenina/Tcf (fator de célula
T) (Kinzler KW et al., 1996; Polakis P, 1997). Além disso, outros mecanismos moleculares
envolvidos na gênese de câncer de cólon abrangem não só a inativação de APC, mas também a
ativação do oncogene Ras e inativação de p53. Neste processo ocorreria uma contínua renovação
das células epiteliais do intestino e desregulação da expressão de genes nas células-tronco das
criptas intestinais (Kinzler KW et al., 1996).

41
Há evidências de que o evento inicial de mutação de APC resulte na indução de
Ciclooxigenase-2 (COX-2), uma enzima de grande importância no desenvolvimento do câncer de
cólon. Estudos epidemiológicos mostram que indivíduos que recebem tratamento com anti-
inflamatórios não esteroidais (NSAID) têm diminuição do risco de desenvolver câncer de cólon e
pólipos adenomatosos (Taketo MM, 1998). Tal fato é suportado pela farmacologia de NSAIDs
em modelos experimentais, onde a inibição específica de COX-2 diminui o potencial metastático
e o crescimento de células de câncer de cólon humano (Tsujii M et al., 1997; Sheng H et al.,
1997).
Ainda confirmando o papel da COX-2 na patogênese do câncer de cólon, na síndrome
FAP em humanos, o nível tissular de prostaglandina E2 (PGE2) está aumentado. No câncer
cóloretal humano, a quantidade de mRNA de COX-2 possui correlação com o tamanho do tumor
e a expressão de COX-2 está aumentada nos pólipos de camundongos APC∆716 e Min, modelos
para FAP humana. Instigantemente, uma mutação nula de COX-2 reduz de forma significativa o
número e tamanho de pólipos intestinais nestes camundongos. Além disso, o tratamento de
camundongos APC∆716 com um inibidor seletivo de COX-2 reduz o número e tamanho dos
pólipos de modo mais efetivo do que o tratamento com sulindac, inibidor das duas isoenzimas
COX (Oshima M et al., 1996; Takaku K et al., 2000).

42
1.5. Fosfolipase A2 e Câncer
Como já exposto no item 1.1, as PLA2 influenciam a produção de PGE2, não somente
liberando o AA dos fosfolipídeos de membrana, mas também regulando a expressão de COX-2.
Logo, há três potenciais mecanismos de ação para as PLA2 em tumores. O primeiro, que será
discutido mais adiante nesta seção, é a liberação de AA, aumentando os níveis intracelulares
deste ácido graxo, induzindo a apoptose das células de câncer de cólon, conferindo às PLA2
função anti-neoplásica (Cao Y et al., 2000). O segundo, onde a PLA2, especialmente a do grupo
V, induz a expressão de COX-2, importante enzima para o desenvolvimento de vários tipos de
câncer, como o coloretal, o de pulmão, o gástrico, o de mama e o pancreático (Gasparini G et al.,
2003; Hull MA, 2005; Chell S et al., 2006). E o terceiro mecanismo, que é a própria produção de
PGE2, que possui várias funções pró-neoplásicas, como indução de fatores pró-angiogênicos,
indução da proliferação e inibição da apoptose das células neoplásicas e aumento da invasividade
(Tsujii M et al., 1998; Liu XH et al., 2002b; Fukuda R et al., 2003; Buchanan FG et al., 2003;
Spinella F et al., 2004; Liu XH et al., 2005; Lev-Ari S et al., 2006).
Em relação ao câncer de cólon, a mutação no gene APC foi identificada na cepa de
camundongos Min (Bilger A et al., 1996), que assim como pacientes portadores da síndrome
FAP, apresenta um grande número de pólipos intestinais que invariavelmente tornam-se
malignos. O fenótipo associado a esta mutação é bastante reduzido em algumas cepas de
camundongos, o que levou à busca do gene modificador de Min (Mom). Estudos de linkage
identificaram uma região distal no cromossomo 4 como sendo o locus do Mom, o qual contém o
gene para a sPLA2 IIA, a primeira PLA2 a ser proposta como tendo um papel no câncer de cólon
(MacPhee M et al., 1995; Cormier RT et al., 2000). O sequenciamento desta região revelou uma
mutação de mudança de quadro de leitura que abolia a expressão da sPLA2IIA em cepas sensíveis

43
à mutação Min. O papel de prevenção ao câncer de cólon da sPLA2IIA foi confirmado quando a
expressão de um transgene para sPLA2IIA reduziu o número de pólipos em cepas de
camundongos sensíveis a Min (Cormier RT et al., 1997). A descoberta de um gene capaz de
suprimir a formação de pólipos em camundongos levou a uma corrida para se identificar
mutações em pacientes de câncer de cólon no gene humano homólogo. Entretanto, não foram
encontradas mutações que pudessem associar alterações no gene de sPLA2IIA com variações
fenotípicas na FAP ou relação entre os níveis de expressão de RNA mensageiro ou proteína de
sPLA2IIA nos pólipos de pacientes com FAP (Dobbie Z et al., 1996; Dimberg J et al., 1998;
Kennedy BP et al., 1998). A participação da sPLA2 IIA como supressora do câncer de cólon
continua não esclarecida. Ainda não foi demonstrado in vivo ou em células primárias que esta
PLA2 participa de fato na produção de eicosanóides. As evidências apontam a COX-2 como
enzima limitante para a produção de prostanóides e promotora de neoplasia, tornando, portanto,
pouco provável que a sPLA2IIA seja a enzima responsável por esta produção. A sPLA2IIA
também é expressa em outras neoplasias, como no adenocarcinoma prostático e no
adenocarcinoma gástrico (Jiang J et al., 2002; Leung SY et al., 2002). Em estudo no ano de 2002,
pesquisadores analisaram os padrões de expressão gênica em câncer gástrico usando
microarranjos de cDNA representando cerca de 30.300 genes. A expressão de sPLA2 IIA estava,
significativamente, correlacionada com a maior sobrevida dos pacientes, sugerindo que a
atividade da enzima pudesse estar suprimindo ou a progressão ou metástase do câncer gástrico
humano. Este efeito protetor poderia estar relacionado com o aumento da liberação de AA, que
induziria a apoptose das células cancerosas, ou com a atividade antibacteriana de sPLA2 IIA, já
que as bactérias possuem papel na patogênese do câncer gástrico (Leung SY et al., 2002). No
mesmo ano, foi feito um estudo imunohistoquímico em várias amostras de pacientes com
neoplasia intraepitelial prostática, com adenocarcinoma prostático ou em tecidos prostáticos

44
benignos. Os pesquisadores observaram que em 45% das amostras de tecido prostático benigno
havia marcação imunohistoquímica para a sPLA2 IIA, enquanto que em 91% das amostras de
neoplasia intraepitelial prostática havia imunomarcação e havia positividade para a enzima em
90% das amostras de adenocarcinomas prostáticos. O estudo também mostrou que as células
malignas tinham marcação immunohistoquímica mais intensa em relação às células benignas,
sugerindo que a desregulação de sPLA2 IIA tenha papel na carcinogênese prostática (Jiang J et
al., 2002). Ainda dentro desta subfamília, a sPLA2 do grupo IID é expressa constitutivamente em
linhagens celulares de carcinoma de cólon humano. Os transcritos desta enzima foram detectados
por RT-PCR nas linhagens HT29, KM12, KM20L2, WiDr e HCT2998, sendo que a expressão
destes transcritos diminuía de maneira tempo-dependente, quando as células eram tratadas com
IL-1β (Murakami M et al., 2002b).
Outra enzima com potencial envolvimento na carcinogênese de cólon é a sPLA2 do grupo
X. Em estudo que comparou a potência de três sPLA2 (grupos IB, IIA e X) para a liberação de
AA entre várias linhagens de câncer de cólon humano (HT-29, Colo320DM, HCT-15 e HCT-
116), observou-se que a enzima com maior potência para a liberação de AA, quando
administrada exogenamente, era a sPLA2 do grupo X. Esta liberação de AA pela sPLA2X levava
à produção de PGE2 dependente de COX-2 pelas células neoplásicas (Morioka Y et al., 2000). O
mesmo foi visto em linhagem celular mielomonocítica (U937) e de leucemia promielocítica
(HL60) (Hanasaki K et al., 1999). Outra evidência de sua participação na carcinogênese é a
análise imunohistológica de adenocarcinomas de cólon humanos, que indica expressão de
sPLA2X nas células neoplásicas juntamente com a COX-2, evidenciando um possível papel para
a fosfolipase na biossíntese de PGE2 durante a tumorigênese de cólon (Morioka Y et al., 2000).

45
Além disso, a sPLA2 atua na proliferação e apoptose de células de câncer de cólon murino
(WB-2054-M4), pois com o uso de inibidor de sPLA2 há redução da proliferação e aumento,
dependente de dose, da taxa de apoptose nestas células. Porém, a produção de PGE2 nesta
linhagem foi inibida pelo tratamento com inibidores de cPLA2 e de sPLA2, sugerindo que a
formação deste mediador lipídico e a inibição da proliferação são processos não correlacionados
(Longo WE et al., 1999).
Uma das mais importantes enzimas da via de metabolismo de AA na carcinogênese de
cólon é a cPLA2. Tal importância foi confirmada com estudos imunohistoquímicos, que
indicaram expressão aumentada da proteína de cPLA2 em adenocarcinomas de intestino delgado e
coloretal (Wendum D et al., 2003; Panel V et al., 2006), em estudos onde a expressão gênica da
enzima também estava em adenocarcinomas coloretais (Dimberg J et al., 1998) e com a
expressão aumentada de cPLA2 em linhagens de câncer de cólon, como na HT-29, na Lovo e na
SW480 (Panel V et al., 2006). Além da observação direta em tumores de cólon, estudos
experimentais em camundongos APC∆716 e camundongos Min, modelos para a FAP humana,
evidenciaram ainda mais o papel da cPLA2 na carcinogênese de cólon. Dois estudos, um em
camundongos APC∆716 e outro em camundongos Min mostraram resultados semelhantes, embora
com abordagens metodológicas distintas. No primeiro, os pesquisadores introduziram uma
mutação no gene da cPLA2 dos camundongos APC∆716 e observaram que nos animais mutantes o
tamanho dos pólipos presentes no intestino delgado estava significativamente reduzido, apesar do
número não apresentar diferenças em relação aos animais não mutantes, sugerindo um papel para
a enzima na expansão dos pólipos do intestino delgado (Takaku K et al., 2000). Já no segundo
estudo, foi feita a disrupção do gene da cPLA2 em camundongos APC Min, reduzindo em 83% o
número e em 20% o tamanho dos tumores no intestino delgado (Hong KH et al., 2001).

46
Juntamente com o fenótipo para o camundongo APC∆716 knockout para COX-2, que apresentam
diminuição do número e tamanho dos pólipos intestinais, tais resultados sugerem que o efeito
protetor da deleção de cPLA2 é atribuído a uma diminuição na geração de AA para COX-2 com
diminuição na síntese de prostaglandinas (PGs) (Oshima M et al., 1996). Entretanto, um estudo
recente em camundongos knockout para cPLA2, onde os animais desenvolviam pólipos no cólon
após a exposição ao carcinógeno organotrópico azoximetano, evidenciou que os animais que não
possuíam o gene de cPLA2 apresentavam maior número de tumores em relação aos animais
selvagens. Ainda, o número de células apoptóticas no epitélio colônico estava significativamente
reduzido nos animais knockout, o que poderia explicar a tumorigênese aumentada resultante da
deleção de cPLA2 apesar da redução da produção de PGE2. Tais resultados sugerem que o papel
pró-apoptótico de cPLA2 no cólon poderia ser mais importante do que sua função na produção de
eicosanóides no desenvolviemnto tumoral (Ilsley JNM et al., 2005). Além do papel de cPLA2 em
câncer de cólon, há publicações demonstrando seu papel em outros tumores. Em linhagens
celulares de câncer de pulmão, a atividade do promotor de cPLA2 é dependente do oncogene Ras
e a enzima é responsável pela produção de eicosanóides e pelo crescimento celular (Heasley LE
et al., 1997). E em linhagens celulares de colangiocarcinoma humano, a cPLA2 é responsável
pela liberação de AA induzida por fator de crescimento de hepatócito (HGF) e por IL-6, dois
importantes mitógenos de células epiteliais biliares. Nestas células a liberação de AA resulta na
produção de PGE2, que induz a proliferação celular (Wu T et al., 2002).
Embora as PLA2s estejam implicadas na patogênese do câncer de cólon como promotoras
do desenvolvimento neoplásico, tais enzimas hidrolisam os fosfolipídeos de membrana gerando
AA livre, que é um conhecido indutor de apoptose, caracterizando, assim, uma função anti-
neoplásica para as fosfolipases. Ao metabolizar o AA livre em PGH2, a COX-2 estaria reduzindo

47
um sinal apoptótico favorecendo o crescimento dos pólipos intestinais. Em adenocarcinomas de
cólon há a superexpressão de uma outra enzima, além de COX-2, que utiliza AA, a ligase 4 de
ácido graxo-CoA (FACL4). Em estudo onde o AA exógeno causou apoptose de linhagens
celulares de carcinoma de cólon, assim como a triacsina C, um inibidor de FACL4 e ainda,
somando-se a estas evidências, a indometacina e o sulindac significantemente potencializaram o
efeito indutor de apoptose da triacsina C, evidenciou-se o mecanismo pelo qual a apoptose é
regulada pelos níveis de AA não esterificado e que COX-2 e FACL4 promovem carcinogênese
pela diminuição destes níveis (Cao Y et al., 2000).
Um dos produtos finais da cascata do AA e, conseqüentemente, da ação das PLA2s é a
PGE2, que pode modular cada aspecto da tumorigênese de cólon, desde o aumento da
proliferação celular e resistência à apoptose até a formação de novos vasos e metástase
(Watanabe K et al., 1999; Tomozawa S et al., 2000; Sheng H et al., 2001; Sonoshita et al., 2001;
Hansen-Petrik MB et al., 2002; Fukuda R et al., 2003; Castellone MD et al., 2005; Wang D et al.,
2006). O aumento na produção deste prostanóide pelos tumores de cólon e linhagens celulares
derivadas deste tipo de neoplasia está associado com a superexpressão de COX-2 (Panel V et al.,
2006), que cataliza a conversão de ácido araquidônico em PGH2, o precursor de prostaglandinas e
tromboxanes. Então, PGH2 é metabolizada por um grupo de enzimas, as PGE sintases, para
produzir PGE2. Entre estas enzimas terminais está a PGE sintase microssomal-1 (mPGE-1), que
também possui sua expressão aumentada em amostras de câncer de cólon. A síntese aumentada
de PGE2, evidenciada pela superexpressão de COX-2, é um fator prognóstico negativo e um alvo
importante para a quimioprevenção e terapia do câncer de cólon. O inibidor específico de COX-2
foi efetivo na redução do tamanho e do número de pólipos intestinais na prevenção da progressão
para câncer, sendo o uso destas drogas, os COXIBs, a maior prova para o papel das enzimas

48
envolvidas na cascata do AA no câncer de cólon (Oshima M et al., 1996; Sheng H et al., 1997;
Taketo MM, 1998; Bertagnolli MM, 1999).
1.6. Angiogênese e Enzimas do Metabolismo do Ácido Araquidônico
Além de processos como inflamação, desregulação da proliferação celular, perda da
diferenciação celular, mutações genéticas e metástase, outro processo é muito importante para o
desenvolvimento tumoral: a angiogênese. A angiogênese é o processo de formação de novos
vasos a partir de outros pré-formados. Na vida adulta a taxa de proliferação das células
endoteliais é muito baixa quando comparada aos outros tipos celulares no corpo. Há situações
fisiológicas, onde há regulação da formação destes vasos, como no caso do ciclo menstrual e da
cicatrização de feridas. Tal processo, quando não regulado, pode resultar em patologias como
artrite reumatóide, retinopatia diabética, psoríase e hemangiomas juvenis, além de ser importante
para o crescimento e metástase de tumores sólidos (Ferrara N e Alitalo K, 1999). Vários modelos
experimentais vêm demonstrando que a inibição da angiogênese evita o crescimento e muitas
vezes causa a regressão tumoral (O'Reilly MS et al., 1996; Parangi S et al., 1996; Kisker O et al.,
2001a; Kisker O et al., 2001b). Nesta seção discutiremos a relação entre as enzimas participantes
do metabolismo do AA e o processo de angiogênese.
O mecanismo da angiogênese pode ser dividido em três fases: iniciação,
proliferação/invasão e maturação. Logo no início da formação dos novos vasos, o controle
transcricional da indução na expressão de fatores angiogênicos em células inflamatórias e/ou
tumorais é ativado pela hipóxia. Este controle se dá, principalmente pelo Fator Indutível por
Hipóxia 1α (HIF-1α), um fator de transcrição que é acumulado no citoplasma durante a hipóxia,

49
ativando a transcrição de genes envolvidos no metabolismo da glicose, angiogênese e
sobrevivência (Harris AL, 2002; Salnikow K et al., 2002). Um destes genes é o Fator de
Crescimento do Endotélio Vascular (VEGF), que estimula a proliferação de células endoteliais.
Além disso, a sinalização via os receptores de VEGF inicia cascatas de transdução de sinal, como
a das MAP quinases, importantes para a regulação da migração das células vasculares (Olsson
AK et al., 2006). Para iniciar a formação de novos capilares, as células endoteliais de vasos
sanguíneos pré-existentes devem degradar a membrana basal e invadir o estroma dos tecidos
vizinhos. Estes processos de invasão e migração de célula endotelial requerem a ação das
metaloproteinases, para a degradação da matriz extracelular existente. Após a degradação
proteolítica da matriz e, em resposta ao VEGF e a outros fatores pro-angiogênicos, as células
endoteliais e os fibroblastos entram em divisão celular e começam a migrar orientados por um
gradiente quimiotático pela nova matriz extracelular. Uma vez estabelecidas nesta matriz, as
células endoteliais agrupam-se para formar um lúmen central, sintetizam nova membrana basal, e
recrutam pericitos e células musculares lisas para envolverem os vasos (Stromblad S e Cheresh
DA, 1996; Brat DJ e Van Meir EG, 2001; Papetti M e Herman IM, 2002).
A íntima relação entre angiogênese e progressão tumoral leva à noção de “switch
angiogênico”, uma etapa durante o início do desenvolvimento neoplásico, na qual a angiogênese
é induzida para permitir a expansão tumoral. Como uma consequência da grande proliferação das
células tumorais e aparecimento de áreas com pouca oxigenação, a hipóxia induziria a expressão
de VEGF pelas células neoplásicas e, consequentemente, da angiogênese (Cheresh DA, 1998;
Papetti M et al., 2002).
A família do VEGF também inclui o Fator de Crescimento Placentário (PGF), VEGF-B,
VEGF-C e VEGF-D. A sinalização pelos receptores destes fatores (Flt-1 e Flk-1 para VEGF)
resulta na ativação da fosfolipase Cγ, levando a um aumento nos níveis intracelulares de inositol

50
fosfato e cálcio. Como em outros tipos celulares, o cálcio induz a ativação da calcineurina e do
Fator Nuclear de Células T Ativadas (NFAT) em células endoteliais, uma via de sinalização que
é bloqueada pela droga imunosupressora ciclosporina A. Em células endoteliais da veia umbilical
humana (HUVECs), o VEGF estimula a defosforilação, translocação para o núcleo e ativação do
NFAT, processo envolvido na regulação da expressão gênica da enzima COX-2 por este fator de
crescimento. Tal enzima está envolvida na regulação da angiogênese por modular a produção de
fatores angiogênicos tanto em células endoteliais como em células de carcinoma de cólon
humano (Tsujii M et al., 1998; Hernandez GL et al., 2001).
Inibidores de COX possuem atividade anti-angiogênica (Skopinska-Rozewska E et al.,
1998). O tratamento com o inibidor seletivo de COX-2 (NS398) em células de câncer de cólon
que superexpressavam COX-2 diminuiu a expressão de VEGF e FGFβ e inibiu a angiogênese
tumoral (Sawaoka H et al., 1999), sugerindo que a COX-2 pode aumentar a expressão destes
fatores angiogênicos em câncer de cólon. Alguns trabalhos sugerem que, enquanto COX-2 regula
a angiogênese em células de câncer de cólon, COX-1 modula a angiogênese em células
endoteliais (Tsujii M et al., 1998; Sawaoka H et al., 1999). Contudo, diversas evidências sugerem
que a COX-2 também está envolvida em processos angiogênicos em células endoteliais.
Primeiro, a expressão de COX-2 foi associada com angiogênese por células endoteliais gástricas
humanas in vitro, e as atividades tanto de COX-1 quanto de COX-2 foram necessárias para
angiogênese durante a cicatrização de úlceras gástricas (Hull MA et al., 1999). Segundo, a
atividade COX-2 em células endoteliais de microvasculatura é requerida para atividade
angiogênica de oncostatina M (Pourtau J et al., 1999). Terceiro, NSAIDs seletivos ou não para
COX-2 inibem a angiogênese indesejada na córnea e câncer (Sawaoka H et al., 1999; Yamada M
et al., 1999).

51
Entre os produtos da atividade de COX, os prostanóides PGE1 e PGE2 são reportados
como fatores promotores da angiogênese (BenEzra D, 1978; Ziche M et al., 1982; Form DM e
Auerbach R, 1983; Pai R et al., 2001; Eibl G et al., 2003; Salcedo R et al., 2003). Em contraste,
15-deoxi-δ12,14-PGJ2, um produto da PGD2, induz apoptose de célula endotelial pela ativação de
PPARγ e inibe a angiogênese (Bishop-Bailey D e Hla T, 1999; Xin X et al., 1999). Parece que os
metabólitos de COX, mais do que os níveis de proteína ou da atividade COX são mais
importantes na regulação da angiogênese (Nie D e Honn KV, 2002).
A ação de COX-2 pode não só ser sobre as células neoplásicas como também sobre as
células endoteliais, independentemente do perfil de expressão de COX no tumor. Em modelo
angiogênico tumoral, onde os tumores experimentais não expressavam COX-2, quando havia
tratamento com celecoxib (inibidor de COX-2) havia inibição significativa da evolução tumoral
pelo bloqueio da proliferação de células endoteliais e indução da apoptose das mesmas células,
sugerindo que a angiogênese é dependente de prostaglandinas (PGs) derivadas de COX-2
expressa no endotélio (Leahy KM et al., 2002).
O papel da COX na regulação da angiogênese induzida pelas células de câncer de cólon
foi avaliado por modelos in vitro de co-cultura de células endoteliais com células de carcinoma
de cólon humano. As células que super expressavam COX-2 produziram prostaglandinas, fatores
proangiogênicos e estimularam a migração endotelial e a formação de tubos vasculares. Tal efeito
era inibido por combinações de anticorpos contra os fatores angiogênicos, por NS-398 (inibidor
específico para COX-2) e por aspirina, indicando que a COX-2 pode modular a produção de
fatores angiogênicos pelas células de câncer de cólon (Tsujii M et al., 1998).
Embora a relação entre PLA2s e angiogênese ainda não esteja bem definida há estudos
demonstrando a ação de sPLA2 IIA e sPLA2 V na angiogênese inflamatória. A PLA2 libera AA

52
de fosfolipídeos, que pode ser usado na produção de Fator Ativador de Plaquetas (PAF), que está
envolvido com o fenótipo angiogênico na inflamação crônica. Isto foi confirmado em estudo
com modelo murino de angiogênese induzida por inflamação, onde o uso de SB203347 (inibidor
das sPLA2s) reduziu o índice vascular pela diminuição dos níveis de PAF. Sendo que o uso de
antagonista de PAF reduzia o índice vascular, sugerindo uma função para o mediador na
formação de novos vasos durante a inflamação (Jackson JR et al., 1998). Ainda em relação à
sPLA2 V foi demonstrado em células endoteliais da veia umbilical humana (HUVECs) e em
células endoteliais de aorta bovina (BAECs) a regulação da síntese de PAF, induzida por VEGF,
por sPLA2V, caracterizando assim uma nova função para esta enzima na indução de angiogênese
e no efeito pro-inflamatório do VEGF (Bernatchez PN et al., 2001).
Há também vários trabalhos demonstrando a regulação da atividade fosfolipásica em
HUVECs e células endoteliais de bovino, caracterizando o envolvimento de cálcio celular,
ativação de PKC e MAPK na estimulação de cPLA2 (Wong JT et al., 1998) e a atividade de
sPLA2 na liberação de AA (Murakami M et al., 1993).
Outra função para as fosfolipases é de indução da motilidade celular. Algumas sPLA2,
como a sPLA2IA e IB, induzem migração de fibroblastos NIH-3T3. Pouco se conhece dos
mecanismos pelos quais a sPLA2 atue sobre a motilidade celular, porém um estudo revelou que o
efeito do Fator de Crescimento de Fibroblasto (bFGF) sobre motilidade de células endoteliais foi
acentuadamente reduzido por inibição farmacológica de PLA2 (Sa G e Fox PL, 1994). Em adição
a isto, a sPLA2 do fluido sinovial de pacientes com artrite induz a migração de células de aorta
bovina (BAEC) e esta indução é dependente da atividade catalítica da enzima (Rizzo MT et al.,
2000).

53
2. OBJETIVOS
Considerando-se o exposto anteriormente fica evidente a importância de se dissecar os
mecanismos pelos quais as PLA2s atuam nos mais variados processos da patogênese do câncer de
cólon, entre eles, proliferação celular, produção de mediadores lipídicos e angiogênese. O estudo
das PLA2s se torna promissor a partir do momento em que há pelo menos quatro grandes sub-
famílias de fosfolipases em mamíferos e suas funções no processo neoplásico ainda não estão
bem estabelecidas, podendo-se identificar novos alvos terapêuticos. Com isso, o objetivo geral
deste trabalho é o de avaliar o papel das PLA2s na ativação de células de câncer de cólon por
estresse microambiental.
Desta forma, tomando por base a relação entre PLA2s, carcinoma de cólon e angiogênese
inflamatória, os objetivos específicos deste trabalho são:
1. Avaliar o papel das diferentes PLA2s na produção de VEGF e PGE2 em linhagem de
carcinoma de cólon humano.
2. Analisar a regulação de VEGF por PGE2 em linhagem de carcinoma de cólon ativada
por diferentes tipos de estresse microambiental.
3. Caracterizar as vias de sinalização envolvidas na produção de VEGF e PGE2 em
linhagem de carcinoma de cólon humano.

54
3. MATERIAIS E MÉTODOS
3.1. Linhagens Celulares
As linhagens celulares de câncer de cólon humano Caco-2 (ATCC, cedidas pelo Dr. José
A. Morgado Díaz, Instituto Nacional de Câncer) e HCT-116 (ATCC, cedidas pelo Dr. Thomas
McIntyre, University of Utah) foram cultivadas em meio Dulbecco modificado por Eagle
(DMEM) (Invitrogen), suplementado com 10% de soro fetal bovino (SFB), NaHCO3
(concentração final 44 mM), NaH2PO4.H2O (concentração final 1 mM), piruvato de sódio
(concentração final 1 mM), HEPES (concentração final 10 mM), solução MEM de vitaminas,
solução MEM de aminoácidos essenciais e não essenciais, L-glutamina (concentração final 2
mM), β-mercaptoetanol (concentração final 55 µM), penicilina (concentração final 100.000 U) e
estreptomicina (concentração final 100 mg/L) (todos estes reagentes da Invitrogen). As células
foram mantidas em garrafas (de 25, 75 e 150 cm2 de área) e placas de 6 poços (962 mm2 de área
por poço) de cultura (Techno Plastic Products). As células Caco-2 eram recolhidas com solução
de 0,25% de Tripsina e 0,38 g/L de EDTA em HBSS sem Ca++ e Mg++ e repicadas após atingir
confluência. As culturas celulares eram incubadas em estufa a 37oC e atmosfera de 5% de CO2.
Todos os experimentos eram realizados quando as células estavam em monocamadas sub-
confluentes em meio de cultura completo, 12 horas após o plaqueamento. As linhagens celulares
eram estimuladas com adição de 100µL de 2M de cloreto de sódio (NaCl) ao meio DMEM 10%
SFB para mimetizar a condição de estresse hipertônico, a osmolaridade final do meio era de
aproximadamente 540 mOsm (versus o normal de 367 mOsm). A osmolaridade do meio foi
mediada por método de congelação utilizando um osmômetro (Advanced Micro Osmometer,

55
Model 3300, da Advanced Instruments). As condições para mimetizar a ativação de HIF-1α
ocorrida na hipóxia eram dadas pelo cultivo das linhagens em meio com concentração final de 1
mM de cloreto de cobalto (CoCl2).
3.2. Reagentes
O cloreto de cobalto (CoCl2) e o cloreto de sódio (NaCl) foram obtidos da Sigma-Aldrich.
O NaCl e o CoCl2 foram dissolvidos em água ultra-pura (Millipore) nas concentrações de 2M e
1M, respectivamente. Para a condição de estresse hipertônico, a solução estoque de NaCl foi
diluída em DMEM estéril para a concentração final desejada (0,01; 0,03 ou 0,1M) nas culturas.
Assim como para a condição de hipóxia, a solução de CoCl2 foi diluída em DMEM estéril para a
concentração final desejada (0,1; 0,3 ou 1mM) nas culturas. O Bromoenol lactona (BEL), o
Araquidonil Trifluorometil Cetona (ATK), o NS-398 e a Prostaglandina E2 (PGE2) foram obtidos
da Cayman Chemical e diluídos em etanol (Merck) ou DMSO (Sigma-Aldrich) nas
concentrações de 100, 30, 100 e 10mM, respectivamente, conforme as recomendações do
fabricante. Os inibidores farmacológicos de p38 MAPK (SB202190), de JNK (SP600125) e de
MEK 1/2 (U0126) foram obtidos da BIOMOL Research Laboratories, PA, USA e diluídos nas
concentrações de 30, 20 e 13mM, respectivamente, conforme as recomendações do fabricante. Os
agonistas dos receptores EP para PGE2 foram o 16,16-dimetil Prostaglandina E2 (agonista para
maioria dos subtipos de receptores EP), o 17-fenil trinor Prostaglandina E2 (agonista para os
receptores EP1 e EP3), Butaproste (agonista para o receptor EP2) e a Sulprostona (agonista para o
receptor EP3) obtidos da Cayman Chemical e diluídos todos na concentração de 10mM em etanol
(Merck), conforme as recomendações do fabricante. O antagonista dos receptores EP e DP
utilizado foi o AH 6809 da Cayman Chemical, diluído a 6,7mM em ácido acético 75%, de acordo

56
com as recomendações do fabricante. O ácido araquidônico marcado com Trítio
([5,6,8,9,11,12,14,15-3H] Ácido Araquidônico) com atividade específica de 214 Ci/mmol
utilizado nos ensaios de liberação de ácido araquidônico foi obtido da Amersham Biosciences. Os
anticorpos monoclonais para os ensaios de Western blot foram o IgG anti-COX-2 de
camundongo (clone 33) da BD Transduction Laboratories, diluído na concentração de 1µg/mL; o
IgG anti-phospho-p44/42 MAPK (Thr202/Tyr204) (clone E10) de camundongo da Cell Signaling
Technology, diluído a 0,25µg/mL, e o IgG anti-GAPDH (clone 6C5) de camundongo da Santa
Cruz Biotechnology, diluído a 0,003µg/mL. Os anticorpos policlonais utilizados foram o IgG
anti-fosfo-p38 (pTpY 180/182) de coelho e o IgG anti-fosfo-JNK1&2 (SAPK) (pTpY 183/185)
de coelho, diluídos a 0,25µg/mL, da BIOMOL Research Laboratories. Os anticorpos secundários
conjugados a HRP, produzidos em cabra, foram o anti-IgG de camundongo da Santa Cruz
Biotechnology, diluído a 0,1µg/mL, e o anti-IgG de coelho da Jackson Immuno Research
Laboratories, diluído a 0,08µg/mL.
3.3. Tratamentos com PGE2 e inibidores farmacológicos
As linhagens celulares HCT116 e Caco-2 eram plaqueadas em placas de 6 poços de fundo
chato em densidade de 5x105/9,6cm2 de área (5x105/poço). As células eram mantidas em 3 mL
de DMEM 10% SFB por 12 horas a 37oC e 5%CO2. Após sua adesão à placa, o meio era trocado
e a PGE2, todos os inibidores farmacológicos, o antagonista e todos os agonistas dos receptores
EP eram adicionados 15 minutos antes da adição de NaCl ou CoCl2 em volume final de 2mL. Os
inibidores das MAP quinases (SB202190, SP600125 e U0126) eram adicionados 30 minutos
antes da exposição ao estímulo.

57
3.4. Enzyme Linked Immuno Sorbent Assay (ELISA) e Enzyme Immuno Assay (EIA)
A produção de VEGF e PGE2 pelas linhagens Caco-2 e HCT116 foi medida por ELISA e
EIA, respectivamente, no sobrenadante da cultura. O sobrenadante era colhido após 24 horas de
ativação celular por estresse hipertônico ou por CoCl2, centrifugado a 100 x g por 5 minutos para
remoção de células e estocado a –70oC. Os sobrenadantes foram testados para VEGF usando o
ELISA para VEGF humano conforme as especificações do fabricante (DuoSet, R&D Systems,
United Kingdom, UK). Para o ELISA, o sistema de revelação usado foi o Immuno Pure TMB
Substrate Kit da Pierce. Os níveis de PGE2 foram medidos usando o EIA para PGE2 conforme as
especificações do fabricante (Prostaglandin E2 EIA Kit-Monoclonal; Cayman Chemical, Ann
Arbor, USA). Para o EIA, o sistema de revelação era fornecido pelo próprio kit e consistia de
Reagente de Elmman. As reações de ELISA foram analisadas em comprimento de onda de 450
nm e as reações de EIA em comprimento de onda de 405 nm, ambas em espectrofotômetro
(Spectra Max 190, Molecular Devices).
3.5. Liberação de Ácido Araquidônico
As células Caco-2 eram plaqueadas em densidade de 5x105/poço em placas de cultura de
6 poços em 3 mL de meio DMEM contendo 10% SFB como descrito previamente. As células
aderiam às placas por 6 horas a 37oC e 5% de CO2 e logo após eram incubadas com 1 µCi/poço
de [3H] ácido araquidônico em 2 mL de DMEM 10% SFB por 12 horas, para a incorporação do
ácido araquidônico marcado aos fosfolipídeos de membrana. Depois da marcação, as células

58
Caco-2 eram lavadas duas vezes com Solução Salina Tamponada com Fosfato (PBS) (KCl 2,67
mM; NaCl 137,93 mM; KH2PO4 1,47 mM; Na2HPO4 8,1 mM; pH 7,4) e, então, era adicionado
meio DMEM completo com os inibidores farmacológicos nas concentrações desejadas por 15
minutos. As células Caco-2 eram ativadas pelos tempos indicados em cada experimento. Em
seguida, o sobrenadante era removido e centrifugado a 1400 x g por 10 minutos a 4oC. As células
eram recolhidas com solução de 0,1% de Triton X-100. Para a leitura da radioatividade, as
amostras eram preparadas adicionando-se 1,6mL de líquido de cintilação Ultima Gold para
amostras aquosas e não aquosas (PerkinElmer Life & Analytical Sciences) em 200µL de amostra.
A radioatividade do sobrenadante e do lisado celular de cada amostra era medida em cintilador
modelo LS6000LL da Beckman. A porcentagem de liberação de ácido araquidônico para cada
tratamento era calculada pela divisão entre a quantidade de radioatividade liberada no
sobrenadante e a quantidade de radioatividade total (encontrada no sobrenadante e no lisado
celular) mutiplicada por 100 e subtraída da liberação observada na ausência de estimulação.
3.6. Western blot
Os lisados celulares eram obtidos pela desadesão das células usando um raspador de
células (COSTAR) e por adição de 100 µL de SDS 10% a cada poço da placa de cultura. Em
seguida, era adicionado 100 µL de tampão de carregamento [Tampão de carregamento 2x e 10%
β-Mercaptoetanol (Solução estoque de tampão de carregamento 5x: Tris Base 460mM; Glicerol
7,5%; Azul de Bromofenol 0,2mM; SDS 690mM; pH 6,8)]. Após a lise celular, todas as amostras
foram imediatamente aquecidas a 100oC por 5 minutos. Os lisados celulares eram, então,
sonicados em Amplitude de 30% e 30 Joules de energia em processador ultra-sônico de alta

59
intensidade. As amostras foram separadas por eletroforese em gel de poliacrilamida SDS-PAGE
10% [Acrilamida/Bis-acrilamida (29:1)] a 29 mA/gel por aproximadamente 1 hora. Foi utilizado
marcador de peso molecular de 10 a 250 kDa (Protein Standards Unstained Broad Range da BIO-
RAD). Após a eletroforese, as proteínas foram transferidas do gel para membranas de
nitrocelulose (porosidade 0,45 µm da BioAgency) a 250 mA por 2 horas a 4oC. Em seguida, as
membranas foram bloqueadas com TBS 1x (Tris 10 mM; NaCl 150 mM pH 7,4) e leite desnatado
em pó a 5% por 12 horas, no caso de marcação para COX-2, ou por 2 horas, para as demais
marcações, sob agitação a temperatura ambiente. As membranas eram lavadas 3 vezes em TTBS
(TBS 1x; Tween-20 0,2%) por 10 minutos sob agitação a temperatura ambiente. A seguir, as
membranas foram incubadas com os anticorpos primários diluídos em TTBS e 0,05% de Azida
Sódica nas diluições recomendadas pelos fabricantes, já mencionadas anteriormente, por 2 ou 12
horas sob agitação a 4oC, dependendo do anticorpo. Após a incubação, as membranas eram
lavadas 3 vezes com TTBS por 15 minutos e incubadas com os anticorpos secundários anti-IgG
conjugados a peroxidase (HRP) nas diluições recomendadas, já mencionadas anteriormente, pelo
fabricante por 1 hora sob agitação a temperatura ambiente. As membranas foram lavadas 3 vezes
em TTBS por 10 minutos sob agitação a temperatura ambiente. Em seguida, a membrana foi
revelada por quimioluminescência com o kit ECLTM Western Blotting Analysis System conforme
especificações do fabricante (Amersham Biosciences). Os filmes de raio-X (Light Film Kodak
BioMax) foram expostos às membranas e revelados em revelador automático (Processador
Kodak X-OMAT 2000).

60
3.7. Análise Estatística
Os dados eram expressos como média e erro padrão da média. Comparações múltiplas
entre os grupos foram feitas pela análise de variância (ANOVA) seguida de teste de Bonferroni
ou de Dunnett. Valores de p < 0,05 foram considerados estatisticamente significativos.

61
4. RESULTADOS
O objetivo geral deste trabalho é avaliar o papel das PLA2s na ativação de células de
câncer de cólon por estresse microambiental. Para tanto, iniciamos uma série de experimentos in
vitro com duas linhagens de câncer de cólon humano, a Caco-2 e a HCT116. A linhagem Caco-2
expressa constitutivamente a COX-2 e possui a maquinaria enzimática necessária para a
produção de prostanóides, como a PGE2, porém não se sabe qual a PLA2 que participa desta
produção. Como contrapartida utilizamos a linhagem HCT-116, que não expressa COX-2,
mesmo após estimulação, portanto, não possui a maquinaria enzimática para a produção do
prostanóide, servindo como controle para a participação de PGE2 nos eventos estudados. Usamos,
então, dois modelos de ativação celular in vitro. O primeiro modelo é por estresse hipertônico,
que mimetiza o ambiente fisiológico intestinal, que consiste na estimulação das células em meio
hipertônico por adição de 100 mM de NaCl (Arbabi S et al., 2001). No segundo modelo, as
células são cultivadas em meio contendo 1mM de CoCl2, que permite a estabilização do fator de
transcrição induzível por hipóxia (HIF)-1α, simulando o que ocorre na hipóxia tecidual. Em
condições de normóxia, na presença de oxigênio, a enzima prolil hidroxilase é ativada e altera
pós-transducionalmente HIF-1α, permitindo sua ligação com o complexo de Von Hippel-Lindau
(VHL), e seu direcionamento para a degradação via proteossoma. Já em condições de hipóxia, na
ausência de oxigênio, a enzima prolil hidroxilase não modifica HIF-1α e a proteína torna-se
estável e se acumula no citoplasma. Então, HIF-1α estabilizado é translocado para o núcleo onde
se liga a elementos responsivos a hipóxia (HRE) e ativa a transcrição de genes alvo, como o
VEGF (Harris AL, 2002). O CoCl2 diminui os níveis intracelulares de ascorbato, que é co-fator
da enzima prolil hidroxilase, tornando-a inativa, impedindo a ligação de HIF-1α com o complexo

62
VHL e sua degradação via proteossoma (Salnikow K et al., 2004). Com isso, HIF-1α é
translocado para o núcleo e ativa a transcrição de VEGF, simulando o que ocorre na hipóxia
tecidual. Com estes dois modelos de ativação nós criamos condições experimentais próximas ao
ambiente fisiopatológico intestinal durante a progressão tumoral.
Todos os experimentos eram realizados em densidade celular de 5x105 células por poço
de 962 mm2 de área com um tempo médio para adesão celular prévia de 12 horas. Antes da
realização dos experimentos o meio era retirado, as células lavadas com PBS uma vez e, então o
meio contendo as drogas ou o estímulo específico era adicionado. O material (sobrenadante e
lisado celular) era recolhido após os tempos determinados para cada experimento.
4.1. O estresse hipertônico induz a liberação de AA, o aumento da expressão de COX-2 e a
produção de PGE2 em Caco-2
Na primeira parte do estudo observou-se que o estresse hipertônico induz a produção de
PGE2 por Caco-2 e que a maior estimulação se dá quando 100 mM de NaCl são adicionados ao
meio (Figura 3B). Levando-se em consideração que este tipo de estímulo influenciava o produto
final da cascata do AA, verificou-se se havia efeito do estresse hipertônico sobre a liberação de
AA e sobre a expressão de COX-2. A análise da liberação de AA demonstrou que houve maior
liberação após 24 horas de ativação (Figura 3A) e que a expressão de COX-2 aumenta à medida
que a concentração de NaCl adicionada ao meio de cultura também aumenta (Figura 3C). Logo,
como a produção de PGE2, a expressão de COX-2 e a liberação de AA eram máximas em 24
horas de ativação com adição de 100 mM de NaCl ao meio, a partir daí os experimentos
seguintes foram realizados sob estas condições de estimulação.

63
Figura 3: O estresse hipertônico induz a liberação de AA, a expressão de COX-2 e a produção de PGE2 em Caco-2. A, Liberação de AA tritiado por Caco-2 em 2-24 horas após a adição de 100mM de NaCl ao meio. B, Produção de PGE2 (pg/ml) por Caco-2 após a adição de 10-100mM de NaCl ao meio. O * significa estatisticamente diferente das células não ativadas por estresse hipertônico (p < 0,05). C, Células Caco-2 foram cultivadas em meio sem NaCl ou com 10-100mM de NaCl. O extrato protéico total foi analisado por Western blot com anticorpo monoclonal anti-COX-2. As barras dos gráficos A e B representam a média e o erro padrão da média de amostras realizadas em triplicata. O gráfico A foi obtido a partir de um experimento. O gráfico B e a figura C representam três experimentos independentes.
0
2000
4000
6000
8000
10000
12000
NaCl (M) - 0.10.03
*
0.01
B
PGE 2
(pg/
ml)
2 4 8 12 18 24
0
10
20
Tempo (h)
*
A
NaCl 0.1M
∆ d
e3 H
AA
libe
rado
(%)
COX-2
NaCl (M)_______________- 0.03 0.10.01
GAPDH
70 kDa
37 kDa
C
COX-2
NaCl (M)_______________- 0.03 0.10.01
GAPDH
70 kDa
37 kDa
COX-2 COX-2 COX-2
NaCl (M)_____________________________________________- 0.03 0.10.01
GAPDH GAPDH GAPDH
70 kDa70 kDa70 kDa
37 kDa37 kDa37 kDa
C

64
4.2. A produção de PGE2 é dependente da atividade de cPLA2 e de COX-2 em Caco-2
estimulada com estresse hipertônico
A produção de PGE2 é dependente da atividade de COX-2 em Caco-2 estimulada com
estresse osmótico, pois o uso de NS-398, um inibidor específico de COX-2, diminui a produção,
de maneira dose-dependente, de PGE2 (Figura 4A). O inibidor de PLA2 citosólica (cPLA2), o
ATK, inibiu a produção de PGE2 (Figura 4B), enquanto que o uso de inibidores de PLA2
secretórias (sPLA2), o LY311727, e de PLA2 independente de cálcio (iPLA2), o BEL, não
modificaram esta produção (Figuras 4C e 4D).
4.3. cPLA2 regula o fornecimento de AA em Caco-2 estimulada com estresse hipertônico
Como já foi demonstrado que a cPLA2 além de ter ação modulatória sobre a liberação de
AA pode também induzir a expressão de COX-2 (Fujishima H et al., 1999; Balsinde J et al.,
1999b), verificamos, então, em qual ponto estava ocorrendo a inibição, se na expressão de COX-
2 ou se no fornecimento do AA. Os resultados mostraram que não há modificação do perfil de
expressão de COX-2 em Caco-2 estimulada com estresse hipertônico após tratamento com ATK,
o inibidor de cPLA2 (Figura 5A). O estresse hipertônico induz máxima liberação de AA com 24
horas de ativação (Figura 3A). Então, verificou-se o efeito da inibição de cPLA2 neste tempo
após a ativação e houve bloqueio da atividade de cPLA2, pelo uso do inibidor ATK, diminuindo
em praticamente 90% a liberação de AA (Figura 5B). Estes resultados indicam que a ação da
cPLA2 é a de regular o fornecimento de AA para a produção de PGE2 e não a de regular a
expressão de COX-2.

65
Figura 4: A produção de PGE2 é dependente da atividade de cPLA2 e de COX-2 em Caco-2 estimulada por estresse hipertônico. Produção de PGE2 (pg/ml) por Caco-2 após tratamento com NS-398 (A), ATK (B), LY311727 (C), BEL(D) e estimulação com adição de 100mM de NaCl ao meio. O símbolo + significa estatisticamente diferente das células não ativadas por estresse hipertônico (p < 0,05) e o * significa estatisticamente diferente das células ativadas por estresse hipertônico (p < 0,05). As barras dos gráficos representam a média e o erro padrão da média de amostras realizadas em triplicata. Os gráficos A e C representam três experimentos independentes, o gráfico B representa dois experimentos independentes e o gráfico D representa quatro experimentos independentes.
0
1000
2000
3000
4000
5000
6000
7000
Estresse Hipertônico --
+ + + +-NS-398 (µM) 0.001
+0.1 10.01
+
*
* **
A
PGE 2
(pg/
ml)
0
2500
5000
7500
10000
Estresse Hipertônico --
+ + + +-ATK (µM) 0.1
+3 101
+
* * *
B
PGE 2
(pg/
ml)
0
2500
5000
7500
10000
Estresse Hipertônico --
+ +-LY311727 (µM) 10 100
+
+
C
PGE 2
(pg/
ml)
0
1000
2000
3000
4000
5000
6000
7000
Estresse Hipertônico --
+ +BEL (µM) - 10
+
D
PGE 2
(pg/
ml)

66
Figura 5: cPLA2 regula o fornecimento de AA em Caco-2 estimulada com estresse hipertônico. A, Expressão de COX-2 em Caco-2 cultivadas em meio com 100mM de NaCl e tratadas com 1-10µM de ATK. O extrato protéico total foi extraído por lise celular e anlisado por Western blot com anticorpo monoclonal anti-COX-2. B, Liberação de AA tritiado por Caco-2 após o tratamento com ATK e estimulação com a adição de 100mM de NaCl ao meio. O símbolo * significa estatisticamente diferente das células ativadas por estresse hipertônico e não tratadas com ATK (p < 0,05). A figura A representa um experimento independente representativo de três experimentos independentes. As barras do gráfico B representam a média e o erro padrão da média de amostras realizadas em triplicata. O gráfico B representa dois experimentos independentes.
Estresse hipertônico +-ATK (10µM) ++
0.0
2.5
5.0
7.5
10.0
*
B
Libe
raçã
o de
3 H A
A (%
)
A
COX-2
ATK (µM) - - 3 10______________NaCl 0,1M
1
GAPDH
70 kDa
37 kDa
A
COX-2
ATK (µM) - - 3 10______________NaCl 0,1M
1
GAPDH
COX-2
ATK (µM) - - 3 10______________NaCl 0,1M
1
GAPDH
70 kDa70 kDa70 kDa
37 kDa37 kDa37 kDa

67
4.4. As quinases ERK 1/2, JNK 1/2/3 e p38 estão envolvidas na produção de PGE2 por Caco-2
estimuladas com estresse hipertônico
A associação entre a produção de PGE2 e o desenvolvimento do câncer de cólon é bem
definida (Oshima M et al., 1996; Sheng H et al., 1997; Tsujii M et al., 1998; Dong M et al., 2003;
Panel V et al., 2006; Buchanan FG e DuBois RN, 2006), entretanto, os estímulos e mecanismos
indutores da expressão de COX-2 em células coloretais não estão, ainda, bem esclarecidos. O
epitélio colônico está em constante contato com um meio muito variado em conteúdos entéricos e
as células respondem às mudanças na pressão osmótica com adaptações moleculares
compensatórias que permitem a elas restabelecer a homeostasia estrutural e funcional (Naftalin
RJ e Pedley KC, 1990; Powell DW, 1995; Arbabi S et al., 2000; Yang T et al., 2000; Thiagarajah
JR et al., 2001; Hubert A et al., 2004; Yang T et al., 2005; Dmitrieva NI e Burg MB, 2005; Lim
WC et al., 2005). Um dos mecanismos pelos quais as células respondem ao estresse hipertônico é
modificando as cascatas das MAP quinases, que são proteínas quinases que compõem
importantes vias intracelulares de transdução de sinal (Han J et al., 1994; Galcheva-Gargova Z et
al., 1994; Kultz D e Burg M, 1998; Yang T et al., 2000). As MAP quinases (MAPKs)
pertencentes a estas vias desempenham importante função na ativação de cPLA2.
Tomando como base a relação entre estresse hipertônico e MAPKs, observamos se as
principais quinases envolvidas na resposta ao estresse em mamíferos estavam modulando a
cascata do AA. Primeiramente, observou-se que as quinases ERK1/2, SAPK/JNK e p38
participavam da produção de PGE2 em Caco-2 estimulada por estresse hipertônico, já que o uso
dos inibidores farmacológicos de cada quinase inibiu a produção do eicosanóide (Figura 6).

68
Figura 6: As quinases ERK1/2, JNK e p38 estão envolvidas na produção de PGE2 por Caco-2 estimulada por estresse hipertônico. Produção de PGE2 por células Caco-2 tratadas com os inibidores de JNK (SP 600125), de p38 (SB 202190), de ERK1/2 (U 0126) e ativadas com a adição de 100mM de NaCl ao meio. O símbolo + significa estatisticamente diferente das células não ativadas por estresse hipertônico (p < 0,05) e o * significa estatisticamente diferente das células ativadas por estresse hipertônico e não tratadas com os inibidores de MAP quinases (p < 0,05). As barras do gráfico representam a média e o erro padrão da média de amostras realizadas em triplicata. O gráfico representa três experimentos independentes.
0
10000
20000
30000
Estresse Hipertônico - + + + +
+
- -SP 600125 (20µM) --
- -SB 202190 (15µM) - -
- -U 0126 (20µM) - -
++
+
*
* *
PGE 2
(pg/
ml)

69
4.5. O estresse hipertônico modula a fosforilação das três MAPKs em Caco-2
Considerando então que todas as 3 subfamílias de quinases participam da produção de PGE2
frente ao estresse hipertônico, verificou-se se este tipo de estímulo por estresse induzia a
fosforilação das quinases e observou-se que o estresse hipertônico aumenta a expressão de
ERK1/2, p38 e JNK, em suas formas fosforiladas, após 0,5 e 2 horas de ativação (Figura 7).
4.6. MAPKs modulam a expressão de COX-2 e a liberação de AA por cPLA2 em Caco-2
estimulada por estresse hipertônico
A próxima etapa do estudo foi verificar se as MAPKs possuíam ação modulatória sobre a
expressão de COX-2 ou sobre a liberação de AA por cPLA2. Os resultados da Figura 8 mostram
que ERK1/2 está envolvida na indução de COX-2 enquanto que p38 e SAPK/JNK possuem ação
inibitória sobre a expressão desta enzima. Em relação à liberação de AA os resultados indicam
que todas as três quinases modulam a atividade de cPLA2 (Figura 9).
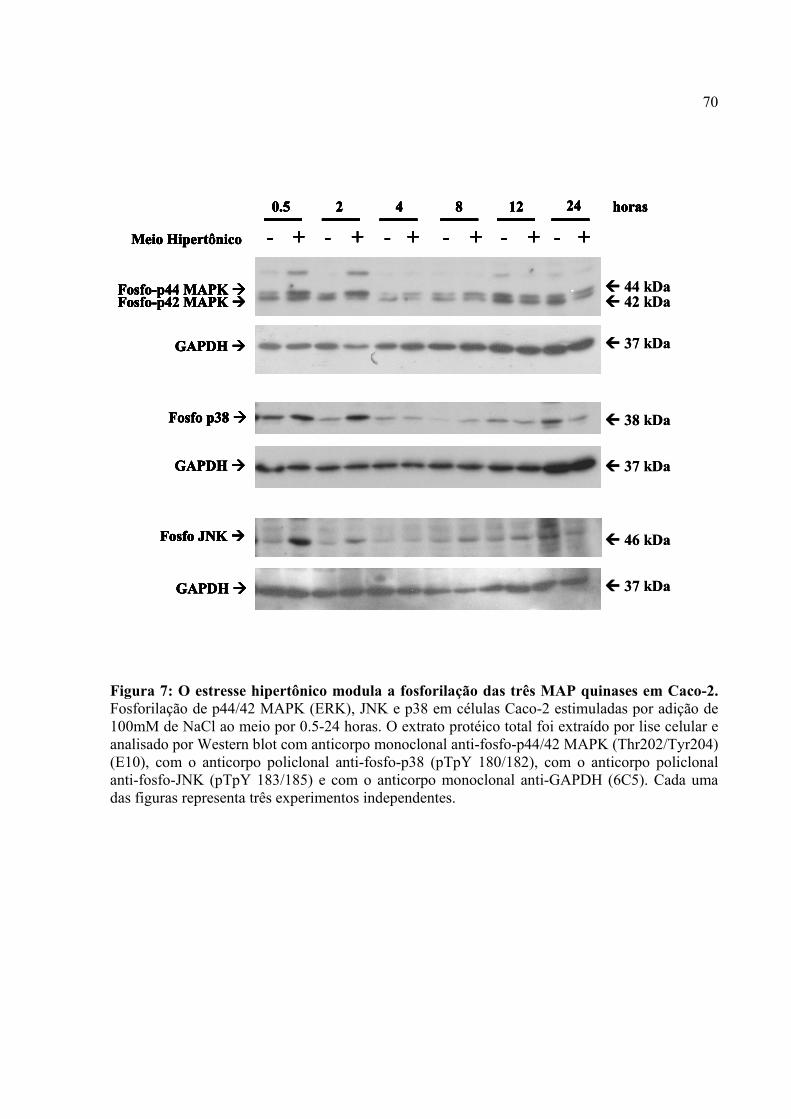
70
Figura 7: O estresse hipertônico modula a fosforilação das três MAP quinases em Caco-2. Fosforilação de p44/42 MAPK (ERK), JNK e p38 em células Caco-2 estimuladas por adição de 100mM de NaCl ao meio por 0.5-24 horas. O extrato protéico total foi extraído por lise celular e analisado por Western blot com anticorpo monoclonal anti-fosfo-p44/42 MAPK (Thr202/Tyr204) (E10), com o anticorpo policlonal anti-fosfo-p38 (pTpY 180/182), com o anticorpo policlonal anti-fosfo-JNK (pTpY 183/185) e com o anticorpo monoclonal anti-GAPDH (6C5). Cada uma das figuras representa três experimentos independentes.
Fosfo-p44 MAPK
GAPDH
Fosfo-p42 MAPK
GAPDH
Fosfo p38
GAPDH
Fosfo JNK
Meio Hipertônico -- -- - -+ + + + + +0.5 2 4 8 12 24 horas
Fosfo-p44 MAPK
GAPDH
Fosfo-p42 MAPK Fosfo-p44 MAPK
GAPDH
Fosfo-p42 MAPK
GAPDH
Fosfo p38
GAPDH
Fosfo p38
GAPDH
Fosfo JNK
GAPDH
Fosfo JNK
Meio Hipertônico -- -- - -+ + + + + +0.5 2 4 8 12 24 horas
Meio Hipertônico -- -- - -+ + + + + +0.5 2 4 8 12 24 horas
42 kDa
37 kDa
38 kDa
46 kDa
37 kDa
37 kDa
44 kDaFosfo-p44 MAPK
GAPDH
Fosfo-p42 MAPK
GAPDH
Fosfo p38
GAPDH
Fosfo JNK
Meio Hipertônico -- -- - -+ + + + + +0.5 2 4 8 12 24 horas
Fosfo-p44 MAPK
GAPDH
Fosfo-p42 MAPK Fosfo-p44 MAPK
GAPDH
Fosfo-p42 MAPK
GAPDH
Fosfo p38
GAPDH
Fosfo p38
GAPDH
Fosfo JNK
GAPDH
Fosfo JNK
Meio Hipertônico -- -- - -+ + + + + +0.5 2 4 8 12 24 horas
Meio Hipertônico -- -- - -+ + + + + +0.5 2 4 8 12 24 horas
42 kDa
37 kDa
38 kDa
46 kDa
37 kDa
37 kDa
44 kDa

71
Figura 8: MAPKs modulam a expressão de COX-2 em Caco-2 estimulada por estresse hipertônico. Expressão de COX-2 e GAPDH em células Caco-2 cultivadas em meio com 100mM de NaCl e tratadas com inibidor de JNK (SP600125), de p38 (SB202190) ou de ERK1/2 (U0126). O extrato protéico total foi extraído por lise celular e analisado por Western blot com anticorpo monoclonal anti-COX-2 e anticorpo monoclonal anti-GAPDH (6C5). Dez (10) µL de lisado celular foram adicionados em cada lane. A figura representa um experimento independente representativo de três experimentos independentes.
- -SP 600125
(20µM)SB 202190
(15µM)U 0126 (20µM)
NaCl 0.1 M
Con
trol
e
COX-2
GAPDH
70 kDa
37 kDa
- -SP 600125
(20µM)SB 202190
(15µM)U 0126 (20µM)
NaCl 0.1 M
Con
trol
e
COX-2
GAPDH
- -SP 600125
(20µM)SB 202190
(15µM)U 0126 (20µM)
NaCl 0.1 M
Con
trol
e
COX-2
GAPDH
70 kDa
37 kDa

72
Figura 9: As três MAP quinases participam da liberação de AA por cPLA2 em Caco-2 estimuladas por estresse hipertônico. Liberação de AA tritiado por Caco-2 após tratamento durante 15 minutos com o inibidor de cPLA2 (ATK) ou por 30 minutos com o inibidor de JNK (SP 600125), de p38 (SB 202190) ou de ERK1/2 (U 0126) e ativação com a adição de 100mM de NaCl ao meio de cultura por 24 horas. O símbolo * significa estatisticamente diferente das células ativadas por estresse hipertônico e não tratadas com os inibidores (p < 0,05). As barras do gráfico representam a média e o erro padrão da média de amostras realizadas em triplicata. O gráfico representa dois experimentos independentes.
0.0
2.5
5.0
7.5
10.0
Estresse Hipertônico + + + +
+
- -SP 600125 (20µM) --
- -SB 202190 (15µM) - -
- -U 0126 (20µM) - -
+
+
+- -ATK (10µM) -+ -
**
**
Libe
raçã
o de
3 H A
A (%
)

73
4.7. O estresse hipertônico induz a produção de VEGF em Caco-2 e PGE2 modula a produção
do fator angiogênico neste tipo de estresse microambiental
A PGE2 tem, entre outras, a função de modular a angiogênese e a produção de VEGF.
Analisamos, então, o papel da PGE2 na regulação da produção de VEGF por Caco-2 durante a
estimulação com o meio hipertônico. E observamos que o estresse hipertônico aumenta a
produção de VEGF por Caco-2 após 24 horas de ativação (Figura 10).
A inibição da produção de PGE2 com os inibidores para COX-2 (NS-398) (Figura 11B) e
cPLA2 (ATK) (Figura 11A) aumentaram a produção de VEGF, sugerindo uma função inibitória
para o prostanóide sobre a produção de VEGF.
Para confirmar se a inibição da produção de VEGF em Caco-2 estimulada com estresse
hipertônico era devido à ação da PGE2 e não conseqüência da ação inespecífica das drogas
utilizadas nos experimentos, utilizou-se uma linhagem de câncer de cólon humano que não
expressa COX-2, a HCT116, e observou-se que não há diferenças na produção de VEGF em
nenhum dos tratamentos utilizados, descartando a possibilidade da interferência dos inibidores
farmacológicos (Figura 12).
Para demonstrar se PGE2 era capaz de inibir a produção do fator angiogênico em Caco-2
no estresse hipertônico foi adicionada PGE2 exógena ao meio e foi observada a inibição da
produção de VEGF à medida que a concentração de PGE2 no meio aumentava (Figura 13A). E o
fenômeno do aumento da produção de VEGF observado quando do tratamento com NS398 ou
ATK foi revertido pela adição de PGE2 ao meio (Figuras 13B e 13C).
Estes resultados indicam que a PGE2 produzida por Caco-2 no estresse hipertônico possui
ação inibitória sobre a produção do fator angiogênico.
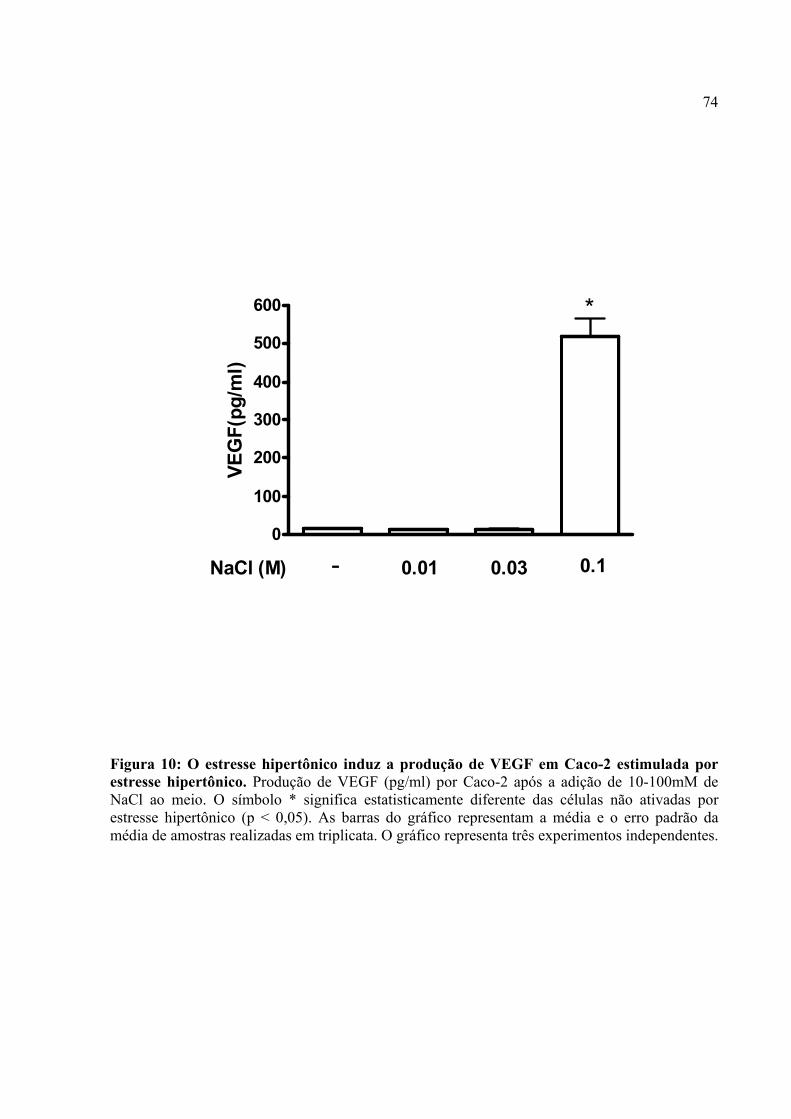
74
Figura 10: O estresse hipertônico induz a produção de VEGF em Caco-2 estimulada por estresse hipertônico. Produção de VEGF (pg/ml) por Caco-2 após a adição de 10-100mM de NaCl ao meio. O símbolo * significa estatisticamente diferente das células não ativadas por estresse hipertônico (p < 0,05). As barras do gráfico representam a média e o erro padrão da média de amostras realizadas em triplicata. O gráfico representa três experimentos independentes.
0
100
200
300
400
500
600
NaCl (M) - 0.10.01 0.03
*
VEG
F(pg
/ml)
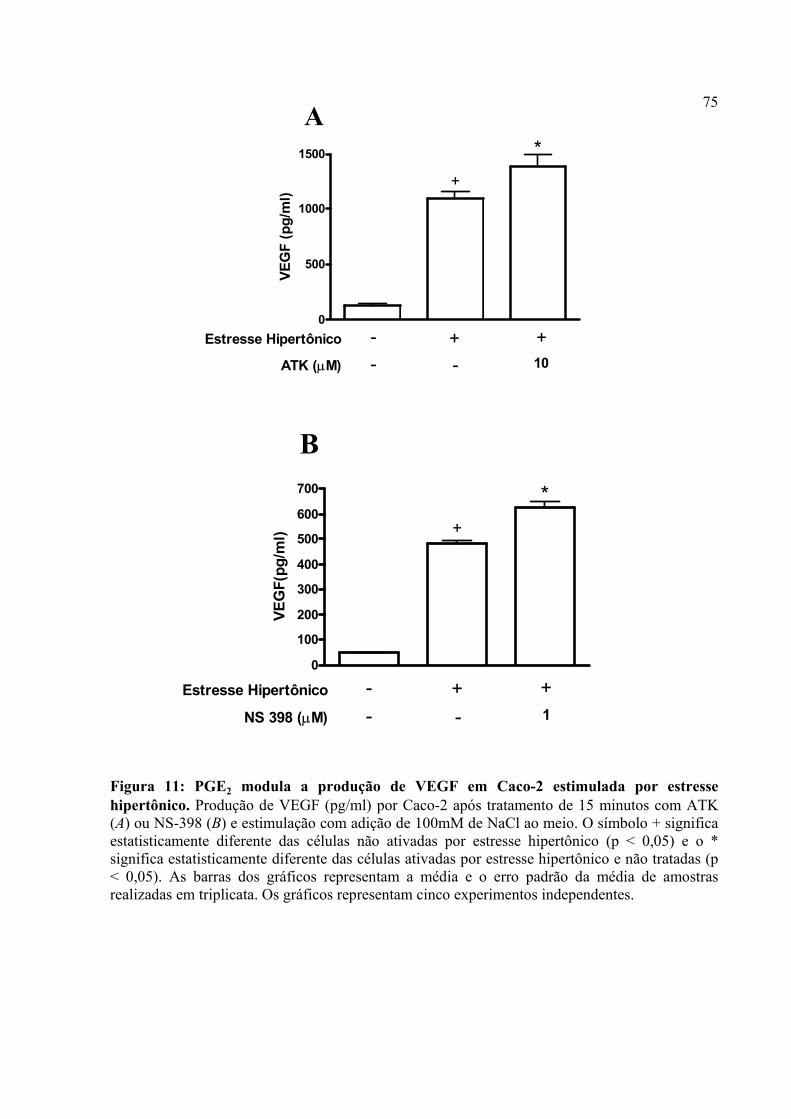
75
Figura 11: PGE2 modula a produção de VEGF em Caco-2 estimulada por estresse hipertônico. Produção de VEGF (pg/ml) por Caco-2 após tratamento de 15 minutos com ATK (A) ou NS-398 (B) e estimulação com adição de 100mM de NaCl ao meio. O símbolo + significa estatisticamente diferente das células não ativadas por estresse hipertônico (p < 0,05) e o * significa estatisticamente diferente das células ativadas por estresse hipertônico e não tratadas (p < 0,05). As barras dos gráficos representam a média e o erro padrão da média de amostras realizadas em triplicata. Os gráficos representam cinco experimentos independentes.
0
500
1000
1500
Estresse Hipertônico --
+-
+10ATK (µM)
+
*A
VEG
F (p
g/m
l)
0
100
200
300
400
500
600
700
Estresse Hipertônico --
+-
+1NS 398 (µM)
+
*
B
VEG
F(pg
/ml)

76
Figura 12: Papel da cascata do AA sobre a produção de VEGF em HCT116 estimulada por estresse hipertônico. Produção de VEGF (pg/ml) por HCT116 após tratamento com ATK (A) ou NS-398 (B) e estimulação com adição de 100mM de NaCl ao meio. As barras dos gráficos A e B representam a média e o erro padrão da média de amostras de quatro experimentos independentes.
0
250
500
750
1000
1250
1500
Estresse Hipertônico --
+-
+10ATK (µM)
+3
+1
+
A
VEG
F (p
g/m
l)
0
500
1000
1500
Estresse Hipertônico --
+-
+1NS 398 (µM)
+ +0.10.001 0.01
+
+
B
VEG
F (p
g/m
l)

77
Figura 13: A adição de PGE2 ao meio reverte o efeito de sua inibição sobre a produção de VEGF em Caco-2 estimulada por estresse hipertônico. A, Produção de VEGF (pg/ml) por Caco-2 após adição de 0.01-1µM de PGE2 ao meio e estimulação com estresse hipertônico (100mM de NaCl). B, Produção de VEGF (pg/ml) por Caco-2 após o tratamento com NS-398 e adição de PGE2 ao meio e estimulação com estresse hipertônico (100mM de NaCl). C, Produção de VEGF (pg/ml) por Caco-2 após o tratamento com ATK e adição de PGE2 ao meio e estimulação com estresse hipertônico (100mM de NaCl). O símbolo + significa estatisticamente diferente das células não ativadas por estresse hipertônico (p < 0,05), o * significa estatisticamente diferente das células ativadas por estresse hipertônico e não tratadas e o ** significa estatisticamente diferente das células ativadas por estresse hipertônico e tratadas com NS398 ou ATK (p < 0,05). As barras do gráfico A representam a média e o erro padrão da média de amostras de quatro experimentos independentes. As barras dos gráficos B e C representam a média e o erro padrão da média de amostras realizadas em triplicata. O gráfico B representa dois experimentos independentes e o gráfico C representa cinco experimentos independentes.
0
50
100
150
200
250
Estresse Hipertônico --
+ + + +-PGE2 (µM) 0.01
+0.1 0.30.03
+1
+
***
A
VEG
F (p
g/m
l)
0
100
200
300
400
500
600
700
Estresse Hipertônico - + + +
- -PGE2 (µM) -- -NS 398 (µM) 1 1
0.1
+
*
**
B
VEG
F(pg
/ml)
0
250
500
750
1000
Estresse Hipertônico - + + +
- -PGE2 (µM) -- -ATK (µM) 10 10
0.1
+
*
**
C
VEG
F (p
g/m
l)

78
4.8. O receptor EP2 está envolvido na regulação da produção de VEGF por PGE2 em Caco-2
estimulada com estresse hipertônico
Para verificar qual o receptor tipo EP de PGE2 envolvido na redução da produção de
VEGF no estresse hipertônico que poderia explicar a alça regulatória existente neste tipo de
estresse microambiental, as células Caco-2 foram ativadas com meio hipertônico por 24 horas e
tratadas com um antagonista para receptores tipo EP, demonstrando que a inibição da sinalização
via receptor EP resultava em aumento da produção de VEGF (Figura 14A). Então, para
identificar qual o receptor específico envolvido no fenômeno as Caco-2 foram ativadas com meio
hipertônico e tratadas com ATK e diversos agonistas de receptor EP. O uso do inbidor de cPLA2
foi para eliminar a interferência da PGE2 produzida pelas células de câncer de cólon e os
receptores tipo EP serem ativados somente pelos respectivos agonistas. A Figura 14B mostra que,
com a inibição da produção de PGE2 pelo uso de ATK, houve um aumento da produção de
VEGF, e este efeito é revertido pela adição de PGE2 ao meio, confirmando a ação inibitória do
prostanóide sobre o VEGF. Esta reversão somente é verificada com o uso dos agonistas 16,16-
dimetil PGE2 para a maioria dos receptores EP, e com o uso do agonista Butaproste, específico
para o receptor EP2. Foram feitos dois experimentos independentes com esta abordagem e a
análise estatística foi idêntica para os dois, com exceção do agonista específico para EP3,
Sulprostona, porém a inibição verificada no outro experimento foi mínima, explicando o efeito
borderline do receptor EP3. Tomados em conjunto, estes resultados indicam que o princiapl
receptor para PGE2 envolvido na produção de VEGF em estresse hipertônico é o do tipo EP2.

79
Figura 14: O receptor EP2 está envolvido na regulação da produção de VEGF por PGE2 em Caco-2 estimulada com estresse hipertônico. A, Produção de VEGF (pg/ml) por Caco-2 após tratamento com inibidor de cPLA2 (ATK) e PGE2, agonista EP (16,16-dimetil PGE2), agonista EP1 e EP3 (17-fenil trinor PGE2), agonista EP2 (Butaproste) e agonista EP3 (Sulprostona) durante 15 minutos e ativação por estresse hipertônico. O símbolo + significa estatisticamente diferente das células não ativadas por estresse hipertônico (p < 0,05), o * significa estatisticamente diferente das células ativadas por estresse hipertônico e não tratadas (p < 0,05) e o ** significa estatisticamente diferente das células ativadas por estresse hipertônico e tratadas com ATK (p < 0,05). B, Produção de VEGF (pg/ml) por Caco-2 após tratamento com antagonista de receptor tipo EP e DP (AH 6809) durante 15 minutos e ativação por estresse hipertônico. O símbolo + significa estatisticamente diferente das células não ativadas por estresse hipertônico (p < 0,05) e o * significa estatisticamente diferente das células ativadas por estresse hipertônico e não tratadas (p < 0,05). As barras dos gráficos representam a média e o erro padrão da média de amostras realizadas em triplicata. Os gráficos representam dois experimentos independentes.
0
250
500
750
1000
Estresse Hipertônico - +
+
- -PGE2 (0.1µM) -
-- -16,16-dimetil PGE2 (0.1µM) -
- -17-fenil trinor PGE2 (0.1µM) - -
+
+- +ATK (10µM)
+
- -Butaproste (0.1µM) - -
- -Sulprostona (0.1µM) - -
+ + + + + +-
-
-
-
-
-
-
-
-
+
-
-
-
-
-
-
-
-
+
***
******
B
+ + + ++
VEG
F (p
g/m
l)
0
100
200
300
400
500
600
Estresse Hipertônico --
+- 10AH 6809 (µM)
+
+
*
A
VEG
F (p
g/m
l)

80
4.9. A adição de CoCl2 ao meio induz a produção de PGE2 e a expressão de COX-2 por Caco-2
Observou-se se o fenômeno de regulação da produção de VEGF por PGE2 se repetia no
estresse por hipóxia. Primeiramente, verificou-se se este tipo de estímulo induzia a produção de
PGE2 e expressão de COX-2 por Caco-2 após 24 horas e se verificou que havia produção do
prostanóide e expressão da enzima após este período (Figura 15). Como a produção máxima de
PGE2 se deu com adição de 1mM de CoCl2 ao meio e com ativação de 24 horas, os experimentos
mimetizando a hipóxia foram realizados sob estas condições.
4.10. A produção de PGE2 é dependente de COX-2 e cPLA2 em Caco-2 estimulada por CoCl2
Avaliamos a produção de PGE2 em Caco-2 estimulada por hipóxia e constatamos que a
produção de PGE2 é dependente de COX-2 e cPLA2 (Figura 16) e que havia a maior estimulação
da produção de VEGF frente a este estímulo após 24 horas na concentração de 1 mM de CoCl2
(Figura 17A). Entretanto, repetindo os mesmos tratamentos com os inibidores farmacológicos
verificamos que a PGE2 não possui papel regulatório sobre a produção de VEGF em Caco-2
ativadas por hipóxia (Figura 17B e 17C), apesar de haver uma tendência à inibição quando do
tratamento com NS-398. Ainda, não houve aumento da produção do fator angiogênico quando do
tratamento com NS398 nas Caco-2 estimuladas com hipóxia em nenhuma das doses testadas.
Tais dados podem ser explicados pela presença de diferentes vias de sinalização nos dois
estímulos, o estresse hipertônico e a hipóxia. Os resultados demonstram que o feedback negativo
de PGE2 sobre a produção de VEGF pela Caco-2 no estresse hipertônico não está presente na
estimulação por hipóxia.

81
Figura 15: A adição de CoCl2 ao meio induz a produção de PGE2 e a expressão de COX-2 em Caco-2. A, Produção de PGE2 (pg/ml) por Caco-2 após a adição de meio com 0.1-1mM de CoCl2. B, Células Caco-2 foram cultivadas em meio sem CoCl2 ou com 0.1-1mM de CoCl2. O extrato protéico total foi analisado por Western blot com anticorpo monoclonal anti-COX-2. O símbolo * significa estatisticamente diferente das células não ativadas por estresse hipertônico (p < 0,05). As barras do gráfico A representam a média e o erro padrão da média de amostras realizadas em triplicata. O gráfico A representa três experimentos independentes e a figura B representa dois experimentos independentes.
0
2500
5000
7500
10000
CoCl2 (mM) - 10.1 0.3
*
*
A
PGE 2
(pg/
ml)
BCoCl2 (mM)
- 0.30.1 1
COX-2
GAPDH
70 kDa
37 kDa
BCoCl2 (mM)
- 0.30.1 1
COX-2COX-2COX-2
GAPDHGAPDHGAPDH
70 kDa70 kDa70 kDa
37 kDa37 kDa37 kDa

82
Figura 16: A produção de PGE2 é dependente de COX-2 e cPLA2 em Caco-2 estimulada por CoCl2. Produção de PGE2 (pg/ml) por Caco-2 após tratamento com NS-398 (A) ou ATK (B) e ativação com CoCl2. O símbolo + significa estatisticamente diferente das células não ativadas por CoCl2 (p < 0,05) e o * significa estatisticamente diferente das células ativadas por CoCl2 e não tratadas (p < 0,05). As barras dos gráficos representam a média e o erro padrão da média de amostras realizadas em triplicata. Os gráficos representam três experimentos independentes.
0
5000
10000
15000
20000
CoCl2 (1mM) --
+ +NS-398 (µM) 1
+
*
-
A
PGE 2
(pg/
ml)
0
2500
5000
7500
10000
12500
CoCl2 (1mM) --
+ +ATK (µM) 10
+
*
B
-
PGE 2
(pg/
ml)

83
Figura 17: A adição de CoCl2 ao meio induz a produção de VEGF em Caco-2 e PGE2 não modula a produção do fator angiogênico neste tipo de estímulo. A, Produção de VEGF (pg/ml) por Caco-2 ativadas com 0.1-1mM de CoCl2. B, Produção de VEGF (pg/ml) por Caco-2 após tratamento com NS-398 e ativação com CoCl2. C, Produção de VEGF (pg/ml) produzidos por Caco-2 após o tratamento com ATK e estimulação com CoCl2. O símbolo + e * significam estatisticamente diferente das células não ativadas por CoCl2 (p < 0,05). As barras dos gráficos representam a média e o erro padrão da média de amostras realizadas em triplicata. Os gráficos A e C representam três experimentos independentes e o gráfico B representa quatro experimentos independentes.
0
250
500
750
1000
CoCl2 (mM) - 10.1 0.3
*
A
VEG
F(pg
/ml)
0
100
200
300
400
500
600
CoCl2 (1mM) --
+-
+NS 398 (µM)
+
1
B
VEG
F(pg
/ml)
0
250
500
750
1000
1250
1500
1750
CoCl2 (1mM) --
+-
+10
+
C
ATK (µM)
VEG
F (p
g/m
l)

84
4.11. ERK1/2, JNK e p38 estão envolvidas na produção de VEGF por Caco-2 estimulada por
CoCl2
Tendo como justificativa as diferentes ações modulatórias das MAPKs sobre a produção
de VEGF na hipóxia tecidual, verificou-se o efeito regulador das quinases sobre a produção deste
fator angiogênico, em Caco-2, frente aos dois tipos de estresse microambiental. E observou-se
que, nas condições que mimetizavam a hipóxia, todas as 3 quinases possuíam papel estimulador
da produção de VEGF em Caco-2 (Figura 18).
4.12. p38 e ERK1/2 estão envolvidas na produção de VEGF por Caco-2 estimulada por estresse
hipertônico
Já em relação ao estresse hipertônico, a SAPK/JNK inibe a produção de VEGF, enquanto
que p38 MAPK estimula a produção do fator angiogênico (Figura 19A). Porém, como há inibição
da produção de PGE2 no estresse hipertônico quando do tratamento de Caco-2 com todos os
inibidores de quinases (Figura 6), não podemos descartar a possibilidade do aumento na produção
de VEGF observado no tratamento com o inibidor de SAPK/JNK ser uma conseqüência do
bloqueio de PGE2 e não da ação da própria MAPK. Logo, para retirar a variável de produção de
PGE2 do sistema, nós tratamos as células com o inibidor de COX-2 (NS398) e observamos que a
inibição da produção de VEGF se mantinha a mesma com a utilização do inibidor de p38 MAPK
e que os níveis do fator angiogênico diminuíam com o uso do inibidor de ERK1/2, indicando que
p38 e ERK1/2 possuem efeito estimulatório sobre a produção de VEGF (Figura 19B). O aumento
observado anteriormente com o inibidor de SAPK/JNK (Figura 19A) parece ser devido à inibição
de PGE2, uma vez que removida esta alça de regulação inibitória, o inibidor SP 600125 deixou de
interferir na produção de VEGF (Figura 19B).

85
Figura 18: ERK1/2, JNK e p38 estão envolvidas no aumento da produção de VEGF em Caco-2 estimulada por CoCl2. Produção de VEGF (pg/ml) por Caco-2 após tratamento com inibidor de JNK (SP600125), de p38 (SB202190) ou de ERK1/2 (U 0126) durante 30 minutos e ativação com CoCl2. O símbolo + significa estatisticamente diferente das células não ativadas por CoCl2 (p < 0,05) e o * significa estatisticamente diferente das células ativadas por CoCl2 e não tratadas (p < 0,05). As barras do gráfico representam a média e o erro padrão da média de amostras realizadas em triplicata. O gráfico representa dois experimentos independentes.
0
100
200
300
400
500
600
700
CoCl2 (1mM) - + + + +
+
- -SP 600125 (20µM) --
- -SB 202190 (15µM) - -
- -U 0126 (20µM) - -
+
+
*
*
*
+
VEG
F (p
g/m
l)

86
Figura 19: p38 e ERK1/2 são as quinases envolvidas na produção de VEGF em Caco-2 estimulada por estresse hipertônico. A, Produção de VEGF (pg/ml) por Caco-2 após tratamento com inibidor de JNK (SP600125), de p38 (SB202190) ou de ERK1/2 (U 0126) durante 30 minutos e ativação por estresse hipertônico. O símbolo + significa estatisticamente diferente das células não ativadas por estresse hipertônico (p < 0,05) e o * significa estatisticamente diferente das células ativadas por estresse hipertônico e não tratadas (p < 0,05). B, Produção de VEGF (pg/ml) por Caco-2 após tratamento com NS 398 e/ou SP600125, SB202190 ou U0126 durante 30 minutos e ativação por estresse hipertônico. O símbolo + significa estatisticamente diferente das células não ativadas por estresse hipertônico (p < 0,05) e o * significa estatisticamente diferente das células ativadas por estresse hipertônico e tratadas com NS-398 (p < 0,05). As barras dos gráficos A e B representam a média e o erro padrão da média de amostras realizadas em triplicata. O gráfico A representa três experimentos independentes e o gráfico B representa dois experimentos independentes.
0
500
1000
1500
2000
2500
Estresse Hipertônico - + + + +
+
- -SP 600125 (20µM) --
- -SB 202190 (15µM) - -
- -U 0126 (20µM) - -
++
+
*
*
A
VEG
F (p
g/m
l)
0250500750
100012501500175020002250
Estresse Hipertônico - + + + +
+
- -SP 600125 (20µM) --
- -SB 202190 (15µM) - -
- -U 0126 (20µM) - -
++
- +NS 398 (1µM) + + +
+
*
*
BVE
GF
(pg/
ml)

87
5. DISCUSSÃO
O papel das PLA2s na ativação por estresse microambiental em câncer de cólon foi
avaliado utilizando-se dois modelos de ativação celular in vitro. O primeiro modelo consiste na
estimulação das células em meio hipertônico por adição de 100mM de NaCl, que mimetiza o
estresse hipertônico observado no ambiente fisiológico intestinal, com absorção de água (Powell
DW, 1995; Thiagarajah JR et al., 2001). A adição de 100mM de NaCl ao meio de cultura resulta
em uma osmolaridade de aproximadamente 540mOsm, o que está de acordo com as referências
encontradas na literatura sobre a osmolaridade final no lúmen do intestino grosso (Yang T et al.,
2000; Thiagarajah JR et al., 2001; Hubert A et al., 2004; Yang T et al., 2005). A osmolaridade
final do meio sem a adição de NaCl é de aproximadamente 367 mOsm, caracterizando um
microambiente isotônico, como referenciado pela literatura (Yang T et al., 2000; Hubert A et al.,
2004). No segundo modelo, as células são cultivadas em meio contendo 1mM de CoCl2, que
permite a estabilização de HIF-1α ativando a transcrição de VEGF, simulando o que ocorre na
hipóxia tecidual (Harris AL, 2002; Salnikow K et al., 2004). O CoCl2 age diminuindo os níveis
de ascorbato intracelular, que é co-fator para a enzima prolilhidroxilase, impedindo sua atividade
(Salnikow K et al., 2004). Esta enzima, ativa, modifica a proteína HIF-1α, permitindo sua ligação
com o complexo de von Hippel-Lindau (VHL) e seu direcionamento para a degradação via
proteossoma (Harris AL, 2002). Então, com estes dois modelos de ativação, as condições
experimentais estavam próximas ao ambiente fisiopatológico intestinal durante a progressão
tumoral, onde as células neoplásicas que se encontram mais próximas ao centro da massa tumoral
estão em um ambiente com baixa tensão de oxigênio e as células que se encontram mais
próximas à periferia do tumor estão em um ambiente hipertônico. De acordo com o objetivo geral

88
deste trabalho, as duas linhagens celulares escolhidas para os estudos in vitro foram a HCT116 e
a Caco-2. A linhagem Caco-2 possui a capacidade de produzir PGE2 e expressa a enzima COX-2,
constitutivamente, o que é aumentado após estimulação (Figura 3C). Logo, a linhagem Caco-2
possui a maquinaria enzimática necessária para a produção de prostanóides, contudo, não se
conhece qual a PLA2 envolvida na produção do eicosanóide. Para comparação utilizamos a
linhagem HCT116, que não expressa COX-2, mesmo após estimulação, portanto, não sendo
capaz de produzir PGE2, servindo como controle da participação de PGE2 nos eventos estudados.
Na primeira parte do estudo observou-se que o estresse hipertônico e o CoCl2 induzem a
expressão de COX-2 (Figuras 3C e 14B) e a produção de PGE2 (Figuras 3B e 14A) em Caco-2
depois de 24 horas de ativação. A expressão de COX-2 e a produção de PGE2 máximas se davam
pela adição de 0,1M de NaCl e 1mM de CoCl2 ao meio. Outros trabalhos na literatura
demonstraram a indução da expressão de COX-2 e produção de prostaciclina (PGI2) em
linhagens celulares ativadas por estresse hipertônico, como em células endoteliais (Arbabi S et
al., 2000), células epiteliais de pulmão (Lim WC et al., 2005), células ductais da medula renal
(Yang T et al., 2005) e em células de câncer de cólon (Arbabi S et al., 2001). Este último
trabalho demonstra a indução do aumento da expressão de COX-2 por Caco-2, com 6 a 8 horas
de ativação após a adição de 100mM de NaCl ao meio de cultura (Arbabi S et al., 2001), o que
reforça nosso resultado, em que observa-se a máxima expressão de COX-2 depois de 24 horas de
estímulo com estresse hipertônico (Figura 3C). Na hipóxia, mimetizada pelo uso do CoCl2,
também há a expressão de COX-2 em linhagens de câncer de próstata (Liu XH et al., 1999), e
produção aumentada de PGE2, um dos principais prostanóides oriundos da atividade de COX-2,
em células sinoviais (Ferguson GM et al., 1988) e em células de carcinoma oral (Schmalz G et
al., 2000). Porém em um trabalho, onde se verificou a relação entre a expressão de COX-2 e a

89
estabilização de HIF-1α em condições de hipóxia, duas linhagens de câncer de cólon, HCT116 e
HT29, foram tratadas com o CoCl2 ou com um quelante de ferro, a desferrioxamina (DFX). Os
resultados mostraram que o CoCl2 induziu o acúmulo da proteína HIF-1α, mas não o aumento da
expressão da proteína COX-2, ao contrário da DFX, que induziu tanto o aumento da expressão de
HIF-1α como de COX-2. A diferença nos resultados é explicada pelos diferentes modos de ação
dos agentes indutores de hipóxia, sugerindo que a quelação do ferro é necessária para a indução
do aumento da expressão de COX-2 (Woo KJ et al., 2006). Nossos resultados corroboram, em
parte, com estes últimos, pois revelam que na linhagem HCT116, o CoCl2 não induziu a
expressão de COX-2 ou a produção de PGE2 (dados não mostrados), e na linhagem Caco-2 há
aumento relativamente pequeno da expressão de COX-2 (Figura 14B) com a adição do CoCl2 à
cultura, talvez pelo fato de, nestas condições, não haver um agente sequestrador de ferro, como a
DFX.
Com o intuito de avaliar quais as PLA2s envolvidas na produção de PGE2 por Caco-2
ativadas com estresse hipertônico, foram usados vários inibidores farmacológicos para cada
enzima, que poderia estar envolvida neste processo. A produção de PGE2 é dependente da
atividade de COX-2 em Caco-2 estimuladas com estresse hipertônico, pois o uso de NS398, um
inibidor específico de COX-2, diminuiu a produção de PGE2 (Figura 4A). O inibidor de PLA2
citosólica (cPLA2), o ATK, inibiu a produção de PGE2 (Figuras 4B), enquanto o uso de
inibidores de PLA2 secretórias (sPLA2), o LY311727, e de PLA2 independente de cálcio (iPLA2),
o BEL, não modificaram esta produção (Figuras 4C e 4D). Logo, constatou-se que a produção de
PGE2 por Caco-2 ativadas com estresse hipertônico é dependente de COX-2 e cPLA2. O uso de
inibidores como o ATK traz algumas desvantagens como a inespecificidade da droga, pois além
de inibir a cPLA2, também inibe a iPLA2 e as ciclooxigenases (Riendeau D et al., 1994; Balsinde

90
J et al., 1999a). Porém, tais desvantagens são contornadas com o uso de inibidores específicos
para COX-2 (NS398) e para iPLA2 (BEL). Outro tipo de desvantagem apresentada pelo inibidor
LY311727 é sua falta de acesso ao interior da célula, por ser impermeável à membrana
citoplasmática (Mounier CM et al., 2004). Então, o resultado que mostra a não participação de
sPLA2 na produção de PGE2 no estresse hipertônico deve ser analisado levando-se em
consideração que tal conclusão é limitada à participação potencial de sPLA2 atuando pelo lado
externo da membrana plasmática não podendo se chegar a uma conclusão sobre sua participação
antes de serem secretadas. Também se utilizou outro inibidor para sPLA2, o 12-epi-escalaradial,
que também não inibiu a produção do prostanóide em nenhum dos dois tipos de estresse
microambiental (dados não mostrados). Porém, não se conhece muito a respeito da
permeabilidade deste inibidor à membrana celular.
A ação da cPLA2 em Caco-2 estimuladas por estresse hipertônico não é a de modular a
expressão de COX-2, como os resultados com o inibidor de cPLA2 (ATK) mostram (Figura 5A).
Tais resultados contrastam com os encontrados em mastócitos derivados de camundongos
deficientes no gene para a cPLA2-α que possuem redução de aproximadamente 70% na
capacidade de induzir COX-2 após estimulação com citocinas e fatores de crescimento
(Fujishima H et al., 1999). Também contrastam com os resultados na linhagem de osteoblasto
MC3T3-E1, onde a cPLA2 possui papel no aumento da expressão de COX-2 durante a produção
de PGE2 em resposta ao Fator de Necrose Tumoral α (TNFα) e à IL-1β (Murakami M et al.,
1997). Em relação ao câncer de cólon, há um estudo mostrando que cerca de 50% dos tumores
de cólon humano (de um total de 65 tumores analisados) apresentavam expressão aumentada de
cPLA2 e COX-2, além da expressão destas duas enzimas em várias linhagens de câncer de cólon.

91
Contudo, não havia nenhum experimento demonstrando a regulação de COX-2 pela cPLA2 nestas
linhagens (Panel V et al., 2006).
Outro passo do metabolismo do AA, além da expressão de COX-2, onde a cPLA2 poderia
estar agindo é na liberação do AA. O estresse hipertônico induz máxima liberação de AA com 24
horas de ativação (Figura 3A) e quando a atividade de cPLA2 foi bloqueada, pelo uso do inibidor
ATK, a liberação de AA foi reduzida em praticamente 90%, indicando que a ação da cPLA2 é
regular o fornecimento de AA para a produção de PGE2 (Figura 5B). Embora não haja estudos
em relação ao papel de cPLA2 em células neoplásicas ativadas por estresse hipertônico, há um
trabalho com células de colangiocarcinoma humano estimuladas com Fator de Crescimento para
Hepatócito (HGF) e IL-6, cujos resultados mostram que a liberação de AA e a produção de PGE2
são mediadas por cPLA2, mas os pesquisadores não verificaram se a expressão de COX-2 era
inibida com o uso do inibidor de cPLA2 (MAFP) (Wu T et al., 2002). Porém, o papel de cPLA2 e
do AA em câncer tem sido amplamente estudado em modelos in vivo, como nos camundongos
knockout para cPLA2 (Ilsley JNM et al., 2005), e em modelos in vitro, com várias linhagens de
câncer (Soydan AS et al., 1996; Lagorce-Pages C et al., 2004). Tais modelos revelam um papel
ambíguo para a cPLA2. A cPLA2 seria a enzima responsável pela liberação do AA, resultando na
produção de PGE2, o que resultaria em aumento da proliferação de células neoplásicas (Oshima
M et al., 1996; Sheng H et al., 2001; Castellone MD et al., 2005). Porém, em alguns modelos
teria o papel de inibir o desenvolvimento tumoral, pois a liberação de AA seria um sinal pró-
apoptótico (Cao Y et al., 2000; Taketo MM et al., 2002; Levine LAWR, 2003; Ilsley JNM et al.,
2005) para as células neoplásicas. Neste estudo, avaliou-se se a PGE2 produzida por Caco-2 no
estresse hipertônico poderia estar modulando a proliferação celular. Então, as células foram
tratadas com o inibidor de COX-2, NS-398, para o bloqueio da produção do prostanóide, ativadas
por estresse hipertônico durante 24 horas e avaliadas quanto à proliferação. Não houve diferenças

92
entre as células tratadas e não tratadas com o inibidor, sugerindo que a PGE2 não induz a
proliferação das células de carcinoma de cólon frente o estresse hipertônico (dados não
mostrados).
Como mencionado anteriormente, um dos mecanismos pelos quais as células respondem
ao estresse microambiental é modificando as cascatas das MAP quinases, que são proteínas
quinases que compõem importantes vias intracelulares de transdução de sinal. Em relação a estas
vias de sinalização, observou-se que as quinases ERK1/2, JNK e p38 participavam da produção
de PGE2 em Caco-2 ativada por estresse hipertônico (Figura 6) e que o estresse hipertônico
possuía ação modulatória sobre a expressão das MAP quinases, com a indução do aumento da
expressão de todas as três quinases com meia hora e duas horas de ativação (Figura 7). Tais
resultados confirmam os resultados encontrados na literatura, que indicam que há envolvimento
de MAPKs no estresse hipertônico (Han J et al., 1994; Arbabi S et al., 2000; Yang T et al., 2000;
Munnik T e Meijer HJG, 2001; Zatechka SDJr e Lou MF, 2002; Yang T et al., 2005). Os
resultados indicaram que as três quinases modulam a atividade de cPLA2 na liberação de AA
(Figura 9), fato que é, em parte, confirmado em células MDCK estimuladas com ATP e
ionomicina e tratadas com um inibidor de ERK1/2 (U0126) com a diminuição da liberação de
AA, independentemente da fosforilação e translocação de cPLA2 nestas células (Evans JH et al.,
2002).
A atividade de cPLA2 depende dos níveis de cálcio intracelular e da ação das MAPKs. O
cálcio tem a função de promover a ligação da enzima com a membrana, que é mediada por um
domínio de ligação do fosfolipídeo com o íon (Channon JY e Leslie CC, 1990; Clark JD et al.,
1991; Nalefski EA et al., 1994). O tratamento de células com agonistas que mobilizam cálcio
induz a ligação de cPLA2 com a membrana nuclear e do retículo endoplasmático (Glover S et al.,
1995; Schievella AR et al., 1995; Sierra-Honigmann MR et al., 1996). E a estimulação de uma

93
variedade de tipos celulares com diversos agonistas que induzem a liberação de AA também
promove a fosforilação de serina na cPLA2, acompanhada por aumento na atividade enzimática
(Lin LL et al., 1992; Qiu ZH et al., 1993; Clark JD et al., 1995). Também foi demonstrado que a
fosforilação da Ser505 por ERK 1/2 é necessária para a liberação de AA por cPLA2 em células de
ovário de hamster chinês (CHO) tratadas com diversos agonistas, indicando função para as MAP
quinases na ativação de cPLA2 (Nemenoff RA et al., 1993). Porém, a importância da fosforilação
de cPLA2 pelas MAPKs para a liberação de AA é questionável. Em sistemas onde não há
aumento de cálcio intracelular, como no caso da indução por ácido ocadaico, a fosforilação da
fosfolipase parece ser mais importante para a liberação de AA do que em situações com aumento
de cálcio intracelular (Ambs P et al., 1995; Kramer RM et al., 1996; de Carvalho MG et al.,
1996). O que parece estar acontecendo no estresse hipertônico é que as MAPKs possuem função
na indução da liberação de AA pela cPLA2 por mecanismo independente da fosforilação da
enzima, já que não foram vistas diferenças na fosforilação de cPLA2 em diferentes tempos de
estimulação com estresse hipertônico. E o tratamento de Caco-2, estimuladas com meio
hipertônico por 8 horas, com os inibidores de quinases não modificou a fosforilação de cPLA2
(dados não mostrados). A cPLA2 é fosforilada sobre a Ser505 in vitro por ERK 1/2 (Nemenoff RA
et al., 1993). Porém, a inibição da via MEK/ERK reduz a liberação de AA independentemente da
fosforilação e translocação de cPLA2 em células MDCK, indicando que as quinases podem
modular a liberação de AA, atuando em outros pontos da via de ativação celular diferentes da
fosforilação da Ser505 e translocação da cPLA2 (Evans JH et al., 2002), o que reforça os
resultados demonstrando o papel das MAPKs na indução da liberação de AA por cPLA2 no
estresse hipertônico. É possível que as MAPKs alterem as propriedades da membrana celular
e/ou a conformação da cPLA2 favorecendo a liberação de AA.

94
Ainda, em relação ao papel de MAPKs no estresse hipertônico, os resultados indicaram
que ERK1/2 está envolvida na indução de COX-2 enquanto que p38 e SAPK/JNK possuem ação
inibitória sobre a expressão desta enzima (Figura 8), diferentemente das células coletoras ductais
de medula renal (IMCD) cultivadas em meio hipertônico, cuja expressão de COX-2 é dependente
de todos os três membros da família MAPK (Yang T et al., 2000; Yang T et al., 2005) e de
monócitos humanos ativados com LPS, onde a p38 possui função na transcrição e estabilização
do mRNA de COX-2 (Dean JL et al., 1999). Além disso, duas publicações do mesmo grupo de
pesquisadores, uma em células endoteliais (Arbabi S et al., 2000) e outra em células de câncer de
cólon (Arbabi S et al., 2001), mostram que o estresse hipertônico induz a expressão de COX-2,
que é dependente somente de p38, o que difere dos resultados do presente estudo. No entanto,
outras publicações demonstram que existe um cross-talk entre os membros da família de
proteínas MAPKs (Berra E et al., 1998; Chan ED e Riches DWH, 2001; Seo M et al., 2002; Lee J
et al., 2002; Ohashi R et al., 2004; House SL et al., 2005), o que evidencia a possibilidade de
algum tipo de regulação entre as MAPKs envolvidas na modulação da expressão de COX-2 no
estresse hipertônico. Há evidências de inibição de ERK por p38, como em modelo de doença
renal crônica em ratos (Ohashi R et al., 2004). A MAPK p38 está envolvida em processos
inflamatórios, e sua ativação é necessária para a regulação da ativação transcripcional de genes de
citocinas incluindo IL-1β e TNF-α (Herlaar E e Brown Z, 1999). Os pesquisadores
demonstraram que, apesar do tratamento com inibidor farmacológico para p38 dos ratos com
ablação parcial dos rins, os animais não apresentavam diminuição do processo inflamatório renal,
com maior proliferação de células tubulares, miofibroblastos e macrófagos. O resultado
inesperado foi devido à ativação de ERK1/2, MAPK associada com a sobrevivência e
proliferação celulares (Ohashi R et al., 2004). Outro estudo em animais transgênicos para fator de

95
crescimento de fibroblasto-2 (FGF-2) demonstrou o papel cardioprotetor do fator durante a
injúria por isquemia-reperfusão, por meio da ativação de ERK1/2 e inibição de p38, levando à
proteção da disfunção contrátil pós-isquêmica e do infarto do miocárdio (House SL et al., 2005).
Além destes modelos in vivo, um estudo com a linhagem HeLa evidenciou a função pró-
apoptótica de p38, pois a inibição da atividade basal de ERK1/2 foi suficiente para induzir a
ativação de p38 e a apoptose das células (Berra E et al., 1998). Já em mioblastos, a ativação de
p38 induz a parada do ciclo celular pela inibição de ERK1/2, possibilitando a diferenciação
muscular (Lee J et al., 2002). Tomando estas evidências como base, pode-se sugerir que ERK1/2
tenha função inibitória sobre JNK e p38 na expressão de COX-2 no estresse hipertônico. Já que o
tratamento de Caco-2 com o inibidor de ERK1/2 diminui a expressão de COX-2 e o tratamento
com os inibidores de JNK e p38 aumentam a expressão da enzima (Figura 8), porém, o estresse
hipertônico induz a expressão de COX-2, evidenciando que a função estimulatória de ERK1/2
sobressai em relação às funções das outras MPAKs (Figura 3C).
A PGE2 tem, entre outras funções, modular a angiogênese e a produção de VEGF (Tsujii
M et al., 1998; Liu XH et al., 2002a; Fukuda R et al., 2003; Chang SH et al., 2004), analisamos,
então, o papel da PGE2 na regulação da produção de VEGF por Caco-2 durante a estimulação
com o meio hipertônico. E observamos que o estresse hipertônico aumenta a produção de VEGF
por Caco-2 (Figura 10). A inibição da produção de PGE2 com os inibidores seletivos para COX-2
(NS398) e cPLA2 (ATK) aumentaram a produção de VEGF (Figura 11), podendo este fenômeno
ser revertido pela adição de PGE2 ao meio (Figura 13). Nossos resultados contrastam com os
resultados de Tsujii e colaboradores (1998), que demonstraram que a transfecção de Caco-2 com
um vetor de superexpressão de COX-2 resultou no aumento da produção basal de VEGF e esta
produção foi inibida com NS398, na concentração de 10 µM (Tsujii M et al., 1998). Esta

96
diferença pode refletir a forma utilizada para se conseguir o aumento de expressão de COX-2,
estimulação fisiológica ou expressão forçada, com consequente modificação do papel de PGE2.
A PGE2 se liga a quatro subtipos de receptor EP, denominados EP1-EP4, que são produtos
de genes separados. Estes receptores possuem funções fisiológicas no trato gastrintestinal, como
a de estimular a motilidade das células musculares da parede intestinal e a secreção de cloreto e
bicarbonato das células epiteliais, além de estarem envolvidos na carcinogênense do cólon. Os
modelos de animais knockout vêm contribuindo para a melhor compreensão da farmacologia e
fisiologia destes receptores (Hull MA et al., 2004). Todos os quatro subtipos de receptores EP
estão envolvidos na carcinogênese de cólon (Watanabe K et al., 1999; Eibl G et al., 2003; Amano
H et al., 2003; Spinella F et al., 2004; Bradbury D et al., 2005; Chang SH et al., 2005). Em estudo
onde os animais eram tratados com carcinógeno, azoximetano, os camundongos knockout para
EP1 apresentavam diminuição de 60% no número de focos de cripta aberrante (ACF) em relação
aos animais selvagens. Neste mesmo estudo, camundongos Min foram tratados com antagonista
de receptor EP1 e apresentaram redução do número de pólipos em 57% (Watanabe K et al.,
1999). Ainda dentro dos modelos in vivo, há publicações comprovando as ações dos receptores
EP2 e EP3 em modelo de angiogênese tumoral (Amano H et al., 2003; Kamiyama M et al., 2006).
Os pesquisadores inocularam Sarcoma-180 em camundongos knockout para EP3 e o crescimento
tumoral foi reduzido em relação aos animais selvagens. O crescimento tumoral e a angiogênese
em animais selvagens foram inibidos pelo tratamento com antagonista de receptor EP3. Além
disto, as marcações imunohistoquímicas revelaram marcação de VEGF menos intensa em células
estromais nos animais EP3-/-, evidenciando um mecanismo para o envolvimento destas células do
hospedeiro na produção do fator angiogênico (Amano H et al., 2003). Já em animais knockout
para o receptor EP2, a PGE2 atuaria estimulando a motilidade e a resistência à apoptose das

97
células endoteliais, por meio da sinalização via EP2, contribuindo para a angiogênese tumoral
(Kamiyama M et al., 2006).
Para verificar qual o receptor de PGE2 envolvido na redução da produção de VEGF no
estresse hipertônico que poderia explicar a alça regulatória existente neste tipo de estresse
microambiental, as Caco-2 foram ativadas e tratadas com vários agonistas de receptores EP e os
resultados indicaram que o receptor envolvido na regulação por PGE2 sobre a diminuição da
produção de VEGF neste tipo de estímulo é o EP2 (Figura 14). Tais resultados contrastam com os
encontrados na literatura que mostram que a produção de VEGF estimulada por PGE2 é
dependente de EP2 em células de câncer pancreático (Eibl G et al., 2003), porém, os
pesquisadores utilizaram como antagonista de receptores EP1 e EP2, o AH 6809, que, na verdade,
é também antagonista para receptores DP (Keery RJ e Lumley P, 1988; Woodward DF et al.,
1995; Abramovitz M et al., 2000) e um antagonista de receptor EP1, o SC-19220, ao invés da
utilização de antagonista com maior especificidade para receptor EP2 para elucidar a função deste
receptor (Eibl G et al., 2003). Outro estudo em células de carcinoma ovariano demonstrou que a
endotelina-1 induzia o aumento na produção de VEGF dependente de PGE2, via receptores EP2 e
EP4 (Spinella F et al., 2004), diferindo dos resultados do presente estudo. Entretanto, as
concentrações de 16,16-dimetil PGE2 e Butaproste utilizadas nestes experimentos foram bem
maiores (20µM) do que as usadas neste estudo (0,1µM) (Figura 14), podendo ocorrer saturação
de receptores EP (Spinella F et al., 2004). Outra publicação mostra resultados similares a estes
em células de musculatura lisa das vias aéreas transfectadas com vetores contendo o promotor de
VEGF, onde PGE2 estimulou a transcrição e a produção do fator angiogênico, com a participação
do fator de transcrição SP-1 por mecanismo dependente de cAMP, envolvendo receptores EP2 e
EP4 (Bradbury D et al., 2005). Finalmente, mais um trabalho em células de câncer de mama com

98
superexpressão de EP2, mostrou que havia indução da produção de VEGF em resposta a agonista
EP2 e PGE2 (Chang SH et al., 2005). Porém, a abordagem metodológica utilizada nestas duas
publicações, a expressão forçada por transfecção vetorial, difere da utilizada neste estudo. Ainda
em relação à ação conjunta dos receptores EP2 e EP4, um estudo em células de Kupffer de
camundongos knockout para os receptores EP2 e EP4 demonstrou que a inibição da produção de
TNF-α por estas células estimuladas com LPS se dava pela ação de PGE2 via EP2 e EP4
(Fennekohl A et al., 2002), evidenciando que a ação concomitante destes receptores pode
também ser inibitória.
Como mencionado na seção 4.12, não se pode desconsiderar a função do receptor EP3 na
inibição da produção de VEGF por PGE2 no estresse hipertônico. Apesar de ter sido mínima a
inibição da produção de VEGF com o uso do inibidor específico para EP3 (Sulprostona),
caracterizando um efeito borderline para o receptor, há a possibilidade do efeito conjunto de EP2
e EP3 na inibição da produção do fator angiogênico no estresse hipertônico. Já foram
identificadas, em humanos, pelo menos oito variantes oriundas de splicing alternativo para o
receptor EP3. Há algumas diferenças entre estas variantes à ativação de vias de transdução de
sinal, atividade constitutiva e tráfico intracelular. Inicialmente foi descrito que o receptor EP3 se
acopla à proteína G inibitória levando à inibição de cAMP, porém, posteriormente, foi
demonstrado que algumas variantes, como a EP3γ, se acoplariam à proteína G estimulatória
resultando no aumento de cAMP (Hata AN e Breyer RM, 2004). Reforçando o efeito inibitório
da ação conjunta de EP2 e EP3 visto na produção de VEGF no estresse hipertônico, há evidências
de inibição da produção da quimiocina CCL27 por queratinócitos via estes dois receptores. O
mecanismo pelo qual a PGE2 inibiu a produção de CCL27 nos queratinócitos foi por inibição da
atividade de NF-κB (Kanda N et al., 2004).

99
Os receptores EP2 são receptores acoplados à proteína G estimulatória, que, por sua vez,
estimula a adenilil ciclase e, conseqüentemente, o aumento nos níveis intracelulares de AMP
cíclico (cAMP), cuja principal função é ativar a proteína quinase dependente de AMP cíclico
(PKA). A PKA fosforila resíduos de serina e treonina em proteínas alvo, que variam, de acordo
com o tipo celular. A ativação do receptor EP2 está implicada na via de sinalização β-
catenina/Tcf, importante no desenvolvimento do câncer de cólon. A estimulação de EP2 leva à
estabilização da β-catenina pela inibição da atividade quinase de GSK-3, de maneira dependente
de PKA, impedindo a degradação da β-catenina (Hull MA et al., 2004; Hata AN et al., 2004). A
relação entre a transcrição de VEGF e a sinalização por receptor EP2 se daria pela ação de PKA,
que fosforila a proteína ligante do elemento de resposta ao AMP cíclico (CREB). A CREB pode
interagir com HIF-1α no núcleo ativando a transcrição de genes como o VEGF (Harris AL,
2002). Porém, não é o que ocorre no presente estudo, onde, provavelmente, a transcrição de
VEGF não é ativada pela sinalização via EP2. Contudo, há vários estudos na literatura
comprovando o papel inibitório do cAMP por meio da ativação de PKA (den Dekker E et al.,
2002; Li R et al., 2004; Flamand N et al., 2004; Aronoff DM et al., 2005; Liu X et al., 2006). O
cAMP ativa a PKA, que fosforila e inativa diversos elementos sinalizadores nas vias de ativação
plaquetária em resposta ao cálcio, tais como o receptor tipo α para tromboxana A2 (TxA2), a
fosfolipase Cβ3 (PLC-β3) e receptores IP3 (Cavallini L et al., 1996; Yue C et al., 1998;
Wojcikiewicz RJ e Luo SG, 1998; Walsh MT et al., 2000). Outros efeitos inibitórios do cAMP,
por via independente de PKA, são vistos na inibição da proliferação de células epiteliais de retina
(Hecquet C et al., 2002), na indução da apoptose de células musculares lisas (Li R et al., 2004) e
na inibição da síntese de colágeno em fibroblastos cardíacos (Liu X et al., 2006), todos efeitos
mediados pela inibição da atividade ERK. Ainda, cAMP inibe a fagocitose em macrófagos via

100
proteína de troca diretamente ativada por cAMP-1 (Epac-1) (Aronoff DM et al., 2005), a
reorganização do citoesqueleto de actina e a motilidade em células CHO (Nagasawa SY et al.,
2005) e a biosíntese de leucotrieno B4 (LTB4) por neutrófilos (Flamand N et al., 2004). Todos
estes resultados reforçam a ação inibitória de EP2, que pode ser devida à ação de cAMP, sobre a
produção de VEGF no estresse hipertônico.
Para observar se o fenômeno de regulação da produção de VEGF por PGE2 se repetia no
estresse por CoCl2, a produção de PGE2 e a expressão de COX-2 foram avaliadas. O meio
contendo 1 mM de CoCl2 foi capaz de induzir a máxima produção do prostanóide após 24 horas
de ativação (Figura 15A) e de induzir a expressão de COX-2 após este período (Figura 15B).
Ainda, constatou-se que a produção de PGE2 é dependente de cPLA2 e COX-2 (Figuras 16A e
16B) e que havia a estimulação da produção de VEGF frente a este estímulo (Figura 17A). Este
último resultado é confirmado pelo uso de CoCl2 mimetizando a hipóxia em fibroblastos
humanos (Poulios E et al., 2006), linhagem de câncer de pulmão (Litz J e Krystal GW, 2006),
epitélio da retina (Cai CM et al., 2006), linhagens de glioma (Newcomb EW et al., 2006), em
linhagens de câncer de próstata (Liu XH et al., 1999) e em astrócitos (Ijichi A et al., 1995),
aumentando a expressão tanto do RNA mensageiro quanto da proteína VEGF. Entretanto,
repetindo os mesmos tratamentos com os inibidores farmacológicos verificamos que a PGE2 não
possui papel regulatório sobre a produção de VEGF em Caco-2 ativadas por CoCl2 (Figuras 17B
e 17C). Nossos resultados mostram que houve maior produção do fator angiogênico nas Caco-2
estimuladas com CoCl2 (1mM) (cerca de 750 pg/ml) (Figura 17A) em comparação ao estímulo
com meio hipertônico (cerca de 500 pg/ml) (Figura 10). Ainda, não houve aumento da produção
do fator angiogênico quando do tratamento com NS-398 nas Caco-2 estimuladas com CoCl2
(Figura 17B), enquanto que, em relação ao estímulo de estresse hipertônico o tratamento com
NS-398 resultou em aumento da produção de VEGF em Caco-2 (Figura 11B). Tais dados podem

101
ser explicados pela presença de diferentes vias de sinalização nos dois estímulos, o estresse
hipertônico e o CoCl2. Entretanto, não podemos descartar a possibilidade de que a concentração
de CoCl2 poderia estar muito elevada saturando a capacidade de produção de VEGF das células
Caco-2, não possibilitando o aumento da sua produção frente ao tratamento com NS398. Logo,
repetimos o mesmo experimento com uma concentração de CoCl2 menor (0,3 mM) mas não
encontramos diferenças entre os resultados obtidos com o uso dos inibidores farmacológicos
(dados não mostrados), demonstrando que o feedback negativo de PGE2 sobre a produção de
VEGF pela Caco-2 do estresse hipertônico não está presente na estimulação por CoCl2. Os
resultados dos experimentos com estresse por CoCl2 indicam que PGE2 não modula a produção
de VEGF por Caco-2, apesar da tendência à diminuição da produção de VEGF quando as células
são tratadas com NS-398, o que contrasta com a maior parte dos trabalhos, que sugerem que a
PGE2 funcionaria como um fator pró-angiogênico. Estes trabalhos apontam a PGE2 como um
mediador químico que estabilizaria HIF-1α e, em conseqüência, aumentaria a produção de
VEGF. Porém, tais estudos somente demonstram a indução da estabilização de HIF-1α ou o
aumento no RNA mensageiro de VEGF e não na sua produção, com o aumento da proteína
secretada (Pai R et al., 2001; Liu XH et al., 2002a; Fukuda R et al., 2003). Com exceção de um
estudo, que mostra o aumento na produção de VEGF, via EP2, por células de câncer pancreático
humano quando estimuladas com PGE2 em concentrações iguais às do presente estudo (0,01-
1µM), sem nenhum outro tipo de estímulo (Eibl G et al., 2003). Foi verificada a ação da PGE2
exógena na produção de VEGF por Caco-2 em condições basais, ou seja, sem a estimulação por
estresse hipertônico, e não houve alteração da produção de VEGF nas células de câncer de cólon
(dados não mostrados), o que difere dos resultados com a linhagem de câncer pancreático (Eibl G
et al., 2003). Tal diferença podeser devido ao tipo celular, pois a linhagem Caco-2 não estimulada

102
produz menos quantidade de fator angiogênico (cerca de 100pg/mL/24 horas) em relação à
linhagem de câncer pancreático (cerca de 800pg/106células/24 horas).
A ação das MAPKs na sinalização para a produção de VEGF por Caco-2 ativadas com os
dois tipos de estresse microambiental também foi verificada. Observou-se que, no estresse por
CoCl2 as três MAP quinases possuíam uma ação indutora da produção de VEGF (Figura 18),
enquanto que no estresse hipertônico, as quinases envolvidas na produção do fator angiogênico
eram a p38 e a ERK (Figura 19B). Não há relatos na literatura relacionando estresse hipertônico,
MAP quinases e produção de VEGF em sistemas celulares. No entanto, há estudos demonstrando
a ação de p38 e JNK na produção de VEGF em várias linhagens de células de glioma maligno
humano sob condições de normóxia estimuladas com IL-1β (Yoshino Y et al., 2006) e a ação
estimulatória de JNK na produção do fator angiogênico em linfoma cutâneo de célula T
(Krejsgaard T et al., 2006). Em relação à condição de hipóxia, um estudo utilizando condrócitos
articulares humanos estimulados sob tais condições ou estimulados com IL-1, demonstrou
diferentes níveis de regulação por parte das MAPKs sobre a produção do VEGF. A secreção de
VEGF induzida por hipóxia era dependente de p38, enquanto que a produção induzida por IL-1
era dependente de JNK (Murata M et al., 2006). Em células endoteliais submetidas à hipóxia, a
MAPK envolvida na produção de VEGF é a p38 (Fan B et al., 2005), já em células de carcinoma
hepatocelular (HCC) cultivadas com meio contendo CoCl2, a MAPK envolvida no aumento da
transcrição do VEGF é a ERK1/2 (Huang GW et al., 2004). Uma revisão publicada em 2000
aborda o papel das MAP quinases no controle da expressão de VEGF na hipóxia. Os autores
fazem uma distinção dos mecanismos de regulação entre as quinases, afirmando que ERK1/2
fosforila diretamente HIF-1α estabilizado, aumentando a dependência do fator de transcrição na
ativação da transcrição de VEGF. Ao contrário de p38 e JNK, que seriam MAPKs ativadas sob

103
várias condições de estresse celular e que estariam contribuindo para o aumento da expressão do
fator angiogênico por estabilizar o seu mRNA (Berra E et al., 2000). Em suma, os resultados que
mostram o envolvimento de todas as três quinases na produção de VEGF por Caco-2 estimulada
por CoCl2, podem apontar para diferentes ações em relação ao aumento da produção de VEGF.
Enquanto que, os resultados obtidos com o estresse hipertônico, evidenciam p38 e ERK1/2
possuindo funções estimulatórias sobre a produção do fator angiogênico. Para elucidar quais as
diferenças entre os mecanismos regulatórios das diferentes MAPKs sobre a produção de VEGF,
futuros experimentos devem ser feitos evidenciando o papel das três quinases sobre a fosforilação
de HIF-1α e sobre a estabilização do mRNA de VEGF.
Tomando como base nossos resultados em Caco-2 e a literatura hipotetizamos dois
modelos de regulação da expressão de COX-2 e produção de VEGF por PLA2s em câncer de
cólon no estresse hipertônico e no estresse por CoCl2 (Figuras 20A e 20B). No estresse por
CoCl2, a produção de PGE2 é dependente de cPLA2 e COX-2 e o prostanóide não possui ação
modulatória sobre a produção de VEGF. Todas três sub-famílias de quinases possuem efeito
estimulatório tanto sobre a produção de PGE2 quanto sobre a produção de VEGF (Figura 20A).
Enquanto que no estresse hipertônico, a cPLA2 fornece ácido araquidônico (AA) como substrato
para a enzima COX-2 resultando na produção de PGE2, que regula negativamente a produção de
VEGF, via receptor EP2. Além desta, há outra alça regulatória abrangendo as MAPKs, onde
ERK1/2, p38 e SAPK/JNK teriam papel na ativação da cPLA2 e ERK1/2 induziria a expressão de
COX-2. A produção de VEGF, por sua vez, é estimulada por p38 e ERK1/2 (Figura 20B).
As respostas celulares são diferenciadas ao longo dos gradientes de tensão de oxigênio e
de acidez encontrados desde a região do tumor mais próxima aos vasos sanguíneos até a região
mais distante dos vasos, no centro da massa tumoral. Há uma complexa regulação da atividade de

104
enzimas que regulam os níveis citoplasmáticos de HIF-1α, como a prolil hidroxilase, e,
consequentemente, há a transcrição de grupos diferentes de genes em diferentes áreas do tumor
(Pouyssegur J et al., 2006). Ainda, um estudo recente revelou que o tumor pode sofrer
modificações morfológicas e fenotípicas decorrentes da pressão seletiva do microambiente. Por
meio de modelo matemático, capaz de considerar múltiplos fatores envolvidos no
desenvolvimento tumoral, os pesquisadores demonstraram que condições adversas, como hipóxia
e heterogenicidade de matriz extracelular, exercem força seletiva sobre o tumor, que cresce como
uma massa formada, predominantemente, por clones de aspecto agressivo e invasivo. Enquanto
que condições menos adversas do microambiente, como normóxia e homogenicidade de matriz
extracelular, permitem que clones de aspecto agressivo co-existam com clones de aspecto menos
agressivo resultando no desenvolvimento de massa tumoral heterogênea e não invasiva
(Anderson ARA et al., 2006). Nossos resultados sugerem que as células de câncer de cólon
respondem de forma diferenciada ao estresse microambiental, pois indicam que, no estresse
hipertônico, a PGE2 inibe a produção de VEGF pelas células neoplásicas, o que não ocorre em
ambiente que mimetiza as condições de hipóxia, caracterizando diferenças na ativação celular
dependendo do tipo de estresse microambiental. Fazendo uma analogia ao que pode estar
acontecendo em um tumor sólido localizado no cólon, as células neoplásicas em contato com o
ambiente hipertônico do lúmen intestinal produziriam PGE2 que, por sua vez, diminuiria a
produção do fator angiogênico VEGF, o que não ocorreria com as células localizadas no centro
da massa tumoral, em condições de hipóxia. Provavelmente, a diferença nas respostas aos
estímulos distintos seria uma forma de adaptação celular às condições externas. As células de
câncer de cólon em um ambiente hipertônico podem estar secretando menos fator angiogênico até
encontrarem condições onde a produção do fator seja de suma importância para a continuação do

105
desenvolvimento tumoral, como na hipóxia. Ainda, a interpretação de nossos resultados deve
levar em consideração a influência da PGE2 sobre outros tipos celulares, além de sua ação sobre
as células neoplásicas. Os resultados na literatura com modelos in vivo demonstrando a
participação de receptores EP na angiogênese tumoral evidenciaram o importante papel de
células do estroma do hospedeiro neste processo (Amano H et al., 2003). Ainda, situações de
estresse microambiental, como, por exemplo, baixa tensão de oxigênio e acidez, são
fundamentais para a transformação celular e para o crescimento tumoral (Harris AL, 2002;
Bedogni B et al., 2005; Webster KA et al., 2005), evidenciando a importância do estudo dos
mecanismos de regulação do pH intracelular e do metabolismo para o desenvolvimento de novas
terapias anti-câncer. Logo, estes resultados geram perspectivas para a realização de novos
experimentos in vitro, utilizando co-culturas com células estromais, para clarificar a função de
PGE2 e VEGF sobre outros tipos celulares presentes em um tumor. E, principalmente,
desenvolver modelos in vivo, que propiciem melhor entendimento do comportamento da célula
neoplásica em diferentes regiões de um mesmo tumor.

106
Figura 20: Modelos de regulação da expressão de COX-2 e produção de PGE2 e VEGF por cPLA2 em células de câncer de cólon. A, No estresse por CoCl2, a produção de PGE2 é dependente de cPLA2 e COX-2 e o prostanóide não possui ação modulatória sobre a produção de VEGF. Todas três sub-famílias de MAP quinases possuem efeito estimulatório tanto sobre a produção de PGE2 quanto sobre a produção de VEGF. B, A cPLA2 fornece ácido araquidônico (AA) como substrato para a enzima COX-2 resultando na produção de PGE2, que regula negativamente a produção de VEGF. Além desta, há outra alça regulatória abrangendo as MAP quinases, onde ERK1/2, p38 e JNK teriam papel na ativação da cPLA2, ERK1/2 induziria a expressão de COX-2, enquanto p38 e JNK inibiriam a expressão da enzima. A produção de VEGF, por sua vez, é estimulada por p38.
p-38 ERK1/2
cPLA2
CÉLULA NEOPLÁSICA
COX-2AA PGE2
p-38 ERK1/2
JNK
ERK1/2
VEGF
ESTRESSE HIPERTÔNICO
p-38 JNK
Receptor EP2
p-38 ERK1/2
cPLA2
CÉLULA NEOPLÁSICA
COX-2AA PGE2
p-38 ERK1/2
JNK
ERK1/2
VEGF
ESTRESSE HIPERTÔNICO
p-38 JNK
cPLA2
CÉLULA NEOPLÁSICA
COX-2AA PGE2
p-38 ERK1/2
JNK
ERK1/2
VEGF
ESTRESSE HIPERTÔNICO
p-38 JNK
Receptor EP2
p38 ERK1/2
JNK
cPLA2
CÉLULA NEOPLÁSICA
COX-2AA
VEGF
CLORETO DE COBALTO
p38 ERK1/2
JNK
PGE2
p38 ERK1/2
JNK
cPLA2
CÉLULA NEOPLÁSICA
COX-2AA
VEGF
CLORETO DE COBALTO
p38 ERK1/2
JNK
p38 ERK1/2
JNK
cPLA2
CÉLULA NEOPLÁSICA
COX-2AA
VEGF
CLORETO DE COBALTO
p38 ERK1/2
JNK
PGE2
A
B

107
6. CONCLUSÕES
• O estresse hipertônico induz a liberação de AA, a expressão de COX-2 e a produção de
PGE2 em Caco-2.
• A produção de PGE2 é dependente da atividade de cPLA2 e de COX-2 em Caco-2
estimulada com estresse hipertônico.
• cPLA2 regula o fornecimento de AA em Caco-2 estimulada com estresse hipertônico.
• As quinases ERK1/2, JNK e p38 estão envolvidas na produção de PGE2 por Caco-2
estimuladas com estresse hipertônico.
• O estresse hipertônico modula a fosforilação de ERK1/2, JNK e p38.
• ERK1/2 induz o aumento da expressão de COX-2 em Caco-2 ativada por estresse
hipertônico.
• A produção de VEGF dependente do estresse hipertônico é reduzida pela produção
concomitante de PGE2.
• O receptor EP2 está envolvido na regulação da produção de VEGF por PGE2 em Caco-2
ativada por estresse hipertônico.
• A adição de CoCl2 ao meio de cultura induz a produção de PGE2 e a expressão de COX-
2 por Caco-2.
• A produção de PGE2 é dependente de COX-2 e cPLA2 em Caco-2 ativada por CoCl2.
• As quinases ERK1/2, JNK e p38 estão envolvidas na produção de VEGF por Caco-2
estimulada por CoCl2.
• ERK1/2 e p38 estão envolvidas na produção de VEGF por Caco-2 ativada por estresse
hipertônico.

108
7. REFERÊNCIAS BIBLIOGRÁFICAS 1. AARSMAN AJ; DE JONG JG; ARNOLDUSSEN E; NEYS FW; VAN WASSENAAR
PD; VAN DEN BOSCH H. Immunoaffinity purification, partial sequence, and subcellular localization of rat liver phospholipase A2. J Biol Chem, v. 264, n. 17, p. 10008-10014, 1989.
2. ABRAMOVITZ M; ADAM M; BOIE Y; CARRIERE MC; DENIS D; GODBOUT C; LAMONTAGNE S; ROCHETTE C; SAWYER N; TREMBLAY NM. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta, v. 1483, n. 2, p. 285-293, 2000.
3. ACKERMANN EJ; CONDE-FRIEBOES K; DENNIS EA. Inhibition of Macrophage Ca[IMAGE]-independent Phospholipase A2 by Bromoenol Lactone and Trifluoromethyl Ketones. J Biol Chem, v. 270, n. 1, p. 445-450, 1995.
4. ALBERTYN J; HOHMANN S; THEVELEIN JM; PRIOR BA. GPD1, which encodes glycerol-3-phosphate dehydrogenase, is essential for growth under osmotic stress in Saccharomyces cerevisiae, and its expression is regulated by the high-osmolarity glycerol response pathway. Mol Cell Biol, v. 14, n. 6, p. 4135-4144, 1994.
5. AMANO H; HAYASHI I; ENDO H; KITASATO H; YAMASHINA S; MARUYAMA T; KOBAYASHI M; SATOH K; NARITA M; SUGIMOTO Y; MURATA T; YOSHIMURA H; NARUMIYA S; MAJIMA M. Host Prostaglandin E2-EP3 Signaling Regulates Tumor-Associated Angiogenesis and Tumor Growth. J Exp Med, v. 197, n. 2, p. 221-232, 2003.
6. AMBS P; BACCARINI M; FITZKE E; DIETER P. Role of cytosolic phospholipase A2 in arachidonic acid release of rat-liver macrophages: regulation by Ca2+ and phosphorylation. Biochem J, v. 311, n. Pt 1, p. 189-195, 1995.
7. ANDERSON ARA; WEAVER AM; CUMMINGS PT; QUARANTA V. Tumor Morphology and Phenotypic Evolution Driven by Selective Pressure from the Microenvironment. Cell, v. 127, n. 5, p. 905-915, 2006.
8. ARBABI S; ROSENGART MR; GARCIA I; JELACIC S; MAIER RV. Epithelial cyclooxygenase-2 expression: a model for pathogenesis of colon cancer. J Surg Res, v. 97, n. 1, p. 60-64, 2001.

109
9. ARBABI S; ROSENGART MR; GARCIA I; MAIER RV. Hypertonic saline solution induces prostacyclin production by increasing cyclooxygenase-2 expression. Surgery, v. 128, n. 2, p. 198-205, 2000.
10. ARONOFF DM; CANETTI C; SEREZANI CH; LUO M; PETERS-GOLDEN M. Cutting Edge: Macrophage Inhibition by Cyclic AMP (cAMP): Differential Roles of Protein Kinase A and Exchange Protein Directly Activated by cAMP-1. J Immunol, v. 174, n. 2, p. 595-599, 2005.
11. ASAKAWA H; MIYAGAWA J; HANAFUSA T; KUWAJIMA M; MATSUZAWA Y. High glucose and hyperosmolarity increase secretion of interleukin-1 [beta] in cultured human aortic endothelial cells. J Diabetes Complicat, v. 11, n. 3, p. 176-179, 1997.
12. ATSUMI G; MURAKAMI M; TAJIMA M; SHIMBARA S; HARA N; KUDO I. The perturbed membrane of cells undergoing apoptosis is susceptible to type II secretory phospholipase A2 to liberate arachidonic acid. Biochim Biophys Acta, v. 1349, n. 1, p. 43-54, 1997.
13. BALBOA MA; BALSINDE J; WINSTEAD MV; TISCHFIELD JA; DENNIS EA. Novel group V phospholipase A2 involved in arachidonic acid mobilization in murine P388D1 macrophages. J Biol Chem, v. 271, n. 50, p. 32381-32384, 1996.
14. BALSINDE J; BALBOA MA; INSEL PA; DENNIS EA. Regulation and inhibition of phospholipase A2. Annu Rev Pharmacol Toxicol, v. 39, p. 175-189, 1999a.
15. BALSINDE J; BARBOUR SE; BIANCO ID; DENNIS EA. Arachidonic acid mobilization in P388D1 macrophages is controlled by two distinct Ca2+-dependent phospholipase A2 enzymes. Proc Natl Acad Sci U S A, v. 91, n. 23, p. 11060-11064, 1994.
16. BALSINDE J ; DENNIS EA. Distinct roles in signal transduction for each of the phospholipase A2 enzymes present in P388D1 macrophages. J Biol Chem, v. 271, n. 12, p. 6758-6765, 1996.
17. BALSINDE J; SHINOHARA H; LEFKOWITZ LJ; JOHNSON CA; BALBOA MA; DENNIS EA. Group V phospholipase A2-dependent induction of cyclooxygenase-2 in macrophages. J Biol Chem, v. 274, n. 37, p. 25967-25970, 1999b.
18. BARTOLI F; LIN HK; GHOMASHCHI F; GELB MH; JAIN MK; APITZ-CASTRO R. Tight binding inhibitors of 85-kDa phospholipase A2 but not 14-kDa phospholipase A2

110
inhibit release of free arachidonate in thrombin- stimulated human platelets. J Biol Chem, v. 269, n. 22, p. 15625-15630, 1994.
19. BEDOGNI B; WELFORD SM; CASSARINO DS; NICKOLOFF BJ; GIACCIA AJ; POWELL MB. The hypoxic microenvironment of the skin contributes to Akt-mediated melanocyte transformation. Cancer Cell, v. 8, n. 6, p. 443-454, 2005.
20. BENEZRA D. Neovasculogenic ability of prostaglandins, growth factors, and synthetic chemoattractants. Am J Ophtalmol, v. 86, n. 4, p. 455-461, 1978.
21. BERNATCHEZ PN; WINSTEAD MV; DENNIS EA; SIROIS MG. VEGF stimulation of endothelial cell PAF synthesis is mediated by group V 14 kDa secretory phospholipase A(2). Br J Pharmacol, v. 134, n. 1, p. 197-205, 2001.
22. BERRA E; AZ-MECO MT; MOSCAT J. The Activation of p38 and Apoptosis by the Inhibition of Erk Is Antagonized by the Phosphoinositide 3-Kinase/Akt Pathway. J Biol Chem, v. 273, n. 17, p. 10792-10797, 1998.
23. BERRA E; PAGES G; POUYSSEGUR J. MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev, v. 19, n. 1-2, p. 139-145, 2000.
24. BERTAGNOLLI MM. Cyclooxygenase-2 as a target for prevention of colorectal cancer. Curr Oncol Rep, v. 1, n. 2, p. 173-178, 1999.
25. BEZZINE S; KODURI RS; VALENTIN E; MURAKAMI M; KUDO I; GHOMASHCHI F; SADILEK M; LAMBEAU G; GELB MH. Exogenously Added Human Group X Secreted Phospholipase A2 but Not the Group IB, IIA, and V Enzymes Efficiently Release Arachidonic Acid from Adherent Mammalian Cells. J Biol Chem, v. 275, n. 5, p. 3179-3191, 2000.
26. BIANCO ID; KELLEY MJ; CROWL RM; DENNIS EA. Identification of two specific lysines responsible for the inhibition of phospholipase A2 by manoalide. Biochim Biophys Acta, v. 1250, n. 2, p. 197-203, 1995.
27. BILGER A; SHOEMAKER AR; GOULD KA; DOVE WF. Manipulation of the mouse germline in the study of Min-induced neoplasia. Semin Cancer Biol, v. 7, n. 5, p. 249-260, 1996.

111
28. BINGHAM CO, I.; MURAKAMI M; FUJISHIMA H; HUNT JE; AUSTEN KF; ARM JP. A heparin-sensitive phospholipase A2 and prostaglandin endoperoxide synthase-2 are functionally linked in the delayed phase of prostaglandin D2 generation in mouse bone marrow-derived mast cells. J Biol Chem, v. 271, n. 42, p. 25936-25944, 1996.
29. BINGHAM COIII; FIJNEMAN RJ; FRIEND DS; GODDEAU RP; ROGERS RA; AUSTEN KF; ARM JP. Low molecular weight group IIA and group V phospholipase A2 enzymes have different intracellular locations in mouse bone marrow-derived mast cells. J Biol Chem, v. 274, n. 44, p. 31476-31484, 1999.
30. BISHOP-BAILEY D ; HLA T. Endothelial Cell Apoptosis Induced by the Peroxisome Proliferator-activated Receptor (PPAR) Ligand 15-Deoxy-Delta 12,14-prostaglandin J2. J Biol Chem, v. 274, n. 24, p. 17042-17048, 1999.
31. BONVENTRE JV; HUANG Z; TAHERI MR; O'LEARY E; LI E; MOSKOWITZ MA; SAPIRSTEIN A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature, v. 390, n. 6660, p. 622-625, 1997.
32. BORSCH-HAUBOLD AG; BARTOLI F; ASSELIN J; DUDLER T; KRAMER RM; PITZ-CASTRO R; WATSON SP; GELB MH. Identification of the Phosphorylation Sites of Cytosolic Phospholipase A2 in Agonist-stimulated Human Platelets and HeLa Cells. J Biol Chem, v. 273, n. 8, p. 4449-4458, 1998.
33. BRADBURY D; CLARKE D; SEEDHOUSE C; CORBETT L; STOCKS J; KNOX A. Vascular Endothelial Growth Factor Induction by Prostaglandin E2 in Human Airway Smooth Muscle Cells Is Mediated by E Prostanoid EP2/EP4 Receptors and SP-1 Transcription Factor Binding Sites. J Biol Chem, v. 280, n. 34, p. 29993-30000, 2005.
34. BRASH AR. Arachidonic acid as a bioactive molecule. J Clin Invest, v. 107, n. 11, p. 1339-1345, 2001.
35. BRAT DJ ; VAN MEIR EG. Glomeruloid microvascular proliferation orchestrated by VPF/VEGF: a new world of angiogenesis research. Am J Pathol, v. 158, n. 3, p. 789-796, 2001.
36. BUCHANAN FG ; DUBOIS RN. Connecting COX-2 and Wnt in cancer. Cancer Cell, v. 9, n. 1, p. 6-8, 2006.

112
37. BUCHANAN FG; WANG D; BARGIACCHI F; DUBOIS RN. Prostaglandin E2 Regulates Cell Migration via the Intracellular Activation of the Epidermal Growth Factor Receptor. J Biol Chem, v. 278, n. 37, p. 35451-35457, 2003.
38. BUCKLAND AG ; WILTON DC. The antibacterial properties of secreted phospholipases A2. Biochim Biophys Acta, v. 1488, n. 1-2, p. 71-82, 2000.
39. CAI CM; SUN BC; LIU XY. Short hairpin RNA targeting vascular endothelial growth factor effectively inhibits expression of vascular endothelial growth factor in human retinal pigment epithelium. Zhonghua Yan Ke Za Zhi, v. 42, n. 4, p. 334-337, 2006.
40. CAO Y; PEARMAN AT; ZIMMERMAN GA; MCINTYRE TM; PRESCOTT SM. Intracellular unesterified arachidonic acid signals apoptosis. Proc Natl Acad Sci U S A, v. 97, n. 21, p. 11280-11285, 2000.
41. CARMAN GM; DEEMS RA; DENNIS EA. Lipid Signaling Enzymes and Surface Dilution Kinetics. J Biol Chem, v. 270, n. 32, p. 18711-18714, 1995.
42. CASTELLONE MD; TERAMOTO H; WILLIAMS BO; DRUEY KM; GUTKIND JS. Prostaglandin E2 Promotes Colon Cancer Cell Growth Through a Gs-Axin-beta-Catenin Signaling Axis. Science, v. 310, n. 5753, p. 1504-1510, 2005.
43. CAVALLINI L; COASSIN M; BOREAN A; ALEXANDRE A. Prostacyclin and Sodium Nitroprusside Inhibit the Activity of the Platelet Inositol 1,4,5-Trisphosphate Receptor and Promote Its Phosphorylation. J Biol Chem, v. 271, n. 10, p. 5545-5551, 1996.
44. CHAN ED ; RICHES DWH. IFN-gamma + LPS induction of iNOS is modulated by ERK, JNK/SAPK, and p38mapk in a mouse macrophage cell line. Am J Physiol, v. 280, n. 3, p. C441-C450, 2001.
45. CHANG J; MUSSER JH; MCGREGOR H. Phospholipase A2: Function and pharmacological regulation. Biochemical Pharmacology, v. 36, n. 15, p. 2429-2436, 1987.
46. CHANG SH; LIU CH; CONWAY R; HAN DK; NITHIPATIKOM K; TRIFAN OC; LANE TF; HLA T. From the Cover: Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc Natl Acad Sci U S A, v. 101, n. 2, p. 591-596, 2004.

113
47. CHANG SH; LIU CH; WU MT; HLA T. Regulation of vascular endothelial cell growth factor expression in mouse mammary tumor cells by the EP2 subtype of the prostaglandin E2 receptor. Prostaglandins Other Lipid Mediat, v. 76, n. 1-4, p. 48-58, 2005.
48. CHANNON JY ; LESLIE CC. A calcium-dependent mechanism for associating a soluble arachidonoyl-hydrolyzing phospholipase A2 with membrane in the macrophage cell line RAW 264.7. J Biol Chem, v. 265, n. 10, p. 5409-5413, 1990.
49. CHELL S; KADI A; CAROLINE WILLIAMS A; PARASKEVA C. Mediators of PGE2 synthesis and signalling downstream of COX-2 represent potential targets for the prevention/treatment of colorectal cancer. Biochim Biophys Acta, v. 1766, n. 1, p. 104-119, 2006.
50. CHEN J; ENGLE SJ; SEILHAMER JJ; TISCHFIELD JA. Cloning and recombinant expression of a novel human low molecular weight Ca(2+)-dependent phospholipase A2. J Biol Chem, v. 269, n. 4, p. 2365-2368, 1994.
51. CHEN Y ; DENNIS EA. Expression and characterization of human group V phospholipase A2. Biochim Biophys Acta, v. 1394, n. 1, p. 57-64, 1998.
52. CHERESH DA. Death to a blood vessel, death to a tumor. Nat Med, v. 4, n. 4, p. 395-396, 1998.
53. CHO W. Membrane Targeting by C1 and C2 Domains. J Biol Chem, v. 276, n. 35, p. 32407-32410, 2001.
54. CLARK JD; LIN LL; KRIZ RW; RAMESHA CS; SULTZMAN LA; LIN AY; MILONA N; KNOPF JL. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca2+-dependent translocation domain with homology to PKC and GAP. Cell, v. 65, n. 6, p. 1043-1051, 1991.
55. CLARK JD; SCHIEVELLA AR; NALEFSKI EA; LIN LL. Cytosolic phospholipase A2. J Lipid Mediat Cell Signal, v. 12, n. 2-3, p. 83-117, 1995.
56. CORMIER RT; BILGER A; LILLICH AJ; HALBERG RB; HONG KH; GOULD KA; BORENSTEIN N; LANDER ES; DOVE WF. The Mom1AKR intestinal tumor resistance region consists of Pla2g2a and a locus distal to D4Mit64. Oncogene, v. 19, n. 28, p. 3182-3192, 2000.

114
57. CORMIER RT; HONG KH; HALBERG RB; HAWKINS TL; RICHARDSON P; MULHERKAR R; DOVE WF; LANDER ES. Secretory phospholipase Pla2g2a confers resistance to intestinal tumorigenesis. Nat Genet, v. 17, n. 1, p. 88-91, 1997.
58. DAS S ; CHO W. Roles of Catalytic Domain Residues in Interfacial Binding and Activation of Group IV Cytosolic Phospholipase A2. J Biol Chem, v. 277, n. 26, p. 23838-23846, 2002.
59. DE CARVALHO MG; MCCORMACK AL; OLSON E; GHOMASHCHI F; GELB MH; YATES JRIII; LESLIE CC. Identification of phosphorylation sites of human 85-kDa cytosolic phospholipase A2 expressed in insect cells and present in human monocytes. J Biol Chem, v. 271, n. 12, p. 6987-6997, 1996.
60. DEAN JL; BROOK M; CLARK AR; SAKLATVALA J. p38 Mitogen-activated Protein Kinase Regulates Cyclooxygenase-2 mRNA Stability and Transcription in Lipopolysaccharide-treated Human Monocytes. J Biol Chem, v. 274, n. 1, p. 264-269, 1999.
61. DEN DEKKER E; GORTER G; HEEMSKERK JWM; AKKERMAN JW. Development of Platelet Inhibition by cAMP during Megakaryocytopoiesis. J Biol Chem, v. 277, n. 32, p. 29321-29329, 2002.
62. DENNIS EA. The growing phospholipase A2 superfamily of signal transduction enzymes. Trends Biochem Sci, v. 22, n. 1, p. 1-2, 1997.
63. DESSEN A. Structure and mechanism of human cytosolic phospholipase A2. Biochim Biophys Acta, v. 1488, n. 1-2, p. 40-47, 2000.
64. DESSEN A; TANG J; SCHMIDT H; STAHL M; CLARK JD; SEEHRA J; SOMERS WS. Crystal structure of human cytosolic phospholipase A2 reveals a novel topology and catalytic mechanism. Cell, v. 97, n. 3, p. 349-360, 1999.
65. DIAZ BL ; ARM JP. Phospholipase A2. Prostaglandins Leukot Essent Fatty Acids, v. 69, n. 2-3, p. 87-97, 2003.
66. DIMBERG J; SAMUELSSON A; HUGANDER A; SODERKVIST P. Gene expression of cyclooxygenase-2, group II and cytosolic phospholipase A2 in human colorectal cancer. Anticancer Res, v. 18, n. 5A, p. 3283-3287, 1998.

115
67. DMITRIEVA NI ; BURG MB. Hypertonic stress response. Mutat Res, v. 569, n. 1-2, p. 65-74, 2005.
68. DOBBIE Z; MULLER H; SCOTT RJ. Secretory phospholipase A2 does not appear to be associated with phenotypic variation in familial adenomatous polyposis. Hum Genet, v. 98, n. 3, p. 386-390, 1996.
69. DOLAN-O'KEEFE M; CHOW V; MONNIER J; VISNER GA; NICK HS. Transcriptional regulation and structural organization of the human cytosolic phospholipase A2 gene. Am J Physiol Lung Cell Mol Physiol, v. 278, n. 4, p. L649-L657, 2000.
70. DONG M; GUDA K; NAMBIAR PR; REZAIE A; BELINSKY GS; LAMBEAU G; GIARDINA C; ROSENBERG DW. Inverse association between phospholipase A2 and COX-2 expression during mouse colon tumorigenesis. Carcinogenesis, v. 24, n. 2, p. 307-315, 2003.
71. DRONADULA N; LIU Z; WANG C; CAO H; RAO GN. STAT-3-dependent Cytosolic Phospholipase A2 Expression Is Required for Thrombin-induced Vascular Smooth Muscle Cell Motility. J Biol Chem, v. 280, n. 4, p. 3112-3120, 2005.
72. DUBOIS RN; ABRAMSON SB; CROFFORD L; GUPTA RA; SIMON LS; VAN DE PUTTE LB; LIPSKY PE. Cyclooxygenase in biology and disease. FASEB J, v. 12, p. 1063-1073, 1998.
73. EIBL G; BRUEMMER D; OKADA Y; DUFFY JP; LAW RE; REBER HA; HINES OJ. PGE2 is generated by specific COX-2 activity and increases VEGF production in COX-2-expressing human pancreatic cancer cells. Biochemical and Biophysical Research Communications, v. 306, n. 4, p. 887-897, 2003.
74. ENOMOTO A; MURAKAMI M; KUDO I. Internalization and degradation of type IIA phospholipase A2 in mast cells. Biochem Biophys Res Commun, v. 276, n. 2, p. 667-672, 2000.
75. EVANS JH; FERGUS DJ; LESLIE CC. Inhibition of the MEK1/ERK pathway reduces arachidonic acid release independently of cPLA2 phosphorylation and translocation. BMC Biochem, v. 3, n. 1, p. 302002.

116
76. EVANS JH; GERBER SH; MURRAY D; LESLIE CC. The Calcium Binding Loops of the Cytosolic Phospholipase A2 C2 Domain Specify Targeting to Golgi and ER in Live Cells. Mol Biol Cell, v. 15, n. 1, p. 371-383, 2004.
77. EVANS JH ; LESLIE CC. The Cytosolic Phospholipase A2 Catalytic Domain Modulates Association and Residence Time at Golgi Membranes. J Biol Chem, v. 279, n. 7, p. 6005-6016, 2004.
78. EVANS JH; SPENCER DM; ZWEIFACH A; LESLIE CC. Intracellular Calcium Signals Regulating Cytosolic Phospholipase A2 Translocation to Internal Membranes. J Biol Chem, v. 276, n. 32, p. 30150-30160, 2001.
79. FAN B; WANG YX; YAO T; ZHU YC. p38 Mitogen-activated protein kinase mediates hypoxia-induced vascular endothelial growth factor release in human endothelial cells. Sheng Li Xue Bao, v. 57, n. 1, p. 13-20, 2005.
80. FENG J; WEHBI H; ROBERTS MF. Role of Tryptophan Residues in Interfacial Binding of Phosphatidylinositol-specific Phospholipase C. J Biol Chem, v. 277, n. 22, p. 19867-19875, 2002.
81. FENNEKOHL A; SUGIMOTO Y; SEGI E; MARUYAMA T; ICHIKAWA A; PUSCHEL GP. Contribution of the two Gs-coupled PGE2-receptors EP2-receptor and EP4-receptor to the inhibition by PGE2 of the LPS-induced TNFalpha-formation in Kupffer cells from EP2-or EP4-receptor-deficient mice. Pivotal role for the EP4-receptor in wild type Kupffer cells. J Hepatol, v. 36, n. 3, p. 328-334, 2002.
82. FERGUSON GM; WATANABE S; GEORGESCU HI; EVANS CH. The synovial production of collagenase and chondrocyte activating factors in response to cobalt. J Orthop Res, v. 6, n. 4, p. 525-530, 1988.
83. FERRARA N ; ALITALO K. Clinical applications of angiogenic growth factors and their inhibitors. Nat Med, v. 5, n. 12, p. 1359-1364, 1999.
84. FLAMAND N; PLANTE H; PICARD S; LAVIOLETTE M; BORGEAT P. Histamine-induced inhibition of leukotriene biosynthesis in human neutrophils: involvement of the H2 receptor and cAMP. Br J Pharmacol, v. 141, n. 4, p. 552-561, 2004.
85. FORM DM ; AUERBACH R. PGE2 and angiogenesis. Proc Soc Exp Biol Med, v. 172, n. 2, p. 214-218, 1983.

117
86. FUJISHIMA H; SANCHEZ MEJIA RO; BINGHAM COIII; LAM BK; SAPIRSTEIN A; BONVENTRE JV; AUSTEN KF; ARM JP. Cytosolic phospholipase A2 is essential for both the immediate and the delayed phases of eicosanoid generation in mouse bone marrow-derived mast cells. Proc Natl Acad Sci U S A, v. 96, n. 9, p. 4803-4807, 1999.
87. FUKUDA R; KELLY B; SEMENZA GL. Vascular Endothelial Growth Factor Gene Expression in Colon Cancer Cells Exposed to Prostaglandin E2 Is Mediated by Hypoxia-inducible Factor 1. Cancer Res, v. 63, n. 9, p. 2330-2334, 2003.
88. GALCHEVA-GARGOVA Z; DERIJARD B; WU IH; DAVIS RJ. An osmosensing signal transduction pathway in mammalian cells. Science, v. 265, n. 5173, p. 806-808, 1994.
89. GASPARINI G; LONGO R; SARMIENTO R; MORABITO A. Inhibitors of cyclo-oxygenase 2: a new class of anticancer agents? Lancet Oncol, v. 4, n. 10, p. 605-615, 2003.
90. GELB MH; JAIN MK; BERG OG. Inhibition of phospholipase A2. FASEB J, v. 8, n. 12, p. 916-924, 1994.
91. GELB MH; JAIN MK; HANEL AM; BERG OG. Interfacial enzymology of glycerolipid hydrolases: lessons from secreted phospholipases A2. Annu Rev Biochem, v. 64, p. 653-688, 1995.
92. GHOMASHCHI F; STEWART A; HEFNER Y; RAMANADHAM S; TURK J; LESLIE CC; GELB MH. A pyrrolidine-based specific inhibitor of cytosolic phospholipase A2[alpha] blocks arachidonic acid release in a variety of mammalian cells. Biochim Biophys Acta, v. 1513, n. 2, p. 160-166, 2001.
93. GHOSH M; TUCKER DE; BURCHETT SA; LESLIE CC. Properties of the Group IV phospholipase A2 family. Prog Lipid Res, v. 45, n. 6, p. 487-510, 2006.
94. GIJON MA; SPENCER DM; KAISER AL; LESLIE CC. Role of phosphorylation sites and the C2 domain in regulation of cytosolic phospholipase A2. J Cell Biol, v. 145, n. 6, p. 1219-1232, 1999.
95. GLINGHAMMAR B ; RAFTER J. Colonic luminal contents induce cyclooxygenase 2 transcription in human colon carcinoma cells. Gastroenterology, v. 120, n. 2, p. 401-410, 2001.

118
96. GLOVER S; BAYBURT T; JONAS M; CHI E; GELB MH. Translocation of the 85-kDa Phospholipase A[IMAGE] from Cytosol to the Nuclear Envelope in Rat Basophilic Leukemia Cells Stimulated with Calcium Ionophore or IgE/Antigen. J Biol Chem, v. 270, n. 25, p. 15359-15367, 1995.
97. GRIFFITHS RJ. Prostaglandins and Inflammation. In: J.L.GALLIN ; R. SNYDERMAN (Eds.). Inflammation: Basic Principles and Clinical Correlates. Philadelphia: Lippincott Williams & Wilkins, 1999. Cap. 22 , p. 349-360.
98. HAN J; LEE JD; BIBBS L; ULEVITCH RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science, v. 265, n. 5173, p. 808-811, 1994.
99. HAN SK; KIM KP; KODURI R; BITTOVA L; MUNOZ NM; LEFF AR; WILTON DC; GELB MH; CHO W. Roles of Trp31 in High Membrane Binding and Proinflammatory Activity of Human Group V Phospholipase A2. J Biol Chem, v. 274, n. 17, p. 11881-11888, 1999.
100. HANASAKI K; ONO T; SAIGA A; MORIOKA Y; IKEDA M; KAWAMOTO K; HIGASHINO K; NAKANO K; YAMADA K; ISHIZAKI J; ARITA H. Purified group X secretory phospholipase A2 induced prominent release of arachidonic acid from human myeloid leukemia cells. J Biol Chem, v. 274, n. 48, p. 34203-34211, 1999.
101. HANSEN-PETRIK MB; MCENTEE MF; JULL B; SHI H; ZEMEL MB; WHELAN J. Prostaglandin E2 Protects Intestinal Tumors from Nonsteroidal Anti-inflammatory Drug-induced Regression in ApcMin/+ Mice. Cancer Res, v. 62, n. 2, p. 403-408, 2002.
102. HARRIS AL. Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer, v. 2, n. 1, p. 38-47, 2002.
103. HASHIMOTO SHU; MATSUMOTO KEN; GON YASU; NAKAYAMA TOMO; TAKESHITA IKUK; HORIE TAKA. Hyperosmolarity-induced Interleukin-8 Expression in Human Bronchial Epithelial Cells through p38 Mitogen-activated Protein Kinase. Am J Respir Crit Care Med, v. 159, n. 2, p. 634-640, 1999.
104. HATA AN ; BREYER RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther, v. 103, n. 2, p. 147-166, 2004.
105. HAZEN SL; ZUPAN LA; WEISS RH; GETMAN DP; GROSS RW. Suicide inhibition of canine myocardial cytosolic calcium-independent phospholipase A2. Mechanism-based

119
discrimination between calcium- dependent and -independent phospholipases A2. J Biol Chem, v. 266, n. 11, p. 7227-7232, 1991.
106. HEASLEY LE; THALER S; NICKS M; PRICE B; SKORECKI K; NEMENOFF RA. Induction of Cytosolic Phospholipase A2 by Oncogenic Ras in Human Non-small Cell Lung Cancer. J Biol Chem, v. 272, n. 23, p. 14501-14504, 1997.
107. HECQUET C; LEFEVRE G; VALTINK M; ENGELMANN K; MASCARELLI F. cAMP inhibits the proliferation of retinal pigmented epithelial cells through the inhibition of ERK1/2 in a PKA-independent manner. Oncogene, v. 21, n. 39, p. 6101-6112, 2002.
108. HEFNER Y; BORSCH-HAUBOLD AG; MURAKAMI M; WILDE JI; PASQUET S; SCHIELTZ D; GHOMASHCHI F; YATES JRIII; ARMSTRONG CG; PATERSON A; COHEN P; FUKUNAGA R; HUNTER T; KUDO I; WATSON SP; GELB MH. Serine 727 Phosphorylation and Activation of Cytosolic Phospholipase A2 by MNK1-related Protein Kinases. J Biol Chem, v. 275, n. 48, p. 37542-37551, 2000.
109. HERLAAR E ; BROWN Z. p38 MAPK signalling cascades in inflammatory disease. Mol Med Today, v. 5, n. 10, p. 439-447, 1999.
110. HERNANDEZ GL; VOLPERT OV; INIGUEZ MA; LORENZO E; MARTINEZ-MARTINEZ S; GRAU R; FRESNO M; REDONDO JM. Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporin A: roles of the nuclear factor of activated T cells and cyclooxygenase 2. J Exp Med, v. 193, n. 5, p. 607-620, 2001.
111. HONG KH; BONVENTRE JC; O'LEARY E; BONVENTRE JV; LANDER ES. Deletion of cytosolic phospholipase A(2) suppresses Apc(Min)-induced tumorigenesis. Proc Natl Acad Sci U S A, v. 98, n. 7, p. 3935-3939, 2001.
112. HOUSE SL; BRANCH K; NEWMAN G; DOETSCHMAN T; SCHULTZ JE. Cardioprotection induced by cardiac-specific overexpression of fibroblast growth factor-2 is mediated by the MAPK cascade. Am J Physiol, v. 289, n. 5, p. H2167-H2175, 2005.
113. HUANG GW; YANG LY; LU WQ. Effects of PI3K and p42/p44 MAPK on overexpression of vascular endothelial growth factor in hepatocellular carcinoma. World J Gastroenterol, v. 10, n. 6, p. 809-812, 2004.

120
114. HUANG Z; PAYETTE P; ABDULLAH K; CROMLISH WA; KENNEDY BP. Functional Identification of the Active-Site Nucleophile of the Human 85-kDa Cytosolic Phospholipase A<sub>2</sub>. Biochemistry, v. 35, n. 12, p. 3712-3721, 1996.
115. HUBERT A; CAULIEZ B; CHEDEVILLE A; HUSSON A; LAVOINNE A. Osmotic stress, a proinflammatory signal in Caco-2 cells. Biochimie, v. 86, n. 8, p. 533-541, 2004.
116. HUGGINS KW; BOILEAU AC; HUI DY. Protection against diet-induced obesity and obesity- related insulin resistance in Group 1B PLA2-deficient mice. Am J Physiol Endocrinol Metab, v. 283, n. 5, p. E994-1001, 2002.
117. HULL MA. Cyclooxygenase-2: How good is it as a target for cancer chemoprevention? Eur J Cancer, v. 41, n. 13, p. 1854-1863, 2005.
118. HULL MA; KO SCW; HAWCROFT G. Prostaglandin EP receptors: Targets for treatment and prevention of colorectal cancer? Mol Cancer Ther, v. 3, n. 8, p. 1031-1039, 2004.
119. HULL MA; THOMSON JL; HAWKEY CJ. Expression of cyclooxygenase 1 and 2 by human gastric endothelial cells. Gut, v. 45, n. 4, p. 529-536, 1999.
120. IJICHI A; SAKUMA S; TOFILON PJ. Hypoxia-induced vascular endothelial growth factor expression in normal rat astrocyte cultures. Glia, v. 14, n. 2, p. 87-93, 1995.
121. ILSLEY JNM; NAKANISHI M; FLYNN C; BELINSKY GS; DE GUISE S; ADIB JN; DOBROWSKY RT; BONVENTRE JV; ROSENBERG DW. Cytoplasmic Phospholipase A2 Deletion Enhances Colon Tumorigenesis. Cancer Res, v. 65, n. 7, p. 2636-2643, 2005.
122. Instituto Nacional de Câncer - MS. (2006). Estimativa de Incidência e Mortalidade por Câncer no Brasil.
123. Instituto Nacional de Câncer - MS. (2002). Estimativa da Incidência e Mortalidade por Câncer no Brasil.
124. JACKSON JR; BOLOGNESE B; MANGAR CA; HUBBARD WC; MARSHALL LA; WINKLER JD. The role of platelet activating factor and other lipid mediators in inflammatory angiogenesis. Biochim Biophys Acta, v. 1392, n. 1, p. 145-152, 1998.

121
125. JIANG J; NEUBAUER BL; GRAFF JR; CHEDID M; THOMAS JE; ROEHM NW; ZHANG S; ECKERT GJ; KOCH MO; EBLE JN; CHENG L. Expression of group IIA secretory phospholipase A2 is elevated in prostatic intraepithelial neoplasia and adenocarcinoma. Am J Pathol, v. 160, n. 2, p. 667-671, 2002.
126. JIMENEZ JL; SMITH GR; CONTRERAS-MOREIRA B; SGOUROS JG; MEUNIER FA; BATES PA; SCHIAVO G. Functional Recycling of C2 Domains Throughout Evolution: A Comparative Study of Synaptotagmin, Protein Kinase C and Phospholipase C by Sequence, Structural and Modelling Approaches. J Mol Biol, v. 333, n. 3, p. 621-639, 2003.
127. KAMIYAMA M; POZZI A; YANG L; DEBUSK LM; BREYER RM; LIN PC. EP2, a receptor for PGE2, regulates tumor angiogenesis through direct effects on endothelial cell motility and survival. Oncogene, v. 25, n. 53, p. 7019-7028, 2006.
128. KANDA N; MITSUI H; WATANABE S. Prostaglandin E2 suppresses CCL27 production through EP2 and EP3 receptors in human keratinocytes. J Allergy Clin Immunol, v. 114, n. 6, p. 1403-1409, 2004.
129. KEERY RJ ; LUMLEY P. AH6809, a prostaglandin DP-receptor blocking drug on human platelets. Br J Pharmacol, v. 94, n. 3, p. 745-754, 1988.
130. KENNEDY BP; PAYETTE P; MUDGETT J; VADAS P; PRUZANSKI W; KWAN M; TANG C; RANCOURT DE; CROMLISH WA. A natural disruption of the secretory group II phospholipase A2 gene in inbred mouse strains. J Biol Chem, v. 270, n. 38, p. 22378-22385, 1995.
131. KENNEDY BP; SORAVIA C; MOFFAT J; XIA L; HIRUKI T; COLLINS S; GALLINGER S; BAPAT B. Overexpression of the nonpancreatic secretory group II PLA2 messenger RNA and protein in colorectal adenomas from familial adenomatous polyposis patients. Cancer Res, v. 58, n. 3, p. 500-503, 1998.
132. KIM KP; RAFTER JD; BITTOVA L; HAN SK; SNITKO Y; MUNOZ NM; LEFF AR; CHO W. Mechanism of human group V phospholipase A2 (PLA2)-induced leukotriene biosynthesis in human neutrophils. A potential role of heparan sulfate binding in PLA2 internalization and degradation. J Biol Chem, v. 276, n. 14, p. 11126-11134, 2001.
133. KINZLER KW ; VOGELSTEIN B. Colorectal Tumors. In: C.R.SCRIVER; A. L. BEAUDET; W. S. SLY; D. VALLE (Eds.). The metabolic and molecular bases of inherited diseases McGraw-Hill Co., 1995. Cap. 12 , p. 643-663.

122
134. ______. Lessons from hereditary colorectal cancer. Cell, v. 87, n. 2, p. 159-170, 1996.
135. KISKER O; BECKER CM; PROX D; FANNON M; D'AMATO R; FLYNN E; FOGLER WE; SIM BK; ALLRED EN; PIRIE-SHEPHERD SR; FOLKMAN J. Continuous administration of endostatin by intraperitoneally implanted osmotic pump improves the efficacy and potency of therapy in a mouse xenograft tumor model. Cancer Res, v. 61, n. 20, p. 7669-7674, 2001a.
136. KISKER O; ONIZUKA S; BANYARD J; KOMIYAMA T; BECKER CM; ACHILLES EG; BARNES CM; O'REILLY MS; FOLKMAN J; PIRIE-SHEPHERD SR. Generation of multiple angiogenesis inhibitors by human pancreatic cancer. Cancer Res, v. 61, n. 19, p. 7298-7304, 2001b.
137. KLIGERMAN J. Estimativas sobre a Incidência e Mortalidade por Câncer no Brasil - 2002. Rev Bras Cancerol, v. 48, n. 2, p. 715-719, 2002.
138. KOMADA M; KUDO I; MIZUSHIMA H; KITAMURA N; INOUE K. Structure of cDNA coding for rat platelet phospholipase A2. J Biochem (Tokyo), v. 106, n. 4, p. 545-547, 1989.
139. KRAMER RM; HESSION C; JOHANSEN B; HAYES G; MCGRAY P; CHOW EP; TIZARD R; PEPINSKY RB. Structure and properties of a human non-pancreatic phospholipase A2. J Biol Chem, v. 264, n. 10, p. 5768-5775, 1989.
140. KRAMER RM; ROBERTS EF; UM SL; BORSCH-HAUBOLD AG; WATSON SP; FISHER MJ; JAKUBOWSKI JA. p38 Mitogen-activated Protein Kinase Phosphorylates Cytosolic Phospholipase A2 (cPLA2) in Thrombin-stimulated Platelets. Evidence that proline-directed phosphorylation is not required for mobilization of arachidonic acid by cPLA2. J Biol Chem, v. 271, n. 44, p. 27723-27729, 1996.
141. KREJSGAARD T; VETTER-KAUCZOK CS; WOETMANN A; LOVATO P; LABUDA T; ERIKSEN KW; ZHANG Q; BECKER JC; ODUM N. Jak3- and JNK-dependent vascular endothelial growth factor expression in cutaneous T-cell lymphoma. Leukemia, v. 20, n. 10, p. 1759-1766, 2006.
142. KUCHERLAPATI R; LIN DP; EDELMANN W. Mouse models for human familial adenomatous polyposis. Semin Cancer Biol, v. 11, n. 3, p. 219-225, 2001.
143. KULTZ D ; BURG M. Evolution of osmotic stress signaling via MAP kinase cascades. J Exp Biol, v. 201, n. Pt 22, p. 3015-3021, 1998.

123
144. KUSUNOKI C; SATOH S; KOBAYASHI M; NIWA M. Structure of genomic DNA for rat platelet phospholipase A2. Biochim Biophys Acta, v. 1087, n. 1, p. 95-97, 1990.
145. LAGORCE-PAGES C; PARAF F; WENDUM D; MARTIN A; FLEJOU JF. Expression of inflammatory secretory phospholipase A2 and cytosolic phospholipase A2 in premalignant and malignant Barrett's oesophagus. Virchows Arch, v. 444, n. 5, p. 426-435, 2004.
146. LAYE JP ; GILL JH. Phospholipase A2 expression in tumours: a target for therapeutic intervention? Drug Discov Today, v. 8, n. 15, p. 710-716, 2003.
147. LEAHY KM; ORNBERG RL; WANG Y; ZWEIFEL BS; KOKI AT; MASFERRER JL. Cyclooxygenase-2 inhibition by celecoxib reduces proliferation and induces apoptosis in angiogenic endothelial cells in vivo. Cancer Res, v. 62, n. 3, p. 625-631, 2002.
148. LEE J; HONG F; KWON S; KIM SS; KIM DO; KANG HS; LEE SJ; HA J; KIM SS. Activation of p38 MAPK induces cell cycle arrest via inhibition of Raf/ERK pathway during muscle differentiation. Biochemical and Biophysical Research Communications, v. 298, n. 5, p. 765-771, 2002.
149. LESLIE CC. Regulation of arachidonic acid availability for eicosanoid production. Biochem Cell Biol, v. 82, n. 1, p. 1-17, 2004a.
150. ______. Regulation of the specific release of arachidonic acid by cytosolic phospholipase A2. Prostaglandins Leukot Essent Fatty Acids, v. 70, n. 4, p. 373-376, 2004b.
151. ______. Properties and regulation of cytosolic phospholipase A2. J Biol Chem, v. 272, n. 27, p. 16709-16712, 1997.
152. LEUNG SY; CHEN X; CHU KM; YUEN ST; MATHY J; JI J; CHAN ASY; LI R; LAW S; TROYANSKAYA OG; TU IP; WONG J; SO S; BOTSTEIN D; BROWN PO. Phospholipase A2 group IIA expression in gastric adenocarcinoma is associated with prolonged survival and less frequent metastasis. Proc Natl Acad Sci U S A, v. 99, n. 25, p. 16203-16208, 2002.
153. LEV-ARI S; MAIMON Y; STRIER L; KAZANOV D; ARBER N. Down-regulation of prostaglandin E2 by curcumin is correlated with inhibition of cell growth and induction of apoptosis in human colon carcinoma cell lines. J Soc Integr Oncol, v. 4, n. 1, p. 21-26, 2006.

124
154. LEVINE LAWR. Does the release of arachidonic acid from cells play a role in cancer chemoprevention? FASEB J, v. 17, n. 8, p. 800-802, 2003.
155. LEWIS RA; SOBERMAN RJ; AUSTEN KF. Leukotrienes and other products of the 5-lipoxygenase pathway. Biochemistry and relation to pathobiology in human diseases. N Engl J Med, v. 323, p. 645-655, 1990.
156. LI R; CINDROVA-DAVIES T; SKEPPER JN; SELLERS LA. Prostacyclin Induces Apoptosis of Vascular Smooth Muscle Cells by a cAMP-Mediated Inhibition of Extracellular Signal-Regulated Kinase Activity and Can Counteract the Mitogenic Activity of Endothelin-1 or Basic Fibroblast Growth Factor. Circ Res, v. 94, n. 6, p. 759-767, 2004.
157. LIM WC; PARK M; BAHN JJ; INOUE H; LEE YJ. Hypertonic sodium chloride induction of cyclooxygenase-2 occurs independently of NF-[kappa]B and is inhibited by the glucocorticoid receptor in A549 cells. FEBS Lett, v. 579, n. 24, p. 5430-5436, 2005.
158. LIN LL; LIN AY; DEWITT DL. Interleukin-1 alpha induces the accumulation of cytosolic phospholipase A2 and the release of prostaglandin E2 in human fibroblasts. J Biol Chem, v. 267, n. 33, p. 23451-23454, 1992.
159. LIN LL; WARTMANN M; LIN AY; KNOPF JL; SETH A; DAVIS RJ. cPLA2 is phosphorylated and activated by MAP kinase. Cell, v. 72, n. 2, p. 269-278, 1993.
160. LIO YC ; DENNIS EA. Interfacial activation, lysophospholipase and transacylase activity of group VI Ca2+-independent phospholipase A2. Biochim Biophys Acta, v. 1392, n. 2-3, p. 320-332, 1998.
161. LIO YC; REYNOLDS LJ; BALSINDE J; DENNIS EA. Irreversible inhibition of Ca2+ independent phospholipase A2 by methyl arachidonyl fluorophosphonate. Biochim Biophys Acta, v. 1302, n. 1, p. 55-60, 1996.
162. LITZ J ; KRYSTAL GW. Imatinib inhibits c-Kit-induced hypoxia-inducible factor-1alpha activity and vascular endothelial growth factor expression in small cell lung cancer cells. Mol Cancer Ther, v. 5, n. 6, p. 1415-1422, 2006.
163. LIU X; SUN SQ; HASSID A; OSTROM RS. cAMP Inhibits Transforming Growth Factor-beta-Stimulated Collagen Synthesis via Inhibition of Extracellular Signal-Regulated Kinase 1/2 and Smad Signaling in Cardiac Fibroblasts. Mol Pharmacol, v. 70, n. 6, p. 1992-2003, 2006.

125
164. LIU XH; KIRSCHENBAUM A; LU M; YAO S; DOSORETZ A; HOLLAND JF; LEVINE AC. Prostaglandin E2 Induces Hypoxia-inducible Factor-1alpha Stabilization and Nuclear Localization in a Human Prostate Cancer Cell Line. J Biol Chem, v. 277, n. 51, p. 50081-50086, 2002a.
165. LIU XH; KIRSCHENBAUM A; LU M; YAO S; KLAUSNER A; PRESTON C; HOLLAND JF; LEVINE AC. Prostaglandin E2 Stimulates Prostatic Intraepithelial Neoplasia Cell Growth through Activation of the Interleukin-6/GP130/STAT-3 Signaling Pathway. Biochemical and Biophysical Research Communications, v. 290, n. 1, p. 249-255, 2002b.
166. LIU XH; KIRSCHENBAUM A; YAO S; STEARNS ME; HOLLAND JF; CLAFFEY K; LEVINE AC. Upregulation of vascular endothelial growth factor by cobalt chloride-simulated hypoxia is mediated by persistent induction of cyclooxygenase-2 in a metastatic human prostate cancer cell line. Clin Exp Metastasis, v. 17, n. 8, p. 687-694, 1999.
167. LIU XH; KIRSCHENBAUM A; YU K; YAO S; LEVINE AC. Cyclooxygenase-2 Suppresses Hypoxia-induced Apoptosis via a Combination of Direct and Indirect Inhibition of p53 Activity in a Human Prostate Cancer Cell Line. J Biol Chem, v. 280, n. 5, p. 3817-3823, 2005.
168. LOMBARDO D ; DENNIS EA. Cobra venom phospholipase A2 inhibition by manoalide. A novel type of phospholipase inhibitor. J Biol Chem, v. 260, n. 12, p. 7234-7240, 1985.
169. LONGO WE; GROSSMANN EM; ERICKSON B; PANESAR N; MAZUSKI JE; KAMINSKI DL. The effect of phospholipase A2 inhibitors on proliferation and apoptosis of murine intestinal cells. J Surg Res, v. 84, n. 1, p. 51-56, 1999.
170. LUCAS KK ; DENNIS EA. Distinguishing phospholipase A2 types in biological samples by employing group-specific assays in the presence of inhibitors. Prostaglandins Other Lipid Mediat, v. 77, n. 1-4, p. 235-248, 2005.
171. MACPHEE M; CHEPENIK KP; LIDDELL RA; NELSON KK; SIRACUSA LD; BUCHBERG AM. The secretory phospholipase A2 gene is a candidate for the Mom1 locus, a major modifier of ApcMin-induced intestinal neoplasia. Cell, v. 81, n. 6, p. 957-966, 1995.
172. MAGNE S; COUCHIE D; PECKER F; PAVOINE C. beta 2-Adrenergic Receptor Agonists Increase Intracellular Free Ca2+ Concentration Cycling in Ventricular Cardiomyocytes through p38 and p42/44 MAPK-mediated Cytosolic Phospholipase A2 Activation. J Biol Chem, v. 276, n. 43, p. 39539-39548, 2001.

126
173. MAYER RJ ; MARSHALL LA. New insights on mammalian phospholipase A2(s); comparison of arachidonoyl-selective and -nonselective enzymes. FASEB J, v. 7, n. 2, p. 339-348, 1993.
174. MCNISH RW ; PETERS-GOLDEN M. Redistribution of 5-lipoxygenase and cytosolic phospholipase A2 to the nuclear fraction upon macrophage activation. Biochem Biophys Res Commun, v. 196, p. 147-153, 1993.
175. MIYASHITA A; CRYSTAL RG; HAY JG. Identification of a 27 bp 5'-flanking region element responsible for the low level constitutive expression of the human cytosolic phospholipase A2 gene. Nucleic Acids Res, v. 23, n. 2, p. 293-301, 1995.
176. MORIOKA Y; IKEDA M; SAIGA A; FUJII N; ISHIMOTO Y; ARITA H; HANASAKI K. Potential role of group X secretory phospholipase A(2) in cyclooxygenase-2-dependent PGE(2) formation during colon tumorigenesis. FEBS Lett, v. 487, n. 2, p. 262-266, 2000.
177. MOUNIER CM; GHOMASHCHI F; LINDSAY MR; JAMES S; SINGER AG; PARTON RG; GELB MH. Arachidonic Acid Release from Mammalian Cells Transfected with Human Groups IIA and X Secreted Phospholipase A2 Occurs Predominantly during the Secretory Process and with the Involvement of Cytosolic Phospholipase A2-{alpha}. J Biol Chem, v. 279, n. 24, p. 25024-25038, 2004.
178. MUKHERJEE AB; MIELE L; PATTABIRAMAN N. Phospholipase A2 enzymes: regulation and physiological role. Biochem Pharmacol, v. 48, n. 1, p. 1-10, 1994.
179. MUNNIK T ; MEIJER HJG. Osmotic stress activates distinct lipid and MAPK signalling pathways in plants. FEBS Lett, v. 498, n. 2-3, p. 172-178, 2001.
180. MURAKAMI M; BINGHAM COIII; MATSUMOTO R; AUSTEN KF; ARM JP. IgE-dependent activation of cytokine-primed mouse cultured mast cells induces a delayed phase of prostaglandin D2 generation via prostaglandin endoperoxide synthase-2. J Immunol, v. 155, n. 9, p. 4445-4453, 1995.
181. MURAKAMI M; KODURI RS; ENOMOTO A; SHIMBARA S; SEKI M; YOSHIHARA K; SINGER A; VALENTIN E; GHOMASHCHI F; LAMBEAU G; GELB MH; KUDO I. Distinct arachidonate-releasing functions of mammalian secreted phospholipase A2s in human embryonic kidney 293 and rat mastocytoma RBL-2H3 cells through heparan sulfate shuttling and external plasma membrane mechanisms. J Biol Chem, v. 276, n. 13, p. 10083-10096, 2001.

127
182. MURAKAMI M ; KUDO I. Diversity and regulatory functions of mammalian secretory phospholipase A2s. Adv Immunol, v. 77:163-94., p. 163-194, 2001.
183. MURAKAMI M; KUDO I; INOUE K. Molecular nature of phospholipases A2 involved in prostaglandin I2 synthesis in human umbilical vein endothelial cells. Possible participation of cytosolic and extracellular type II phospholipases A2. J Biol Chem, v. 268, n. 2, p. 839-844, 1993.
184. MURAKAMI M; KUWATA H; AMAKASU Y; SHIMBARA S; NAKATANI Y; ATSUMI G; KUDO I. Prostaglandin E2 amplifies cytosolic phospholipase A2- and cyclooxygenase-2-dependent delayed prostaglandin E2 generation in mouse osteoblastic cells. Enhancement by secretory phospholipase A2. J Biol Chem, v. 272, n. 32, p. 19891-19897, 1997.
185. MURAKAMI M; SHIMBARA S; KAMBE T; KUWATA H; WINSTEAD MV; TISCHFIELD JA; KUDO I. The functions of five distinct mammalian phospholipase A2S in regulating arachidonic acid release. Type IIa and type V secretory phospholipase A2S are functionally redundant and act in concert with cytosolic phospholipase A2. J Biol Chem, v. 273, n. 23, p. 14411-14423, 1998.
186. MURAKAMI M; YOSHIHARA K; SHIMBARA S; LAMBEAU G; GELB MH; SINGER AG; SAWADA M; INAGAKI N; NAGAI H; ISHIHARA M; ISHIKAWA Y; ISHII T; KUDO I. Cellular Arachidonate-releasing Function and Inflammation-associated Expression of Group IIF Secretory Phospholipase A2. J Biol Chem, v. 277, n. 21, p. 19145-19155, 2002a.
187. MURAKAMI M; YOSHIHARA K; SHIMBARA S; SAWADA M; INAGAKI N; NAGAI H; NAITO M; TSURUO T; MOON TC; CHANG HW; KUDO I. Group IID heparin-binding secretory phospholipase A2 is expressed in human colon carcinoma cells and human mast cells and up-regulated in mouse inflammatory tissues. Eur J Biochem, v. 269, n. 11, p. 2698-2707, 2002b.
188. MURATA M; YUDOH K; NAKAMURA H; KATO T; INOUE K; CHIBA J; NISHIOKA K; MASUKO-HONGO K. Distinct signaling pathways are involved in hypoxia- and IL-1-induced VEGF expression in human articular chondrocytes. J Orthop Res, v. 24, n. 7, p. 1544-1554, 2006.
189. MURRAY D ; HONIG B. Electrostatic Control of the Membrane Targeting of C2 Domains. Mol Cell, v. 9, n. 1, p. 145-154, 2002.

128
190. MUTHALIF MM; HEFNER Y; CANAAN S; HARPER J; ZHOU H; PARMENTIER JH; AEBERSOLD R; GELB MH; MALIK KU. Functional Interaction of Calcium-/Calmodulin-dependent Protein Kinase II and Cytosolic Phospholipase A2. J Biol Chem, v. 276, n. 43, p. 39653-39660, 2001.
191. NAFTALIN RJ ; PEDLEY KC. Video enhanced imaging of the fluorescent Na+ probe SBFI indicates that colonic crypts absorb fluid by generating a hypertonic interstitial fluid. FEBS Lett, v. 260, n. 2, p. 187-194, 1990.
192. NAGASAWA SY; TAKUWA N; SUGIMOTO N; MABUCHI H; TAKUWA Y. Inhibition of Rac activation as a mechanism for negative regulation of actin cytoskeletal reorganization and cell motility by cAMP. Biochem J, v. 385, n. Pt 3, p. 737-744, 2005.
193. NALEFSKI EA ; FALKE JJ. The C2 domain calcium-binding motif: Structural and functional diversity. Protein Sci, v. 5, n. 12, p. 2375-2390, 1996.
194. NALEFSKI EA; SULTZMAN LA; MARTIN DM; KRIZ RW; TOWLER PS; KNOPF JL; CLARK JD. Delineation of two functionally distinct domains of cytosolic phospholipase A2, a regulatory Ca(2+)-dependent lipid-binding domain and a Ca(2+)-independent catalytic domain. J Biol Chem, v. 269, n. 27, p. 18239-18249, 1994.
195. NEELI I; LIU Z; DRONADULA N; MA ZA; RAO GN. An Essential Role of the Jak-2/STAT-3/Cytosolic Phospholipase A2 Axis in Platelet-derived Growth Factor BB-induced Vascular Smooth Muscle Cell Motility. J Biol Chem, v. 279, n. 44, p. 46122-46128, 2004.
196. NEMENOFF RA; WINITZ S; QIAN NX; VAN PUTTEN V; JOHNSON GL; HEASLEY LE. Phosphorylation and activation of a high molecular weight form of phospholipase A2 by p42 microtubule-associated protein 2 kinase and protein kinase C. J Biol Chem, v. 268, n. 3, p. 1960-1964, 1993.
197. NEMETH ZH; DEITCH EA; SZABO C; HASKO G. Hyperosmotic Stress Induces Nuclear Factor-{kappa}B Activation and Interleukin-8 Production in Human Intestinal Epithelial Cells. Am J Pathol, v. 161, n. 3, p. 987-996, 2002.
198. NEWCOMB EW; LUKYANOV Y; SCHNEE T; ALI MA; LAN L; ZAGZAG D. Noscapine inhibits hypoxia-mediated HIF-1alpha expression andangiogenesis in vitro: a novel function for an old drug. Int J Oncol, v. 28, n. 5, p. 1121-1130, 2006.

129
199. NIE D ; HONN KV. Cyclooxygenase, lipoxygenase and tumor angiogenesis. Cell Mol Life Sci, v. 59, n. 5, p. 799-807, 2002.
200. O'REILLY MS; HOLMGREN L; CHEN C; FOLKMAN J. Angiostatin induces and sustains dormancy of human primary tumors in mice. Nat Med, v. 2, n. 6, p. 689-692, 1996.
201. OHASHI R; NAKAGAWA T; WATANABE S; KANELLIS J; ALMIREZ RG; SCHREINER GF; JOHNSON RJ. Inhibition of p38 Mitogen-Activated Protein Kinase Augments Progression of Remnant Kidney Model by Activating the ERK Pathway. Am J Pathol, v. 164, n. 2, p. 477-485, 2004.
202. OHTO T; UOZUMI N; HIRABAYASHI T; SHIMIZU T. Identification of Novel Cytosolic Phospholipase A2s, Murine cPLA2 delta, epsilon, and zeta, Which Form a Gene Cluster with cPLA2 beta. J Biol Chem, v. 280, n. 26, p. 24576-24583, 2005.
203. OLSSON AK; DIMBERG A; KREUGER J; CLAESSON-WELSH L. VEGF receptor signalling in control of vascular function. Nat Rev Mol Cell Biol, v. 7, n. 5, p. 359-371, 2006.
204. ONO T; TOJO H; KURAMITSU S; KAGAMIYAMA H; OKAMOTO M. Purification and characterization of a membrane-associated phospholipase A2 from rat spleen. Its comparison with a cytosolic phospholipase A2 S-1. J Biol Chem, v. 263, n. 12, p. 5732-5738, 1988.
205. OSHIMA M; DINCHUK JE; KARGMAN SL; OSHIMA H; HANCOCK B; KWONG E; TRZASKOS JM; EVANS JF; TAKETO MM. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell, v. 87, n. 5, p. 803-809, 1996.
206. OSTERSTROM A; DIMBERG J; FRANSEN K; SODERKVIST P. Expression of cytosolic and group X secretory phospholipase A2 genes in human colorectal adenocarcinomas. Cancer Letters, v. 182, n. 2, p. 175-182, 2002.
207. PAI R; SZABO IL; SOREGHAN BA; ATAY S; KAWANAKA H; TARNAWSKI AS. PGE2 Stimulates VEGF Expression in Endothelial Cells via ERK2/JNK1 Signaling Pathways. Biochemical and Biophysical Research Communications, v. 286, n. 5, p. 923-928, 2001.

130
208. PANEL V; BOELLE PY; AYALA-SANMARTIN J; JOUNIAUX AM; HAMELIN R; MASLIAH J; TRUGNAN G; FLEJOU JF; WENDUM D. Cytoplasmic phospholipase A2 expression in human colon adenocarcinoma is correlated with cyclooxygenase-2 expression and contributes to prostaglandin E2 production. Cancer Letters, v. 243, n. 2, p. 255-263, 2006.
209. PAPETTI M ; HERMAN IM. Mechanisms of normal and tumor-derived angiogenesis. Am J Physiol Cell Physiol, v. 282, n. 5, p. C947-C970, 2002.
210. PARANGI S; O'REILLY M; CHRISTOFORI G; HOLMGREN L; GROSFELD J; FOLKMAN J; HANAHAN D. Antiangiogenic therapy of transgenic mice impairs de novo tumor growth. Proc Natl Acad Sci U S A, v. 93, n. 5, p. 2002-2007, 1996.
211. PICKARD RT; CHIOU XG; STRIFLER BA; DEFELIPPIS MR; HYSLOP PA; TEBBE AL; YEE YK; REYNOLDS LJ; DENNIS EA; KRAMER RM; SHARP JD. Identification of essential residues for the catalytic function of 85-kDa cytosolic phospholipase A2. Probing the role of histidine, aspartic acid, cysteine, and arginine. J Biol Chem, v. 271, n. 32, p. 19225-19231, 1996.
212. PISANI P; PARKIN DM; BRAY F; FERLAY J. Estimates of the worldwide mortality from 25 cancers in 1990. Int J Cancer, v. 83, n. 1, p. 18-29, 1999.
213. PLESNIAK LA; BOEGEMAN SC; SEGELKE BW; DENNIS EA. Interaction of phospholipase A2 with thioether amide containing phospholipid analogues. Biochemistry, v. 32, n. 19, p. 5009-5016, 1993.
214. PLESNIAK LA; YU L; DENNIS EA. Conformation of micellar phospholipid bound to the active site of phospholipase A2. Biochemistry, v. 34, n. 15, p. 4943-4951, 1995.
215. POLAKIS P. The adenomatous polyposis coli (APC) tumor suppressor. Biochim Biophys Acta, v. 1332, n. 3, p. F127-F147, 1997.
216. POULIOS E; TROUGAKOS IP; GONOS ES. Comparative effects of hypoxia on normal and immortalized human diploid fibroblasts. Anticancer Res, v. 26, n. 3A, p. 2165-2168, 2006.
217. POURTAU J; MIRSHAHI F; LI H; MURAINE M; VINCENT L; TEDGUI A; VANNIER JP; SORIA J; VASSE M; SORIA C. Cyclooxygenase-2 activity is necessary for the angiogenic properties of oncostatin M. FEBS Lett, v. 459, n. 3, p. 453-457, 1999.

131
218. POUYSSEGUR J; DAYAN F; MAZURE NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature, v. 441, n. 7092, p. 437-443, 2006.
219. POWELL DW. Dogma destroyed: colonic crypts absorb. J Clin Invest, v. 96, n. 5, p. 2102-2103, 1995.
220. QIU ZH; DE CARVALHO MS; LESLIE CC. Regulation of phospholipase A2 activation by phosphorylation in mouse peritoneal macrophages. J Biol Chem, v. 268, n. 32, p. 24506-24513, 1993.
221. REDDY ST ; HERSCHMAN HR. Prostaglandin Synthase-1 and Prostaglandin Synthase-2 Are Coupled to Distinct Phospholipases for the Generation of Prostaglandin D2 in Activated Mast Cells. J Biol Chem, v. 272, n. 6, p. 3231-3237, 1997.
222. REYNOLDS LJ; MIHELICH ED; DENNIS EA. Inhibition of venom phospholipases A2 by manoalide and manoalogue. Stoichiometry of incorporation. J Biol Chem, v. 266, n. 25, p. 16512-16517, 1991.
223. RICHMOND BL; BOILEAU AC; ZHENG S; HUGGINS KW; GRANHOLM NA; TSO P; HUI DY. Compensatory phospholipid digestion is required for cholesterol absorption in pancreatic phospholipase A(2)-deficient mice. Gastroenterology, v. 120, n. 5, p. 1193-1202, 2001.
224. RIENDEAU D; GUAY J; WEECH PK; LALIBERTE F; YERGEY J; LI C; DESMARAIS S; PERRIER H; LIU S; NICOLL-GRIFFITH D. Arachidonyl trifluoromethyl ketone, a potent inhibitor of 85-kDa phospholipase A2, blocks production of arachidonate and 12- hydroxyeicosatetraenoic acid by calcium ionophore-challenged platelets. J Biol Chem, v. 269, n. 22, p. 15619-15624, 1994.
225. RIZZO MT; NGUYEN E; ALDO-BENSON M; LAMBEAU G. Secreted phospholipase A2 induces vascular endothelial cell migration. Blood, v. 96, n. 12, p. 3809-3815, 2000.
226. ROBERTS MF; DEEMS RA; MINCEY TC; DENNIS EA. Chemical modification of the histidine residue in phospholipase A2 (Naja naja naja). A case of half-site reactivity. J Biol Chem, v. 252, n. 7, p. 2405-2411, 1977.
227. RUIS H ; SCHULLER C. Stress signaling in yeast. Bioessays, v. 17, n. 11, p. 959-965, 1995.

132
228. SA G ; FOX PL. Basic fibroblast growth factor-stimulated endothelial cell movement is mediated by a pertussis toxin-sensitive pathway regulating phospholipase A2 activity. J Biol Chem, v. 269, n. 5, p. 3219-3225, 1994.
229. SALCEDO R; ZHANG X; YOUNG HA; MICHAEL N; WASSERMAN K; MA WH; MARTINS-GREEN M; MURPHY WJ; OPPENHEIM JJ. Angiogenic effects of prostaglandin E2 are mediated by up-regulation of CXCR4 on human microvascular endothelial cells. Blood, v. 102, n. 6, p. 1966-1977, 2003.
230. SALNIKOW K; DONALD SP; BRUICK RK; ZHITKOVICH A; PHANG JM; KASPRZAK KS. Depletion of Intracellular Ascorbate by the Carcinogenic Metals Nickel and Cobalt Results in the Induction of Hypoxic Stress. J Biol Chem, v. 279, n. 39, p. 40337-40344, 2004.
231. SALNIKOW K; KLUZ T; COSTA M; PIQUEMAL D; DEMIDENKO ZN; XIE K; BLAGOSKLONNY MV. The regulation of hypoxic genes by calcium involves c-Jun/AP-1, which cooperates with hypoxia-inducible factor 1 in response to hypoxia. Mol Cell Biol, v. 22, n. 6, p. 1734-1741, 2002.
232. SAWAOKA H; TSUJI S; TSUJII M; GUNAWAN ES; SASAKI Y; KAWANO S; HORI M. Cyclooxygenase inhibitors suppress angiogenesis and reduce tumor growth in vivo. Lab Invest, v. 79, n. 12, p. 1469-1477, 1999.
233. SCHEVITZ RW; BACH NJ; CARLSON DG; CHIRGADZE NY; CLAWSON DK; DILLARD RD; DRAHEIM SE; HARTLEY LW; JONES ND; MIHELICH ED; ET AL.; . Structure-based design of the first potent and selective inhibitor of human non-pancreatic secretory phospholipase A2. Nat Struct Biol, v. 2, n. 6, p. 458-465, 1995.
234. SCHIEVELLA AR; REGIER MK; SMITH WL; LIN LL. Calcium-mediated Translocation of Cytosolic Phospholipase A2 to the Nuclear Envelope and Endoplasmic Reticulum. J Biol Chem, v. 270, n. 51, p. 30749-30754, 1995.
235. SCHMALZ G; SCHWEIKL H; HILLER KA. Release of prostaglandin E2, IL-6 and IL-8 from human oral epithelial culture models after exposure to compounds of dental materials. Eur J Oral Sci, v. 108, n. 5, p. 442-448, 2000.
236. SEILHAMER JJ; PRUZANSKI W; VADAS P; PLANT S; MILLER JA; KLOSS J; JOHNSON LK. Cloning and recombinant expression of phospholipase A2 present in rheumatoid arthritic synovial fluid. J Biol Chem, v. 264, n. 10, p. 5335-5338, 1989.

133
237. SENO K; OKUNO T; NISHI K; MURAKAMI Y; WATANABE F; MATSUURA T; WADA M; FUJII Y; YAMADA M; OGAWA T; OKADA T; HASHIZUME H; KII M; HARA S; HAGISHITA S; NAKAMOTO S; YAMADA K; CHIKAZAWA Y; UENO M; TESHIROGI I; ONO T; OHTANI M. Pyrrolidine inhibitors of human cytosolic phospholipase A2. J Med Chem, v. 43, n. 6, p. 1041-1044, 2000.
238. SEO M; LEE YIL; CHO CH; BAE CD; KIM IH; JUHNN YS. Bi-directional Regulation of UV-induced Activation of p38 Kinase and c-Jun N-terminal Kinase by G Protein beta gamma -Subunits. J Biol Chem, v. 277, n. 27, p. 24197-24203, 2002.
239. SERHAN CN ; OLIW E. Unorthodox routes to prostanoid formation: new twists in cyclooxygenase-initiated pathways. J Clin Invest, v. 107, n. 12, p. 1481-1489, 2001.
240. SHAPIRO L ; DINARELLO CA. Osmotic Regulation of Cytokine Synthesis in vitro. Proc Natl Acad Sci U S A, v. 92, n. 26, p. 12230-12234, 1995.
241. SHARP JD; PICKARD RT; CHIOU XG; MANETTA JV; KOVACEVIC S; MILLER JR; VARSHAVSKY AD; ROBERTS EF; STRIFLER BA; BREMS DN. Serine 228 is essential for catalytic activities of 85-kDa cytosolic phospholipase A2. J Biol Chem, v. 269, n. 37, p. 23250-23254, 1994.
242. SHENG H; SHAO J; KIRKLAND SC; ISAKSON P; COFFEY RJ; MORROW J; BEAUCHAMP RD; DUBOIS RN. Inhibition of Human Colon Cancer Cell Growth by Selective Inhibition of Cyclooxygenase-2. J Clin Invest, v. 99, n. 9, p. 2254-2259, 1997.
243. SHENG H; SHAO J; WASHINGTON MK; DUBOIS RN. Prostaglandin E2 Increases Growth and Motility of Colorectal Carcinoma Cells. J Biol Chem, v. 276, n. 21, p. 18075-18081, 2001.
244. SIERRA-HONIGMANN MR; RADLEY JR; PROBER JS. "Cytosolic" phospholipase A2 is in the nucleus of subconfluent endothelial cells but confined to the cytoplasm of confluent endothelial cells and redistributes to the nuclear envelope and cell junctions upon histamine stimulation. Lab Invest, v. 74, n. 3, p. 684-695, 1996.
245. SIX DA ; DENNIS EA. The expanding superfamily of phospholipase A(2) enzymes: classification and characterization. Biochim Biophys Acta, v. 1488, n. 1-2, p. 1-19, 2000.
246. SKOPINSKA-ROZEWSKA E; PIAZZA GA; SOMMER E; PAMUKCU R; BARCZ E; FILEWSKA M; KUPIS W; CABAN R; RUDZINSKI P; BOGDAN J; MLEKODAJ S; SIKORSKA E. Inhibition of angiogenesis by sulindac and its sulfone metabolite (FGN-

134
1): a potential mechanism for their antineoplastic properties. Int J Tissue React, v. 20, n. 3, p. 85-89, 1998.
247. SMANI T; ZAKHAROV SI; CSUTORA P; LENO E; TREPAKOVA ES; BOLOTINA VM. A novel mechanism for the store-operated calcium influx pathway. Nat Cell Biol, v. 6, n. 2, p. 113-120, 2004.
248. SMART BP; OSLUND RC; WALSH LA; GELB MH. The First Potent Inhibitor of Mammalian Group X Secreted Phospholipase A<sub>2</sub>: Elucidation of Sites for Enhanced Binding. J Med Chem, v. 49, n. 10, p. 2858-2860, 2006.
249. SMITH WL. Prostanoid biosynthesis and mechanisms of action. Am J Physiol, v. 263, p. F181-F191, 1992.
250. SONOSHITA, M.; TAKAKU, K.; SASAKI, N.; SUGIMOTO, Y.; USHIKUBI, F.; NARUMIYA, S.; OSHIMA, M.; TAKETO, M. M. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in APC∆716 knockout mice. Nat Med, v. 7, n. 9, p. 1048-1051, 2001.
251. SOYDAN AS; TAVARES IA; WEECH PK; TEMBLAY NM; BENNETT A. High molecular weight phospholipase A2 and fatty acids in human colon tumours and associated normal tissue. Eur J Cancer, v. 32A, n. 10, p. 1781-1787, 1996.
252. SPINELLA F; ROSANO L; DI CASTRO V; NATALI PG; BAGNATO A. Endothelin-1-induced Prostaglandin E2-EP2, EP4 Signaling Regulates Vascular Endothelial Growth Factor Production and Ovarian Carcinoma Cell Invasion. J Biol Chem, v. 279, n. 45, p. 46700-46705, 2004.
253. STREET IP; LIN HK; LALIBERTE F; GHOMASHCHI F; WANG Z; PERRIER H; TREMBLAY NM; HUANG Z; WEECH PK; GELB MH. Slow- and tight-binding inhibitors of the 85-kDa human phospholipase A2. Biochemistry, v. 32, n. 23, p. 5935-5940, 1993.
254. STROMBLAD S ; CHERESH DA. Cell Adhesion and Angiogenesis. Trends Cell Biol, v. 6, p. 462-468, 1996.
255. TAKAKU K; SONOSHITA M; SASAKI N; UOZUMI N; DOI Y; SHIMIZU T; TAKETO MM. Suppression of intestinal polyposis in Apc(delta 716) knockout mice by an additional mutation in the cytosolic phospholipase A(2) gene. J Biol Chem, v. 275, n. 44, p. 34013-34016, 2000.

135
256. TAKETO MM. Cyclooxygenase-2 inhibitors in tumorigenesis (Part II). J Natl Cancer Inst, v. 90, n. 21, p. 1609-1620, 1998.
257. TAKETO MM ; SONOSHITA M. Phospolipase A2 and apoptosis. Biochim Biophys Acta, v. 1585, n. 2-3, p. 72-76, 2002.
258. TAY A; SIMON JS; SQUIRE J; HAMEL K; JACOB HJ; SKORECKI K. Cytosolic phospholipase A2 gene in human and rat: chromosomal localization and polymorphic markers. Genomics, v. 26, n. 1, p. 138-141, 1995.
259. TAY HK ; MELENDEZ AJ. Fc gamma RI-triggered Generation of Arachidonic Acid and Eicosanoids Requires iPLA2 but Not cPLA2 in Human Monocytic Cells. J Biol Chem, v. 279, n. 21, p. 22505-22513, 2004.
260. THIAGARAJAH JR; JAYARAMAN S; NAFTALIN RJ; VERKMAN AS. In vivo fluorescence measurement of Na+ concentration in the pericryptal space of mouse descending colon. Am J Physiol Cell Physiol, v. 281, n. 6, p. C1898-C1903, 2001.
261. TISCHFIELD JA. A reassessment of the low molecular weight phospholipase A2 gene family in mammals. J Biol Chem, v. 272, n. 28, p. 17247-17250, 1997.
262. TISCHFIELD JA; XIA YR; SHIH DM; KLISAK I; CHEN J; ENGLE SJ; SIAKOTOS AN; WINSTEAD MV; SEILHAMER JJ; ALLAMAND V; GYAPAY G; LUSIS AJ. Low-molecular-weight, calcium-dependent phospholipase A2 genes are linked and map to homologous chromosome regions in mouse and human. Genomics, v. 32, n. 3, p. 328-333, 1996.
263. TOMOZAWA S; TSUNO NH; SUNAMI E; HATANO K; KITAYAMA J; OSADA T; SAITO S; TSURUO T; SHIBATA Y; NAGAWA H. Cyclooxygenase-2 overexpression correlates with tumour recurrence, especially haematogenous metastasis, of colorectal cancer. Br J Cancer, v. 83, n. 3, p. 324-328, 2000.
264. TSUJII M; KAWANO S; DUBOIS RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci U S A, v. 94, n. 7, p. 3336-3340, 1997.
265. TSUJII M; KAWANO S; TSUJI S; SAWAOKA H; HORI M; DUBOIS RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell, v. 93, n. 5, p. 705-716, 1998.

136
266. UOZUMI N; KUME K; NAGASE T; NAKATANI N; ISHII S; TASHIRO F; KOMAGATA Y; MAKI K; IKUTA K; OUCHI Y; MIYAZAKI J; SHIMIZU T. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature, v. 390, p. 618-622, 1997.
267. VADAS P; SCOTT K; SMITH G; RAJKOVIC I; STEFANSKI E; SCHOUTEN BD; SINGH R; PRUZANSKI W. Serum phospholipase A2 enzyme activity and immunoreactivity in a prospective analysis of patients with septic shock. Life Sci, v. 50, n. 11, p. 807-811, 1992.
268. VALENTIN E ; LAMBEAU G. Increasing molecular diversity of secreted phospholipases A2 and their receptors and binding proteins. Biochim Biophys Acta, v. 1488, n. 1-2, p. 59-70, 2000.
269. WALSH MT; FOLEY JF; KINSELLA BT. The alpha , but Not the beta , Isoform of the Human Thromboxane A2 Receptor Is a Target for Prostacyclin-mediated Desensitization. J Biol Chem, v. 275, n. 27, p. 20412-20423, 2000.
270. WANG D; WANG H; BROWN J; DAIKOKU T; NING W; SHI Q; RICHMOND A; STRIETER R; DEY SK; DUBOIS RN. CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J Exp Med, v. 203, n. 4, p. 941-951, 2006.
271. WATANABE K; KAWAMORI T; NAKATSUGI S; OHTA T; OHUCHIDA S; YAMAMOTO H; MARUYAMA T; KONDO K; USHIKUBI F; NARUMIYA S; SUGIMURA T; WAKABAYASHI K. Role of the prostaglandin E receptor subtype EP1 in colon carcinogenesis. Cancer Res, v. 59, n. 20, p. 5093-5096, 1999.
272. WEBSTER KA; GRAHAM RM; BISHOPRIC NH. BNip3 and signal-specific programmed death in the heart. J Mol Cell Cardiol, v. 38, n. 1, p. 35-45, 2005.
273. WENDUM D; SVRCEK M; RIGAU V; BOELLE PY; SEBBAGH N; PARC R; MASLIAH J; TRUGNAN G; FLEJOU JF. COX-2, inflammatory secreted PLA2, and cytoplasmic PLA2 protein expression in small bowel adenocarcinomas compared with colorectal adenocarcinomas. Mod Pathol, v. 16, n. 2, p. 130-136, 2003.
274. WOJCIKIEWICZ RJ ; LUO SG. Phosphorylation of Inositol 1,4,5-Trisphosphate Receptors by cAMP-dependent Protein Kinase. Type I, II, and III receptors are differentially susceptible to phosphorylation and are phosphorylated in intact cells. J Biol Chem, v. 273, n. 10, p. 5670-5677, 1998.

137
275. WONG JT; TRAN K; PIERCE GN; CHAN AC; O K; CHOY PC. Lysophosphatidylcholine stimulates the release of arachidonic acid in human endothelial cells. J Biol Chem, v. 273, n. 12, p. 6830-6836, 1998.
276. WOO KJ; LEE TJ; PARK JW; KWON TK. Desferrioxamine, an iron chelator, enhances HIF-1[alpha] accumulation via cyclooxygenase-2 signaling pathway. Biochemical and Biophysical Research Communications, v. 343, n. 1, p. 8-14, 2006.
277. WOODWARD DF; PEPPERL DJ; BURKEY TH; REGAN JW. 6-isopropoxy-9-oxoxanthene-2-carboxylic acid (AH 6809), a human EP2 receptor antagonist. Biochemical Pharmacology, v. 50, n. 10, p. 1731-1733, 1995.
278. WU T; HAN C; LUNZ JGIII; MICHALOPOULOS G; SHELHAMER JH; DEMETRIS AJ. Involvement of 85-kd cytosolic phospholipase A2 and cyclooxygenase-2 in the proliferation of human cholangiocarcinoma cells. Hepatology, v. 36, n. 2, p. 363-373, 2002.
279. WU T; IKEZONO T; ANGUS CW; SHELHAMER JH. Characterization of the promoter for the human 85 kDa cytosolic phospholipase A2 gene. Nucleic Acids Res, v. 22, n. 23, p. 5093-5098, 1994.
280. XIN X; YANG S; KOWALSKI J; GERRITSEN ME. Peroxisome Proliferator-activated Receptor gamma áLigands Are Potent Inhibitors of Angiogenesis in Vitro and in Vivo. J Biol Chem, v. 274, n. 13, p. 9116-9121, 1999.
281. YAMADA M; KAWAI M; KAWAI Y; MASHIMA Y. The effect of selective cyclooxygenase-2 inhibitor on corneal angiogenesis in the rat. Curr Eye Res, v. 19, n. 4, p. 300-304, 1999.
282. YAMADA Y; ENDO S; KAMEI Y; MINATO T; YOKOYAMA M; TANIGUCHI S; NAKAE H; INADA K; OGAWA M. Plasma levels of type II phospholipase A2 and nitrite/nitrate in patients with burns. Burns, v. 24, n. 6, p. 513-517, 1998.
283. YANG T; HUANG Y; HEASLEY LE; BERL T; SCHNERMANN JB; BRIGGS JP. MAPK Mediation of Hypertonicity-stimulated Cyclooxygenase-2 Expression in Renal Medullary Collecting Duct Cells. J Biol Chem, v. 275, n. 30, p. 23281-23286, 2000.
284. YANG T; ZHANG A; HONEGGAR M; KOHAN DE; MIZEL D; SANDERS K; HOIDAL JR; BRIGGS JP; SCHNERMANN JB. Hypertonic induction of COX-2 in

138
collecting duct cells by reactive oxygen species of mitochondrial origin. J Biol Chem, v. 280, n. 41, p. 34966-34973, 2005.
285. YASSAD A; LAVOINNE A; BION A; DAVEAU M; HUSSON A. Glutamine accelerates interleukin-6 production by rat peritoneal macrophages in culture. FEBS Lett, v. 413, n. 1, p. 81-84, 1997.
286. YEDGAR S; LICHTENBERG D; SCHNITZER E. Inhibition of phospholipase A2 as a therapeutic target. Biochim Biophys Acta, v. 1488, n. 1-2, p. 182-187, 2000.
287. YOKOTA Y; HIGASHINO K; NAKANO K; ARITA H; HANASAKI K. Identification of group X secretory phospholipase A2 as a natural ligand for mouse phospholipase A2 receptor. FEBS Lett, v. 478, n. 1-2, p. 187-191, 2000.
288. YOSHINO Y; AOYAGI M; TAMAKI M; DUAN L; MORIMOTO T; OHNO K. Activation of p38 MAPK and/or JNK contributes to increased levels of VEGF secretion in human malignant glioma cells. Int J Oncol, v. 29, n. 4, p. 981-987, 2006.
289. YU L ; DENNIS EA. Effect of polar head groups on the interactions of phospholipase A2 with phosphonate transition-state analogues. Biochemistry, v. 32, n. 38, p. 10185-10192, 1993.
290. YUAN W; QUINN DM; SIGLER PB; GELB MH. Kinetic and inhibition studies of phospholipase A2 with short-chain substrates and inhibitors. Biochemistry, v. 29, n. 25, p. 6082-6094, 1990.
291. YUE C; DODGE KL; WEBER G; SANBORN BM. Phosphorylation of Serine 1105 by Protein Kinase A Inhibits Phospholipase C beta 3 Stimulation by G alpha q. J Biol Chem, v. 273, n. 29, p. 18023-18027, 1998.
292. ZATECHKA SDJR ; LOU MF. Studies of the mitogen-activated protein kinases and phosphatidylinositol-3 kinase in the lens. 1. The mitogenic and stress responses. Exp Eye Res, v. 74, n. 6, p. 703-717, 2002.
293. ZICHE M; JONES J; GULLINO PM. Role of prostaglandin E1 and copper in angiogenesis. J Natl Cancer Inst, v. 69, n. 2, p. 475-482, 1982.

139
8. ANEXO Gentile LB, Diaz BL (2007). Role of Cytosolic PLA2 and Kinase signaling pathways on PGE2 generation by a human colon cancer cell line in hypertonic stress. Manuscrito a submeter.

140
Role of Cytosolic PLA2 and Kinase signaling pathways on PGE2 generation by a human colon cancer cell line in hypertonic stress Gentile, L.B.1,2, Diaz, B.L.1,3 1Divisão de Biologia Celular, Coordenação de Pesquisa, Instituto Nacional de Câncer; 2Programa de Pós-Graduação em Ciências Morfológicas, Instituto de Ciências Biomédicas, 3Programa de Imunobiologia, Instituto de Biofísica, Universidade Federal do Rio de Janeiro, Rio de Janeiro-RJ, Brasil Cyclooxygenase (COX)-2-derived prostaglandin (PG)E2 controls every aspect of colon cancer development, modulating from cell proliferation and resistance to apoptosis to angiogenesis, invasion, and metastasis. Here we investigate the role of different PLA2s in supplying AA for COX-2-dependent PGE2 generation and the signaling pathways that lead to PGE2 generation after activation by a physiologically relevant stimulus. Human colon cancer cell line Caco-2 was activated in complete DMEM medium with addition of NaCl (100 mM) to emulate the colon hypertonic environment. AA release and PGE2 generation were analyzed in the cell culture supernatants, while, COX-2 expression, cPLA2 and kinases phosphorylated were analyzed in cell lysates by western blotting. Pharmacological inhibitors of cPLA2 (ATK), calcium-independent (i)PLA2 (BEL), sPLA2 (Scalaradial), COX-2 (NS-398), p38 (SB202190), JNK (SP600125), ERK1/2 (U0126) were added 15-30 min before Caco-2 activation. Human colon cancer cell line Caco-2 exposed to a hypertonic environment responded with marked AA release, COX-2 induction and PGE2 generation. sPLA2 (scalaradial) and iPLA2 (BEL) inhibitors did not modify, while COX-2 (NS-398) and cPLA2 (ATK) inhibitors each completely inhibited PGE2 generation. ATK also abolished stimulated AA release, but did not modify COX-2 expression. All three kinases inhibited AA release and completely abolished PGE2 generation, however only inhibition of ERK1/2 reduced COX-2 induction. Our results indicate that PGE2 generation by Caco-2 cells exposed to hyperosmotic stress depends on cPLA2 and COX-2 activity. Kinases participate in PGE2 generation, however ERK1/2 is involved in cPLA2 activation and COX-2 induction, while ERK1/2, p38 and JNK modulate cPLA2 activity.

141
INTRODUCTION PGE2 can modulate every aspect of colon tumorigenesis, ranging from increased cell
proliferation and resistence to apoptosis to the formation of new blood vessels and metatstic potential (1-8). The increased production of PGE2 by colon tumors and colon cancer-derived cell lines is associated with overexpression of cyclooxygenase (COX)-2 (9;10), which catalyses the conversion of arachidonic acid in PGH2, the committed precursor of prostaglandins and thromboxanes (11). PGH2 is further metabolized by a group of PGE synthases to produce PGE2, of which mPGES-1 is also overexpressed in colon cancer samples. Heightened PGE2 synthesizing capacity as indicated by COX-2 overexpression is a negative prognostic factor and is a prime target for colon cancer chemoprevention and therapy. Specific inhibitor of COX-2 proved to be effective in reducing intestinal polyps in both number and size and in the prevention of progression to cancer. Despite the problems about their cardiovascular safety profile that ultimately prompted for their market withdraw, the COXIBs are a major proof of principle for the role of the arachidonic acid cascade in colon cancer (12-15).
The release of arachidonic acid (AA) sterified to membrane phospholipids is the first step in PGE2 synthesis and is performed by one or more enzymes belonging to the Phospholipase (PL)A2 family. This family of enzymes can be divided into four sub families based on molecular weight, calcium requirement, cellular localization and sequence homology: cytosolic (group IV); secretory (groups II, III, V, X XII); calcium-independent (group VI); and PAF acetyl-hydrolases (groups VII and VIII) (for review of PLA2 nomenclature and classification please see…) (16;17). Cytosolic (c)PLA2α (group IVA) is regarded as the major enzyme involved in AA mobilization for eicosanoid generation. However, other PLA2s, particularly secretory PLA2s, can participate in AA mobilization either alone or in cooperation with cPLA2α. cPLA2 presents constitutive catalytic activity towards monomeric substrates that is calcium independent, but in an intact cell setting its stimulus-initiated AA releasing capacity is dependent on calcium dependent translocation to target membranes (18). cPLA2 activity can also be augmented by phosphorylation of serine residues by MAP Kinases (19;20). A definitive role for cPLA2 on colon cancer has been difficult to ascribe since its activity supply free AA as substrate for PGE2 synthesis and as a bona fide pro-apoptotic signal, suggesting both pro- and anti-cancer potential roles (21-26).
Although there is a plethora of evidence on the role of COX-2 in PGE2 synthesis and the intracellular events that enhance its expression here is little evidence on the extra cellular signals and how they couple to intracellular signaling cascades triggering PLA2 activation and AA mobilization. Epithelial cells from the colonic epithelium are in constant contact with a a variable environment of enteric contents and respond to changes in osmotic pressure with compensatory molecular adaptations that allow them to reestablish normal cellular structure and function. Hypertonic induction of COX-2 in mouse in mouse inner medullary collecting duct cell line (mIMCD-K2), human colon cancer cell line (Caco-2) and human pulmonary epithelial cell line (A549) (27-30) suggest that AA cascade may be modulated by such environmental stress.
One of the mechanisms for which the cells respond to stress is modifying MAPKinases cascades, that are important proteins in transduction signaling (28;31-33). The MAP Kinases are formed by three major subgroups, extracellular-signal-regulated kinases (ERK), stress-activated protein kinases or N-terminal c-jun kinases (JNK or SAPK) and the MAPK14 or p38 (34;35). These signaling cascades lead to changes in PGE2 synthesis in colon cancer by increasing COX-2

142
and PGE synthases expression and enhancing cPLA2α activity. Based on the previously exposed, we sought to determine the effects of different PLA2 in supplying AA for PGE2 generation in colorectal carcinoma cells and the signaling pathways involved after the activation by hypertonic stress.
MATERIALS AND METHODS Reagents. Sodium chloride (NaCl) was obtained from Sigma-Aldrich. NaCl was dissolved in sterile pyrogen-free water (Millipore) at concentration of 2M. For hypertonic stress condition, the stock NaCl solution was diluted in sterile DMEM to the desired final concentration (0,01; 0,03 or 0,1M) in the cultures. Bromoenol lactone (BEL), Arachidonyl Trifluoromethyl Ketone (ATK), NS-398 and Prostaglandin E2 (PGE2) were obtained from Cayman Chemical and diluted in ethanol (Merck) or DMSO (Sigma-Aldrich) at concentration of 100, 30, 100 and 10 Mm, respectively, according manufacturer recommendation. p38 (SB202190), JNK (SP600125) and ERK1/2 (U0126) pharmacological inhibitors were obtained from BIOMOL Research Laboratories and diluted at concentration of 30, 20 and 13mM, respectively, according manufacturer recommendation. The [5,6,8,9,11,12,14,15-3H] Arachidonic Acid with specific activity of 214 Ci/mmol used to arachidonic acid releasing assays was obtained from Amersham Biosciences. Monoclonal antibodies for immunoblot assays were anti-COX-2 IgG mouse (33) from BD Transduction Laboratories, diluted at 1µg/mL; anti-phospho-p44/42 MAPK (Thr202/Tyr204) (E10) IgG mouse from Cell Signaling Technology, diluted at 0,25µg/mL, and anti-GAPDH (6C5) IgG mouse from Santa Cruz Biotechnology, diluted at 0,003µg/mL. Polyclonal antibodies for immunoblot assays were anti-phospho-p38 (pTpY 180/182) IgG rabbit and anti-phospho-JNK1&2 (SAPK) (pTpY 183/185) IgG rabbit, diluted at 0,25µg/mL, from BIOMOL Research Laboratories. The goat HRP-linked secondary antibodies were anti-IgG mouse from Santa Cruz Biotechnology, diluted at 0,1µg/mL, and anti-IgG rabbit from Jackson Immuno Research Laboratories, diluted at 0,08µg/mL.
Cell Culture and Culture Conditions. Caco-2 (ATCC, gift of Dr. José Morgado Díaz, National Institute of Cancer) cell line was maintained in Dulbecco modified Eagle medium (DMEM) (Invitrogen), supplemented with 10% fetal bovine serum (FBS), NaHCO3 (final concentration 44 mM), NaH2PO4.H2O (final concentration 1mM), piruvato de sódio (final concentration 1 mM), HEPES (final concentration 10 mM), MEM vitamins solution, MEM essential and non-essential amino acids solution, L-glutamin (final concentration 2 mM), β-mercaptoethanol (final concentration 55 µM), penicillin (final concentration 100.000 U) e streptomycin (final concentration 100 mg/L) (all reagents from Invitrogen). Cells were mainted in culture bottles (with 25, 75 and 150 cm2) and 6-well flat-bottom plates (area of 962 mm2/well) (Techno Plastic Products). Cells were collected by 0,25% trypsin and 0,38 g/L EDTA in HBSS without Ca++ and Mg++ and plated after reaching confluency. Cells were grown in culture medium at 37oC in a 5% CO2 atmosphere and all experiments were performed with subconfluent monolayers in full culture medium, after 12 hours of plating. Cell line was stimulated with addition of 100µL of sodium chloride (NaCl) 2M to DMEM medium containing 10% SFB to mimic hypertonic environment, the final osmolarity of medium was approximately 540 mOsm

143
(versus 367 mOsm to the common medium). The osmolarity of the medium was measured by freezing method using a osmometer (Advanced Micro Osmometer, Model 3300, da Advanced Instruments). PGE2 and Pharmacological Inhibitosr Treatments. Caco-2 cell line were plated in 6-well flat-bottom plates at a density of 5x105/9.6cm2 (5x105/well). Cells were maintained in 3 mL of DMEM 10% FBS for 12 hours at 37oC and 5%CO2. After cellular adhesion, the medium was changed and PGE2 or the pharmacological inhibitors were added 15 minutes before the addition of NaCl at final volume of 2 mL. The MAP Kinases inhibitors (SB202190, SP600125 and U0126) were added 30 minutes before exposure to stimuli. Enzyme Immuno Assay (EIA). PGE2 production by Caco-2 cell line was measured by EIA in culture supernatant. Medium was collected after 24h of cellular activation by hypertonic stress and centrifuged at 100 x g for 5 minutes to remove floating cells and frozen at –70oC. PGE2 levels were assayed using a human PGE2-specific EIA Kit (Prostaglandin E2 EIA Kit-Monoclonal; Cayman Chemical, Ann Arbor, USA). The development of the reaction was performed by Elmman’s reagent provide by the kit. The reading of plate was performed at a wavelength of 405 nm in a plte reader (Spectra Max 190, Molecular Devices). Arachidonic Acid Release. Caco-2 cells were collected and plated at 5x105 cells per well in 6-well tissue culture plates in 3 mL of DMEM containing 10% FBS as previously described. Cells were allowed to attach for 6 hours at 37oC and 5% CO2 atmosphere and then we were labeled overnight with 1 µCi/well [3H] arachidonic acid in 2 mL of DMEM containing 10%FBS. After labeling, Caco-2 cells were washed twice with PBS (KCl 2,67 mM; NaCl 137,93 mM; KH2PO4 1,47 mM; Na2HPO4 8,1 mM; pH 7,4) and full medium DMEM containing the pharmacological inhibitors at desired concentration was additioned for 15 minutes. Caco-2 were stimulated for the indicated time at each experiment. After that, the supernatant was removed and centrifuged at 1400 x g for 10 minutes at 4oC. The cells were collected by 0,1% Triton X-100 solution. For measuring the radioactivity, samples were prepared by addition of 1,6 mL of liquid scintillation Ultima Gold for aquous and non-aquous samples (PerkinElmer Life & Analytical Sciences) in 200µL of sample. The radioactivity released into the supernatant and into cellular lysate was measured by LS6000LL model scintillator from Beckman. counted and calculated as the percentage of total radioactivity (cellular and released). The percentage of arachidonic acid release for each treatment was calculated by the division between the amount of released radioactivity into supernatant and the amount of total radioactivity (supernatant radioactivity plus cellular lysate radioactivity) multiplied by 100 and subtracted from releasing observed in non-activated cells. Immunoblot Analysis. Cells were collected by cell scraper (COSTAR) and by addition of 100 µL of SDS 10% per well of culture plate to obtain the cellular lysate. After that, 100 µL of loading buffer was added [Loading buffer 2x and 10% β-Mercaptoethanol (Loading buffer stock solution 5x: 4,2g of Tris Base; 10mL of glycerol; 10ml of 0,1% Bromophenol blue; 15g of SDS in final volume of 75mL; pH 6,8)]. Then, cellular lysates were immediately warmed at 100oC for

144
5 minutes. Cellular lysates were sonicated at 30% of amplitude and 30 Joules of energy in a high intensity ultra-sonic processor. Samples were resolved by electrophoresis on a SDS-PAGE 10% polyacrylamide gel [Acrylamide/Bis-acrylamide (29:1)] at 29 mA/gel for 1 hour. After electrophoresis, the proteins were transferred to nitrocellulose membranes (BioAgency) at 250 mA for 2 hours at 4oC. The membranes were blocked with TBS 1x (Tris 10 mM; NaCl 150 mM pH 7,4) and 5% of dry milk for 12 hours, to COX-2 labeling, or for 2 hours, to another labeling, under shaking at room temperature. The membranes were washed three times with TTBS (TBS 1x; Tween-20 0,2%) for 10 minutes at room temperature. The membranes were incubated with primary antibodies diluted in TTBS and 0,05% of sodium azide at concentration recommended by manufacturer, as previously mentioned, for 2 or 12 hours at 4oC. After incubation, membranes were washed three times with TTBS for 15 minutes and incubated with HRP-linked secondary antibodies anti-IgG at concentration recommended by manufacturer, as previously mentioned, for 1 hour at room temperature. The membranes were washed three times with TTBS for 10 minutes and developed by luminescence using the ECLTM Western Blotting Analysis System (Amersham Biosciences), according manufacturer’s instructions. The X-ray films (Light Film Kodak BioMax) were exposed to the membranes and developed by automatic developer (Kodak X-OMAT 2000 Processor). Statistical Analysis. Data are expressed as means and standard error mean (SEM). Multiple comparisons among groups were performed by one-way ANOVA and Bonferroni’s or Dunnett’s test. P values < 0.05 were considered statistically significant. RESULTS
Hypertonic Stress induces AA releasing, COX-2 expression and PGE2 production in Caco-2—To study the role of PLA2s for COX-2 dependent PGE2 generation in Caco2 activated by hypertonic stress we initially examined if the addition of 0,1M of sodium chloride (NaCl) to the medium DMEM 10%FBS have stimulated COX-2 expression and PGE2 production in Caco2. As shown in Fig.1, the AA release was measured at 2, 4, 8, 12, 18 and 24 hours of hypertonic stress and the level of AA started to increase at 12 hours (above 5%) and have maximal release at 24 hours (approximately 18%). EIA and Immunoblotting revealed that hypertonic stress induces maximum COX-2 expression and PGE2 production in Caco-2 stimulated with 100mM of NaCl after 24 hour. Then, the next experiments were performed with 24 hours of stimulation and addition of 100 mM of NaCl to medium because in these conditions we have maximal AA releasing, COX-2 expression and PGE2 production by Caco-2. The PGE2 production depends on cPLA2 and COX-2 activities in Caco2 stimulated by osmotic stress—The use of NS 398, a COX-2 specific inhibitor, and ATK, a cPLA2 inhibitor, have decreased the PGE2 production in Caco2 (Figures 2A and 2B). However, the use of sPLA2 inhibitor, LY311727, and iPLA2 inhibitor, BEL, have not effect on this production (Fig. 2C and 2D).

145
cPLA2 regulates AA supplying in Caco-2 stimulated by hypertonic stress — It was shown previously that cPLA2 can modulate AA release and COX-2 expression (36;37). Then, we verified if cPLA2 was regulating PGE2 production in Caco-2 by COX-2 expression or by AA supplying. Caco-2 were stimulated by hypertonic stress and treated with ATK at 1, 3 and 10 µM and we did not verify any changes in COX-2 expression, suggesting the role of cPLA2 is not in regulating the COX-2 expression (Fig. 3A). Because we have shown that the maximal AA release was at 24 hours, the effect of ATK, a cPLA2 inhibitor, in the AA release was observed at that time. ATK inhibited ~90% of AA release by Caco-2 showing that cPLA2 plays a role in AA supplying to PGE2 production by Caco-2 in hypertonic stress (Fig. 3B). ERK1/2, JNK and p38 participate in PGE2 generation by Caco2 stimulated with hypertonic stress— Considering that cells respond to hypertonic stress modifying MAPKs signaling (28;33;38;39), we observe if the main kinases involved in the response to stress in mammals were modulating the AA pathway. First, we observed that ERK1/2, SAPK/JNK and p38 play a role in PGE2 production in Caco2 stimulated by hypertonic stress, because the use of MAPK inhibitors decreased this production (Fig. 4). Hypertonic stress modulates MAPKs expression in Cao-2— As shown in Fig. 4 all three subgroups of kinases influence the PGE2 production in hypertonic stress. So, we verified if this type of stimulation could modulate the MAPKs expression and we observed that hypertonic stress induces phosphorylation of ERK1/2, JNK and p38 at 30 minutes and 2 hours after stimulation (Fig. 5). MAPKs modulate the COX-2 expression and AA supplying by cPLA2 in Caco-2 activated by hypertonic stress— To investigate the role of MPAKs on AA metabolism we tested if the kinases were modulating the COX-2 expression or the AA supplying in Caco-2. The role of MAPKs on COX-2 expression induced by hypertonic stress was assayed by immunoblotting that revealed only inhibition of ERK1/2 on COX-2 induction. Otherwise, p38 and JNK inhibit the expression of this enzyme (Fig. 6). cPLA2 is activated by increase of intracellular calcium level and by phosphorylation of serine residues, allowing to its translocation from the cytosol to the nuclear envelope (40-43) and MAPKs can phosphorylate cPLA2 at Ser505, integrating this enzyme in major signaling pathways triggered by activation of a diversity of receptors (43). To study if MAPKs modulate the AA supplying by cPLA2 we treated Caco-2 with kinases inhibitors and our results indicate that all three kinases modulate the activity of cPLA2. All MPAKs pharmacological inhibitors were able to inhibit in ~50-90% the AA releasing in Caco-2 stimulated by hypertonic stress (Figure 7).

146
DISCUSSION
The additon of 100 mM of NaCl to culture medium mimics the hypertonic stress found in intestinal physiological environment, with water absorption (44;45). The osmolartity of this hypertonic medium is approximately 540 mOsm, this data is in agreement with those found in the literature about final osmolariy in colon lumen (27;27;28;44;46). The final osmolarity in medium without addition of NaCl is about 367 mOsm, which characterizes an isotonic environment (28;46). Caco-2 cell line can produce PGE2 and express constitutively COX-2, which is augmented by stimulation with NaCl (Fig. 1C). Then, Caco-2 cell line has the enzymatic profile necessary to produce protanoids, however, we do not know which PLA2 is involved on PGE2 production in this type of environmental stress. We observed that hypertonic stress induces the maximal COX-2 expression and PGE2 production (Fig.1) by Caco-2 after 24 hours of activation. Hypertonic medium induces COX-2 expression and prostacyclin (PGI2) in other cell lines, such as endothelial cells (47), lung epithelial cells (30), kidney medullar cells (27) and colon cancer cells (29). This last study demonstrated that the addition of 100 mM of NaCl to culture medium induced the augment of COX-2 expression by Caco-2 after 6 to 8 hours, which is similar to our results with 24 hous of activation by hypertonic stress.
Using pharmacological approach to evaluate what PLA2 was involved in PGE2 production in hypertonic stress, we observed that the PGE2 production by Caco-2 stimulated with hypertonic stress depends on COX-2 and cPLA2 activity. The use of inhibitors like ATK presents some disadvantages as the unspecificity of the pharmacological inhibitor, because ATK inhibit cPLA2, iPLA2 and cyclooxygenases (16;48). Nevertheless, these disadvantages can be reduced with the use of specific inhibitors for COX-2 (NS-398) and for iPLA2 (BEL). Another kind of disadvantage presents by inhibitor LY311727 is its unpermeability to cytoplasmic membrane (49). Then, the result shows sPLA2 do not participate on PGE2 production in hypertonic stress (Fig. 2C) must be analysed considering that this is related to action of sPLA2 on the outside of cytopalsmic membrane and we do not conclude about the action before their secretion. We tested also other sPLA2 inhibitor, 12-epi-escalaradial, which did not inhibit the prostanoid production in hypertonic stress (data not shown), but there is not much information about the permeability of this inhibitor.
The results with the inhibitor ATK show cPLA2 do not play a role on COX-2 expression in Caco-2 (Fig. 5A). In others cell types the cPLA2 modulates the COX-2 expression, like bone marrow-derived mast cells in cPLA2α-deficient mice that show decreased ability to induce the expression of COX-2 in approximately 70% after stimulation by growth factors and cytokines (37). Furthermore, kinetic and pharmacological studies in a osteoblastic cell line showed that delayed PGE2 generation by this cell line in response to interleukin (IL)-1β and tumor necrosis factor (TNF) α was dependent upon cPLA2 and COX-2. Expression of these two enzymes was reduced by cPLA2 or COX-2 inhibitors and restored by adding exogenous arachidonic acid or PGE2, indicating that PGE2 produced by these cells acted as an autocrine amplifier of delayed PGE2 generation through enhanced cPLA2 and COX-2 expression (50). In colon cancer, there is a study that shows increase of cPLA2 and COX-2 expression in 50% of the 65 human colon tumors analysed and in several colon cancer cell lines. However, authors did not demonstrate regulation of COX-2 by cPLA2 in these cell lines (9).
The other step of the AA metabolism, besides COX-2 expression, where cPLA2 could be modulating is AA releasing. Hypertonic stress induces maximal AA release after 24 hours of

147
activation (Fig. 1A) and, with the block of cPLA2 activity, AA releasing was reduced about 90%, showing that cPLA2 regulates the AA supplying to PGE2 production (Fig. 3B). Although there are not studies about the role of cPLA2 in neoplasic cells activated by hypertonic stress, a study in human cholangiocarcinoma cells stimulated by Hepatocyte growth factor (HGF) and IL-6 revealed that cPLA2 plays a role on AA supplying to PGE2 generation (51). The role of AA and cPLA2 in cancer has been broadly studied with in vivo models, like in cPLA2 knockout mice (23), and with in vitro models, using cell lines (52;53). These models revealed a ambiguous role for cPLA2. cPLA2 could be responsible for AA releasing in the production of PGE2, which would increase proliferation of neoplastic cells (1;5;12). However, some studies show that cPLA2 would inhibit the tumoral development because AA would be a proapoptotic signal for cancer cells (23-26). The possibility that PGE2 produced by Caco-2 in hypertonic stress could modulate cellular proliferation was tested. Then, we treated cells with COX-2 inhibitor (NS-398) for blocking PGE2 generation and activated them by hypertonic stress during 24 hours and we did not verify inhibition of cellular proliferation, suggesting that PGE2 has not a proproliferative role in hypertonic stress (data not shown).
Mammals cells respond to environmental stress modifying MAPKs cascades to adapt to new conditions (28;33;38;39). Under hypertonic stress the phosphorylation of ERK1/2, SAPK and p38 ocurred at 30 minutes and 2 hours and these activation lead to PGE2 production by Caco-2 (Figs. 6 and 7). Our results are in agreement with others indicating the involvement of MAPKs on PGI2 production by endothelial cells, on augment of expression of COX-2 by colon cancer cells and on cellular survival of renal medullary cells in response to hypertonic conditions (27-29;47;54). Using pharmacological blockade of MAPKs we have demonstrated that ERK1/2, JNK and p38 modulate the cPLA2 activity on AA releasing (Fig. 9), this result is confirmed in part by findings in MDCK cells stimulated by ATP and ionomycin and treated with ERK1/2 inhibitor that had presented a reduction of AA releasing in a phosphorylation and translocation cPLA2 independent way (55).
cPLA2 protein is regulated by phosphorylation and intracellular influx of calcium. Calcium promotes the binding of enzyme to the membrane through a calcium binding loop (CBL) found in the C2 domain (40-42). The treatment of cells with calcium mobilizing agonists induces the binding of cPLA2 to the nuclear membrane and to the endoplasmic reticulum (56-58). Moreover, these agonists induce AA releasing and phosphorylation on serine residues of cPLA2, increasing the enzymatic activity (59-61). Then, MAPKs play a role on cPLA2 activation, it has been demonstrated that the phosphorylation of Ser505 by ERK1/2 is necessary for AA releasing by cPLA2 in CHO cells treated with different agonists (43). The role of MAPKs is controversy because in some systems these kinases are important to phosphorylate of cPLA2 but in other systems acting by different way on AA releasing in conditions where there is not increase of intracellular clacium like induction by okadaic acid (62-64). Probably MAPKs have been acting on the induction of AA releasing by cPLA2 in hypertonic stress through a phosphorylation cPLA2 independent mechanism because we did not verify any modification on phosphorylation of cPLA2 at several stimulation times. In addition, the treatment of Caco-2 with MAPKs inhibitors and stimulated by hypertonic medium during 8 hours did not modify the phosphorylation cPLA2 (data nor shown). cPLA2 is phosphorylated on Ser505 in vitro by ERK1/2 (43). However, the inhibiton of ERK1/2 reduces the AA releasing through a phosphorylation cPLA2 independent mechanism in MDCK cells (55), this fact strengths our results demonstrating the role of MAPKs on induction of AA releasing by cPLA2 in hypertonic stress.
We postulated that MAPKs could modulate the COX-2 expression in Caco-2 activated by hypertonic stress and our results have indicated ERK1/2 is involved on COX-2 induction whereas

148
p38 and JNK inhibit this enzyme expression (Fig. 8). These results differ from results in renal medullary collecting duct cells grown in hypertonic medium, wich induction COX-2 expression is dependent of ERK1/2, JNK and p38 (27;28) and in human monocytes activated by LPS, which transcription and stabilization of COX-2 mRNA depends on p38 (65). In addition, studies with endothelial (47) and colon cancer cells (29) show that the hypertonic stress induces the COX-2 expression dependent of p38, differing from ours results.
Taken together our data show that hypertonic stress induces AA releasing, COX-2 expression and PGE2 production by Caco-2. The PGE2 depends on cPLA2 and COX-2 activities and cPLA2 regulates the AA supplying in hypertonic environment. MPAKs participate in PGE2 generation, however ERK1/2 is involved in COX-2 induction, while ERK1/2, JNK and p38 modulate cPLA2 activity.
REFERENCES
1. Sheng H, Shao J, Washington MK, and DuBois RN (2001) J Biol Chem 276, 18075-18081
2. Fukuda R, Kelly B, and Semenza GL (2003) Cancer Res 63, 2330-2334
3. Tomozawa S, Tsuno NH, Sunami E, Hatano K, Kitayama J, Osada T, Saito S, Tsuruo T, Shibata Y, and Nagawa H (2000) Br J Cancer 83, 324-328
4. Wang D, Wang H, Brown J, Daikoku T, Ning W, Shi Q, Richmond A, Strieter R, Dey SK, and DuBois RN (2006) J Exp Med 203, 941-951
5. Castellone MD, Teramoto H, Williams BO, Druey KM, and Gutkind JS (2005) Science 310, 1504-1510
6. Sonoshita, M., Takaku, K., Sasaki, N., Sugimoto, Y., Ushikubi, F., Narumiya, S., Oshima, M., and Taketo, M. M. (2001) Nat Med 7, 1048-1051
7. Hansen-Petrik MB, McEntee MF, Jull B, Shi H, Zemel MB, and Whelan J (2002) Cancer Res 62, 403-408
8. Watanabe K, Kawamori T, Nakatsugi S, Ohta T, Ohuchida S, Yamamoto H, Maruyama T, Kondo K, Ushikubi F, Narumiya S, Sugimura T, and Wakabayashi K (1999) Cancer Res 59, 5093-5096
9. Panel V, Boelle PY, Ayala-Sanmartin J, Jouniaux AM, Hamelin R, Masliah J, Trugnan G, Flejou JF, and Wendum D (2006) Cancer Letters 243, 255-263
10. Eibl G, Bruemmer D, Okada Y, Duffy JP, Law RE, Reber HA, and Hines OJ (2003) Biochemical and Biophysical Research Communications 306, 887-897
11. Diaz BL and Arm JP (2003) Prostaglandins Leukot Essent Fatty Acids 69, 87-97

149
12. Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, and Taketo MM (1996) Cell 87, 803-809
13. Taketo MM (1998) J Natl Cancer Inst 90, 1609-1620
14. Bertagnolli MM (1999) Curr Oncol Rep 1, 173-178
15. Sheng H, Shao J, Kirkland SC, Isakson P, Coffey RJ, Morrow J, Beauchamp RD, and DuBois RN (1997) J Clin Invest 99, 2254-2259
16. Balsinde J, Balboa MA, Insel PA, and Dennis EA (1999) Annu Rev Pharmacol Toxicol 39, 175-189
17. Yedgar S, Lichtenberg D, and Schnitzer E (2000) Biochim Biophys Acta 1488, 182-187
18. Leslie CC (1997) J Biol Chem 272, 16709-16712
19. Magne S, Couchie D, Pecker F, and Pavoine C (2001) J Biol Chem 276, 39539-39548
20. Hefner Y, Borsch-Haubold AG, Murakami M, Wilde JI, Pasquet S, Schieltz D, Ghomashchi F, Yates JRIII, Armstrong CG, Paterson A, Cohen P, Fukunaga R, Hunter T, Kudo I, Watson SP, and Gelb MH (2000) J Biol Chem 275, 37542-37551
21. Takaku K, Sonoshita M, Sasaki N, Uozumi N, Doi Y, Shimizu T, and Taketo MM (2000) J Biol Chem 275, 34013-34016
22. Hong KH, Bonventre JC, O'Leary E, Bonventre JV, and Lander ES (2001) Proc Natl Acad Sci U S A 98, 3935-3939
23. Ilsley JNM, Nakanishi M, Flynn C, Belinsky GS, De Guise S, Adib JN, Dobrowsky RT, Bonventre JV, and Rosenberg DW (2005) Cancer Res 65, 2636-2643
24. Levine LAWR (2003) FASEB J 17, 800-802
25. Taketo MM and Sonoshita M (2002) Biochim Biophys Acta 1585, 72-76
26. Cao Y, Pearman AT, Zimmerman GA, McIntyre TM, and Prescott SM (2000) Proc Natl Acad Sci U S A 97, 11280-11285
27. Yang T, Zhang A, Honeggar M, Kohan DE, Mizel D, Sanders K, Hoidal JR, Briggs JP, and Schnermann JB (2005) J Biol Chem 280, 34966-34973
28. Yang T, Huang Y, Heasley LE, Berl T, Schnermann JB, and Briggs JP (2000) J Biol Chem 275, 23281-23286
29. Arbabi S, Rosengart MR, Garcia I, Jelacic S, and Maier RV (2001) J Surg Res 97, 60-64
30. Lim WC, Park M, Bahn JJ, Inoue H, and Lee YJ (2005) FEBS Lett 579, 5430-5436

150
31. Ruis H and Schuller C (1995) Bioessays 17, 959-965
32. Albertyn J, Hohmann S, Thevelein JM, and Prior BA (1994) Mol Cell Biol 14, 4135-4144
33. Kultz D and Burg M (1998) J Exp Biol 201, 3015-3021
34. Johnson GL and Lapadat R (2002) Science 298, 1911-1912
35. Rennefahrt U, Janakiraman M, Ollinger R, and Troppmair J (2005) Cancer Letters 217, 1-9
36. Balsinde J, Shinohara H, Lefkowitz LJ, Johnson CA, Balboa MA, and Dennis EA (1999) J Biol Chem 274, 25967-25970
37. Fujishima H, Sanchez Mejia RO, Bingham COIII, Lam BK, Sapirstein A, Bonventre JV, Austen KF, and Arm JP (1999) Proc Natl Acad Sci U S A 96, 4803-4807
38. Galcheva-Gargova Z, Derijard B, Wu IH, and Davis RJ (1994) Science 265, 806-808
39. Han J, Lee JD, Bibbs L, and Ulevitch RJ (1994) Science 265, 808-811
40. Channon JY and Leslie CC (1990) J Biol Chem 265, 5409-5413
41. Clark JD, Lin LL, Kriz RW, Ramesha CS, Sultzman LA, Lin AY, Milona N, and Knopf JL (1991) Cell 65, 1043-1051
42. Nalefski EA, Sultzman LA, Martin DM, Kriz RW, Towler PS, Knopf JL, and Clark JD (1994) J Biol Chem 269, 18239-18249
43. Nemenoff RA, Winitz S, Qian NX, Van Putten V, Johnson GL, and Heasley LE (1993) J Biol Chem 268, 1960-1964
44. Thiagarajah JR, Jayaraman S, Naftalin RJ, and Verkman AS (2001) Am J Physiol Cell Physiol 281, C1898-C1903
45. Powell DW (1995) J Clin Invest 96, 2102-2103
46. Hubert A, Cauliez B, Chedeville A, Husson A, and Lavoinne A (2004) Biochimie 86, 533-541
47. Arbabi S, Rosengart MR, Garcia I, and Maier RV (2000) Surgery 128, 198-205
48. Riendeau D, Guay J, Weech PK, Laliberte F, Yergey J, Li C, Desmarais S, Perrier H, Liu S, and Nicoll-Griffith D (1994) J Biol Chem 269, 15619-15624
49. Mounier CM, Ghomashchi F, Lindsay MR, James S, Singer AG, Parton RG, and Gelb MH (2004) J Biol Chem 279, 25024-25038

151
50. Murakami M, Kuwata H, Amakasu Y, Shimbara S, Nakatani Y, Atsumi G, and Kudo I (1997) J Biol Chem 272, 19891-19897
51. Wu T, Han C, Lunz JGIII, Michalopoulos G, Shelhamer JH, and Demetris AJ (2002) Hepatology 36, 363-373
52. Lagorce-Pages C, Paraf F, Wendum D, Martin A, and Flejou JF (2004) Virchows Arch 444, 426-435
53. Soydan AS, Tavares IA, Weech PK, Temblay NM, and Bennett A (1996) Eur J Cancer 32A, 1781-1787
54. Zatechka SDJr and Lou MF (2002) Exp Eye Res 74, 703-717
55. Evans JH, Fergus DJ, and Leslie CC (2002) BMC Biochem 3, 30
56. Schievella AR, Regier MK, Smith WL, and Lin LL (1995) J Biol Chem 270, 30749-30754
57. Sierra-Honigmann MR, Radley JR, and Prober JS (1996) Lab Invest 74, 684-695
58. Glover S, Bayburt T, Jonas M, Chi E, and Gelb MH (1995) J Biol Chem 270, 15359-15367
59. Clark JD, Schievella AR, Nalefski EA, and Lin LL (1995) J Lipid Mediat Cell Signal 12, 83-117
60. Qiu ZH, de Carvalho MS, and Leslie CC (1993) J Biol Chem 268, 24506-24513
61. Lin LL, Lin AY, and DeWitt DL (1992) J Biol Chem 267, 23451-23454
62. Ambs P, Baccarini M, Fitzke E, and Dieter P (1995) Biochem J 311, 189-195
63. Kramer RM, Roberts EF, Um SL, Borsch-Haubold AG, Watson SP, Fisher MJ, and Jakubowski JA (1996) J Biol Chem 271, 27723-27729
64. de Carvalho MG, McCormack AL, Olson E, Ghomashchi F, Gelb MH, Yates JRIII, and Leslie CC (1996) J Biol Chem 271, 6987-6997
65. Dean JL, Brook M, Clark AR, and Saklatvala J (1999) J Biol Chem 274, 264-269

152
Figure 1: Hypertonic stress induces AA release, COX-2 expression and PGE2 production in Caco-2. A, Net AA released by Caco-2 at 2-24 hours after the addition of 100mM of NaCl. B, PGE2 production (pg/ml) by Caco-2 after the addition of 100mM of NaCl. The * means p < 0,05 as compared to non stimulated cells by hypertonic stress. C, Caco-2 cells were growth in medium without or with 10-100mM of NaCl. Total cellular protein was obtained by cellular lysis and Western blot analysis was performed with monoclonal anti-COX-2 antibody. Graph bars show mean and SEM from triplicate samples. The graph A represents one experiment. The graph B and the figure C represent three independent experiments.
0
2000
4000
6000
8000
10000
12000
NaCl (M) - 0.10.03
*
0.01
B
PGE 2
(pg/
ml)
2 4 8 12 18 24
0
10
20
Tempo (h)
*
A
NaCl 0.1M
Net
AA
rele
ase
(%)
C
COX-2
NaCl (M)_______________- 0.03 0.10.01
GAPDH
70 kDa
37 kDa
COX-2 COX-2 COX-2
NaCl (M)_____________________________________________- 0.03 0.10.01
GAPDH GAPDH GAPDH
70 kDa70 kDa70 kDa
37 kDa37 kDa37 kDa
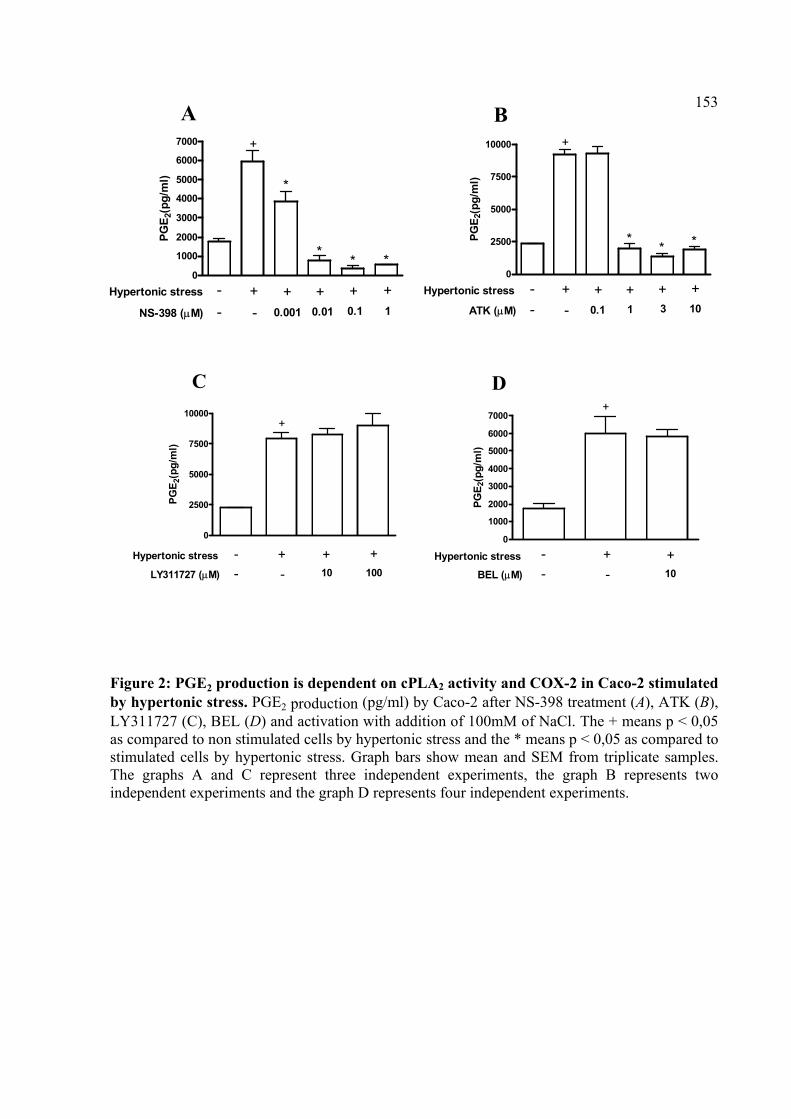
153
Figure 2: PGE2 production is dependent on cPLA2 activity and COX-2 in Caco-2 stimulated by hypertonic stress. PGE2 production (pg/ml) by Caco-2 after NS-398 treatment (A), ATK (B), LY311727 (C), BEL (D) and activation with addition of 100mM of NaCl. The + means p < 0,05 as compared to non stimulated cells by hypertonic stress and the * means p < 0,05 as compared to stimulated cells by hypertonic stress. Graph bars show mean and SEM from triplicate samples. The graphs A and C represent three independent experiments, the graph B represents two independent experiments and the graph D represents four independent experiments.
0
1000
2000
3000
4000
5000
6000
7000
Hypertonic stress --
+ + + +-NS-398 (µM) 0.001
+0.1 1
+
*
* **
A
0.01
PGE 2
(pg/
ml)
0
2500
5000
7500
10000
Hypertonic stress --
+ + + +-ATK (µM) 0.1
+3 10
+
* * *
B
1
PGE 2
(pg/
ml)
0
2500
5000
7500
10000
Hypertonic stress --
+ +-LY311727 (µM) 10 100
+
C
+
PGE 2
(pg/
ml)
0
1000
2000
3000
4000
5000
6000
7000
Hypertonic stress --
+ +BEL (µM) -
+
D
10
PGE 2
(pg/
ml)

154
Figure 3: cPLA2 regulates AA supply in Caco-2 stimulated by hypertonic stress. A, COX-2 expression in Caco-2 stimulated with 100mM of NaCl and treated with 1-10µM of ATK. Total protein extract was extracted by cellular lysis and analysed by Western blot with monoclonal anti-COX-2 antibody. B, Net AA released by Caco-2 after ATK treatment and activation with addition of 100mM of NaCl to the medium. The * means p < 0,05 as compared to activated cells by hypertonic stress and non treated with ATK. The figure A represents three independent experiments. Graph bars show mean and SEM from triplicate samples. The graph B represents two independent experiments.
Hypertonic stress +-ATK (10µM) +
0.0
2.5
5.0
7.5
10.0
*
B
+
Net
3 H A
A r
elea
se (%
)
COX-2
ATK (µM) - - 3 10______________NaCl 0,1M
1
GAPDH
70 kDa
37 kDa
COX-2
ATK (µM) - - 3 10______________NaCl 0,1M
1
GAPDH
COX-2
ATK (µM) - - 3 10______________NaCl 0,1M
1
GAPDH
70 kDa70 kDa70 kDa
37 kDa37 kDa37 kDa
A

155
Figure 4: Hypertonic stress modulates the phosphorylation of MAPKs in Caco-2. Phosphorylation of p44/42 MAPK (ERK), JNK and p38 in Caco-2 stimulated by addition of 100mM of NaCl to the medium for 0.5-24 hours. Total protein extract was obtained by cellular lysis and analyzed by Western blot with monoclonal anti-fosfo-p44/42 MAPK (Thr202/Tyr204) (E10), polyclonal anti-fosfo-p38 (pTpY 180/182), polyclonal anti-fosfo-JNK (pTpY 183/185) and monoclonal anti-GAPDH (6C5) antibodies. Each figure represents three independent experiments.
GAPDH
GAPDH
GAPDH
JNK
-- -- - -+ + + + + +0.5 2 4 8 12 24
GAPDH GAPDH
GAPDH
p38
GAPDH
p38
GAPDH
JNK
GAPDH
JNK
-- -- - -+ + + + + +0.5 2 4 8 12 24
-- -- - -+ + + + + +0.5 2 4 8 12 24
42 kDa
37 kDa
38 kDa
46 kDa
37 kDa
37 kDa
44 kDa
GAPDH
GAPDH
GAPDH
JNK
-- -- - -+ + + + + +0.5 2 4 8 12 24
GAPDH
Phospho-p44 MAPK
GAPDH
Phospho-p42 MAPK
GAPDH GAPDH
Phospho
GAPDH
JNK
GAPDH
PhosphoJNK
Hypertonic stress -- -- - -+ + + + + +0.5 2 4 8 12 24
-- -- - -+ + + + + +0.5 2 4 8 12 24 hours
42 kDa
37 kDa
38 kDa
46 kDa
37 kDa
37 kDa
44 kDa
GAPDH
GAPDH
GAPDH
JNK
-- -- - -+ + + + + +0.5 2 4 8 12 24
GAPDH GAPDH
GAPDH
p38
GAPDH
p38
GAPDH
JNK
GAPDH
JNK
-- -- - -+ + + + + +0.5 2 4 8 12 24
-- -- - -+ + + + + +0.5 2 4 8 12 24
42 kDa
37 kDa
38 kDa
46 kDa
37 kDa
37 kDa
44 kDa
GAPDH
GAPDH
GAPDH
JNK
-- -- - -+ + + + + +0.5 2 4 8 12 24
GAPDH
Phospho-p44 MAPK
GAPDH
Phospho-p42 MAPK
GAPDH GAPDH
Phospho
GAPDH
JNK
GAPDH
PhosphoJNK
Hypertonic stress -- -- - -+ + + + + +0.5 2 4 8 12 24
-- -- - -+ + + + + +0.5 2 4 8 12 24 hours
42 kDa
37 kDa
38 kDa
46 kDa
37 kDa
37 kDa
44 kDa

156
Figure 5: ERK1/2, JNK and p38 kinases play a role in PGE2 production by Caco-2 stimulated by hypertonic stress. PGE2 production in Caco-2 trested with kinases inhibitors to JNK (SP 600125), to p38 (SB 202190), to ERK1/2 (U 0126) and activated with addition of 100mM of NaCl to the medium. The + means p < 0,05 as compared to non activated cells by hypertonic stress and the * means p < 0,05 as compared to activated cells by hypertonic stress and non treated with MAPKs inhibitors. Graph bars represent mean and SEM of samples in triplicate. The graph represents three independent experiments.
0
10000
20000
30000
Hypertonic stress - + + + +
+
- -SP 600125 (20µM) --
- -SB 202190 (15µM) - -
- -U 0126 (20µM) - -
+
+
*
* *
+
PGE 2
(pg/
ml)

157
Figure 6: MAPKs modulate COX-2 expression in Caco-2 stimulated by hypertonic stress. COX-2 and GAPDH expression in Caco-2 activated with 100mM of NaCl and treated with JNK (SP600125), p38 (SB202190) or ERK1/2 (U0126) inhibitors. Total protein extract was obtained by cellular lysis and analysed by Western blot with monoclonal anti-COX-2 and monoclonal anti-GAPDH (6C5) antibodies. The figure represents three independent experiments.
- -SP 600125
(20µM)SB 202190
(15µM)U 0126 (20µM)
NaCl 0.1 M
Con
trol
COX-2
GAPDH
70 kDa
37 kDa
- -SP 600125
(20µM)SB 202190
(15µM)U 0126 (20µM)
NaCl 0.1 M
Con
trol
COX-2
GAPDH
- -SP 600125
(20µM)SB 202190
(15µM)U 0126 (20µM)
NaCl 0.1 M
Con
trol
COX-2
GAPDH
70 kDa
37 kDa

158
Figure 7: ERK1/2, JNK and p38 play a role in AA release by cPLA2 in Caco-2 stimulated by hypertonic stress. Net AA released by Caco-2 after treatment with cPLA2 inhibitor (ATK) or JNK (SP 600125), p38 (SB 202190) or ERK1/2 (U 0126) inhibitors and activation with the addition of 100mM of NaCl to the medium during 24 hours. The * means p < 0,05 as compared to activated cells by hypertonic stress and non treated with inhibitors. Graph bars represent mean and SEM from samples in triplicate. The graph represents two independent experiments.
0.0
2.5
5.0
7.5
10.0
Hypertonic stress + + + +
+
- -SP 600125 (20µM) --
- -SB 202190 (15µM) - -
- -U 0126 (20µM) - -
+
+
+- -ATK (10µM) -+
**
**
-
Net
3 H A
A r
elea
se (%
)

Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas

Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo