Embed Size (px)
Citation preview

Teresa Cristina Raposo Löwen
Desenvolvimento e Validação da Metodologia de Análise do Teor de
Lamivudina e do Ensaio Limite do Enantiômero (+)BCH-189 em
Comprimidos de Lamivudina.
PPGVS/INCQS
FIOCRUZ
2003

Desenvolvimento e Validação da Metodologia de Análise do Teor de
Lamivudina e do Ensaio Limite do Enantiômero (+)BCH-189 em
Comprimidos de Lamivudina.
Teresa Cristina Raposo Löwen
Programa de Pós-Graduação em Vigilância Sanitária
Instituto Nacional de Controle de Qualidade em Saúde
Fundação Oswaldo Cruz
Orientadores: Dra. Elizabeth Moreira dos Santos
Dra. Tereza Cristina dos Santos
Rio de Janeiro
2003
ii

Desenvolvimento e Validação da Metodologia de Análise do Teor de Lamivudina e
do Ensaio Limite do Enantiômero (+)BCH-189 em Comprimidos de Lamivudina.
Teresa Cristina Raposo Löwen
Dissertação submetida à Comissão Examinadora composta pelo corpo docente do
Programa de Pós-Graduação em Vigilância Sanitária do Instituto Nacional de Controle de
Qualidade em Saúde da Fundação Oswaldo Cruz e por professores convidados de outras
instituições, como parte dos requisitos necessários à obtenção do grau de Mestre.
Aprovado:
Prof. _______________________________________
Dra. Elizabeth Moreira dos Santos
Prof. _______________________________________
Dra. Elizabete Pereira dos Santos
Prof. _______________________________________
Dra. Sheila Garcia
Orientador: ________________________________________
Dra. Elizabeth Moreira dos Santos
Orientador: ________________________________________
Dra. Tereza Cristina dos Santos
Rio de Janeiro
2003
iii

Löwen, Teresa Cristina Raposo
Desenvolvimento e Validação da Metodologia de Análisedo Teor de Lamivudina e do Ensaio Limite do Enantiômero(+)BCH-189 em Comprimidos de Lamivudina./ Teresa CristinaRaposo Löwen. Rio de Janeiro: INCQS/ FIOCRUZ, 2003. xiv, 81 p., il., tab. Dissertação em Vigilância Sanitária, Prog. Pós-Graduação emVigilância Sanitária/ INCQS, 2001. Orientadores: Dra. ElizabethMoreira dos Santos; Dra. Tereza Cristina dos Santos. 1. Lamivudina e (+)BCH-189. 2. Antiretroviral. 3. Validação.4. Cromatografia líquida de alta eficiência.
I. Desenvolvimento e Validação da Metodologia de Análise doTeor de Lamivudina e do Ensaio Limite do Enantiômero(+)BCH-189 em Comprimidos de Lamivudina.
iv

Aos meus Pais, Avelina Raposo Löwen e Manoel
Antonio de Almeida Löwen “ in memoriam”
e ao meu irmão Luiz Claudio Raposo Löwen.
v

“ Em relação a todos os atos de iniciativa e de criação existe uma
verdade elementar: no momento em que nos compromissamos a
Providência também se põe em movimento. Todo um fluir de
acontecimentos surge a nosso favor. Como resultado da decisão,
seguem todas as formas de coincidências, encontros e ajuda que
nenhum homem jamais poderia ter sonhado encontrar. Qualquer
coisa que você possa fazer ou sonhar, você pode começar .
A coragem contém, em si mesma, o poder, o gênio e a magia.”
Goethe
vi

AGRADECIMENTOS
Gostaria de agradecer em primeiro lugar à minha orientadora Dra. Elizabeth Moreira dos
Santos, por ter lançado a semente, colocando a minha qualificação como uma condição
para o meu crescimento profissional, e sobretudo pelo carinho e afeto com o qual sempre
fui tratada durante todo o nosso convívio.
Agradeço especialmente à minha orientadora e amiga Dra. Tereza Cristina dos Santos
pelo estimulo, confiança, paciência e muitas vezes pela cobrança.
À Dra. Paula Fernandes de Aguiar, por sempre ter encontrado tempo para solucionar as
minhas dúvidas relativas a cálculos estatísticos, o meu muitíssimo obrigada.
À minha amiga e companheira de mestrado Antonia Maria Cavalcanti de Oliveira, pelas
muitas e proveitosas discussões, o incentivo e acima de tudo pela sua preciosa amizade.
À Andréia Nilza Diogo, amiga conseguida ao longo do curso de mestrado, com a qual
também compartilhei as dificuldades e as alegrias desta jornada.
Às secretárias da pós-graduação Simone e Gisele pela atenção, ajuda e principalmente
pelo carinho.
Aos meus amigos e colegas de trabalho do Instituto Vital Brazil, que sempre me
apoiaram e incentivaram, especialmente ao Rosemberg Bernardes Moure, Iara Coutinho e
Flávia Willi pelo auxilio como analistas, e a toda a equipe do Controle da Qualidade em
especial a Isabella Piazza, Celina Filgueiras e Joseane Zaja Almada que me substituíam no
trabalho quando necessitava me ausentar.
Enfim a todos os meus amigos sem o apoio dos quais eu não teria conseguido.
vii

RESUMO
O crescimento do número de casos de aids no Brasil, obrigou o governo a
desenvolver uma política de medicamentos que possibilitasse melhorar e prolongar a
qualidade de vida dos indivíduos infectados pelo HIV. Esta política inclui, entre várias
outras iniciativas, um programa de acesso universal e gratuito aos medicamentos
antiretrovirais. O principal objetivo da terapia antiretroviral é retardar a progressão da
imunodeficiência e/ou restaurar, tanto quanto possível, a imunidade, aumentando o tempo e
a qualidade de vida da pessoa infectada.
Ao mesmo tempo na Política Nacional de Medicamentos, que visa "garantir a
necessária segurança, eficácia e qualidade destes produtos, a promoção do uso racional e o
acesso da população àqueles considerados essenciais", estão destacadas as seguintes
diretrizes:
1) o incentivo ao desenvolvimento científico e tecnológico na área de produção de
fármacos;
2) o incentivo a produção nacional utilizando a capacidade instalada dos laboratórios
oficiais para atender as demandas do SUS;
3) destaca-se também o importante papel desenvolvido pelos laboratórios oficiais no que
tange ao domínio tecnológico de processos de produção de medicamentos de interesse
em saúde pública e como uma das instâncias favorecedoras do monitoramento de
preços no mercado.
Sendo um dos objetivos da Política Nacional de Medicamentos do Ministério da
Saúde assegurar o acesso da população a medicamentos seguros, eficazes e de qualidade,
fundamentando-se no cumprimento da regulamentação sanitária e nas boas práticas de
fabricação e controle, a validação de métodos analíticos, parte integrante das boas práticas
de laboratório surge como uma peça chave para assegurar a confiabilidade dos resultados
obtidos e assim garantir a qualidade aos produtos e serviços.
O presente trabalho propôs uma metodologia padrão para a validação do método
analítico de controle da qualidade do produto Lamivudina comprimidos, com enfoque na
dosagem do principio ativo Lamivudina e no ensaio limite do enantiômero (+)BCH-189
comprovadamente mais citotóxico em linfócitos não infectados, que a Lamivudina.
viii

ABSTRACT
The raise of the number of AIDS cases in Brazil, obliged the government to develop
a medicine policy which make possible improve and length the life quality of HIV infected
individual. This policy includes, among a variety of other initiatives, a universal and free
access program to antiretroviral medicine. The main objective of the antiretroviral therapy
is to retard the immunodeficiency progression and/or repair, as soon as possible, the
immunity, raising the time and the quality of life of the infected person.
At the same time at the Medicine National Policy, that look at “warrant the
necessary security, effectiveness and quality of these products, the promotion of the rational
use and the access of the population to the ones considered essentials”, are detached the
following purpose:
1) the incentive to the scientific and technological development in drugs production
area;
2) the incentive to the national production using the installed capacity of the official
laboratories to attend the SUS needs;
3) the important role developed by the official laboratories is also detached referring to
the technological domain of production processes of interest medicine in public health and
with one of the favour instances of the market prices monitoring.
Being one of the objectives of the Medicine National Policy from the Health
Ministry assert the access of the population to secure, effective and quality medicines,
founding in the accomplishment of the sanitary regulation and in the good manufacture
practices and control, the validation of analytical methods, an integrant part of the good
laboratory practices, surges as a principal tool to assert the trustworthy of obtained results
and thus guarantee the quality to products and services.
The present work proposes a pattern methodology to the validation of the analytical
method of Lamivudine tablets quality control, focusing the dosage of Lamivudine active
principle and the limit assay of the enatiomer (+)BCH-189 proved to be more cytotoxic in
non-infected lymphocytes than Lamivudine.
ix

SIGLAS E ABREVIATURAS
a: Coeficiente linear
α: Nível de significância
ABIA: Associação Brasileira Interdisciplinar de aids
aids: Acquired immune deficiency syndrome ou síndrome da imunodeficiência adquirida
ANOVA: Análise de variância
ANVISA: Agência Nacional de Vigilância Sanitária
ARV: Antiretrovirais
AZT: Zidovudina
b: Coeficiente angular
(+)BCH-189: Enantiômero dextrorotatório da lamivudina
BCH-189: Mistura racêmica
β-L(-): Configuração levorotatória do enantiômero β
β-D(+): Configuração dextrorotatória do enantiômero β
º C: Graus Celcius
CDC: Center for Disease Control and Prevention ou Centro de Controle e Prevenção de
Doenças
CD4: Linfócitos T ou células CD4
CLAE: Cromatografia líquida de alta eficiência
CMP/dCMP-K: Citidilato/2' desoxicitidilato quinase
D(+): Configuração dextrorotatória
DAD: Detector de arranjo de fotodiodos
dCK-2': Desoxicitidina quinase
ddC: Zalcitabina
ddI: Didanosina
DNA: Ácido desoxiribonucleico
DP: Desvio padrão
DPR: Desvio padrão relativo
d4T: Estavudina
DOU: Diário Oficial da União
x

DST: Doenças Sexualmente Transmissíveis
E: Exponencial
EUA: Estados Unidos da América
Fcalc. : F estatístico calculado (Teste de F de Snedecor)
Fcrit. : F estatístico tabelado (Teste de F de Snedecor)
FDA: Food and Drug Administration
Gcalc. : G estatístico calculado (Teste de Grubbs)
Gcrit. : G estatístico tabelado (Teste de Grubbs)
gl: Graus de liberdade
h: Hora
H0: Hipótese nula
H1: Hipótese alternativa
HBV: Vírus da hepatite B
HCl: Ácido clorídrico
HIV-1: Human Immunodeficiency Virus ou Vírus da Imunodeficiência Humana tipo 1
HIV-2: Human Immunodeficiency Vírus ou Vírus da Imunodeficiência Humana tipo 2
H2O2: Peróxido de hidrogênio
HTLV-III: Human T-Lymphotrophic Virus ou Vírus T- Limfotrópico Humano tipo III
IC: Intervalo de confiança
ICH: International Conference on Harmonization ou Conferencia Internacional de Harmonização INCA: Instituto Nacional do Câncer
INCQS: Instituto Nacional de Controle de Qualidade em Saúde
IP: Inibidor da protease
ITRN: Inibidores da transcriptase reversa análogos de nucleosídeo
ITRNN: Inibidor da transcriptase reversa não-análogo de nucleosídeo
IVB: Instituto Vital Brazil
k': Fator de capacidade
L(-): Configuração levorotatória
LAV: Lymphadenopathy Associated Virus ou Vírus Associado à Linfadenopatia
M: Concentração molar
µ: Micra
xi

mcg: Micrograma
min: Minuto
mL: Mililitro
µL: Microlitro
MQ: Média quadrática
MS: Ministério da Saúde
N: Número de pratos teóricos
N: Concentração normal
n: Número de replicatas
NaOH: Hidróxido de sódio
NDP-K: Nucleosídeo 5' difosfato quinase
ng: Nanograma
nm: Nanometro
OMS: Organização Mundial da Saúde
OPAS: Organização Pan-americana da Saúde
pH: Potencial de hidrogênio iônico
PI: Precisão intermediária
Poli γ: Polimerase gamma
r: Coeficiente de correlação
R: Resolução
% R - Porcentagem de recuperação
RE: Resolução (legislação)
RNA: Ácido ribonucleico
S2: Variância
SQ : Soma quadrática
SQR: Substância química de referencia
SUS: Sistema Único de Saúde
T: Fator de cauda ou assimetria
3TC: Lamivudina, (-)BCH-189
3TC-DP: Lamivudina-difosfato
3TC-MP: Lamivudina-monofosfato
xii

3TC-TP: Lamivudina-trifosfato
TR: Tempo de retenção
TRIPS: Trade-related Aspects of Intellectual Property Rights ou Aspectos dos direitos de
propriedade intelectual
USA: United States of America ou Estados Unidos da América
USP 24: The United States Pharmacopeia ou Farmacopeia Americana
UV/Vis: Ultravioleta / visível
v/v: Volume por volume
VISA: Vigilância Sanitária
X: Média
xiii

ESQUEMAS
Pág.
Esquema 1: Mecanismo de ativação da Lamivudina (KEWN, 1997)........................ 18
GRÁFICOS
Pág.
Gráfico 1: Evolução do número de casos de aids no Brasil de 1982 a 2000 segundo
dados do Boletim Epidemiológico..............................................................................
07
Gráfico 2: Evolução do número de óbitos por aids no Brasil de 1982 a 2000
segundo dados do Boletim Epidemiológico...............................................................
07
Gráfico 3: Evolução do número de casos de aids em mulheres no Brasil de 1982 a
2000 segundo dados do Boletim Epidemiológico......................................................
08
Gráfico 4: Evolução do número de óbitos de Mulheres por aids no Brasil de 1982
a 2000 segundo dados do Boletim Epidemiológico....................................................
08
xiv

FIGURAS
Pág.
Figura 1: Esquema simplificado do ciclo de vida do vírus. (The HIV Life Cycle,
2001)............................................................................................................................
5
Figura 2: Estrutura química da ddC e do BCH-189....................................................
16
Figura 3: Estrutura química da Lamivudina e do (+)BCH-189..................................
17
Figura 4: Exemplo de cromatograma do método de análise do teor de Lamivudina.
27
Figura 5: Espectro Ultravioleta da Lamivudina - Máximo de absorção a 270nm......
27
Figura 6: Exemplo de cromatograma do ensaio limite do (+)BCH-189....................
28
Figura 7: Amostra de Lamivudina, padrão secundário, submetida a estresse com
H2O2 3% por 24 h........................................................................................................
37
Figura 8: Amostra de Lamivudina, padrão secundário, submetida a estresse
com HCl 0,1 M por 24 h............................................................................................
37
Figura 9: Amostra de Lamivudina, padrão secundário, submetida a estresse com
NaOH 0,1 M por 24 h................................................................................................
38
Figura 10: Amostra de Lamivudina, padrão secundário, submetida a estresse com
temperatura de 1500C por 24 h....................................................................................
38
xv

Figura 11: Curva de calibração – Linearidade............................................................
42
Figura 12: Plotagem de resíduos..................................................................................
45
Figura 13: Plotagem de ajuste de linha........................................................................
45
Figura 14: Especificidade do método de ensaio limite do enantiômero
(+)BCH-189.................................................................................................................
60
Figura 15: Especificidade do método de ensaio limite do enantiômero (+)BCH-189
na concentração de trabalho.........................................................................................
61
Figura 16: Cromatograma do produto ( Princípio ativo + Excipientes)......................
61
Figura 17: Cromatograma da amostra de Lamivudina submetida ao estresse com
HCl 0,1 N por 24h......................................................................................................
62
Figura 18: Cromatograma da amostra de Lamivudina submetida ao estresse com
H2O2 3% por 24h.........................................................................................................
62
Figura 19: Cromatograma da amostra de Lamivudina submetida ao estresse com
NaOH 0,1 N por 24h...................................................................................................
63
Figura 20: Cromatograma da amostra de Lamivudina submetida ao estresse com a
temperatura de 150°C por 24 h....................................................................................
63
Figura 21: Cromatograma do limite de detecção conc. 1 = 862,4 ng/mL...................
64
Figura 22: Cromatograma do limite de detecção conc. 2 =86,2 ng/mL.....................
65
Figura 23: Cromatograma do limite de detecção conc. 3A = 17,2 ng/mL................. 65
xvi
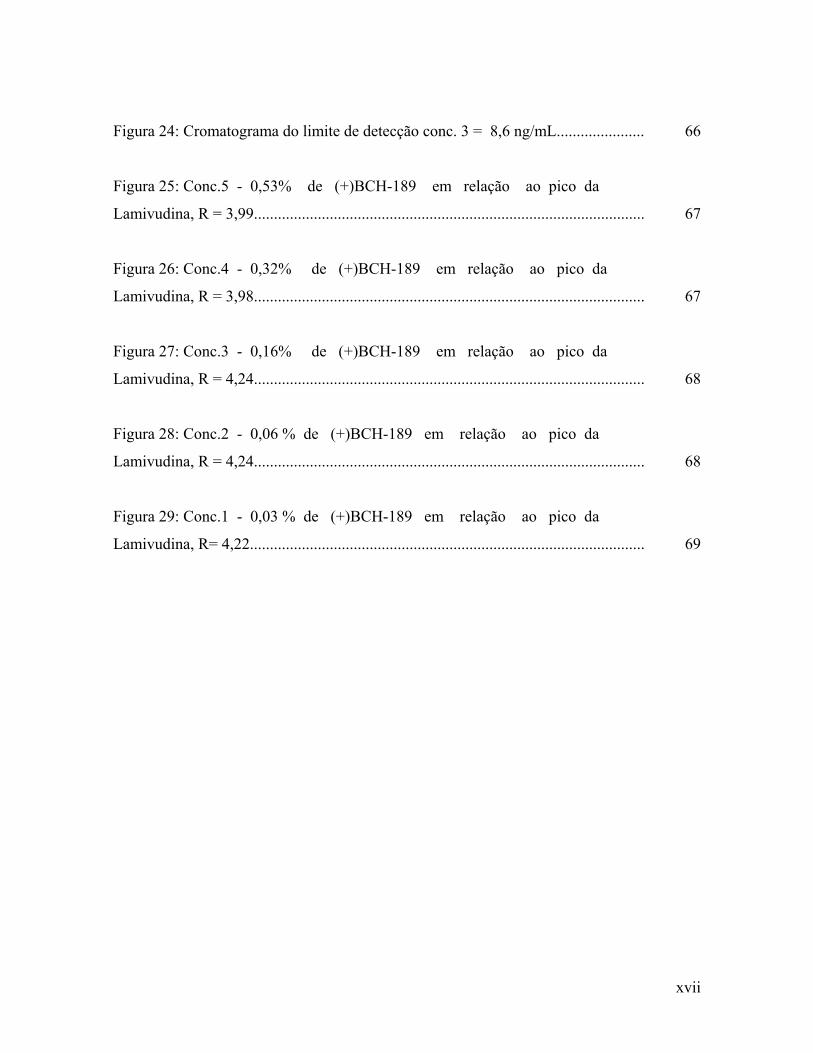
Figura 24: Cromatograma do limite de detecção conc. 3 = 8,6 ng/mL......................
66
Figura 25: Conc.5 - 0,53% de (+)BCH-189 em relação ao pico da
Lamivudina, R = 3,99..................................................................................................
67
Figura 26: Conc.4 - 0,32% de (+)BCH-189 em relação ao pico da
Lamivudina, R = 3,98..................................................................................................
67
Figura 27: Conc.3 - 0,16% de (+)BCH-189 em relação ao pico da
Lamivudina, R = 4,24..................................................................................................
68
Figura 28: Conc.2 - 0,06 % de (+)BCH-189 em relação ao pico da
Lamivudina, R = 4,24..................................................................................................
68
Figura 29: Conc.1 - 0,03 % de (+)BCH-189 em relação ao pico da
Lamivudina, R= 4,22...................................................................................................
69
xvii

TABELAS
pág.
Tabela 1: Características requeridas para a validação pela USP..............................
20
Tabela 2: Características requeridas para a validação pelo ICH..............................
20
Tabela 3: Parâmetros e recomendações para o teste de verificação da
adequação do sistema..............................................................................................
35
Tabela 4: Resultados do teste de adequação do sistema para o método de análise
do teor de Lamivudina..............................................................................................
36
Tabela 5: Resultados do teste de adequação do sistema para o ensaio limite para
o (+)BCH-189.........................................................................................................
36
Tabela 6: Dados de concentração e área para o cálculo da Linearidade..................
39
Tabela 7: Repetitividade das injeções da Concentração 100%...............................
40
Tabela 8: Verificação da Homocedasticidade..........................................................
41
Tabela 9: Representação das médias da concentração real em cada nível de
concentração.............................................................................................................
41
Tabela 10: Confirmação da linearidade por ANOVA.............................................
43
Tabela 11: Dados para o cálculo do intervalo de confiança dos coeficientes da
reta............................................................................................................................
43
xviii

Tabela 12: Resultado dos resíduos...........................................................................
44
Tabela 13: Resultados da exatidão expressos em concentração...............................
46
Tabela 14: Resultados da Repetitividade .................................................................
48
Tabela 15: Precisão intermediária do laboratório A por analistas diferentes..........
49
Tabela 16: Resultados do teste de F entre os analistas 1 e 2 do laboratório A........
49
Tabela 17: Precisão intermediária do laboratório B por analistas diferentes...........
50
Tabela 18: Resultados do teste de F entre os analistas 2 e 1 do laboratório B.........
50
Tabela 19: Precisão intermediária do Laboratório C por analistas diferentes.........
51
Tabela 20: Resultados do teste de F entre os analistas 2 e 1 do laboratório C.........
51
Tabela 21: Precisão intermediária do laboratório A com equipamentos diferentes.
53
Tabela 22: Resultados do teste de F do laboratório A - equipamentos diferentes...
53
Tabela 23: Precisão intermediária do laboratório B com equipamentos diferentes.
54
Tabela 24: Resultados do teste de F do laboratório B - equipamentos diferentes...
54
Tabela 25: Precisão intermediária do laboratório C com equipamentos diferentes.
55
Tabela 26: Resultados do teste de F do laboratório C - equipamentos diferentes ..
55
Tabela 27: Precisão intermediária lab. A - analistas e equipamentos diferentes..... 56
xix

Tabela 28: Resultados do teste de F lab. A - analistas e equipamentos diferentes..
56
Tabela 29: Precisão intermediária lab. B - analistas e equipamentos diferentes.....
57
Tabela 30: Resultados do teste de F lab. B - analistas e equipamentos diferentes..
57
Tabela 31: Precisão intermediária lab. C - analistas e equipamentos diferentes.....
58
Tabela 32: Resultados do teste de F lab. C - analistas e equipamentos diferentes..
58
xx

SUMÁRIO
pág.
1. INTRODUÇÃO
1
1.1. Apresentação
1
1.2. Breve Histórico da Evolução da Legislação e dos Sistemas de Regulação Sanitária
1
1.3. Breve Histórico da aids no Brasil
4
1.4. Evolução da Política de Medicamentos Antiretrovirais do Ministério da Saúde
9
1.5. Fármacos Antiretrovirais
12
1.6. A Terapia Tríplice
13
2. HISTÓRICO DA LAMIVUDINA
15
2.1. Propriedades Físico-químicas
17
2.3. Mecanismo de Ação da Lamivudina
18
3. VALIDAÇÃO NO CONTROLE DA QUALIDADE
19
3.1. Linearidade
21
3.2. Exatidão
21
3.3. Especificidade
22
3.4. Limite de Detecção e Quantificação
22
3.5. Precisão 3.5.1. Repetitividade 3.5.2. Precisão Intermediária 3.5.3. Reprodutibilidade
22
23
23
23
xxi

3.6. Faixa
23
3.7. Teste de verificação da adequação do sistema (System Suitability Testing)
23
4. OBJETIVOS
25
4.1. Objetivo geral
25
4.2. Objetivos específicos
25
5. PARTE EXPERIMENTAL
26
5.1. Metodologia de Análise
26
5.1.1. Materiais e Reagentes
28
5.2. Ensaio do Teor de Lamivudina
29
5.2.1. Sistema Cromatográfico
29
5.2.2. Preparo da Solução Tampão de Acetato de Amônio 0,1 M
29
5.2.3. Preparo da Fase Móvel
29
5.2.4. Preparo da Solução Padrão
30
5.2.5. Preparo da Solução Amostra
30
5.3. Ensaio Limite para a Impureza Enantiomérica (+) BCH-189
30
5.3.1. Sistema Cromatográfico
30
5.3.2. Preparo da Solução de Acetato de Amônia 0,1M
31
5.3.3. Preparo da Fase Móvel
31
5.3.4. Preparo da Solução para a Avaliação da Resolução
31
5.3.5. Preparo da Amostra
31
5.4. Metodologia para a Validação da Analise de Teor da Lamivudina.
32
5.4.1. Teste de Verificação da Adequação do Sistema (System Suitability Testing)
32
xxii

5.4.2. Especificidade
32
5.4.3. Linearidade
32
5.4.4. Exatidão
33
5.4.5. Precisão
33
5.5. Metodologia para a Validação do Ensaio Limite do Enantiômero (+)BCH 189
33
5.5.1. Especificidade
33
5.5.2. Limite de Detecção
34
5.6. Metodologia para a Pesquisa Bibliográfica sobre a Toxicidade do (+)BCH-189
34
6. RESULTADOS E DISCUSSÃO
35
6.1. Resultado do Teste de Adequação do Sistema (System Suitability Testing)
35
6.2. Resultados da Especificidade para o Método de Análise do Teor
36
6.3. Resultados da Linearidade
39
6.4. Resultados da Exatidão
46
6.5. Resultados da Precisão
47
6.5.1. Resultados da Repetitividade
47
6.5.2. Resultados da Precisão Intermediária
48
6.6. Resultados da Faixa
59
6.7. Discussão da Validação do Método de Análise do Teor da Lamivudina Comprimidos
59
6.8. Resultados da Especificidade do Ensaio Limite para o Enantiômero (+)BCH 189
60
6.9. Resultados do Limite de Detecção para o Enantiômero (+)BCH 189
64
6.10. Discussão da Validação do Ensaio Limite do Enantiômero (+)BCH-189
69
6.11. Resultado da Pesquisa Bibliográfica sobre a Toxicidade do (+)BCH-189
70
xxiii

7. CONCLUSÃO
71
8. BIBLIOGRAFIA
73
xxiv

1. INTRODUÇÃO
1.1. Apresentação
A Lamivudina (3TC) é uma droga quiral, com atividade antiretroviral, inibidora da
transcriptase reversa, que faz parte da terapia tríplice utilizada no tratamento da aids
(síndrome da imunodeficiência adquirida).
Dentre todos os antiretrovirais (ARV), análogos de nucleosídeos, aprovados pelo
Food and Drug Administration (FDA) para o uso na clínica, a Lamivudina é o único que
possui a configuração não natural β-L(-). Esta configuração parece ser a responsável pela
baixa toxicidade em cultura de células, quando comparada a do seu enantiômero β-D(+),
também conhecido como (+)BCH-189.
Este estudo apresenta um método de análise, validado, para o produto Lamivudina
comprimido, no que se refere ao ensaio de teor e ao ensaio limite para o seu enantiômero
(+)BCH-189. É importante salientar que, tendo em vista a necessidade de fornecer
medicamentos seguros e de qualidade é necessário dispor de metodologias de Controle da
Qualidade atualizadas e validadas de acordo com parâmetros fornecidos pelos órgãos
reguladores.
Na fase final desta dissertação foi publicada a RDC 150 de 17 de junho de 2003
que aprova o fascículo 4 da parte II, da 4ª edição da Farmacopéia Brasileira (FB), onde
encontra-se a monografia da Lamivudina comprimido (BRASIL, 2003b).
Realizando uma comparação entre a monografia proposta nesta dissertação e a
apresentada pela Farmacopéia Brasileira verificou-se que a segunda não preconiza nenhum
ensaio de pureza para o comprimido de Lamivudina. Este estudo ressalta a importância da
realização do ensaio limite para o (+)BCH-189 em comprimidos de Lamivudina, tendo em
vista que este enantiômero é citotóxico.
1.2. Breve Histórico da Evolução da Legislação e dos Sistemas de Regulação Sanitária
Para contextualizar a situação atual do Brasil frente a pandemia da aids, e enfatizar
a importância de dispor de metodologias de análise atualizadas e validadas para garantir a .

qualidade dos medicamentos fornecidos a população, será necessário uma retrospectiva de
alguns fatos que tiveram importância para a formação do quadro atual.
Historicamente fatos negativos tendem a impulsionar o desenvolvimento científico
e tecnológico no sentido de procurar soluções preventivas para evitar que estes tornem a
ocorrer. No caso dos medicamentos podemos destacar alguns acontecimentos trágicos. O
primeiro ocorreu nos anos 30 nos Estados Unidos da América (EUA) e ficou conhecido
como o caso da sulfanilamida, quando foi utilizado dietilenoglicol como excipiente na
preparação de xarope o que ocasionou mais de 100 mortes. Este fato levou a alterações na
legislação norte americana que passou a exigir estudos de segurança e eficácia para o
registro de medicamentos (COSTA, 2001). O segundo episódio foi a tragédia da
talidomida, nos anos 60 esta droga foi produzida e comercializada como uma mistura
racêmica. Infelizmente, tarde demais foi descoberto que o efeito farmacológico desejado,
sedativo e hipnótico, só é encontrado no enantiômero D(+), o enantiômero L(-) é
teratogênico, o que ocasionou um número expressivo de casos de focomegalia em muitos
países, exceto nos EUA onde a legislação já estava mais restritiva no que diz respeito ao
registro de medicamentos (GUIMARÃES, 2002).
Este fato promoveu, em diversos países, o desenvolvimento de novas leis, a
exemplo dos EUA, exigindo que a segurança e a eficácia dos medicamentos fosse
demonstrada por ensaios não clínicos e clínicos antes do registro, além de ter sido
determinante para o desenvolvimento de duas áreas muito importantes a estereoquímica,
mais especificamente o estudo dos fármacos quirais (BEESLEY, 1998) e a
farmacovigilância, que é o conjunto de métodos e técnicas que tem por objetivo a
identificação e a avaliação dos efeitos do uso, agudo ou crônico, do tratamento
farmacológico no conjunto da população ou em subgrupos de pacientes expostos a
tratamentos específicos (ROZENFELD apud TOGNONI, 1989 ; GUIMARÃES, 2002).
Em relação as drogas quirais, atualmente são realizados estudos de toxicidade dos
isômeros individualmente antes do registro pois os mesmos podem possuir ações
farmacológicas diferentes e até opostas (FDA, 1992).
Com isso ficou evidente a necessidade do acompanhamento dos eventos adversos
dos medicamentos após o registro para determinar a freqüência e os fatores de risco além
da detecção precoce de reações novas e graves. Os estudos realizados antes do registro
envolvem um número limitado de pacientes, aqueles com complicações ou em uso de
2

outras terapias são excluídos bem como os muito idosos ou muito jovens que não
participam. Portanto, certos efeitos menos comuns ou restritos a um certo grupo podem
não ser detectados, pois tais condições não correspondem ao cotidiano da terapia
farmacológica. A partir do momento que o medicamento é liberado para o uso e
consumido por um grande número de pacientes o gerenciamento da informação de seus
eventos adversos pode evidenciar efeitos até então desconhecidos (ROSENFELD, 1998).
No Brasil, além dos fatos citados anteriormente, outros eventos também geraram
reações importantes. Podemos citar a crise do sistema de saúde e o derrame de
medicamentos falsificados como fatos que impulsionaram a edição de muitas normas e a
mudança do sistema de regulação sanitária com a criação em 26 de janeiro de 1999, pela
Lei 9.782, da Agência Nacional de Vigilância Sanitária (ANVISA) (COSTA, 2001). A
ANVISA, tem por finalidade institucional promover a proteção da saúde da população, por
intermédio do controle sanitário da produção e da comercialização de produtos e serviços
submetidos à vigilância sanitária, inclusive dos ambientes, dos processos, dos insumos e
das tecnologias a eles relacionados, bem como o controle dos portos, aeroportos e
fronteiras (BRASIL, 1999a).
O derrame de medicamentos falsificados também motivou a criação dos genéricos e
de toda a sua legislação especifica, que busca garantir a eficácia, a segurança e a qualidade
destes produtos através de estudos de equivalência farmacêutica, bioequivalência e
biodisponibilidade, realizados por instituições credenciadas e com metodologias analíticas
validadas (BRASIL, 1999b).
Para que a ANVISA cumpra sua atribuição de proteção da saúde da população
através da avaliação da qualidade dos medicamentos disponíveis no mercado é necessário,
entre outras providências, a atualização e validação das metodologias de análise dos
medicamentos.
Por último deve-se citar a pandemia da aids que vem demonstrando a fragilidade da
situação de acesso aos antiretrovirais. Entre as alternativas para viabilizar o acesso,
destaca-se a inadiável necessidade de fortalecer os laboratórios nacionais produtores de
insumos farmacêuticos. Os laboratórios farmacêuticos oficiais devem ser apoiados devido
ao seu papel estratégico em relação ao desenvolvimento tecnológico de processos de
produção de medicamentos de interesse em saúde pública bem como, ao seu papel como
regulador de mercado no que se refere a custos e principalmente ao seu papel social como
3

fornecedor de medicamentos essenciais para o Sistema Único de Saúde (SUS), que
possuam eficácia, segurança e qualidade (BERMUDEZ, 2002 ; SILVA, 2003).
1.3. Breve Histórico da aids no Brasil
A síndrome da imunodeficiência adquirida (aids) é causada por um retrovírus com
genoma RNA, da família Lentiviridae. Pertencente ao grupo dos retrovírus citopáticos e
não-oncogênicos que necessitam, da enzima transcriptase reversa para sua multiplicação.
Os primeiros casos de aids foram descritos no ano de 1981, nos Estados Unidos da
América, e em 1982, no Brasil, a partir da identificação de homossexuais masculinos
adultos que apresentavam sarcoma de Kaposi, pneumonia por Pneumocystis carinii e
comprometimento do sistema imune. (MS, 2002a)
O Vírus da Imunodeficiência Humana tipo1 (HIV-1) foi isolado pelos
pesquisadores Luc Montaigner na França e Robert Gallo nos Estados Unidos da América
em 1983, recebendo respectivamente os nomes de Vírus Associado à Linfadenopatia
(LAV) e Vírus T- Linfotrópico Humano tipo III (HTLV-III). Em 1986 foi identificado um
segundo agente etiológico, denominado HIV-2. Em 1986 um comitê internacional
recomendou o termo HIV para denominá-los reconhecendo-os capazes de infectar seres
humanos (MS, 2002A).
O HIV age ligando-se aos receptores CD4 da célula hospedeira (Fig.1-item 1,
pág. 5) através dos quais consegue, fundindo o seu envelope com a parede celular,
introduzir o seu material genético, RNA, na célula hospedeira. O seu principal alvo são os
linfócitos T. O RNA viral sofre a ação da enzima transcriptase reversa e é convertido em
DNA, o filamento simples de DNA é duplicado pela ação da DNA polimerase (Fig.1-item
2, pág. 5). O DNA resultante entra no núcleo da célula e através da ação da enzima
integrase se incorpora ao genoma celular (Fig.1-item 3, pág. 5). Nesta fase é possível ao
HIV permanecer em estado latente por vários anos. A ativação da célula hospedeira resulta
na transcrição do DNA viral em RNA mensageiro (Fig.1-item 4, pág.5) o qual é traduzido
em proteína viral (Fig.1-item 5, pág. 5). O RNA viral produzido será o material genético da
nova geração de vírus (Fig.1-item 6, pág. 5) (THE HIV Life Cycle, 2001).
4

Figura 1 : Esquema simplificado do ciclo de vida do vírus (The HIV Life Cycle, 2001).
O perfil da aids no Brasil modificou-se com o tempo. No seu início, anos 80,
retratava um fenômeno restrito aos grandes centros urbanos, tendo como categorias de
exposição preponderantes os homossexuais masculinos, os hemofílicos e as demais
pessoas que receberam sangue ou hemoderivados. Em meados da década, já surgia outro
segmento que passou a ocupar lugar de destaque, os usuários de drogas injetáveis. No final
dos anos 90 a freqüência dos casos entre mulheres decorrente de transmissão heterossexual
cresceu substancialmente, assumindo um importante papel. Atualmente o perfil da
epidemia é de uma doença na qual há o predomínio da transmissão heterossexual, que não
está mais restrita a segmentos populacionais e vai se disseminando na população em geral
com predominância entre municípios pequenos e mais pobres (SZWARCWALD, 2000;
GUIMARÃES, 2000).
As tendências atuais da pandemia de aids no Brasil são a heterossexualização, a
feminização, a interiorização e a pauperização.
"A heterossexualização é, evidentemente, um reflexo do comportamento
sociossexual da população, que em sua grande maioria é heterossexual. Já a
5

feminização da aids parece envolver, além da maior vulnerabilidade
biológica da mulher ao HIV, uma desigualdade claramente observável na
distribuição de poder entre os gêneros. (...) Quanto ao processo de
pauperização, a escolaridade é uma das variáveis indiretas utilizadas para
mostrar o crescimento neste grupo de baixa renda. Até 1982, 100% dos
casos entre pessoas com escolaridade conhecida eram daquelas que tinham
nível superior ou até 11 anos de estudo. De lá para cá, a situação se
inverteu. Hoje, mais de 60% dos casos de aids são registrados entre
analfabetos ou pessoas com até 8 anos de estudo" (GRANGEIRO, 2002).
O crescimento do número de casos de aids, no Brasil, obrigou o governo a
desenvolver uma política de medicamentos que possibilitasse melhorar e prolongar a
qualidade de vida dos indivíduos infectados pelo HIV.
Os gráficos 2 e 4, páginas 7 e 8, mostram o impacto da introdução da terapia
antiretroviral para a diminuição dos óbitos devido a aids.
No item 1.4, Evolução da Política de Medicamentos Antiretrovirais do Ministério
da Saúde, os eventos relevantes relacionados com a epidemia de aids no Brasil são
enumerados cronologicamente.
6

Gráfico 1: Evolução do número de casos de aids no Brasil de 1982 a 2000 segundo dados do
Boletim Epidemiológico (GALVÃO, 2002)
05 0 0 0
1 0 0 0 01 5 0 0 02 0 0 0 02 5 0 0 0
n º d e c a s o s
1982
1985
1988
1991
1994
1997
2000
a n o
E vo lu ção d o n º d e caso s d e a id s n o B ras il d e 1982 a 2000 .
Gráfico 2: Evolução do número de óbitos por aids no Brasil de 1982 a 2000 segundo dados do
Boletim Epidemiológico (GALVÃO, 2002)
02000400060008000
1000012000
Nº de óbitos
1982
1984
1986
1988
1990
1992
1994
1996
1998
2000
anos
Número de Óbitos por aids no Brasil de 1982 a 2000
7

Gráfico 3: Evolução do número de casos de aids em mulheres no Brasil de 1982 a 2000
segundo dados do Boletim Epidemiológico (GALVÃO, 2002)
010002000300040005000600070008000
Nº de Casos19
82
1984
1986
1988
1990
1992
1994
1996
1998
2000
anos
Número de Casos de aids em Mulheres no Brasil de 1982 a 2000
Gráfico 4: Evolução do número de óbitos de Mulheres por aids no Brasil de 1982 a 2000
segundo dados do Boletim Epidemiológico (GALVÃO, 2002)
0500
10001500200025003000
Nº de óbitos
1982
1984
1986
1988
1990
1992
1994
1996
1998
2000
anos
Número de Óbitos de Mulheres por aids no Brasil de 1982 a 2000
8

1.4. Evolução da Política de Medicamentos Antiretrovirais do Ministério da Saúde
Como citado anteriormente os primeiros casos de aids no Brasil foram descritos
em 1982, neste mesmo ano foram relatados dez casos de aids e dez óbitos, todos em
indivíduos do sexo masculino. No ano seguinte já eram trinta e nove casos e trinta e oito
óbitos. Foram relatados ainda os dois primeiros casos em mulheres (GALVÃO, 2002).
Em 1984 o número de casos já chegava a cento e quarenta, o crescimento
acelerado levou o governo a tomar algumas medidas (GALVÃO, 2002).
A partir deste ano o Ministério da Saúde começou a obrigar os hospitais a
informar todos os casos de aids e a incidência de todas as condições associadas.
Em 1985, a portaria do Ministério da Saúde n° 236 de 02 de maio, estabelece
diretrizes para o programa da aids (BRASIL, 1985). Em 1986 a aids passa a ser uma
doença de notificação compulsória.
Em 1988 é criado o programa de aids no âmbito do Ministério da Saúde e devido a
elevada incidência, de infeções oportunistas, neste mesmo ano tem início a distribuição,
pelo sistema público de saúde, de medicamentos para tais infeções. Ainda neste ano a
Constituição estabelece a saúde como direito fundamental, destaca as atribuições da
Vigilância Sanitária como obrigação do estado e é criado o Sistema Único de Saúde (SUS).
Em 1990 são editadas as Leis 8.078 de 11 de setembro de 1990, código de defesa
do consumidor, que reforça a legislação de proteção e defesa da saúde, reafirmando a
responsabilidade do produtor pela qualidade do produto e do serviço e lhe impondo
atividades de informação ao consumidor (BRASIL, 1990a). Ainda é promulgada a Lei
8.080 de 19 de setembro de 1990, Lei orgânica da saúde, que destaca a abrangência das
ações de vigilância, ao incluir no SUS a vigilância de produtos, de serviços, dos
ambientes e dos processos de trabalho além de atribuir a vigilância o papel de coordenar
a rede nacional de laboratórios para a qualidade da saúde. No seu artigo 6° estabelece
como campo de atuação do SUS a formulação da política de medicamentos de interesse
para a saúde (BRASIL, 1990b).
Em 1991 o número de casos de aids registrados já ultrapassava dez mil. Tem
início então a distribuição da Zidovudina (AZT). Em 1993 o AZT começa a ser fabricado
no Brasil e a Didanosina (ddI) começa a ser distribuída (GALVÃO, 2002).
9

Em 14 de maio de 1996 é assinada a Lei 9.279 de propriedade industrial que
coloca a legislação brasileira em concordância com a “Trade-related aspects of
intellectual Property rights” (TRIPS) (BRASIL, 1996a). Em 13 de novembro do mesmo
ano é emitida a Lei 9.313, de acordo com a qual é da responsabilidade do Governo, a
disponibilização do tratamento mais adequado aos pacientes infectados pelo HIV, dentro
de parâmetros técnicos e científicos definidos pelo Ministério da Saúde, por intermédio
da Coordenação Nacional de Doenças Sexualmente Transmissíveis e aids (Coordenação
Nacional de DST e AIDS) (BRASIL, 1996b). Assim, neste ano tem início, na rede
pública de saúde, a implementação nacional da distribuição universal e gratuita dos ARV
(GALVÃO, 2002).
A política do Ministério da Saúde para a assistência aos indivíduos infectados
pelo HIV inclui, entre várias outras iniciativas, a disponibilização de modalidades de
assistência que visam à redução das internações hospitalares, além de estabelecer
recomendações técnicas consensuais para utilização de medicamentos antiretrovirais
além de um programa de acesso universal e gratuito aos mesmos. Estas iniciativas
determinaram um impacto semelhante ao verificado nos países desenvolvidos no que diz
respeito à redução das mortes causadas pela aids, da ocorrência de infeções oportunistas e
das internações hospitalares (MS, 2002b). Neste período são distribuídos além do AZT e
da ddI, a Lamivudina (3TC), o Saquinavir e o Ritonavir.
A partir do início da distribuição gratuita do "coquetel anti-aids", associação, de
três antiretrovirais, o número de óbitos começou a diminuir, como podemos observar nos
gráficos 2 e 4, páginas 7 e 8.
Em 1997 o Brasil começa a produzir a Zalcitabina (ddC) e a Estavudina (d4T) e
inicia-se a distribuição do Indinavir e da d4T.
Em 30 de outubro de 1998 é emitida a portaria 3.916 do Ministério da Saúde, que
aprova a Política Nacional de Medicamentos, visando "garantir a necessária segurança,
eficácia e qualidade destes produtos, a promoção do uso racional e o acesso da população
àqueles considerados essenciais". Estão destacados nas diretrizes desta política:
a) o incentivo ao desenvolvimento científico e tecnológico na área de
produção de fármacos;
b) o incentivo a produção nacional utilizando a capacidade instalada dos
laboratórios oficiais para atender as demandas do SUS;
10

c) destaca-se também o importante papel desenvolvido pelos laboratórios
oficiais no que tange ao domínio tecnológico de processos de produção de
medicamentos de interesse em saúde pública e como uma das instâncias
favorecedoras do monitoramento de preços no mercado (BRASIL, 1998).
Em 1998 os laboratórios oficiais iniciam a produção de ddI e o sistema público
inicia a distribuição de Nelfinavir, Nevirapina e da Delavirdina. Ainda neste ano a
IVB-Lamivudina é desenvolvida no Instituto Vital Brazil, por pesquisa interna sendo
posteriormente comparada às formulações de outros laboratórios. O seu registro no
Ministério da Saúde foi concedido em agosto de 1998.
Em 1999 a Lei 9.782 dispõe sobre o Sistema Nacional de Vigilância Sanitária e
cria a Agência Nacional de Vigilância Sanitária (ANVISA) (BRASIL, 1999a).
A resolução 391 de 9 de agosto de 1999 detalha o procedimento para a implantação
dos medicamentos genéricos, uma prioridade da política de medicamentos do Ministério da
Saúde. O objetivo da Política de Genéricos visa o oferecimento de medicamentos
intercambiáveis com os de marca com qualidade, segurança e eficácia comprovadas
através de estudos de equivalência farmacêutica e bioequivalência. Os medicamentos
genéricos surgiram para facilitar o acesso da população aos medicamentos e funcionar
como reguladores de mercado (BRASIL, 1999b).
Também neste ano tem início a produção da 3TC pelo Instituto Vital Brazil e do
AZT + 3TC por outro laboratório nacional.
Em 6 de outubro o decreto presidencial n.º 3201 "dispõe sobre a concessão de
ofício de licença compulsória nos casos de emergência nacional e de interesse público de
que trata o artigo 71 da Lei n.º 972, de 14 de maio de 1996" (BRASIL, 1999c).
Em 2000 teve início a produção nacional de Indinavir e Nevirapina.
Em fevereiro de 2001 o governo ameaçou quebrar a patente dos medicamentos
Nelfinavir, fabricado pela Roche, e Efavirenz, fabricado pela Merck. Inicia-se, também, a
distribuição do Amprenavir pelo sistema público de saúde. Em agosto, o Ministério da
Saúde anunciou a licença compulsória da patente do medicamento Nelfinavir e em seguida
a Merck fez um acordo baixando o preço do Efavirenz para evitar o licenciamento
compulsório. A partir da produção nacional e da política de negociação de preços com os
laboratórios detentores das patentes, alguns antiretrovirais tiveram seus preços reduzidos
em mais de 80% entre 1996 e 2001 (GALVÃO, 2002).
11

"A produção de muitas drogas utilizadas no coquetel anti-aids permite ao
Brasil continuar o controle da disseminação da aids. A industria
farmacêutica vê isso como um ato de guerra. Nós vemos isso como um ato
de vida." (MS, 2003).
Também em 2001 a IVB-Lamivudina foi aprovada no ensaio de equivalência
farmacêutica, realizado pela Universidade Federal de Minas Gerais, e em 2002 foi
aprovada no teste de bioequivalência realizado no Instituto Nacional do Câncer (INCA).
Atualmente, o Brasil produz sete dos quinze antiretrovirais que fazem parte do
consenso de tratamento brasileiro. Assumindo-se que todos os antiretrovirais fossem
importados o programa de distribuição gratuita e universal se tornaria inviável devido ao
alto custo (MS, 2002c; MS, 2002d).
Tal fato evidencia a necessidade de fomento à industria nacional de insumos e aos
laboratórios farmacêuticos nacionais, em especial aos oficiais devido ao seu importante
papel como facilitadores da internalização por transferência de tecnologia, reguladores de
mercado, como referência de custos e qualidade.
1.5. Fármacos Antiretrovirais
O impacto social da pandemia da aids levou a um aumento das pesquisas a cerca
de fármacos que possam atuar inibindo ou bloqueando a replicação do HIV. Diante da
urgência da situação a aprovação por parte do FDA realizou-se através de um
procedimento abreviado (FDA, 2002b).
O desafio da industria farmacêutica é desenvolver fármacos que consigam
bloquear as atividades do vírus sem lesionar a célula hospedeira. Com o conhecimento do
ciclo de replicação do vírus foi possível determinar os prováveis alvos para a atuação
destes fármacos, a interferência em qualquer um dos passos deste ciclo impede a
multiplicação e/ou a liberação de novos vírus.
Existem duas classes principais de antiretrovirais aprovados para o uso, os
inibidores da transcriptase reversa e os inibidores da protease.
12

Os inibidores da transcriptase reversa são fármacos que inibem a replicação do
HIV bloqueando a ação da enzima transcriptase reversa que age convertendo o RNA viral
em DNA, eles dividem-se em análogos de nucleosídeos, que possuem similaridade
estrutural com os 2’-desoxinucleotideos naturais com os quais competem, e os não
análogos de nucleosídeos, que não requerem ativação prévia e atuam diretamente sobre a
transcriptase reversa (FLORES, 1997; SILVA, 1998).
A transcriptase reversa é um alvo importante para ação dos antiretrovirais pois é
uma enzima crucial para a replicação do vírus e não é conhecida nenhuma enzima similar
na célula hospedeira (GRAY, 1995).
Os inibidores da protease são drogas que agem impedindo a ação da enzima
protease que é fundamental para a clivagem das cadeias protéicas produzidas pela célula
infectada em proteínas virais estruturais e enzimas que formarão o novo HIV.
Existem outros fármacos, ainda em fase de estudos, que deverão agir nas demais
etapas do ciclo de reprodução do HIV.
O processo de desenvolvimento de novos fármacos antiretrovirais está em contínuo
desenvolvimento. A partir da descoberta de que a configuração não natural β-L(-) da
Lamivudina pode ser a responsável por sua baixa toxicidade em células hospedeiras não
infectadas, novas pesquisas vem sendo realizadas utilizando esta configuração como
modelo para o desenvolvimento de novos fármacos mais potentes e menos tóxicos.
1.6. A Terapia Tríplice
O principal objetivo da terapia antiretroviral é retardar a progressão da
imunodeficiência e/ou restaurar, tanto quanto possível, a imunidade, aumentando o tempo
e a qualidade de vida da pessoa infectada. Entretanto, a evolução natural da infeção pelo
HIV caracteriza-se por intensa e contínua replicação viral, que resulta, principalmente, na
destruição e/ou disfunção de células CD4 e de outras células do sistema imune levando à
imunodeficiência, que em sua forma mais grave manifesta-se por meio da ocorrência de
infeções oportunistas e neoplasias que caracterizam a Síndrome de Imunodeficiência
Adquirida. Dessa forma, a supressão da intensa e continuada replicação viral é
fundamental para que seja possível diminuir ou reverter o dano imunológico (MS, 2002b).
13

No Consenso sobre Terapia Antiretroviral em Adultos (MS, 1996), a Lamivudina
foi incluída como alternativa para a combinação com o AZT devido a sua baixa toxicidade.
No Consenso sobre Terapia Antiretroviral para Adultos e Adolescentes infectados pelo
HIV (MS, 1997), foi introduzida a terapia tríplice para pacientes que faziam uso da terapia
dupla e que apresentavam critérios de falha terapêutica. Já o consenso sobre a Infeção pelo
HIV em Adultos e Adolescentes (MS, 1999), indica que a terapia inicial deve ser
composta por dois inibidores da transcriptase reversa análogos de nucleosídeos (ITRN),
associado a um inibidor da transcriptase reversa não-análogo de nucleosídeo (ITRNN) ou a
um inibidor da protease (IP) em situações específicas.
Estudos demonstraram que com a utilização do "coquetel anti-aids", terapia
combinada de antiretrovirais, havia um aumento na atividade antiretroviral, com elevação
dos níveis de linfócitos T e redução nos títulos plasmáticos de RNA-HIV e redução da
replicação viral por potencializar o efeito terapêutico ou por sinergismo de ação em sítios
diferentes do ciclo de replicação viral. Além de evidenciar a redução da incidência de
cepas multirresistentes (MS, 2002a).
A terapia antiretroviral é uma área complexa, sujeita a constantes mudanças para
incorporar os novos conhecimentos gerados pelos ensaios clínicos, por isso as
recomendações preconizadas pelos consensos são revistas periodicamente.
A utilização do "coquetel anti-aids", vem mostrando resultados positivos em todos
os países nos quais os pacientes infectados pelo HIV têm tido acesso a esses
medicamentos. Estudos realizados por vários pesquisadores nos E.U.A, Europa e Brasil
têm demonstrado uma redução significativa nos óbitos secundários à aids e na ocorrência
de infeções oportunistas.
O impacto da terapia antiretroviral no Brasil de 1995 a 2001 pode ser resumido
com os seguintes dados: Redução da mortalidade de 40 a 70%, redução da morbidade de
60 a 80%, Redução de hospitalização 358.000 internações evitadas, economia de U$ 1,1
bilhão de dólares (GRANGEIRO, 2002).
14

2. HISTÓRICO DA LAMIVUDINA
A Lamivudina, também conhecida como 3TC ou (-)-BCH-189, e um nucleosídeo
análogo sintético; inibidor da transcriptase reversa, com atividade contra os vírus 1 e 2 da
imunodeficiência humana (HIV-1 e HIV-2) e o vírus da hepatite B (HBV) (SOUDEYNS,
1991; BENHAMOU, 1995; BLANEY, 1995).
Esta molécula foi desenvolvida pelo Dr. Bernard Belleau, em cooperação com os
pesquisadores da Universidade McGill (Montreal, Quebec, Canada) e da BiChem Pharma.
As pesquisas foram iniciadas em 1987 e visavam descobrir novos fármacos com ação na
transcriptase reversa. Uma análise da estereoquímica e de parâmetros eletrônicos levou a
conclusão de que a atividade e a especificidade dos nucleosídeos análogos como inibidores
da transcriptase reversa eram dependentes da forma do anel desoxiribose tanto quanto do
ambiente eletrônico na região do carbono 3’. Por esta razão, os pesquisadores planejaram e
sintetizaram nucleosídeos análogos com um anel pentose isostérico no qual o carbono 3’
foi substituído por um heteroátomo. O resultado desta modificação foi que o par de
elétrons disponível no heteroátomo participaria da formação de pontes de hidrogênio entre
átomos do centro catalítico da enzima, logo a ligação entre o nucleosídeo e a enzima seria
mais forte. Este tipo de modificação simularia o efeito do grupo hidroxila presente no
desoxinucleosideo natural na formação da cadeia (DIONNE, 1999).
Entre os novos compostos sintetizados o racêmico BCH-189 foi considerado
potente como inibidor da replicação do HIV “in vitro”, ele foi obtido pela substituição do
carbono 3' do anel ribose da 2',3'-didesoxicitidina (ddC) por um átomo de enxofre.
O BCH 189 possui dois centros quirais, localizados nos carbonos 1' e 4' do anel
oxatiolan, portanto possui quatro isômeros óticos sendo um par de diasteroisômeros (α e
β) com dois enantiômeros cada.
15

O BCH-189
dois foram conside
polimerase humana
portanto muito imp
(COATES, 1992; S
Em contrap
uma surpresa que
nucleosídeos biolo
possuíam a configu
A causa da
a inibição ou pert
adquirida pode ser
a) Inibição
b) Termino
mitocon
c) Alteraçã
d) A persi
ineficien
e) Alguma
Dados cient
análogos esta relac
3’
ddC BCH-189
Figura 2: Estrutura química da ddC e do BCH-189
é a mistura dos enantiômeros β-D(+) natural e o β-L(-) não natural, os
rados equipotentes contra o HIV, mais apenas o β-L(+) inibe a DNA-
, fato associado com a toxicidade dos fármacos nucleosídeos, sendo
ortante a realização do ensaio limite para determinar a sua presença.
CHINAZI, 1992; SOMMADOSSI, 1992; BLANEY, 1995)
artida a Lamivudina ou β-L(-) é livre desta toxicidade associada. Foi
o enantiômero β-L(-) apresentasse atividade biológica pois todos os
gicamente ativos anteriormente conhecidos como agentes terapêuticos
ração natural β-D(+).
toxicidade mitocondrial dos antiretrovirais análogos de nucleosídeos é
urbação da síntese de DNA mitocondrial. A toxicidade mitocondrial
causada por:
direta da DNA polimerase gamma (poli γ) sem incorporação.
da cadeia pela incorporação do nucleosídeo análogo no DNA
drial pela poli γ.
o da fidelidade da síntese de DNA pela poli γ.
stência do nucleosídeo análogo no DNA mitocondrial devido a
te remoção, ou
das combinações anteriores
íficos indicam que o aumento da toxicidade celular dos nucleosídeos
ionada com a facilidade da sua incorporação ao DNA mitocondrial pela
16

DNA polimerase humana e a dificuldade da sua remoção, a Lamivudina trifosfato
(3TC-TP) é um dos nucleosídeo análogo menos freqüentemente incorporados a DNA
polimerase humana e um dos mais eficientemente removidos. Estes dois fatos combinados
podem explicar a baixa toxicidade mitocondrial induzida pela Lamivudina in vivo. A baixa
incorporação da Lamivudina também pode ser atribuída a sua forma isomérica (-)
(HART,1992; JOHNSON, 2001; LIM, 2001).
A Lamivudina foi aprovada em 1995 pelo Food and Drug Administration para o
uso no tratamento da aids (FDA, 1995).
2.1. Propriedades Físico-químicas
A Lamivudina é um sólido cristalino branco ou quase branco com ponto de fusão
entre 160 e 162º C, solubilidade de aproximadamente 70 mg/mL em água a 20ºC. Seu
nome químico é (2R,cis)-4-amino-1-(2-hidroximetil-1,3-oxatiolan-5-il)-(1H)-pirimidin-2-
ona, também citada como (-)2',3'-didesoxi-3'-tiocitidina. Sua fórmula molecular é
C8H11N3O3S, seu peso molecular é 229,26 (THE MERCK INDEX, 1996).
As estruturas químicas da Lamivudina e do seu enantiômero (+)BCH-189 são
mostradas na figura 3, pág. 17.
É importante lembrar que o (+)BCH-189 apesar de também possuir atividade
inibitória contra o HIV-1 e HIV-2 é significativamente mais citotóxico em linfócitos não
infectados ( COATES,1992; BLANEY, 1995).
' 4'
Figura 3: Estrutura
Lamivudina
química da Lam
1
(+) BCH-189
ivudina e do (+)BCH-189
17

2.2. Mecanismo de Ação da Lamivudina
Os nucleosídeos análogos inibidores da transcriptase reversa são drogas que
compõe uma classe de antiretrovirais, na qual se inclui a Lamivudina, que necessitam ser
fosforilados intracelularmente mediante a ação das enzimas celulares que atuam nos
nucleotídeos naturais, dando origem a forma ativa 5' trifosfato que possui similaridade
estrutural com os 2' desoxinucleotídeos naturais com os quais compete.
No caso da Lamivudina, após a fosforilação a forma lamivudina-trifosfato
(3TC-TP) atua tanto como inibidora competitiva da transcriptase reversa, competindo com
os nucleotídeos naturais para se unir a transcriptase reversa, quanto como finalizador da
cadeia ao se incorporar ao DNA pró-viral, já que não possui o grupo hidroxila na posição
3' para formar ponte diester e assim prolongar a cadeia (FLORES, 1997).
3TC 3TC-MP 3TC-DP 3TC-TP Inibição competitiva da dCK-2' CMP/dCMP-K NDP-K transcriptase reversa e incorporação induzindo ao termino da cadeia. dCK-2'- desoxicitidina quinase
CMP/dCMP-K - citidilato/2' desoxicitidilato quinase
NDP-K - nucleosídeo 5' difosfato quinase
Esquema 1: Mecanismo de ativação da Lamivudina (KEWN, 1997)
18

3. VALIDAÇÃO NO CONTROLE DA QUALIDADE
Como citado anteriormente, um dos objetivos da Política Nacional de
Medicamentos do Ministério da Saúde é assegurar o acesso da população a medicamentos
seguros, eficazes e de qualidade, fundamentando-se no cumprimento da regulamentação
sanitária e nas boas práticas de fabricação e controle. A validação de métodos analíticos,
parte integrante das boas práticas de laboratório surge como uma peça chave para
assegurar a confiabilidade dos resultados obtidos e assim garantir a qualidade aos produtos.
No Brasil só foi dado à validação a devida importância a partir da edição, pela
Agência Nacional de Vigilância Sanitária, da resolução 391 de 9 de agosto de 1999. Esta
resolução normatiza, disciplina e regulamenta critérios para registro de medicamentos
genéricos no Ministério da Saúde, qualificando a validação de métodos analíticos como um
dos pré-requisito para o registro (BRASIL, 1999b). Desde então foram emitidas e
revogadas varias legislações sempre com o intuito de complementar e aprofundar as
legislações anteriores. A legislação atualmente em vigor no Brasil é a resolução -
RE n° 899, de 29 de maio de 2003, segundo a qual a validação deve garantir, por meio de
estudos experimentais, que o método atenda às exigências das aplicações analíticas,
assegurando a confiabilidade dos resultados (BRASIL, 2003a).
Como este estudo foi iniciado antes da emissão desta resolução ele baseou-se nos
parâmetros de validação especificados na Farmacopéia Americana (USP 24, 2000) e na
Comissão Internacional de Harmonização (ICH, 1994; ICH, 1996) apresentados nas
tabelas 1 e 2 pág. 20.
19
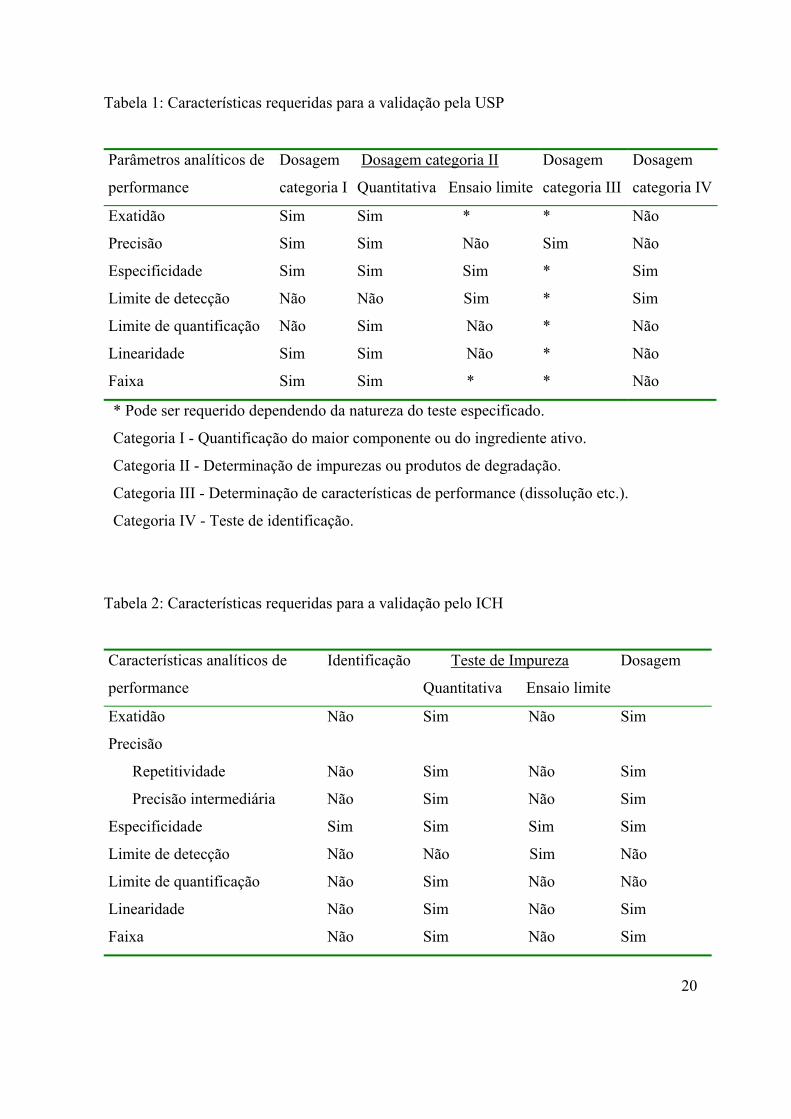
Tabela 1: Características requeridas para a validação pela USP
Parâmetros analíticos de
performance
Dosagem
categoria I
Dosagem categoria II
Quantitativa Ensaio limite
Dosagem
categoria III
Dosagem
categoria IV
Exatidão Sim Sim * * Não
Precisão Sim Sim Não Sim Não
Especificidade Sim Sim Sim * Sim
Limite de detecção Não Não Sim * Sim
Limite de quantificação Não Sim Não * Não
Linearidade Sim Sim Não * Não
Faixa Sim Sim * * Não
* Pode ser requerido dependendo da natureza do teste especificado.
Categoria I - Quantificação do maior componente ou do ingrediente ativo.
Categoria II - Determinação de impurezas ou produtos de degradação.
Categoria III - Determinação de características de performance (dissolução etc.).
Categoria IV - Teste de identificação.
Tabela 2: Características requeridas para a validação pelo ICH
Características analíticos de
performance
Identificação Teste de Impureza
Quantitativa Ensaio limite
Dosagem
Exatidão Não Sim Não Sim
Precisão
Repetitividade Não Sim Não Sim
Precisão intermediária Não Sim Não Sim
Especificidade Sim Sim Sim Sim
Limite de detecção Não Não Sim Não
Limite de quantificação Não Sim Não Não
Linearidade Não Sim Não Sim
Faixa Não Sim Não Sim
20

De acordo com as tabelas 1 e 2, diferentes métodos de análise requerem diferentes
esquemas de validação, e estão divididos em quatro categorias. No presente trabalho
utilizamos as categorias I e II que se referem a métodos analíticos para quantificação do
maior componente da substância ou ingrediente ativo em produtos farmacêuticos acabados
e a métodos analíticos para determinação de impurezas da substância ou compostos de
degradação em produtos farmacêuticos acabados; respectivamente.
Na categoria I é recomendada a avaliação dos seguintes parâmetros: especificidade,
linearidade, exatidão, precisão, e faixa (range).
Na categoria II (ensaio limite) os parâmetros recomendados são os seguintes:
especificidade e limite de detecção.
3.1. Linearidade
A linearidade de um método analítico é a capacidade de obter resultados
diretamente proporcionais a concentração da substância de interesse, dentro de uma faixa
definida. Para determinação da linearidade é recomendado o uso de no mínimo 5
concentrações abrangendo a faixa especificada. No caso da determinação quantitativa do
maior componente ou substância ativa estas concentrações devem ter o alcance mínimo de
80 a 120% da concentração teórica do teste (ICH, 1994; ICH, 1996; USP 24, 2000).
3.2. Exatidão
A exatidão de um método analítico é a proximidade dos resultados obtidos
experimentalmente, em relação ao valor verdadeiro ou de referência. É medido como a
porcentagem de substância recuperada na análise, ao se contaminar a matriz, mistura dos
excipientes da formulação, com diferentes quantidades conhecidas da substância em
estudo, em níveis acima e abaixo do valor normal esperado na amostra.
A International Conference on Harmonization (ICH) recomenda que sejam
efetuadas pelo menos 9 determinações com no mínimo três concentrações, em triplicata,
cobrindo a faixa especificada.
A exatidão de um método analítico é normalmente expressa como a media das
porcentagem de recuperação (ICH, 1994; ICH, 1996; USP 24, 2000).
21

3.3. Especificidade
A especificidade de um método analítico é a habilidade de determinar com exatidão
e especificamente a substância de interesse na presença de outros componentes tais como:
impurezas, produtos de degradação e excipientes.
A especificidade de um método analítico para determinação de impurezas (ensaio
limite) é avaliada contaminando a matéria-prima ou produto com quantidades apropriadas
de impureza e demonstrando que estas impurezas podem ser isoladas dos demais
componentes (ICH, 1994; ICH, 1996; USP 24, 2000).
A especificidade de um método analítico para determinação quantitativa do maior
componente da substância ou ingrediente ativo é avaliada demonstrando que os resultados
da análise não são afetados pela presença de impurezas e/ou produtos de degradação.
Para métodos cromatográficos a especificidade é demonstrada através do próprio
cromatograma que deve apresentar resolução maior ou igual a dois entre os sinais da
substância ativa e o da impureza (SHABIR, 2003).
3.4. Limite de Detecção e Quantificação
O limite de detecção de um método analítico é a menor quantidade da substância de
interesse na amostra que pode ser detectada mas não necessariamente quantificada como
um valor exato.
O limite de quantificação de um método analítico é a menor quantidade da
substância de interesse que pode ser determinada com precisão e exatidão aceitáveis (ICH,
1994; ICH, 1996; USP 24, 2000).
3.5. Precisão
A precisão de um método analítico é o grau de concordância de uma série de
resultados obtidos de múltiplas análises, de uma mesma amostra homogênea.
Para a análise deste parâmetro devemos levar em consideração a repetitividade, a
precisão intermediária e a reprodutibilidade, sendo que este último item só é necessário
quando o método for submetido a avaliação com vistas a inclusão na Farmacopéia (ICH,
1994; ICH, 1996; USP 24, 2000).
22

3.5.1 - Repetitividade (precisão entre ensaios) refere-se ao uso do procedimento analítico
em um laboratório durante um curto período de tempo usando o mesmo analista e o mesmo
equipamento, isto é, nas mesmas condições.
O ICH recomenda que sejam efetuadas no pelo menos 9 determinações com no
mínimo três concentrações, em triplicata, cobrindo a faixa especificada ou 6
determinações a 100% da concentração do teste (ICH, 1994; ICH, 1996; USP 24, 2000).
3.5.2 - Precisão intermediária refere-se a variações dentro do mesmo laboratório tais
como: Método executado em dias diferentes, por analistas diferentes, com equipamentos
diferentes (ICH, 1994; ICH, 1996; USP 24, 2000).
3.5.3 - Reprodutibilidade (ensaio interlaboratorial) refere-se ao uso do procedimento
analítico em diferentes laboratórios (ICH, 1994; ICH, 1996; USP 24, 2000).
A precisão de um método analítico é normalmente expressa como a estimativa do
desvio padrão ou o desvio padrão relativo (coeficiente de variação) de uma série de
medidas e deve incluir os estudos da media, do desvio padrão, desvio padrão relativo e
intervalo de confiança.
3.6. Faixa
A faixa de um método analítico é o intervalo entre as concentrações inferior e
superior da substância de interesse na amostra para o qual o método tem um adequado
nível de precisão, exatidão e linearidade (ICH, 1994; ICH, 1996; USP 24, 2000).
3.7. Teste de Verificação da Adequação do Sistema (System Suitability Testing)
O teste de verificação da adequação do sistema pode ser descrito como um sistema
de avaliação do equipamento, partes eletrônicas, operações analíticas e amostras a serem
analisadas como um todo. Em cromatografia líquida de alta eficiência pode ser verificada
através do desvio padrão relativo das áreas obtidas por varias injeções do padrão,
23

calculando a reprodutibilidade das injeções calcula-se também resolução, fator de
capacidade, eficiência da coluna e fator de cauda.
24

4. OBJETIVOS
4.1. Objetivo Geral
Este projeto tem por objetivo propor uma metodologia padrão para a validação do
método analítico de controle da qualidade do produto Lamivudina comprimido, com
enfoque na dosagem do principio ativo Lamivudina (-)2',3'-didesoxi-3'-tiocitidina e no
ensaio limite do enantiômero (+)BCH-189 comprovadamente mais citotóxico em
linfócitos não infectados, que a forma L(-).
4.2. Objetivos Específicos
a) Propor e validar a metodologia para análise do teor do comprimido de Lamivudina
segundo os parâmetros do International Conference on Harmonization (ICH) e da
Farmacopéia Americana (USP 24).
b) Propor e validar a metodologia para ensaio limite para o enantiômero (+) BCH-189
segundo os parâmetros do ICH e da USP 24.
c) Efetuar pesquisa bibliográfica sobre a ação farmacológica e a toxicidade do
enantiômero (+)BCH-189.
25

5. PARTE EXPERIMENTAL
5.1. Metodologia de Análise
O método utilizado foi a análise por cromatografia líquida de alta eficiência
(CLAE) com detecção por ultravioleta tanto para a quantificação do maior componente,
Lamivudina, quanto para o ensaio limite do enantiômero (+)BCH-189.
A metodologia para análise do teor do produto IVB-Lamivudina foi previamente
avaliada através de um estudo preliminar realizado em 2000, usando como referência a
resolução n° 391 de 9 agosto de 1999.
Para este estudo foram selecionadas varias metodologias de análise, fornecidas
pelos fabricantes da matéria-prima, por laboratórios oficiais e retiradas de artigos
científicos, estas foram testadas e escolhida a metodologia que apresentou os melhores
resultados nos parâmetros de adequação do sistema, este método foi validado através da
verificação dos seguintes parâmetros: linearidade, especificidade, precisão (repetitividade)
e exatidão.
Posteriormente verificou-se a necessidade de complementar este estudo de acordo
com os requisitos do ICH e da USP 24 além de realizar a validação do ensaio limite para o
enantiômero (+)BCH-189.
Na otimização do método de análise do teor de Lamivudina foram avaliados alguns
parâmetros de robustez tais como: variação de pH, proporção da fase móvel, fluxo, lotes
das colunas e concentração da amostra, sendo escolhido o método que apresentou os
melhores resultados nos parâmetros de adequação do sistema.
O cromatograma obtido está representado na figura 4, pág. 27, e , apresentou os
seguintes resultados T = 1,48 ; N = 2286 e k’ = 657.
Foi efetuada a confirmação do comprimento de onda máximo de absorção
utilizando espectrofotometria UV/Vis, com detector de arranjo de fotodiodos (DAD).
O espectro obtido esta representado na figura 5, pág. 27. Nesta fase também foram
avaliadas a estabilidade da solução padrão e do eluente considerando-se estáveis por 24 h e
sete dias respectivamente.
26

2.71
3.12
6.58
, LA
MIVU
DINA
0 1 2 3 4 5 6 7 8 9
Retention Time (min)
0
200
400
600
800
Intensity (mV)
Figura 4: Exemplo de cromatograma do método de análise do teor de Lamivudina.
Figura 5: Espectro Ultravioleta da Lamivudina - Máximo de absorção a 270nm.
27

A metodologia de análise desenvolvida para o ensaio limite do enantiômero
(+)BCH-189, fig.6 pág.28, foi selecionada testando metodologias de análise, fornecidas
pelos fabricantes da matéria-prima e por laboratórios oficiais, considerando a metodologia
que apresentou os melhores resultados nos parâmetros de adequação do sistema, onde a
Lamivudina apresentou N = 4385 ; T = 1,82 ; k’= 804 e o (+)BCH-189 apresentou
N = 5697 ; T = 1,05 ; k’ = 1031 e a resolução entre os picos (R) foi igual a 4,39.
8.05
, La
mivu
dina
10.3
2, (
+) B
CH 1
89
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Intensity (mV)
Figura 6: Exemplo de cromatograma do ensaio limite do (+)BCH-189
Após a definição das condições analíticas os métodos foram submetidos a
validação.
5.1.1. Materiais e Reagentes
A substância química de referência (Lamivudina) utilizada no estudo preliminar
pertence ao lote 981010, Teor – 100,0%, sintetizada na Universidade da Geórgia - USA.
No estudo complementar a substância química de referência utilizada foi a
Lamivudina - controle L1, Teor – 100,2%, fornecida pelo INCQS e com ela foi
padronizada uma matéria-prima que foi utilizada como padrão secundário nas análises.
A mistura racêmica utilizada foi fornecida pelo laboratório Cristália, constituída,
segundo o fabricante, por 51% de Lamivudina e 49% de (+)BCH-189.
28

Os comprimidos de Lamivudina utilizados para a avaliação da precisão foram
fabricados por laboratórios oficiais.
Todos os reagentes utilizados eram de grau pró análise ou cromatográfico e todos
os equipamentos estavam devidamente qualificados e/ou calibrados.
A balança analítica utilizada foi da marca Sartórius e possui quatro casas decimais.
Para a execução das analises foram utilizados dois Sistema LaChrom (Merck-
Hitachi) para cromatografia líquida de alta eficiência, cada um composto por uma bomba
modelo L-7100, um detector UV modelo L-7400 e um injetor Rheodyne manual modelo
L-7725i, acoplados a um computador Pentium.
Para obter o espectro da Lamivudina foi utilizado um cromatógrafo líquido de alta
eficiência da marca Shimadzu série LC-10 A, equipado com detector de arranjo de
fotodiodo.
5.2. Ensaio do Teor de Lamivudina
5.2.1. Sistema Cromatográfico
A cromatografia foi executada nas seguintes condições: coluna fase reversa
C-18 (5µm), 250 x 4mm, temperatura ambiente, fluxo de 1,0 mL/min detecção no
comprimento de onda de 270 nm, o volume de injeção foi de 20,0 µL, utilizando como fase
móvel a mistura da solução de acetato de amônio 0,1 M e metanol na proporção de 85:15
v/v, com pH ajustado a 5,0 com ácido acético glacial PA.
5.2.2. Preparo da Solução Tampão de Acetato de Amônio 0,1 M
A solução tampão foi preparada, pesando 7,67 g de acetato de amônio transferindo
para balão volumétrico de 1000,0 mL acrescentando 500 mL de água purificada,
adicionando 1,0 mL de ácido acético glacial, levando ao ultra-som por 15 minutos e
completando o volume.
5.2.3. Preparo da Fase Móvel
A fase móvel foi preparada misturando 850 mL do tampão de acetato de amônio
0,1 M com 150 mL de metanol grau CLAE , corrigindo o pH para 5,0 com ácido acético
29

glacial, filtrando por membrana 0,45 µ e desaerando por 15 minutos, utilizando o ultra-
som.
5.2.4. Preparo da Solução Padrão
O padrão foi preparado pesando cuidadosamente cerca de 20,0 mg de Lamivudina
padrão secundário, transferindo para balão volumétrico de 50,0 mL, adicionando 25 mL da
fase móvel e colocando no banho de ultra-som por aproximadamente 15 minutos. Após
esfriar, completando o volume com a fase móvel, homogeneizando e filtrando através de
membrana 0,45 µ.
5.2.5. Preparo da Solução Amostra
Inicialmente foi determinado o peso médio de no mínimo 20 comprimidos que em
seguida foram triturados.
A solução da amostra foi preparada pesando o equivalente a 20,0 mg de
Lamivudina e transferindo para balão volumétrico de 50,0 mL, dissolvendo com 25 mL de
fase móvel e colocando no banho de ultra-som por aproximadamente 15 minutos, após
esfriar, completando o volume com a fase móvel, homogeneizando e filtrando através de
membrana 0,45 µ. Foram preparadas triplicatas da amostra.
O procedimento seguinte foi a injeção no cromatógrafo, de 20,0 µL de cada solução
(padrão e amostra) sendo efetuadas no mínimo 3 injeções de padrão.
O cálculo de teor do comprimido foi executado a partir da comparação das médias
das áreas dos cromatogramas dos padrões com as áreas dos cromatogramas das amostras.
5.3. Ensaio Limite para a Impureza Enantiomérica (+) BCH-189
5.3.1. Sistema Cromatográfico
A cromatografia foi executada nas seguintes condições: coluna quiral (fase
estacionária β ciclodextrina) - 5µm, 250 x 4,6mm., a temperatura ambiente, fluxo de
1,0 mL/min, detecção no comprimento de onda de 270 nm, o volume de injeção foi de 20,0
µL, utilizando como fase móvel a mistura da solução de acetato de amônio 0,1 M e
metanol na proporção de 91:9 v/v.
30

5.3.2. Preparo da Solução de Acetato de Amônio 0,1M
A solução de acetato de amônio 0,1 M foi preparada, pesando 7,67 g de acetato de
amônio transferindo para balão volumétrico de 1000,0 mL, acrescentando 500 mL de água
purificada, levando ao ultra-som por 15 minutos e completando o volume.
5.3.3. Preparo da Fase Móvel
A fase móvel foi preparada misturando 910 mL da solução de acetato de amônio
0,1 M com 90 mL de Metanol grau CLAE, filtrando por membrana 0,45µ e desaerando por
15 minutos, utilizando o ultra-som.
5.3.4. Preparo da Solução para a Avaliação da Resolução
Foi preparada uma solução da mistura racêmica de (+)BCH-189 e Lamivudina em
fase móvel, onde a concentração dos enantiômeros era cerca de 75,0 mcg/mL Procedeu-se
a injeção no cromatógrafo de 20 µL desta solução e verificou-se que a resolução entre a
Lamivudina e o (+)BCH-189 foi maior que 1,5.
5.3.5. Preparo da Amostra
Inicialmente foi determinado o peso médio de no mínimo 20 comprimidos que em
seguida foram triturados.
A solução da amostra foi preparada pesando o equivalente a 25,0 mg de
Lamivudina e transferindo para balão volumétrico de 100,0mL, dissolvendo com 50 mL de
fase móvel e colocando no banho de ultra-som por aproximadamente 15 minutos. Após
esfriar, completando o volume com a fase móvel, homogeneizando e filtrando através de
membrana 0,45 µ. Foram preparadas triplicatas da amostra.
O procedimento seguinte foi a injeção no cromatógrafo, 20 µL da solução amostra.
O cálculo do ensaio limite de (+)BCH-189 no comprimido foi executado
relacionando a área do picos de (+)BCH-189 com a área do pico de Lamivudina no
cromatograma da amostra.
31
Segundo a Pharmacopeia Forum o limite máximo permitido de (+)BCH-189 na
matéria-prima Lamivudina é de 0,3% em relação ao pico principal, portanto este mesma
especificação foi utilizada para o ensaio limite de (+)BCH-189 no comprimido de

Lamivudina, já que não há especificação de limite de (+)BCH-189 neste produto em
nenhum compêndio oficial (PHARMACOPEIAL Forum, 2001).
5.4. Metodologia para a Validação da Análise de Teor da Lamivudina
Foram avaliados o teste de verificação da adequação do sistema e os seguintes
parâmetros: especificidade, linearidade, exatidão, precisão e faixa.
5.4.1.Teste de Verificação da Adequação do Sistema (System Suitability Testing)
O teste de verificação da adequação do sistema foi executado para assegurar a
performance do sistema de cromatografia líquida de alta eficiência antes e durante a
realização das análises provendo assim dados de qualidade aceitável.
O teste é baseado no conceito de que o equipamento, partes eletrônicas, operações
analíticas e amostras constituem um sistema integral que pode ser avaliado como um todo.
O teste de verificação da adequação do sistema foi executado avaliando os
seguintes parâmetros: número de pratos teóricos, fator de cauda e repetitividade das
injeções. O parâmetro repetitividade das injeções foi avaliado na concentração 100% do
valor rotulado através do cálculo do desvio padrão relativo da área de seis injeções de
padrão.
5.4.2. Especificidade
Para a determinação da especificidade do método amostras de Lamivudina padrão
secundário, foram submetidas a condições de estresse tais como: exposição a solução de
HCl 0,1 N por 24 h, a solução de NaOH 0,1 N por 24 h, a solução de H2O2 3% por 24 h e
a temperatura de 150° C por 24 h. Foi analisada também uma solução com todos os
excipientes.
Os cromatogramas foram avaliados quanto a interferência dos produtos de
degradação e dos excipientes, utilizando os mesmos parâmetros de comprimento de onda,
fluxo, temperatura e fase móvel especificados para a Lamivudina.
5.4.3. Linearidade
Para a verificação da linearidade, foi preparada uma solução mãe de Lamivudina
padrão secundário e a partir desta, diluições a fim de obtermos soluções em cinco níveis
32

de concentração, como recomendado em literatura. As concentrações utilizadas no estudo
foram 50 %, 75 %, 100 %, 125 % e 150 %, do valor rotulado.
A análise foi conduzida com triplicata de injeção em cada nível de concentração.
5.4.4. Exatidão
A Exatidão foi avaliada, efetuando-se nove determinações em triplicata de injeção
em cada nível de concentração, 75%, 100% e 125% do valor rotulado de Lamivudina.
Foi preparada uma matriz, mistura dos excipientes da formulação, e calculada a quantidade
a ser adicionada ao principio ativo para se obter comprimidos de peso médio constante nas
concentração preestabelecidas.
A analise foi realizada pelo mesmo analista no mesmo dia.
5.4.5. Precisão
A precisão foi verificada nos níveis repetitividade e precisão intermediária
efetuando 6 (seis) determinações com duplicata de injeção a 100% do valor rotulado,
através da analise de 3 (três) amostras produzidas por diferentes laboratórios.
No nível repetitividade as seis análises, de cada uma das amostras, foram analisadas
no mesmo dia e pelo mesmo analista, com duplicata de injeção.
Para o parâmetro precisão intermediária foram executados três tipos de
comparação. No primeiro, as mesmas três amostras de laboratórios diferentes foram
analisadas da mesma forma descrita acima por dois analistas diferentes. Além disso elas
também foram analisadas pelo mesmo analista utilizando dois equipamentos diferentes e
finalmente a terceira comparação foi realizada entre analistas diferentes com equipamentos
diferentes, esta última comparação pode ser usada para sugerir a resistência do método.
5.5. Metodologia para a Validação do Ensaio Limite do Enantiômero (+)BCH-189
Além dos parâmetros teste de adequação do sistema e da especificidade, descritos
anteriormente também foi determinado o limite de detecção.
5.5.1. Especificidade
Para a determinação da especificidade do ensaio limite do enantiômero
(+)BCH-189 foram utilizadas as mesmas amostras que para a especificidade do método de
análise do teor, item 5.4.2, pág. 32.
33

5.5.2. Limite de Detecção
O limite de detecção foi avaliado experimentalmente analisando-se soluções com
baixas concentrações de (+)BCH-189 e determinando a concentração mínima em que este
pode ser detectado.
Com a mistura racêmica fornecida pelo laboratório Cristália foi preparada uma
solução mãe e a partir desta foram preparadas soluções de concentrações decrescentes.
Como a especificação do limite de (+)BCH-189, é dada em relação a concentração
de Lamivudina, foi avaliada a detecção de (+)BCH-189 em presença de Lamivudina, na
concentração de 250 mcg/ml, que é a concentração do ensaio. Nestas condições foi
avaliada também a resolução entre os picos (Pharmacopeial Forum, 2001).
5.6. Metodologia para a Pesquisa Bibliográfica sobre a Toxicidade do (+)BCH-189
A pesquisa bibliográfica sobre a farmacologia e a toxicidade do enantiômero
(+)BCH-189 foi realizada nos bancos de dados medline e science direct. A análise dos
artigos buscava informações sobre a farmacologia; estudos experimentais do tipo "in vivo"
"in vitro"; toxicidade de antiretrovirais em células do hospedeiro; mecanismo molecular
de inibição e toxicidade dos nucleosídeos análogos; toxicidade mitocondrial;
citotoxicidade e estudos comparativos da toxicidade das drogas antiretrovirais, mais
especificamente do (+)BCH-189. Foram utilizadas as seguintes palavras chave na
pesquisa: (+) 3TC toxicity; (+)BCH-189; toxicity; mitochondrial toxicity; cytotoxicity.
34

6. RESULTADOS E DISCUSSÃO
A análise estatística dos dados foi realizada usando o software Excel.
6.1. Resultados do Teste de Adequação do Sistema (System Suitability Testing)
O teste de adequação do sistema foi realizado antes de todas as corridas
cromatográficas e todos os resultados obtidos para os critérios: número de pratos teóricos,
assimetria, fator de capacidade e resolução; foram considerados satisfatórios segundo as
especificações apresentadas na tabela 3, pág.35.
Tabela 3: Parâmetros e recomendações para o teste de verificação da adequação do
sistema (SHABIR, 2003)
Parâmetros Recomendações
Fator de capacidade ( k') O pico deve estar bem resolvido de outros
picos e do volume morto k' > 2,0.
Repetitividade É desejável um DPR ≤ 1% para N ≥ 5
Tempo de retenção relativo Não é essencial se a resolução é declarada
Resolução ( R ) R > 2 entre o pico de interesse e o potencial
interferente que elui mais próximo
Fator de cauda ( T ) T ≤ 2
Número de pratos teóricos ( N ) Em geral deve ser > 2000
Os resultados do teste de adequação do sistema para o método de análise do teor de
Lamivudina, tabela 4 pág.36 referem-se ao cromatograma apresentado na fig. 4 pág. 27.
35

Tabela 4: Resultados do teste de adequação do sistema para o método de análise do teor de
Lamivudina
Nome TR k’ T N
Lamivudina 6,58 657 1,48 2286
Os resultados do teste de adequação do sistema para o ensaio limite para o
(+)BCH-189, tabela 5 pág.36 referem-se ao cromatograma apresentado na fig. 6 pág. 28.
Tabela 5: Resultados do teste de adequação do sistema para o ensaio limite para o
(+)BCH-189
Nome TR k’ T N R
Lamivudina 8,05 804 1,82 4385 -
(+)BCH-189 10,32 1031 1,05 5697 4,39
6.2. Resultados da Especificidade para o Método de Análise do Teor
Os cromatogramas obtidos com as amostras submetidas a condições de estresse e com a
solução contendo os excipientes, item 5.4.2 pág. 32, foram avaliados e verificado que
nenhum dos componentes, sejam eles produtos de degradação, impurezas ou excipientes,
possui o mesmo tempo de retenção da Lamivudina. Portanto os resultados obtidos não são
afetados pela presença destas substâncias.
A amostra de Lamivudina na concentração de trabalho, isto é 20,0 mg para 50,0 mL
(400mcg/mL), estressada com H2O2 3% por 24 h não sofre interferência dos produtos de
degradação, como observa-se na figura 7 pág. 37.
36

2.13
2.61
3.24
6.98
, LA
MIVU
DINA
0 1 2 3 4 5 6 7 8 9
Retention Time (min)
0
200
400
600
800
1000
1200Intensity (mV)
Figura 7: Amostra de Lamivudina, padrão secundário, submetida a estresse com H2O2 3%
por 24 h.
Verifica-se na figura 8 pág. 37 que a amostra de Lamivudina na concentração de
trabalho, estressada com HCl 0,1M por 24 h, não sofreu interferência dos produtos de
degradação.
2.09
2.82
3.20
7.00
, LA
MIVU
DINA
0 1 2 3 4 5 6 7 8 9
Retention Time (min)
0
200
400
600
800
Intensity (mV)
Figura 8: Amostra de Lamivudina, padrão secundário, submetida a estresse com HCl
0,1 M por 24 h.
A amostra de Lamivudina na concentração de trabalho, estressada com NaOH
0,1 M por 24 h não sofre interferência dos produtos de degradação como verifica-se na
figura 9 pág. 38.
37
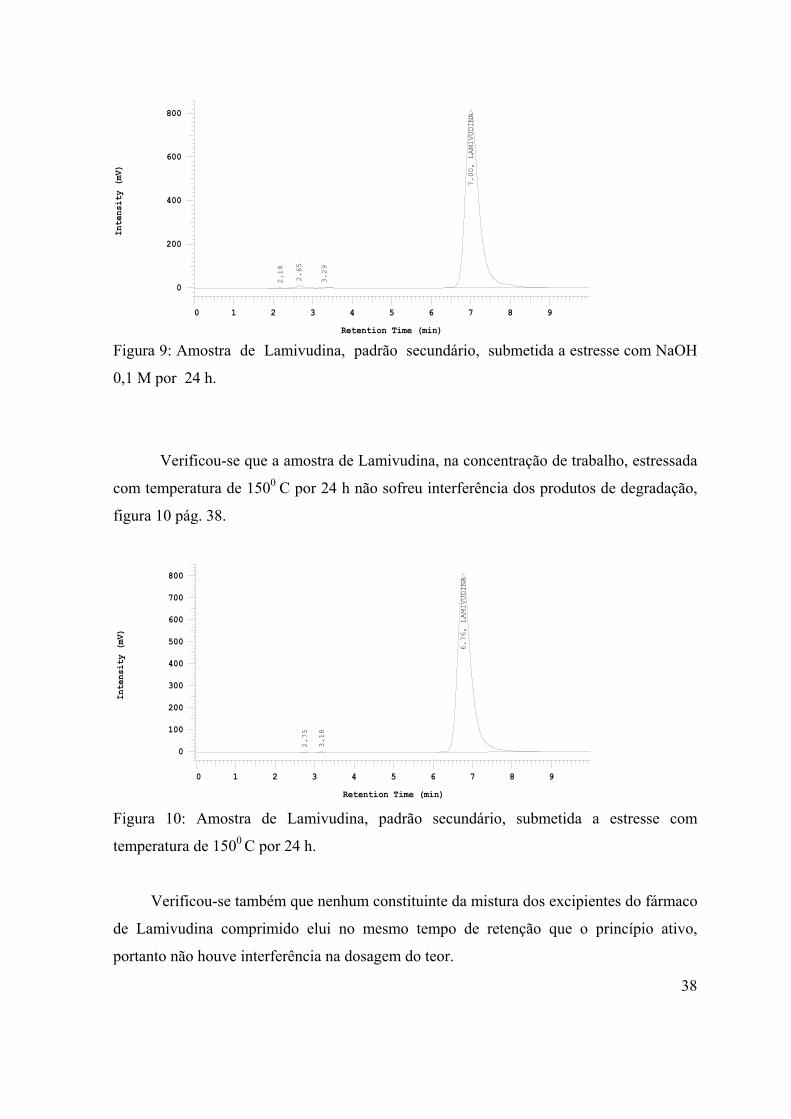
2.18
2.65
3.29
7.00
, LA
MIVU
DINA
0 1 2 3 4 5 6 7 8 9
Retention Time (min)
0
200
400
600
800Intensity (mV)
Figura 9: Amostra de Lamivudina, padrão secundário, submetida a estresse com NaOH
0,1 M por 24 h.
Verificou-se que a amostra de Lamivudina, na concentração de trabalho, estressada
com temperatura de 1500 C por 24 h não sofreu interferência dos produtos de degradação,
figura 10 pág. 38.
2.75
3.18
6.76
, LA
MIVU
DINA
0 1 2 3 4 5 6 7 8 9
Retention Time (min)
0
100
200
300
400
500
600
700
800
Intensity (mV)
Figura 10: Amostra de Lamivudina, padrão secundário, submetida a estresse com
temperatura de 1500 C por 24 h.
Verificou-se também que nenhum constituinte da mistura dos excipientes do fármaco
de Lamivudina comprimido elui no mesmo tempo de retenção que o princípio ativo,
portanto não houve interferência na dosagem do teor.
38

6.3. Resultados da Linearidade
A linearidade foi verificada traçando-se a curva de calibração (gráfico das
respostas, eixo y, em função da concentração, eixo x), onde x corresponde à concentração
da Lamivudina padrão e y é a área da Lamivudina obtida no cromatograma, em cinco
concentrações da solução padrão: 50, 75, 100, 125 e 150 %.
Os resultados obtidos para as cinco concentrações da solução padrão estão contidos
na tabela 6, pág. 39.
Tabela 6: Dados de concentração para o cálculo da Linearidade
Amostra
1
Concentração
2
3
Média
DP
DPR
Am.1 – 50% 50,09 50,02 50,09 50,07 0,04 0,08
Am.2 – 75% 75,13 75,05 75,11 75,10 0,04 0,05
Am.3 – 100% 100,03 99,94 100,03 100,00 0,05 0,05
Am.4 – 125% 124,00 123,99 123,93 123,97 0,04 0,03
Am.5 – 150% 148,91 148,67 148,80 148,79 0,12 0,08
Inicialmente suspeitou-se que o valor 148,67, referente a amostra 5 – 150%, era
aberrante, isto é não pertencia a população, portanto aplicou-se o teste de Grubbs, para o
nível de significância de 0,05.
G = (yij - y) / sij
Onde :
yij = valor suspeito de ser aberrante
yi = média dos valores obtidos para uma determinada concentração i
sij = desvio padrão dos valores obtidos
G calculado = 148,67 – 148,79 / 0,12 = -1,00
Em função do número de replicatas (n)
Se G calculado < G tabelado ou crítico, o valor suspeito não é considerado aberrante.
Se G calculado > G tabelado ou crítico, o valor suspeito é considerado aberrante.
Para n = 3 ; G crítico = 1,155
G calculado é menor que o G crítico, logo o valor suspeito não é aberrante.
39

Na concentração de 100% foram realizadas 6 injeções para verificar a
repetitividade.
Tabela 7: Repetitividade das injeções da concentração 100%
Concentração 100% Concentração das seis injeções
de padrão a 100 %
Injeção A 100,03
Injeção B 99,95
Injeção C 100,04
Injeção D 99,94
Injeção E 100,00
Injeção F 100,04
Média 100,00
DP 0,05
DPR 0,05
Para um número de injeções maior ou igual a cinco, é recomendável um
DPR < 1% (SHABIR, 2003). O desvio padrão relativo encontrado entre as seis injeções da
concentração 100% foi 0,05 sendo portanto considerado satisfatório.
Foi verificada a homocedasticidade do método, isto é, que as respostas (leituras do
equipamento) são independentes da variância da concentração da substância de interesse,
através do método de Cochran. A curva de calibração foi calculada pelo método dos
mínimos quadrados, que estima qual a melhor reta que passa pelos pontos obtidos
experimentalmente a partir da curva de calibração. Foram calculados os valores dos
coeficientes linear, (a) e angular (b) da reta e o coeficiente de correlação (r), e os intervalos
de confiança para os coeficientes da reta.
40
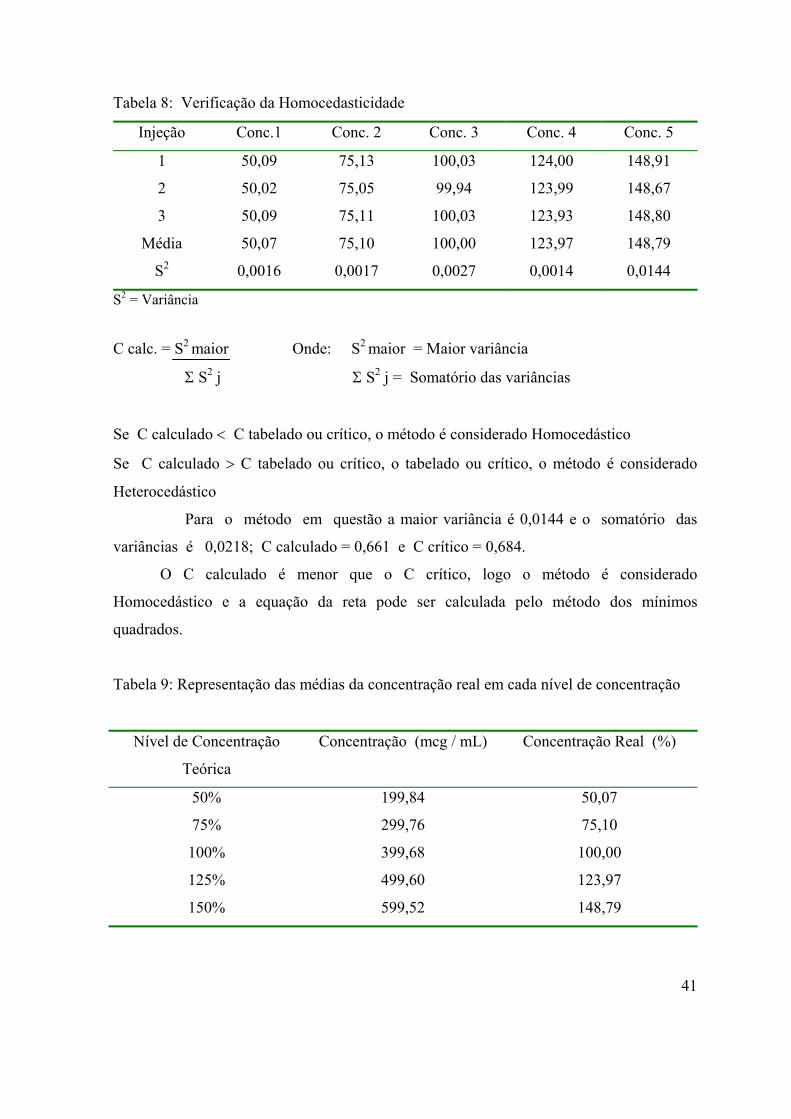
Tabela 8: Verificação da Homocedasticidade
Injeção Conc.1 Conc. 2 Conc. 3 Conc. 4 Conc. 5
1 50,09 75,13 100,03 124,00 148,91
2 50,02 75,05 99,94 123,99 148,67
3 50,09 75,11 100,03 123,93 148,80
Média 50,07 75,10 100,00 123,97 148,79
S2 0,0016 0,0017 0,0027 0,0014 0,0144
S2 = Variância
C calc. = S2 maior Onde: S2 maior = Maior variância
Σ S2 j Σ S2 j = Somatório das variâncias
Se C calculado < C tabelado ou crítico, o método é considerado Homocedástico
Se C calculado > C tabelado ou crítico, o tabelado ou crítico, o método é considerado
Heterocedástico
Para o método em questão a maior variância é 0,0144 e o somatório das
variâncias é 0,0218; C calculado = 0,661 e C crítico = 0,684.
O C calculado é menor que o C crítico, logo o método é considerado
Homocedástico e a equação da reta pode ser calculada pelo método dos mínimos
quadrados.
Tabela 9: Representação das médias da concentração real em cada nível de concentração
Nível de Concentração
Teórica
Concentração (mcg / mL) Concentração Real (%)
50% 199,84 50,07
75% 299,76 75,10
100% 399,68 100,00
125% 499,60 123,97
150% 599,52 148,79
41
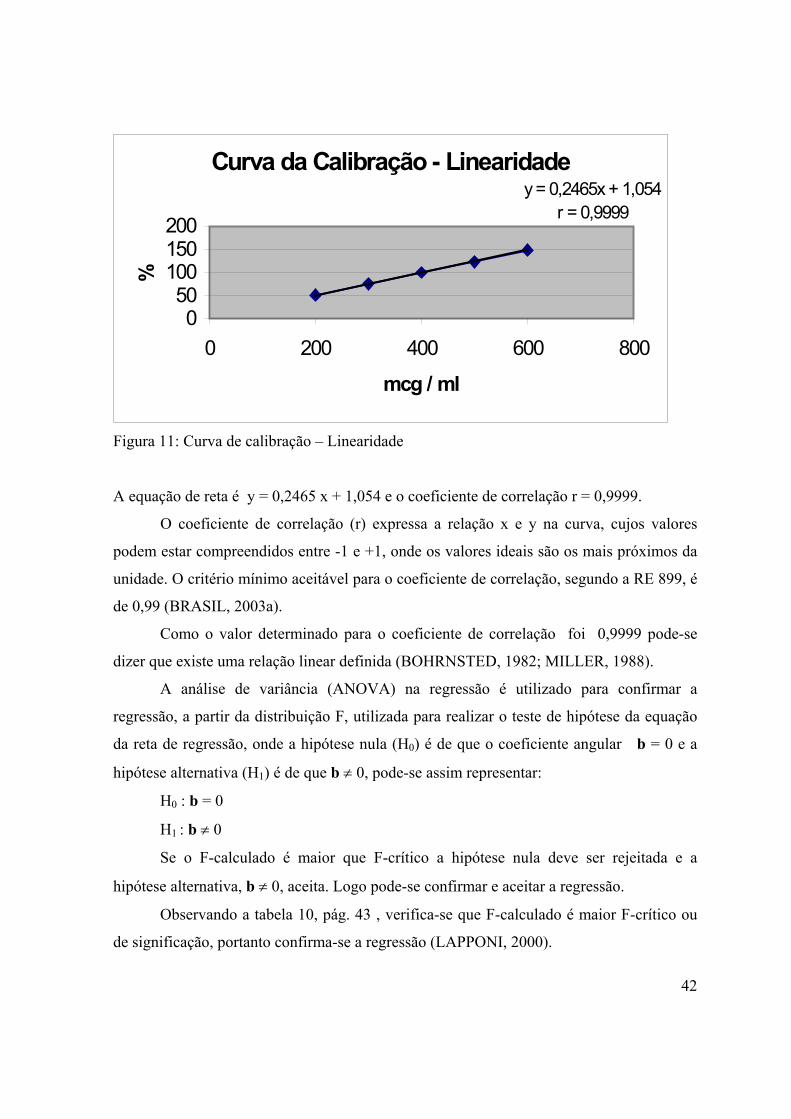
Curva da Calibração - Linearidadey = 0,2465x + 1,054
r = 0,9999
050
100150200
0 200 400 600 800
mcg / ml
%
Figura 11: Curva de calibração – Linearidade
A equação de reta é y = 0,2465 x + 1,054 e o coeficiente de correlação r = 0,9999.
O coeficiente de correlação (r) expressa a relação x e y na curva, cujos valores
podem estar compreendidos entre -1 e +1, onde os valores ideais são os mais próximos da
unidade. O critério mínimo aceitável para o coeficiente de correlação, segundo a RE 899, é
de 0,99 (BRASIL, 2003a).
Como o valor determinado para o coeficiente de correlação foi 0,9999 pode-se
dizer que existe uma relação linear definida (BOHRNSTED, 1982; MILLER, 1988).
A análise de variância (ANOVA) na regressão é utilizado para confirmar a
regressão, a partir da distribuição F, utilizada para realizar o teste de hipótese da equação
da reta de regressão, onde a hipótese nula (H0) é de que o coeficiente angular b = 0 e a
hipótese alternativa (H1) é de que b ≠ 0, pode-se assim representar:
H0 : b = 0
H1 : b ≠ 0
Se o F-calculado é maior que F-crítico a hipótese nula deve ser rejeitada e a
hipótese alternativa, b ≠ 0, aceita. Logo pode-se confirmar e aceitar a regressão.
Observando a tabela 10, pág. 43 , verifica-se que F-calculado é maior F-crítico ou
de significação, portanto confirma-se a regressão (LAPPONI, 2000).
42

Como as concentrações utilizadas para a avaliação da linearidade variaram de 200
a 600 mcg / mL pode-se afirmar que o método é linear neste intervalo.
Tabela 10: Confirmação da linearidade por ANOVA
ANOVA
gl SQ MQ F F de significação
Regressão 1 18203,54 18203,54 235729,04 4,53E-29
Resíduo 13 1,00 0,08
Total 14 18204,54
gl = graus de liberdade; SQ = soma quadrática; MQ = média quadrática; F = F calculado; E =.exponencial
A tabela 11, pág. 43 contém os coeficientes da reta e os seus valores máximo e
mínimo para um nível de significância (α) igual a 0,05%, a partir dos quais foram
calculados os intervalos de confiança para os coeficientes da reta .
IC (a) = 1,52 - 1,05 = 0,47
IC (b) = 0,25 - 0,25 = 0
Tabela 11: Dados para o cálculo do intervalo de confiança dos coeficientes da reta
Coeficientes 95% inferiores 95% superiores
Interseção 1,05 0,59 1,52
Variável X 0,25 0,25 0,25
Foram calculados os resíduos, tabela 12, pág. 44, e com eles traçado um gráfico,
fig.12, pág.45, que mostra a sua distribuição. A população dos resíduos normalmente
distribuídos apresenta média igual a zero (MILLER, 1988).
43

Tabela 12: Resultado de resíduos
Concentração Y previsto Resíduos
1 50,32 -0,23
2 50,32 -0,30
3 50,32 -0,23
4 74,95 0,18
5 74,95 0,10
6 74,95 0,16
7 99,59 0,44
8 99,59 0,35
9 99,59 0,44
10 124,22 -0,22
11 124,22 -0,23
12 124,22 -0,29
13 148,85 0,06
14 148,85 -0,18
15 148,85 -0,05
44
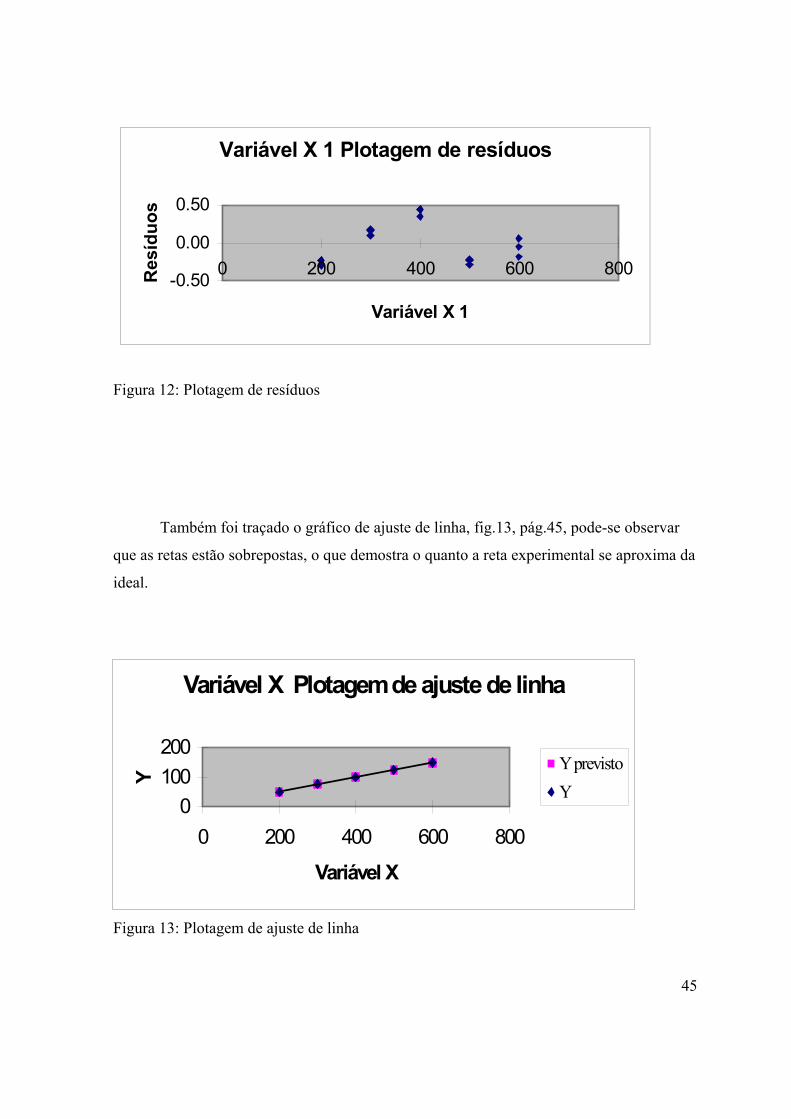
Variável X 1 Plotagem de resíduos
-0.50
0.00
0.50
0 200 400 600 800
Variável X 1
Res
íduo
s
Figura 12: Plotagem de resíduos
Também foi traçado o gráfico de ajuste de linha, fig.13, pág.45, pode-se observar
que as retas estão sobrepostas, o que demostra o quanto a reta experimental se aproxima da
ideal.
Variável X Plotagem de ajuste de linha
0100200
0 200 400 600 800
Variável X
Y
Y previstoY
Figura 13: Plotagem de ajuste de linha
45
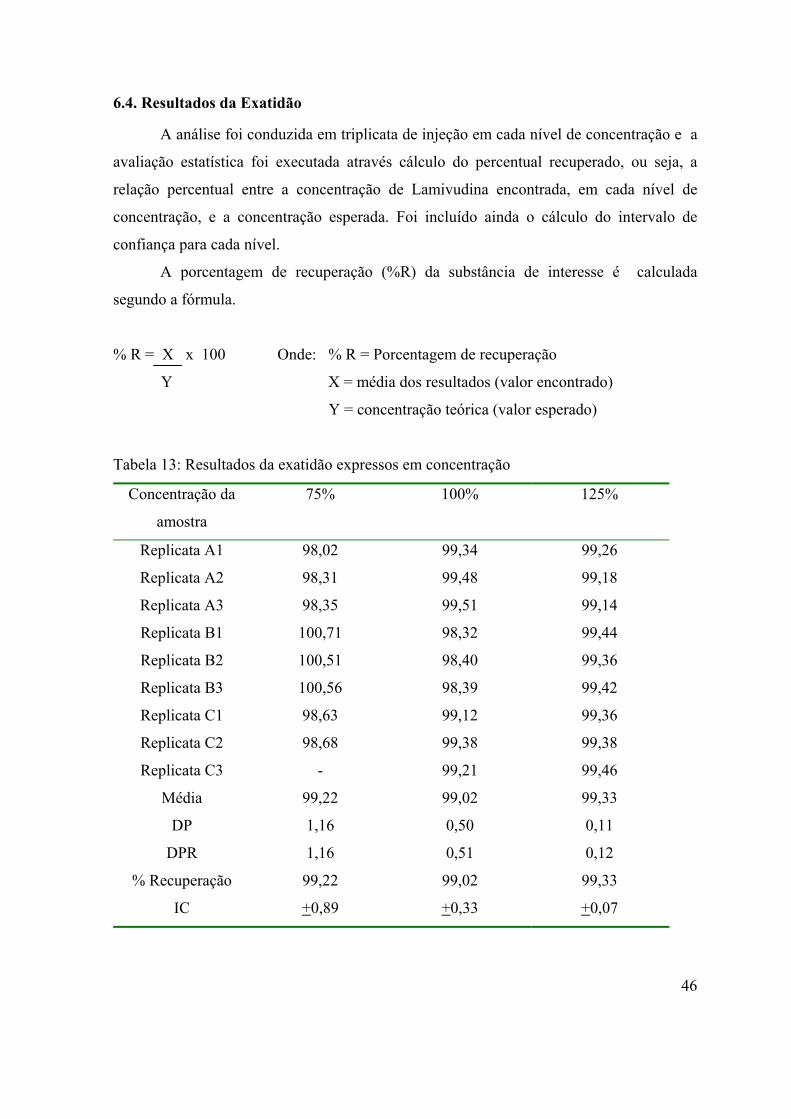
6.4. Resultados da Exatidão
A análise foi conduzida em triplicata de injeção em cada nível de concentração e a
avaliação estatística foi executada através cálculo do percentual recuperado, ou seja, a
relação percentual entre a concentração de Lamivudina encontrada, em cada nível de
concentração, e a concentração esperada. Foi incluído ainda o cálculo do intervalo de
confiança para cada nível.
A porcentagem de recuperação (%R) da substância de interesse é calculada
segundo a fórmula.
% R = X x 100 Onde: % R = Porcentagem de recuperação
Y X = média dos resultados (valor encontrado)
Y = concentração teórica (valor esperado)
Tabela 13: Resultados da exatidão expressos em concentração
Concentração da
amostra
75% 100% 125%
Replicata A1 98,02 99,34 99,26
Replicata A2 98,31 99,48 99,18
Replicata A3 98,35 99,51 99,14
Replicata B1 100,71 98,32 99,44
Replicata B2 100,51 98,40 99,36
Replicata B3 100,56 98,39 99,42
Replicata C1 98,63 99,12 99,36
Replicata C2 98,68 99,38 99,38
Replicata C3 - 99,21 99,46
Média 99,22 99,02 99,33
DP 1,16 0,50 0,11
DPR 1,16 0,51 0,12
% Recuperação 99,22 99,02 99,33
IC +0,89 +0,33 +0,07
46

Suspeitou-se que o valor 96,24; referente a replicata C3 da concentração 75%, era
aberrante, portanto foi aplicado o teste de Grubbs:
G = (yij - y) / sij
G = 99,22 - 96,24 / 1,16 = 1,158
Foi utilizado o n = 3 pois se trata de triplicata de injeção;
G crítico = 1,155
G calculado é maior que o G crítico, logo o valor suspeito é aberrante e foi retirado dos
cálculos. Os resultados apresentados na tabela 13, pág.46 já não consideram o valor da
replicata C3 da concentração 75%.
De acordo com a bibliografia, as médias dos valores de recuperação do método
analítico devem ser iguais a 100 % + 2 do valor rotulado (SHABIR, 2003). Portanto podem
variar de 98 a 102 %. Na tabela 13, pág.46 estão representadas as médias obtidas para cada
nível de concentração 75, 100 e 125%. Os valores obtidos foram respectivamente 99,22 %;
99,02 % e 99,33 %, sendo assim os valores de recuperação do método analítico foram
considerados satisfatórios.
6.5. Resultados da Precisão
A Precisão foi avaliada nos níveis Repetitividade e Precisão intermediária.
6.5.1. Resultados da Repetitividade
Os resultados foram avaliados através da análise dos desvios padrões relativos para
cada determinação, sendo considerado satisfatório o desvio padrão relativo menor ou igual
a 2 % pois segundo a literatura o valor máximo aceitável deve ser definido de acordo com
a metodologia empregada, a concentração da amostra, o tipo de matriz e a finalidade do
método, não se admitindo valores superiores a 5% (BRASIL, 2003a).
47
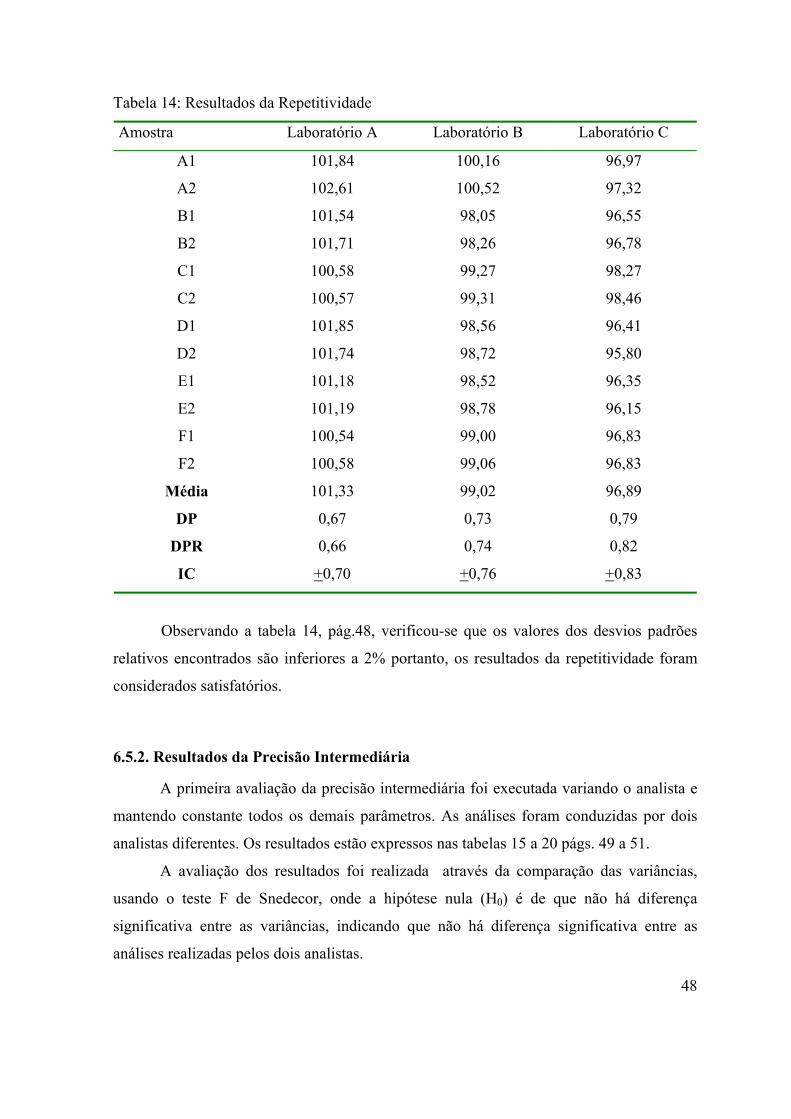
Tabela 14: Resultados da Repetitividade
Amostra Laboratório A Laboratório B Laboratório C
A1 101,84 100,16 96,97
A2 102,61 100,52 97,32
B1 101,54 98,05 96,55
B2 101,71 98,26 96,78
C1 100,58 99,27 98,27
C2 100,57 99,31 98,46
D1 101,85 98,56 96,41
D2 101,74 98,72 95,80
E1 101,18 98,52 96,35
E2 101,19 98,78 96,15
F1 100,54 99,00 96,83
F2 100,58 99,06 96,83
Média 101,33 99,02 96,89
DP 0,67 0,73 0,79
DPR 0,66 0,74 0,82
IC +0,70 +0,76 +0,83
Observando a tabela 14, pág.48, verificou-se que os valores dos desvios padrões
relativos encontrados são inferiores a 2% portanto, os resultados da repetitividade foram
considerados satisfatórios.
6.5.2. Resultados da Precisão Intermediária
A primeira avaliação da precisão intermediária foi executada variando o analista e
mantendo constante todos os demais parâmetros. As análises foram conduzidas por dois
analistas diferentes. Os resultados estão expressos nas tabelas 15 a 20 págs. 49 a 51.
A avaliação dos resultados foi realizada através da comparação das variâncias,
usando o teste F de Snedecor, onde a hipótese nula (H0) é de que não há diferença
significativa entre as variâncias, indicando que não há diferença significativa entre as
análises realizadas pelos dois analistas.
48
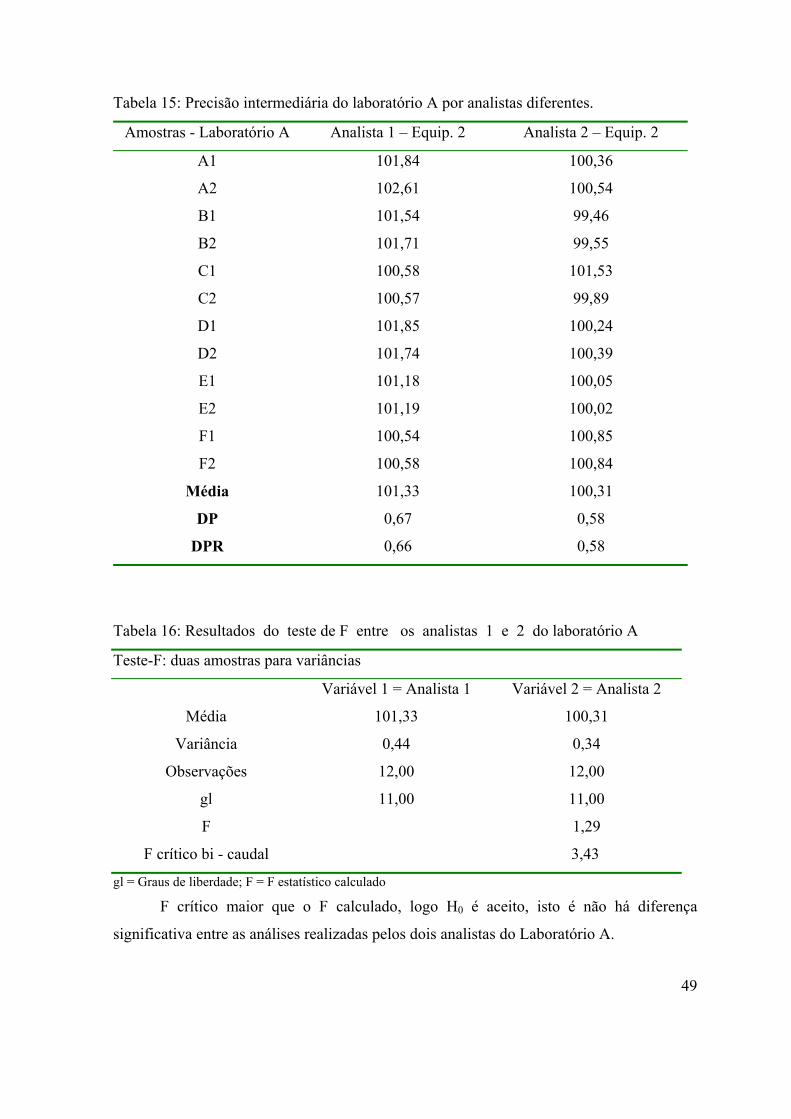
Tabela 15: Precisão intermediária do laboratório A por analistas diferentes.
Amostras - Laboratório A Analista 1 – Equip. 2 Analista 2 – Equip. 2
A1 101,84 100,36
A2 102,61 100,54
B1 101,54 99,46
B2 101,71 99,55
C1 100,58 101,53
C2 100,57 99,89
D1 101,85 100,24
D2 101,74 100,39
E1 101,18 100,05
E2 101,19 100,02
F1 100,54 100,85
F2 100,58 100,84
Média 101,33 100,31
DP 0,67 0,58
DPR 0,66 0,58
Tabela 16: Resultados do teste de F entre os analistas 1 e 2 do laboratório A
Teste-F: duas amostras para variâncias
Variável 1 = Analista 1 Variável 2 = Analista 2
Média 101,33 100,31
Variância 0,44 0,34
Observações 12,00 12,00
gl 11,00 11,00
F 1,29
F crítico bi - caudal 3,43
gl = Graus de liberdade; F = F estatístico calculado
F crítico maior que o F calculado, logo H0 é aceito, isto é não há diferença
significativa entre as análises realizadas pelos dois analistas do Laboratório A.
49

Tabela 17: Precisão intermediária do laboratório B por analistas diferentes.
Amostras - Laboratório B Analista 1 – Equip. 2 Analista 2 – Equip. 2
A1 100,16 100,80
A2 100,52 100,84
B1 98,05 98,15
B2 98,26 98,14
C1 99,27 99,75
C2 99,31 99,78
D1 98,56 99,36
D2 98,72 99,20
E1 98,52 99,39
E2 98,78 99,56
F1 99,00 99,20
F2 99,06 99,17
Média 99,02 99,44
DP 0,73 0,83
DPR 0,74 0,83
Tabela 18: Resultados do teste de F entre os analistas 2 e 1 do laboratório B
Teste-F: duas amostras para variâncias
Variável 1 = Analista 2 Variável 2 =Analista 1
Média 99,45 99,02
Variância 0,69 0,53
Observações 12,00 12,00
gl 11,00 11,00
F 1,30
F crítico bi - caudal 3,43
gl = Graus de liberdade; F = F estatístico calculado
F crítico maior que o F calculado, logo H0 é aceito, isto é não há diferença
significativa entre as análises realizadas pelos dois analistas do Laboratório B.
50
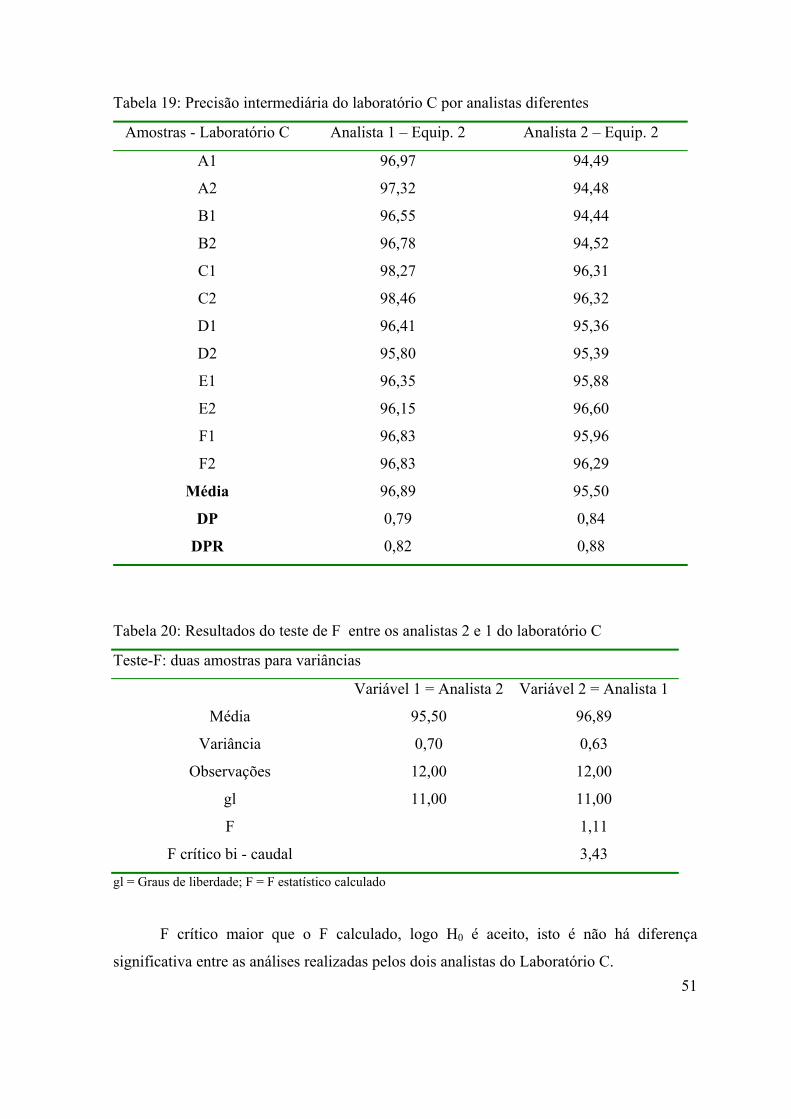
Tabela 19: Precisão intermediária do laboratório C por analistas diferentes
Amostras - Laboratório C Analista 1 – Equip. 2 Analista 2 – Equip. 2
A1 96,97 94,49
A2 97,32 94,48
B1 96,55 94,44
B2 96,78 94,52
C1 98,27 96,31
C2 98,46 96,32
D1 96,41 95,36
D2 95,80 95,39
E1 96,35 95,88
E2 96,15 96,60
F1 96,83 95,96
F2 96,83 96,29
Média 96,89 95,50
DP 0,79 0,84
DPR 0,82 0,88
Tabela 20: Resultados do teste de F entre os analistas 2 e 1 do laboratório C
Teste-F: duas amostras para variâncias
Variável 1 = Analista 2 Variável 2 = Analista 1
Média 95,50 96,89
Variância 0,70 0,63
Observações 12,00 12,00
gl 11,00 11,00
F 1,11
F crítico bi - caudal 3,43
gl = Graus de liberdade; F = F estatístico calculado
F crítico maior que o F calculado, logo H0 é aceito, isto é não há diferença
significativa entre as análises realizadas pelos dois analistas do Laboratório C. 51

A avaliação dos resultados da precisão intermediária, para comparar os resultados
das análises realizadas pelo mesmo analista em equipamentos diferentes e por analistas
diferentes em equipamentos diferentes, foi executada através da comparação das
variâncias, usando o teste F de Snedecor, onde a hipótese nula (H0) é de que não há
diferença significativa entre as variâncias, indicando que não há diferença significativa
entre as análises realizadas. Os resultados estão expressos nas tabelas 21 a 32 págs. 53 a
58.
52
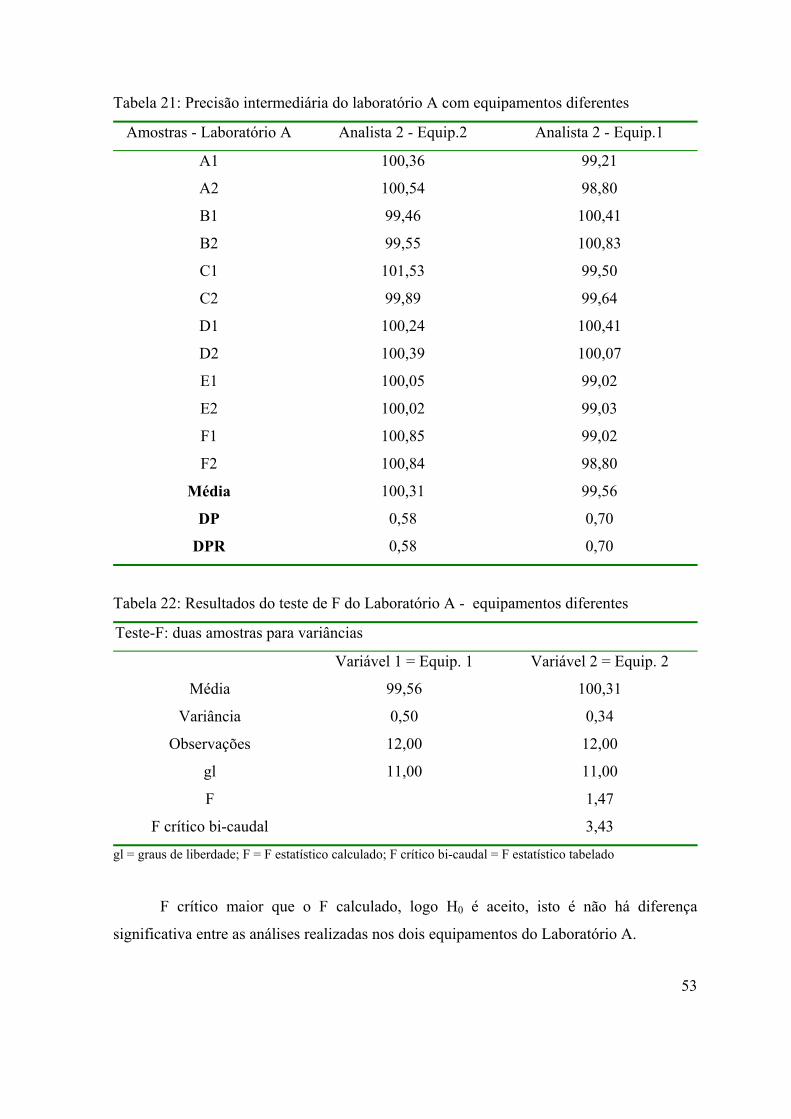
Tabela 21: Precisão intermediária do laboratório A com equipamentos diferentes
Amostras - Laboratório A Analista 2 - Equip.2 Analista 2 - Equip.1
A1 100,36 99,21
A2 100,54 98,80
B1 99,46 100,41
B2 99,55 100,83
C1 101,53 99,50
C2 99,89 99,64
D1 100,24 100,41
D2 100,39 100,07
E1 100,05 99,02
E2 100,02 99,03
F1 100,85 99,02
F2 100,84 98,80
Média 100,31 99,56
DP 0,58 0,70
DPR 0,58 0,70
Tabela 22: Resultados do teste de F do Laboratório A - equipamentos diferentes
Teste-F: duas amostras para variâncias
Variável 1 = Equip. 1 Variável 2 = Equip. 2
Média 99,56 100,31
Variância 0,50 0,34
Observações 12,00 12,00
gl 11,00 11,00
F 1,47
F crítico bi-caudal 3,43
gl = graus de liberdade; F = F estatístico calculado; F crítico bi-caudal = F estatístico tabelado
F crítico maior que o F calculado, logo H0 é aceito, isto é não há diferença
significativa entre as análises realizadas nos dois equipamentos do Laboratório A.
53
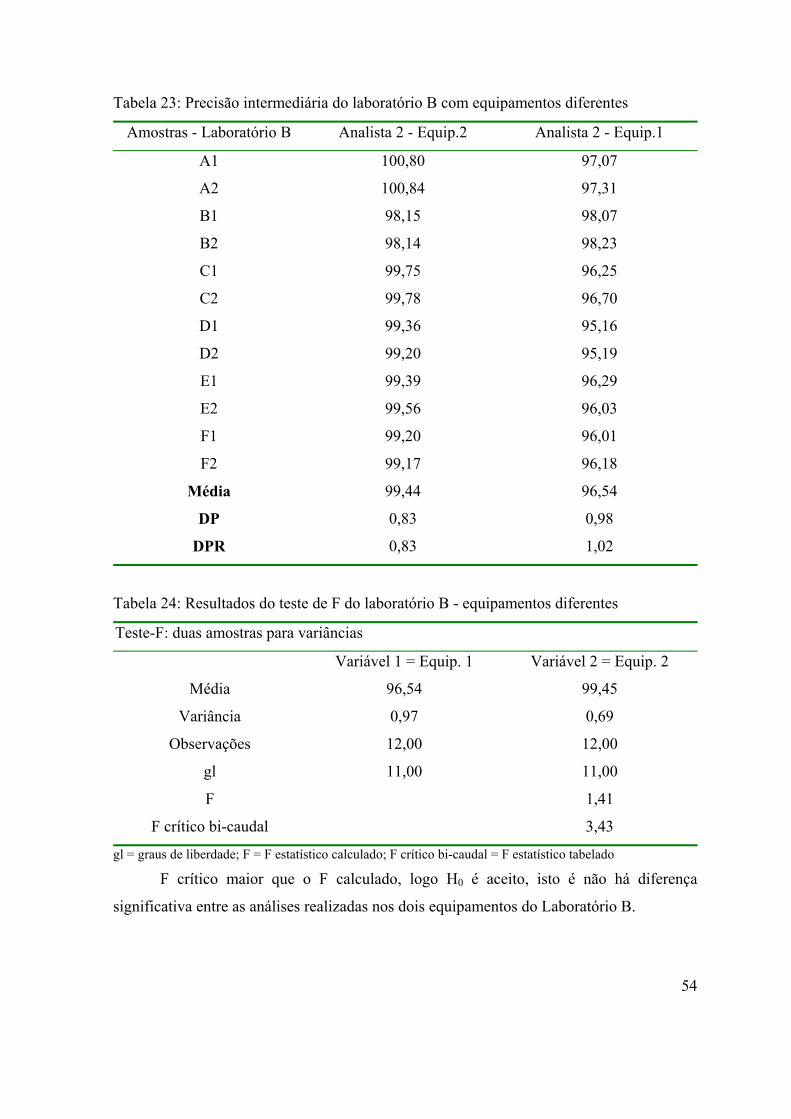
Tabela 23: Precisão intermediária do laboratório B com equipamentos diferentes
Amostras - Laboratório B Analista 2 - Equip.2 Analista 2 - Equip.1
A1 100,80 97,07
A2 100,84 97,31
B1 98,15 98,07
B2 98,14 98,23
C1 99,75 96,25
C2 99,78 96,70
D1 99,36 95,16
D2 99,20 95,19
E1 99,39 96,29
E2 99,56 96,03
F1 99,20 96,01
F2 99,17 96,18
Média 99,44 96,54
DP 0,83 0,98
DPR 0,83 1,02
Tabela 24: Resultados do teste de F do laboratório B - equipamentos diferentes
Teste-F: duas amostras para variâncias
Variável 1 = Equip. 1 Variável 2 = Equip. 2
Média 96,54 99,45
Variância 0,97 0,69
Observações 12,00 12,00
gl 11,00 11,00
F 1,41
F crítico bi-caudal 3,43
gl = graus de liberdade; F = F estatístico calculado; F crítico bi-caudal = F estatístico tabelado
F crítico maior que o F calculado, logo H0 é aceito, isto é não há diferença
significativa entre as análises realizadas nos dois equipamentos do Laboratório B.
54
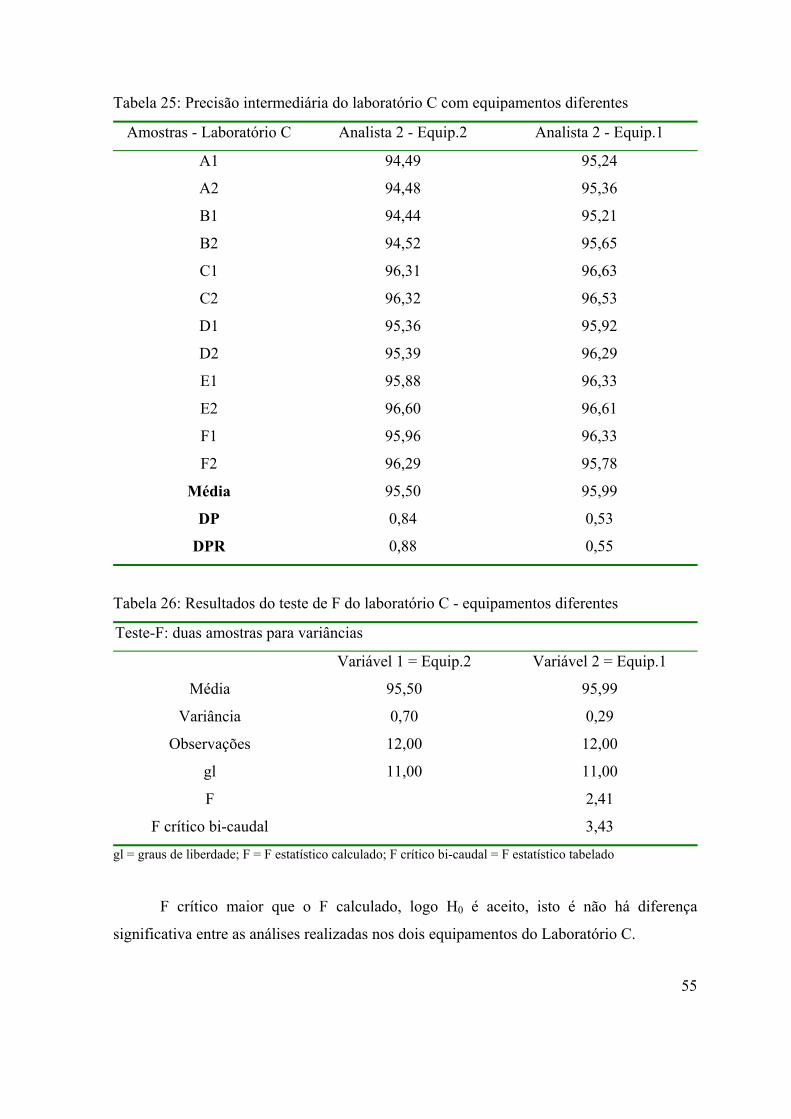
Tabela 25: Precisão intermediária do laboratório C com equipamentos diferentes
Amostras - Laboratório C Analista 2 - Equip.2 Analista 2 - Equip.1
A1 94,49 95,24
A2 94,48 95,36
B1 94,44 95,21
B2 94,52 95,65
C1 96,31 96,63
C2 96,32 96,53
D1 95,36 95,92
D2 95,39 96,29
E1 95,88 96,33
E2 96,60 96,61
F1 95,96 96,33
F2 96,29 95,78
Média 95,50 95,99
DP 0,84 0,53
DPR 0,88 0,55
Tabela 26: Resultados do teste de F do laboratório C - equipamentos diferentes
Teste-F: duas amostras para variâncias
Variável 1 = Equip.2 Variável 2 = Equip.1
Média 95,50 95,99
Variância 0,70 0,29
Observações 12,00 12,00
gl 11,00 11,00
F 2,41
F crítico bi-caudal 3,43
gl = graus de liberdade; F = F estatístico calculado; F crítico bi-caudal = F estatístico tabelado
F crítico maior que o F calculado, logo H0 é aceito, isto é não há diferença
significativa entre as análises realizadas nos dois equipamentos do Laboratório C.
55

Tabela 27: Precisão intermediária lab. A com analistas e equipamentos diferentes
Amostras - Laboratório A Analista 2 - Equip.1 Analista 1 - Equip.2
A1 99,21 101,84
A2 98,80 102,61
B1 100,41 101,54
B2 100,83 101,71
C1 99,50 100,58
C2 99,64 100,57
D1 100,41 101,85
D2 100,07 101,74
E1 99,02 101,18
E2 99,03 101,19
F1 99,02 100,54
F2 98,80 100,58
Média 99,56 101,33
DP 0,70 0,67
RSD 0,70 0,66
Tabela 28: Resultados do teste de F lab. A para analistas e equipamentos diferentes
Teste-F: duas amostras para variâncias
Variável 1 = Ana.2/Equip.1 Variável 2 = Ana.1/Equip.2
Média 99,56 101,33
Variância 0,50 0,44
Observações 12,00 12,00
gl 11,00 11,00
F 1,14
F crítico bi-caudal 3,43
gl = graus de liberdade; F = F estatístico calculado; F crítico bi-caudal = F estatístico tabelado
F crítico maior que o F calculado, logo H0 é aceito, isto é não há diferença
significativa entre as análises realizadas com analistas e equipamentos diferentes.
56
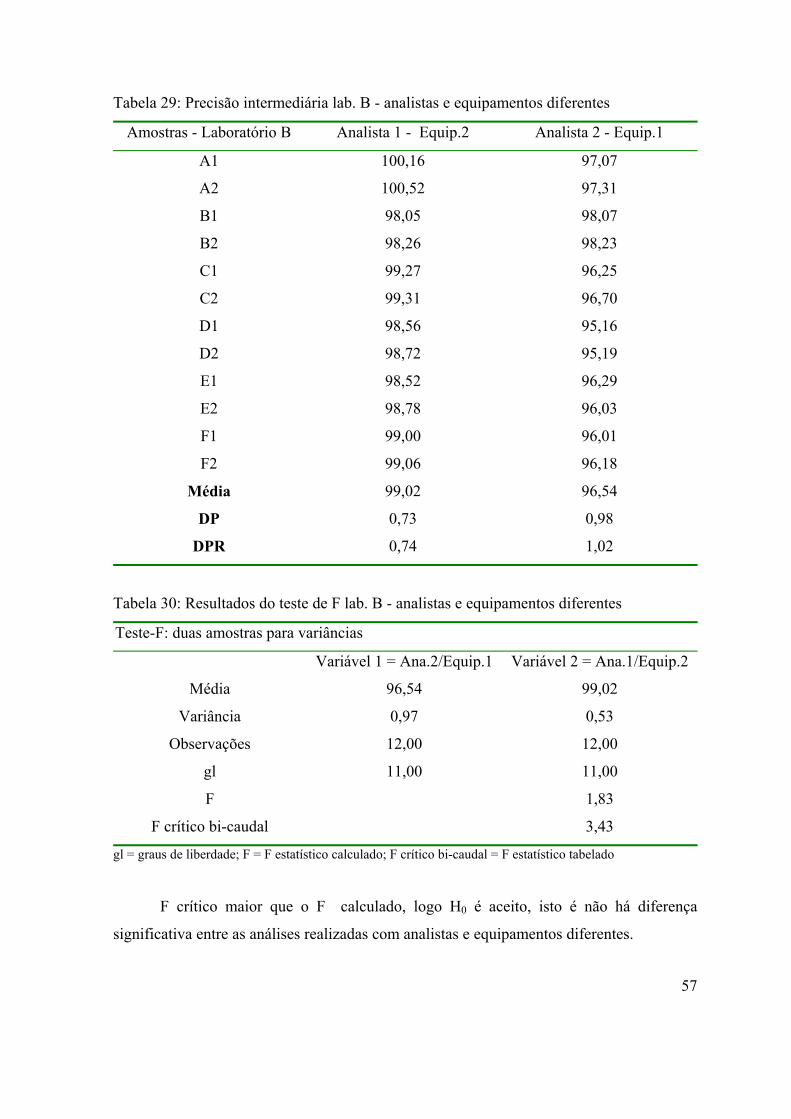
Tabela 29: Precisão intermediária lab. B - analistas e equipamentos diferentes
Amostras - Laboratório B Analista 1 - Equip.2 Analista 2 - Equip.1
A1 100,16 97,07
A2 100,52 97,31
B1 98,05 98,07
B2 98,26 98,23
C1 99,27 96,25
C2 99,31 96,70
D1 98,56 95,16
D2 98,72 95,19
E1 98,52 96,29
E2 98,78 96,03
F1 99,00 96,01
F2 99,06 96,18
Média 99,02 96,54
DP 0,73 0,98
DPR 0,74 1,02
Tabela 30: Resultados do teste de F lab. B - analistas e equipamentos diferentes
Teste-F: duas amostras para variâncias
Variável 1 = Ana.2/Equip.1 Variável 2 = Ana.1/Equip.2
Média 96,54 99,02
Variância 0,97 0,53
Observações 12,00 12,00
gl 11,00 11,00
F 1,83
F crítico bi-caudal 3,43
gl = graus de liberdade; F = F estatístico calculado; F crítico bi-caudal = F estatístico tabelado
F crítico maior que o F calculado, logo H0 é aceito, isto é não há diferença
significativa entre as análises realizadas com analistas e equipamentos diferentes.
57

Tabela 31: Precisão intermediária lab. C - analistas e equipamentos diferentes
Amostras - Laboratório C Analista 1 - Equip.2 Analista 2 - Equip.1
A1 96,97 95,24
A2 97,32 95,36
B1 96,55 95,21
B2 96,78 95,65
C1 98,27 96,63
C2 98,46 96,53
D1 96,41 95,92
D2 95,80 96,29
E1 96,35 96,33
E2 96,15 96,61
F1 96,83 96,33
F2 96,83 95,78
Média 96,89 95,99
DP 0,79 0,53
DPR 0,82 0,55
Tabela 32: Resultados do teste de F lab. C - analistas e equipamentos diferentes
Teste-F: duas amostras para variâncias
Variável 1 Variável 2
Média 96,89 95,99
Variância 0,63 0,29
Observações 12,00 12,00
gl 11,00 11,00
F 2,17
F crítico bi-caudal 3,43
gl = graus de liberdade; F = F estatístico calculado; F crítico bi-caudal = F estatístico tabelado
F crítico maior que o F calculado, logo H0 é aceito, isto é não há diferença
significativa entre as análises realizadas com analistas e equipamentos diferentes.
58

6.6. Resultados da Faixa
Foi determinado como faixa de trabalho o intervalo de 75 a 125% do valor
rotulado, pois o mesmo apresentou resultados satisfatórios nos parâmetros precisão,
exatidão e linearidade.
6.7. Discussão da Validação do Método de Análise do Teor da Lamivudina
Comprimidos
Os resultados da validação provaram que o método de análise do teor da
Lamivudina é:
a) Específico, isto é, os resultados não foram afetados pela presença de produtos de
degradação, impurezas ou excipientes;
b) Linear, apresentando a seguinte equação da reta: y = 0,2465 x + 1,0545 e um
coeficiente de correlação (r) igual a 0,9999;
c) Exato, já que os valores da recuperação para cada nível de concentração foram: para
75% - 99,22 + 0,89 ; para 100% - 99,02 + 0,33 e para 125% - 99,33 + 0,07 ;
d) Preciso, nos itens:
Repetitividade todos os desvios padrões encontrados para cada determinação foram
abaixo de 2%;
Precisão intermediária, variando apenas o analista, somente o equipamento e ao
mesmo tempo analista e equipamento; usando o teste de F de Snedecor para cada tipo de
comparação, não houve diferença significativa entre as variâncias em nenhuma das
comparações realizadas, portanto não houve diferença significativa entre as análises.
Foi então determinado como faixa de trabalho o intervalo de 75 a 125 % do valor
rotulado, pois o mesmo apresentou resultados satisfatórios nos parâmetros precisão,
exatidão e linearidade.
Portanto a validação do método de análise do teor de Lamivudina comprimidos foi
considerada satisfatória.
59

6.8. Resultados da Especificidade do Ensaio Limite para o Enantiômero
(+) BCH-189
As amostras utilizadas na especificidade do ensaio limite para o enantiômero
(+)BCH-189, foram estressadas da mesma forma que as utilizadas na especificidade para o
método de análise de teor, item 6.2 pág. 36.
Os cromatogramas obtidos foram avaliados e verificou-se que nenhum dos
componentes, sejam eles produtos de degradação ou excipientes, possui o mesmo tempo de
retenção do (+)BCH-189 e que a resolução obtida entre os sinais dos picos foi sempre
maior que dois, portanto os resultados obtidos não são afetados pela presença destas
substâncias.
A figura 14, pág. 60, refere-se a mistura racêmica de (+)BCH-189 e Lamivudina na
concentração de 44 mcg/mL.
O cromatograma apresentou uma resolução de 4,09 entre os picos e os tempos de
retenção da Lamivudina e do (+)BCH-189 foram respectivamente 8,30 min e 10,29 min.
8.30
, La
mivu
dina
10.2
9, (
+) B
CH 1
89
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Intensity (mV)
Figura 14: Especificidade do método de ensaio limite do enantiômero (+)BCH-189
A figura 15, pág. 61, refere-se a uma amostra que contém 250 mcg de Lamivudina
e 0,75 mcg de (+)BCH-189 por mililitro, que corresponde ao limite máximo permitido de
(+)BCH-189 em Lamivudina matéria-prima. Os tempos de retenção obtidos para a
60
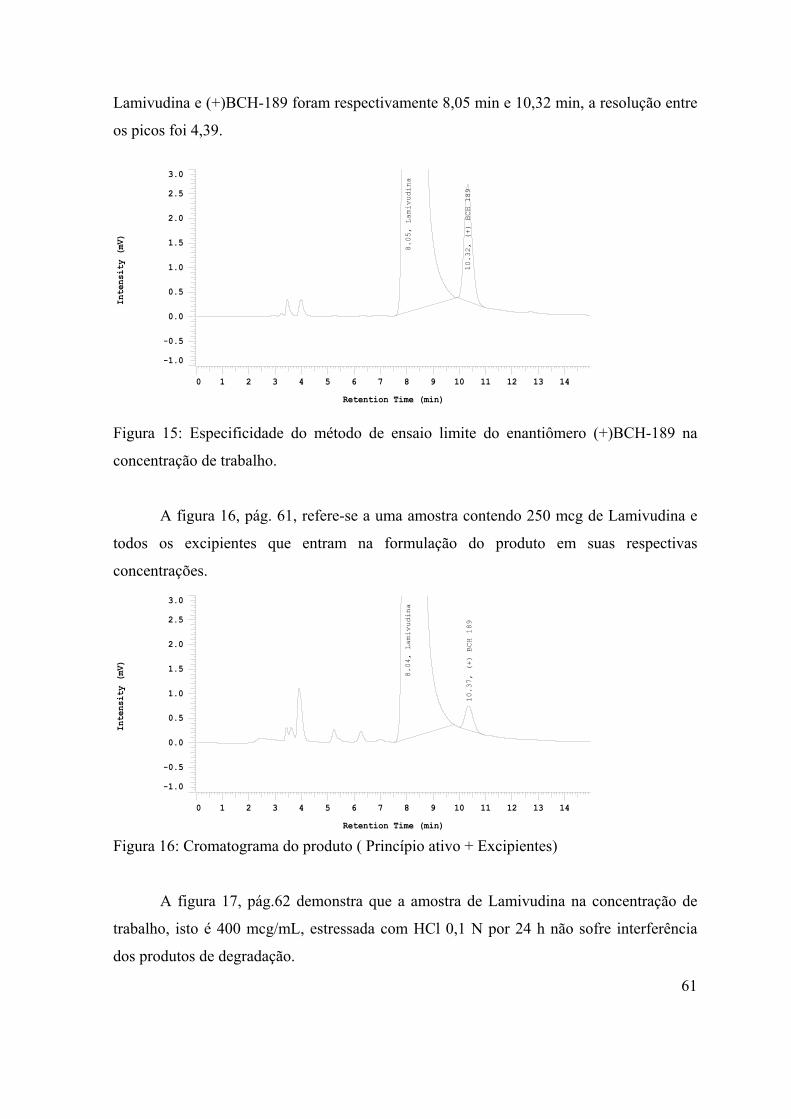
Lamivudina e (+)BCH-189 foram respectivamente 8,05 min e 10,32 min, a resolução entre
os picos foi 4,39.
8.
05,
Lami
vudi
na
10.3
2, (
+) B
CH 1
89
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Intensity (mV)
Figura 15: Especificidade do método de ensaio limite do enantiômero (+)BCH-189 na
concentração de trabalho.
A figura 16, pág. 61, refere-se a uma amostra contendo 250 mcg de Lamivudina e
todos os excipientes que entram na formulação do produto em suas respectivas
concentrações.
8.04
, La
mivu
dina
10.3
7, (
+) B
CH 1
89
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Intensity (mV)
Figura 16: Cromatograma do produto ( Princípio ativo + Excipientes)
A figura 17, pág.62 demonstra que a amostra de Lamivudina na concentração de
trabalho, isto é 400 mcg/mL, estressada com HCl 0,1 N por 24 h não sofre interferência
dos produtos de degradação.
61

7.73
, La
mivu
dina
9.90
, (+
) BC
H 18
9
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Intensity (mV)
Figura 17: Cromatograma amostra de Lamivudina submetida ao estresse com HCl 0,1 N
por 24h.
A figura 18, pág. 62, demonstra que a amostra de Lamivudina na concentração de
trabalho, isto é 400mcg/mL, estressada com H2O2 3% por 24 h não sofre interferência dos
produtos de degradação.
Intensity (mV)
8.01
, La
mivu
dina
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Figura 18: Cromatograma amostra de Lamivudina submetida ao estresse com H2O2 3% por
24h.
A amostra de Lamivudina na concentração de trabalho, isto é 400 mcg/mL,
estressada com NaOH 0,1 N por 24 h não sofre interferência dos produtos de degradação,
como verifica-se na figura 19 pág. 63.
62
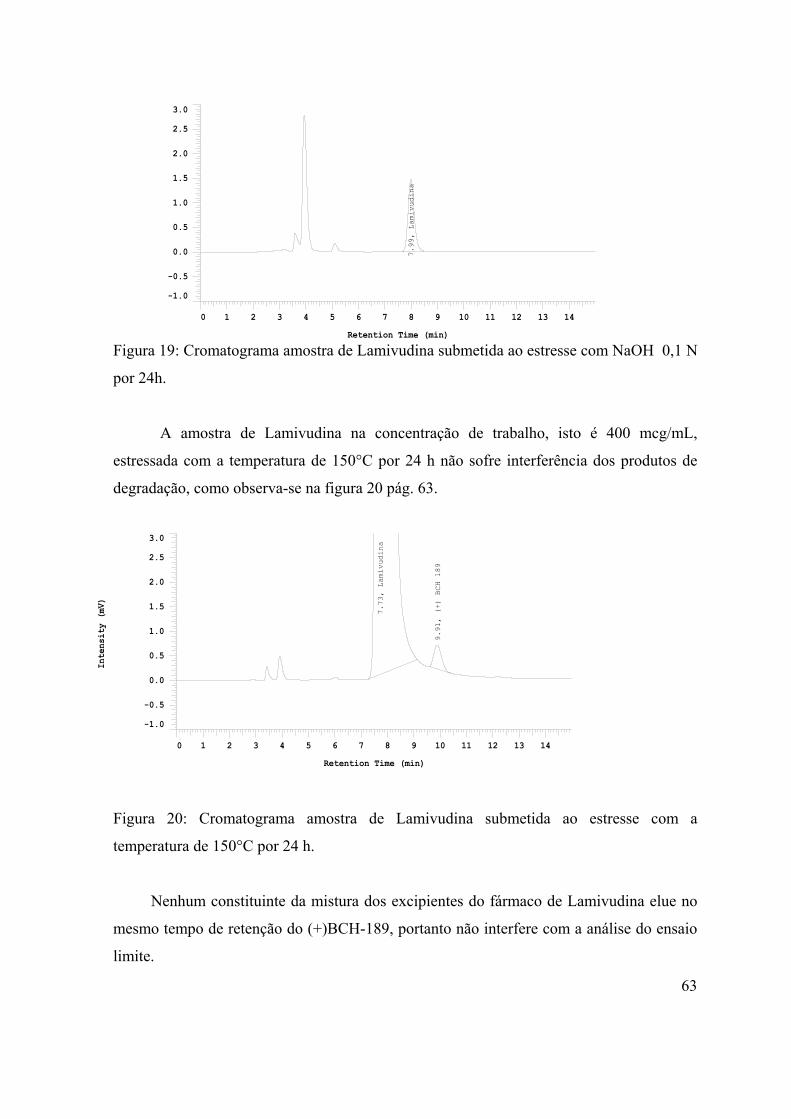
Intensity (mV)
Figura 19: Cromatograma amostra de Lamivudina submetida ao estresse com NaOH 0,1 N
por 24h.
7.99
, La
mivu
dina
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
A amostra de Lamivudina na concentração de trabalho, isto é 400 mcg/mL,
estressada com a temperatura de 150°C por 24 h não sofre interferência dos produtos de
degradação, como observa-se na figura 20 pág. 63.
7.73
, La
mivu
dina
9.91
, (+
) BC
H 18
9
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Intensity (mV)
Figura 20: Cromatograma amostra de Lamivudina submetida ao estresse com a
temperatura de 150°C por 24 h.
Nenhum constituinte da mistura dos excipientes do fármaco de Lamivudina elue no
mesmo tempo de retenção do (+)BCH-189, portanto não interfere com a análise do ensaio
limite.
63

6.9. Resultados do Limite de Detecção para o Enantiômero (+) BCH-189
Foram preparadas soluções da mistura racêmica contendo as seguintes
concentrações de (+)BCH-189:
conc.1 = 862,4 ng/mL , conc.2 = 86,2 ng/mL e conc.3 = 8,6 ng/mL.
Os cromatogramas estão apresentados nas figuras 21 a 26 págs. 64 a 66.
Como na concentração 3 não foi obtida detecção, preparou-se uma solução de
concentração intermediária 17,2 ng/mL na qual pôde ser detectada a substância de
interesse sendo portanto considerado como o limite de detecção experimental.
7.88
, La
mivu
dina
9.75
, (+
) BC
H 18
9
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Intensity (mV)
Figura 21: Cromatograma do limite de detecção conc.1 = 862,4 ng/mL
64

7.86
, La
mivu
dina
9.74
, (+
) BC
H 18
9
0 1 2 3 4 5 6 7 8 9 10 11
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Intensity (mV)
Figura 22: Cromatograma do limite de detecção conc.2 = 86,2 ng/mL
7.90
, La
mivu
dina
9.78
, (+
) BC
H 18
9
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Intensity (mV)
Figura 23: Cromatograma do limite de detecção conc.3 A = 17,2 ng/mL
65

0 1 2 3 4 5 6 7 8 9 10 11
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Intensity (mV)
Figura 24: Cromatograma do limite de detecção conc.3 = 8,6 ng/mL
Foi avaliada a interferência da concentração de Lamivudina na detecção de
pequenas concentrações de (+)BCH-189, preparando cinco soluções contendo uma
concentração constante de cerca de 250,0 mcg/mL de Lamivudina e concentrações
decrescentes de (+)BCH-189 (1,32; 0,8; 0,4; 0,16 e 0,08 mcg/mL), que correspondem
respectivamente a 0,53%; 0,32%; 0,16%; 0,06%; 0,03% de (+)BCH-189 em relação a
Lamivudina.
A partir dos cromatogramas obtidos foi determinada a resolução entre os picos e
verificou-se que apesar da baixa concentração de (+)BCH-189 em relação a Lamivudina a
sua detecção não foi afetada, até a concentração de 0,03%.
Os cromatogramas estão apresentados nas figuras 25 a 29 págs. 67 a 69.
66

7.46
, La
mivu
dina
9.42
, (+
) BC
H 18
9
0 1 2 3 4 5 6 7 8 9 10 11 12
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0Intensity (mV)
Figura 25: Conc.5 - 0,53% de (+)BCH-189 em relação ao pico da Lamivudina, R= 3,99
7.50
, La
mivu
dina
9.48
, (+
) BC
H 18
9
0 1 2 3 4 5 6 7 8 9 10 11 12
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Intensity (mV)
Figura 26: Conc.4 - 0,32% de (+)BCH-189 em relação ao pico da Lamivudina, R = 3,98
67
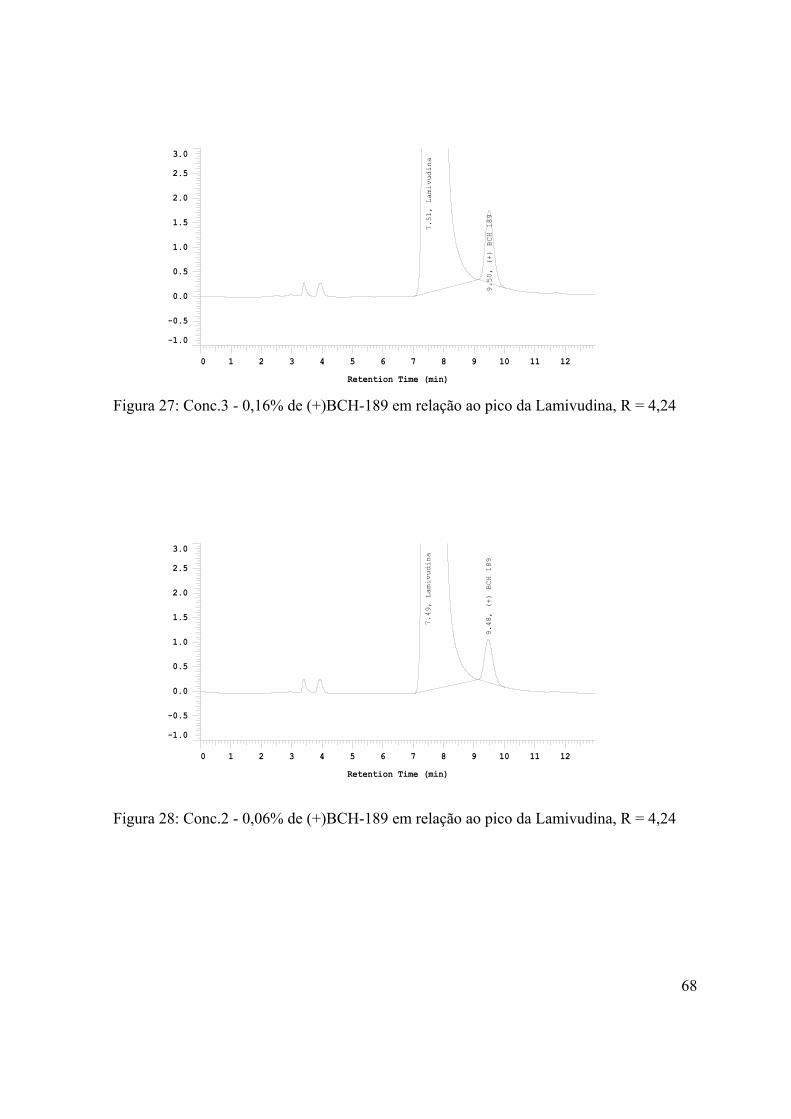
Intensity (mV) 7.51
, La
mivu
dina
9.50
, (+
) BC
H 18
9
0 1 2 3 4 5 6 7 8 9 10 11 12
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Figura 27: Conc.3 - 0,16% de (+)BCH-189 em relação ao pico da Lamivudina, R = 4,24
Intensity (mV) 7.49
, La
mivu
dina
9.48
, (+
) BC
H 18
9
0 1 2 3 4 5 6 7 8 9 10 11 12
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Figura 28: Conc.2 - 0,06% de (+)BCH-189 em relação ao pico da Lamivudina, R = 4,24
68
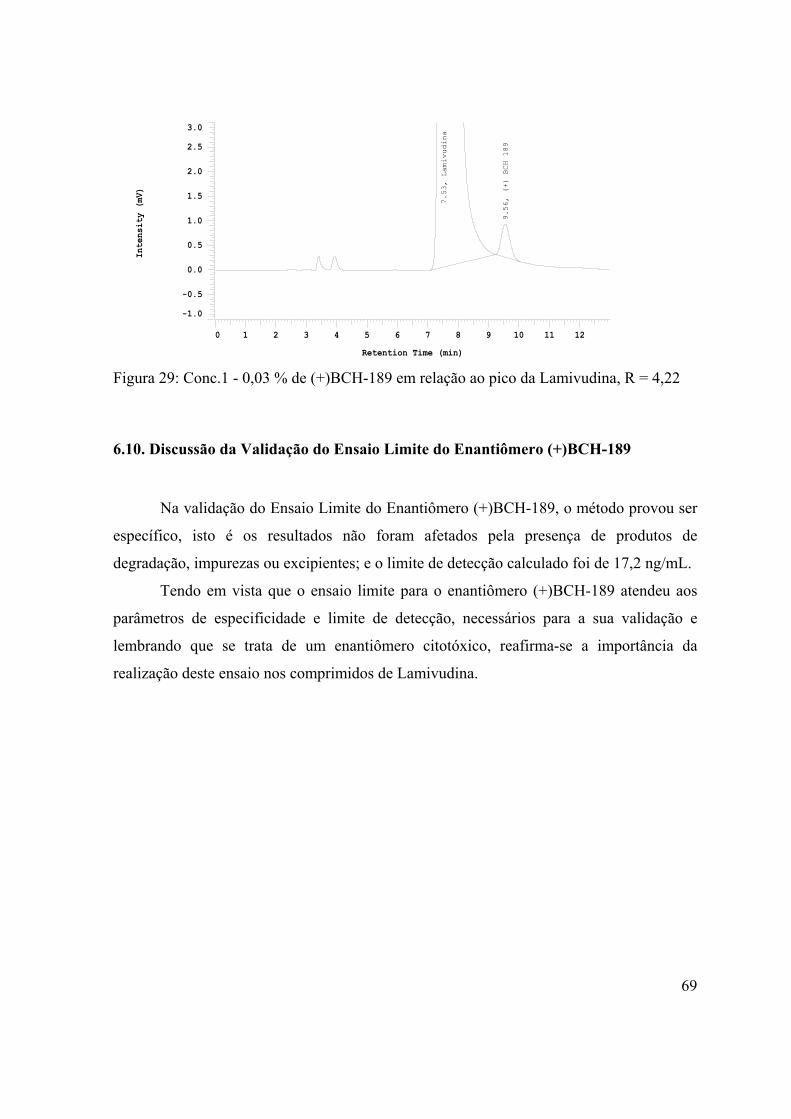
7.53
, La
mivu
dina
9.56
, (+
) BC
H 18
9
0 1 2 3 4 5 6 7 8 9 10 11 12
Retention Time (min)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0Intensity (mV)
Figura 29: Conc.1 - 0,03 % de (+)BCH-189 em relação ao pico da Lamivudina, R = 4,22
6.10. Discussão da Validação do Ensaio Limite do Enantiômero (+)BCH-189
Na validação do Ensaio Limite do Enantiômero (+)BCH-189, o método provou ser
específico, isto é os resultados não foram afetados pela presença de produtos de
degradação, impurezas ou excipientes; e o limite de detecção calculado foi de 17,2 ng/mL.
Tendo em vista que o ensaio limite para o enantiômero (+)BCH-189 atendeu aos
parâmetros de especificidade e limite de detecção, necessários para a sua validação e
lembrando que se trata de um enantiômero citotóxico, reafirma-se a importância da
realização deste ensaio nos comprimidos de Lamivudina.
69

6.11. Resultado da Pesquisa Bibliográfica sobre a Toxicidade do (+)BCH-189
A pesquisa bibliográfica sobre a ação farmacológica e a toxicidade do enantiômero
(+) BCH-189 revelou que a causa da toxicidade mitocondrial dos antiretrovirais análogos
de nucleosídeos é a inibição ou perturbação da síntese de DNA mitocondrial. E também
que o aumento da toxicidade celular dos nucleosídeos análogos esta relacionada com a
facilidade da sua incorporação ao DNA mitocondrial pela DNA polimerase humana e a
dificuldade da sua remoção (LIM, 2001).
A Lamivudina e o (+)BCH-189 são equipotentes contra o HIV, mais apenas o
(+)BCH-189 inibe a DNA-polimerase humana, fato associado com a toxicidade dos
fármacos nucleosídeos. A Lamivudina trifosfato (3TC-TP) é um dos nucleosídeo análogo
menos freqüentemente incorporados a DNA polimerase humana e um dos mais
eficientemente removidos. Estes dois fatos combinados podem explicar a sua baixa
toxicidade mitocondrial in vivo. A baixa incorporação da Lamivudina também pode ser
atribuída a sua forma isomérica β-L (-) (FENG, 1999; FENG, 2001; JOHNSON, 2001;
LIM, 2001; ANDERSON, 2002).
Devido ao fato do (+)BCH-189 ser significativamente mais citotóxico em linfócitos
não infectados é muito importante a realização do ensaio limite para determinar a sua
presença em comprimidos de Lamivudina.
70

7. CONCLUSÃO
É relevante ressaltar a importância da validação dos métodos analíticos como forma
de assegurar a confiabilidade dos resultados obtidos e assim garantir que os medicamentos
distribuídos são seguros e de qualidade; capazes de retardar a progressão da
imunodeficiência, melhorar e prolongar a qualidade de vida dos indivíduos infectados.
Os resultados da validação provaram que o método de análise do teor da
Lamivudina é específico, linear, exato e preciso, na faixa de trabalho de 75 a 125 % do
valor rotulado. E a validação do ensaio limite do enantiômero (+)BCH-189, provou que o
método é específico, e o limite de detecção foi determinado em 17,2 ng/mL. Sendo as
validações do método de análise do teor de Lamivudina e do ensaio limite do enantiômero
(+)BCH-189 consideradas satisfatórias.
A validação das metodologias de análise dos medicamentos é importante para que
o Ministério da Saúde através das VISAS cumpra sua atribuição de proteção da saúde da
população avaliando a qualidade dos medicamentos disponíveis no mercado.
A Lamivudina e o (+)BCH-189 são equipotentes contra o HIV, mas apenas o
(+)BCH-189 inibe a DNA-polimerase humana, fato associado com a toxicidade dos
fármacos nucleosídeos. Devido ao fato do (+)BCH-189 ser significativamente mais
citotóxico em linfócitos não infectados que a Lamivudina é muito importante a realização
do ensaio limite para determinar a sua presença em comprimidos de Lamivudina.
Na fase final desta dissertação foi publicada a RDC 150 de 17 de junho de 2003
que aprova o fascículo 4 da parte II, da 4ª edição da Farmacopéia Brasileira (FB), onde
encontram-se as monografias da Lamivudina matéria-prima e Lamivudina comprimido. Ao
comparar a monografia para Lamivudina comprimido proposta nesta dissertação com a
apresentada na Farmacopéia Brasileira verificou-se que esta não preconiza nenhum teste
de pureza em comprimidos de Lamivudina.
A especificação para o ensaio limite do (+)BCH-189 sugerida nesta dissertação,
0,3% em relação a Lamivudina, foi baseada na especificação para o ensaio limite do
(+)BCH-189 em Lamivudina matéria-prima constante na Farmacopeial Forum, vol.27, jan-
fev 2001. Na Farmacopéia Brasileira o ensaio de pureza preconizado para a Lamivudina
matéria-prima é o de substâncias relacionadas onde é avaliado o somatório das impurezas,
sem identificar o (+)BCH-189, e cujo limite máximo é 1,0% em relação a Lamivudina. 71

Sendo assim, o ensaio de pureza, substâncias relacionadas, apresentado na
Farmacopéia Brasileira pode aprovar uma matéria-prima com limite de (+)BCH-189
superior a 0,3% em relação a Lamivudina, ou seja uma matéria-prima reprovada pelos
parâmetros da Farmacopeial Forum.
É importante lembrar a metodologia preconizada na Farmacopéia Brasileira deve
contemplar todos os testes necessários para assegurar a qualidade do produto, no caso a
Lamivudina comprimidos.
Sendo assim, quando a análise é realizada por um laboratório que não possui as
informações a respeito da qualidade da matéria-prima utilizada na fabricação do produto,
torna-se indispensável a realização de um ensaio de pureza que identifique e determine um
limite máximo para o (+)BCH-189 nos comprimidos de Lamivudina.
72

8. BIBLIOGRAFIA
ANDERSON, K. S. Perspectives on the molecular mechanism of inhibition and toxicity of
nucleoside analogs that target HIV-1 reverse transcriptase. Biochimica et Biophisica
Acta. v.1587, p 296-299, 2002.
BEESLEY, T.E. et al. Chiral Chromatography, Chichester, 1998.
BENHAMOU,Y. E. et al. Efficacy of lamivudine on replication of hepatitis B virus in
HIV-infected patients. The Lancet, v. 345, n. 8, p. 396 - 397, 1995.
BERMUDEZ, J. A. Z. Formulação, Implementação e Componentes de uma Política de
Medicamentos. SEMINÁRIO INTERNACIONAL ACESSO A MEDICAMENTOS:
DIREITO FUNDAMENTAL E PAPEL DO ESTADO. Rio de Janeiro Outubro de 2002.
Disponível em: http://www.opas.org.br/medicamentos/seminar/apresent/bermude1.pdf
Acesso em: 25 de junho de 2003.
BLANEY, S. M. et al. Pharmacokinetis of lamivudine and BCH-189 in plasma and
cerebrospinal fluid of no human primates. Antimicrobial Agents and Chemotherapy, v.
39, n.12, p. 2779-2782, 1995.
BOHRNSTEDT, G.W.; KNOKE, D. Statistics for Social Data Analysis, Itasca Illinois:
F.E. Peacock Publishers, Inc. 1982.
BRASIL 1990a. Lei n° 8.078 de 11 de setembro de 1990. Código de Defesa do
Consumidor : dispõe sobre a proteção do consumidor e dá outras providências. Diário
Oficial [da] República Federativa do Brasil, Brasília, DF, Disponível em:
http://www.anvisa.gov.br/legis/leis/index.htm. Acesso em: 20 de set. de 2002.
73

BRASIL 1990b. Lei n° 8.080 de 19 de setembro de 1990. Dispõe sobre as condições para a
promoção, proteção e recuperação da saúde, a organização e o funcionamento dos serviços
correspondentes, e dá outras providências. Diário Oficial [da] República Federativa do
Brasil, Brasília, DF, Disponível em: http://www.anvisa.gov.br/legis/leis/index.htm. Acesso
em: 20 de set. de 2002.
BRASIL, Portaria nº 236 de 02 de maio de 1995. Estabelece as diretrizes para o programa
de controle da Síndrome de Imunodeficiência Adquirida, SIDA ou AIDS, no âmbito do
território nacional, atribui à divisão de Dermatologia Sanitária, da Secretaria Nacional de
Programas Especiais de Saúde, a coordenação do mesmo. Diário Oficial [da] Republica
Federativa do Brasil, Brasília, DF, Disponível: www.aids.gov.br/c_geral/eciv03.htm .
Acesso em: 03 de jun. de 2003.
BRASIL 1996a. Lei n0 9.279 de 14 de maio de 1996. Regula direitos e obrigações
relativos à propriedade industrial. Diário Oficial [da] República Federativa do Brasil,
Brasília, DF, Disponível em: http://www.anvisa.gov.br/legis/lei9279.htm. Acesso em: 27
de dez. de 2000.
BRASIL 1996b. Lei n0 9.313 de 13 de novembro de 1996. Dispõe sobre a distribuição
gratuita de medicamentos aos portadores do HIV e doentes de Aids. Diário Oficial [da]
República Federativa do Brasil, Brasília, DF, 14 de novembro de 1996. Disponível em:
http://www.aids.gov.br/assistencia/lei9313.htm . Acesso em: 27 de dez. de 2000.
BRASIL 1998. Portaria n0 3.916 de 30 de outubro de 1998. Aprova a Política Nacional de
Medicamentos. Diário Oficial [da] Republica Federativa do Brasil, Brasília, DF, 10 de
novembro de 1998. Disponível em: http://www.anvisa.gov.br/legis/portarias/index98.htm
Acesso em: 20 de set. de 2002.
74

BRASIL 1999a. Lei n° 9.782 de 26 de janeiro de 1999. Define o Sistema Nacional de
Vigilância Sanitária, cria a Agência Nacional de Vigilância Sanitária, e dá outras
providências. Diário Oficial [da] Republica Federativa do Brasil, Brasília, DF, (*)Tem
dispositivos alterados pela Medida Provisória n° 2.190/01. Disponível em:
http://www.anvisa.gov.br/legis/leis/index.htm . Acesso em: 20 de set. de 2002.
BRASIL 1999 b. Portaria n0 391 de 9 de agosto de 1999. Aprova o regulamento técnico
para medicamento genérico. Diário Oficial [da] Republica Federativa do Brasil,
Brasília, DF, (*) Revogada pela RDC n.º 10 de 2 de janeiro de 2001, que por sua vez foi
revogada pela Resolução nº 475 de 19 de março de 2002, que por sua vez foi
revogada pela Resolução n.º 899 de 29 de maio de 2003. Disponível em:
http://www.anvisa.gov.br/legis/resolução/index_99.htm Acesso em: 20 de set. de 2002.
BRASIL 1999 c. Decreto n° 3.201 de 06 de outubro de 1999. Dispõe sobre a concessão, de
ofício, de licença compulsória nos casos de emergência nacional e de interesse público de
que trata o art. 71 da Lei n° 9.279, de 14 de maio de 1996. Diário Oficial [da] República
Federativa do Brasil, Brasília, DF, Disponível em:
http://www.anvisa.gov.br/legis/decretos/index.htm. Acesso em: 20 de set. de 2002.
BRASIL 2003a. Resolução (RE) n0 899, de 29 de maio de 2003. Determina a publicação
do "Guia para validação de métodos analíticos e bioanalíticos". Diário Oficial [da]
Republica Federativa do Brasil, Brasília, DF, 02 de junho de 2003. Disponível em:
http://www.anvisa.gov.br/legis/resol/2003/index_re_mai.htm. Acesso em: 03 de jun. de
2003.
BRASIL 2003b. Resolução (RDC) n° 150, de 17 de junho de 2003. Aprova o “Fascículo 4
da Parte II, da 4ª Edição da Farmacopéia Brasileira”. Diário Oficial [da] Republica
Federativa do Brasil, Brasília, DF, 20 de junho de 2003. Disponível em:
http://www.anvisa.gov.br/legis/resol/2003/index_rdc_jun.htm. Acesso em: 04 de agosto
de 2003.
75

COATES, J.A. et al. The separated of 2', 3'-dideoxy-3'-thiacytidine (BCH-189) both inhibit
human immunodeficiency virus replication in vitro. Antimicrobial Agents and
Chemotherapy, v. 36, n.1, p. 202-205, 1992.
COSTA, E. A.; ROSENFELD, S. Constituição da Vigilância Sanitária no Brasil
Fundamentos da Vigilância Sanitária. Rio de Janeiro, v. 1, p. 15-40, 2000.
COSTA, E. A. Vigilância Sanitária - saúde e cidadania. Cadernos de Saúde; Belo
Horizonte, v.4, p. 15-27. 2001.
DIONE, G. 3TC: A Canadian Scientific Success. McGill Journal of Medicine, v. 5, n.1,
p. 60-65, 1999. Disponível em: http;//www.Mjm.mcgill.ca. Acesso em: 02 de março de
2003.
FDA 1992 (FOOD AND DRUGS ADMINISTRATION). FDA's Policy statement for the
development of new stereoisomeric drugs. Data de publicação 5/1/92
[on-line], Disponível em: http:/www.fda.gov/cder/guidance/stereo.htm . Acesso em: 26 de
abril de 2002.
FDA 1995 (FOOD AND DRUGS ADMINISTRATION). FDA grants accelerated approval
for 3TC with AZT to treat AIDS. [on-line], Disponível em: http://www.fda.gov . Acesso
em: 8 de maio de 2002.
FENG, J.Y.; ANDERSON, K. S., Mechanistic studies comparing the incorporation of (+)
and (-) isomers of 3TC-TP by HYV reverse transcriptase. Biochemistry. v. 38, p. 55-63,
1999.
FENG, J.Y. et al. Insights into the Molecular Mechanism of Mitochondrial Toxicity by
AIDS Drugs. The Journal of Biological Chemistry. v. 276, n. 26, p. 23832-23837, 2001.
FLORES, Jesuz. Farmacologia Humana: 3 ed. Barcelona: Masson, 1997, p. 1187-1211.
76

GALVÃO, J. 1980-2001 Uma cronologia da epidemia de HIV/AIDS no Brasil e no
mundo. Coleção ABIA. Políticas Públicas, n.2, 2002.
GRANGEIRO, A. Aids in Brazil - National Response. Ministério da Saúde, Secretaria de
Políticas de Saúde Coordenação Nacional de DST/ Aids. 2002.
GRAY, N.M. et al, The intracellular phosphorylation of (-)-2’-deoxy-3’-thiacytidine (3TC)
and the incorporation of 3TC 5’-monophosphate into DNA by HIV-1 reverse transcriptase
and human DNA polimerase γ. Biochemical Pharmacology, v. 50, n.7, p.1043-1051,
1995.
GUIMARÃES, M.D.C. et al. Estudo temporal das doenças associadas à AIDS no Brasil
1980-1999. Cadernos de Saúde Pública, Rio de Janeiro, v.16, s.1, p.21-36, 2000.
GUIMARÃES, M.C.L et al. O Registro Sanitário do Medicamento Similar no Brasil e
suas Implicações na Saúde Pública: A Responsabilidade do Estado Frente à
Vulnerabilidade da População. Pharmacia Brasileira, Brasília, n.34, p.76-81, out./nov.
2002.
HART, G. J., et al. Effects of (-)- 2- deoxy-3-thiacytidine (3TC) - 5- triphosphate on
human immunodeficiency virus reverse transcriptase and mammalian DNA polymerase
alpha, beta and gamma. Antimicrobial Agents and Chemotherapy, v. 36, p.1688-1694,
1992.
ICH (INTERNATIONAL Conference on Harmonization) Harmonized Tripartite
Guidelines. Text on Validation of Analytical Procedures, October 1994. [on-line],
Disponível em: http://www.ich.org/pdfICH/Q2A.pdf Acesso em: 05 de janeiro de 2001.
ICH (INTERNATIONAL Conference on Harmonization) Harmonized Tripartite
Guidelines. Validation of Analytical Procedures: Methodology, November 1996. [on-line],
Disponível em: http://www.ich.org/pdfICH/Q2B.pdf Acesso em: 05 de janeiro de 2001.
77

JOHNSON, A. A. et all, Toxicity of Antiviral Nucleoside Analogs and the Human
Mitochondrial DNA polymerase. The Journal of Biological Chemistry, v. 276, ed. 44, p.
40847-40857, 2001.
KEWN, S. et al, Lamivudine (3TC) Phosphorylation and Drug Interactions In Vitro.
Biochemical Pharmacology, v. 54, p. 589-595, 1997.
LAPPONI, J.C. Estatística usando Excel. São Paulo: Lapponi treinamento e editora,
2000.
LIM, S.E.; COPELAND, W.C. Differential Incorporation and Removal of Antiviral
Deoxynucleotides by Human DNA polimerase γ. The Journal of Biological Chemistry,
v.276, ed. 26, p. 23616-23623, 2001.
MILLER, J.C.; MILLER, J.N. Statistics for Analytical Chemistry. 2 ed. Chichester: Ellis
Horwood Limited, 1988.
MS (MINISTÉRIO DA SAÚDE), Secretaria de Assistência à Saúde. Programa Nacional
de DST e Aids. Consenso sobre Terapia antiretroviral em Adultos. Brasília, 1996.
MS (MINISTÉRIO DA SAÚDE), Secretaria de Assistência à Saúde. Programa Nacional
de DST e Aids. Consenso sobre Terapia antiretroviral para Adultos e Adolescentes
infectados pelo HIV. Brasília, 1997.
MS (MINISTÉRIO DA SAÚDE), Secretaria de Assistência à Saúde. Programa Nacional
de DST e Aids. Infeção pelo HIV em Adultos e Adolescentes. Brasília, 1999.
MS (MINISTÉRIO DA SAÚDE), Secretaria de Assistência à Saúde, Programa Nacional
de DST e Aids, Recomendações para Terapia antiretroviral em Adultos e Adolescentes.
Brasília, 2001.
78

MS (MINISTÉRIO DA SAÚDE) 2002a, Secretaria de Assistência à Saúde, Coordenação
Nacional de DST e Aids, Unidade de assistência. "Aids: Etiologia, clínica, diagnóstico e
tratamento", 2002. [on line] Disponível em: http://www.aids.gov.br. Acesso em 10 de abr.
de 2002.
MS (MINISTÉRIO DA SAÚDE) 2002b , Secretaria de Assistência à Saúde, Coordenação
Nacional de DST e Aids, Unidade de assistência. Acesso Universal e gratuito. [on line]
Disponível em: http://www.aids.gov.br . Acesso em 10 de abr. de 2002.
MS (MINISTÉRIO DA SAÚDE) 2002c, Programa Nacional de DST/AIDS, National
AIDS Drug Policy. Special Edition, Brasília, jun. 2002.
MS (MINISTÉRIO DA SAÚDE) 2002d, Coordenação Nacional de DST/AIDS, A
Experiência do Programa Brasileiro de Aids. Brasília. 2002.
MS (MINISTÉRIO DA SAÚDE), Coordenação Nacional de DST/AIDS, Resposta + (A
Experiência do Programa Brasileiro de Aids). [on line] Disponível em:
http://www.aids.gov.br. Acesso em: 02 de setembro de 2003.
PHARMACOPEIAL Forum. USP Monographs, v. 27, n. 1, Jan-Feb, 2001.
ROSENFELD, S. Farmacovigilância: Elementos para a discussão e perspectivas. Caderno
de Saúde Pública, Rio de Janeiro, v.14, n.2, abr-jun, 1998.
SCHINAZI, R. F. et al. A. Activities of the four optical isomers of 2’, 3’-dideoxy-3’-
thiacytidine (BCH-189) against human immunodeficiency virus type 1 in human
lymphocytes. Antimicrobial Agents and Chemotherapy, v. 36, n. 3, p. 672-676, 1992.
79

SHABIR, G. A. Validation of high-performance liquid chromatography methods for
pharmaceutical analysis Understanding the differences and similarities between validation
requirements of the US Food and Drug Administration, the US Pharmacopoeia and the
International Conference on Harmonization. Journal of Chromatography A, 987, p. 57-
66, 2003. Disponível em http://www.sciencedirect.com , Acesso em: 16 de abril de 2003.
SILVA, J. C. EDITORIAL ABIQUIF 2003. Disponível em: http://www.abquif.org.br
Acesso em 03 de abril de 2003.
SILVA, P. Farmacologia Humana. 5ed. p.1187-1211. Rio de Janeiro. Guanabara
Koogan, 1998.
SOMMADOSSI, J. P., et al. Comparison of cytotoxity of the (-)- and (+)-enantiomer of 2’,
3’-dideoxy- 3’-thiacytidine in normal human bone marrow progenitor cells. Biochemical
Pharmacology, v. 44, n.10, p.1922-1925, 1992.
SOUDEYNS, H., et al. Anti-human immunodeficiency virus type 1 activity and in vitro
toxicity of 2’-deoxy-3’-thiacytidine (BCH-189), a novel heterocyclic nucleoside analog.
Antimicrobial Agents and Chemotherapy, v. 35, n. 7, p. 1386-1390, 1991.
SWARTZ, M. E. , KRULL, I. S. Validação de Métodos Cromatográficos. Pharmaceutical
Technology, São Paulo, jun., 1998.
SZWARCWALD, C.L. et al. A Disseminação da epidemia de AIDS no Brasil, no período
de 1987-1996: Uma análise espacial. Cadernos de Saúde Pública, Rio de Janeiro, v.16,
s.1, p.7-19, 2000.
THE HIV Life Cycle. [on line] Disponível em:
http://www.aidsmeds.com/lessons/LifeCycleIntro.htm Acesso em 30 de agosto de 2001.
80

THE MERCK Index an encyclopedia of chemicals, drugs, and biologicals. 12 ed., p.914,
Merck Research Laboratories Division of Merck & Co., Inc. Whitehouse Station, U.S.A.
1996.
THE UNITED States Pharmacopoeia. 24 ed. Rockville, 2000. CD-ROM.
81




![CÓDIGOS BCH BINÁRIOS - cesarkallas.net · 3_BCH_V2011_Rev3 - Geraldo Gil R. Gomes 3.1 3.1. INTRODUÇÃO [1] Os códigos BCH (Bose, Chaudhuri e Hocquenghen) são uma importante e](https://img.document.onl/doc/110x75/5ba2971c09d3f210318c5723/codigos-bch-binarios-3bchv2011rev3-geraldo-gil-r-gomes-31-31-introducao.jpg)








![elarscan.ru · JIHCTOB, I'lPMBeneHHOro (þ01)MaTa A4, BCH pa60ra flPOB011VW]aCb Ha í1pw,eM OC06eHHO flPHHTH02 Hamero, poccwäc.l{oro HPOH3BOAH- (9TIAP CKAMAKC 3000 A3). Ha flJ1aHCžçaH](https://img.document.onl/doc/110x75/5e032c38d9e2ea2f204218a0/jihctob-ilpmbenehhoro-01mata-a4-bch-pa60ra-flpob011vwacb-ha-1pwem-oc06ehho.jpg)













![Resumo [ BCH-UFC]](https://img.document.onl/doc/110x75/62b6eeca37065b0c8b431c93/resumo-bch-ufc.jpg)

