Embed Size (px)
Citation preview

RENATA FONTES PRADO FARACO
REAÇÕES DE CARBOCICLIZAÇÃO RADICALAR DE
META-IODOBENZAMIDA DERIVADA DE D-GALACTOSE
VISANDO À OBTENÇÃO DE MACROLACTAMAS,
POTENCIAIS AGENTES BIOATIVOS
Belo Horizonte
Faculdade de Farmácia/UFMG
2007

RENATA FONTES PRADO FARACO
REAÇÕES DE CARBOCICLIZAÇÃO RADICALAR DE
META-IODOBENZAMIDA DERIVADA DE D-GALACTOSE
VISANDO À OBTENÇÃO DE MACROLACTAMAS,
POTENCIAIS AGENTES BIOATIVOS
Dissertação apresentada ao Curso de Pós-
Graduação, em Ciências Farmacêuticas, como
requisito parcial à obtenção do grau de Mestre.
ORIENTADOR
Prof. Dra. Maria Auxiliadôra Fontes Prado
LINHA DE PESQUISA
Linha II – Fármacos e Medicamentos
ÁREA DE CONHECIMENTO
1.06.01.00-7 – Síntese Orgânica

Faraco, Renata Fontes Prado
F219r
Reações de Carbociclização Radicalar de meta-Iodobenzaminda derivada de D-Galactose visando à obtenção de Macrolactamas, Potencias Agentes Bioativos / Renata Fontes Prado Faraco. – 2007.
152 f.: il.
Orientadora: Profa. Maria Auxiliadora Fontes Prado Dissertação (Mestrado) - Universidade Federal de Minas Gerais.
Faculdade de Farmácia. Programa de Pós-Graduação em Ciências Farmacêuticas.
1. Medicamentos – Teses. 2. Fármacos – Teses. I. Prado, Maria
Auxiliadora Fontes. Universidade Federal de Minas Gerais. Faculdade de Farmácia.
CDD 615.19


“Não sei curar o desespero, doutor Breuer. Apenas eu o estudo. O
desespero é o preço pago pela autoconsciência. Olhe profundamente
para dentro de si e sempre encontrará o desespero.”
Irvin D. Yalom, em Quando Nietzsche Chorou
“No fim tudo dá certo. Se não deu certo é porque ainda não chegou
o fim.”
(Anônimo)

Dedico este trabalho:
Aos meus pais, Guilherme e Dôra, exemplos de dedicação e perseverança e os
primeiros a me mostrarem a beleza da ciência.
Aos meus irmãos, Lívia, Paula, Laura, Fábio e Andréia, companheiros de toda uma vida.
À família Faraco, pela sempre carinhosa acolhida e amor.
Ao meu e-terno amor, marido, amigo e companheiro, André, que esteve comigo
desde os primeiros passos no Laboratório de QF, onde nos conhecemos e iniciamos
nossa história...
Ao meu filho Gabriel, que desde o início deu novo sentido à minha vida.

No início era preciso ver, depois, bastava ouvir, hoje, basta pensarmos para sentirmos
todo o amor que nos une. Quando o Gabriel nascer, simplesmente sentiremos o AMOR.
Anotar sentimentos não é nada fácil...
Nada poderia ser melhor.
Desde que nos conhecemos, passamos por diferentes situações em nossa história!
Razão.
Emoção.
E tudo vivido com muita intensidade...
Ganhamos experiência, nos conhecemos mais.
Agora bastam os olhares, às vezes, nem isto!
Brincar ainda é sua especialidade, que continue sendo...
Rapidamente nossas vidas mudarão!
Iniciaremos uma nova fase, talvez mais complexa, mas com certeza mais rica.
Ele, nosso neném, está chegando!
Lar terá um novo significado... Amo vocês!!!

AGRADECIMENTOS ESPECIAIS
À Dôra, minha orientadora e mãe, pela dedicação, empenho, profissionalismo, ensinamentos e
discussões (mesmo que durante tarde da noite e aos domingos!), não somente durante a
execução deste trabalho, mas desde o Centro Pedagógico, Coltec e Iniciações Científicas.
Ao André, meu marido, amigo e companheiro de todos os momentos, que me incentivou, apoiou e
deu condições para que eu retornasse ao meio acadêmico, que me confortou em momentos de
decepção, que não me deixou parar em momentos de desânimo...
À Ana Paula, pela inestimável ajuda na execução da fase final deste trabalho, quando não pude
mais me dedicar integralmente à parte experimental, mas tive a Ana realizando tudo com carinho,
eficiência e dedicação.

AGRADECIMENTOS
Ao professor Délio Soares Raslan, meu primeiro professor de Química Orgânica, que com muita
paciência e entusiasmo me mostrou o encanto desta disciplina.
À professora Rosemeire Brondi Alves (DQ/UFMG), sempre disponível, pela grande ajuda na
realização de reações realizadas por aquecimento de microondas e na aquisição de dados de
rotação específica, pelos ensinamentos e incentivo.
Ao professor José Dias de Souza Filho (DQ/UFMG), Peixe, pelos ensinamentos no espectrômetro
de RMN e de processamento de espectros e realização de alguns experimentos.
À Mara, Inácio e Gustavo, do Laboratório de Química Orgânica do Departamento de
Química/UFMG, pela ajuda sempre que eu os “visitava”.
Ao professor Marcos N. Eberlin e à doutoranda Patrícia Verardi Abdelnur (IQ/UNICAMP), pela
aquisição dos espectros de massas.
À professora Elzíria de Aguiar Nunan e à estagiária Gabriela Aires Martins (FaFar/UFMG), pela
realização dos testes de atividade antimicrobiana.
Aos professores e funcionários do DQ/UFMG (em especial do LAREMAR e Secretaria de Pós-
Graduação), que me acolheram como se eu fosse aluna deste Departamento.
Ao professor Márcio Matos Coelho, pela dedicação incondicional à Pós-Graduação da Faculdade
de Farmácia.
À Rose e Karen, secretárias do PPGCF, sempre incentivando e ajudando.
Aos professores Basílio, Ricardo e Thaís pelo incentivo e ensinamentos.
À Soninha, secretária do PFA, pelos momentos de alegria.
À Lavina e Raquel, pelos cafés e dedicação ao Laboratório, mesmo em momentos de estresse.
À Dani, companheira de bancada, pela ajuda no meu retorno à QF, pelos desabafos e dicas
durante a execução deste trabalho.
Aos colegas do Laboratório de Química Farmacêutica, pela convivência.
Aos professores e funcionários da FaFar, pelos momentos de convivência.
Ao CNPq, pela bolsa concedida.
À DEUS, pelas oportunidades que tive.

SUMÁRIO
LISTA DE FIGURAS ................................................................................................................... i
LISTA DE TABELAS ............................................................................................................... viii
LISTA DE ABREVIATURAS, SIGLAS E SÍMBOLOS .......................................................... ix
RESUMO ........................................................................................................................... xi
ABSTRACT ...................................................................................................................... xii
1 INTRODUÇÃO ........................................................................................................................ 1
1.1 MACROLACTAMAS NATURAIS E SEMI-SINTÉTICAS COM ATIVIDADE BIOLÓGICA ............................. 2
1.2 MACROLACTAMAS SINTÉTICAS COM ATIVIDADE BIOLÓGICA ...................................................... 12
1.3 SÍNTESE DE MACROCICLOS POR REAÇÃO DE CARBOCICLIZAÇÃO RADICALAR MEDIADA POR
HIDRETO DE TRI-N-BUTILESTANHO ......................................................................................... 16
1.4 NOVAS METODOLOGIAS PARA REAÇÃO DE CICLIZAÇÃO RADICALAR ......................................... 41
2 JUSTIFICATIVA, OBJETIVOS E PLANO DE SÍNTESE .................................................. 43
2.1 JUSTIFICATIVA E OBJETIVOS .................................................................................................... 43
2.2 PLANO DE SÍNTESE .................................................................................................................. 46
3 MATERIAL E MÉTODOS ..................................................................................................... 48
3.1 MÉTODOS GERAIS ................................................................................................................... 48
3.1.1 Aparelhagem utilizada ...................................................................................................... 48
3.1.2 Cromatografia ................................................................................................................... 48
3.1.3 Purificação de solventes .................................................................................................. 49
3.1.4 Reveladores ...................................................................................................................... 49
3.1.5 Procedimentos Gerais ...................................................................................................... 49

3.2 SÍNTESES ............................................................................................................................... 50
3.2.1 Síntese de 4,6-O-benzilideno-αααα-D-galactopiranosídeo de metila – GA2 ....................... 50
3.2.2 Síntese de 2,3-di-O-benzil-4,6-O-benzilideno-αααα-D-galactopiranosídeo de metila – GA3 ... 52
3.2.3 Síntese de 2,3-di-O-benzil-αααα-D-galactopiranosídeo de metila – GA4 ............................ 54
3.2.4 Síntese de 2,3-di-O-benzil-6-desoxi-6-iodo-αααα-D-galactopiranosídeo de metila – GA5 .... 56
3.2.5 Síntese de 6-azido-2,3-di-O-benzil-6-desoxi-αααα-D-galactopiranosídeo de metila – GA6 .. 59
3.2.6 Síntese de 4-O-alil-6-azido-2,3-di-O-benzil-6-desoxi-αααα-D-galactopiranosídeo de
metila – GA7 ..................................................................................................................... 61
3.2.7 Síntese de 4-O-alil-6-amino-2,3-di-O-benzil-6-desoxi-αααα-D-galactopiranosídeo de
metila (GA8), de Cloreto de 3-iodobenzoíla e de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-
(3-iodobenzoilamino)-αααα-D-galactopiranosídeo de metila – GAX ................................... 63
3.2.7.1 Síntese de 4-O-alil-6-amino-2,3-di-O-benzil-6-desoxi-αααα-D-galactopiranosídeo de
metila (GA8) .................................................................................................................. 64
3.2.7.2 Síntese de Cloreto de 3-iodobenzoíla .......................................................................... 64
3.2.7.3 Síntese de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-αααα-D-
galactopiranosídeo de metila – GAX ........................................................................... 65
3.2.8 Reação de macrociclização radicalar ............................................................................. 69
3.3 TESTES DE ATIVIDADE ANTIBACTERIANA E ANTIFÚNGICA .......................................................... 75
4 RESULTADOS E DISCUSSÃO .......................................................................................... 77
4.1 SÍNTESE DE 4,6-O-BENZILIDENO-αααα-D-GALACTOPIRANOSÍDEO DE METILA – GA2 ......................... 77
4.2 SÍNTESE DE 2,3-DI-O-BENZIL-4,6-O-BENZILIDENO-αααα-D-GALACTOPIRANOSÍDEO DE METILA – GA3 .. 83
4.3 SÍNTESE DE 2,3-DI-O-BENZIL-αααα-D-GALACTOPIRANOSÍDEO DE METILA – GA4 .............................. 87
4.4 SÍNTESE DE 2,3-DI-O-BENZIL-6-DESOXI-6-IODO-αααα-D-GALACTOPIRANOSÍDEO DE METILA – GA5 ... 91
4.5 SÍNTESE DE 6-AZIDO-2,3-DI-O-BENZIL-6-DESOXI-αααα-D-GALACTOPIRANOSÍDEO DE METILA – GA6 .... 95
4.6 SÍNTESE DE 4-O-ALIL-6-AZIDO-2,3-DI-O-BENZIL-6-DESOXI-αααα-D-GALACTOPIRANOSÍDEO DE
METILA – GA7 ....................................................................................................................... 98

4.7 SÍNTESE DE 4-O-ALIL-6-AMINO-2,3-DI-O-BENZIL-6-DESOXI-αααα-D-GALACTOPIRANOSÍDEO DE
METILA (GA8), DE CLORETO DE 3-IODOBENZOÍLA E DE 4-O-ALIL-2,3-DI-O-BENZIL-6-DESOXI-6-
(3-IODOBENZOILAMINO)-αααα-D-GALACTOPIRANOSÍDEO DE METILA – GAX ................................... 104
4.7.1 Síntese de 4-O-alil-6-amino-2,3-di-O-benzil-6-desoxi-αααα-D-galactopiranosídeo de
metila (GA8) ................................................................................................................... 104
4.7.2 Síntese de Cloreto de 3-iodobenzoíla ........................................................................... 105
4.7.3 Síntese de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-αααα-D-galactopiranosídeo
de metila – GAX .............................................................................................................. 105
4.8 REAÇÃO DE MACROCICLIZAÇÃO RADICALAR .......................................................................... 114
4.9 TESTES DE ATIVIDADE ANTIBACTERIANA E ANTIFÚNGICA ......................................................... 136
5 CONCLUSÃO ............................................................................................................. 137
6 REFERÊNCIAS BIBLIOGRÁFICAS ................................................................................. 138

i
LISTA DE FIGURAS
Figura 1 – Estruturas químicas de fármacos macrocíclicos. .......................................................... 1
Figura 2 – Estruturas químicas de depsipeptídeos com atividade biológica. ................................. 2
Figura 3 – Estruturas químicas das lingbiabelinas A e B. .............................................................. 3
Figura 4 – Estruturas químicas das macrolactamas indolactama-V e benzolactama-V8. .............. 3
Figura 5 – Estrutura química da vicenistatina. ............................................................................... 3
Figura 6 – Estruturas químicas de fluvirucinas. ............................................................................. 4
Figura 7 – Estruturas químicas da ascomicina e de seu derivado hidroxilado em C-33. ................ 5
Figura 8 – Estruturas químicas de substâncias com atividade imunomoduladora. ........................ 7
Figura 9 – Estrutura química da leinamicina. ................................................................................. 8
Figura 10 – Estruturas químicas de ansamicinas. ......................................................................... 8
Figura 11 – Estruturas químicas de produtos de fermentação de cepas deTü 6239. ..................... 9
Figura 12 – Estruturas químicas da maitansina e de seu derivado contendo o grupo dissulfeto. ...... 10
Figura 13 – Estrutura química da mixovirescina A1. ..................................................................... 10
Figura 14 – Estruturas químicas de macrolactamas contendo dímero de tirosina. ...................... 11
Figura 15 – Estruturas químicas de ustiloxinas e de fomopsinas. ................................................ 11
Figura 16 – Estruturas químicas de macrolactamas diméricas e tetramérica. ............................. 12
Figura 17 – Estruturas químicas de macrociclos taxóides e do paclitaxel. ................................... 13
Figura 18 – Estruturas químicas de “conjugados quinolona-macrociclo” e de um derivado de
cadeia aberta da ofloxacina. ................................................................................... 14
Figura 19 – Estruturas químicas de macrolactamas com atividade antitumoral. .......................... 15
Figura 20 – Estruturas químicas da migrastatina e de seu análogo sintético. .............................. 15
Figura 21 – Inibidores de β-secretase. ........................................................................................ 16
Figura 22 – Mecanismos envolvidos na reação radicalar mediada por Bu3SnH/AIBN. . ............... 18
Figura 23 – Rotâmeros e radicais formados por transferência 1,5 em orto-iodobenzamidas. ...... 19
Figura 24 – Rotâmeros e produtos de ciclização de iodobenzamidas. ........................................ 19
Figura 25 – Macrociclos obtidos a partir de iodoenonas. ............................................................. 20
Figura 26 – Macrolactonas sintetizadas a partir de ésteres acrilato e fumarato. .......................... 21

ii
Figura 27 – Macrociclizações em substratos ativados para ciclização exo. ................................. 22
Figura 28 – Síntese de precursor da roseofilina por macrociclização 13-endo. ........................... 23
Figura 29 – Síntese de macrolactona precursora da micotoxina zearalenona. ............................ 23
Figura 30 – Síntese de trienonas cíclicas de 14 membros. .......................................................... 24
Figura 31 – Macrociclização de iodoenonas cis e trans. .............................................................. 24
Figura 32 – Síntese de macrolactonas e éteres macrocíclicos por reação radicalar. ................... 25
Figura 33 – Ciclizações radicalares de iodotrienona. ................................................................... 26
Figura 34 – Síntese de macrociclos de 22 membros. ................................................................... 26
Figura 35 – Mecanismo da formação de macrolactonas a partir de 2-(tri-n-
butilestanilmetilpropanoatos) de ω-fenilselenoalquila. .............................................. 27
Figura 36 – Síntese de macrolactamas precursoras da lenoxamina. ........................................... 28
Figura 37 – Síntese de macrociclo 10-endo, precursor da periplanona B. ................................... 28
Figura 38 – Síntese de precursor do paclitaxel. ........................................................................... 29
Figura 39 – Síntese de macrolactamas estereoisoméricas por carbociclização radicalar. ........... 29
Figura 40 – Macrolactonas sintetizadas a partir de ω-fenilselenoésteres. .................................... 30
Figura 41 – Síntese do anel furanocembranóide presente na lofotoxina. .................................... 30
Figura 42 – Ciclizações 9-endo a partir de bromoalil e bromobutenilcicloexanóis. ....................... 31
Figura 43 – Síntese de ciclos de seis a nove membros por reação radicalar. .............................. 31
Figura 44 – Síntese de triciclos derivados da D-glicose. .............................................................. 32
Figura 45 – Síntese de triciclos derivados da D-manose. ............................................................ 32
Figura 46 – Síntese de ciclopeptídeos por reação de carbociclização radicalar. ......................... 33
Figura 47 – Macrolactamas obtidas por reação de aliloxi-orto-iodobenzamidas com Bu3SnH. .... 35
Figura 48 – orto-Iodobenzamidas sem unidade sacarídica submetidas à reação de ciclização
radicalar e respectivos resultados. .......................................................................... 37
Figura 49 – orto-Iodobenzamidas e produtos de reação com Bu3SnH. ........................................ 38
Figura 50 – Aliloxi-orto-iodobenzoatos e seus produtos da reação com Bu3SnH. ........................ 39
Figura 51 – Amidoésteres e amidoéteres submetidos à reação com Bu3SnH. ............................ 40
Figura 52 – Amida alquílica e macrolactama obtida da reação com Bu3SnH. .............................. 41
Figura 53 – Macrolactamas possíveis de serem formadas a partir da benzamida GAX. .............. 45

iii
Figura 54 – orto-Iodobenzamidas com e sem unidade sacarídica e produtos das reações de
macrociclização radicalar. ....................................................................................... 45
Figura 55 – Retrossíntese da benzamida GAX. ........................................................................... 46
Figura 56 – Rota de síntese planejada para obtenção da iodobenzamida GAX. ......................... 47
Figura 57 – Substâncias submetidas aos testes de atividade antibacteriana e antifúngica. .......... 75
Figura 58 – Primeira etapa da rota de síntese – Formação do acetal benzilidênico. ................... 77
Figura 59 – Produtos de formação do acetal benzilidênico: 1,3-dioxano (anel de seis membros)
e 1,3-dioxalano (anel de cinco membros). .............................................................. 78
Figura 60 – Mecanismo de isomerização de acetal benzilidênico catalisada por ácido. .............. 78
Figura 61 – Mecanismo para a formação de GA2. ...................................................................... 79
Figura 62 – Reação química de eliminação do excesso de benzaldeído. .................................... 79
Figura 63 – Espectro no IV (ATR) de 4,6-O-benzilideno-α-D-galactopiranosídeo de metila – GA2. ..... 80
Figura 64 – Espectro de RMN de 1H (200 MHz, CDCl3) de 4,6-O-benzilideno-α-D-
galactopiranosídeo de metila – GA2. ...................................................................... 81
Figura 65 – Mapa de contornos COSY (200 MHz, CDCl3) de 4,6-O-benzilideno-α-D-
galactopiranosídeo de metila – GA2. ...................................................................... 81
Figura 66 – Espectro de RMN de 13C e DEPT 135 (50 MHz, CDCl3) de 4,6-O-benzilideno-α-D-
galactopiranosídeo de metila – GA2. ...................................................................... 82
Figura 67 – Mapa de contornos HMQC (200 MHz, CDCl3) de 4,6-O-benzilideno-α-D-
galactopiranosídeo de metila – GA2. ...................................................................... 83
Figura 68 – Segunda etapa da rota de síntese – O-benzilação das hidroxilas em C-2 e C-3 de GA2. ... 83
Figura 69 – Espectro no IV (ATR) de 2,3-di-O-benzil-4,6-O-benzilideno-α-D-
galactopiranosídeo de metila – GA3. .................................................................... 85
Figura 70 – Espectro de RMN de 1H (200 MHz, CDCl3) de 2,3-di-O-benzil-4,6-O-benzilideno-α-
D-galactopiranosídeo de metila – GA3. ................................................................... 86
Figura 71 – Espectro de RMN de 13C e DEPT 135 (50 MHz, CDCl3) de 2,3-di-O-benzil-4,6-O-
benzilideno-α-D-galactopiranosídeo de metila – GA3. ............................................. 87
Figura 72 – Terceira etapa da rota de síntese – Remoção do grupo acetal benzilidênico de GA3. ..... 87
Figura 73 – Fotografia de aparelho de microondas doméstico adaptado utilizado na remoção do
grupo acetal benzilidênico de GA3 (Departamento de Química/ICEx/UFMG). .......... 88
Figura 74 – Espectro no IV (ATR) de 2,3-di-O-benzil-α-D-galactopiranosídeo de metila – GA4. .... 89

iv
Figura 75 – Espectro de RMN de 1H (200 MHz, CDCl3) de 2,3-di-O-benzil-α-D-
galactopiranosídeo de metila – GA4. ...................................................................... 90
Figura 76 – Espectro de RMN de 13C e DEPT 135 (50 MHz, CDCl3) de 2,3-di-O-benzil-α-D-
galactopiranosídeo de metila – GA4. ....................................................................... 91
Figura 77 – Quarta etapa da rota de síntese – Substituição seletiva da hidroxila de C-6 de GA4
por iodo. .................................................................................................................. 91
Figura 78 – Mecanismo de substituição regiosseletiva da hidroxila de C-6 de GA4 por iodo. ...... 92
Figura 79 – Espectro no IV (ATR) de 2,3-di-O-benzil-6-desoxi-6-iodo-α-D-galactopiranosídeo
de metila – GA5. .................................................................................................... 93
Figura 80 – Espectro de RMN de 1H (200 MHz, CDCl3) de 2,3-di-O-benzil-6-desoxi-6-iodo-α-D-
galactopiranosídeo de metila – GA5. ........................................................................ 94
Figura 81 – Espectro de RMN de 13C e DEPT 135 (50 MHz, CDCl3) de 2,3-di-O-benzil-6-desoxi-
6-iodo-α-D-galactopiranosídeo de metila – GA5. ........................................................ 95
Figura 82 – Quinta etapa da rota de síntese – Substituição do átomo de iodo em C-6 de GA5
pelo grupo azido. .................................................................................................... 95
Figura 83 – Espectro no IV (ATR) de 6-azido-2,3-di-O-benzil-6-desoxi-α-D-
galactopiranosídeo de metila – GA6. .................................................................... 96
Figura 84 – Espectro de RMN de 1H (200 MHz, CDCl3) de 6-azido-2,3-di-O-benzil-6-desoxi-α-D-
galactopiranosídeo de metila – GA6. ......................................................................... 97
Figura 85 – Espectro de RMN de 13C e DEPT 135 (50 MHz, CDCl3) de 6-azido-2,3-di-O-benzil-6-
desoxi-α-D-galactopiranosídeo de metila – GA6. ........................................................ 98
Figura 86 – Sexta etapa da rota de síntese – Alilação da hidroxila em C-4 de GA6. ................... 98
Figura 87 – Equilíbrios envolvidos na O-alilação de GA6 por reação de catálise de
transferência de fase. ............................................................................................ 99
Figura 88 – Espectro no IV (ATR) de 4-O-alil-6-azido-2,3-di-O-benzil-6-desoxi-α-D-
galactopiranosídeo de metila – GA7. .................................................................... 100
Figura 89 – Espectro de RMN de 1H (200 MHz, CDCl3) de 4-O-alil-6-azido-2,3-di-O-benzil-6-
desoxi-α-D-galactopiranosídeo de metila – GA7. .................................................. 101
Figura 90 – Seção expandida na região de δ 6,2 – 4,0 do espectro de RMN de 1H (200
MHz, CDCl3) de 4-O-alil-6-azido-2,3-di-O-benzil-6-desoxi-α-D-
galactopiranosídeo de metila – GA7. .................................................................. 101

v
Figura 91 – Mapa de contornos COSY (200 MHz, CDCl3) de 4-O-alil-6-azido-2,3-di-O-benzil-6-
desoxi-α-D-galactopiranosídeo de metila – GA7. ................................................... 102
Figura 92 – Espectro de RMN de 13C e DEPT 135 (50 MHz, CDCl3) de 4-O-alil-6-azido-2,3-di-
O-benzil-6-desoxi-α-D-galactopiranosídeo de metila – GA7. ................................. 103
Figura 93 – Mapa de contornos HMQC (200 MHz, CDCl3) de 4-O-alil-6-azido-2,3-di-O-benzil-
6-desoxi-α-D-galactopiranosídeo de metila – GA7. ............................................... 103
Figura 94 – Redução do grupo azido de GA7. .......................................................................... 104
Figura 95 – Obtenção do cloreto de 3-iodobenzoíla a partir do ácido 3-iodobenzóico. .............. 105
Figura 96 – Síntese da iodobenzamida GAX por reação entre GA8 e cloreto de 3-iodobenzoíla. ... 105
Figura 97 – Espectro no IV (ATR) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-α-D-
galactopiranosídeo de metila – GAX. ........................................................................ 106
Figura 98 – Espectro de RMN de 1H (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-
iodobenzoilamino)-α-D-galactopiranosídeo de metila – GAX. ..................................... 108
Figura 99 – Seção expandida na região de δ 8,1 – 7,1 do espectro de RMN de 1H (400 MHz,
CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-α-D-
galactopiranosídeo de metila – GAX. ................................................................... 108
Figura 100 – Seção expandida na região de δ 6,8 – 4,1 do espectro de RMN de 1H (400 MHz,
CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-α-D-
galactopiranosídeo de metila – GAX. ..................................................................... 109
Figura 101 – Seção expandida na região de δ 4,1 – 3,2 do espectro de RMN de 1H (400 MHz, CDCl3)
de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-α-D-galactopiranosídeo de
metila – GAX. ....................................................................................................... 109
Figura 102 – Mapa de contornos COSY (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-
6-(3-iodobenzoilamino)-α-D-galactopiranosídeo de metila – GAX. ............................. 110
Figura 103 – Espectro de RMN de 13C e DEPT 135 (100 MHz, CDCl3) de 4-O-alil-2,3-di-O-
benzil-6-desoxi-6-(3-iodobenzoilamino)-α-D-galactopiranosídeo de metila – GAX. .... 111
Figura 104 – Mapa de contornos HMQC (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-
6-(3-iodobenzoilamino)-α-D-galactopiranosídeo de metila – GAX. ............................. 112
Figura 105 – Espectros de massas [ESI(+)-MS] e análise seqüencial [ESI(+)-MS/MS] de 4-
O-alil-2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-α-D-galactopiranosídeo
de metila – GAX. ................................................................................................ 113

vi
Figura 106 – Proposta de fragmentação para o íon de m/z 644 após análise seqüencial de 4-
O-alil-2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-α-D-galactopiranosídeo
de metila – GAX. ................................................................................................. 113
Figura 107 – Reação de macrociclização radicalar de GAX e seus possíveis produtos. ............ 114
Figura 108 – Mecanismo de formação das macrolactamas GAXA e GAXB e do produto de
hidrogenólise GAXC, por reação radicalar com hidreto de tri-n-butilestanho. ...... 116
Figura 109 – Espectro no IV (ATR) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-benzoilamino-α-D-
galactopiranosídeo de metila – GAXC. ................................................................ 118
Figura 110 – Espectro de RMN de 1H (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-
benzoilamino-α-D-galactopiranosídeo de metila – GAXC. .................................... 119
Figura 111 – Seção expandida na região de δ 7,9 – 7,1 do espectro de RMN de 1H (400 MHz,
CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-benzoilamino-α-D-
galactopiranosídeo de metila – GAXC. ............................................................... 119
Figura 112 – Mapa de contornos COSY (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-
6-benzoilamino-α-D-galactopiranosídeo de metila – GAXC. ................................... 120
Figura 113 – Espectro de RMN de 13C e DEPT 135 (100 MHz, CDCl3) de 4-O-alil-2,3-di-O-
benzil-6-desoxi-6-benzoilamino-α-D-galactopiranosídeo de metila – GAXC. .......... 121
Figura 114 – Mapa de contornos HMQC (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-
6-benzoilamino-α-D-galactopiranosídeo de metila – GAXC. ................................... 121
Figura 115 – Espectros de massas [ESI(+)-MS] e análise seqüencial [ESI(+)-MS/MS] de 4-
O-alil-2,3-di-O-benzil-6-desoxi-6-benzoilamino-α-D-galactopiranosídeo de
metila – GAXC. ................................................................................................... 122
Figura 116 – Proposta de fragmentação para o íon de m/z 518 após análise seqüencial de 4-
O-alil-2,3-di-O-benzil-6-desoxi-6-benzoilamino-α-D-galactopiranosídeo de
metila – GAXC. ................................................................................................... 122
Figura 117 – Produto obtido da segunda reação radicalar de GAX: 4-O-alil-2,3-di-O-benzil-6-
desoxi-6-(3-fenilbenzoilamino)-α-D-galactopiranosídeo de metila (GAXD). ............ 123
Figura 118 – Proposta de mecanismo para a formação de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-
fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD. .............................. 124
Figura 119 – Espectro de RMN de 1H (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-
(3-fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD. ......................... 125

vii
Figura 120 – Seção expandida na região de δ 8,0 – 7,0 do espectro de RMN de 1H (400 MHz,
CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-fenilbenzoilamino)-α-D-
galactopiranosídeo de metila – GAXD. .............................................................. 126
Figura 121 – Mapa de contornos COSY (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-
6-(3-fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD......................... 126
Figura 122 – Espectro de RMN de 13C (50 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-
fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD. ................................ 128
Figura 123 – Espectro de DEPT 135 (50 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-
fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD. ............................... 128
Figura 124 – Mapa de contornos HMQC (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-
6-(3-fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD. ....................... 129
Figura 125 – Mapa de contornos HMBC (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-
6-(3-fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD. ....................... 129
Figura 126 – Seção expandida do mapa de contornos HMBC (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-
benzil-6-desoxi-6-(3-fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD. ..... 130
Figura 127 – Espectros de massas [ESI(+)-MS] e análise seqüencial [ESI(+)-MS/MS] de 4-O-
alil-2,3-di-O-benzil-6-desoxi-6-(3-fenilbenzoilamino)-α-D-galactopiranosídeo de
metila – GAXD. .................................................................................................... 131
Figura 128 – Proposta de fragmentação para o íon de m/z 594 após análise seqüencial de 4-O-
alil-2,3-di-O-benzil-6-desoxi-6-(3-fenilbenzoilamino)-α-D-galactopiranosídeo de
metila – GAXD. .................................................................................................... 131
Figura 129 – Conformações syn e anti de 7 e dos respectivos radicais, conformação 7a’’
estabilizada por ligação de hidrogênio e formação do macrociclo 8. .................... 133
Figura 130 – Conformações syn e anti de 18 e dos respectivos radicais, conformação de 18a’
estabilizada por ligação de hidrogênio e formação do macrociclo 19. ................... 134
Figura 131 – Conformações syn e anti de GAX e dos respectivos radicais e formação de GAXC
e GAXD. ................................................................................................................ 135

viii
LISTA DE TABELAS
Tabela 1 – Quantidade de reagentes utilizados na reação de obtenção de GA2 ......................... 50
Tabela 2 – Dados físico-químicos e espectrométricos de GA2 .................................................... 51
Tabela 3 – Quantidade de reagentes utilizados na reação de obtenção de GA3 .......................... 52
Tabela 4 – Dados físico-químicos e espectrométricos de GA3 .................................................... 53
Tabela 5 – Quantidade de reagentes utilizados na reação de obtenção de GA4 ......................... 55
Tabela 6 – Dados físico-químicos e espectrométricos de GA4 .................................................... 55
Tabela 7 – Quantidade de reagentes utilizados na reação de obtenção de GA5 ......................... 57
Tabela 8 – Dados físico-químicos e espectrométricos de GA5 .................................................... 57
Tabela 9 – Quantidade de reagentes utilizados na reação de obtenção de GA6 ......................... 59
Tabela 10 – Dados físico-químicos e espectrométricos de GA6 .................................................. 59
Tabela 11 – Quantidade de reagentes utilizados na reação de obtenção de GA7 ....................... 61
Tabela 12 – Dados físico-químicos e espectrométricos de GA7 .................................................. 62
Tabela 13 – Quantidade de reagentes utilizados na reação de obtenção de GAX ...................... 66
Tabela 14 – Dados físico-químicos e espectrométricos de GAX ................................................. 66
Tabela 15 – Condições empregadas nas reações radicalares e rendimentos de produtos isolados ... 70
Tabela 16 – Dados físico-químicos e espectrométricos de GAXC ............................................... 70
Tabela 17 – Dados físico-químicos e espectrométricos de GAXD ............................................... 73
Tabela 18 – Microorganismos utilizados para os testes de atividade antimicrobiana ................... 76

ix
LISTA DE ABREVIATURAS, SIGLAS E SÍMBOLOS
[α]D rotação específica
AIBN 2,2-azobisisobutironitrila
ATR Attenuated Total Reflectance
Bn Benzila
Bu3SnH Hidreto de tri-n-butilestanho
oC graus Celsius
CCD Cromatografia em Camada Delgada
CCS Cromatografia em Coluna de Sílica
C ipso Carbono aromático sem hidrogênio
COSY homonuclear COrelation SpectroscopY
δ deslocamento químico (RMN) / deformação angular (IV)
d dupleto
dd dupleto duplo
ddd dupleto duplo duplo
dddd dupleto duplo duplo duplo
ddl dupleto duplo largo
DEPT Distortionless Enhancement by Polarization Transfer
dl dupleto largo
DMF N-N-Dimetilformamida
DMSO Dimetilsulfóxido
F.F. Faixa de Fusão
F.M. Fórmula Molecular
HMBC Heteronuclear Multiple Bond Coherence
HMQC Heteronuclear Multiple Quantum Coherence
IV Infravermelho

x
J constante de acoplamento escalar spin nuclear-spin nuclear
m multipleto
M.M. Massa Molar
m/m massa por massa
MO Irradiação microondas
m/v massa por volume
ν número de onda
ν deformação axial
p. página
qd quarteto duplo
RMN de 13C Ressonância Magnética Nuclear de Carbono 13
RMN de 1H Ressonância Magnética Nuclear de Hidrogênio
s simpleto
sl sinal largo
t tripleto
t.a. temperatura ambiente
td tripleto duplo
tdd tripleto duplo duplo
THF Tetraidrofurano
TMS Tetrametilsilano
tt tripleto triplo
Vol. Volume
v/v volume por volume

xi
RESUMO
Os macrociclos apresentam importantes atividades biológicas e muitos deles, ou seus derivados,
são usados como fármacos (eritromicina, ciclosporina, vancomicina e anfotericina B). Entre os
macrociclos que apresentam atividades biológicas encontram-se as macrolactamas, como a
vicenistatina, ascomicina, pimecrolimus, entre outros.
Embora a síntese de macrociclos seja considerada um dos maiores desafios da síntese orgânica,
a diversidade estrutural e o potencial bioativo são estímulos para diversos grupos de pesquisa
investigarem a síntese dessas substâncias. Diversas metodologias são usadas para a síntese de
macrociclos, entre as quais a carbociclização radicalar mediada por hidreto de tri-n-butilestanho
(Bu3SnH).
Com o intuito de se verificar o papel de carboidratos e de sua estereoquímica em
macrociclizações mediadas por hidreto de tri-n-butilestanho, assim como as influências dos
grupos envolvidos diretamente na reação de ciclização e de suas posições relativas na unidade
sacarídica, o grupo do Laboratório de Química Farmacêutica da Faculdade de Farmácia/UFMG,
em parceria com o Departamento de Química do ICEx/UFMG (QF/DQ/UFMG), vem
desenvolvendo um programa de pesquisa de síntese de benzomacrolactamas por reação de
carbociclização radicalar mediada por hidreto de tri-n-butilestanho.
No âmbito deste programa, foi sintetizada a m-aliloxiiodobenzamida inédita 4-O-alil-2,3-di-O-
benzil-6-desoxi-6-(3-iodobenzoilamino)-α-D-galactopiranosídeo de metila (GAX), que foi submetida
à reação de carbociclização radicalar com Bu3SnH. Foram realizadas quatro reações de GAX com
Bu3SnH em diferentes condições (solvente, diluição e tempo de adição dos reagentes). De
nenhuma das reações isolaram-se macrolactamas. O produto acíclico reduzido, 4-O-alil-2,3-di-O-
benzil-6-desoxi-6-benzoilamino-α-D-galactopiranosídeo de metila (GAXC), foi isolado de três
reações. Em uma das reações, em que a diluição e o tempo de adição dos reagentes foram
maiores, isolou-se o produto inédito e inesperado, 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-
fenilbenzoilamino)-α-D-galactopiranosídeo de metila (GAXD), formado por reação entre o radical
arila proveniente de GAX e benzeno, o solvente da reação.
As estruturas dos produtos obtidos foram elucidadas pelos seus espectros no IV, de RMN de 1H e
de 13C e experimentos de RMN 2D, além de massas de alta resolução e exatidão, em alguns casos.
A m-aliloxiiodobenzamida GAX, os produtos isolados das reações radicalares (GAXC e GAXD) e
dois intermediários (GA6 e GA7) foram submetidos a testes de atividade antimibacteriana e
antifúngica e não demonstraram atividade.
Palavras-chave: carbociclização radicalar, benzomacrolactamas, meta-iodobenzamida

xii
ABSTRACT
Macrocycles display remarkable biologic activities and many of these compounds, or their
derivatives, are used as drugs, among them, erythromycin, ciclosporin, vancomycin and
anphotericin B. Among macrocycles with biological activities, there are the macrolactams, as
vicenistatin, ascomycin, pimecrolimus, and others.
Although macrocycles synthesis is considered one of the greatest challenges in organic synthesis,
the immense chemical diversity and the bioactive potential of these compounds have been
stimulating their synthesis investigation. Different methodologies can be used for macrocycles
synthesis, among them, the tri-n-butyltin hydride-mediated radical carbocyclization reaction.
To investigate the carbohydrates role and their stereochemistry in macrocyclizations by tri-n-
butyltin hydride, as well as the influence of the groups directly involved in the cyclization reaction
and their relative position in the carbohydrate, the research group of “Laboratório de Química
Farmacêutica/Faculdade de Farmácia/UFMG”, in partnership with “Departamento de Química/
ICEx/UFMG” (QF/DQ/UFMG), have been developing researches related to benzomacrolactams
synthesis by tri-n-butyltin hydride-mediated radical carbocyclization reaction.
Carrying on this research program, it was synthesized the m-allyloxyiodobenzamide methyl 4-O-
allyl-2,3-di-O-benzyl-6-deoxy-6-(3-iodobenzoylamino)-α-D-galactopyranoside (GAX), which was
undergone to Bu3SnH-mediated radical carbocyclization reactions. Four reactions of GAX with
Bu3SnH were carried out, in different conditions (reagents concentration and addition time,
solvent). Macrolactams were not isolated from any reaction. The acyclic reduction product, methyl
4-O-allyl-2,3-di-O-benzyl-6-deoxy-6-benzoylamino-α-D-galactopyranoside (GAXC) was isolated from
three reactions. In one of the reactions, which the reagents dilution and addition time were bigger
than in other reactions, it was got an unexpected and unheard product, methyl 4-O-allyl-2,3-di-O-
benzyl-6-deoxy-6-(3-phenylbenzoylamino)-α-D-galactopyranoside (GAXD). This product was formed
by attack of aryl radical from GAX on benzene, the reaction solvent.
The structures of the synthesized compounds were elucidated by their IR and NMR spectra (1H, 13C and 2D experiments). It was also obtained the high resolution mass spectra of GAX, GAXC
and GAXD.
The m-allyloxyiodobenzamide GAX, the products isolated from radical reactions (GAXC and
GAXD), GA6 and GA7 were undergone to antibacterial and antifungal tests and did not show any
activity.
Keywords: radical carbocyclization, benzomacrolactams, meta-iodobenzamide
INTRODUÇÃO
1
1 INTRODUÇÃO
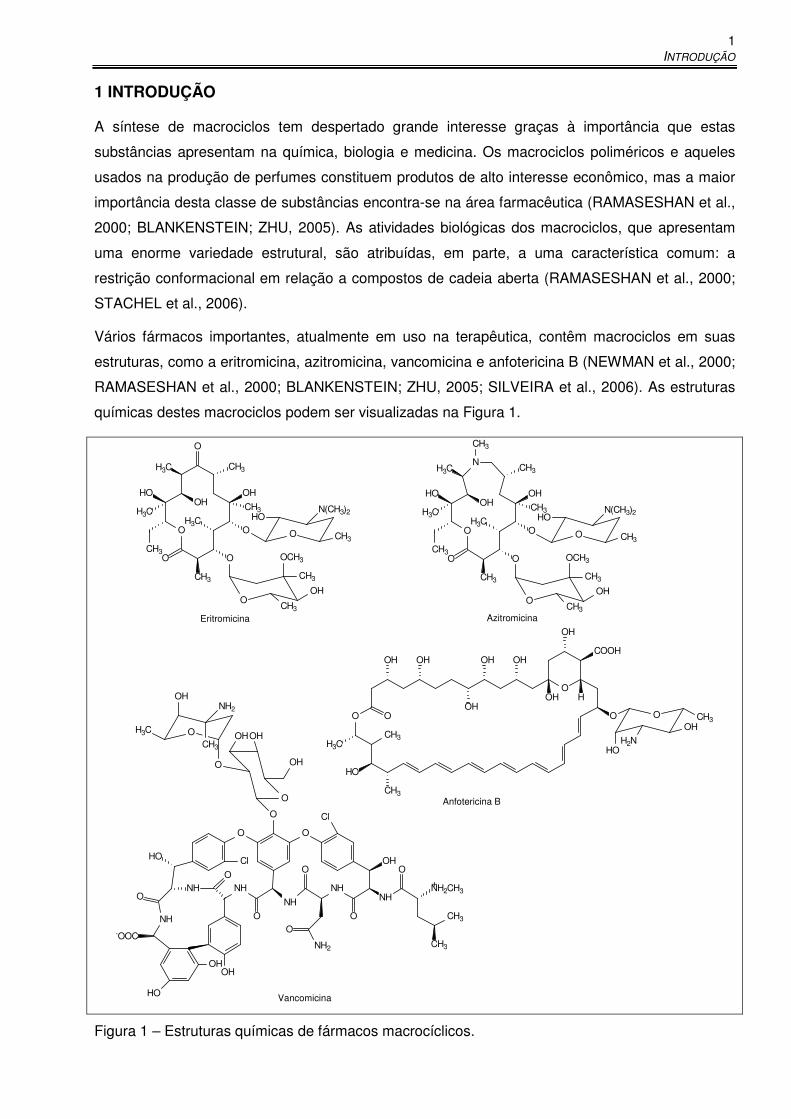
A síntese de macrociclos tem despertado grande interesse graças à importância que estas
substâncias apresentam na química, biologia e medicina. Os macrociclos poliméricos e aqueles
usados na produção de perfumes constituem produtos de alto interesse econômico, mas a maior
importância desta classe de substâncias encontra-se na área farmacêutica (RAMASESHAN et al.,
2000; BLANKENSTEIN; ZHU, 2005). As atividades biológicas dos macrociclos, que apresentam
uma enorme variedade estrutural, são atribuídas, em parte, a uma característica comum: a
restrição conformacional em relação a compostos de cadeia aberta (RAMASESHAN et al., 2000;
STACHEL et al., 2006).
Vários fármacos importantes, atualmente em uso na terapêutica, contêm macrociclos em suas
estruturas, como a eritromicina, azitromicina, vancomicina e anfotericina B (NEWMAN et al., 2000;
RAMASESHAN et al., 2000; BLANKENSTEIN; ZHU, 2005; SILVEIRA et al., 2006). As estruturas
químicas destes macrociclos podem ser visualizadas na Figura 1.
Eritromicina
O
HOO
CH3H3C
OH
CH3
OH3C
O
CH3
OCH3
H3C
HO
OCH3
OH
CH3
OCH3
N(CH3)2
CH3
OH
O
Azitromicina
O
HOO
NCH3H3C
CH3
OH
CH3
OH3C
O
CH3
OCH3
H3C
HO
OCH3
OH
CH3
OCH3
N(CH3)2
CH3
OH
+
O
OH
OHOHO
O
OH
H3C
NH2
CH3
O
OO
Cl
Cl
NH
O
NH
O
NH
O
OH
NH
HO
NH2
NH
CH3
CH3
OH
-OOC
O
HO
NHO
O
OHO
NH2CH3
Vancomicina
O
HOH2N
OHCH3
CH3
O
O
OH H
COOH
OH
OHOH
OH
OHOH
O
H3C
O
HO
CH3Anfotericina B
Figura 1 – Estruturas químicas de fármacos macrocíclicos.

INTRODUÇÃO
2
Entre os macrociclos que são agentes bioativos encontram-se as macrolactamas, cujas atividades
biológicas podem ser atribuídas, em parte, ao fato de elas serem macrociclos e amidas. Em um
trabalho publicado recentemente, no qual são apresentados dados relativos a 128 candidatos a
fármacos que se encontram em desenvolvimento em três grandes indústrias farmacêuticas, é
relatado que 12% das reações realizadas na síntese das 128 substâncias são de acilação,
especialmente N-acilações. Os autores consideram que este dado não surpreende, uma vez que
as amidas estão presentes em um grande número de fármacos. Além disso, de 53 fármacos que,
em 2003, foram responsáveis pela arrecadação acima de um bilhão de dólares, nove apresentam
o grupo amida e três o grupo sulfonamida (CAREY et al., 2006).
Tendo em vista sua importância, as macrolactamas vêm sendo amplamente estudadas sob vários
aspectos: isolamento e elucidação estrutural de produtos naturais, síntese, modificações
moleculares, estudos de relação estrutura química-atividade biológica, mecanismo de ação e
triagem clínica.
1.1 MACROLACTAMAS NATURAIS E SEMI-SINTÉTICAS COM ATIVIDADE BIOLÓGICA
Diversos depsipeptídeos macrocíclicos apresentam atividade biológica: criptoficina A (atividade
antitumoral – PATEL et al., 1999), hapalosina (atividade citotóxica leve – WAGNER et al., 1999) e
estevastelina B (inibe ativação de células T e B – KOHYAMA; YAMAMOTO, 2001) (FIGURA 2).
O NH
NO
O
O
H
O
O
HH3C
O Cl
OCH3
Criptoficina A
O NH
NHN
O
OH
C13H27
OO
OH
AcO
O
H
Estevastelina B
O
O N
O
OO
CH3
OH
Hapalosina
Figura 2 – Estruturas químicas de depsipeptídeos com atividade biológica.
Depsipeptídeos cíclicos foram isolados de cianobactéria marinha (Lyngbya majuscula), em 2000,
e foram denominados lingbiabelinas A e B (FIGURA 3). Ambas apresentam atividade contra

INTRODUÇÃO
3
células tumorais, sendo a atividade in vitro da lingbiabelina A ligeiramente superior à da
lingbiabelina B (LUESCH et al., 2000; MILLIGAN et al., 2000; YOKOKAWA et al, 2002).
O
ONS
HN HN
N
S
OO Cl Cl
HO
O
O H
Lingbiabelina A
O
ONS
HN HN
N
S
OO Cl Cl
HO
O
O
Lingbiabelina B
Figura 3 – Estruturas químicas das lingbiabelinas A e B.
A indolactama-V e benzolactama-V8 (FIGURA 4), macrolactamas de 9 e 8 membros,
respectivamente, ligam-se à proteína quinase C, uma enzima fundamental envolvida na
transdução celular, ativando-a para a promoção de tumores. Esta atividade parece estar
diretamente relacionada à presença do hidrogênio da amida, uma vez que lactonas análogas não
exibiram esta atividade (NAKAGAWA et al., 2001).
(-) Indolactama-V
N
H
NN
OH
O
H
NN
O
H
OH
Benzolactama -V8
Figura 4 – Estruturas químicas das macrolactamas indolactama-V e benzolactama-V8.
A vicenistatina (FIGURA 5), uma macrolactama de 20 membros ligada a um aminoaçúcar
(vicenisamina) e produzida por Streptomyces halstedii HC-34, apresenta atividade antitumoral
(SHINDO et al., 1993).
O
OH
H3CHN
CH3
O N
H
O
Vicenistatina
Figura 5 – Estrutura química da vicenistatina.
INTRODUÇÃO
4
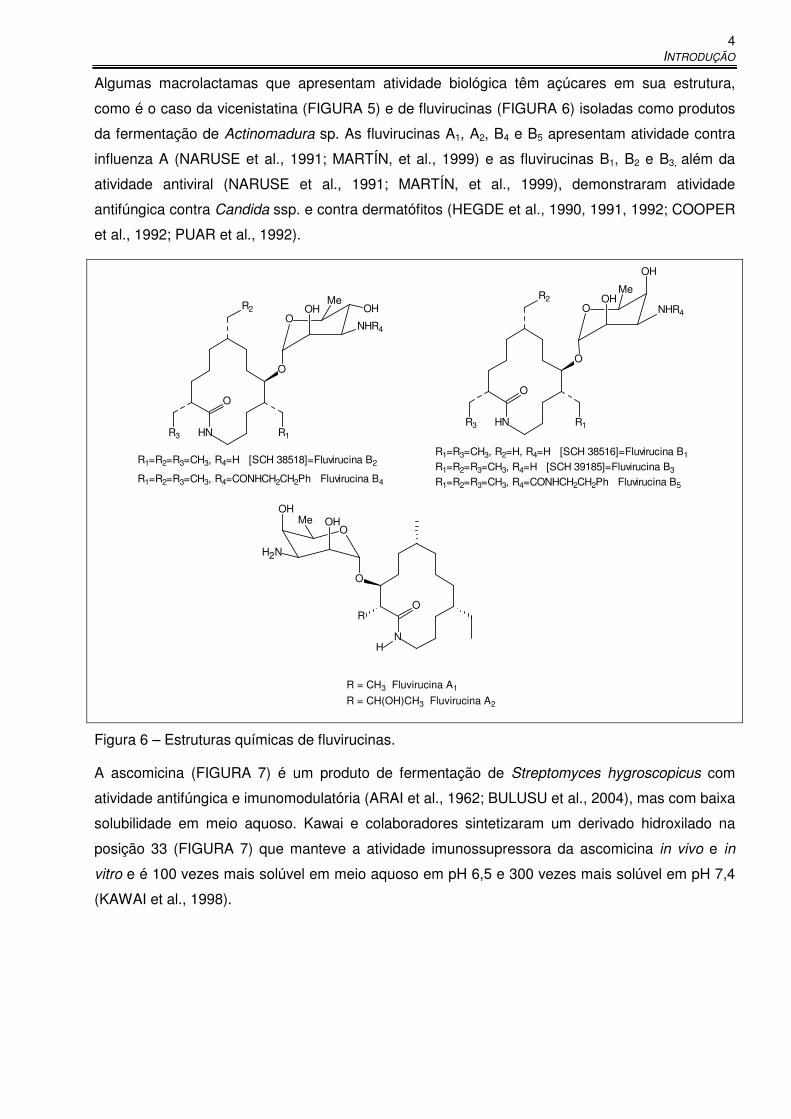
Algumas macrolactamas que apresentam atividade biológica têm açúcares em sua estrutura,
como é o caso da vicenistatina (FIGURA 5) e de fluvirucinas (FIGURA 6) isoladas como produtos
da fermentação de Actinomadura sp. As fluvirucinas A1, A2, B4 e B5 apresentam atividade contra
influenza A (NARUSE et al., 1991; MARTÍN, et al., 1999) e as fluvirucinas B1, B2 e B3, além da
atividade antiviral (NARUSE et al., 1991; MARTÍN, et al., 1999), demonstraram atividade
antifúngica contra Candida ssp. e contra dermatófitos (HEGDE et al., 1990, 1991, 1992; COOPER
et al., 1992; PUAR et al., 1992).
R = CH3 Fluvirucina A1
R = CH(OH)CH3 Fluvirucina A2
HN
O
R3 R1
O
OOH
Me
OH
NHR4
R2
R1=R3=CH3, R2=H, R4=H [SCH 38516]=Fluvirucina B1
R1=R2=R3=CH3, R4=H [SCH 39185]=Fluvirucina B3
R1=R2=R3=CH3, R4=CONHCH2CH2Ph Fluvirucina B5
HN
O
R3 R1
O
OOH
Me
NHR4
OHR2
R1=R2=R3=CH3, R4=H [SCH 38518]=Fluvirucina B2
R1=R2=R3=CH3, R4=CONHCH2CH2Ph Fluvirucina B4
N
O
H
R
O
OOH
H2N
OHMe
Figura 6 – Estruturas químicas de fluvirucinas.
A ascomicina (FIGURA 7) é um produto de fermentação de Streptomyces hygroscopicus com
atividade antifúngica e imunomodulatória (ARAI et al., 1962; BULUSU et al., 2004), mas com baixa
solubilidade em meio aquoso. Kawai e colaboradores sintetizaram um derivado hidroxilado na
posição 33 (FIGURA 7) que manteve a atividade imunossupressora da ascomicina in vivo e in
vitro e é 100 vezes mais solúvel em meio aquoso em pH 6,5 e 300 vezes mais solúvel em pH 7,4
(KAWAI et al., 1998).

INTRODUÇÃO
5
O
O
O
OH
HO
O
O
O
OH
O
NO
O
OH
O
O
O
OH
HO
O
O
O
OH
O
NO
O
Derivado hidroxilado em C-33Ascomicina
Figura 7 – Estruturas químicas da ascomicina e de seu derivado hidroxilado em C-33.
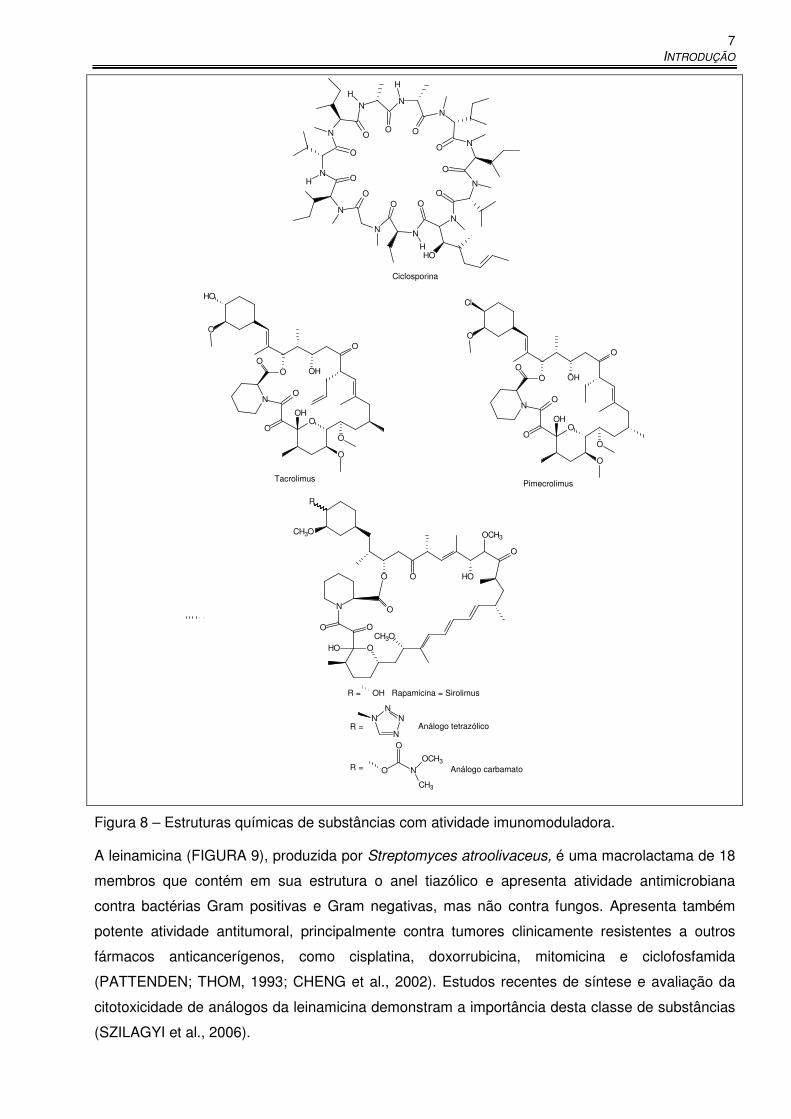
A atividade imunomoduladora de certas substâncias, como a apresentada pela ascomicina, é de
grande interesse pela variedade de enfermidades que podem ser tratadas com estes compostos.
Diversas macrolactamas com atividade imunomoduladora têm sido utilizadas como alternativa aos
glicocorticóides no tratamento tópico de doenças inflamatórias crônicas de pele. A ciclosporina
(FIGURA 8), produzida pelo fungo Tolypocladium inflatum, embora apresente atividade
imunossupressora, não é utilizada topicamente em função de sua reduzida penetração na pele. É
de uso sistêmico e apresenta algumas desvantagens relacionadas a efeitos imunossupressores
inespecíficos, além de efeitos adversos como hipertensão e nefrotoxicidade. Os efeitos
imunomodulatórios estão relacionados também à inibição de mecanismos de defesa contra
tumores, e a incidência de câncer de pele é maior em pacientes tratados com ciclosporina
(BORNHOVD et al., 2001).
O tacrolimus (FK506, FIGURA 8) foi isolado em 1984 de uma cultura de Streptomyces
tsukubaensis e apresenta atividade imunossupressora semelhante à da ciclosporina in vivo e in
vitro. Quimicamente, o tacrolimus é um análogo da ascomicina (FIGURA 7). É altamente
hidrofóbico e, assim como a ciclosporina, atua inibindo a ativação e maturação de células T. O uso
oral do tacrolimus para profilaxia e tratamento de rejeição após transplante de fígado e rim é
comum em diversos países e o uso sistêmico para tratamento de psoríase foi primeiramente
descrito em 1992. O tacrolimus é eficaz no tratamento sistêmico de eczema atópico, alopecia
areata, síndrome de Sézary e pioderma gangrenoso. Os efeitos adversos do tacrolimus, quando
utilizado de forma sistêmica, são semelhantes aos da ciclosporina. Sua penetração tópica é
altamente variável, dependendo da concentração, veículo e integridade da pele (a absorção é
maior em peles com processo inflamatório). Na forma de pomadas, o tacrolimus, em geral, atinge
a concentração terapêutica para tratamento de dermatite atópica. O efeito adverso mais comum,

INTRODUÇÃO
6
no caso de uso tópico, é sensação de queimação e calor após administração do fármaco
(DUMONT, 2000; BORNHOVD et al., 2001).
O pimecrolimus (32-epicloroascomicina, SDZ ASM 981, Elidel®, FIGURA 8) é outro derivado da
ascomicina e foi desenvolvido especificamente para uso tópico. Seus efeitos clínicos são
semelhantes aos do tacrolimus e sua eficácia no tratamento da dermatite atópica foi demonstrada
após um estudo duplo cego realizado em 1998 (GRASSBERGER et al., 1999; BORNHOVD et al.,
2001; BULUSU et al., 2004).
Uma outra macrolactama imunomoduladora é o sirolimus (FIGURA 8), também chamado de
rapamicina. É um metabólito secundário fúngico e interfere no ciclo celular em uma fase posterior
à interferência verificada no caso da ciclosporina, tacrolimus e pimecrolimus. Foi aprovado para
uso oral nos EUA em 1999 para prevenção de rejeição no caso de transplante de fígado e há
estudos indicando sucesso no caso de transplante renal, mas ainda não há dados com relação ao
seu uso por via tópica (BORNHOVD et al., 2001; WAGNER, et al., 2005). A rapamicina apresenta
um tempo de meia-vida extremamente longo (63 horas), e no caso de exposição prolongada de
pacientes a este fármaco há grande risco de toxicidade. Em 2005, Wagner e colaboradores
relataram a síntese de análogos da rapamicina (substituição da hidroxila em C-40 por um tetrazol
e um carbamato), que apresentaram atividade imunossupressora, mas menor tempo de meia-vida
que a rapamicina (WAGNER et al., 2005).
INTRODUÇÃO
7
OCH3
O O HO
CH3O
N
OHO
O
R
CH3O
O
O O
R = N
NN
NAnálogo tetrazólico
R =
O NOCH3
CH3
O
Análogo carbamato
TacrolimusPimecrolimus
Ciclosporina
N
N
N
N
N
NN
N
N
N
N
OO O
O
O
O
O
O
OO O
HO
HH
H
H
O
O
O
OH
HO
O
O
O
OH
O
NO
O
O
O
O
OH
Cl
O
O
O
OH
O
NO
O
R = OH Rapamicina = Sirolimus
Figura 8 – Estruturas químicas de substâncias com atividade imunomoduladora.
A leinamicina (FIGURA 9), produzida por Streptomyces atroolivaceus, é uma macrolactama de 18
membros que contém em sua estrutura o anel tiazólico e apresenta atividade antimicrobiana
contra bactérias Gram positivas e Gram negativas, mas não contra fungos. Apresenta também
potente atividade antitumoral, principalmente contra tumores clinicamente resistentes a outros
fármacos anticancerígenos, como cisplatina, doxorrubicina, mitomicina e ciclofosfamida
(PATTENDEN; THOM, 1993; CHENG et al., 2002). Estudos recentes de síntese e avaliação da
citotoxicidade de análogos da leinamicina demonstram a importância desta classe de substâncias
(SZILAGYI et al., 2006).

INTRODUÇÃO
8
S
NNH O
S S
OH
O
O
OH
O
Leinamicina
Figura 9 – Estrutura química da leinamicina.
Nishio e colaboradores isolaram dois novos antibióticos do tipo trieno-ansamicinas de cepa de
Streptomyces sp. (amostra de solo), que foram denominados TCM-135 A e B (FIGURA 10). Os
dois compostos apresentaram também potente atividade antitumoral contra diversos tipos de
células tumorais, sendo o TCM-135 A cerca de 10 vezes mais potente que o TCM-135 B (NISHIO
et al., 2000). Na Figura 10 encontram-se as estruturas de outras ansamicinas com atividade
antitumoral: a geldanamicina, que possui derivados em triagem clínica para tratamento de câncer,
a trienomicina A e a reblastatina (TAKATSU et al., 2000; STEAD, et al., 2000; LEMARCHAND;
BACH, 2004; PENG; BLAGG, 2006).
N
MeO
MeO
OCONH2
OHO
O
MeOO
H
Geldanamicina
N
MeO
MeO
OCONH2
OH
MeOO
H
OH
Reblastatina
S
HN
O
NH
CH3O
OCH3O
HO
H3C
OH
N
H
O
O
TCM-135A
NH
CH3O
OCH3O
HO
H3C
OH
N
H
O
O
NH
S
O
TCM-135B
Trienomicina A
NH
CH3O
OCH3O
HO
H3C
N
H
O
O
OH
CH3
Figura 10 – Estruturas químicas de ansamicinas.

INTRODUÇÃO
9
Três macrolactamas foram isoladas entre os produtos de fermentação de cepas de Tü 6239, uma
nova espécie de Streptomyces obtida de amostras de solo em São José do Rio Preto: ripromicina,
ikarugamicina e um epóxido da ikarugamicina (FIGURA 11). A ikarugamicina tem atividade
antiprotozoária e as três substâncias demonstraram atividade antibiótica contra bactérias Gram
positivas e efeito citostático sobre diferentes tipos de células tumorais humanas (BERTASSO et
al., 2003).
NH
HN
O
O
HO
O
H3C
H3C
H H
H H
H
H
H
Ikarugamicina
H3C
H3C
OH3C
O
NH
HN
O
O
HO
O
Ripromicina
NH
HN
O
O
HO
O
H3C
H3C
O
H
H
H
H
H
H
H
Epóxido da ikarugamicina
Figura 11 – Estruturas químicas de produtos de fermentação de cepas deTü 6239.
A maitansina (FIGURA 12) é um potente agente antitumoral da classe dos benzoansamacrolídeos
maitansinóides, que foi isolada originalmente em 1972 de cascas do arbusto africano Maytenus
ovatus (KUPCHAN et al., 1972). Apesar de a maitansina apresentar atividade antimitótica muitas
vezes superior à da vincristina e vinblastina em testes in vitro, esta substância demonstrou baixo
índice terapêutico nas triagens clínicas de fase I e II e se tornou um dos muitos agentes
citotóxicos que não chegaram à fase de uso terapêutico (YU; FLOSS, 2005; WIDDISON et al.,
2006). No entanto, a maitansina passou a ser um protótipo para a obtenção de agentes
antitumorais mais potentes, como o maitansinóide com um grupo metilditiopropanoíla (FIGURA
12) em substituição ao grupo acetila presente na maitansina (WIDDISON et al., 2006).

INTRODUÇÃO
10
Derivado da MaitansinaMaitansina
N
O
N
O
CH3 H3COOH H
O
HCH3
ON CH3
O
CH3
CH3
O
OH3CCl
CH3O
N
O
N
O
CH3 H3COOH H
O
HCH3
O
O
OH3CCl
CH3O
N
CH3 O
SS
CH3
CH3
Figura 12 – Estruturas químicas da maitansina e de seu derivado contendo o grupo dissulfeto.
As mixovirescinas são macrolactamas de 28 membros isoladas da fermentação de Myxococcus
virescin e o principal componente é a mixovirescina A1 (FIGURA 13), que inibe o crescimento de
E. coli e de outras enterobactérias (TROWITZSCH et al., 1982; ONISHI et al., 1984; WILLIAMS;
LI, 1994).
O
HN
O
OCH3CH3
CH3
OH
OH
OH
CH3
O
CH2OCH3
Mixovirescina A1
Figura 13 – Estrutura química da mixovirescina A1.
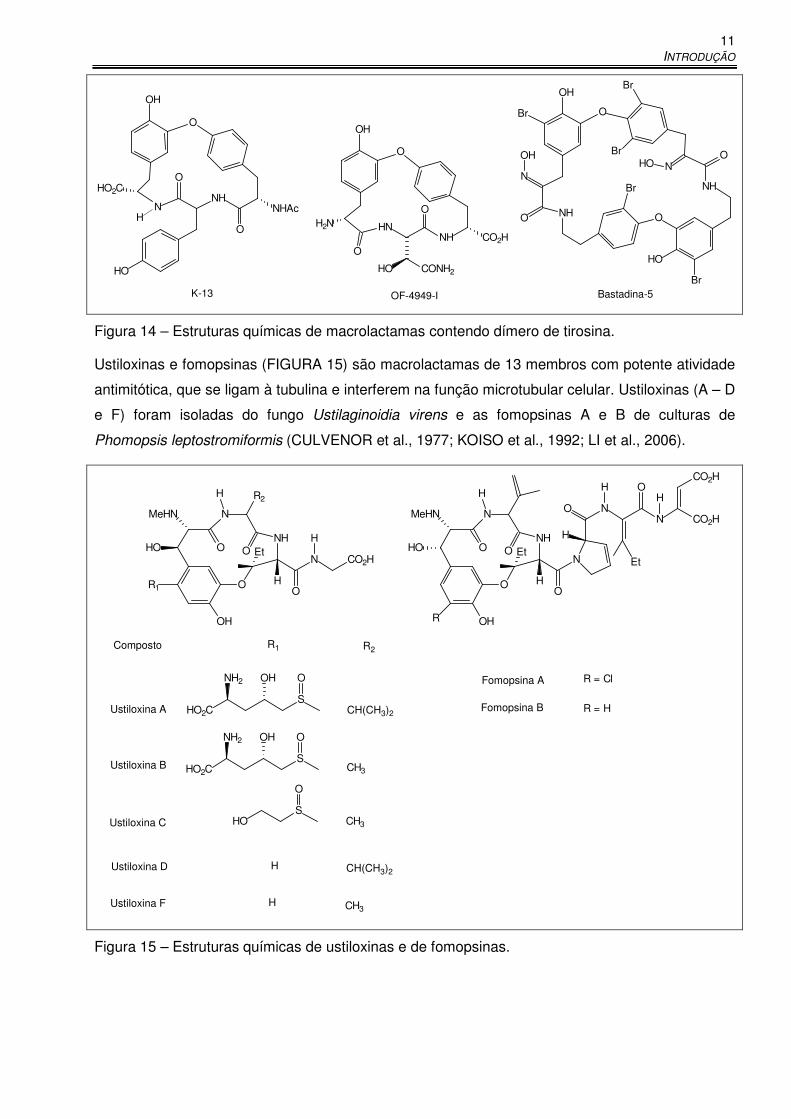
Macrolactamas contendo um dímero de tirosina em suas estruturas químicas (FIGURA 14)
apresentam importantes atividades biológicas. O composto K-13, produzido por Micromonospora
halophytica, é um potente inibidor não competitivo da enzima conversora da angiotensina (ECA) e
apresenta atividade anti-hipertensiva. A substância OF-4949-I, assim como outras produzidas pelo
fungo Penicilium rugulosum, é inibidora da aminopeptidase B. A bastadina-5, produzida por
Ianthella basta, bloqueia canais de cálcio do retículo sarcoplasmático e é usada no tratamento de
enfermidades relacionadas a músculos esquelético e cardíaco (MACK et al., 1994; BAILEY et al.,
1999; MASUNO et al., 2006). Masuno e colaboradores relataram a síntese de uma série de
macrolactamas de 14 e 18 membros, análogos simplificados da bastadina-5, potenciais agentes
para o tratamento de arritmias e falhas cardíacas (MASUNO et al., 2006).
INTRODUÇÃO
11
HO
NHN
OH
O
O
O
NHAc
HO2C
H
K-13
OH
O
CO2HH2N
NHHN
O
O
HO CONH2
OF-4949-I
OH
O
NH
ONH
Br
HO
Br
O
N
OH
Br
Br
Br
ONHO
Bastadina-5
Figura 14 – Estruturas químicas de macrolactamas contendo dímero de tirosina.
Ustiloxinas e fomopsinas (FIGURA 15) são macrolactamas de 13 membros com potente atividade
antimitótica, que se ligam à tubulina e interferem na função microtubular celular. Ustiloxinas (A – D
e F) foram isoladas do fungo Ustilaginoidia virens e as fomopsinas A e B de culturas de
Phomopsis leptostromiformis (CULVENOR et al., 1977; KOISO et al., 1992; LI et al., 2006).
N
O
ONH
HO
H
N
O
Et
OH
MeHN
O
H
R
NN
CO2H
CO2H
OH
H
Et
O
H
Fomopsina A
Fomopsina B
R = Cl
R = H
N
O
ONH
HO
H
N CO2H
H
O
Et
OH
MeHN
O
H R2
R1
CH3HUstiloxina F
CH(CH3)2HUstiloxina D
CH3HOS
O
Ustiloxina C
CH3HO2CS
ONH2 OH
Ustiloxina B
CH(CH3)2Ustiloxina A HO2CS
ONH2 OH
R1 Composto R2
Figura 15 – Estruturas químicas de ustiloxinas e de fomopsinas.

INTRODUÇÃO
12
1.2 MACROLACTAMAS SINTÉTICAS COM ATIVIDADE BIOLÓGICA
Embora a síntese de macrociclos seja considerada um dos maiores desafios da síntese orgânica,
diversos grupos de pesquisa têm se dedicado à síntese desses potenciais agentes bioativos
(BLANKENSTEIN; ZHU, 2005), inclusive de macrolactamas.
Gentile e colaboradores relataram a síntese de duas macrolactamas diméricas e uma tetramérica
(FIGURA 16). Os três macrociclos apresentaram moderada atividade citotóxica em testes in vitro
(GENTILE et al., 2000).
Macrolactamas diméricas
N
N
OO
H
H
O
OO
O
N
N
OO
H
H
O
O
OPh
O
OH
NHO
O
Macrolactama tetramérica
N
N
NO
O
O
N
O
O
O
O
O
O
O
O
O
H
H
H
H
Figura 16 – Estruturas químicas de macrolactamas diméricas e tetramérica.
Ojima e colaboradores descreveram a síntese de uma série de macrociclos taxóides, derivados
conformacionalmente restritos do paclitaxel (Taxol®, quimioterápico de ampla utilização), e
relataram os resultados dos testes de citotoxicidade dos novos compostos. Dos produtos obtidos,
uma lactama e um carbamato (FIGURA 17) destacaram-se quanto à atividade citotóxica (OJIMA
et al., 2002).

INTRODUÇÃO
13
Macrociclos taxóides
O
NH
O
O
O
O
HO
O
OHH
OHOAcO
OAc
OO
NH
O O
O
HO
O
OHH
OHOAcO
OAc
O
Paclitaxel
OH
OH
OH
OAcO
OBz OAc
OPh
O
OH
NHBz
Figura 17 – Estruturas químicas de macrociclos taxóides e do paclitaxel.
Considerando a necessidade de se obter novos derivados de quinolonas em função do alarmante
surgimento de bactérias resistentes a esta importante classe de agentes antibacterianos e que
macrociclos de 14 membros são capazes de inibir o processo de translação bacteriana, Jefferson
e colaboradores sintetizaram e avaliaram a atividade antibacteriana de uma série de “conjugados
quinolona-macrociclo”. Observou-se que conjugados derivados do ácido nalidíxico (FIGURA 18)
foram menos potentes que a quinolona de origem e que dois conjugados, um derivado da
ofloxacina e outro da ciprofloxacina (FIGURA 18), apresentaram atividade contra E. coli e S.
aureus semelhante às das quinolonas. Com o objetivo de verificar se o conjugado da ofloxacina
era um pró-fármaco, ou seja, se a atividade antibacteriana se devia à liberação da quinolona por
hidrólise da ligação amídica, foi sintetizado um análogo de cadeia aberta (FIGURA 18), que
também seria susceptível à hidrólise e, portanto apresentaria atividade semelhante. No entanto, o
derivado de cadeia aberta foi inativo, indicando que a presença da unidade macrocíclica,
conformacionalmente restrita, é importante para a atividade biológica (JEFFERSON et al., 2003).
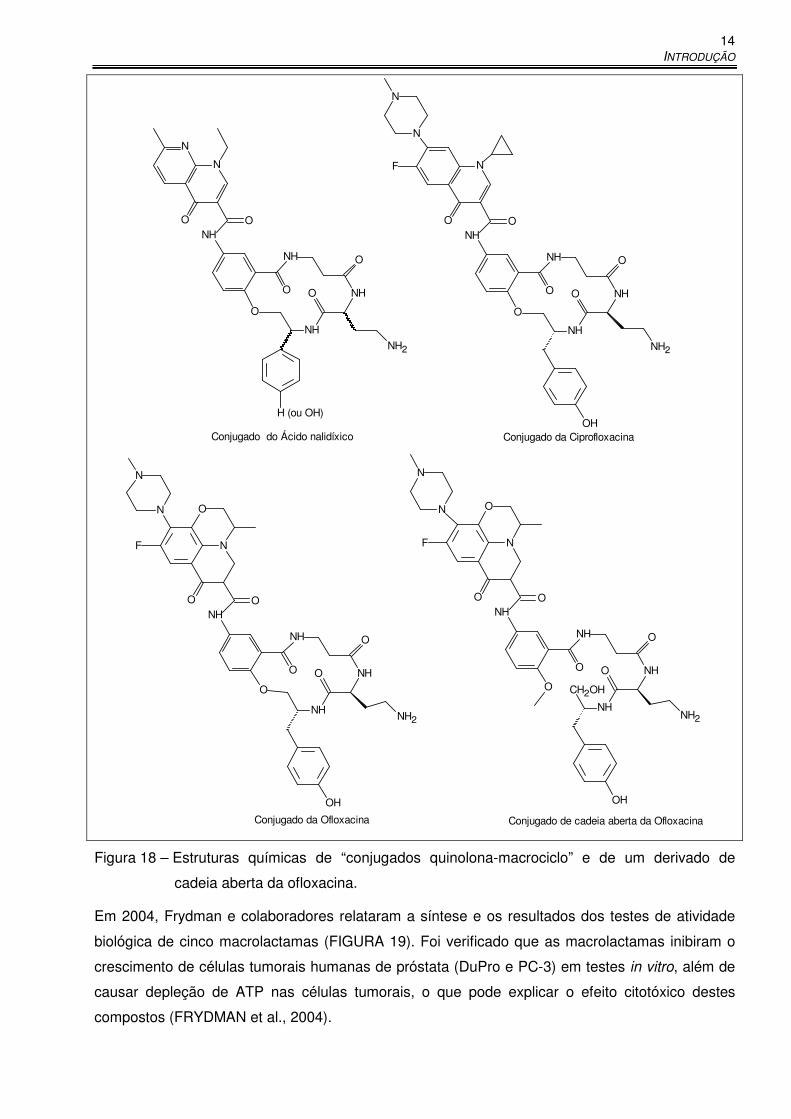
INTRODUÇÃO
14
NH
NH
NH
O
O
OO
NH
N
O
N
OH
NH2
O
F
N
NH
NH
CH2OH
NH
O
OO
NH
N
O
OH
NH2
O
F
N
N
O
O
Conjugado de cadeia aberta da Ofloxacina
NH
NH
NH
O
OO
NH
N
O
OH
NH2
O
F
N
N
O
O
Conjugado da Ofloxacina
Conjugado da Ciprofloxacina
NH
NH
NH
O
O
OO
NH
N
N
O O
H (ou OH)
NH2
Conjugado do Ácido nalidíxico
Figura 18 – Estruturas químicas de “conjugados quinolona-macrociclo” e de um derivado de
cadeia aberta da ofloxacina.
Em 2004, Frydman e colaboradores relataram a síntese e os resultados dos testes de atividade
biológica de cinco macrolactamas (FIGURA 19). Foi verificado que as macrolactamas inibiram o
crescimento de células tumorais humanas de próstata (DuPro e PC-3) em testes in vitro, além de
causar depleção de ATP nas células tumorais, o que pode explicar o efeito citotóxico destes
compostos (FRYDMAN et al., 2004).

INTRODUÇÃO
15
( )
H
HH
H
N N
NN
O
n
n = 1, 2 ou 3
H
HH
H 4 HCl
N N
NN
O
NH
NH
NH
R
O
NH
R = n-C13H27
3 HCl
2 HCl
Figura 19 – Estruturas químicas de macrolactamas com atividade antitumoral.
Shan e colaboradores testaram a atividade antitumoral de uma macrolactama sintética (FIGURA
20) análoga da migrastatina, uma macrolactona de 14 membros inicialmente isolada de cultura de
Streptomyces, e verificaram potente inibição de metástase em testes in vitro (SHAN et al., 2005).
O
NH
OCH3
OH
O
O
O
O
Migrastatina
N
OCH3
OH
OH
Análogo sintético da Migrastatina
Figura 20 – Estruturas químicas da migrastatina e de seu análogo sintético.
O principal componente de placas de amilóide, associadas à Doença de Alzheimer, é um
fragmento peptídico neurotóxico, que é formado a partir da proteína precursora de amilóide (PPA).
A fragmentação da PPA é catalisada por enzimas proteolíticas, entre as quais a β-secretase. Por
isso, a β-secretase é um alvo terapêutico atrativo para o tratamento e prevenção da Doença de
Alzheimer (STACHEL et al., 2006).
Stachel e colaboradores relataram que a macrociclização de um inibidor fraco de β-secretase
conduziu a uma macrolactama de 15 membros com maior potência. Os autores consideraram que
a maior atividade inibitória da macrolactama em relação ao análogo de cadeia aberta é
conseqüência da redução da liberdade conformacional. Outras macrolactamas de 15 e 14
membros foram sintetizadas e avaliadas quanto à atividade inibidora de β-secretase. Uma
macrolactama de 14 membros apresentou alta atividade inibidora de β-secretase in vitro e reduziu
a formação de amilóide in vivo (STACHEL et al., 2006). As estruturas do inibidor de cadeia aberta
e das macrolactamas encontram-se representadas na Figura 21.

INTRODUÇÃO
16
NH
O
HN
O
OH
N
O
SOO
NH
O
HN
O
OH
N
O
SOO
NH
O
HN
O
OMe
N
O
SOO
O
NH
O
HN
O
N
N
O
SOO
H
N
O
H
HN
O
N
NS
OO
H
N
O
H
HN
O
OH
NS
OO
HN
O
OMe
NS
OO
O
Inibidor mais potente
Figura 21 – Inibidores de β-secretase.
1.3 SÍNTESE DE MACROCICLOS POR REAÇÃO DE CARBOCICLIZAÇÃO RADICALAR MEDIADA POR
HIDRETO DE TRI-N-BUTILESTANHO
O sucesso na síntese de macrociclos depende fundamentalmente da tendência de os grupos do
substrato que reagem entre si adquirirem disposição espacial adequada. Fatores importantes que
restrigem a liberdade rotacional e podem favorecer uma conformação apropriada para a ciclização
em detrimento de outras, são as interações intramoleculares, como as eletrostáticas e ligações de
hidrogênio, e as forças de repulsão estérica e eletrônica. O substrato a ser ciclizado pode
apresentar previamente, por suas características estruturais, a pré-organização necessária para a
ciclização ou a introdução de elemento(s) que a favoreça pode ser planejada (BLANKENSTEIN;
ZHU, 2005).
Diferentes métodos de síntese têm sido empregados para a obtenção de macrociclos e, na
maioria deles, são preconizadas condições de alta diluição, a fim de se evitar oligomerizações
resultantes de reações intermoleculares (BLANKENSTEIN; ZHU, 2005).
Algumas das estratégias usadas para a síntese de macrociclos são a lactonização e lactamização
a partir de ω-hidroxiácidos e ω-aminoácidos, respectivamente, reação de Diels-Alder
intramolecular, expansão de anéis, acoplamento com paládio (reação de Heck), metátese de
alquenos e reações de carbociclização radicalar (PORTER; CHANG, 1987; ROXBURGH, 1995;
SHAN et al., 2005; STACHEL et al., 2006).

INTRODUÇÃO
17
As reações radicalares passaram a ser ferramenta importante na síntese orgânica, a partir do
século XX, especialmente as de carbociclização mediadas por hidretos de tri-organoestanho, que
têm sido extensivamente aplicadas para a construção de anéis de cinco e seis membros
(BALRAJU et al., 2005) e cujos fatores estéricos e eletrônicos que as regem já são bem
conhecidos (BALDWIN, 1976; GIESE, 1983; GIESE, 1986; CURRAN, 1988; HANDA;
PATTENDEN, 1997). Até a primeira metade dos anos 80, as reações de carbociclização radicalar
só eram usadas para a síntese de ciclos de cinco e seis membros e os primeiros trabalhos de
síntese de macrociclos por ciclização radicalar foram publicados a partir de 1986 (PORTER et al.,
1986; PORTER; CHANG, 1987; PORTER et al., 1988; COX et al., 1989; HITCHCOCK;
PATTENDEN, 1990), o que justifica o fato de o conhecimento dessas reações ser limitado em
relação ao de penta e hexacarbociclizações.
Por meio de levantamento bibliográfico realizado no banco de dados SciFinder Scholar® e Web of
Science®, foi possível verificar que o reagente de escolha para a síntese de compostos cíclicos,
inclusive macrociclos, por reação radicalar, é o hidreto de tri-n-butilestanho (Bu3SnH), o que é
corroborado por citações em artigos publicados (PORTER et al., 1988; CURRAN, 1988;
BOWMAN et al., 2000; ALLIN et al., 2002; BOWMAN et al., 2002; JESSOP et al., 2003). Foi
possível constatar também que a formação de ligação C-C ocorre por ataque intramolecular de
um carbono radicalar (alquil, alquenil, acil, aril) ao carbono de uma ligação C-C múltipla (alqueno
ou alquino).
As principais características das reações com Bu3SnH, responsáveis pela ampla aplicação na
obtenção de compostos cíclicos, são a relativa simplicidade do ponto de vista operacional, o fato
de que a ciclização ocorre sem alteração da configuração dos estereocentros de precursores
quirais e que a maioria dos grupos funcionais é inerte ao reagente (MARCO-CONTELLES et al.,
1998; MARTÍNEZ-GRAU; MARCO-CONTELLES, 1998). No entanto, o Bu3SnH apresenta alguns
inconvenientes: toxicidade, dificuldade em eliminar os resíduos de estanho dos produtos obtidos e
redução do radical formado antes que a ciclização ocorra (BERGE; ROBERTS, 1979; SALOMON
et al., 2000; CLYNE; ALDABBAGH, 2006).
Os mecanismos envolvidos nas reações com Bu3SnH encontram-se bem discutidos (WALLING,
1985; CURRAN, 1988; ALLIN et al., 2002; BECKWITH et al., 2004) e estão representados para o
7-iodo-2-hepteno na Figura 22. A etapa de iniciação envolve a formação do radical tributilestanila
por reação com um iniciador radicalar, como Azobisisobutironitrila (AIBN). O radical tributilestanila
(Bu3Sn.) abstrai um grupo abandonador do substrato, formando um novo radical. O intermediário
radicalar pode seguir alguns caminhos, entre os quais: i) abstração de um hidrogênio do Bu3SnH,
levando a um produto de redução (hidrogenólise); ii) ciclização por reação de adição
intramolecular a uma ligação múltipla levando a um radical ciclizado (endo - ciclo maior; exo - ciclo
menor), que em seguida abstrai um hidrogênio do Bu3SnH; iii) abstração de um hidrogênio do
próprio substrato (transferência 1,5 ou 1,6), alterando a posição do radical, que pode ciclizar ou

INTRODUÇÃO
18
abstrair um hidrogênio do Bu3SnH para levar ao produto de redução. O caminho seguido depende
das constantes de transferência de hidrogênio e de ciclização das espécies envolvidas, além da
concentração do substrato e do hidreto de tri-n-butilestanho.
Exo
++ Bu3Sn HH3CH2C
Exo
Bu3Sn
C N N2+280 oCN C N N C N
AIBN
Bu3SnC N C N++Bu3Sn H
Bu3Sn
H3C
+ Bu3SnI+
H3C
I
+
H3C
Bu3Sn H+ +
+Bu3Sn H+
H3C
Endo Exo
Endo Endo
H3C
Bu3Sn
Bu3Sn
Hidrogenólise
HH3C
H3C
H3C
H3CHC
H3CHC
Figura 22 – Mecanismos envolvidos na reação radicalar mediada por Bu3SnH/AIBN.
Em 1990, Snieckus e colaboradores demonstraram a importância da transferência de hidrogênio
1,5 de grupo α-benzamidoíla para radicais arila em reações radicalares de orto-iodobenzamidas
mediadas por hidreto de tri-n-butilestanho (FIGURA 23). Os resultados obtidos indicam a
importância do rotâmero preferencial do substrato na definição do curso da reação radicalar. A
velocidade de giro de ligações C-N é inferior ao tempo de meia vida de radicais arila e, deste
modo, a relação de produtos formados é diretamente proporcional à quantidade de cada rotâmero
no início da reação (SNIECKUS et al., 1990).

INTRODUÇÃO
19
N
O
CO2Et
NCOPh
CO2Et
R
N R2
R1X
R O
Bu3Sn N R2
R1
R O
H
N R2
R1
R O
H
Produtos
Rotação da amida
N R1
R2X
R O
X = Br, I
ProdutosN R1
R2
R O
H
N R1
R2
R O
H
Bu3Sn
Figura 23 – Rotâmeros e radicais formados por transferência 1,5 em orto-iodobenzamidas.
Na Figura 24 observam-se as iodobenzamidas que foram submetidas à reação de ciclização
radicalar e os respectivos produtos formados (SNIECKUS et al., 1990). No substrato em que R =
Ph não ocorreu transferência de hidrogênio 1,5, uma vez que predomina amplamente o rotâmero
syn. Por outro lado, nos outros substratos, como há quantidades apreciáveis do rotâmero anti,
foram isolados produtos de ciclização resultantes do ataque do radical α-amidoíla, formado por
transferência de hidrogênio 1,5, à ligação dupla.
I
N
O
R
CO2EtI
N
O
R
CO2Et
Anti Syn
R Proporção Anti/Syn
Produtos Rendimento (%)
Ph 10/90 36
C6H11 33/67 27
C4H9 50/50 43
CH2Ph 50/50 38
(CH2)4CH=CHCO2Et - 82
Figura 24 – Rotâmeros e produtos de ciclização de iodobenzamidas.

INTRODUÇÃO
20
Embora os estudos sobre macrociclização radicalar com Bu3SnH sejam restritos quando
comparados com aqueles de obtenção de ciclos de cinco e seis membros (BALRAJU et al., 2005),
já é de conhecimento que macrociclos podem ser obtidos com bons rendimentos por reação
radicalar se precursores adequados forem utilizados e se certas condições de reação forem
seguidas.
Com base nos estudos de macrociclização radicalar mediada por Bu3SnH envolvendo radicais
alquila como nucleófilos (doadores de radical) e alquenos como eletrófilos (aceptores de radical)
desenvolvidos por Porter e colaboradores (PORTER et al., 1986, PORTER; CHANG, 1987;
PORTER et al., 1988) foram estabelecidos os seguintes critérios:
i) Como a maioria dos radicais localizados em carbono são nucleofílicos, a ativação de ligação
C=C que sofrerá adição do radical com um grupo retirador de elétrons é importante para que
ocorra a ciclização;
ii) Condições de alta diluição melhoram o rendimento de produtos macrocíclicos, uma vez que
diminui a probabilidade da ocorrência de processos bimoleculares, como redução direta do radical
formado pelo hidreto de tri-n-butilestanho e reações de dimerização e polimerização; a condição
ideal encontrada foi concentração do precursor de 3 a 7 mmol/L e adição lenta do Bu3SnH;
iii) Em geral, precursores iodados conduzem a melhores rendimentos de produtos ciclizados do
que precursores bromados;
iv) Em geral, as macrociclizações endo são preferenciais em relação às exo.
Porter e colaboradores relataram a obtenção de macrociclos provenientes de ciclização pelo modo
endo com rendimentos bons e moderados, a partir de ω-iodoenonas (FIGURA 25) (PORTER et
al., 1986).
Produto de redução
+(CH2)n
O
(CH2)n
O
I
3-6 mmol/L
(CH2)n
O
Bu3SnH, AIBN
Benzeno, refluxo 3 h
Endo
n
Tamanho do anel
Rendimento (%) Macrociclo Produto de redução 1 10 15 27 5 14 63 22 9 18 54 16
Figura 25 – Macrociclos obtidos a partir de iodoenonas.
Continuando os estudos de macrociclização radicalar empregando-se Bu3SnH/AIBN, Porter e
Chang demonstraram que reação radicalar intramolecular em ésteres acrilatos e fumaratos
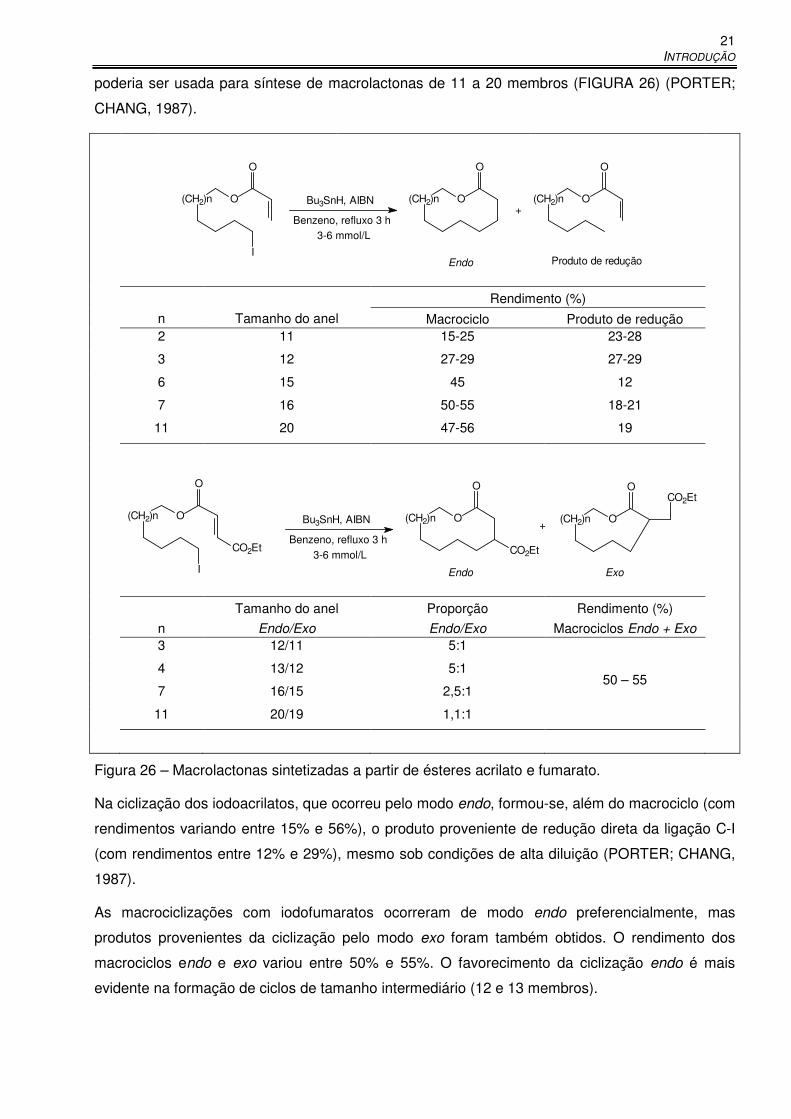
INTRODUÇÃO
21
poderia ser usada para síntese de macrolactonas de 11 a 20 membros (FIGURA 26) (PORTER;
CHANG, 1987).
Produto de redução
+O(CH2)n
O
O(CH2)n
O
I
3-6 mmol/L
O(CH2)n
O
Bu3SnH, AIBN
Benzeno, refluxo 3 h
Endo
n
Tamanho do anel
Rendimento (%)
Macrociclo Produto de redução
2 11 15-25 23-28
3 12 27-29 27-29
6 15 45 12
7 16 50-55 18-21
11 20 47-56 19
+O(CH2)n
OCO2Et
O(CH2)n
O
I
CO2Et3-6 mmol/L
O(CH2)n
O
CO2Et
Bu3SnH, AIBN
Benzeno, refluxo 3 h
Endo Exo
n
Tamanho do anel
Endo/Exo
Proporção
Endo/Exo
Rendimento (%)
Macrociclos Endo + Exo
3 12/11 5:1
50 – 55
4 13/12 5:1
7 16/15 2,5:1
11 20/19 1,1:1
Figura 26 – Macrolactonas sintetizadas a partir de ésteres acrilato e fumarato.
Na ciclização dos iodoacrilatos, que ocorreu pelo modo endo, formou-se, além do macrociclo (com
rendimentos variando entre 15% e 56%), o produto proveniente de redução direta da ligação C-I
(com rendimentos entre 12% e 29%), mesmo sob condições de alta diluição (PORTER; CHANG,
1987).
As macrociclizações com iodofumaratos ocorreram de modo endo preferencialmente, mas
produtos provenientes da ciclização pelo modo exo foram também obtidos. O rendimento dos
macrociclos endo e exo variou entre 50% e 55%. O favorecimento da ciclização endo é mais
evidente na formação de ciclos de tamanho intermediário (12 e 13 membros).

INTRODUÇÃO
22
Porter e colaboradores observaram que a partir de substratos em que o ataque exo seria
favorecido eletronicamente não ocorreu ciclização (FIGURA 27). Somente precursores altamente
ativados por dois substituintes ciano levaram a produtos de ciclização exo (FIGURA 27) (PORTER
et al., 1988).
X = H, Y = COOEt X = Y = COOEt X = H, Y = NO2X = COOMe, Y = SO2Ph
(CH2)n
I Y
X
(CH2)n
X
Y
+X (CH2)n
Y
X
(CH2)n
I Y
X
2-10 mmol/L
(CH2)n
X
Y
Bu3SnH, AIBN
Benzeno, refluxo 3 h
ExoX = Y = CN
n Tamanho do anel Rendimento (%)
3 12 < 5
7 16 35
11 20 50
Figura 27 – Macrociclizações em substratos ativados para ciclização exo.
Os resultados dos trabalhos de Porter e colaboradores (PORTER et al., 1986, PORTER; CHANG,
1987; PORTER et al., 1988) indicam que a adição do radical a alquenos se dá preferencialmente
na região menos impedida estericamente, ocorrendo normalmente pelo modo endo. O modo exo
pode ser forçado com a inclusão de dois substituintes altamente retiradores de elétrons (CN) e a
ciclização exo tem sua eficácia aumentada em ciclos maiores, observação que sugere que a
macrociclização exo é geometricamente mais exigente que a macrociclização endo, sendo
necessários anéis maiores no estado de transição de exociclos.
A roseofilina, um antibiótico citotóxico isolado de Streptomyces griseoviridis em 1992, despertou o
interesse em diversos grupos de pesquisa. Neste contexto, Robertson e Hatley relataram a
síntese de um precursor da roseofilina (FIGURA 28) e uma das etapas envolveu reação de
macrocilização radicalar com Bu3SnH. Para que ocorresse preferencialmente a ciclização 13-
endo, em detrimento da redução do radical alquila, foi necessária alta diluição (2,5 mmol/L) e
adição lenta do hidreto de tri-n-butilestanho (7 horas) (ROBERTSON; HATLEY, 1999).

INTRODUÇÃO
23
N
i-Pr
O
OCH3
N
H
Cl
Roseofilina
13-Endo
(CH2)6I
O
i-Pr
O
O
O
i-Pr
Bu3SnH, AIBN
Benzeno
35 - 50%
Figura 28 – Síntese de precursor da roseofilina por macrociclização 13-endo.
Como parte dos estudos visando à síntese da micotoxina zearalenona (FIGURA 29), Hitchcock e
Pattenden relataram a síntese de um E-cetomacrolídeo resultante de ciclização 14-endo, com 61%
de rendimento, a partir do benzoato de 5-oxohept-6-enil-2-(E-3-bromoprop-1-enil) (FIGURA 29). A
reação foi desenvolvida em benzeno, sob refluxo e atmosfera de nitrogênio, a solução de Bu3SnH
e quantidade catalítica de AIBN foi adicionada lentamente (8 horas) e a concentração final do
substrato foi de 2,5 mmol/L (HITCHCOCK; PATTENDEN, 1992).
O
O
O
O
O
O
Br14-Endo
O
OOH
HO
OH
Zearalenona
Bu3SnH, AIBN
Benzeno, 80 oC
61%
2,5 mmol/L
Figura 29 – Síntese de macrolactona precursora da micotoxina zearalenona.
A síntese de cetonas macrocíclicas de 14 membros de configuração E e Z (FIGURA 30) foi
descrita por Cox e colaboradores. A reação do iodeto alílico (3 mmol/L em benzeno) com
Bu3SnH/AIBN levou, por ciclização 14-endo, às duas trienonas com 52% de rendimento e
proporção de 3:1 entre os isômeros E:Z (COX et al., 1992).

INTRODUÇÃO
24
O
I
O
+
O
Bu3SnH, AIBN
Benzeno, refluxo 3 h
14-Endo (E) 14-Endo (Z)
Figura 30 – Síntese de trienonas cíclicas de 14 membros.
Robertson e colaboradores observaram diferenças na ciclização de iodoenonas cujos substituintes
envolvidos na reação de ciclização apresentam relação cis e trans (FIGURA 31), sugerindo
estereosseletividade para reações de macrociclização radicalar. Além de o rendimento do
macrociclo proveniente da iodoenona trans ter sido mais baixo, as condições empregadas
precisaram ser de maior diluição (5 mmol/L para enona cis, 2 mmol/L para enona trans) e maior
tempo de adição do Bu3SnH (6 horas para enona cis, 10 horas para enona trans). O macrociclo
derivado da iodoenona trans sofreu reação de isomerização com a sílica empregada no processo
de purificação e o produto isolado diferiu daquele identificado por RMN no bruto da reação
(ROBERTSON et al., 1997).
I
O O
SiO2
O
13-Endo37%
13-Endo
I
OO
Bu3SnH, AIBN
Benzeno, refluxo
2 mmol/L
Bu3SnH, AIBN
Benzeno, refluxo
5 mmol/L
51%
Figura 31 – Macrociclização de iodoenonas cis e trans.
A síntese de macrolactonas e éteres macrocíclicos a partir de ω-iodopolioxaalquilacrilatos e
compostos relacionados (FIGURA 32) foi relatada por Beckwith e colaboradores que empregaram
adição lenta de hidreto de tri-n-butilestanho e AIBN sobre o acrilato em benzeno, sob refluxo. Os
substratos b-f, k e l levaram a macrociclos provenientes exclusivamente da ciclização endo. Os
precursores g-j, com um substituinte que ativa o ataque exo, conduziram a produtos de ciclização
endo e exo. Foram determinadas as constantes de ciclização dos substratos b e c que são,

INTRODUÇÃO
25
respectivamente, 30 e 10 vezes maiores que as de seus análogos contendo carbono em
substituição aos oxigênios da cadeia. Os autores atribuíram as diferenças de velocidade de
ciclização a uma menor tensão existente no estado de transição dos substratos contendo oxigênio
ao invés de carbono (BECKWITH et al., 1997).
Substrato Produto Tamanho do anel Rendimento (%)
a - - - b Endo 12 78 c Endo 15 72 d Endo 18 70 e Endo 21 63 f Endo 24 30 g Endo/Exo 12/11 37/21 h Endo/Exo 12/11 43/57 i Endo/Exo 15/14 36/36 j Endo/Exo 15/14 29/29 k Endo 12 64 l Endo 15 87
Figura 32 – Síntese de macrolactonas e éteres macrocíclicos por reação radicalar.
Reação de macrociclização-transanulação com Bu3SnH/AIBN foi descrita empregando-se o
precursor iodotrienona (FIGURA 33) na concentração de 3 mmol/L, em benzeno, sob refluxo e
adição lenta de 1,1 equivalentes de Bu3SnH e quantidade catalítica de AIBN. O radical resultante
da ciclização radicalar 13-endo sofreu, em seguida, ciclizações 5 e 6-exo (FIGURA 33) (BEGLEY
et al., 1996).
+Bu3SnH, AIBN
Benzeno, refluxo
R OO
OI
O
( )n
a) n=0, R=Hb) n=1, R=Hc) n=2, R=Hd) n=3, R=He) n=4, R=Hf) n=5, R=H
g) n=1, R=E-CO2Eth) n=1, R=Z-CO2Eti) n=2, R=E-CO2Etj) n=2, R=Z-CO2t-Bu
)n
Endo
O O
O
O
R
( (
Exo
)n
O O
O
OCH2R
Benzeno, refluxo
Bu3SnH, AIBN( )
nO
OO
I
CO2Et
k) n=1l) n=2 Endo
)( n
O O
O
CO2Et

INTRODUÇÃO
26
.Bu3SnH, AIBN
Benzeno, refluxo 3 h
I
OO
O
13-Endo
5-Exo, 6-Exo
Figura 33 – Ciclizações radicalares de iodotrienona.
Shea e colaboradores sintetizaram macrociclos de 22 membros pelo tratamento de biscetais
derivados do estireno com hidreto de tri-n-butilestanho, em benzeno, sob refluxo (FIGURA 34). O
radical nos substratos é gerado por ataque do radical tributilestanila ao carbono mais externo de
um dos grupos alquenila e o centro radicalar formado promove ataque endo à ligação dupla da
outra unidade do biscetal (SHEA et al., 1992).
O OO O
Ar
O OO O
Ar
Bu3Sn
Ar = 74%
Ar = 54%
Bu3SnH
Benzeno, refluxo
Figura 34 – Síntese de macrociclos de 22 membros.
Baldwin e colaboradores desenvolveram reações de carbociclização radicalar mediada por
Bu3SnH utilizando 2-(tri-n-butilestanilmetilpropanoatos) de ω-fenilselenoalquila como substratos
(FIGURA 35). Os autores consideraram que os critérios estruturais que favorecem a
macrociclização endo estavam atendidos no substrato: o carbono mais interno da ligação dupla
encontra-se impedido estericamente, a função éster ativa eletronicamente o ataque endo do
radical alquila e o grupo tributilestanila possibilita a formação do radical tributilestanila in situ pela
fragmentação da ligação C-Sn, o que permite usar quantidades catalíticas de Bu3SnH e evita a

INTRODUÇÃO
27
formação dos produtos de redução indesejáveis. Além disso, a presença de fenilselênio em
substituição a um halogênio no precursor facilita a purificação dos produtos, uma vez que
tributilestanhofenilselenetos são mais fáceis de separar do que os análogos halogenados. Deste
modo, utilizando quantidades catalíticas de Bu3SnH e de AIBN, alta diluição (5 mmol/L), benzeno
como solvente e refluxo foram obtidas macrolactonas provenientes de ciclização endo (FIGURA
35) com bons rendimentos e não se formaram produtos de ciclização exo e de hidrogenólise
(BALDWIN et al., 1991).
Bu3SnH (cat), AIBN (cat)
Benzeno, refluxo5 mmol/L
Bu3SnBu3SnSePh
Bu3Sn O
O
( )n Endo
Bu3Sn O
O
( )n
O
O
( )n
Bu3Sn O
O
SePh
( )n
n Tamanho do anel Rendimento (%)
5 10 54
6 11 46
7 12 61
8 13 50
9 14 80
10 15 72
Figura 35 – Mecanismo da formação de macrolactonas a partir de 2-(tri-n-
butilestanilmetilpropanoatos) de ω-fenilselenoalquila.
Mesmo tendo conhecimento de que a formação de “macrociclos pequenos” (10-12 membros) por
reação radicalar é desfavorecida quando comparada com a formação de ciclos maiores, Lamas e
colaboradores utilizaram a reação radicalar mediada por hidreto de tri-n-butilestanho para obter
precursores do alcalóide lenoxamina a partir de substratos arilbromoacetilenos (FIGURA 36). As
benzomacrolactamas de configuração E e Z resultantes de ciclização 10-endo foram obtidas com
rendimentos de 45% e 60% e os autores atribuíram o sucesso da reação de ciclização ao fato de
que os arilbromoacetilenos apresentam restrição conformacional que favorece a ciclização
(LAMAS et al., 1992).
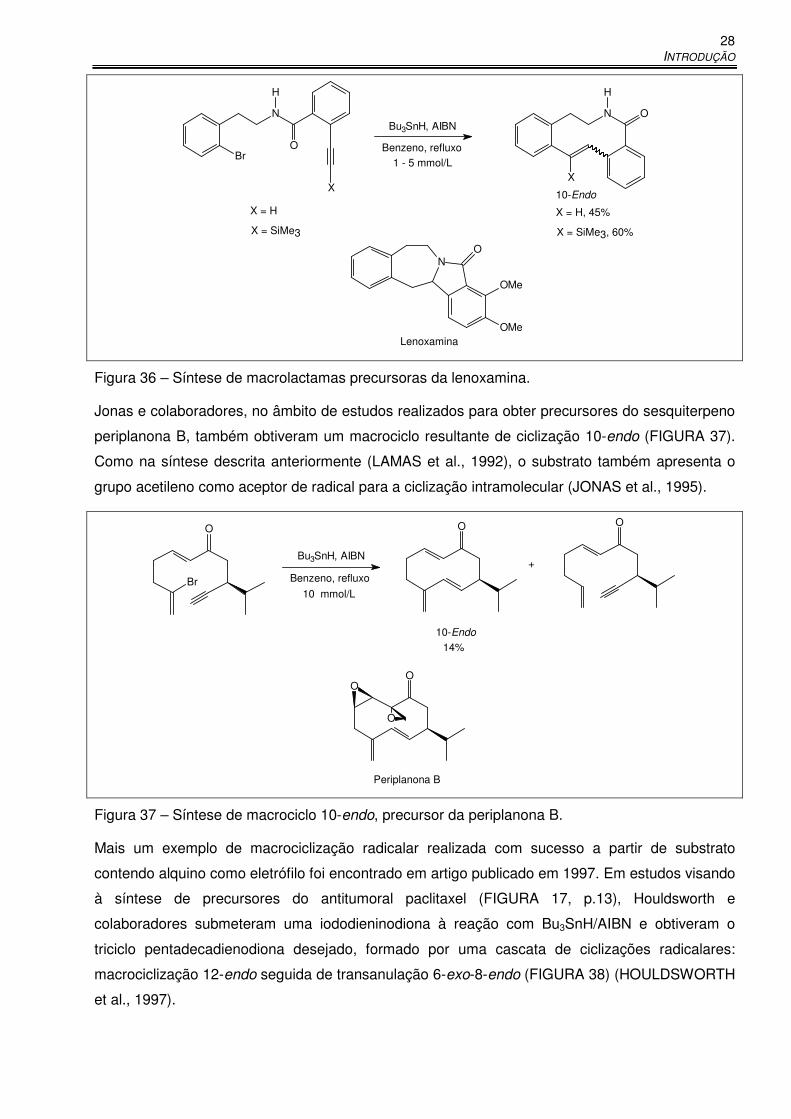
INTRODUÇÃO
28
Br
N
H
O
X
N
H
O
X
10-Endo
OMe
OMe
NO
Lenoxamina
Bu3SnH, AIBN
Benzeno, refluxo1 - 5 mmol/L
X = H
X = SiMe3
X = H, 45%
X = SiMe3, 60%
Figura 36 – Síntese de macrolactamas precursoras da lenoxamina.
Jonas e colaboradores, no âmbito de estudos realizados para obter precursores do sesquiterpeno
periplanona B, também obtiveram um macrociclo resultante de ciclização 10-endo (FIGURA 37).
Como na síntese descrita anteriormente (LAMAS et al., 1992), o substrato também apresenta o
grupo acetileno como aceptor de radical para a ciclização intramolecular (JONAS et al., 1995).
10-Endo
Br
O O
+
O
OO
O
Periplanona B
10 mmol/L
Bu3SnH, AIBN
Benzeno, refluxo
14%
Figura 37 – Síntese de macrociclo 10-endo, precursor da periplanona B.
Mais um exemplo de macrociclização radicalar realizada com sucesso a partir de substrato
contendo alquino como eletrófilo foi encontrado em artigo publicado em 1997. Em estudos visando
à síntese de precursores do antitumoral paclitaxel (FIGURA 17, p.13), Houldsworth e
colaboradores submeteram uma iododieninodiona à reação com Bu3SnH/AIBN e obtiveram o
triciclo pentadecadienodiona desejado, formado por uma cascata de ciclizações radicalares:
macrociclização 12-endo seguida de transanulação 6-exo-8-endo (FIGURA 38) (HOULDSWORTH
et al., 1997).

INTRODUÇÃO
29
I
OO
Bu3SnH, AIBN
Benzeno, refluxo
45-60%
OH
OH
6-Exo, 8-Endo
O
O .
12-Endo
Figura 38 – Síntese de precursor do paclitaxel.
Rodríguez e colaboradores sintetizaram os estereoisômeros E e Z de uma macrolactama de 11
membros (FIGURA 39), por meio da adição intramolecular de um radical arila ao grupo
trimetilsililacetileno. A concentração inicial da fenilacetamida precursora em benzeno foi de 7 mmol/L
e a solução de Bu3SnH/AIBN em benzeno foi adicionada sob refluxo (RODRÍGUEZ et al., 1999).
11-Endo
Bu3SnH, AIBN
Benzeno, refluxo
7 mmol/L
MeO
MeO Br
N
H
O
Me3Si
N
H
O
MeO
MeO
SiMe3
E - 50%Z - 25%
Figura 39 – Síntese de macrolactamas estereoisoméricas por carbociclização radicalar.
Reações de macrociclização com radicais acila foram primeiramente estudadas por Boger e
Mathvink. Uma série de ω-fenilselenoésteres acrilatos conduziram a macrociclos de 11 a 20
membros (FIGURA 40), com bons rendimentos, quando tratados com Bu3SnH e AIBN em
condições de alta diluição. Não foram identificados produtos de redução direta do radical acila ou
de redução do radical alquila produzido por descarbonilação do radical acila (BOGER;
MATHVINK, 1990).

INTRODUÇÃO
30
O
O
(CH2)n
O
SePh
Bu3SnH, AIBN
Benzeno, refluxo5-6 mmol/L
O
O
(CH2)n
O
Endo
n Tamanho do anel Rendimento (%)
2 11 47 3 12 46 5 14 55 7 16 68 11 20 57
Figura 40 – Macrolactonas sintetizadas a partir de ω-fenilselenoésteres.
Em 1992, Astley e Pattenden utilizaram um substrato com características estruturais semelhantes
aos utilizados por Boger e Mathvink para sintetizar o anel furanocembranóide encontrado na
lofotoxina, uma potente substância neurotóxica isolada de corais. O ataque do radical acila gerado
a partir do derivado fenilselenoéster conduziu ao macrociclo proveniente de ciclização 14-endo
com 40% de rendimento (FIGURA 41) (ASTLEY; PATTENDEN, 1992).
14-Endo
PhSeO
O
OO
O
Bu3SnH, AIBN
Benzeno, refluxo
40%
TsOH, CHCl3
O
O
O
OO
OAc
Lofotoxina
Refluxo
50%
Figura 41 – Síntese do anel furanocembranóide presente na lofotoxina.
As ciclizações radicalares mediadas por Bu3SnH envolvendo o ataque intramolecular de radicais
arila a um grupo alquenila vêm sendo amplamente utilizadas para a construção de ciclos fundidos
a anéis aromáticos (BECKWITH; GARA, 1975; BOWMAN et al.,1988; JONES; WILKINSON, 1992;
GHOSH; GHATAK, 1995; GIBSON et al., 1997; ISHIBASHI et al., 1997; ROSA et al., 1997;
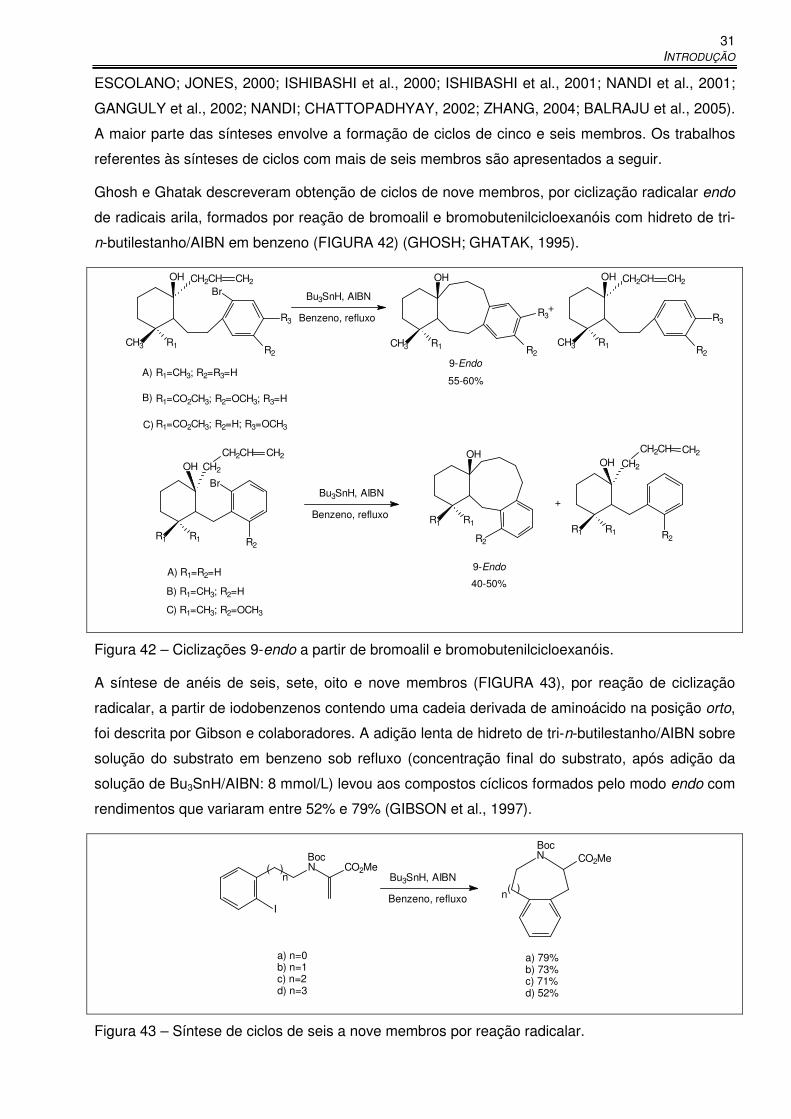
INTRODUÇÃO
31
ESCOLANO; JONES, 2000; ISHIBASHI et al., 2000; ISHIBASHI et al., 2001; NANDI et al., 2001;
GANGULY et al., 2002; NANDI; CHATTOPADHYAY, 2002; ZHANG, 2004; BALRAJU et al., 2005).
A maior parte das sínteses envolve a formação de ciclos de cinco e seis membros. Os trabalhos
referentes às sínteses de ciclos com mais de seis membros são apresentados a seguir.
Ghosh e Ghatak descreveram obtenção de ciclos de nove membros, por ciclização radicalar endo
de radicais arila, formados por reação de bromoalil e bromobutenilcicloexanóis com hidreto de tri-
n-butilestanho/AIBN em benzeno (FIGURA 42) (GHOSH; GHATAK, 1995).
CH3 R1
OH
R2
R3
A) R1=CH3; R2=R3=H
R1=CO2CH3; R2=OCH3; R3=H
R1=CO2CH3; R2=H; R3=OCH3
CH3 R1
OH CH2CH
R2
R3
BrCH2
9-Endo
Bu3SnH, AIBN
Benzeno, refluxo+
CH3 R1
OH CH2CH CH2
R2
R3
55-60%
40-50%
9-EndoA) R1=R2=H
B) R1=CH3; R2=H
C) R1=CH3; R2=OCH3
R1 R1
OH CH2
R2
CH2CH CH2
+
R1 R1
OH
R2
Bu3SnH, AIBN
Benzeno, refluxo
R1 R1
OH CH2
R2
CH2CH
Br
CH2
C)
B)
Figura 42 – Ciclizações 9-endo a partir de bromoalil e bromobutenilcicloexanóis.
A síntese de anéis de seis, sete, oito e nove membros (FIGURA 43), por reação de ciclização
radicalar, a partir de iodobenzenos contendo uma cadeia derivada de aminoácido na posição orto,
foi descrita por Gibson e colaboradores. A adição lenta de hidreto de tri-n-butilestanho/AIBN sobre
solução do substrato em benzeno sob refluxo (concentração final do substrato, após adição da
solução de Bu3SnH/AIBN: 8 mmol/L) levou aos compostos cíclicos formados pelo modo endo com
rendimentos que variaram entre 52% e 79% (GIBSON et al., 1997).
Bu3SnH, AIBN
Benzeno, refluxo
nN CO2Me
I
Boc( )
a) n=0b) n=1c) n=2d) n=3
(
N CO2Me
)n
Boc
a) 79%b) 73%c) 71%d) 52%
Figura 43 – Síntese de ciclos de seis a nove membros por reação radicalar.
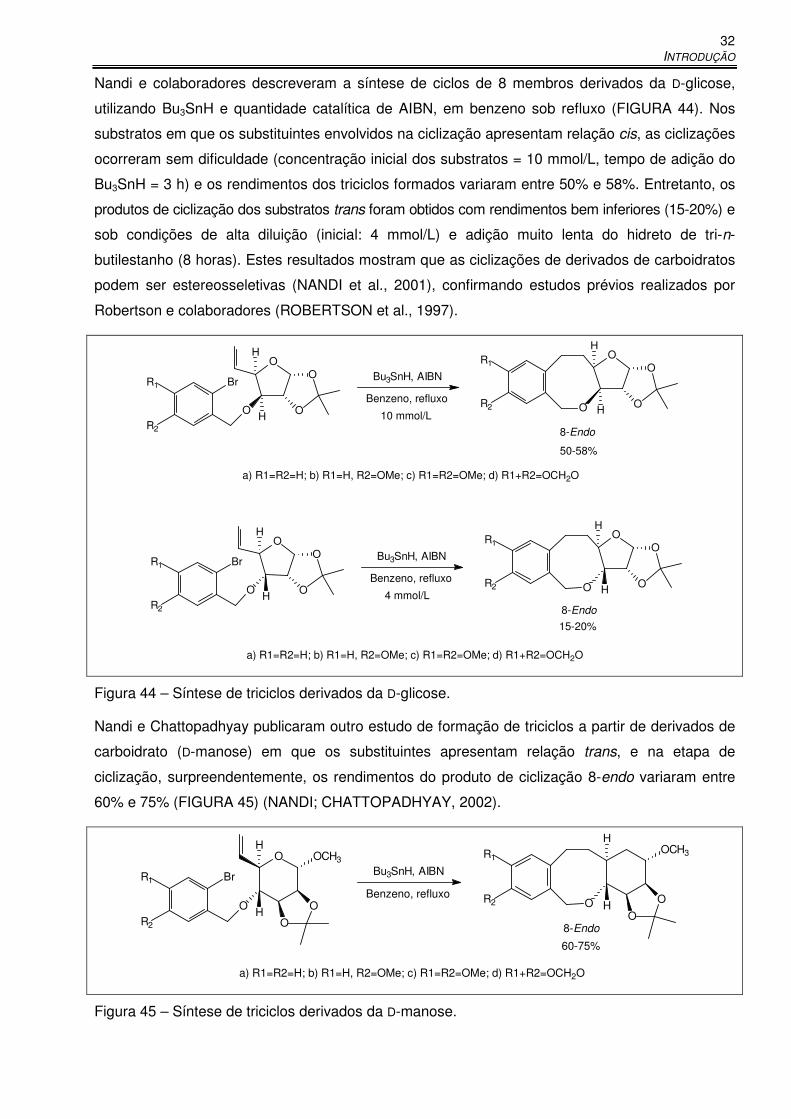
INTRODUÇÃO
32
Nandi e colaboradores descreveram a síntese de ciclos de 8 membros derivados da D-glicose,
utilizando Bu3SnH e quantidade catalítica de AIBN, em benzeno sob refluxo (FIGURA 44). Nos
substratos em que os substituintes envolvidos na ciclização apresentam relação cis, as ciclizações
ocorreram sem dificuldade (concentração inicial dos substratos = 10 mmol/L, tempo de adição do
Bu3SnH = 3 h) e os rendimentos dos triciclos formados variaram entre 50% e 58%. Entretanto, os
produtos de ciclização dos substratos trans foram obtidos com rendimentos bem inferiores (15-20%) e
sob condições de alta diluição (inicial: 4 mmol/L) e adição muito lenta do hidreto de tri-n-
butilestanho (8 horas). Estes resultados mostram que as ciclizações de derivados de carboidratos
podem ser estereosseletivas (NANDI et al., 2001), confirmando estudos prévios realizados por
Robertson e colaboradores (ROBERTSON et al., 1997).
8-Endo
OO
OH
H
O
R1
R2
OO
O
H
O
R1
R2 H
15-20%
Bu3SnH, AIBN
Benzeno, refluxo
4 mmol/L
a) R1=R2=H; b) R1=H, R2=OMe; c) R1=R2=OMe; d) R1+R2=OCH2O
OO
O
H
HO
BrR1
R2 8-Endo
OO
OH
H
O
BrR1
R2
a) R1=R2=H; b) R1=H, R2=OMe; c) R1=R2=OMe; d) R1+R2=OCH2O
Bu3SnH, AIBN
Benzeno, refluxo
10 mmol/L
50-58%
Figura 44 – Síntese de triciclos derivados da D-glicose.
Nandi e Chattopadhyay publicaram outro estudo de formação de triciclos a partir de derivados de
carboidrato (D-manose) em que os substituintes apresentam relação trans, e na etapa de
ciclização, surpreendentemente, os rendimentos do produto de ciclização 8-endo variaram entre
60% e 75% (FIGURA 45) (NANDI; CHATTOPADHYAY, 2002).
a) R1=R2=H; b) R1=H, R2=OMe; c) R1=R2=OMe; d) R1+R2=OCH2O
8-EndoH
O
BrR1
R2
O OCH3
O
O
H
O
R1
R2 H
HOCH3
O
O
60-75%
Bu3SnH, AIBN
Benzeno, refluxo
Figura 45 – Síntese de triciclos derivados da D-manose.
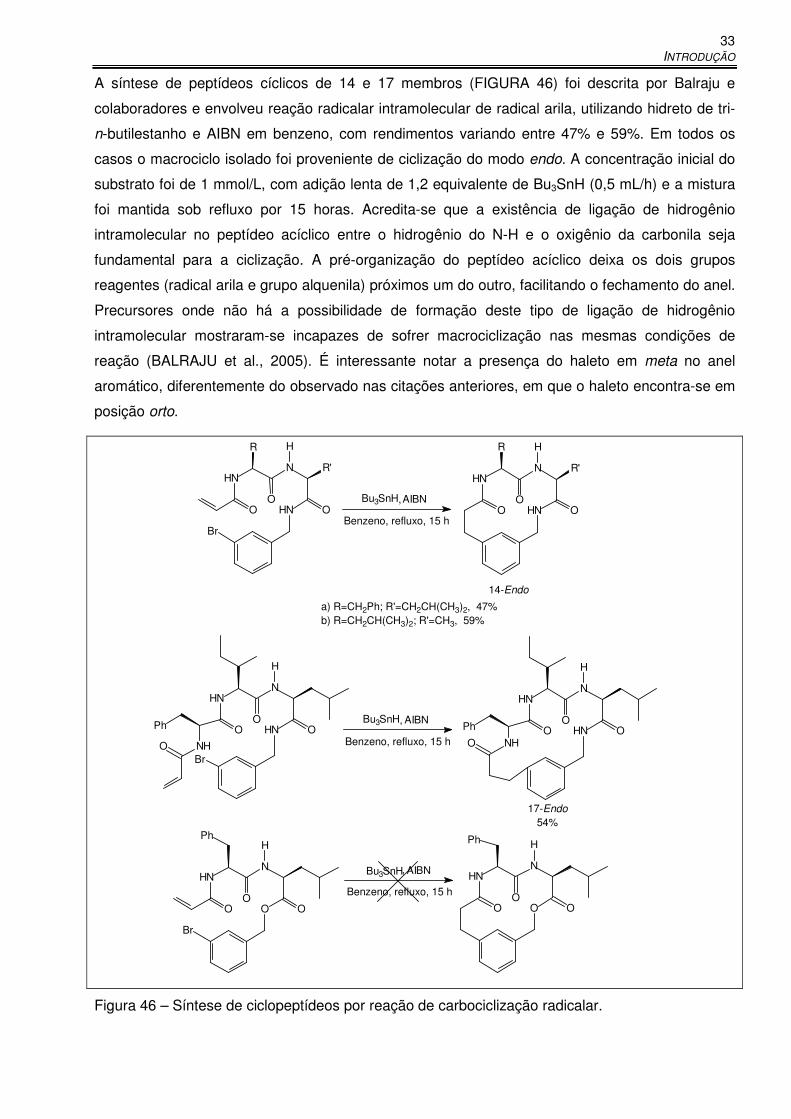
INTRODUÇÃO
33
A síntese de peptídeos cíclicos de 14 e 17 membros (FIGURA 46) foi descrita por Balraju e
colaboradores e envolveu reação radicalar intramolecular de radical arila, utilizando hidreto de tri-
n-butilestanho e AIBN em benzeno, com rendimentos variando entre 47% e 59%. Em todos os
casos o macrociclo isolado foi proveniente de ciclização do modo endo. A concentração inicial do
substrato foi de 1 mmol/L, com adição lenta de 1,2 equivalente de Bu3SnH (0,5 mL/h) e a mistura
foi mantida sob refluxo por 15 horas. Acredita-se que a existência de ligação de hidrogênio
intramolecular no peptídeo acíclico entre o hidrogênio do N-H e o oxigênio da carbonila seja
fundamental para a ciclização. A pré-organização do peptídeo acíclico deixa os dois grupos
reagentes (radical arila e grupo alquenila) próximos um do outro, facilitando o fechamento do anel.
Precursores onde não há a possibilidade de formação deste tipo de ligação de hidrogênio
intramolecular mostraram-se incapazes de sofrer macrociclização nas mesmas condições de
reação (BALRAJU et al., 2005). É interessante notar a presença do haleto em meta no anel
aromático, diferentemente do observado nas citações anteriores, em que o haleto encontra-se em
posição orto.
Br
O
N
H
O
HN
O O
Ph
O
N
H
O
HN
O O
Ph
a) R=CH2Ph; R'=CH2CH(CH3)2, 47%b) R=CH2CH(CH3)2; R'=CH3, 59%
Br
HN
N
H
O
HN
R
R'
O O
HN
N
H
O
HN
O O
NHOBr
Ph
AIBN,Bu3SnH
Benzeno, refluxo, 15 h
Benzeno, refluxo, 15 h
Bu3SnH AIBN,HN
N
H
O
HN
R
R'
O O
14-Endo
54%
Benzeno, refluxo, 15 h
Bu3SnH AIBN,
17-Endo
HN
N
H
O
HN
O ONHO
Ph
Figura 46 – Síntese de ciclopeptídeos por reação de carbociclização radicalar.

INTRODUÇÃO
34
Com o intuito de se verificar o papel de carboidratos e de sua estereoquímica em
macrociclizações mediadas por hidreto de tri-n-butilestanho, assim como as influências dos
grupos envolvidos diretamente na reação de ciclização e de suas posições relativas na unidade
sacarídica, o grupo do Laboratório de Química Farmacêutica da Faculdade de Farmácia/UFMG,
em parceria com o Departamento de Química do ICEx/UFMG (QF/DQ/UFMG) vem
desenvolvendo projetos de pesquisa relacionados à síntese de macrociclos a partir de
carboidratos.
O estímulo para o desenvolvimento deste trabalho é o desafio de sintetizar macrociclos,
reconhecidamente um dos grandes problemas da síntese orgânica (BLANKENSTEIN; ZHU,
2005), aliado à possibilidade de obter substâncias com atividade biológica. Especialmente as
benzomacrolactamas derivadas de carboidratos, maioria das substâncias sintetizadas no âmbito
deste programa de pesquisa, são potenciais agentes biológicos, pois tratam-se de macrociclos,
cujas atividades biológicas são em parte atribuídas à maior restrição conformacional em relação
aos compostos de cadeia aberta (RAMASESHAN et al., 2000; STACHEL et al., 2006), além de
apresentarem quiralidade (conferida pela presença da unidade sacarídica), anel aromático e
grupo amida. Carey e colaboradores relatam que, de 128 candidatos a fármacos que se
encontram em desenvolvimento em três grandes indústrias farmacêuticas, mais de 90% contém
anel aromático, mais de 90% apresenta átomo de nitrogênio, 69 substâncias (54%) são quirais e
que em 27 (74%) a quiralidade é proveniente do material de partida usado na síntese. Além disso,
12% das reações realizadas para a síntese das 128 substâncias são de acilação, especialmente
N-acilações (CAREY et al., 2006).
Uma parte do programa de síntese de macrociclos desenvolvido pelo grupo de pesquisa
QF/DQ/UFMG envolveu a síntese de aliloxi-orto-iodobenzamidas e reação com Bu3SnH. Na Figura 47
podem-se observar as condições de reação e os rendimentos dos produtos isolados das reações
de carbociclização radicalar desenvolvidas com cinco aliloxi-orto-iodobenzamidas (PRADO et al.,
2000; BINATTI et al, 2002; DIAS et al., 2006; PIRES et al., 2006).

INTRODUÇÃO
35
151413
11-Endo
O
OCH3
OBn
BnO
ON O
H
O
OCH3
OBnBnO
NH
O
O
I
33%
O
OCH3
OBnBnO
NO
OH
+
22%
Bu3SnH, AIBN
Benzeno, refluxo
BINATTI et al., 2002
+
11-Endo17% 48%
Benzeno, refluxo
Bu3SnH, AIBN
DIAS et al., 2006
O
O
OCH3
OBnBnO
N
H
O
IO
O
OCH3
OBnBnO
N
H
O
O
O
OCH3
OBn
BnO
N
H
O
11-Endo
8%
+
30%
Bu3SnH, AIBN
Benzeno, refluxo
PIRES et al., 2006
O
OCH3
OBnBnO
ON
H
OO
OCH3
OBnBnO
ON
H
O
I
O
OCH3
OBnBnO
ON
H
O
121110
987
654
I
N
OH
O
N
OH
O
N
OH
O
85%14%
+Bu3SnH, AIBN
Benzeno, refluxo
11-Endo
321
N
HO
O
OCH3
OBnBnO
O
I
N
HO
O
OCH3
OBnBnO
O+ N
HO
O
OCH3
OBnBnO
O
11-Endo
40% 42%
Peróxido de Benzoíla
Benzeno, refluxo
Bu3SnH
PRADO et al., 2000
Figura 47 – Macrolactamas obtidas por reação de aliloxi-orto-iodobenzamidas com Bu3SnH.
Inicialmente, a o-iodobenzamida 1 derivada do α-D-glicopiranosídeo de metila foi submetida à
reação de ciclização radicalar e conduziu à macrolactama 11-endo 2 com 40% de rendimento. A
macrolactama 5 foi obtida pela reação de ciclização radicalar 11-endo da o-iodobenzamida 4,
desprovida de unidade sacarídica, com 14% de rendimento (PRADO et al., 2000). A formação dos
macrociclos pelo modo endo já era prevista uma vez que Porter e colaboradores verificaram que

INTRODUÇÃO
36
as macrociclizações endo são preferenciais em relação às exo (PORTER et al., 1986, PORTER;
CHANG, 1987; PORTER et al., 1988). Além de se confirmar a preferência de ciclização pelo modo
endo, o rendimento superior da macrolactama 2 em relação à lactama desprovida da unidade
sacarídica (5), indicou que a presença do ciclo de seis membros do anel piranosídico presente em
1 favoreceu a ciclização, possivelmente por conferir restrição conformacional ao substrato
(PRADO et al., 2000). Em um estudo subseqüente, a reação de ciclização radicalar foi
desenvolvida com a benzamida de configuração galacto 7, epímero de 1. Pretendia-se verificar a
importância da configuração do açúcar na reação de macrociclização radicalar. Foi obtida a
macrolactama 8, proveniente da ciclização 11-endo com 33% de rendimento, o que indicou que a
configuração de C-4 não interfere no modo de ciclização e nem, significativamente, no rendimento
do produto de macrociclização (glico 40% versus galacto 33%) (BINATTI et al., 2002).
Para se verificar a importância da posição relativa dos grupos 2-iodobenzoilamino e O-aliloxila no
carboidrato para a macrocilização radicalar, sintetizaram-se as benzamidas 10 e 13, isômeros de
posição de 1 e 7, respectivamente. Da reação radicalar desenvolvida com a o-iodobenzamida 10
foi obtido o macrociclo 11 proveniente de ciclização 11-endo com 17% de rendimento (versus 40%
de 2), indicando a importância da posição relativa dos grupos O-aliloxila e 2-iodobenzoilamino na
porção sacarídica da iodobenzamida de configuração glico (DIAS et al, 2006). O mesmo foi
verificado quando se inverteram as posições relativas dos dois grupos no derivado de
configuração galacto: o rendimento da benzomacrolactama 11-endo 14 foi de 8% (PIRES et al.,
2006), versus 33% de 8 (BINATTI et al., 2002).
Duas outras o-iodobenzamidas sem unidade sacarídica e com grupo aliloxila como eletrófilo, 16 e
18, foram submetidas à reação de ciclização radicalar para obtenção de macrolactamas (FIGURA
48). O substrato 18 levou à macrolactama de 12 membros (rendimento de 19%), mas da reação
de 16, que poderia conduzir a um ciclo de 10 membros pelo modo endo, só foi obtido o produto de
hidrogenólise 17. Estes resultados confirmaram que a ciclização pelo modo endo é preferencial
em relação ao modo exo. Além disso, considerando que as benzamidas 4 (FIGURA 47, p. 35) e
18, homólogas de 16, conduziram a produtos de ciclização endo, pode-se supor que as
macrociclizações 11- e 12-endo sejam mais favorecidas que a 10-endo (FARACO et al., 2003).

INTRODUÇÃO
37
16 17
76%
+ 10 -EndoBu3SnH, AIBN
Benzeno, refluxoI
N
O
H
ON
O
H
O
18 19 20
33%
I
N
O
H O
Bu3SnH, AIBN
Benzeno, refluxo+
N
O
H O
N
O H
O
19%
12-Endo
Figura 48 – orto-Iodobenzamidas sem unidade sacarídica submetidas à reação de ciclização
radicalar e respectivos resultados.
Na expectativa de que a presença de uma unidade de carboidrato pudesse conferir restrição
conformacional ao substrato e favorecer a ciclização 10-endo, sintetizou-se a aliloxi-o-
iodobenzamida 21 (FIGURA 49). Da reação de 21 com Bu3SnH foi isolada a dilactama macrocíclia
de 20 membros 22, formada por ataque intermolecular do radical arila ao carbono terminal do
alqueno, seguido de macrociclização 20-endo (FARACO et al., 2004). Embora a ciclização 10-
endo não tenha ocorrido na reação radicalar da benzamida 21 de configuração glico em que os
grupos O-alil e 2-iodobenzoilamino encontram-se nos carbonos 2 e 3, considerando que foi obtida
uma macrolactama dimérica de 20 membros (22), optou-se por submeter duas aliloxi-orto-
iodobenzamidas análogas de 21 à reação radicalar: 24 e 26, isômero de posição (O-alil em C-2 e
2-iodobenzoilamino em C-3) e diastereoisômero (configuração altro) de 21, respectivamente
(FIGURA 49). Das reações radicalares foram isolados apenas os respectivos produtos reduzidos
não ciclizados 25 e 27 (BINATTI, 2005a). A princípio estes resultados confirmariam a dificuldade
de ocorrência de ciclização 10-endo, que não ocorreu mesmo na presença das unidades de
açúcar nos substratos. Vale observar, no entanto, que os substituintes envolvidos na ciclização
apresentam relação trans, o que pode ter contribuído para o insucesso das ciclizações, além da
reconhecida dificuldade de formação de ciclos de 10 membros. Embora haja relatos de ciclização
realizada com sucesso em derivados de carboidratos com substituintes trans (PRADO et al., 2000;
NANDI; CHATTOPADHYAY, 2002; DIAS et al., 2006), em outros substratos observou-se
estereosseletividade nas ciclizações, que foram mais favorecidas nos substratos com substituintes
cis (NANDI et al., 2001).

INTRODUÇÃO
38
21 22 23
+
27%
Benzeno, refluxo
Bu3SnH, AIBN
O
OCH3
NHO
BnO
O
I
BnO
O
OCH3
NHO
BnO
O
BnO
40%
O
HN
OOBn
OBn
ONH
O
OCH3
BnO
BnO
OCH3
O
O
Benzeno, refluxo
Bu3SnH, AIBN
26
BnOO
OCH3
BnO
O
NH
O
I
27
BnOO
OCH3
BnO
O
NH
O
19%
Benzeno, refluxo
Bu3SnH, AIBN
24
BnOO
OCH3
ONH
BnO
O
I
BnOO
OCH3O
NH
BnO
O
2538%
Figura 49 – orto-Iodobenzamidas e produtos de reação com Bu3SnH.
Com o objetivo de avaliar as possíveis diferenças das ciclizações radicalares entre as aliloxi-o-
iodobenzamidas 1 e 7, que conduziram às benzomacrolactamas 11-endo 2 (40%) e 8 (33%)
(FIGURA 47, p. 35), e seus análogos contendo oxigênio em substituição ao NH, os aliloxi-o-
iodobenzoatos 28 e 30 (FIGURA 50) foram sintetizados e submetidos à reação de ciclização
radicalar. Da reação do benzoato de configuração glico 28 não foi isolado produto de ciclização e
só foi isolado o produto não ciclizado 29. O substrato de configuração galacto 30 (500 mg)
conduziu a 30 mg de uma mistura contendo a macrolactama 11-endo 31 e ao produto de
hidrogenólise 32 (FIGURA 50). As diferenças observadas foram atribuídas à menor restrição
conformacional em 28 e 30 em relação a 1 e 7, tanto pelo fato de as rotações serem mais restritas
nas amidas que nos ésteres, quanto pela possível formação em 1 e 7 de uma ligação de
hidrogênio entre o hidrogênio amídico e o oxigênio do grupo aliloxila (BINATTI et al., 2005b).

INTRODUÇÃO
39
+
30
O
OCH3
OBnBnO
O
O
O
I
Bu3SnH, AIBN
Benzeno, refluxo
O
O
O
OCH3
OBnBnO
O
29
30%
O
O
O
OCH3
OBnBnO
O
I
28
Bu3SnH, AIBN
Benzeno, refluxoO
OCH3
OBnBnO
O
O
O
19%
32traços
11-Endo31
O
OCH3
OBn
BnO
O
O
O
Figura 50 – Aliloxi-orto-iodobenzoatos e seus produtos da reação com Bu3SnH.
Em outros trabalhos realizados foram sintetizados dois amidoésteres (33 e 35) e quatro
amidoéteres (37, 39, 40 e 41) contendo o grupo cinamila como alvo de ataque do radical arila
(FIGURA 51), diferentemente dos trabalhos até então realizados, em que os substratos eram
derivados alílicos. Esperava-se que quando submetidos à reação com Bu3SnH os substratos
cinamilados levassem a macrociclos 11-endo, em geral mais favorecidos (PORTER; CHANG,
1987), e/ou 10-exo. A ciclização 10-exo poderia ocorrer tanto pelo fato de os dois carbonos da
ligação dupla serem substituídos, portanto do ponto de vista estérico serem aproximadamente
equivalentes, quanto em função da estabilidade do radical benzílico que seria formado no caso do
ataque 10-exo. Além disso, qualquer que fosse o modo de ciclização, 10-exo ou 11-endo, haveria
formação de um novo centro estereogênico e seria possível verificar a estereosseletividade da
ciclização. Da reação dos amidoésteres derivados do ácido cinâmico 33 e 35 com Bu3SnH foram
isolados as imidas inesperadas 34 e 36 e os produtos de hidrogenólise (FIGURA 51) (OLIVEIRA
et al., 2004). Os amidoéteres 37, 39, 40 e 41 conduziram aos respectivos produtos de
hidrogenólise e da reação desenvolvida com 37 foi isolado também o derivado arilestanho 38
(FIGURA 41) (PINTO; PRADO, 2005; ROCHA et al., 2005; QUEIROGA; PRADO, 2006; OLIVEIRA
et al., 2007). De nenhuma das reações radicalares dos substratos amidoéteres cinamílicos (39,
37, 40 e 41) foram isolados produtos de ciclização, ao contrário do que ocorreu com as
respectivas benzamidas O-aliladas (1, 7, 10 e 13). Estes resultados indicam que o grupo fenila,
por impedimento estérico, dificulta o ataque do radical arila ao carbono mais externo da ligação
dupla e não facilita o ataque exo, reconhecidamente mais difícil (PORTER; CHANG, 1987).

INTRODUÇÃO
40
OLIVEIRA et al., 2004
NH
O
OCH3
OMe
MeO
OPh
O
O
I
O
OCH3
OMe
MeOHO
NO
OH
O
34 11%33
Benzeno, refluxo
Bu3SnH, AIBN
OLIVEIRA et al., 2004
O
OCH3
OMeMeO
HO
N
O
O
36 22%
N
O
OCH3
OMeMeO
O
H
Ph
I
O
O
35
Benzeno, refluxo
Bu3SnH, AIBN
OLIVEIRA et al., 2007
O
OCH3
OBn
BnO
NH
O
O
Bu3Sn
Ph
3829%
Bu3SnH, AIBN
Benzeno, refluxo
O
OCH3
OBn
BnO
NH
O
O
I
Ph
37
PRADO et al., 2000; ROCHA et al., 2005;BINATTI et al., 2002; OLIVEIRA et al., 2007
N
HO
O
OCH3
OBn
BnO
O
I
R
1 R = H, 39 R = Ph
O
OCH3
OBn
BnO
NH
O
O
I
R
7 R = H, 37 R = Ph
DIAS et al., 2006; PINTO; PRADO, 2005;PIRES et al., 2006; QUEIROGA; PRADO, 2006
IO
O
OCH3
OBnBnO
N
H
O
R
10 R = H, 40 R = Ph
O
OCH3
OBnBnO
ON
IH
O
R
13 R = H, 41 R = Ph
Figura 51 – Amidoésteres e amidoéteres submetidos à reação com Bu3SnH.

INTRODUÇÃO
41
Considerando que a aliloxi-o-iodobenzamida 1 conduziu à macrolactama 11-endo 2 com bom
rendimento (40%, FIGURA 47, p. 35) (PRADO et al., 2000) optou-se por estudar a ciclização
radicalar de seu análogo 42 (FIGURA 52), que apresenta o grupo 3-iodopropanoilamino em
substituição ao 2-iodobenzoilamino em C-6. A reação de 42 com Bu3SnH conduziu
majoritariamente ao produto de hidrogenólise e à macrolactama 43, proveniente do ataque 11-
endo do radical alquila à ligação dupla do grupo aliloxila, que foi isolada com rendimento de
apenas 4% (ROCHA; PRADO, 2006). Este resultado, comparado com o da reação radicalar da
benzamida 1, demonstra que o ataque do radical arila é mais favorecido que o do radical alquila,
possivelmente devido à maior liberdade conformacional do grupo alquila.
N
O
OCH3
OBnBnO
O
H
O
I
4%
N
HO
O
OCH3
OBnBnO
O
42
Benzeno, refluxo
Bu3SnH, AIBN
11-Endo43
Figura 52 – Amida alquílica e macrolactama obtida da reação com Bu3SnH.
1.4 NOVAS METODOLOGIAS PARA REAÇÃO DE CICLIZAÇÃO RADICALAR
Uma metodologia que tem sido testada, mas que ainda está pouco desenvolvida, refere-se à
condução de reações radicalares de moléculas pequenas em fase sólida, onde polímeros, como
poliestireno, suportam precursores radicalares (WATANABE et al., 1999; ZHU et al., 1999;
WENDEBORN et al., 2000; CADDICK et al., 2002; CURRAN et al., 2002).
Em função de alguns inconvenientes que há na utilização do hidreto de tri-n-butilestanho, como
toxicidade, dificuldade em separação de resíduos de estanho do produto formado e redução do
radical do substrato antes que ocorra a ciclização (BERGE; ROBERTS, 1979; SALOMON et al.,
2000; CLYNE; ALDABBAGH, 2006), alguns autores têm proposto utilização de reagentes
alternativos para reações radicalares, como salofeno de sódio e cobalto (I), iodeto de samário (II)
(SmI2), tris(trimetilsilil)silano ((Me3Si)3SiH) e tris(trimetilsilil)germano ((Me3Si)3GeH) (BOWMAN et
al., 2002; HARROWVEN et al., 2003).
Outra alternativa que tem sido estudada mais recentemente envolve formação de radicais
induzidos por radiação ultravioleta (gerada, por exemplo, por lâmpada de mercúrio de alta pressão
150 W) para ciclização de radicais arila e piridila, conduzindo a anéis de 5 e 6 membros (PARK et
al., 1997, 2001).

INTRODUÇÃO
42
Clyne e Aldabbagh realizaram um estudo mostrando rendimentos superiores na ciclização
fotoquímica de radicais imidazólicos em relação à reação mediada por hidreto de tri-n-
butilestanho/AIBN para obtenção de ciclos de 6 membros. As principais vantagens da ciclização
fotoquímica são: i) eliminação de resíduos tóxicos de estanho; ii) eliminação do uso de iniciadores
azo (AIBN), que são considerados perigosos; iii) não há necessidade de adição lenta de
Bu3SnH/AIBN, conseqüentemente o tempo total de reação é menor no caso da reação
fotoquímica; iv) procedimento de elaboração e purificação é bem mais simples na reação
fotoquímica, levando a bons rendimentos do produto ciclizado (CLYNE; ALDABBAGH, 2006).
Ainda há poucos relatos de síntese de macrociclos utilizando reações fotoquímicas (GRIESBECK
et al., 2000; YOON et al., 2002), sendo este um campo em aberto para pesquisas que podem ser
promissoras e de grande utilidade para a química orgânica sintética.

JUSTIFICATIVA, OBJETIVOS E PLANO DE SÍNTESE
43
2 JUSTIFICATIVA, OBJETIVOS E PLANO DE SÍNTESE
2.1 JUSTIFICATIVA E OBJETIVOS
Considerando o desafio que constitui a síntese de macrociclos, a possibilidade de obter
substâncias com atividade biológica e a constatação de que o método de escolha para a síntese
de macrociclos por carbociclização radicalar é aquele em que se utiliza o hidreto de tri-n-
butilestanho (Bu3SnH), o grupo de pesquisa do Laboratório de Química Farmacêutica da
Faculdade de Farmácia/UFMG e do Departamento de Química do ICEx/UFMG (QF/DQ/UFMG)
vem desenvolvendo projetos de síntese de macrociclos por reação de carbociclização radicalar
mediada por hidreto de tri-n-butilestanho.
Como foi relatado na Introdução, o grupo de pesquisa QF/DQ/UFMG utilizou, como precursores
dos macrociclos, substâncias contendo carbono ligado a iodo (arílico ou alquílico), no qual seria
formado o radical, e uma ligação dupla carbono-carbono, local do ataque intramolecular do radical
formado inicialmente. As primeiras reações radicalares foram desenvolvidas com iodobenzamidas
derivadas de carboidratos substituídas na posição orto com uma cadeia lateral contendo um grupo
alquenila. Para a escolha deste tipo de precursor foram avaliados os aspectos estruturais que
favoreceriam tanto a ciclização, quanto a atividade biológica dos macrociclos eventualmente
obtidos. A presença da unidade sacarídica se justifica tanto pela premissa de que ela confere
restrição conformacional necessária para a ciclização (BLANKENSTEIN; ZHU, 2005; STACHEL et
al., 2006), quanto pelo fato de que o macrociclo a ser obtido seria quiral, característica de muitos
agentes bioativos (BARREIRO et al., 1997; LIMA, 1997; CAREY et al., 2006). Além disso, anéis
aromáticos e grupos amida são fragmentos estruturais presentes na maioria das moléculas
bioativas (CAREY et al., 2006). Para a síntese das iodobenzamidas utilizaram-se, como materiais
de partida, monossacarídeos disponíveis comercialmente.
Os carboidratos são considerados atrativos como materiais de partida em síntese orgânica, por
serem, em geral, facilmente acessíveis e baratos, além de serem obtidos com pureza
enantiomérica e com configuração absoluta conhecida. Além disso, as hidroxilas destes
compostos poliidroxilados apresentam reatividade diferente, em função de os anéis sacarídicos
adotarem conformações preferenciais, o que possibilita desenvolver transformações
regiosseletivas de suas hidroxilas (BINKLEY, 1988; COLLINS; FERRIER, 1995; FERREIRA,
1995).
Os resultados das reações de carbociclização radicalar até então desenvolvidas pelo grupo
QF/DQ/UFMG, aliados àqueles descritos na literatura, encontram-se relatados na Introdução e
foram utilizados para o planejamento deste trabalho. Citam-se as observações que foram
fundamentais neste planejamento:
� as ciclizações 11- e 12-endo são favorecidas em relação às 10- e 11-exo, respectivamente
(PRADO et al., 2000; FARACO et al., 2003; DIAS et al., 2006; BINATTI et al., 2002);

JUSTIFICATIVA, OBJETIVOS E PLANO DE SÍNTESE
44
� a restrição conformacional imposta pela presença de um ciclo no substrato, como o da
unidade de açúcar, favorece a reação de ciclização (PRADO et al., 2000);
� a configuração de C-4 do açúcar (glico ou galacto) não afeta o modo de ciclização e nem,
significativamente, o rendimento do produto ciclizado (BINATTI et al., 2002);
� a ciclização 12-endo ocorre mesmo que o substrato não contenha a porção sacarídica
(FARACO et al., 2003);
� a reação de ciclização é mais favorecida quando o grupo 2-iodobenzoilamino encontra-se
ligado ao C-6 do açúcar e o alqueno encontra-se no substituinte de C-4 do que no caso de os
dois grupos estarem em posições trocadas (DIAS et al., 2006; PIRES et al., 2006);
� a presença de um grupo fenila em substituição a um hidrogênio no carbono terminal do
alqueno prejudica (ou impede) a ciclização em substratos contendo um anel piranosídico
(PINTO; PRADO, 2005; ROCHA et al., 2005; QUEIROGA; PRADO, 2006; OLIVEIRA et al.,
2007);
� a ciclização é mais favorecida quando os radicais arila encontram-se orto ao grupo amida do
que ao grupo éster, em substratos que contém açúcar em suas estruturas (BINATTI et al.,
2005b);
� a ciclização é mais favorecida no caso de radicais arila do que de radicais alquila (ROCHA;
PRADO, 2006);
� é possível obter macrociclos utilizando-se substratos aromáticos em que o halogênio encontra-
se em posição meta em relação ao substituinte que sofre o ataque do radical arila (BALRAJU
et al., 2005).
Estas observações possibilitaram determinar os requisitos estruturais de um precursor que teria
possibilidade de conduzir a um novo macrociclo de 12 membros: uma meta-iodobenzamida
derivada de um carboidrato contendo o grupo O-aliloxila em C-4 e o grupo benzoilamino em C-6.
Assim, planejou-se sintetizar o precursor 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-
α-D-galactopiranosídeo de metila (GAX) (FIGURA 53), que seria submetida à reação de
carbociclização radicalar com Bu3SnH para conduzir a uma benzomacrolactama de 12 membros
(GAXA) pelo modo endo. Embora os resultados obtidos nos trabalhos realizados anteriormente no
âmbito do programa de pesquisa do grupo QF/DQ/UFMG e os registros da literatura (PORTER et
al., 1986; PORTER; CHANG, 1987; PORTER; CHANG, 1988; HITCHCOCK; PATTENDEN, 1992;
COX et al., 1992; GHOSH; GHATAK, 1995; JONAS et al., 1995; HOULDSWORTH et al., 1997;
ROBERTSON; HATLEY, 1999; NANDI; CHATTOPADHYAY, 2002; BALRAJU et al., 2005)
indiquem que a ciclização 12-endo seria a preferencial, não se poderia descartar a possibilidade
de ocorrência de ciclização 11-exo e, conseqüentemente, a formação da macrolactama GAXB
(FIGURA 53).

JUSTIFICATIVA, OBJETIVOS E PLANO DE SÍNTESE
45
12-exo12-endoGAX
O
OCH3
OBn
BnO
ON
HO
I
GAXB
E/OUO
OCH3
OBn
BnO
O NOH
GAXA
Refluxo
AIBN, Bu3SnH O
OCH3
OBn
BnO
O NOH
H3C
Figura 53 – Macrolactamas possíveis de serem formadas a partir da benzamida GAX.
Além de a reação de macrociclização possibilitar a obtenção de uma ou duas
benzomacrolactamas inéditas (GAXA e /ou GAXB), os resultados seriam comparados com
aqueles obtidos nas reações desenvolvidas com o 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(2-
iodobenzoilamino)-α-D-galactopiranosídeo de metila (7, isômero orto de GAX) (BINATTI et al.,
2002), e com a N-(4-aliloxi-n-butil)-2-iodobenzamida (18), sem a unidade sacarídica, que conduziu
à macrolactama de 12 membros 19 (FARACO et al., 2003) (FIGURA 54).
7 8 911-Endo 33 %
+
22 %
18 19 20
33 %
I
N
O
H O+
N
O
H O
N
O H
O
19 %12-Endo
O
OCH3
OBn
BnO
ON
HO
I
O
OCH3
OBn
BnO
ON
HO
O
OCH3
OBn
BnO
ON
HOBu3SnH, AIBN
Benzeno, refluxo
Bu3SnH, AIBN
Benzeno, refluxo
Figura 54 – orto-Iodobenzamidas com e sem unidade sacarídica e produtos das reações de
macrociclização radicalar.
Assim, são objetivos deste trabalho:
� sintetizar, purificar e caracterizar os intermediários e a benzamida GAX;
� desenvolver reações de carbociclização radicalar mediada por Bu3SnH com GAX;
� purificar e caracterizar eventuais produtos obtidos das reações de carbociclização radicalar;
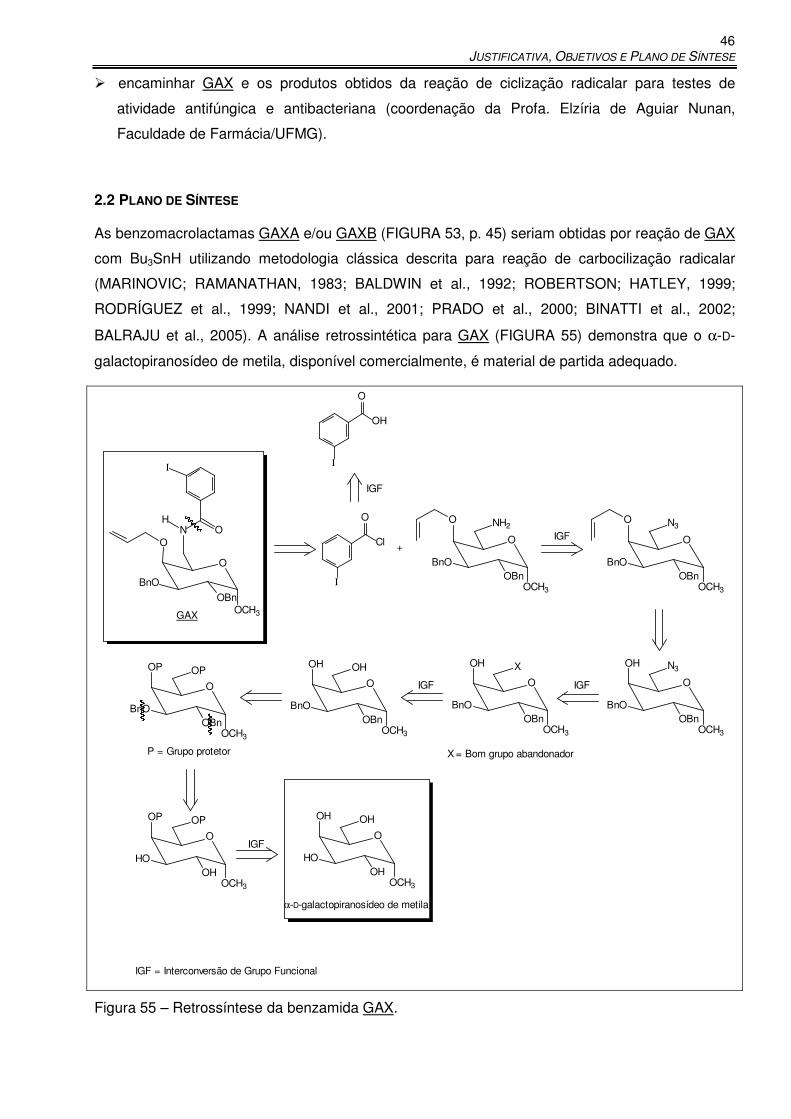
JUSTIFICATIVA, OBJETIVOS E PLANO DE SÍNTESE
46
� encaminhar GAX e os produtos obtidos da reação de ciclização radicalar para testes de
atividade antifúngica e antibacteriana (coordenação da Profa. Elzíria de Aguiar Nunan,
Faculdade de Farmácia/UFMG).
2.2 PLANO DE SÍNTESE
As benzomacrolactamas GAXA e/ou GAXB (FIGURA 53, p. 45) seriam obtidas por reação de GAX
com Bu3SnH utilizando metodologia clássica descrita para reação de carbocilização radicalar
(MARINOVIC; RAMANATHAN, 1983; BALDWIN et al., 1992; ROBERTSON; HATLEY, 1999;
RODRÍGUEZ et al., 1999; NANDI et al., 2001; PRADO et al., 2000; BINATTI et al., 2002;
BALRAJU et al., 2005). A análise retrossintética para GAX (FIGURA 55) demonstra que o α-D-
galactopiranosídeo de metila, disponível comercialmente, é material de partida adequado.
α-D-galactopiranosídeo de metila
X = Bom grupo abandonadorP = Grupo protetor
Cl
I
O
+
OH
I
O
O
OCH3
OBnBnO
NH2O
O
OCH3
OBn
BnO
ON
HO
I
GAX
IGF
O
OCH3
OBnBnO
N3O
IGF
O
OCH3
OBnBnO
N3OH
O
OCH3
OBnBnO
XOH
IGFIGFO
OCH3
OBnBnO
OHOH
O
OCH3
OBnBnO
OPOP
O
OCH3
OHHO
OPOP
O
OCH3
OHHO
OHOH
IGF = Interconversão de Grupo Funcional
IGF
Figura 55 – Retrossíntese da benzamida GAX.

JUSTIFICATIVA, OBJETIVOS E PLANO DE SÍNTESE
47
Com base na análise retrossintética, foi planejada a rota para a obtenção da iodobenzamida GAX
(FIGURA 56), em oito etapas de síntese, todas envolvendo reações relativamente simples e
amplamente utilizadas na química de carboidratos. Inicialmente as hidroxilas de C-4 e C-6 do α-D-
galactopiranosídeo de metila seriam protegidas na forma de acetal benzilidênico (HALL, 1980),
conduzindo a GA2, e as hidroxilas de C-2 e C-3 seriam benziladas (FRÉCHET; BAER, 1975),
levando a GA3. Em seguida, o grupo acetal benzilidênico de GA3 seria removido (BELL;
LORDER, 1940) e a hidroxila de C-6 seria substituída, regiosseletivamente, por iodo (GAREG;
SAMUELSSON, 1980), um bom grupo abandonador. Subseqüente substituição do átomo de iodo
pelo grupo azido (UMEZAWA et al., 1974) levaria a GA6 e O-alilação da hidroxila livre de C-4 de
GA6 (PIETRASZKIEWICZ; JURCZAK, 1984) conduziria a GA7. Redução quimiosseletiva do grupo
azido a amino (DUFOUR et al., 1992) e reação do derivado 6-amino (GA8) com cloreto de 3-
iodobenzoíla (BEAK et al., 1988) conduziria à benzamida GAX. O cloreto de 3-iodobenzoíla seria
obtido por reação do ácido 3-iodobenzóico com cloreto de tionila (BEAK et al., 1988).
i) PhCHO, ZnCl2; ii) BnCl, KOH, 100 oC; iii) HCl 1 mol/L, CH3COCH3, 70
oC; iv) I2, PPh3, imidazol, 110
oC; v) NaN3, DMF, 50 oC;
vi) CH2=CHCH2Br, Bu4NBr, NaOH 50 % p/v, CH2Cl2; vii) LiAlH4, THF; viii) SOCl2, 80 oC; ix) NaOH 2 mol/L, CH2Cl2, banho de gelo
i ii iii
iv
vvivii
viiiix
GAX
GA5 GA6 GA7 GA8
OH
I
O
Cl
I
O
O
OCH3
OH
HO
O
O
Hφ
GA1 GA2 GA3 GA4
O
OCH3
OHHO
OHOH
O
OCH3
OBn
BnO
O
O
H
φ
O
OCH3
OBn
BnO
OHOH
O
OCH3
OBn
BnO
IOH
O
OCH3
OBn
BnO
N3OH
O
OCH3
OBn
BnO
N3O
O
OCH3
OBn
BnO
NH2O
O
OCH3
OBn
BnO
ON
HO
I
Figura 56 – Rota de síntese planejada para obtenção da iodobenzamida GAX.

MATERIAL E MÉTODOS
48
3 MATERIAL E MÉTODOS
3.1 MÉTODOS GERAIS
3.1.1 Aparelhagem utilizada
As faixas de fusão foram determinadas em aparelho Microquímica MQAPF 301 (Laboratório de
Química Farmacêutica, FaFar, UFMG) e não foram corrigidas.
As medidas de rotação específica, [α]D, foram realizadas em polarímetro ADP 200, Bellingham +
Stanley Ltd. (Laboratório de Química Farmacêutica, FaFar, UFMG) e Perkin Elmer 341
(Laboratório de Química Orgânica, Departamento de Química, ICEx, UFMG).
As reações realizadas sob aquecimento de microondas foram conduzidas em aparelho de
microondas doméstico adaptado, Panasonic Junior Smart (Laboratório de Química Orgânica sob
coordenação da Profa. Rosemeire Brondi Alves, Departamento de Química, ICEx, UFMG).
Os espectros na região do infravermelho foram registrados em espectrômetro ATR-IR, Spectrum
One, Perkin Elmer (Laboratório de Química Farmacêutica, FaFar, UFMG).
Os espectros de RMN de 1H e de RMN de 13C foram registrados nos aparelhos BRUKER
AVANCE DPX-200 e BRUKER AVANCE DRX-400 (Laremar, Departamento de Química, ICEx,
UFMG), utilizando-se sonda dual 13C e 1H de detecção direta de 5 mm para os experimentos
unidimensionais de RMN de 1H, de 13C e DEPT 135 e sonda de detecção inversa de 5 mm
equipada com bobina para o emprego de pulsos de gradiente de campo para os experimentos
bidimensionais (COSY e HMQC). Como referência interna utilizou-se o tetrametilsilano (TMS).
Os espectros de massas foram adquiridos em um espectrômetro de massas Micromass Q-TOF
(Laboratório Thomson de Espectrometira de Massas, sob a coordenação do Prof. Marcos N.
Eberlin, Instituto de Química, UNICAMP), de alta resolução e alta exatidão (5 ppm). As amostras
foram analisadas por ionização por elétronspray (modo positivo), através de infusão direta (bomba
Harvard, fluxo 5µL/minuto). Para todas as amostras utilizou-se voltagem capilar de +3000 V com
temperatura de dessolvatação de 100 oC. As voltagens do cone foram +15 V (GAX) e + 30 V
(GAXC e GAXD). Na dissociação induzida por colisão (CID) a energia de colisão foi otimizada
para cada componente, variando ente 0 e 20 eV. As amostras foram solubilizadas em uma
solução a 50% v/v de Metanol (grau HPLC) / Água deionizada com 0,1% de ácido fórmico.
3.1.2 Cromatografia
Para cromatografia em camada delgada (CCD) foi utilizada sílica gel 60 G Merck, sobre lâmina de
vidro, numa espessura de 0,25 mm.
Para cromatografia em coluna de sílica (CCS) utilizou-se sílica gel 60 G Merck (0,063-0,200 mm /
70 – 230 mesh ASTM).

MATERIAL E MÉTODOS
49
3.1.3 Purificação de solventes
• Tolueno (VOGEL, 1981)
Adicionaram-se 100 mL de ácido sulfúrico 95% m/m em 1 L de tolueno, sob banho de gelo.
Manteve-se sob agitação magnética por 30 minutos e em seguida transferiu-se para um funil de
separação. Separou-se o ácido sulfúrico (fase inferior) e lavou-se a fase toluênica com água
destilada e em seguida com solução saturada de bicarbonato de sódio, até cessar liberação de
gás carbônico. O tolueno foi deixado em contato com cloreto de cálcio por 24 horas, filtrou-se a
mistura e destilou-se o tolueno utilizando coluna de vigreux.
• Dimetilformamida (ARMAREGO; PERRIN, 1996)
Manteve-se a dimetilformamida em contato com pastilhas de hidróxido de potássio por 24 horas,
filtrou-se a mistura e destilou-se a dimetilformamida utilizando coluna de vigreux.
• Benzeno (ARMAREGO; PERRIN, 1996)
Ao benzeno, adicionaram-se, cuidadosamente, lascas de sódio metálico até cessar a liberação de
hidrogênio. Em seguida adicionou-se benzofenona e manteve-se sob refluxo até permanência de
coloração azul. O solvente foi destilado e utilizado imediatamente.
3.1.4 Reveladores
• Vapor de Iodo (revelador 1)
• Solução etanólica de ácido sulfúrico (revelador 2)
Solução a 15% v/v de ácido sulfúrico em etanol, seguido de aquecimento em estufa
• Solução etanólica de ninhidrina (revelador 3)
Solução a 0,5% m/v de ninhidrina em etanol, seguido de aquecimento em estufa
3.1.5 Procedimentos Gerais
• “Elaboração usual” – O termo elaboração usual refere-se ao seguinte procedimento: reuniram-
se as fases orgânicas de uma extração, lavou-se com água destilada, manteve-se a solução
orgânica em contato com sulfato de sódio anidro, filtrou-se e eliminou-se o solvente por
destilação sob pressão reduzida, usando evaporador rotativo.
• “Ambiente isento de umidade” – O termo ambiente isento de umidade significa que foi utilizado
tubo secante contendo cloreto de cálcio anidro conectado ao balão de reação ou ao
condensador de refluxo, quando for o caso.

MATERIAL E MÉTODOS
50
• Para o desenvolvimento de todas as reações, salvo quando descrição diferente é relatada,
utilizou-se balão de fundo redondo e a mistura foi mantida sob agitação magnética.
• Todas as reações foram realizadas mais de uma vez e as condições descritas referem-se à
reação na qual foi obtido o melhor rendimento.
3.2 SÍNTESES
3.2.1 Síntese de 4,6-O-benzilideno-αααα-D-galactopiranosídeo de metila – GA2 (HALL, 1980)
GA2
O
OCH3
OH
HO
O
O
φ
O
OCH3
OH
HO
OHOH
GA1
t.a.
PhCHO, ZnCl2
48%
Em ambiente isento de umidade, colocaram-se 9 mL (88,7 mmol) de benzaldeído e 3,5 g (25,7 mmol)
de cloreto de zinco anidro. Aguardaram-se cerca de 15 minutos ou até a formação de pasta
esbranquiçada. Adicionaram-se então o α-D-galactopiranosídeo de metila GA1 (2,0 g; 10,3 mmol)
e quantidade adicional de benzaldeído (6,5 mL; 64,1 mmol). O desenvolvimento da reação foi
acompanhado por CCD (eluente: clorofórmio/metanol 9,5:0,5; reveladores: 1 e 2) e após 4,5 horas
evidenciou-se o término da reação. Adicionaram-se 16,4 g de bissulfito de sódio dissolvidos em
água (cerca de 50 mL) e manteve-se sob agitação magnética por 20 minutos. Filtrou-se o sólido
formado, sob vácuo, lavou-se com água destilada e, em seguida, extraiu-se o filtrado com
diclorometano (3 x 60 mL). Elaboração usual levou a um sólido branco grumoso que após
lavagem com éter de petróleo conduziu a 1,39 g (4,9 mmol) de GA2, com 48% de rendimento.
Tabela 1 – Quantidade de reagentes utilizados na reação de obtenção de GA2
Reagentes Quantidade Relação molar
Benzaldeído 9 mL + 6,6 mL (152,8 mmol) 14,8
Cloreto de Zinco 3,5 g (25,7 mmol) 2,5
α-D-galactopiranosídeo de metila (GA1) 2,0 g (10,3 mmol) 1
12
3
4 5
6
7
GA2
O
OCH3
OH
HO
O
O
φ

MATERIAL E MÉTODOS
51
Tabela 2 – Dados físico-químicos e espectrométricos de GA2
F.M.: C14H18O6
M.M.: 282,29 g / mol
F.F.: 166,6-168,6 oC
F.F. Literatura: (ROBERTSON; LAMB, 1934) 170,0-172,0 oC, recristalizado em éter de petróleo/etanol absoluto 2/1
[α]D: +141,3 (c 1,38, CHCl3)
[α]D Literatura: (ROBERTSON; LAMB, 1934) +166,5 (c 1,38, CHCl3)
IVATR: Figura 63, p. 80
ν máx (cm-1) Atribuição
3471 ν O-H
3068 ν C-H aromático
2908 ν C-H alifático
1452 ν C=C aromático
742, 695 δ C-H aromático monossubstituído
RMN de 1H (CDCl3, 200 MHz): Figura 64, p. 81
δ Multiplicidade Integral Atribuição J (Hz)
7,50 – 7,33 m 5 H H aromáticos -
5,51 s 1 H H-7 -
4,88 d 1 H H-1 J1-2 = 2,5
4,25 dd 1 H H-6 J6-6’ = 12,5
J6-5 = 1,3
4,19 sl 1 H H-4 -
4,02 dd 1 H H-6’ J6’-6 = 12,5
J6’-5 = 1,5
3,91 – 3,89 m 2 H H-2 e H-3 -
3,62 sl 1 H H-5 -
3,42 s 3 H OCH3 -
3,09 sl 2 H OH -
RMN de 13C (CDCl3, 50 MHz): Figura 66, p. 82
δ Atribuição
137,55 C ipso
129,01; 128,09; 126,23 C aromáticos
101,04 C-7
100,23 C-1
75,91 C-4

MATERIAL E MÉTODOS
52
Tabela 2 (conclusão)
69,33; 69,22 C-2 e C-3
69,18 C-6
62,59 C-5
55,52 OCH3
3.2.2 Síntese de 2,3-di-O-benzil-4,6-O-benzilideno-αααα-D-galactopiranosídeo de metila – GA3
(FRÉCHET; BAER, 1975)
O
OCH3
OBn
BnO
O
O
φ
GA3GA2
O
OCH3
OH
HO
O
O
φ
100 oC
BnCl, KOH
95%
Em um sistema isento de umidade e acoplado a um condensador de refluxo, adicionaram-se 1,05 g
(3,7 mmol) de 4,6-O-benzilideno-α-D-galactopiranosídeo de metila (GA2), 1,9 g (34,2 mmol) de
hidróxido de potássio pulverizado e 11,5 mL (12,6 g; 99,7 mmol) de cloreto de benzila. Manteve-se
a mistura sob aquecimento (100 oC) por 8 horas, quando observou-se o término da reação. O
desenvolvimento da reação foi acompanhado por CCD (eluente: hexano/acetato de etila 1:1;
reveladores: 1 e 2). A mistura foi resfriada à temperatura ambiente e transferida para um funil de
separação. Adicionaram-se 30 mL de água destilada e procedeu-se à extração com diclorometano
(3 x 30 mL). Elaboração usual levou a um resíduo oleoso que após adição de hexano e atrito
conduziu a um sólido branco, que foi filtrado sob vácuo O sólido branco foi caracterizado como
sendo o produto planejado, GA3 (1,64 g; 3,5 mmol; 95% de rendimento).
Tabela 3 – Quantidade de reagentes utilizados na reação de obtenção de GA3
Reagentes Quantidade Relação molar
4,6-O-benzilideno-α-D-galactopiranosídeo de metila (GA2)
1,05 g (3,7 mmol) 1
Hidróxido de potássio pulverizado 1,9 g (34,2 mmol) 9,2
Cloreto de benzila 11,5 mL (12,6 g; 99,7 mmol) 27
12
3
4 5
6
7
O
OCH3
OBnBnO
O
O
φ
GA3

MATERIAL E MÉTODOS
53
Tabela 4 – Dados físico-químicos e espectrométricos de GA3
F.M.: C28H30O6
M.M.: 462,54 g / mol
F.F.: 174,7-175,7 oC
F.F. Literatura: (FRÉCHET; BAER, 1975) 176,0-177,0 oC, recristalizado em éter de petróleo
[α]D: +76,2 (c 2,40, CHCl3)
[α]D Literatura: (FRÉCHET; BAER, 1975) +77,0 (c 2,40, CHCl3)
IVATR: Figura 69, p. 85
ν máx (cm-1) Atribuição
3028 ν C-H aromático
2901 ν C-H alifático
1497, 1455 ν C=C aromático
737, 695 δ C-H aromático monossubstituído
RMN de 1H (CDCl3, 200 MHz): Figura 70, p. 86
δ Multiplicidade Integral Atribuição J (Hz)
7,51 – 7,24 m 15 H H aromáticos -
5,48 s 1 H H-7 -
4,89 d 1 H H benzílico Jgem = 12,1
4,84 d 1 H H benzílico Jgem = 12,1
4,77 sl 1 H H-1 -
4,74 d 1 H H benzílico Jgem = 11,9
4,68 d 1 H H benzílico Jgem = 11,9
4,24 sl 1 H H açúcar -
4,18 sl 1 H H açúcar -
4,10 – 3,94 m 3 H H açúcar -
3,57 sl 1 H H açúcar -
3,38 s 3 H OCH3 -
RMN de 13C (CDCl3, 50 MHz): Figura 71, p. 87
δ Atribuição
138,73; 138,51 C ipso dos grupos benzila
137,77 C ipso
128,81; 128,27; 128,06; 128,01; 127,64; 127,58; 127,50; 126,29
C aromáticos
101,04 C-7
99,42 C-1
75,95; 75,38; 74,71; 62,39 C-2, C-3, C-4, C-5

MATERIAL E MÉTODOS
54
Tabela 4 (conclusão)
73,77; 72,15 C benzílicos
69,36 C-6
55,45 OCH3
3.2.3 Síntese de 2,3-di-O-benzil-αααα-D-galactopiranosídeo de metila – GA4 (BELL; LORBER,
1940; COURI et al., 2005)
CH3COOH/H2O 1:1
81%MO
HCl1 mol/L, CH3COCH3Refluxo
79%OU
GA4
O
OCH3
OBnBnO
OHOH
O
OCH3
OBnBnO
O
O
φ
GA3
Método I (BELL; LORBER, 1940)
Em um sistema acoplado a um condensador de refluxo, adicionaram-se 2,0 g (4,3 mmol) de 2,3-
di-O-benzil-4,6-O-benzilideno-α-D-galactopiranosídeo de metila (GA3), 105 mL de acetona, 7,5 mL
de água destilada e 2,6 mL de solução de ácido clorídrico 1 mol/L. A mistura foi mantida sob
refluxo (cerca de 70 oC) por 7 horas. O desenvolvimento da reação foi acompanhado por CCD
(eluente: hexano/acetato de etila 1:1; reveladores: 1 e 2). A mistura foi resfriada à temperatura
ambiente, adicionaram-se 2,9 g (14,5 mmol) de carbonato de bário e deixou-se sob agitação
magnética por 16 horas, até neutralização. Filtrou-se, lavou-se o resíduo com acetona e eliminou-
se o solvente por destilação sob pressão reduzida, em evaporador rotativo, utilizando etanol como
codestilante. O resíduo obtido foi purificado por CCS (eluente: diclorometano/metanol 97:3),
conduzindo a GA4 (1,28 g; 3,4 mmol) com 79% de rendimento.
Método II (COURI et al., 2005)
Em um balão acoplado a um condensador de refluxo adaptado ao aparelho de microondas,
adicionaram-se 1,48 g (3,2 mmol) de 2,3-di-O-benzil-4,6-O-benzilideno-α-D-galactopiranosídeo de
metila (GA3), 20 mL de água destilada e 20 mL de ácido acético. A mistura foi mantida sob
aquecimento em microondas por 18 minutos, em uma potência de aproximadamente 250 W. O
desenvolvimento da reação foi acompanhado por CCD (eluente: hexano/acetato de etila 1:1;
reveladores: 1 e 2). Eliminou-se a água por destilação sob pressão reduzida, usando evaporador

MATERIAL E MÉTODOS
55
rotativo, e o resíduo obtido foi purificado por CCS. O produto planejado GA4 foi eluído com
diclorometano/metanol 97:3, com 81% de rendimento (0,97 g; 2,6 mmol).
Tabela 5 – Quantidade de reagentes utilizados na reação de obtenção de GA4
Reagentes Quantidade
Método I
2,3-di-O-benzil-4,6-O-benzilideno-α-D-galactopiranosídeo de metila (GA3)
2,0 g (4,3 mmol)
Acetona 105 mL
Água destilada 7,5 mL
Solução de HCl 1 mol/L 2,6 mL
Método II
2,3-di-O-benzil-4,6-O-benzilideno-α-D-galactopiranosídeo de metila (GA3)
1,48 g (3,2 mmol)
Água destilada 20 mL
Ácido acético 20 mL
6
54
3
21
O
OCH3
OBnBnO
OHOH
GA4
Tabela 6 – Dados físico-químicos e espectrométricos de GA4
F.M.: C21H26O6
M.M.: 374,43 g / mol
F.F.: 80,8-84,8 oC
F.F. Literatura: (KISS; BURKHARDT, 1970) 87,0-88,0 oC, recristalizado em éter isopropílico
[α]D: +48,5 (c 0,78, CHCl3)
[α]D Literatura: (KISS; BURKHARDT, 1970) +45,4 (c 0,82, CHCl3)
IVATR: Figura 74, p. 89
ν máx (cm-1) Atribuição
3438 ν O-H
3028 ν C-H aromático
2876 ν C-H alifático
1498, 1455 ν C=C aromático
743, 696 δ C-H aromático monossubstituído

MATERIAL E MÉTODOS
56
Tabela 6 (conclusão)
RMN de 1H (CDCl3, 200 MHz): Figura 75, p. 90
δ Multiplicidade Integral Atribuição J (Hz)
7,38 – 7,26 m 10 H H aromáticos -
4,81 d 1 H H benzílico Jgem = 12,2
4,71 – 4,66 m 3 H H benzílicos, H-1 -
4,66 d 1 H H benzílico Jgem = 12,2
4,04 sl 1 H H-açúcar -
3,91 – 3,85 m 1 H H-açúcar -
3,91 d 1 H H-6 J6-6’ = 12,4
3,87 d 1 H H-2 J2-1 = 3,2
3,78 – 3,72 m 1 H H-açúcar -
3,77 d 1 H H-6’ J6’-6 = 12,4
3,37 s 3 H OCH3 -
2,57 sl 2 H OH -
RMN de 13C (CDCl3, 50 MHz): Figura 76, p. 91
δ Atribuição
138,21; 137,94 C ipso dos grupos benzila
128,47; 128,36; 127,98; 127,90; 127,79 C aromáticos
98,55 C-1
77,28; 75,56; 69,04; 68,86 C-2, C-3, C-4, C-5
73,45; 72,87 C benzílicos
62,89 C-6
55,29 OCH3
3.2.4 Síntese de 2,3-di-O-benzil-6-desoxi-6-iodo-αααα-D-galactopiranosídeo de metila – GA5
(GAREGG; SAMUELSSON, 1980)
73%
PhCH3, Refluxo
I2, PPh3, imidazol
GA5
O
OCH3
OBn
BnO
OH I
GA4
O
OCH3
OBnBnO
OHOH
Em um sistema isento de umidade e acoplado a um condensador de refluxo, adicionaram-se 1,65 g
(4,4 mmol) de 2,3-di-O-benzil-α-D-galactopiranosídeo de metila (GA4), 65 mL de tolueno anidro,
0,60 g (8,8 mmol) de imidazol, 1,16 g (4,4 mmol) de trifenilfosfina e 1,11 g (4,4 mmol) de iodo. A
mistura foi mantida sob refluxo (cerca de 110 oC) e após 1 hora de reação, adicionaram-se mais

MATERIAL E MÉTODOS
57
0,30 g (4,4 mmol) de imidazol, 0,57 g (2,2 mmol) de trifenilfosfina e 0,56 g (2,2 mmol) de iodo. O
desenvolvimento da reação foi acompanhado por CCD (eluente: hexano/acetato de etila 7:3;
reveladores: 1 e 2) e verificou-se que após 2 horas da segunda adição a reação havia
completado. A mistura foi resfriada à temperatura ambiente e lavou-se com 70 mL de solução
saturada de bicarbonato de sódio. A fase orgânica foi separada e adicionou-se iodo pulverizado
até permanência de coloração castanho-amarelada, característica de iodo. Em seguida,
adicionou-se solução de tiossulfato de sódio 20% m/v, sob agitação, até desaparecimento da
coloração castanho-amarelada. Separou-se novamente a fase orgânica e extraiu-se a fase
aquosa com diclorometano (3 x 20 mL) e procedeu-se à elaboração usual. O resíduo obtido foi
filtrado em coluna de sílica utilizando como eluente tolueno/acetato de etila 2:1 (200 mL). O novo
resíduo obtido foi submetido à CCS (eluente: hexano/acetato de etila 8:2) e o derivado iodado
GA5 (1,54 g; 3,2 mmol) foi obtido com 73% de rendimento.
Tabela 7 – Quantidade de reagentes utilizados na reação de obtenção de GA5
Reagentes Quantidade Relação molar
2,3-di-O-benzil-α-D-galactopiranosídeo de metila (GA4)
1,65 g (4,4 mmol) 1
Tolueno anidro 65 mL -
Imidazol 0,60 g (8,8 mmol) + 0,30 g (4,4 mmol) 2 + 1
Trifenilfosfina 1,16 g (4,4 mmol + 0,57 g (2,2 mmol) 1 + 0,5
Iodo 1,11 g (4,4 mmol) + 0,56 g (2,2 mmol) 1 + 0,5
6
54
3
21
O
OCH3
OBn
BnO
OH I
GA5
Tabela 8 – Dados físico-químicos e espectrométricos de GA5
F.M.: C21H25IO5
M.M.: 484,33 g / mol
F.F.: 97,0-103,0 oC
F.F. Literatura: (BINATTI et al., 2002) 103,9-105,5 oC
[α]D: +44,6 (c 2,40, CHCl3)
[α]D Literatura: (BINATTI et al., 2002) +44,7 (c 2,40, CHCl3)

MATERIAL E MÉTODOS
58
Tabela 8 (conclusão)
IVATR: Figura 79, p. 93
ν máx (cm-1) Atribuição
3453 ν O-H
3027 ν C-H aromático
2922 ν C-H alifático
1498, 1454 ν C=C aromático
739, 701 δ C-H aromático monossubstituído
RMN de 1H (CDCl3, 200 MHz): Figura 80, p. 94
δ Multiplicidade Integral Atribuição J (Hz)
7,38 – 7,28 m 10 H H aromáticos -
4,82 d 1 H H benzílico Jgem = 11,5
4,81 d 1 H H benzílico Jgem = 12,2
4,69 d 1 H H benzílico Jgem = 11,5
4,64 d 1 H H benzílico Jgem = 12,2
4,63 d 1 H H-1 J1-2 = 3,3
4,11 – 4,09 m 1 H H-açúcar -
3,91 – 3,81 m 3 H H-açúcar -
3,43 s 3 H OCH3 -
3,32 d 2 H H-6 J6-5 = 7,3
2,44 sl 1 H OH -
RMN de 13C (CDCl3, 50 MHz): Figura 81, p. 95
δ Atribuição
138,17; 137,90 C ipso dos grupos benzila
128,54; 128,42; 128,03; 127,99; 127,87; 127,82 C aromáticos
98,55 C-1
77,36; 75,25; 70,15; 68,38 C-2, C-3, C-4, C-5
73,53; 73,04 C benzílicos
55,68 OCH3
3,01 C-6

MATERIAL E MÉTODOS
59
3.2.5 Síntese de 6-azido-2,3-di-O-benzil-6-desoxi-αααα-D-galactopiranosídeo de metila – GA6
(UMEZAWA et al., 1974)
GA6
O
OCH3
OBn
BnO
OH N3
GA5
O
OCH3
OBn
BnO
OH I
50 oC
NaN3, DMF
87%
Em um sistema isento de umidade, adicionaram-se 1,81 g (3,7 mmol) de 2,3-di-O-benzil-6-desoxi-6-
iodo-α-D-galactopiranosídeo de metila (GA5), 113 mL de dimetilformamida anidra e 1,13 g (17,4 mmol)
de azida de sódio. A mistura de reação foi aquecida a 50 oC por 34 horas. O desenvolvimento da
reação foi acompanhado por CCD (eluente: hexano/acetato de etila 7:3; reveladores: 1 e 2). A
dimetilformamida foi eliminada por destilação à pressão reduzida, adicionaram-se 50 mL de água
destilada ao resíduo, extraiu-se com diclorometano (3 x 80 mL) e procedeu-se à elaboração usual.
O resíduo obtido foi purificado por CCS (eluente: hexano/acetato de etila 8:2), conduzindo a GA6
(1,29 g; 3,2 mmol) com 87% de rendimento.
Tabela 9 – Quantidade de reagentes utilizados na reação de obtenção de GA6
Reagentes Quantidade Relação molar
2,3-di-O-benzil-6-desoxi-6-iodo-α-D-galactopiranosídeo de metila (GA5)
1,81 g (3,7 mmol) 1
Dimetilformamida 113 mL -
Azida de Sódio 1,13 g (17,4 mmol) 4,7
6
54
3
21
GA6
O
OCH3
OBn
BnO
OH N3
Tabela 10 – Dados físico-químicos e espectrométricos de GA6
F.M.: C21H25N3O5
M.M.: 399,44 g / mol
F.F.: 65,0-67,5 oC
F.F. Literatura: (BINATTI et al., 2002) Óleo
[α]D: +21,3 (c 2,72, CHCl3)
[α]D Literatura: (BINATTI et al., 2002) +96,9 (c 2,72, CHCl3)
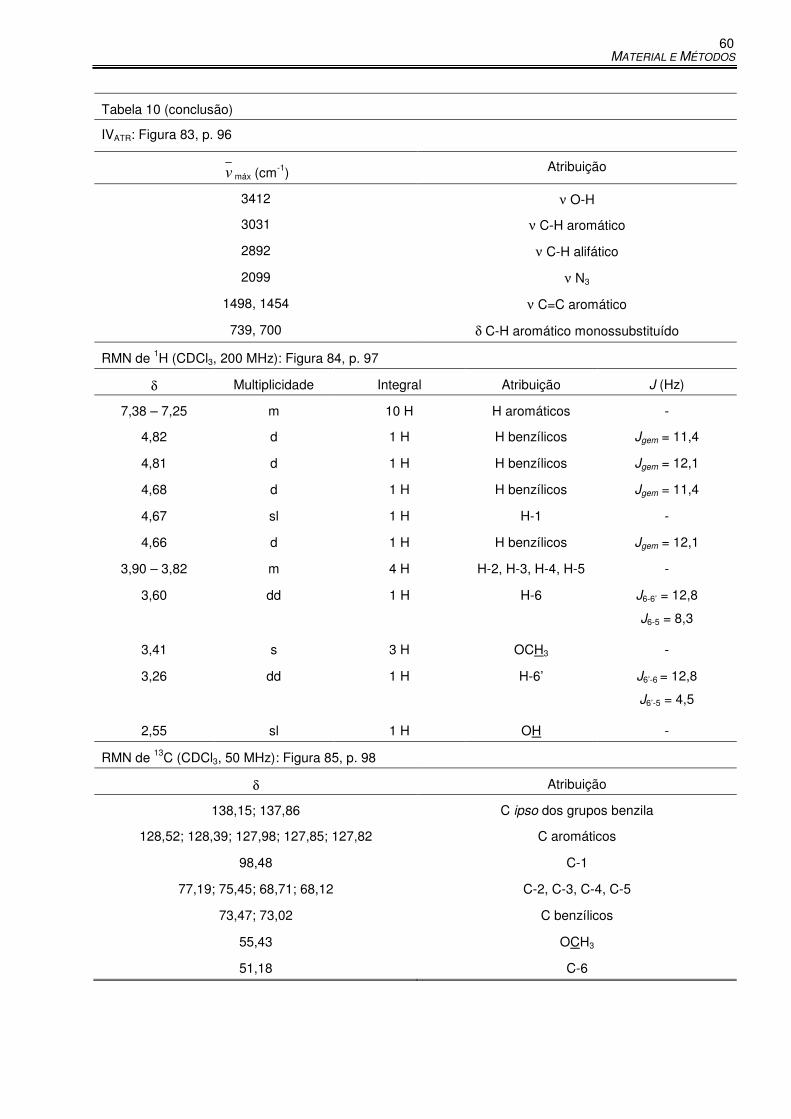
MATERIAL E MÉTODOS
60
Tabela 10 (conclusão)
IVATR: Figura 83, p. 96
ν máx (cm-1) Atribuição
3412 ν O-H
3031 ν C-H aromático
2892 ν C-H alifático
2099 ν N3
1498, 1454 ν C=C aromático
739, 700 δ C-H aromático monossubstituído
RMN de 1H (CDCl3, 200 MHz): Figura 84, p. 97
δ Multiplicidade Integral Atribuição J (Hz)
7,38 – 7,25 m 10 H H aromáticos -
4,82 d 1 H H benzílicos Jgem = 11,4
4,81 d 1 H H benzílicos Jgem = 12,1
4,68 d 1 H H benzílicos Jgem = 11,4
4,67 sl 1 H H-1 -
4,66 d 1 H H benzílicos Jgem = 12,1
3,90 – 3,82 m 4 H H-2, H-3, H-4, H-5 -
3,60 dd 1 H H-6 J6-6’ = 12,8
J6-5 = 8,3
3,41 s 3 H OCH3 -
3,26 dd 1 H H-6’ J6’-6 = 12,8
J6’-5 = 4,5
2,55 sl 1 H OH -
RMN de 13C (CDCl3, 50 MHz): Figura 85, p. 98
δ Atribuição
138,15; 137,86 C ipso dos grupos benzila
128,52; 128,39; 127,98; 127,85; 127,82 C aromáticos
98,48 C-1
77,19; 75,45; 68,71; 68,12 C-2, C-3, C-4, C-5
73,47; 73,02 C benzílicos
55,43 OCH3
51,18 C-6

MATERIAL E MÉTODOS
61
3.2.6 Síntese de 4-O-alil-6-azido-2,3-di-O-benzil-6-desoxi-αααα-D-galactopiranosídeo de
metila – GA7 (PIETRASZKIEWICZ; JURCZAK, 1984)
GA7
O
OCH3
OBnBnO
O N3
GA6
O
OCH3
OBn
BnO
OH N3
NaOH 50% m/v, CH2Cl2, t.a.
CH2=CHCH2Br, Bu4NBr
83%
À uma solução do derivado azidoálcool GA6 (0,75 g; 1,9 mmol) em 15 mL de diclorometano foram
adicionados brometo de tetrabutilamônio (0,90 g; 2,8 mmol) e 5,3 mL de solução de hidróxido de
sódio 50% m/v. Após 10 minutos, adicionaram-se 0,72 mL (1,0 g; 8,3 mmol) de brometo de alila. O
desenvolvimento da reação foi acompanhado por CCD (eluente: hexano/acetato de etila 8:2;
reveladores: 1 e 2) e após 40 horas de reação, adicionaram-se 10 mL de água destilada.
Separou-se a fase orgânica, extraiu-se a fase aquosa com diclorometano (3 x 60 mL) e procedeu-
se à elaboração usual. O resíduo obtido foi purificado por CCS (eluente: hexano/acetato de etila
8:2), obtendo-se um óleo incolor que cristalizou após atrito. O produto planejado GA7 (0,69 g;
1,6 mmol) foi obtido com 83% de rendimento.
Tabela 11 – Quantidade de reagentes utilizados na reação de obtenção de GA7
Reagentes Quantidade Relação molar
6-azido-2,3-di-O-benzil-6-desoxi-α-D-galactopiranosídeo de metila – GA6
0,75 g (1,9 mmol) 1
Diclorometano 15 mL -
Brometo de tetrabutilamônio 0,90 g (2,8 mmol) 1,5
Solução de hidróxido de sódio 50% m/v 5,3 mL -
Brometo de alila 0,72 mL (1,0 g; 8,3 mmol) 4,4
6
54
3
21
O
OCH3
OBnBnO
O N3
GA7
7
8
9
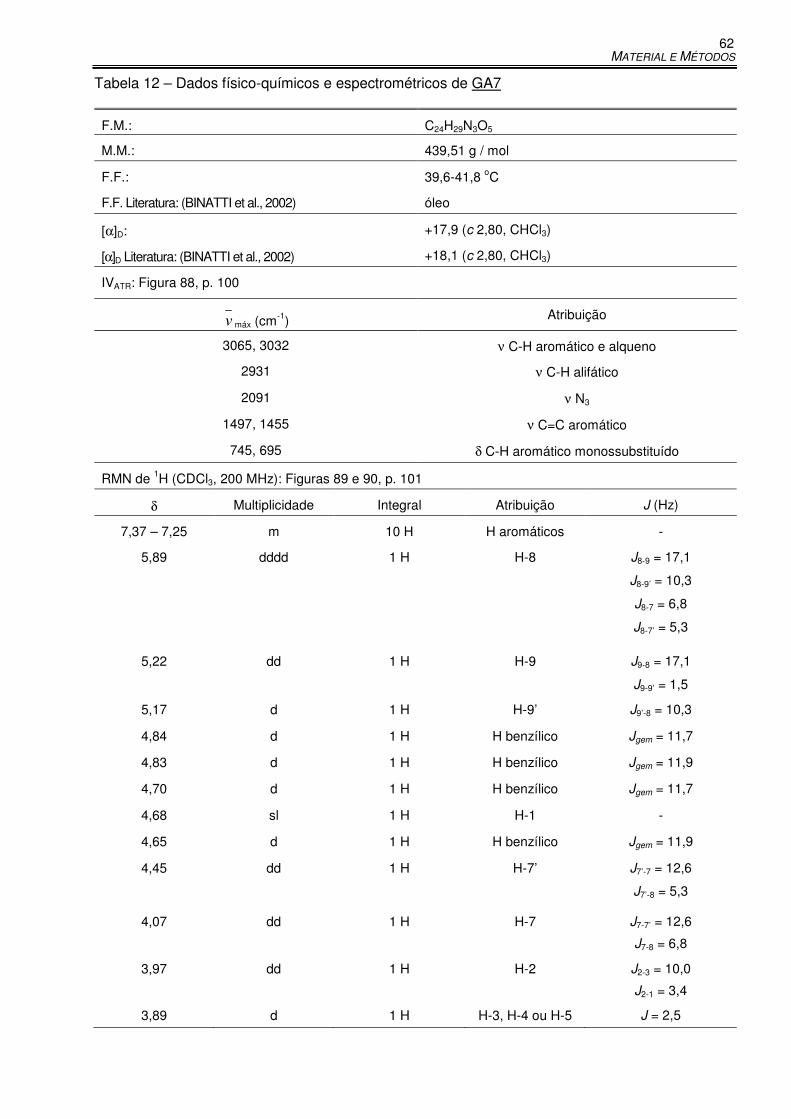
MATERIAL E MÉTODOS
62
Tabela 12 – Dados físico-químicos e espectrométricos de GA7
F.M.: C24H29N3O5
M.M.: 439,51 g / mol
F.F.: 39,6-41,8 oC
F.F. Literatura: (BINATTI et al., 2002) óleo
[α]D: +17,9 (c 2,80, CHCl3)
[α]D Literatura: (BINATTI et al., 2002) +18,1 (c 2,80, CHCl3)
IVATR: Figura 88, p. 100
ν máx (cm-1) Atribuição
3065, 3032 ν C-H aromático e alqueno
2931 ν C-H alifático
2091 ν N3
1497, 1455 ν C=C aromático
745, 695 δ C-H aromático monossubstituído
RMN de 1H (CDCl3, 200 MHz): Figuras 89 e 90, p. 101
δ Multiplicidade Integral Atribuição J (Hz)
7,37 – 7,25 m 10 H H aromáticos -
5,89 dddd 1 H H-8 J8-9 = 17,1
J8-9’ = 10,3
J8-7 = 6,8
J8-7’ = 5,3
5,22 dd 1 H H-9 J9-8 = 17,1
J9-9’ = 1,5
5,17 d 1 H H-9’ J9’-8 = 10,3
4,84 d 1 H H benzílico Jgem = 11,7
4,83 d 1 H H benzílico Jgem = 11,9
4,70 d 1 H H benzílico Jgem = 11,7
4,68 sl 1 H H-1 -
4,65 d 1 H H benzílico Jgem = 11,9
4,45 dd 1 H H-7’ J7’-7 = 12,6
J7’-8 = 5,3
4,07 dd 1 H H-7 J7-7’ = 12,6
J7-8 = 6,8
3,97 dd 1 H H-2 J2-3 = 10,0
J2-1 = 3,4
3,89 d 1 H H-3, H-4 ou H-5 J = 2,5

MATERIAL E MÉTODOS
63
Tabela 12 (conclusão)
3,85 – 3,79 m 1 H H-3, H-4 ou H-5 -
3,70 sl 1 H H-3, H-4 ou H-5 -
3,57 dd 1 H H-6 J6-6’ = 12,4
J6-5= 8,1
3,40 s 3 H OCH3 -
3,18 dd 1 H H-6’ J6’-6 = 12,4
J6’-5 = 5,0
RMN de 13C (CDCl3, 50 MHz): Figura 92, p. 103
δ Atribuição
138,54; 138,37 C ipso dos grupos benzila
134,84 C-8
128,36; 128,33; 128,00; 127,72; 127,57; 127,49 C aromáticos
117,65 C-9
98,77 C-1
78,62 C-2
76,28; 75,35; 69,61 C-3, C-4, C-5
73,90 C-7
73,62; 73,41 C benzílicos
55,45 OCH3
51,33 C-6
3.2.7 Síntese de 4-O-alil-6-amino-2,3-di-O-benzil-6-desoxi-αααα-D-galactopiranosídeo de metila
(GA8), de Cloreto de 3-iodobenzoíla e de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-
iodobenzoilamino)-αααα-D-galactopiranosídeo de metila – GAX
O
OCH3
OBnBnO
O N3
60% 37%
Cl
O
I
NaOH 2 mol/L Et3N, banho de gelo (2) , OU
(1) LiAlH4, THF, N2, t.a.O
OCH3
OBn
BnO
ON
HO
I
GAXGA7

MATERIAL E MÉTODOS
64
3.2.7.1 Síntese de 4-O-alil-6-amino-2,3-di-O-benzil-6-desoxi-αααα-D-galactopiranosídeo de metila
(GA8) (DUFOUR et al., 1992)
GA7
LiAlH4, THF, N2, t.a.O
OCH3
OBnBnO
O N3
GA8
O
OCH3
OBnBnO
O NH2
Em um balão bitubulado, acoplado em uma das extremidades a um condensador conectado a um
sistema de escape de gás (tubo contendo óleo mineral), adicionou-se 0,0366 g (0,96 mmol) de
hidreto de lítio e alumínio. A outra extremidade do balão foi fechada com rolha de saia e o sistema
foi mantido sob atmosfera de nitrogênio. Com o auxílio de uma seringa, foram adicionados,
lentamente, 1,6 mL de tetraidrofurano (grau HPLC) e uma solução de GA7 (0,21 g; 0,48 mmol) em
4,0 mL de tetraidrofurano (grau HPLC). O desenvolvimento da reação foi acompanhado por CCD
(eluente: hexano/acetato de etila 8:2; reveladores: (a) 1 e 2; (b) 1 e 3) e após 2,5 horas
adicionaram-se, cuidadosamente, 1 mL de água destilada, 1 mL de solução de hidróxido de sódio
2 mol/L e mais 4 mL de água destilada. Separou-se a camada orgânica, extraiu-se a fase aquosa
com diclorometano (3 x 15 mL) e procedeu-se à elaboração usual. O resíduo obtido na forma de
um óleo (GA8) foi utilizado no mesmo dia na reação subseqüente, sem purificação adicional.
3.2.7.2 Síntese de Cloreto de 3-iodobenzoíla (BEAK et al., 1988)
Cl
O
I
OH
O
I
Refluxo
SOCl2
A um sistema isento de umidade acoplado a condensador de refluxo, foram adicionados 0,24 g
(0,97 mmol) de ácido 3-iodobenzóico e 1,4 mL (2,3 g; 19,3 mmol) de cloreto de tionila. A mistura
foi mantida sob refluxo (aproximadamente 90 oC) por 3 horas. O excesso de cloreto de tionila foi
removido por co-destilação com benzeno, usando evaporador rotativo, sob pressão reduzida.

MATERIAL E MÉTODOS
65
3.2.7.3 Síntese de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-αααα-D-galactopiranosídeo
de metila – GAX (BEAK et al., 1988; DUFOUR et al., 1992)
NaOH 2 mol/L OU Et3N
Cl
O
IO
OCH3
OBnBnO
O NH2
GA8 GAX
O
OCH3
OBn
BnO
ON
HO
I
Método I (BEAK et al., 1988)
À solução de cloreto de 3-iodobenzoíla, obtido conforme descrito no item 3.2.7.2, em 1,6 mL de
diclorometano, foram adicionados, sob resfriamento em banho de gelo, 1,2 mL de solução de
hidróxido de sódio 2 mol/L e solução da amina GA8, obtida conforme descrito no item 3.2.7.1, em
3,8 mL de diclorometano. O desenvolvimento da reação foi acompanhado por CCD (eluente:
hexano/acetato de etila 1:1; reveladores: (a) 1 e 2; (b) 1 e 3) e após 45 minutos adicionaram-se
10 mL de água destilada e 10 mL de diclorometano. Separou-se a camada orgânica, extraiu-se a
fase aquosa com diclorometano (3 x 15 mL) e procedeu-se à elaboração usual. O resíduo obtido
foi purificado por CCS (eluente: hexano/acetato de etila 6:4), conduzindo a GAX (0,19 g; 0,29 mmol)
com 60% de rendimento, calculado em relação a GA7.
Método II (DUFOUR et al., 1992)
Em um sistema isento de umidade, à solução da amina GA8, obtida conforme descrito no item
3.2.7.1, em 4,5 mL de diclorometano (grau HPLC), foram adicionados, sob resfriamento em banho
de gelo, 0,07 mL (0,05 g; 0,5 mmol) de trietilamina e solução de cloreto de 3-iodobenzoíla obtido
conforme descrito no item 3.2.7.2 em 2,5 mL de diclorometano (grau HPLC). O desenvolvimento
da reação foi acompanhado por CCD (eluente: hexano/acetato de etila 1:1; reveladores: (a) 1 e 2;
(b) 1 e 3) e após 1,5 horas adicionaram-se 10 mL de água destilada. Separou-se a camada
orgânica, extraiu-se a fase aquosa com diclorometano (3 x 15 mL) e procedeu-se à elaboração
usual. O resíduo obtido foi purificado por CCS (eluente: hexano/acetato de etila 6:4), conduzindo a
GAX (0,12 g; 0,18 mmol) com 37% de rendimento, calculado em relação a GA7.

MATERIAL E MÉTODOS
66
Tabela 13 – Quantidade de reagentes utilizados na reação de obtenção de GAX
Reagentes Quantidades Relação molar
Síntese de GA8
6-azido-2,3-di-O-benzil-4-O-alil-6-desoxi-α-D-galactopiranosídeo de metila – GA7
0,21 g (0,48 mmol) 1
Tetraidrofurano (para GA7) 4,0 mL -
Hidreto de lítio e alumínio 0,0366 g (0,96 mmol) 2
Tetraidrofurano (para LiAlH4) 1,6 mL -
Síntese de Cloreto de 3-iodobenzoíla
Ácido 3-iodobenzóico 0,24 g (0,97 mmol) 2
Cloreto de tionila 1,4 mL (2,3 g; 19,3 mmol) 40
Síntese de GAX
Método I
Diclorometano (para GA8) 1,6 mL -
Solução de hidróxido de sódio 2 mol/L 1,2 mL -
Diclorometano (para cloreto de 3-iodobenzoíla)
3,8 -
Método II
Diclorometano (para GA8) 4,5 mL -
Trietilamina 0,07 mL (0,05 g; 0,5 mmol) 1
Diclorometano (para cloreto de 3-iodobenzoíla)
2,5 mL -
GAX
O
OCH3
OBn
BnO
ON
HO
I
f
ed
c
ba
9
8
7
12
3
4 5
6
Tabela 14 – Dados físico-químicos e espectrométricos de GAX
F.M.: C31H34INO6
M.M.: 643,51 g / mol
F.F.: 109,0-110,4 oC
[α]D: +45,5 (c 1,41, CHCl3)

MATERIAL E MÉTODOS
67
Tabela 14 (continuação)
IVATR: Figura 97, p. 106
ν máx (cm-1) Atribuição
3293 ν N-H amida
3058 ν C-H aromático
2927 ν C-H alifático
1637 ν C=O amida (banda de amida I)
1553 δ N-H (banda de amida II)
1497, 1454 ν C=C aromático
RMN de 1H (CDCl3, 400 MHz): Figuras 98 – 101, p. 108 – 109
δ Multiplicidade Integral Atribuição J (Hz)
8,07 t 1 H H-b Jb-d = 1,6
Jb-f = 1,6
7,81 ddd 1 H H-d Jd-e = 7,8
Jd-b = 1,6
Jd-f = 1,1
7,67 ddd 1 H H-f Jf-e = 7,8
Jf-b = 1,6
Jf-d = 1,1
7,39 – 7,25 m 10 H H aromáticos -
7,15 t 1 H H-e Je-d = 7,8
Je-f = 7,8
6,62 dd 1 H NH JNH-6 = 7,0
JNH-6’ = 4,0
5,93 dddd 1 H H-8 J8-9 = 17,3
J8-9’ = 10,3
J8-7 = 6,9
J8-7’ = 5,2
5,24 qd 1 H H-9 J9-8 = 17,3
J9-9’ = 1,5
J9-7 = 1,5
J9-7’ = 1,5
5,20 dd 1 H H-9’ J9’-8 = 10,3
J9’-9 = 1,5
4,84 d 1 H H benzílico Jgem = 12,1
4,84 d 1 H H benzílico Jgem = 11,7
4,71 d 1 H H benzílico Jgem = 11,7

MATERIAL E MÉTODOS
68
Tabela 14 (continuação)
4,67 d 1 H H-1 J1-2 = 3,7
4,66 d 1 H H benzílico Jgem = 12,1
4,49 tdd 1 H H-7’ J7’-7 = 12,4
J7’-8 = 5,2
J7’-9= 1,5
J7’-9’= 1,5
4,11 dd 1 H H-7 J7-7’ = 12,4
J7-8 = 6,9
3,98 dd 1 H H-2 J2-3 = 10,1
J2-1 = 3,7
3,94 – 3,87 m 3 H H-3, H-6 e H-4 ou H-5
-
3,80 d 1 H H-5 ou H-4 J = 2,3
3,42 – 3,38 m 1 H H-6’ -
3,30 s 3 H OCH3 -
RMN de 13C (CDCl3, 100 MHz): Figura 103, p. 111
δ Atribuição
165,94 C=O
140,38 C-d
138,53; 138,44 C ipso dos grupos benzila
136,31 C-a (ipso)
135,96 C-b
134,88 C-8
130,22 C-e
128,40; 128,33; 128,01; 127,72; 127,63; 127,54 C aromáticos
125,94 C-f
118,00 C-9
98,88 C-1
94,26 C-I (ipso)
78,72; 68,27 C-3, C-4 ou C-5
76,45 C-5 ou C-4
76,35 C-2
73,94 C-7
73,67; 73,47 C benzílicos
55,31 OCH3
41,21 C-6

MATERIAL E MÉTODOS
69
Tabela 14 (conclusão)
Massas de alta resolução (TOF MS ES; TOF MSMS ES): Figura 105, p. 113
Íon m/z
Calculado Encontrado
[M + H]+ 644,1509 644,1534
[M + Na]+ 666,1329 666,1451
[M + K]+ 682,1068 682,1188
[M + H]+ – CH3OH 612,1247 612,1757
[M + H]+ – CH3OH – PhCH2OH 504,0672 504,0809
3.2.8 Reação de macrociclização radicalar
GAX
O
OCH3
OBn
BnO
ON
HO
I
GAXB
E/OUO
OCH3
OBn
BnO
O NOH
GAXARefluxo
AIBN, Bu3SnH
ou ciclo-hexano
O
OCH3
OBn
BnO
O NOH
H3C
Foram realizadas quatro reações de macrociclização radicalar, utilizando-se o procedimento geral
descrito a seguir. As condições específicas de cada reação, assim como os produtos isolados e
seus respectivos rendimentos, encontram-se apresentados na Tabela 15 (p. 70).
Para a reação de macrociclização radicalar, foi utilizado um balão tritubulado, adaptado em uma
das extremidades a um condensador de refluxo conectado a um sistema de escape de gás (tubo
contendo óleo mineral). Outra extremidade foi fechada com rolha de saia por onde se introduziu o
nitrogênio (a mistura de reação foi mantida sob atmosfera de nitrogênio). A última extremidade do
balão foi adaptada a um funil de adição fechado com rolha de saia.
A uma solução da m-iodobenzamida GAX (0,2 g; 0,31 mmol) em solvente anidro, sob agitação
magnética e refluxo (temperatura cerca de 90 oC), adicionou-se, lentamente, por meio do funil de
adição, solução de AIBN (10 mg; 0,062 mmol) e hidreto de tri-n-butilestanho (0,13 mL; 0,14 g;
0,47 mmol), em solvente anidro. Uma hora após o término da adição, eliminou-se o solvente por
destilação sob pressão reduzida, usando evaporador rotativo, e o resíduo obtido foi submetido à
CCS (eluente: hexano/acetato de etila).
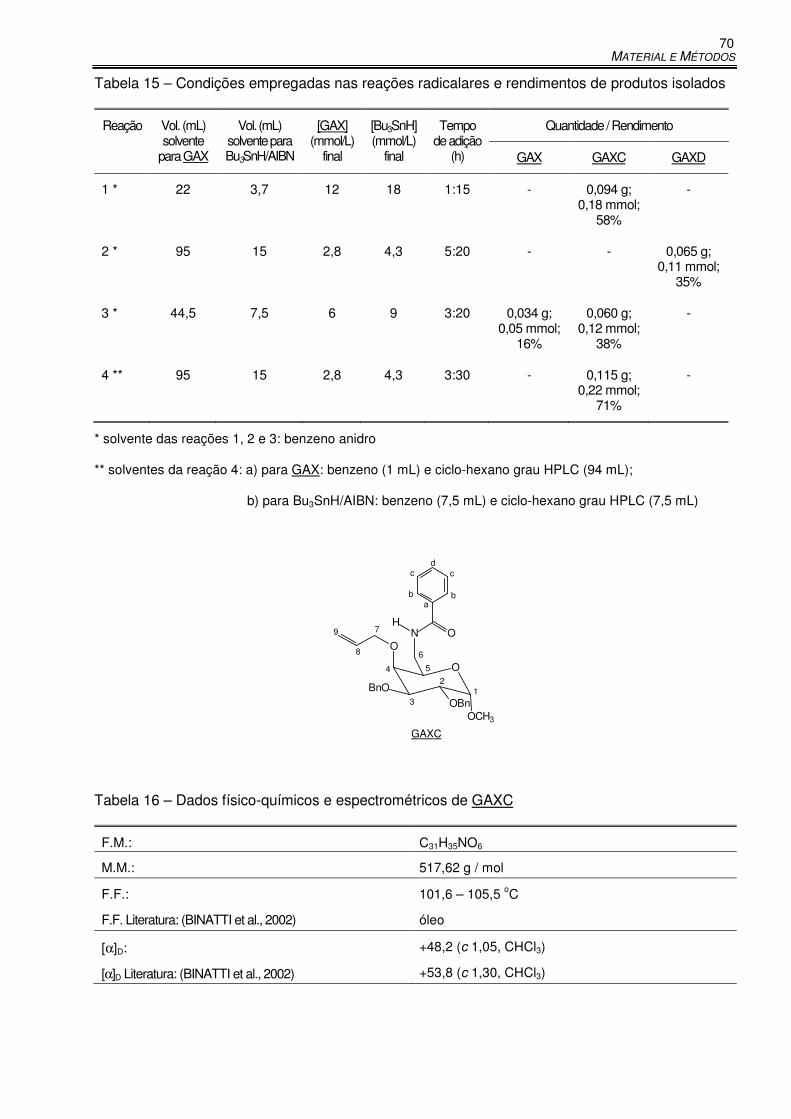
MATERIAL E MÉTODOS
70
Tabela 15 – Condições empregadas nas reações radicalares e rendimentos de produtos isolados
Reação Vol. (mL) solvente
para GAX
Vol. (mL) solvente para Bu3SnH/AIBN
[GAX] (mmol/L)
final
[Bu3SnH] (mmol/L)
final
Tempo de adição
(h)
Quantidade / Rendimento
GAX GAXC GAXD
1 * 22 3,7 12 18 1:15 - 0,094 g; 0,18 mmol;
58%
-
2 * 95 15 2,8 4,3 5:20 - - 0,065 g; 0,11 mmol;
35%
3 * 44,5 7,5 6 9 3:20 0,034 g; 0,05 mmol;
16%
0,060 g; 0,12 mmol;
38%
-
4 ** 95 15 2,8 4,3 3:30 - 0,115 g; 0,22 mmol;
71%
-
* solvente das reações 1, 2 e 3: benzeno anidro
** solventes da reação 4: a) para GAX: benzeno (1 mL) e ciclo-hexano grau HPLC (94 mL);
b) para Bu3SnH/AIBN: benzeno (7,5 mL) e ciclo-hexano grau HPLC (7,5 mL)
6
54
3
21
7
8
9
ab
cd
O
OCH3
OBn
BnO
ON
HO
GAXC
b
c
Tabela 16 – Dados físico-químicos e espectrométricos de GAXC
F.M.: C31H35NO6
M.M.: 517,62 g / mol
F.F.: 101,6 – 105,5 oC
F.F. Literatura: (BINATTI et al., 2002) óleo
[α]D: +48,2 (c 1,05, CHCl3)
[α]D Literatura: (BINATTI et al., 2002) +53,8 (c 1,30, CHCl3)

MATERIAL E MÉTODOS
71
Tabela 16 (continuação)
IVATR: Figura 109, p. 118
ν máx (cm-1) Atribuição
3315 ν N-H amida
3030 ν C-H aromático
2927 ν C-H alifático
1639 ν C=O amida (banda de amida I)
1538 δ N-H (banda de amida II)
1490, 1454 ν C=C aromático
RMN de 1H (CDCl3, 400 MHz): Figuras 110 e 111, p. 119
δ Multiplicidade Integral Atribuição J (Hz)
7,73 dd 2 H H-b Jb-c = 6,9
Jb-d = 1,6
7,48 tt 1 H H-d Jd-c = 7,3
Jd-b = 1,6
7,43 – 7,41 m 2 H H-c -
7,36 – 7,25 m 10 H H aromáticos -
6,65 dd 1 H NH JNH-6 = 7,2
J NH-6’ = 3,4
5,92 dddd 1 H H-8 J8-9 = 17,2
J8-9’ = 10,2
J8-7 = 6,9
J8-7’ = 5,3
5,24 qd 1 H H-9 J9-8 = 17,2
J9-9’ = 1,5
J9-7 = 1,5
J9-7’ = 1,5
5,18 dd 1 H H-9’ J9’-8 = 10,2
J9’-9 = 1,5
4,84 d 1 H H benzílico Jgem = 12,0
4,83 d 1 H H benzílico Jgem = 11,6
4,71 d 1 H H benzílico Jgem = 11,6
4,67 d 1 H H-1 J1-2 = 3,5
4,66 d 1 H H benzílico Jgem = 12,0

MATERIAL E MÉTODOS
72
Tabela 16 (continuação)
4,47 tdd 1 H H-7’ J7’-7 = 12,5
J7’-8 = 5,3
J7’-9= 1,5
J7’-9’= 1,5
4,12 dd 1 H H-7 J7-7’ = 12,5
J7-8 = 6,9
3,98 dd 1 H H-2 J2-3 = 10,1
J2-1 = 3,5
3,92 – 3,88 m 3 H H-3, H-6 e H-4 ou H-5
-
3,80 d 1 H H-5 ou H-4 J = 2,3
3,43 – 3,36 m 1 H H-6’ -
3,29 s 3 H OCH3 -
RMN de 13C (CDCl3, 100 MHz): Figura 113, p. 121
δ Atribuição
167,57 C=O
138,53; 138,45 C ipso dos grupos benzila
134,92 C-8
134,27 C-a (ipso)
131,49 C-d
128,56 C-c
128,37; 128,29; 127,99; 127,68; 127,59; 127,52 C aromáticos
126,76 C-b
117,84 C-9
98,83 C-1
78,73; 68,53 C-3, C-4 ou C-5
76,36 C-2
76,24 C-5 ou C-4
73,94 C-7
73,63; 73,39 C benzílicos
55,20 OCH3
41,01 C-6
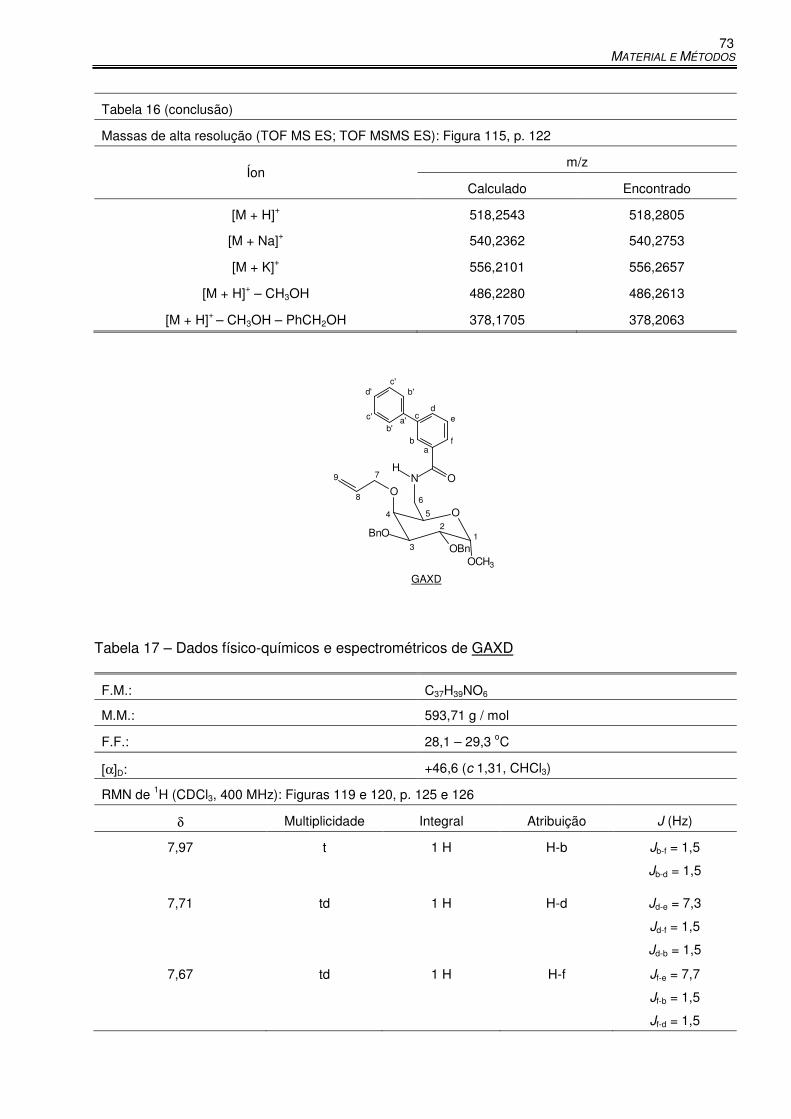
MATERIAL E MÉTODOS
73
Tabela 16 (conclusão)
Massas de alta resolução (TOF MS ES; TOF MSMS ES): Figura 115, p. 122
Íon m/z
Calculado Encontrado
[M + H]+ 518,2543 518,2805
[M + Na]+ 540,2362 540,2753
[M + K]+ 556,2101 556,2657
[M + H]+ – CH3OH 486,2280 486,2613
[M + H]+ – CH3OH – PhCH2OH 378,1705 378,2063
6
54
3
21
7
8
9
a
cd
e
f
GAXD
b
O
OCH3
OBn
BnO
ON
HO
c'
b'
d'
a'
b'
c'
Tabela 17 – Dados físico-químicos e espectrométricos de GAXD
F.M.: C37H39NO6
M.M.: 593,71 g / mol
F.F.: 28,1 – 29,3 oC
[α]D: +46,6 (c 1,31, CHCl3)
RMN de 1H (CDCl3, 400 MHz): Figuras 119 e 120, p. 125 e 126
δ Multiplicidade Integral Atribuição J (Hz)
7,97 t 1 H H-b Jb-f = 1,5
Jb-d = 1,5
7,71 td 1 H H-d Jd-e = 7,3
Jd-f = 1,5
Jd-b = 1,5
7,67 td 1 H H-f Jf-e = 7,7
Jf-b = 1,5
Jf-d = 1,5

MATERIAL E MÉTODOS
74
Tabela 17 (continuação)
7,58 d 2 H H-b’ Jb’-c’ = 7,0
7,49 – 7,43 m 3 H H-e + 2 H aromáticos
-
7,39 – 7,25 m 11 H H aromáticos -
6,69 – 6,68 m 1 H NH -
5,93 dddd 1 H H-8 J8-9 = 17,2
J8-9’ = 10,2
J8-7 = 6,6
J8-7’ = 5,1
5,23 dd 1 H H-9 J9-8 = 17,2
J9-9’ = 1,5
5,16 dl 1 H H-9’ J9’-8 = 10,2
4,84 d 1 H H benzílico Jgem = 12,1
4,83 d 1 H H benzílico Jgem = 12,3
4,71 d 1 H H benzílico Jgem = 12,1
4,67 d 1 H H-1 J1-2 = 3,4
4,66 d 1 H H benzílico Jgem = 12,3
4,48 ddl 1 H H-7’ J7’-7 = 12,4
J7’-8 = 5,1
4,13 ddl 1 H H-7 J7-7’ = 12,4
J7-8 = 6,6
3,99 dd 1 H H-2 J2-3 = 10,0
J2-1 = 3,4
3,95 – 3,89 m 3 H H-3, H-6 e H-4 ou H-5
-
3,81 d 1 H H-5 ou H-4 J = 2,6
3,46 – 3,37 m 1 H H-6’ -
3,30 s 3 H OCH3 -
RMN de 13C (CDCl3, 50 MHz): Figuras 122 e 123, p. 128
δ Atribuição
167,49 C=O
141,71 C-c (ipso)
140,13 C-a’ (ipso)
138,53; 138,44 C ipso dos grupos benzila
134,92 C-8
130,15 C-d
129,68 C-a (ipso)

MATERIAL E MÉTODOS
75
Tabela 17 (conclusão)
128,99; 128,85; 128,37; 128,30; 127,99; 127,73; 127,69; 127,59; 127,53; 127,09
C aromáticos
125,74 C-b
125,37 C-f
117,85 C-9
98,83 C-1
78,74; 68,53 C-3, C-4 ou C-5
76,31 C-2
76,21 C-5 ou C-4
73,95 C-7
73,64; 73,40 C benzílicos
55,23 OCH3
41,03 C-6
Massas de alta resolução (TOF MS ES; TOF MSMS ES): Figura 127, p. 131
Íon m/z
Calculado Encontrado
[M + H]+ 594,2856 594,2786
[M + Na]+ 616,2675 616,2792
[M + K]+ 632,2414 632,2346
[M + H]+ – CH3OH 562,2593 562,2767
[M + H]+ – CH3OH – PhCH2OH 454,2018 454,2053
3.3 TESTES DE ATIVIDADE ANTIBACTERIANA E ANTIFÚNGICA
Foram realizados testes de atividade antibacteriana e antifúngica com GA6 e GA7 e com as
substâncias inéditas GAX, GAXC e GAXD (FIGURA 57), no Laboratório de Controle de Qualidade
Microbiológico (Departamento de Produtos Farmacêuticos, Faculdade de Farmácia, UFMG), sob a
coordenação da Profa. Elzíria de Aguiar Nunan.
GAXD
O
OCH3
OBn
BnO
ON
HO
GAXC
O
OCH3
OBn
BnO
ON
HO
O
OCH3
OBn
BnO
ON
HO
I
GAXGA7
O
OCH3
OBnBnO
O N3
GA6
O
OCH3
OBn
BnO
OH N3
Figura 57 – Substâncias submetidas aos testes de atividade antibacteriana e antifúngica.

MATERIAL E MÉTODOS
76
A atividade antimicrobiana das substâncias foi determinada contra os fungos e bactérias
especificados na Tabela 18.
Tabela 18 – Microorganismos utilizados para os testes de atividade antimicrobiana
Bactérias Gram-positivas Bactérias Gram-negativas Fungos
Bacillus subtilis ATCC 6633 Escherichia coli ATCC 8739 Candida albicans ATCC 10231
Micrococcus luteus ATCC 9341 Pseudomonas aeruginosa ATCC 9027 Aspergillus niger ATCC 16404
Staphylococcus aureus ATCC 6538P - Saccharomyces cerevisae ATCC 2601
Foram utilizados como padrões internos: cloranfenicol (1 mg/mL) para as bactérias, exceto para P.
aeruginosa, gentamicina (1 mg/mL) para P. aeruginosa e anfotericina B (2 mg/mL) para fungos.
Prepararam-se inóculos a 0,05% v/v, a partir de culturas de 24 horas, suspensas em solução
salina 0,9% estéril. Os inóculos foram padronizados em espectrofotômetro a 40% de transmitância
e a um comprimento de onda de 580 nm, empregando-se solução salina como branco. Para B.
subtilis e fungos acrescentou-se à salina Tween 80 a 0,2% v/v.
As amostras foram ensaiadas em triplicata, empregando-se o método convencional de difusão em
ágar (FARMACOPÉIA..., 1988). Discos de papéis estéreis (diâmetro de 6 mm) foram impregnados
com 20 µL das soluções das substâncias GA6, GA7, GAX, GAXC e GAXD (1 mg/mL em
clorofórmio). Como controle positivo, os discos foram impregnados com 10 µL das soluções de
cloranfenicol, gentamicina e anfotericina B, em triplicata. Como prova em branco, os discos foram
impregnados com clorofórmio, em triplicata.
As placas foram preparadas adicionando-se 20 mL de meio-base e, após solidificação deste, 5 mL
de meio superfície contendo inóculos dos microorganismos. Os discos impregnados com as
substâncias testadas, padrões internos e clorofórmio foram depositados sobre os meios de cultura
nas placas. Depois de uma hora, as placas foram incubadas a 36 °C por 24 horas. Os halos de
inibição foram medidos (em mm) com o auxílio de um paquímetro.

RESULTADOS E DISCUSSÃO
77
4 RESULTADOS E DISCUSSÃO
Conforme foi descrito no item 2 (p. 43), o substrato da reação de carbociclização radicalar
mediada por Bu3SnH, que levaria às benzomacrolactamas GAXA e/ou GAXB, foi o 4-O-alil-2,3-di-
O-benzil-6-desoxi-6-(3-iodobenzoilamino)-α-D-galactopiranosídeo de metila (GAX). Para a síntese
de GAX, planejou-se uma rota de oito etapas (FIGURA 56, p. 47) utilizando-se o glicosídeo
disponível comercialmente, α-D-galactopiranosídeo de metila, como material de partida. As
reações desenvolvidas são corriqueiras da química de carboidratos: proteção regiosseletiva de
grupos hidroxila (1a etapa), O-alquilação (2a e 6a tapas), remoção de grupo protetor (3a etapa),
substituição regiosseletiva de hidroxila por grupo abandonador (4a etapa), substituição nucleofílica
do grupo abandonador por grupo azido (5a etapa), redução quimiosseletiva do grupo azido a
amino (7a etapa) e N-acilação (8a etapa).
Para todas as substâncias obtidas foram determinados os pontos de fusão e rotação específica e
foram obtidos os espectros no IV e de RMN de 1H e de 13C. Quando se fez necessário para a
atribuição dos sinais de RMN e para as substâncias inéditas foram obtidos espectros de RMN
bidimensionais (COSY, HMQC e HMBC). Para a atribuição das bandas no IV e dos sinais de RMN
utilizaram-se como referência Silverstein e colaboradores e Pavia e colaboradores (SILVERSTEIN
et al., 1994; PAVIA et al., 2001). Para as substâncias inéditas foram também obtidos espectros de
massas de alta resolução e exatidão.
4.1 SÍNTESE DE 4,6-O-BENZILIDENO-αααα-D-GALACTOPIRANOSÍDEO DE METILA – GA2
A primeira etapa da rota de síntese envolveu a proteção das hidroxilas em C-4 e C-6 do α-D-
galactopiranosídeo de metila na forma de acetal benzilidênico (FIGURA 58).
GA2
O
OCH3
OH
HO
O
O
φ
O
OCH3
OH
HO
OHOH
GA1
t.a.
PhCHO, ZnCl2
48%
Figura 58 – Primeira etapa da rota de síntese – Formação do acetal benzilidênico.
Em sínteses envolvendo transformações em carboidratos, a formação de acetais é amplamente
utilizada na proteção seletiva de um par de hidroxilas (COLLINS; FERRIER, 1995). A conversão
de carboidratos em ésteres e éteres é outra alternativa freqüente (BINKLEY, 1988). O acetal
benzilidênico é geralmente o grupo de escolha para proteção das hidroxilas de C-4 e C-6 em D-

RESULTADOS E DISCUSSÃO
78
glicosídeos, devido à facilidade de sua formação e posterior remoção (HALL, 1980). Assim, optou-
se por esta estratégia para a proteção das hidroxilas em C-4 e C-6 do α-D-galactopiranosídeo.
Como a formação de acetais em carboidratos é reversível, os produtos formados nesta reação
são controlados termodinamicamente, ou seja, o produto formado preferencialmente é aquele
mais estável. Os acetais benzilidênicos dos D-galactopiranosídeos tendem a se formar como anéis
de seis membros, entre as hidroxilas de C-4 e C-6, com o grupo fenila ocupando a posição
equatorial. Esta preferência se deve à maior estabilidade do anel de seis membros (1,3-dioxano)
que se forma entre as hidroxilas de C-4 e C-6, em comparação com o anel de cinco membros
(1,3-dioxalano) que é formado entre as hidroxilas de C-3 e C-4, como pode ser observado na
Figura 59 (BINKLEY, 1988; COLLINS; FERRIER, 1995).
O
OCH3
OH
HO
O
O
φ
1,3-dioxano
O
OCH3
OH
O
O OHφ
1,3-dioxolano
Figura 59 – Produtos de formação do acetal benzilidênico: 1,3-dioxano (anel de seis membros) e
1,3-dioxolano (anel de cinco membros).
Do ponto de vista cinético, a hidroxila primária em C-6 é a que apresenta maior poder nucleofílico
por ser a menos impedida estericamente, de modo que a formação do 4,6-O-benzilideno-α-D-
galactopiranosídeo de metila ocorre mais rapidamente, sendo este o produto cinético. Assim,
mesmo que as condições de reação permitam a isomerização do produto cinético (FIGURA 60),
os produtos de isomerização não são isolados, uma vez que o acetal 4,6 é também o produto
termodinâmico (BINKLEY, 1988).
O
OCH3
OH
HO
O
O
H
φ +
O
OCH3
OH
O
O
O
H
H
φ
+O
OCH3
OH
HO
O
O
φH+
O
OCH3
OH
HO
O
O
φH+
Figura 60 – Mecanismo de isomerização catalisada por ácido dos acetais benzilidênicos.
Neste trabalho, o acetal benzilidênico GA2 foi obtido por reação do α-D-galactopiranosídeo de
metila com benzaldeído e cloreto de zinco, seguindo metodologia descrita por Hall (HALL, 1980).
Misturaram-se, inicialmente, o cloreto de zinco e o benzaldeído, sob agitação magnética vigorosa,
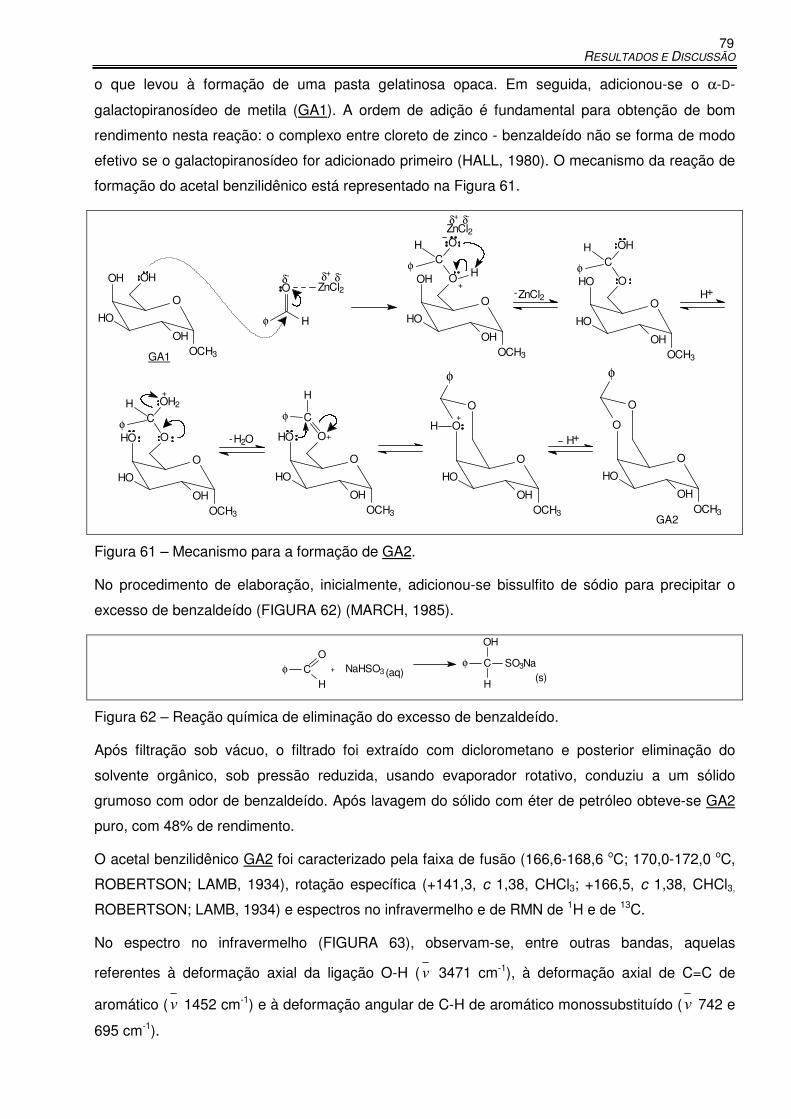
RESULTADOS E DISCUSSÃO
79
o que levou à formação de uma pasta gelatinosa opaca. Em seguida, adicionou-se o α-D-
galactopiranosídeo de metila (GA1). A ordem de adição é fundamental para obtenção de bom
rendimento nesta reação: o complexo entre cloreto de zinco - benzaldeído não se forma de modo
efetivo se o galactopiranosídeo for adicionado primeiro (HALL, 1980). O mecanismo da reação de
formação do acetal benzilidênico está representado na Figura 61.
H2O
+
O
OCH3
OH
HO
O
O
φ
H
O
OCH3
OH
HO
OH O
Cφ
H O
H+
ZnCl2δ+
δ-
δ+
ZnCl2δ-
O
OCH3
OH
HO
OH OH δ-
φ H
OO
OCH3
OH
HO
HO O
Cφ
H OH
H+
H+
O
OCH3
OH
HO
HO O
C
H
φ
+
+
O
OCH3
OH
HO
HO O
Cφ
H OH2
-ZnCl2
GA2
O
OCH3
OH
HO
O
O
φ
-
GA1
Figura 61 – Mecanismo para a formação de GA2.
No procedimento de elaboração, inicialmente, adicionou-se bissulfito de sódio para precipitar o
excesso de benzaldeído (FIGURA 62) (MARCH, 1985).
(s)C
OH
SO3Na
H
φ
(aq)NaHSO3+CO
H
φ
Figura 62 – Reação química de eliminação do excesso de benzaldeído.
Após filtração sob vácuo, o filtrado foi extraído com diclorometano e posterior eliminação do
solvente orgânico, sob pressão reduzida, usando evaporador rotativo, conduziu a um sólido
grumoso com odor de benzaldeído. Após lavagem do sólido com éter de petróleo obteve-se GA2
puro, com 48% de rendimento.
O acetal benzilidênico GA2 foi caracterizado pela faixa de fusão (166,6-168,6 oC; 170,0-172,0 oC,
ROBERTSON; LAMB, 1934), rotação específica (+141,3, c 1,38, CHCl3; +166,5, c 1,38, CHCl3,
ROBERTSON; LAMB, 1934) e espectros no infravermelho e de RMN de 1H e de 13C.
No espectro no infravermelho (FIGURA 63), observam-se, entre outras bandas, aquelas
referentes à deformação axial da ligação O-H ( ν 3471 cm-1), à deformação axial de C=C de
aromático (ν 1452 cm-1) e à deformação angular de C-H de aromático monossubstituído (ν 742 e
695 cm-1).

RESULTADOS E DISCUSSÃO
80
Figura 63 – Espectro no IV (ATR) de 4,6-O-benzilideno-α-D-galactopiranosídeo de metila – GA2.
No espectro de RMN de 1H de GA2 (FIGURA 64), os seguintes sinais foram identificados: um
multipleto em δ 7,50 – 7,33, com integral de cinco hidrogênios, correspondente aos hidrogênios
aromáticos; um simpleto em δ 5,51, com integral de um hidrogênio, correspondente ao hidrogênio
benzilidênico (H-7); um dupleto em δ 4,88, com integral de um hidrogênio e constante de
acoplamento de 2,5 Hz, característica de hidrogênio anomérico (H-1) de D-glicosídeos de
configuração α; dois dupletos duplos em δ 4,25 e 4,02, cada um com integral de um hidrogênio e
com constante de acoplamento entre si de 12,5 Hz (típica de J gem), correspondentes a H-6 e H-6’;
um simpleto em δ 3,42, com integral de três hidrogênios, referente aos hidrogênios metoxílicos; e
um sinal largo em δ 3,09, com integral de dois hidrogênios, referente aos hidrogênios das duas
hidroxilas.
A atribuição dos hidrogênios do açúcar H-2, H-3, H-4 e H-5 foi possível graças à análise do mapa
de contornos homonuclear COSY (FIGURA 65). O ponto de partida foi o sinal referente ao
hidrogênio anomérico (H-1). Pôde-se então identificar, por meio da mancha de H-1 x H-2, que o
sinal de H-2 encontra-se sobreposto ao de outro hidrogênio no multipleto em δ 3,91 – 3,89.
Partindo-se dos sinais de H-6 e H-6’ (δ 4,25 e 4,02), atribuiu-se H-5 (δ 3,62), e pela mancha de
correlação H-5 x H-4 foi possível identificar o sinal de H-4, em δ 4,19. Por exclusão, o hidrogênio
cujo sinal encontra-se sobreposto ao de H-2 é H-3, o que é compatível com as manchas de
correlação H-2 x H-3 e H-3 x H-4.
12
3
4 5
6
7
GA2
O
OCH3
OH
HO
O
O
φ

RESULTADOS E DISCUSSÃO
81
Figura 64 – Espectro de RMN de 1H (200 MHz, CDCl3) de 4,6-O-benzilideno-α-D-
galactopiranosídeo de metila – GA2.
Figura 65 – Mapa de contornos COSY (200 MHz, CDCl3) de 4,6-O-benzilideno-α-D-
galactopiranosídeo de metila – GA2.
H-1 x H-2
H-4 x H-5 e
H-5 x H-6 H-5 x H-6’
H-6’ X H-6
12
3
4 5
6
7
GA2
O
OCH3
OH
HO
O
O
φ
H-1
H aromáticos
H-7
OCH3
H-6 H-6’
OH
H açúcar
12
3
4 5
6
7
GA2
O
OCH3
OH
HO
O
O
φ
H-4
H-2 e H-3 H-5
2.15
47
3.06
29
1.00
00
0.99
83
2.08
29
1.08
81
2.07
96
1.05
35
3.02
08
2.04
57
Inte
gral
7.50
207.
4824
7.47
177.
3644
7.34
487.
3316
5.50
82
4.88
464.
8719
4.27
994.
2736
4.21
744.
2111
4.19
224.
1846
4.05
844.
0514
3.99
593.
9883
3.91
453.
9031
3.89
243.
6235
3.41
65
3.09
40
(ppm)
3.03.23.43.63.84.04.24.44.64.85.05.25.45.65.86.06.26.46.66.87.07.27.47.6
4.27
994.
2736
4.21
744.
2111
4.19
224.
1846
4.05
844.
0514
3.99
593.
9883
3.91
453.
9031
3.89
24
3.62
35
(ppm)
3.63.73.83.94.04.14.24.3

RESULTADOS E DISCUSSÃO
82
A análise do espectro de RMN de 13C, auxiliada pelo espectro de DEPT 135 (FIGURA 66),
permitiu a atribuição segura dos seguintes sinais de ressonância: em δ 137,55, sinal do carbono
ipso; em δ 129,01, 128,09 e 126,23, sinais referentes a carbonos aromáticos; em δ 69,18 sinal
referente a C-6 e, finalmente, em δ 55,52 sinal correspondente ao carbono metoxílico.
Atribuição inequívoca dos demais sinais foi possível graças ao mapa de contornos heteronuclear
HMQC (FIGURA 67). Como todos os hidrogênios haviam sido previamente atribuídos
inequivocamente, observação direta das manchas de correlação permitiu atribuição dos seguintes
sinais: de C-7 em δ 101,04; de C-1 em δ 100,23; de C-4 em δ 75,91; de C-2 e C-3 em δ 69,33 e
69,22 (não sendo possível definir qual sinal corresponde a qual carbono, pois os sinais de H-2 e
H-3 encontram-se sobrepostos) e o de C-5 em δ 62,59.
Figura 66 – Espectro de RMN de 13C e DEPT 135 (50 MHz, CDCl3) de 4,6-O-benzilideno-α-D-
galactopiranosídeo de metila – GA2.
137.
5523
129.
0119
128.
0977
126.
2311
101.
0442
100.
2290
77.6
400
77.0
000
76.3
677
75.9
105
69.3
281
69.2
215
69.1
757
62.5
857
55.5
157
(ppm)
556065707580859095100105110115120125130135140
C ipso
C-7 C-1
OCH3
C-6
12
3
4 5
6
7
GA2
O
OCH3
OH
HO
O
O
φ
C aromáticos
C-4 C-5
C-2 e C-3

RESULTADOS E DISCUSSÃO
83
Figura 67 – Mapa de contornos HMQC (200 MHz, CDCl3) de 4,6-O-benzilideno-α-D-
galactopiranosídeo de metila – GA2.
4.2 SÍNTESE DE 2,3-DI-O-BENZIL-4,6-O-BENZILIDENO-αααα-D-GALACTOPIRANOSÍDEO DE METILA – GA3
Nesta etapa de síntese procedeu-se à O-benzilação (formação de éter benzílico) das hidroxilas
em C-2 e C-3 (FIGURA 68).
O
OCH3
OBn
BnO
O
O
φ
GA3GA2
O
OCH3
OH
HO
O
O
φ
100 oC
BnCl, KOH
95%
Figura 68 – Segunda etapa da rota de síntese – O-benzilação das hidroxilas em C-2 e C-3 de GA2.
Esta etapa tem como objetivo proteger as hidroxilas em C-2 e C-3 para que estes dois centros não
fossem alvo das reações subseqüentes previstas para transformações em C-4 e C-6 (FIGURA 56,
p. 47). O grupo benzila foi o grupo protetor escolhido pela facilidade de introduzi-lo e por
apresentar estabilidade em meio ácido e básico e resistir às condições das reações subseqüentes
H-6 x C-6
H-6’ x C-6
H-5 x C-5
H-2+H-3 x
C-2+C-3
H-4 x C-4
H-1 x C-1
H-7 x C-7
OCH3 x OCH3
12
3
4 5
6
7
GA2
O
OCH3
OH
HO
O
O
φ

RESULTADOS E DISCUSSÃO
84
(BINKLEY, 1988; COLLINS; FERRIER, 1995). Além disso, grupo benziloxila em C-2 e C-3 faz
parte da estrutura dos macrociclos desejados.
A metodologia clássica de síntese de éteres assimétricos é conhecida como “Síntese de
Williamson”: o álcool a ser alquilado é transformado em alcóxido por reação com uma base e o
alcóxido formado promove ataque nucleofílico em um haleto de alquila. Trata-se de reação de
substituição nucleofílica bimolecular (SN2), que é favorecida quando é desenvolvida em solventes
polares e apróticos, como a DMF e DMSO. A escolha da base para formar o alcóxido depende da
acidez da hidroxila a ser alquilada, assim como da estabilidade do produto formado frente à base.
Como os álcoois são, em geral, ácidos fracos, normalmente são escolhidas bases muito fortes
para a formação do alcóxido, como os hidretos, amidetos e alcóxidos (HENDRICKSON et al.,
1970; MARCH, 1985; SOLOMONS; FRYHLE, 2000). Para o desenvolvimento das reações são
necessários cuidados especiais, como adição lenta e sob resfriamento da solução do álcool sobre
a base, uma vez que se forma hidrogênio nesta reação. Além desses cuidados, há necessidade
de se utilizar solvente anidro para evitar o consumo do hidreto pela água. Outro fator que contribui
para tornar essa metodologia de síntese de éteres lenta e trabalhosa é a dificuldade de eliminação
do solvente, necessária para o isolamento do produto. Devido ao alto ponto de ebulição, o
procedimento mais recomendado para eliminação do solvente, sem que haja degradação do
produto sintetizado, é a destilação sob pressão reduzida.
Embora o equilíbrio das reações entre álcoois e bases mais fracas, como hidróxidos de metais
alcalinos, não desloque no sentido de formação dos alcóxidos, é possível obter éteres benzílicos
por reação de álcoois com hidróxido de potássio ou de sódio pulverizados com cloreto de benzila
em DMF ou DMSO (COLLINS; FERRIER, 1995) ou utilizando o próprio cloreto de benzila como
solvente (FRÉCHET; BAER, 1975).
Uma metodologia alternativa e mais simples, que começou a ser utilizada em meados do século
XX, é a catálise de transferência de fase (LUCCHESE; MARZORATI, 2000; LANG; COMASSETO,
1988). A discussão sobre catálise em transferência de fase encontra-se na página 99.
A metodologia clássica de O-alquilação (hidreto de sódio e DMF) e a catálise em transferência de
fase foram empregadas para a síntese de GA3, mas não foi possível obter o produto desejado
(BINATTI, 2001). Assim, neste trabalho, a O-benzilação de GA2 foi realizada conforme
metodologia descrita por Fréchet e Baer (1975). O cloreto de benzila foi utilizado como agente
alquilante e solvente e o hidróxido de potássio como base. Após 8 horas de reação, sob agitação
e temperatura de 100 oC, procedeu-se à elaboração: adição de água, extração com
diclorometano, eliminação do solvente e precipitação do produto por adição de hexano. O produto
di-O-benzilado GA3 foi obtido com 95% de rendimento.
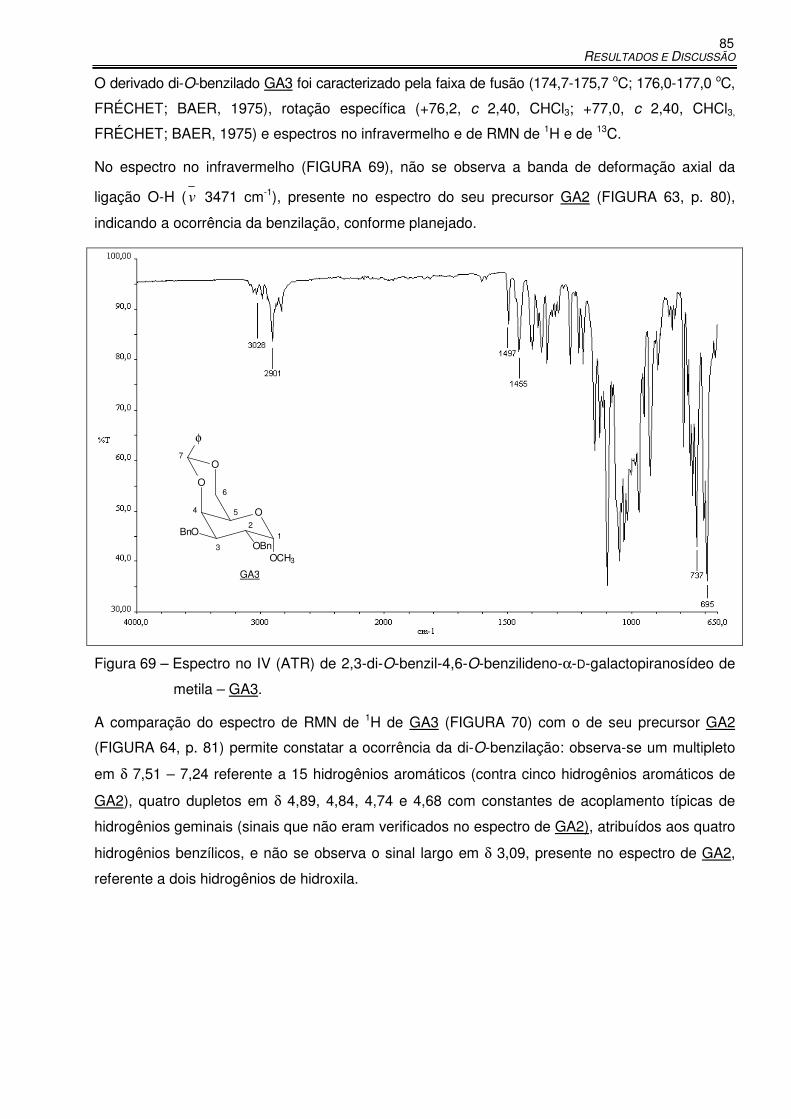
RESULTADOS E DISCUSSÃO
85
O derivado di-O-benzilado GA3 foi caracterizado pela faixa de fusão (174,7-175,7 oC; 176,0-177,0 oC,
FRÉCHET; BAER, 1975), rotação específica (+76,2, c 2,40, CHCl3; +77,0, c 2,40, CHCl3,
FRÉCHET; BAER, 1975) e espectros no infravermelho e de RMN de 1H e de 13C.
No espectro no infravermelho (FIGURA 69), não se observa a banda de deformação axial da
ligação O-H (ν 3471 cm-1), presente no espectro do seu precursor GA2 (FIGURA 63, p. 80),
indicando a ocorrência da benzilação, conforme planejado.
Figura 69 – Espectro no IV (ATR) de 2,3-di-O-benzil-4,6-O-benzilideno-α-D-galactopiranosídeo de
metila – GA3.
A comparação do espectro de RMN de 1H de GA3 (FIGURA 70) com o de seu precursor GA2
(FIGURA 64, p. 81) permite constatar a ocorrência da di-O-benzilação: observa-se um multipleto
em δ 7,51 – 7,24 referente a 15 hidrogênios aromáticos (contra cinco hidrogênios aromáticos de
GA2), quatro dupletos em δ 4,89, 4,84, 4,74 e 4,68 com constantes de acoplamento típicas de
hidrogênios geminais (sinais que não eram verificados no espectro de GA2), atribuídos aos quatro
hidrogênios benzílicos, e não se observa o sinal largo em δ 3,09, presente no espectro de GA2,
referente a dois hidrogênios de hidroxila.
12
3
4 5
6
7
O
OCH3
OBnBnO
O
O
φ
GA3

RESULTADOS E DISCUSSÃO
86
Figura 70 – Espectro de RMN de 1H (200 MHz, CDCl3) de 2,3-di-O-benzil-4,6-O-benzilideno-α-D-
galactopiranosídeo de metila – GA3.
Comparando-se o espectro de RMN de 13C de GA3 (FIGURA 71) com o espectro de GA2
(FIGURA 66, p. 82) notam-se as seguintes modificações principais: presença de dois sinais a mais
referentes a carbonos ipso em δ 138,73 e 138,51 (dos dois grupos benzila), mais sinais
correspondentes a carbonos aromáticos e dois sinais em δ 73,77 e 72,15 referentes aos carbonos
benzílicos.
15.4
71
1.00
00
5.00
44
2.04
29
3.05
39
1.01
39
3.00
27
Inte
gral
7.50
587.
4963
7.42
947.
4010
7.39
227.
3562
7.34
107.
3303
7.30
897.
2981
7.24
38
5.47
79
4.91
554.
8713
4.85
554.
8101
4.76
654.
7072
4.64
72
4.24
014.
2345
4.17
774.
1026
4.08
614.
0521
4.03
574.
0262
4.01
864.
0041
3.98
833.
9637
3.95
493.
9378
3.57
303.
3786
(ppm)
3.23.64.04.44.85.25.66.06.46.87.27.6
4.91
55
4.87
134.
8555
4.81
01
4.76
65
4.70
72
4.64
72
(ppm)
4.704.804.90
4.24
014.
2345
4.17
77
4.10
264.
0861
4.05
214.
0357
4.02
624.
0186
4.00
413.
9883
3.96
373.
9549
3.93
78
(ppm)
4.004.104.20
H aromáticos
H-7 OCH3 H açúcar H benzílicos
e H-1
12
3
4 5
6
7
O
OCH3
OBnBnO
O
O
φ
GA3 φ

RESULTADOS E DISCUSSÃO
87
Figura 71 – Espectro de RMN de 13C e DEPT 135 (50 MHz, CDCl3) de 2,3-di-O-benzil-4,6-O-
benzilideno-α-D-galactopiranosídeo de metila – GA3.
4.3 SÍNTESE DE 2,3-DI-O-BENZIL-αααα-D-GALACTOPIRANOSÍDEO DE METILA – GA4
Na terceira etapa de síntese realizou-se a remoção do grupo acetal benzilidênico (FIGURA 72),
para que pudessem ser realizadas as transformações planejadas em C-4 e C-6.
CH3COOH/H2O 1:1
81%MO
HCl1 mol/L, CH3COCH3Refluxo
79%OU
GA4
O
OCH3
OBnBnO
OHOH
O
OCH3
OBnBnO
O
O
φ
GA3
Figura 72 – Terceira etapa da rota de síntese – Remoção do grupo acetal benzilidênico de GA3.
A remoção do grupo acetal benzilidênico pode ser realizada com facilidade por meio de hidrólise
ácida, em condições brandas. Condições mais drásticas e/ou maior tempo de reação podem levar
também à hidrólise da ligação glicosídica (BINKLEY, 1988; COLLINS; FERRIER, 1995).
138.
7256
138.
5122
137.
7732
128.
8138
128.
2729
128.
0596
128.
0139
127.
6406
127.
5796
127.
5034
126.
2921
101.
0365
99.4
214
77.6
323
77.0
000
76.3
600
75.9
486
75.3
772
74.7
068
73.7
697
72.1
470
69.3
586
62.3
876
55.4
547
(ppm)
556065707580859095100105110115120125130135140
C ipso
C aromáticos
C-7 C-1 C-6
* C açúcar
OCH3 C benzílicos
12
3
4 5
6
7
O
OCH3
OBnBnO
O
O
φ
GA3
* *

RESULTADOS E DISCUSSÃO
88
Inicialmente, a hidrólise do acetal benzilidênico foi realizada utilizando-se mistura de acetona,
água e solução de ácido clorídrico 1 mol/L, sob refluxo por cerca de 7 horas (BELL; LORBER,
1940). A elaboração envolveu adição de carbonato de bário sobre a mistura de reação até
neutralização, filtração lenta e co-destilação do filtrado com etanol. Posterior CCS conduziu a GA4
com 79% de rendimento.
Devido ao tempo relativamente longo de reação, à demora nas etapas de neutralização (o
carbonato de bário permanece insolúvel no meio dificultando a reação com o ácido) e de filtração
(os sólidos, BaCO3 e BaCl2, são finamente divididos) optou-se por obter GA4 utilizando uma
metodologia mais simples e rápida.
Couri e colaboradores descrevem metodologia para remoção de acetais benzilidênicos em
carboidratos, utilizando irradiação microondas (COURI et al., 2005). Pequenas modificações na
metodologia original permitiram obtenção de GA4 de modo mais rápido. Utilizou-se aparelho de
microondas doméstico adaptado, cuja fotografia pode ser visualizada na Figura 73.
Figura 73 – Fotografia de aparelho de microondas doméstico adaptado utilizado na remoção do
grupo acetal benzilidênico de GA3 (Departamento de Química/ICEx/UFMG).
O intermediário GA3 foi misturado a uma solução de ácido acético/água destilada 1:1 v/v e a
solução foi aquecida por meio de irradiação microondas durante 18 minutos, em uma potência de
aproximadamente 250 W. Posterior eliminação da água por destilação à pressão reduzida e
CCS conduziram a GA4 com 81% de rendimento.
O desenvolvimento da primeira reação realizada foi acompanhado, como de costume, por CCD.
Entretanto, observou-se que havia uma relação direta entre o consumo de GA3 e a formação de
GA4, verificados por CCD, com o aspecto da mistura de reação. Como GA3 é insolúvel em meio
aquoso e GA4 é solúvel, no início há grande quantidade de sólido no meio de reação. À medida
que a reação se desenvolve, nota-se que a mistura vai se tornando mais límpida e o término da
reação é evidenciado pela transformação completa da mistura heterogênea em homogênea.
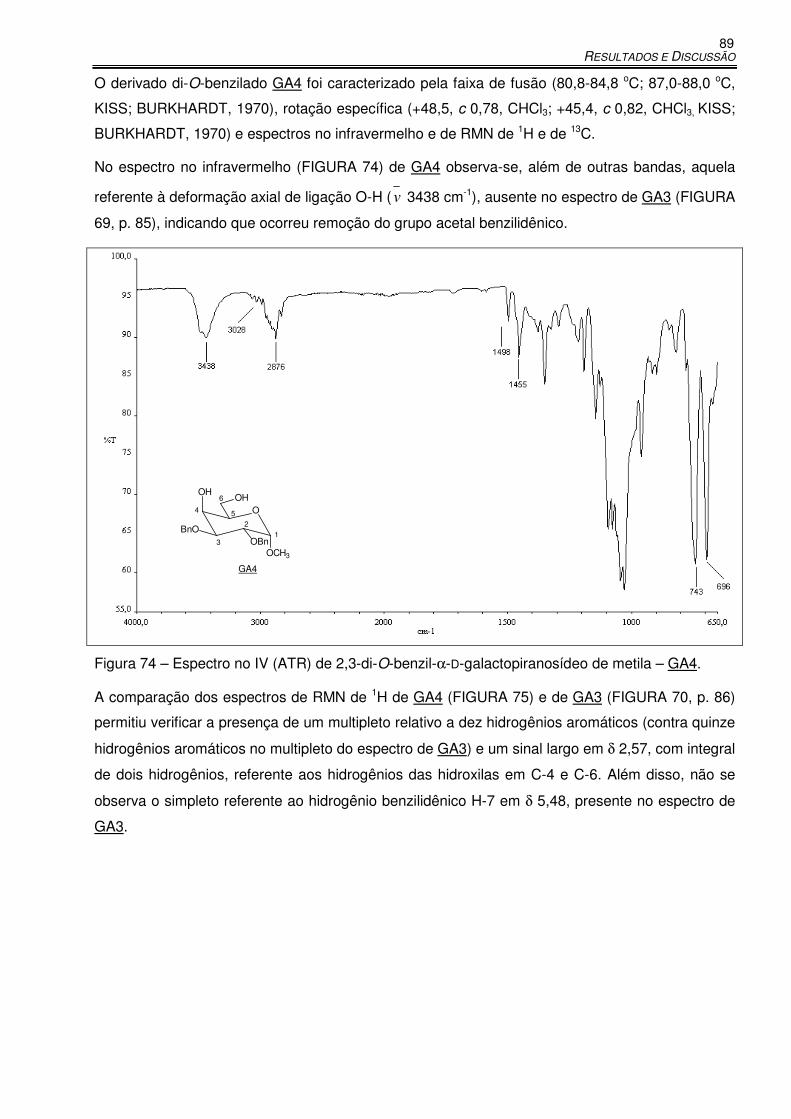
RESULTADOS E DISCUSSÃO
89
O derivado di-O-benzilado GA4 foi caracterizado pela faixa de fusão (80,8-84,8 oC; 87,0-88,0 oC,
KISS; BURKHARDT, 1970), rotação específica (+48,5, c 0,78, CHCl3; +45,4, c 0,82, CHCl3, KISS;
BURKHARDT, 1970) e espectros no infravermelho e de RMN de 1H e de 13C.
No espectro no infravermelho (FIGURA 74) de GA4 observa-se, além de outras bandas, aquela
referente à deformação axial de ligação O-H (ν 3438 cm-1), ausente no espectro de GA3 (FIGURA
69, p. 85), indicando que ocorreu remoção do grupo acetal benzilidênico.
Figura 74 – Espectro no IV (ATR) de 2,3-di-O-benzil-α-D-galactopiranosídeo de metila – GA4.
A comparação dos espectros de RMN de 1H de GA4 (FIGURA 75) e de GA3 (FIGURA 70, p. 86)
permitiu verificar a presença de um multipleto relativo a dez hidrogênios aromáticos (contra quinze
hidrogênios aromáticos no multipleto do espectro de GA3) e um sinal largo em δ 2,57, com integral
de dois hidrogênios, referente aos hidrogênios das hidroxilas em C-4 e C-6. Além disso, não se
observa o simpleto referente ao hidrogênio benzilidênico H-7 em δ 5,48, presente no espectro de
GA3.
6
54
3
21
O
OCH3
OBnBnO
OHOH
GA4

RESULTADOS E DISCUSSÃO
90
Figura 75 – Espectro de RMN de 1H (200 MHz, CDCl3) de 2,3-di-O-benzil-α-D-galactopiranosídeo
de metila – GA4.
No espectro de RMN de 13C de GA4 (FIGURA 76) não se observam os sinais referentes ao
carbono benzilidênico (δ 101,04) e a um carbono ipso (δ 137,77) presentes no espectro de GA3
(FIGURA 71, p. 87). Além disso, o número de sinais de carbonos aromáticos é menor.
10.0
84
5.00
00
1.01
29
3.00
91
2.00
77
3.00
39
2.02
11
Inte
gral
7.37
707.
3644
7.34
807.
3354
7.32
787.
3227
7.30
897.
3007
7.29
437.
2558
4.84
294.
7817
4.71
484.
6977
4.68
764.
6573
4.62
71
4.04
264.
0363
3.94
293.
9088
3.88
103.
8652
3.85
893.
8514
3.79
833.
7775
3.75
293.
7472
3.73
653.
7163
3.37
04
2.56
50
(ppm)
2.02.42.83.23.64.04.44.85.25.66.06.46.87.27.68.0
4.84
29
4.78
17
4.71
484.
6977
4.68
764.
6573
4.62
71(ppm)
4.604.704.804.90
4.04
264.
0363
3.94
293.
9088
3.88
103.
8652
3.85
893.
8514
3.79
833.
7775
3.75
293.
7472
3.73
653.
7163
(ppm)
3.83.94.0
H aromáticos OCH3
H açúcar
H benzílicos e H-1
OH
6
54
3
21
O
OCH3
OBnBnO
OHOH
GA4
H-6 H-6’ H-2

RESULTADOS E DISCUSSÃO
91
Figura 76 – Espectro de RMN de 13C e DEPT 135 (50 MHz, CDCl3) de 2,3-di-O-benzil-α-D-
galactopiranosídeo de metila – GA4.
4.4 SÍNTESE DE 2,3-DI-O-BENZIL-6-DESOXI-6-IODO-αααα-D-GALACTOPIRANOSÍDEO DE METILA – GA5
Na quarta etapa da rota de síntese promoveu-se a substituição regiosseletiva da hidroxila de C-6
por iodo (FIGURA 77).
73%
PhCH3, Refluxo
I2, PPh3, imidazol
GA5
O
OCH3
OBn
BnO
OH I
GA4
O
OCH3
OBnBnO
OHOH
Figura 77 – Quarta etapa da rota de síntese – Substituição seletiva da hidroxila de C-6 de GA4 por
iodo.
Segundo Binkley, a transformação de hidroxila em grupo abandonador, seguida de substituição
por grupo azido, constitui a estratégia mais adequada para a obtenção de desoxiaminoaçúcares
(BINKLEY, 1988), como é o caso de GA7. Optou-se por substituir a hidroxila de C-6 por iodo, o
halogênio mais facilmente deslocado por um nucleófilo como o íon azida (ANISUZZAMAN et
al.,1978).
138.
2075
137.
9408
128.
4710
128.
3567
127.
9758
127.
8996
127.
7929
98.5
453
77.6
323
77.2
819
77.0
000
76.3
600
75.5
601
73.4
498
72.8
707
69.0
386
68.8
558
62.8
904
55.2
871
(ppm)
556065707580859095100105110115120125130135140
C ipso
C aromáticos
C-1
OCH3
C-6
* C açúcar
C benzílicos
** * *
6
54
3
21
O
OCH3
OBnBnO
OHOH
GA4

RESULTADOS E DISCUSSÃO
92
Uma variedade de reagentes são utilizados para a substituição regiosseletiva da hidroxila em C-6
de carboidratos por halogênio, entre eles cloreto de metanossulfonila em DMF, N-halosuccinimida
e trifenilfosfina em DMF e tetra-haleto de carbono e triarilfosfina em piridina (ANISUZZAMAN et
al.,1978).
A estratégia clássica para obtenção de 6-desoxi-iodo-açúcares consiste na transformação da
hidroxila de C-6 em ésteres sulfônicos (tosilato e mesilato) e subseqüente substituição dos grupos
O-tosila e O-mesila por iodeto (GAREGG; SAMUELSSON, 1980). Um método alternativo para a
substituição de hidroxila por iodo, que vem sendo amplamente utilizado na química de
carboidratos e foi utilizado neste trabalho para a síntese de GA5 foi descrito por Garegg e
Samuelsson. Os reagentes são imidazol, trifenilfosfina e iodo e o solvente é o tolueno. Na
ausência de imidazol, o aduto formado entre trifenilfosfina e iodo é pouco solúvel em tolueno, o
que dificulta a ocorrência da reação. Adição do imidazol leva à formação de um complexo
parcialmente solúvel que rapidamente reage com o álcool. O mecanismo proposto para a reação
de substituição de hidroxila por iodo (SN2) (GAREGG; SAMUELSSON, 1980) está representado
na Figura 78.
+
GA4
O
OCH3
OBn
BnO
OH OH
NN Pφ3 I
+
- N
N
H
O
OCH3
OBn
BnO
OH I
GA5
I
φ3P O
+
O
OCH3
OBn
BnO
OH O Pφ3
φ3P + I I φ3P I I+
N
N
H
+- HI N
N Pφ3 I+
Figura 78 – Mecanismo de substituição regiosseletiva da hidroxila de C-6 de GA4 por iodo.
É possível obter o produto de substituição regiosseletiva pelo fato de a hidroxila primária (de C-6)
apresentar maior poder nucleofílico para o ataque ao átomo de fósforo que a hidroxila secundária
(de C-4), em função do menor impedimento estérico. No entanto, para evitar que o produto
diiodado seja formado em quantidades apreciáveis é preciso que o desenvolvimento da reação
seja controlado cuidadosamente por CCD.
Após 3 horas de reação sob refluxo, sucederam-se processos de partição, e a eliminação do
solvente orgânico conduziu a um resíduo contendo o produto, trifenilfosfina e óxido de
trifenilfosfina. O derivado monoiodado GA5 foi obtido puro, após purificação por duas CCS
consecutivas, com 73% de rendimento e foi caracterizado pela faixa de fusão (97,0-103,0 oC;
103,9-105,5 oC, BINATTI et al., 2002), rotação específica (+44,6, c 2,40, CHCl3; +44,7, c 2,40,
CHCl3, BINATTI et al., 2002) e pelos espectros no infravermelho e de RMN de 1H e de 13C.

RESULTADOS E DISCUSSÃO
93
No espectro no infravermelho de GA5 (FIGURA 79) não foram observadas alterações
significativas em relação ao de GA4 (FIGURA 74, p. 89), pois GA5 também apresenta hidroxila
livre, em C-4. A presença de banda referente à deformação axial da ligação C-I não pode ser
observada, uma vez que a absorção desta ligação ocorre em ν mais baixo que o detectado pelo
espectrômetro utilizado.
Figura 79 – Espectro no IV (ATR) de 2,3-di-O-benzil-6-desoxi-6-iodo-α-D-galactopiranosídeo de
metila – GA5.
No espectro de RMN de 1H de GA5 (FIGURA 80), observa-se o sinal referente aos H-6 mais
próximo do sinal do TMS (δ 3,32) do que os sinais de H-6 e H-6’ no espectro de GA4 (FIGURA 75,
p. 90), em função do menor efeito retirador de elétrons do iodo em relação à hidroxila, com
conseqüente menor desproteção de H-6. Além disso, o sinal largo em δ 2,44 é referente a um
hidrogênio, indicando que somente uma hidroxila foi substituída.
6
54
3
21
O
OCH3
OBn
BnO
OH I
GA5

RESULTADOS E DISCUSSÃO
94
Figura 80 – Espectro de RMN de 1H (200 MHz, CDCl3) de 2,3-di-O-benzil-6-desoxi-6-iodo-α-D-
galactopiranosídeo de metila – GA5.
Comparando-se os espectros de 13C e DEPT 135 de GA5 (FIGURA 81) com os de GA4 (FIGURA
76, p. 91) identifica-se o sinal referente a C-6 mais próximo do sinal do TMS (δ 3,01), em função
do efeito do átomo pesado (iodo).
9.98
36
5.01
84
1.00
00
3.01
00
3.00
701.
9950
1.01
30
Inte
gral
7.38
407.
3726
7.35
687.
3448
7.33
797.
3278
7.32
347.
3101
7.30
257.
2950
7.28
747.
2836
4.84
804.
8429
4.79
054.
7817
4.72
244.
6731
4.66
494.
6378
4.62
144.
6125
4.10
954.
1019
4.09
434.
0887
3.91
073.
9006
3.88
543.
8747
3.86
903.
8514
3.83
683.
8223
3.80
653.
4297
3.34
013.
3363
3.30
35
2.44
26
(ppm)
2.02.53.03.54.04.55.05.56.06.57.07.58.0
4.84
804.
8429
4.79
054.
7817
4.72
244.
6731
4.66
494.
6378
4.62
144.
6125
4.10
954.
1019
4.09
434.
0887
(ppm)
4.24.44.64.8
3.34
013.
3363
3.30
35
(ppm)
3.30
H aromáticos
OCH3
H-6 e H-6’
H açúcar
H benzílicos e H-1
6
54
3
21
O
OCH3
OBn
BnO
OH I
GA5
OH
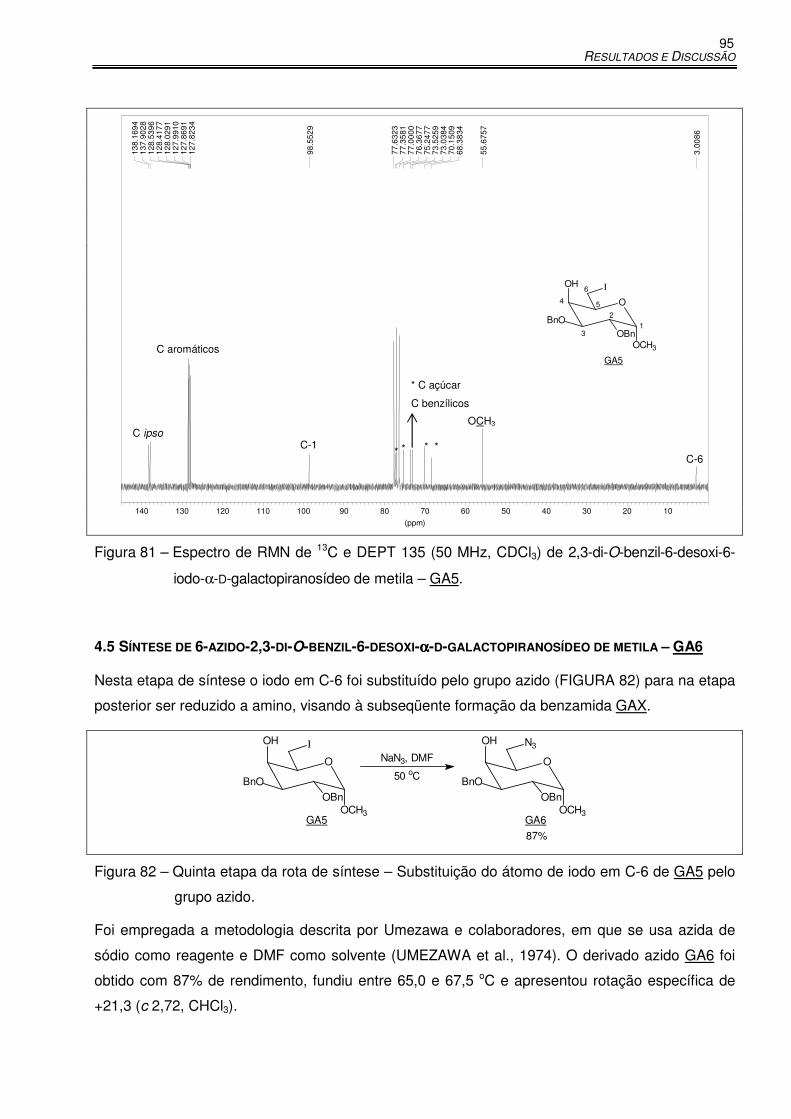
RESULTADOS E DISCUSSÃO
95
Figura 81 – Espectro de RMN de 13C e DEPT 135 (50 MHz, CDCl3) de 2,3-di-O-benzil-6-desoxi-6-
iodo-α-D-galactopiranosídeo de metila – GA5.
4.5 SÍNTESE DE 6-AZIDO-2,3-DI-O-BENZIL-6-DESOXI-αααα-D-GALACTOPIRANOSÍDEO DE METILA – GA6
Nesta etapa de síntese o iodo em C-6 foi substituído pelo grupo azido (FIGURA 82) para na etapa
posterior ser reduzido a amino, visando à subseqüente formação da benzamida GAX.
GA6
O
OCH3
OBn
BnO
OH N3
GA5
O
OCH3
OBn
BnO
OH I
50 oC
NaN3, DMF
87%
Figura 82 – Quinta etapa da rota de síntese – Substituição do átomo de iodo em C-6 de GA5 pelo
grupo azido.
Foi empregada a metodologia descrita por Umezawa e colaboradores, em que se usa azida de
sódio como reagente e DMF como solvente (UMEZAWA et al., 1974). O derivado azido GA6 foi
obtido com 87% de rendimento, fundiu entre 65,0 e 67,5 oC e apresentou rotação específica de
+21,3 (c 2,72, CHCl3).
138.
1694
137.
9028
128.
5396
128.
4177
128.
0291
127.
9910
127.
8691
127.
8234
98.5
529
77.6
323
77.3
581
77.0
000
76.3
677
75.2
477
73.5
259
73.0
384
70.1
509
68.3
834
55.6
757
3.00
86
(ppm)
102030405060708090100110120130140
C ipso
C aromáticos
C-1
OCH3
C-6
C benzílicos
* * * *
* C açúcar
6
54
3
21
O
OCH3
OBn
BnO
OH I
GA5

RESULTADOS E DISCUSSÃO
96
A presença de uma banda intensa em ν 2099 cm-1, no espectro no infravermelho (FIGURA 83),
atribuída à deformação axial das ligações N=N=N, foi de fundamental importância para se
constatar que ocorreu a substituição do átomo de iodo pelo grupo azido.
Figura 83 – Espectro no IV (ATR) de 6-azido-2,3-di-O-benzil-6-desoxi-α-D-galactopiranosídeo de
metila – GA6.
No espectro de RMN de 1H de GA6 (FIGURA 84) não se observam sinais que evidenciem a
presença do grupo azido. No entanto, a separação dos sinais dos hidrogênios de C-6, o que não
se observa no espectro de GA5 (FIGURA 80, p. 94) permitiu constatar que houve a substituição
do átomo de iodo.
6
54
3
21
GA6
O
OCH3
OBn
BnO
OH N3
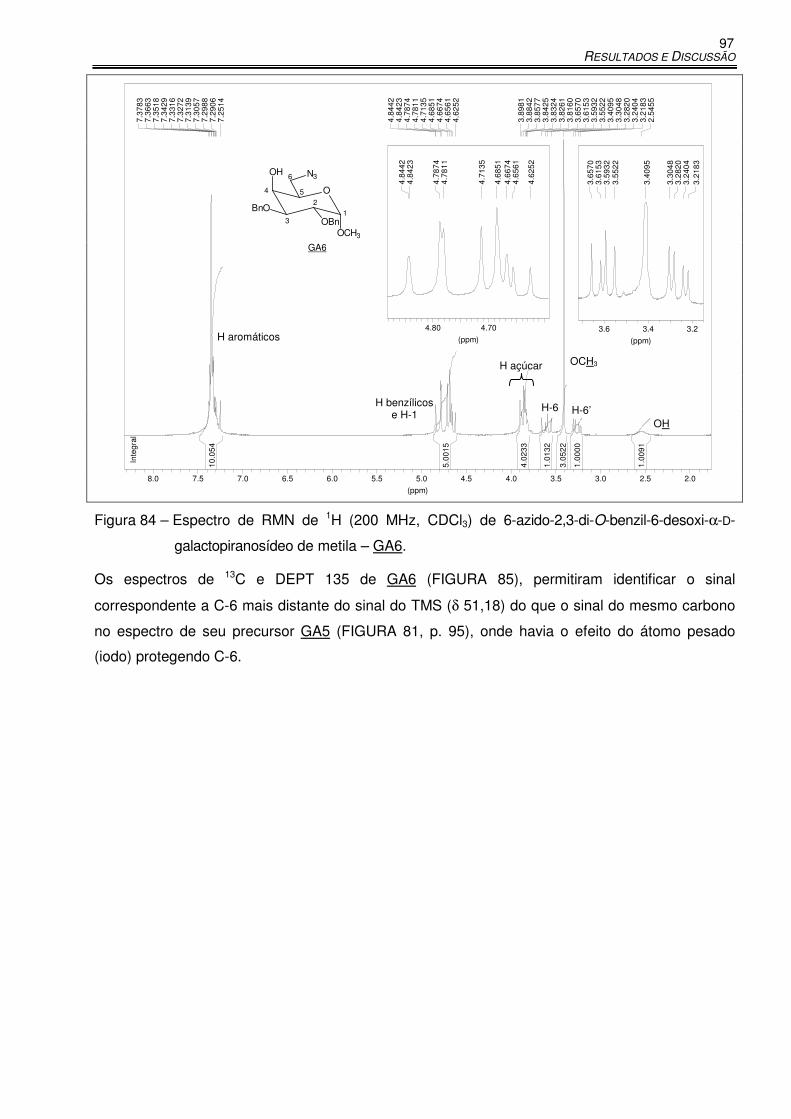
RESULTADOS E DISCUSSÃO
97
Figura 84 – Espectro de RMN de 1H (200 MHz, CDCl3) de 6-azido-2,3-di-O-benzil-6-desoxi-α-D-
galactopiranosídeo de metila – GA6.
Os espectros de 13C e DEPT 135 de GA6 (FIGURA 85), permitiram identificar o sinal
correspondente a C-6 mais distante do sinal do TMS (δ 51,18) do que o sinal do mesmo carbono
no espectro de seu precursor GA5 (FIGURA 81, p. 95), onde havia o efeito do átomo pesado
(iodo) protegendo C-6.
10.0
54
5.00
15
4.02
33
1.01
32
3.05
22
1.00
00
1.00
91
Inte
gral
7.37
837.
3663
7.35
187.
3429
7.33
167.
3272
7.31
397.
3057
7.29
887.
2906
7.25
14
4.84
424.
8423
4.78
744.
7811
4.71
354.
6851
4.66
744.
6561
4.62
52
3.89
813.
8842
3.85
773.
8425
3.83
243.
8261
3.81
603.
6570
3.61
533.
5932
3.55
223.
4095
3.30
483.
2820
3.24
043.
2183
2.54
55
(ppm)
2.02.53.03.54.04.55.05.56.06.57.07.58.0
4.84
424.
8423
4.78
744.
7811
4.71
35
4.68
514.
6674
4.65
61
4.62
52
(ppm)
4.704.80
3.65
703.
6153
3.59
323.
5522
3.40
95
3.30
483.
2820
3.24
043.
2183
(ppm)
3.23.43.6H aromáticos
OH
OCH3
H-6 H-6’
H açúcar
H benzílicos e H-1
6
54
3
21
GA6
O
OCH3
OBn
BnO
OH N3
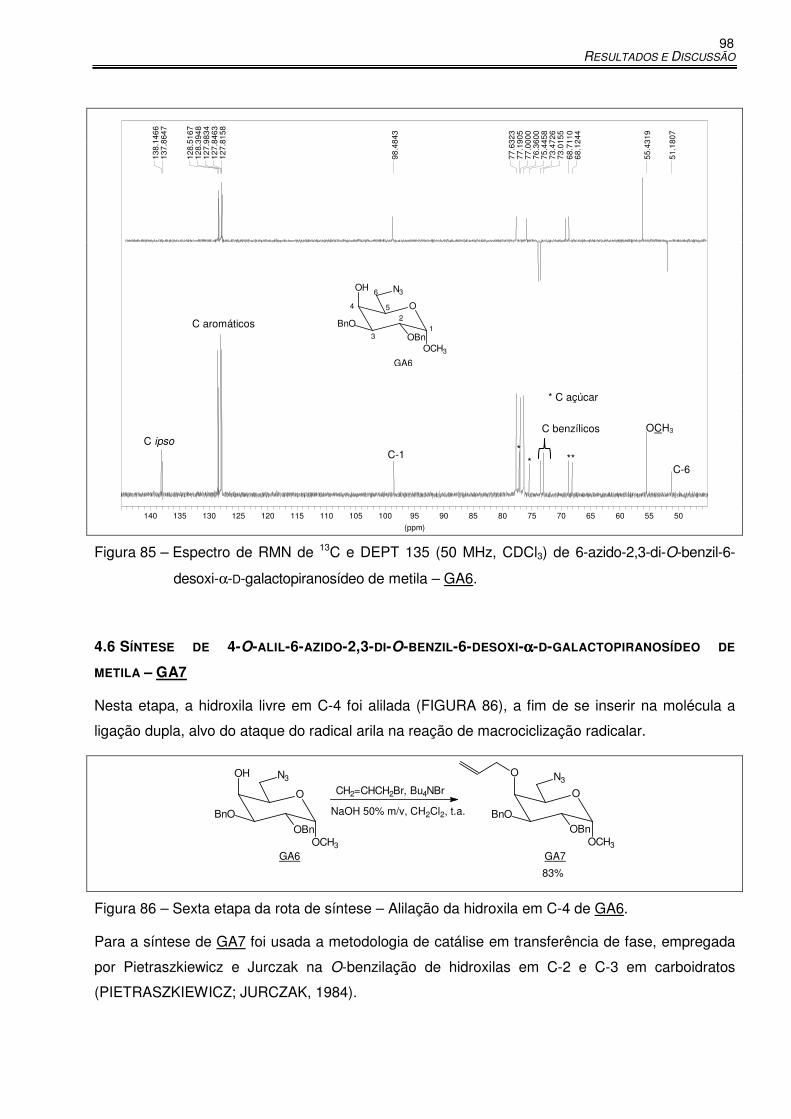
RESULTADOS E DISCUSSÃO
98
Figura 85 – Espectro de RMN de 13C e DEPT 135 (50 MHz, CDCl3) de 6-azido-2,3-di-O-benzil-6-
desoxi-α-D-galactopiranosídeo de metila – GA6.
4.6 SÍNTESE DE 4-O-ALIL-6-AZIDO-2,3-DI-O-BENZIL-6-DESOXI-αααα-D-GALACTOPIRANOSÍDEO DE
METILA – GA7
Nesta etapa, a hidroxila livre em C-4 foi alilada (FIGURA 86), a fim de se inserir na molécula a
ligação dupla, alvo do ataque do radical arila na reação de macrociclização radicalar.
GA7
O
OCH3
OBnBnO
O N3
GA6
O
OCH3
OBn
BnO
OH N3
NaOH 50% m/v, CH2Cl2, t.a.
CH2=CHCH2Br, Bu4NBr
83%
Figura 86 – Sexta etapa da rota de síntese – Alilação da hidroxila em C-4 de GA6.
Para a síntese de GA7 foi usada a metodologia de catálise em transferência de fase, empregada
por Pietraszkiewicz e Jurczak na O-benzilação de hidroxilas em C-2 e C-3 em carboidratos
(PIETRASZKIEWICZ; JURCZAK, 1984).
138.
1466
137.
8647
128.
5167
128.
3948
127.
9834
127.
8463
127.
8158
98.4
843
77.6
323
77.1
905
77.0
000
76.3
600
75.4
458
73.4
726
73.0
155
68.7
110
68.1
244
55.4
319
51.1
807
(ppm)
50556065707580859095100105110115120125130135140
C ipso
C aromáticos
C-1
OCH3
C-6
6
54
3
21
GA6
O
OCH3
OBn
BnO
OH N3
C benzílicos
* * **
* C açúcar

RESULTADOS E DISCUSSÃO
99
Na catálise de transferência de fase são utilizados um solvente orgânico (imiscível com a água),
água, hidróxido ou carbonato de sódio como base e um transferidor de fase. O reagente iônico
(hidróxido ou carbonato de sódio) permanece na fase aquosa e os reagentes orgânicos (haleto de
alquila e álcool) no solvente orgânico. O transferidor de fase, solúvel tanto em água como em
solvente orgânico, é o responsável pela transferência da base para a fase orgânica, possibilitando
assim a formação do alcóxido, que reage com o haleto de alquila levando à formação do éter
(FIGURA 87) (SOLOMONS; FRYHLE, 2000; LANG; COMASSETO, 1988; LUCCHESE;
MARZORATI, 2000). Para que esta metodologia seja empregada com sucesso um fator
determinante é a agitação vigorosa, que permite maior contato entre as fases orgânica e aquosa
(LANG; COMASSETO, 1988).
ROH GA6
RO GA7
=
=
+ +
+ + H2O
++
Na+ -OH(aq)
Na+Br-(aq)(org/aq) (org/aq)
ROH (org) (org)[n-Bu4N
+ -OR](org/aq)
(org/aq)Br (org) (org)
RO
[n-Bu4N+ Br-]
[n-Bu4N+ -OH]
[n-Bu4N+ Br-]
(org)[n-Bu4N
+ -OR]
[n-Bu4N+ -OH]
Figura 87 – Equilíbrios envolvidos na O-alilação de GA6 por reação de catálise de transferência de
fase.
Dentre as vantagens desta técnica, em relação à metodologia clássica (hidreto de sódio como
base e solvente polar aprótico) estão a utilização de solventes sem a necessidade de tratamento
prévio, uso de bases mais fracas, como hidróxidos ou carbonatos, no lugar de bases como
hidretos, amidetos e alcóxidos, além da simplicidade operacional (LUCCHESE; MARZORATI,
2000). Por estes motivos esta foi a técnica escolhida para realizar a O-alilação da hidroxila em C-4
de GA6.
Utilizou-se diclorometano como solvente orgânico, brometo de tetrabutilamônio como transferidor
de fase, solução aquosa de hidróxido de sódio 50% m/v e brometo de alila como agente
alquilante.
O derivado GA7 foi obtido com 83% de rendimento, apresentou faixa de fusão de 39,6-41,8 oC e
rotação específica de +17,9 (c 2,80, CHCl3).
No espectro no infravermelho (FIGURA 88), não se observou a banda de deformação axial da
ligação O-H, presente no espectro de GA6 (FIGURA 83, p. 96), indicando que a O-alilação
ocorreu com sucesso.

RESULTADOS E DISCUSSÃO
100
Figura 88 – Espectro no IV (ATR) de 4-O-alil-6-azido-2,3-di-O-benzil-6-desoxi-α-D-
galactopiranosídeo de metila – GA7.
A presença de sinais entre δ 5,99 e 4,03, característicos de hidrogênios olefínicos, no espectro de
RMN de 1H do produto isolado da reação (FIGURAS 89 e 90) e a ausência de sinal relativo a
hidrogênio de hidroxila, presente no espectro de GA6 (FIGURA 84, p. 97), indicaram a formação
de GA7.
A atribuição mais detalhada dos sinais dos hidrogênios do grupo alila e de alguns hidrogênios do
açúcar foi possível graças à análise do mapa de contornos homonuclear COSY (FIGURA 91).
6
54
3
21
O
OCH3
OBnBnO
O N3
GA7
7
8
9

RESULTADOS E DISCUSSÃO
101
Figura 89 – Espectro de RMN de 1H (200 MHz, CDCl3) de 4-O-alil-6-azido-2,3-di-O-benzil-6-desoxi-α-D-galactopiranosídeo de metila – GA7.
Figura 90 – Seção expandida na região de δ 6,2 – 4,0 do espectro de RMN de 1H (200 MHz, CDCl3)
de 4-O-alil-6-azido-2,3-di-O-benzil-6-desoxi-α-D-galactopiranosídeo de metila – GA7.
10.0
06
1.00
00
2.01
33
5.00
28
1.00
45
1.03
34
2.00
95
1.04
75
1.01
23
0.99
72
3.00
51
1.01
06
Inte
gral
7.37
207.
3480
7.32
537.
3133
7.28
307.
2489
5.99
105.
9891
5.96
265.
9575
5.93
865.
9304
5.91
155.
9045
5.87
865.
8723
5.85
285.
8439
5.82
635.
8199
5.79
415.
7928
5.26
585.
2582
5.19
895.
1806
5.17
305.
1478
4.86
884.
8618
4.81
074.
8019
4.73
314.
6820
4.67
444.
6226
4.49
514.
4680
4.43
144.
4061
4.12
344.
0887
4.06
034.
0256
4.00
353.
9864
3.95
303.
9366
3.90
123.
8886
3.85
143.
8368
3.81
473.
7895
3.70
683.
6992
3.61
723.
5768
3.55
533.
5149
3.39
823.
2221
3.19
753.
1602
3.13
50
(ppm)
2.83.23.64.04.44.85.25.66.06.46.87.27.68.0
3.98
643.
9530
3.93
663.
9012
3.88
863.
8514
3.83
683.
8147
3.78
95
3.70
683.
6992
3.61
72
3.57
683.
5553
3.51
49
3.39
82
3.22
213.
1975
3.16
023.
1350
(ppm)
3.13.23.33.43.53.63.73.83.9
H aromáticos
H-7
OCH3
H-6 H-6’
H açúcar
H-2
H-8
H benzílicos e H-1
9
8
7
GA7
O
OCH3
OBnBnO
O N3
12
3
4 5
6
H-9 e H9’
H-7’
H-3, H4, H-5 1.
0000
2.01
33
5.00
28
1.00
45
1.03
34
2.00
95
Inte
gral
5.95
755.
9386
5.93
045.
9115
5.90
455.
8786
5.87
235.
8528
5.84
395.
8263
5.81
995.
7941
5.79
28
5.26
585.
2582
5.19
895.
1806
5.17
305.
1478
4.86
884.
8618
4.81
074.
8019
4.73
31
4.68
204.
6744
4.62
26
4.49
514.
4680
4.43
144.
4061
4.12
344.
0887
4.06
034.
0256
(ppm)
4.14.24.34.44.54.64.74.84.95.05.15.25.35.45.55.65.75.85.96.06.1
5.99
105.
9891
5.96
265.
9575
5.93
865.
9304
5.91
155.
9045
5.87
865.
8723
5.85
285.
8439
5.82
635.
8199
5.79
415.
7928
(ppm)
5.805.845.885.925.966.00
H-7
H-8 H-7’ H-9 e
H-9’
H benzílicos e H-1
9
8
7
GA7
O
OCH3
OBnBnO
O N3
12
3
4 5
6

RESULTADOS E DISCUSSÃO
102
Figura 91 – Mapa de contornos COSY (200 MHz, CDCl3) de 4-O-alil-6-azido-2,3-di-O-benzil-6-
desoxi-α-D-galactopiranosídeo de metila – GA7.
As principais evidências de que foi sintetizado o produto desejado GA7, obtidas nos espectros de
RMN de 13C e DEPT 135 (FIGURA 92) foram os três sinais em δ 134,84, 117,65 e 73,90,
referentes aos carbonos do grupo alila. Atribuição inequívoca de alguns sinais foi possível graças
ao mapa de contornos heteronuclear HMQC (FIGURA 93). A observação de manchas de
correlação permitiu a atribuição dos seguintes sinais: o de C-8 em δ 134,84; o de C-9 em δ 117,65; o
de C-2 em δ 78,62; o de C-7 em δ 73,90 e os dos carbonos benzílicos em δ 73,62 e 73,41. Os
outros sinais puderam ser atribuídos sem maiores dificuldades pelas análises dos espectros de
RMN de 13C e DEPT 135.
H-7 x H-8
H-8 x H-9 e
H-8 x H-9’
H-7’ x H-8 H-7’ x H-9
H-7 x H-9
H-1 x H-2 H-7 x H-7’
6
54
3
21
O
OCH3
OBnBnO
O N3
GA7
7
8
9
OCH2φ x OCH2φ

RESULTADOS E DISCUSSÃO
103
Figura 92 – Espectro de RMN de 13C e DEPT 135 (50 MHz, CDCl3) de 4-O-alil-6-azido-2,3-di-O-
benzil-6-desoxi-α-D-galactopiranosídeo de metila – GA7.
Figura 93 – Mapa de contornos HMQC (200 MHz, CDCl3) de 4-O-alil-6-azido-2,3-di-O-benzil-6-
desoxi-α-D-galactopiranosídeo de metila – GA7.
138.
5427
138.
3675
134.
8401
128.
3643
128.
3262
127.
9986
127.
7167
127.
5720
127.
4882
117.
6450
98.7
738
78.6
228
77.6
400
77.0
000
76.3
677
76.2
762
75.3
544
73.8
993
73.6
174
73.4
117
69.6
100
55.4
471
51.3
331
(ppm)
50556065707580859095100105110115120125130135140
128.
3643
128.
3262
127.
9986
127.
7167
127.
5720
127.
4882
(ppm)
127.0128.0129.078
.622
8
77.6
400
77.0
000
76.3
677
76.2
762
75.3
544
73.8
993
73.6
174
73.4
117
(ppm)
747678
C ipso
C-7
C-1
OCH3
C-6 C-9
C-8
C aromáticos
C-2
9
8
7
GA7
O
OCH3
OBnBnO
O N3
12
3
4 5
6
** *
* C açúcar
C benzílicos
6
54
3
21
O
OCH3
OBnBnO
O N3
GA7
7
8
9
H-8 x C-8
H-9 + H-9’ x C-9
H-2 x C-2
H-7 x C-7
H-7’ x C-7
H-1 x C-1
OCH2φ xOCH2φ
H-6 x C-6
H-6’ x C-6
OCH3 x OCH3

RESULTADOS E DISCUSSÃO
104
4.7 SÍNTESE DE 4-O-ALIL-6-AMINO-2,3-DI-O-BENZIL-6-DESOXI-αααα-D-GALACTOPIRANOSÍDEO DE METILA
(GA8), DE CLORETO DE 3-IODOBENZOÍLA E DE 4-O-ALIL-2,3-DI-O-BENZIL-6-DESOXI-6-(3-
IODOBENZOILAMINO)-αααα-D-GALACTOPIRANOSÍDEO DE METILA – GAX
Amidas podem ser obtidas de várias maneiras: a partir de haleto de acila, de anidridos de ácidos
carboxílicos, de ésteres e de ácidos carboxílicos utilizando-se dicicloexilcarbodiimida (DCC) como
ativante. Todos estes métodos envolvem reações de substituição nucleofílica entre a amina e o
carbono carbonílico (HENDRICKSON et al., 1970; MARCH, 1985; SOLOMONS; FRYHLE, 2000).
Para a síntese da amida GAX optou-se por utilizar a reação entre a amina GA7 e o cloreto de acila
adequado (cloreto de 3-iodobenzoíla).
4.7.1 Síntese de 4-O-alil-6-amino-2,3-di-O-benzil-6-desoxi-αααα-D-galactopiranosídeo de
metila – GA8
A sétima etapa da rota de síntese planejada para a obtenção da benzamida GAX envolve a
redução quimiosseletiva do grupo azido de GA7 a amino (FIGURA 94).
GA7
LiAlH4, THF, N2, t.a.O
OCH3
OBnBnO
O N3
GA8
O
OCH3
OBnBnO
O NH2
Figura 94 – Redução do grupo azido de GA7.
O grupo azido é susceptível à redução por diversos reagentes, tais como hidrogênio em presença
de Pd/C, sódio em presença de álcool, hidreto de lítio e alumínio e trifenilfosfina em THF e água
(HENDRICKSON et al., 1970; MARCH, 1985; SOLOMONS; FRYHLE, 2000). Entretanto, quando
se deseja redução quimiosseletiva do grupo azido na presença de ligação dupla carbono-carbono,
duas opções são utilizar hidreto de lítio e alumínio (DUFOUR et al., 1992) e trifenilfosfina em THF
e água (reação de Staudinger) (VAULTIER et al., 1983; DUFOUR et al., 1992).
Neste trabalho optou-se por utilizar o hidreto de lítio e alumínio como agente redutor, seguindo a
metodologia descrita por Dufour e colaboradores: GA7 e o agente redutor em THF anidro, sob
atomosfera de nitrogênio. Após adição de solução de hidróxido de sódio, seguiram-se extração e
eliminação do solvente (DUFOUR et al., 1992). O produto recém-obtido foi utilizado na etapa
seguinte sem purificação ou caracterização por métodos espectrométricos. No entanto, por CCD
verificou-se a formação da amina (revelação com solução alcoólica de ninhidrina) e o consumo de
GA7.

RESULTADOS E DISCUSSÃO
105
4.7.2 Síntese de Cloreto de 3-iodobenzoíla
O cloreto de 3-iodobenzoíla foi obtido a partir do ácido 3-iodobenzóico, por reação clássica de
formação de cloretos de ácido, usando cloreto de tionila (FIGURA 95) (MARCH, 1985).
Cl
O
I
OH
O
I
Refluxo
SOCl2
Figura 95 – Obtenção do cloreto de 3-iodobenzoíla a partir do ácido 3-iodobenzóico.
Seguiu-se a técnica descrita por Beak e colaboradores: ácido 3-iodobenzóico e excesso de cloreto
de tionila, sob refluxo, e remoção do excesso do reagente por codestilação com benzeno, sob
pressão reduzida, usando evaporador rotativo (BEAK et al., 1988). O cloreto de ácido recém-
sintetizado foi utilizado sem purificação adicional.
4.7.3 Síntese de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-αααα-D-galactopiranosídeo de
metila – GAX
A síntese da iodobenzamida GAX se deu por reação entre o derivado amino GA8 e o cloreto de 3-
iodobenzoíla (FIGURA 96).
NaOH 2 mol/L OU Et3N
Cl
O
IO
OCH3
OBnBnO
O NH2
GA8 GAX
O
OCH3
OBn
BnO
ON
HO
I
Figura 96 – Síntese da iodobenzamida GAX por reação entre GA8 e cloreto de 3-iodobenzoíla.
Um dos métodos mais utilizados para a preparação de amidas consiste no tratamento de cloretos
de acila com aminas. A reação é altamente exotérmica e, por este motivo, há a necessidade de
controle cuidadoso da temperatura utilizando-se banho de gelo durante o desenvolvimento da
reação (MARCH, 1985). Para que a reação seja efetiva a adição de base se faz necessária, uma
vez que há liberação de cloreto de hidrogênio. A base utilizada pode ser orgânica, como a
trietilamina (DUFOUR et al., 1992), e inorgânica, como hidróxido de sódio em solução aquosa
(BEAK et al, 1988). O procedimento em que se utiliza solução aquosa de hidróxido de sódio é
denominado Schotten–Bowmann (MARCH, 1985).
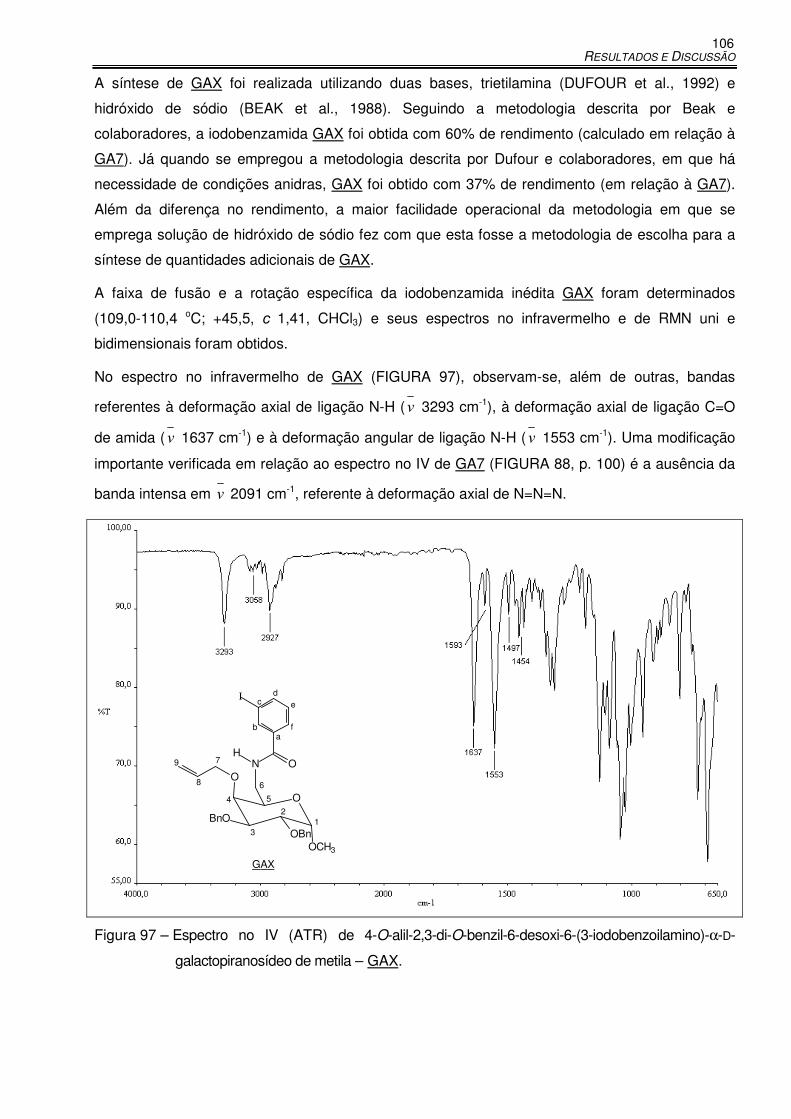
RESULTADOS E DISCUSSÃO
106
A síntese de GAX foi realizada utilizando duas bases, trietilamina (DUFOUR et al., 1992) e
hidróxido de sódio (BEAK et al., 1988). Seguindo a metodologia descrita por Beak e
colaboradores, a iodobenzamida GAX foi obtida com 60% de rendimento (calculado em relação à
GA7). Já quando se empregou a metodologia descrita por Dufour e colaboradores, em que há
necessidade de condições anidras, GAX foi obtido com 37% de rendimento (em relação à GA7).
Além da diferença no rendimento, a maior facilidade operacional da metodologia em que se
emprega solução de hidróxido de sódio fez com que esta fosse a metodologia de escolha para a
síntese de quantidades adicionais de GAX.
A faixa de fusão e a rotação específica da iodobenzamida inédita GAX foram determinados
(109,0-110,4 oC; +45,5, c 1,41, CHCl3) e seus espectros no infravermelho e de RMN uni e
bidimensionais foram obtidos.
No espectro no infravermelho de GAX (FIGURA 97), observam-se, além de outras, bandas
referentes à deformação axial de ligação N-H (ν 3293 cm-1), à deformação axial de ligação C=O
de amida ( ν 1637 cm-1) e à deformação angular de ligação N-H ( ν 1553 cm-1). Uma modificação
importante verificada em relação ao espectro no IV de GA7 (FIGURA 88, p. 100) é a ausência da
banda intensa em ν 2091 cm-1, referente à deformação axial de N=N=N.
Figura 97 – Espectro no IV (ATR) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-α-D-
galactopiranosídeo de metila – GAX.
GAX
O
OCH3
OBn
BnO
ON
HO
I
f
ed
c
ba
9
8
7
12
3
4 5
6

RESULTADOS E DISCUSSÃO
107
A análise comparativa dos espectros de RMN de 1H do produto obtido (FIGURAS 98, 99, 100 e
101) e de GA7 (FIGURAS 89 e 90, p. 101) permitiu verificar diferenças que indicaram a obtenção
de GAX: presença de sinais relativos a 14 hidrogênios aromáticos (contra dez hidrogênios
aromáticos no espectro de GA7) e um dupleto duplo em δ 6,65 referente ao hidrogênio do grupo
NH (ausente no espectro de GA7).
É importante observar que três dos quatro sinais dos hidrogênios do anel aromático do grupo
iodobenzoíla de GAX encontram-se mais distantes do sinal do TMS que os dez hidrogênios
aromáticos dos grupos benzila, em função dos efeitos de desproteção em orto causados pelo iodo
e carbonila. Pelas multiplicidades e valores de constantes de acoplamento dos sinais dos quatro
hidrogênios do grupo iodobenzoíla foi possível atribuir os sinais de H-b e H-e e essas atribuições
puderam ser confirmadas analisando-se as manchas de correlação observadas no mapa de
contornos homonuclear COSY (FIGURA 102). No entanto, não foi possível distinguir os sinais de
H-d e H-f, que só puderam ser atribuídos após análises posteriores.
GAX
O
OCH3
OBn
BnO
ON
HO
I
f
ed
c
ba
9
8
7
12
3
4 5
6
δ 8,07 t 1 H H-b Jb-d = 1,6 Hz
Jb-f = 1,6 Hz
δ 7,81 ddd 1 H H-d ou H-f Jorto = 7,8 Hz
Jmeta = 1,6 Hz
Jmeta = 1,1 Hz
δ 7,67 ddd 1 H H-f ou H-d Jorto = 7,8 Hz
Jmeta = 1,6 Hz
Jmeta = 1,1 Hz
δ 7,39 – 7,25 m 10 H H aromáticos -
δ 7,15 t 1 H H-e Je-d = 7,8 Hz
Je-f = 7,8 Hz
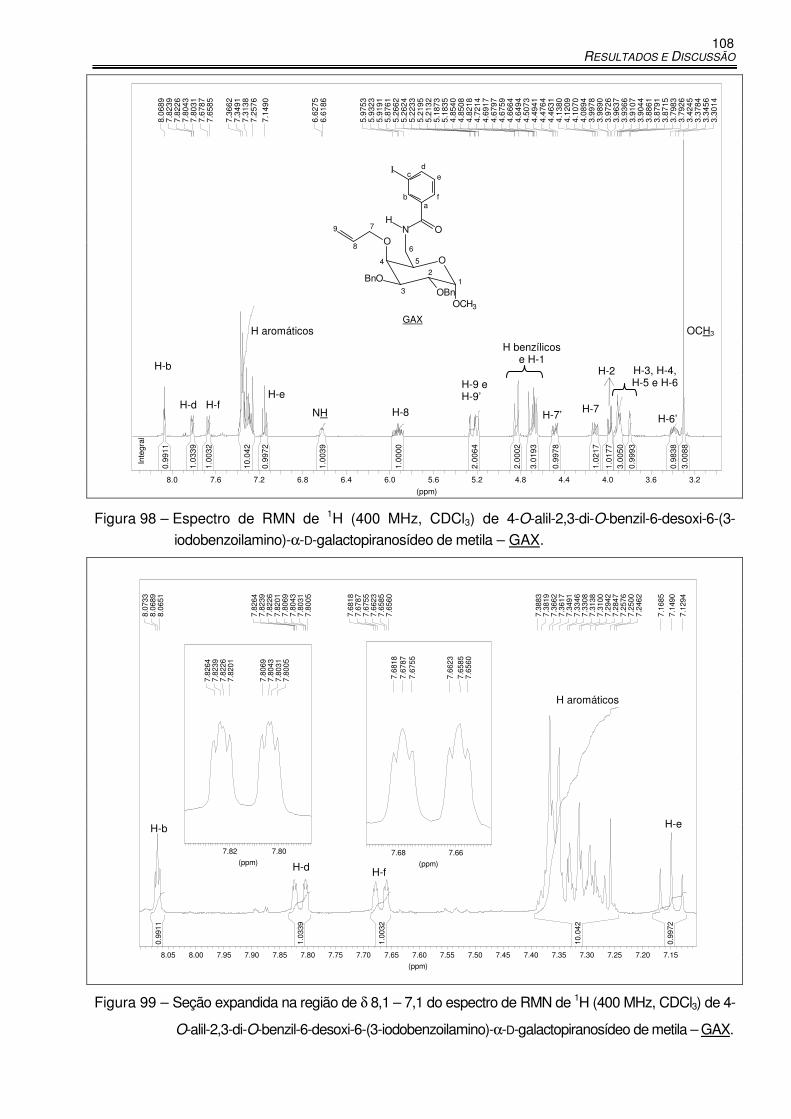
RESULTADOS E DISCUSSÃO
108
Figura 98 – Espectro de RMN de 1H (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-
iodobenzoilamino)-α-D-galactopiranosídeo de metila – GAX.
Figura 99 – Seção expandida na região de δ 8,1 – 7,1 do espectro de RMN de 1H (400 MHz, CDCl3) de 4-
O-alil-2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-α-D-galactopiranosídeo de metila – GAX.
H aromáticos
H-b
H-d
H-e
H-f
0.99
11
1.03
39
1.00
32
10.0
42
0.99
72
8.07
338.
0689
8.06
51
7.82
647.
8239
7.82
267.
8201
7.80
697.
8043
7.80
317.
8005
7.68
187.
6787
7.67
557.
6623
7.65
857.
6560
7.38
837.
3819
7.36
627.
3617
7.34
917.
3346
7.33
087.
3138
7.31
007.
2942
7.28
477.
2576
7.25
007.
2462
7.16
85
7.14
907.
1294
(ppm)
7.157.207.257.307.357.407.457.507.557.607.657.707.757.807.857.907.958.008.05
7.82
647.
8239
7.82
267.
8201
7.80
697.
8043
7.80
317.
8005
(ppm)
7.807.82
7.68
187.
6787
7.67
55
7.66
237.
6585
7.65
60
(ppm)
7.667.68
0.99
11
1.03
39
1.00
32
10.0
42
0.99
72
1.00
39
1.00
00
2.00
64
2.00
02
3.01
93
0.99
78
1.02
17
1.01
773.
0050
0.99
93
0.98
383.
0088
Inte
gral
8.06
897.
8239
7.82
267.
8043
7.80
317.
6787
7.65
85
7.36
627.
3491
7.31
387.
2576
7.14
90
6.62
756.
6186
5.97
535.
9323
5.91
915.
8761
5.26
625.
2624
5.22
335.
2195
5.21
325.
1873
5.18
354.
8540
4.85
084.
8218
4.72
144.
6917
4.67
974.
6759
4.66
644.
6494
4.50
734.
4941
4.47
644.
4631
4.13
804.
1209
4.10
704.
0894
3.99
783.
9890
3.97
263.
9637
3.93
663.
9107
3.90
443.
8861
3.87
913.
8715
3.79
833.
7926
3.42
453.
3784
3.34
563.
3014
(ppm)
3.23.64.04.44.85.25.66.06.46.87.27.68.0
H aromáticos
H-7
OCH3
H-6’
H-3, H-4, H-5 e H-6
H-2
H-8
H benzílicos e H-1
H-b
H-d H-f H-e
H-9 e H-9’
NH H-7’
GAX
O
OCH3
OBn
BnO
ON
HO
I
f
ed
c
ba
9
8
7
12
3
4 5
6

RESULTADOS E DISCUSSÃO
109
Figura 100 – Seção expandida na região de δ 6,8 – 4,1 do espectro de RMN de 1H (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-α-D-galactopiranosídeo de metila – GAX.
Figura 101 – Seção expandida na região de δ 4,1 – 3,2 do espectro de RMN de 1H (400 MHz, CDCl3) de 4-O-
alil-2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-α-D-galactopiranosídeo de metila – GAX.
H-7 H-8
H benzílicos e H-1
H-9 e H-9’ NH H-7’
1.00
39
1.00
00
2.00
64
2.00
02
3.01
93
0.99
78
1.02
17
Inte
gral
6.63
576.
6275
6.61
866.
6098
5.97
535.
9620
5.95
765.
9494
5.94
505.
9361
5.93
235.
9191
5.91
465.
9064
5.90
145.
8932
5.88
885.
8761
5.27
005.
2662
5.26
245.
2587
5.22
715.
2233
5.21
955.
2157
5.21
325.
2107
5.20
945.
1873
5.18
545.
1835
4.85
404.
8508
4.82
184.
7214
4.69
174.
6797
4.67
594.
6664
4.64
944.
5105
4.50
734.
5042
4.49
794.
4941
4.49
094.
4795
4.47
644.
4726
4.46
634.
4631
4.46
004.
1380
4.12
094.
1070
4.08
94
(ppm)
4.24.44.64.85.05.25.45.65.86.06.26.46.6
6.63
576.
6275
6.61
866.
6098
(ppm)
6.60
5.97
53
5.96
205.
9576
5.94
945.
9450
5.93
615.
9323
5.91
915.
9146
5.90
645.
9014
5.89
325.
8888
5.87
61
(ppm)
5.885.925.96
5.26
625.
2624
5.25
875.
2271
5.22
335.
2195
5.21
575.
2132
5.21
075.
2094
5.18
735.
1854
5.18
35
(ppm)
5.205.24
4.49
094.
4795
4.47
644.
4726
4.46
634.
4631
4.46
00
(ppm)
4.50
4.13
804.
1209
4.10
704.
0894
(ppm)
4.1
1.01
77
3.00
50
0.99
93
0.98
38
3.00
88
Inte
gral
3.99
783.
9890
3.97
263.
9637
3.93
663.
9265
3.91
073.
9044
3.89
873.
8861
3.87
913.
8709
3.79
833.
7926
3.42
453.
4151
3.40
243.
3936
3.38
793.
3784
3.30
14
(ppm)
3.303.353.403.453.503.553.603.653.703.753.803.853.903.954.00
3.99
783.
9890
3.97
263.
9637
3.93
663.
9265
3.91
073.
9044
3.89
873.
8861
3.87
913.
8709
3.79
833.
7926
(ppm)
3.803.853.903.95
3.42
45
3.41
51
3.40
24
3.39
36
3.38
79
3.37
84
(ppm)
3.383.403.42OCH3
H-3, H-6 e H-4 ou H-5 H-2
H-6’ H-5 ou H-4

RESULTADOS E DISCUSSÃO
110
Figura 102 – Mapa de contornos COSY (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-
iodobenzoilamino)-α-D-galactopiranosídeo de metila – GAX.
A análise do espectro de RMN de 13C, auxiliada pelo DEPT 135 (FIGURA 103), permitiu confirmar
que foi obtida a benzamida GAX. Observam-se, além de outros, o sinal referente ao carbono
carbonílico e quatro sinais de carbonos ipso (dois dos grupos benzila, um do carbono ligado à
carbonila e um do carbono ligado a iodo).
Foi possível fazer a atribuição de todos os sinais de carbonos não hidrogenados apenas com
base nos valores de deslocamentos químicos tabelados ou calculados: C=O, C-I (mais próximo do
TMS devido ao efeito do átomo pesado), carbonos ipso dos grupos benzila e, por exclusão, o
carbono ligado à carbonila (C-a). No entanto, a atribuição inequívoca de outros sinais de carbonos
aromáticos de GAX só foi possível graças ao mapa de contornos heteronuclear HMQC (FIGURA 104).
Observação das manchas de correlação entre os sinais de hidrogênios aromáticos, previamente
atribuídos, e dos sinais de carbono permitiu a atribuição dos sinais de C-b e C-e. Verificou-se
correlação entre os sinais atribuídos a H-d ou H-f com os sinais de carbono em δ 140,38 e
125,94. Como o iodo exerce forte efeito de desproteção sobre os carbonos orto, foi possível
concluir que os sinais em δ 140,38 e 125,94 referem-se a C-d e C-f, respectivamente. Dessa
forma, os sinais dos hidrogênios H-d e H-f também puderam ser distinguidos.
NH x H-6
H-e x H-d H-e x H-f
H-7 x H-8
H-8 x H-9 e H-8 x H-9’
H-1 x H-2
H-7 x H-7’
H-6 x H-6’
H-b x H-d
H-b x H-f
OCH2φ x OCH2φ
GAX
O
OCH3
OBn
BnO
ON
HO
I
f
ed
c
ba
9
8
7
12
3
4 5
6

RESULTADOS E DISCUSSÃO
111
GAX
O
OCH3
OBn
BnO
ON
HO
I
f
ed
c
ba
9
8
7
12
3
4 5
6
Figura 103 – Espectro de RMN de 13C e DEPT 135 (100 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-
desoxi-6-(3-iodobenzoilamino)-α-D-galactopiranosídeo de metila – GAX.
δ 165,94 C=O δ 128,40 – 127,54 C aromáticos
δ 140,38 C-d δ 125,94 C-f
δ 138,53; 138,44 C ipso Bn δ 94,26 C-I (ipso)
δ 136,31 C-a (ipso) δ 7,81 (ddd) H-d
δ 135,96 C-b δ 7,67 (ddd) H-f
δ 130,22 C-e
165.
9386
140.
3790
138.
5289
138.
4416
136.
3136
135.
9642
134.
8764
130.
2155
128.
3972
128.
3336
128.
0081
127.
7222
127.
6270
127.
5396
125.
9357
118.
0034
98.8
833
94.2
621
78.7
230
77.3
176
77.0
000
76.6
824
76.4
521
76.3
489
73.9
430
73.6
730
73.4
666
68.2
737
55.3
073
41.2
054
(ppm)
405060708090100110120130140150160
128.
3972
128.
3336
128.
0081
127.
7222
127.
6270
127.
5396
(ppm)
127.6128.0128.4
77.3
176
77.0
000
76.6
824
76.4
521
76.3
489
73.9
430
73.6
730
73.4
666
(ppm)
7374757677
C-2
C-e
C-9
GAX
O
OCH3
OBn
BnO
ON
HO
I
f
ed
c
ba
9
8
7
12
3
4 5
6
C ipso
C aromáticos C-7
C-1 OCH3 C-6
* C açúcar
C benzílicos
C-d C-b C-f
C-I
C-8 * *
C-4 ou C-5
C=O

RESULTADOS E DISCUSSÃO
112
Figura 104 – Mapa de contornos HMQC (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-
iodobenzoilamino)-α-D-galactopiranosídeo de metila – GAX.
Análise dos espectros de massas de GAX confirmou a obtenção da amida desejada. Na Figura 105
observam-se os picos relativos a [M+H]+ de m/z 644,1534 (calculado: 644,1509), [M+Na]+ de m/z
666,1451 e [M+K]+ de m/z 682,1188. No espectro tandem MS/MS (FIGURA 105), observam-se,
além do pico correspondente a [M+H]+, dois outros picos de m/z 612,1757 e m/z 504,0809,
relativos aos fragmentos [M+H]+ - MeOH e [M+H]+ - MeOH - BnOH, respectivamente. Inicialmente
ocorre perda de metanol, o que leva à formação de uma ligação dupla entre C-1 e C-2, e em
seguida, a perda de álcool benzílico, favorecida pela formação de duplas conjugadas (FIGURA
106).
H-d x C-d
H-b x C-b
H-f x C-f
H-e x C-e
H-9 + H-9’ x C-9
H-1 x C-1
H-2 x C-2
H-7 x C-7
H-7’ x C-7 OCH2φ x OCH2φ
H-8 x C-8
H-6 x C-6 H-6’ x C-6
OCH3 x OCH3
GAX
O
OCH3
OBn
BnO
ON
HO
I
f
ed
c
ba
9
8
7
12
3
4 5
6

RESULTADOS E DISCUSSÃO
113
Figura 105 – Espectros de massas [ESI(+)-MS] e análise seqüencial [ESI(+)-MS/MS] de 4-O-alil-
2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-α-D-galactopiranosídeo de metila – GAX.
O
BnO
O
OBn
OCH3
N
O
I
H
H
+
O
BnO
O
OBn
N
O
I
H
+O
BnO
O
OBn
N
OI
H
H+
O
BnO
O
OBn
N
OI
H
H
+
OO
OBn
N
OI
H
H+
- CH3OH
m/z 644m/z 612
m/z 504 m/z 612
m/z 612
- BnOH
Figura 106 – Proposta de fragmentação para o íon de m/z 644 após análise seqüencial de 4-O-alil-
2,3-di-O-benzil-6-desoxi-6-(3-iodobenzoilamino)-α-D-galactopiranosídeo de metila – GAX.
O
BnO
O
OBn
N
OI
H
H+
[M + H]+
100 200 300 400 500 600 700 800 900 1000 1100 1200 1300 1400m/z0
100
%
gax 39 (1.318) Cm (20:63) TOF MS ES+ 1.95e3195.0298
97.0863644.1534
289.2856
318.3112
645.1611
667.1699
[M + H]+ [M + Na]+
[M + K]+
605 610 615 620 625 630 635 640 645 650 655 660 665 670 675 680 685 690 695 700 705 710 715 720m/z0
100
%
gax 39 (1.318) Cm (20:63) TOF MS ES+ 1.19e3644.1534
645.1611666.1451
646.1697 667.1699682.1188
50 100 150 200 250 300 350 400 450 500 550 600 650m/z0
100
%
gax_644 29 (0.968) Sb (7,50.00 ); Cm (9:51) TOF MSMS 644.10ES+ 105612.1757
504.0809181.1529 644.1534
[M + H]+
OO
OBn
N
OI
H
H+

RESULTADOS E DISCUSSÃO
114
4.8 REAÇÃO DE MACROCICLIZAÇÃO RADICALAR
A última etapa da rota de síntese planejada envolveu a reação da iodobenzamida GAX com hidreto
de tri-n-butilestanho, visando à obtenção das macrolactamas GAXA e/ou GAXB (FIGURA 107).
+ +O
OCH3
OBn
BnO
ON
HO
I
GAX
ou ciclo-hexano
AIBN, Bu3SnH
Refluxo
GAXA
O
OCH3
OBn
BnO
O NOH
O
OCH3
OBn
BnO
O NOH
H3C
GAXB
O
OCH3
OBn
BnO
ON
HO
H
GAXC
Figura 107 – Reação de macrociclização radicalar de GAX e seus possíveis produtos.
Conforme encontra-se detalhado na Introdução, Porter e colaboradores estabeleceram que as
macrociclizações de modo endo são preferenciais ou exclusivas em relação às macrociclizações
de modo exo (PORTER et al., 1986; PORTER; CHANG, 1987; PORTER et al., 1988), o que foi
confirmado posteriormente (ROBERTSON; HATLEY, 1999; HITCHCOCK; PATTENDEN, 1992;
COX et al., 1992; ROBERTSON et al., 1997; BECKWITH et al., 1997; BEGLEY et al., 1996; SHEA
et al., 1992; BALDWIN et al., 1991; LAMAS et al., 1992; JONAS et al., 1995; HOULDSWORTH et
al., 1997; RODRÍGUEZ et al., 1999; BOGER; MATHVINK, 1990; ASTLEY; PATTENDEN, 1992;
GHOSH; GHATAK, 1995; GIBSON et al., 1997; NANDI et al., 2001; NANDI; CHATTOPADHYAY,
2002; BALRAJU et al., 2005). Além disso, os produtos ciclizados isolados das reações realizadas
no âmbito do programa de síntese de macrociclos do grupo QF/DQ/UFMG foram provenientes de
ciclização endo (PRADO et al., 2000; BINATTI et al., 2002; FARACO et al., 2003; FARACO et al.,
2004; BINATTI et al., 2005; DIAS et al., 2006; PIRES et al., 2006; ROCHA; PRADO, 2006). Era de
se esperar, portanto que da reação de GAX com Bu3SnH fosse obtida a macrolactama de 12
membros GAXA. No entanto, não se poderia descartar a possibilidade de formação do produto de
ciclização 11-exo, GAXB. A formação do produto não ciclizado reduzido, GAXC (FIGURA 107),
também poderia ser prevista, uma vez que os produtos de hidrogenólise, nos quais o átomo de
halogênio do precursor é substituído por hidrogênio, são formados nas reações mediadas por
Bu3SnH (WALLING, 1985; CURRAN, 1988; BECKWITH et al., 2004; ALLIN et al., 2002; JESSOP
et al., 2003).
A formação das macrolactamas GAXA e GAXB, além do produto de hidrogenólise GAXC, por
reação radicalar com hidreto de tri-n-butilestanho ocorreria pelo mecanismo representado na
Figura 108. Por meio de aquecimento, forma-se o radical isobutironitrila, a partir de 2,2’-
azobisisobutironitrila (AIBN), que ao abstrair o átomo de hidrogênio do hidreto de tri-n-butilestanho
conduz ao radical tributilestanila. Este radical, por sua vez, abstrai o átomo de iodo produzindo um
radical arila (C), que precisa ter um tempo de vida suficiente para, por reação intramolecular,

RESULTADOS E DISCUSSÃO
115
formar novos radicais (A – endo e/ou B - exo). Os tempos de vida dos radicais A, B e C dependem
da velocidade com que estes radicais captam o hidrogênio do hidreto de tri-n-butilestanho, de
modo que a concentração do hidreto é fundamental no curso que a reação irá seguir. Quanto
maior a concentração de hidreto, menor será o tempo de vida do radical C, e consequentemente,
o produto de hidrogenólise (GAXC) irá formar preferencialmente aos radicais A e B. Por outro
lado, baixas concentrações do hidreto devem favorecer a transformação de C em A e B, que
captam o hidrogênio do Bu3SnH levando aos respectivos produtos ciclizados (GAXA e GAXB).
Baixas concentrações do hidreto de tri-n-butilestanho podem ser alcançadas em reações
desenvolvidas em altas diluições, entre 10-1 e 10-2 mol/L, e quando o reagente é adicionado
lentamente, em torno de 1,5 horas (MARINOVIC; RAMANATHAN, 1983; WALLING, 1985;
BECKWITH et al., 1997).

RESULTADOS E DISCUSSÃO
116
+Bu3Sn H
Bu3Sn H +
+
+
B
AIBN
A GAXA
N C N N C N 80 oC2 + N2
Bu3Sn H + + C N
+ Bu3SnI+
e/ou
+Bu3Sn H +
C N
C N
Bu3Sn
Bu3Sn
Bu3Sn
GAXB
NH
O
O
NH
O
O
GAXC
NH
O
OBu3Sn
Bu3Sn
C
NH
O
O
A
NH
O
O
C
NH
O
O
I
NH
O
O
NH
O
O
C
NH
O
O
NH
O
O
B
NH
O
O
A
B
Figura 108 – Mecanismo de formação das macrolactamas GAXA e GAXB e do produto de
hidrogenólise GAXC, por reação radicalar com hidreto de tri-n-butilestanho.
A primeira reação foi realizada seguindo a metodologia e as condições descritas por Marinovic e
Ramanathan (MARINOVIC; RAMANATHAN, 1983) e utilizadas na síntese de macrolactamas no
âmbito do programa de pesquisa do grupo QF/DQ/UFMG (PRADO et al., 2000; BINATTI et al.,
2002; FARACO et al., 2003; FARACO et al., 2004; DIAS et al., 2006; ROCHA; PRADO, 2006). A

RESULTADOS E DISCUSSÃO
117
uma solução de GAX em benzeno, sob refluxo e atmosfera de nitrogênio, foi adicionada,
lentamente (uma hora e quinze minutos), solução de Bu3SnH/AIBN em benzeno. As
concentrações finais de GAX e de Bu3SnH foram de 12 mmol/L e 18 mmol/L, respectivamente.
Após o término da adição, a solução foi mantida sob refluxo por mais uma hora e quinze minutos.
O benzeno foi eliminado sob pressão reduzida, usando evaporador rotativo, e o resíduo obtido foi
submetido à CCS.
Cromatografia em camada delgada do resíduo obtido após eliminação do benzeno indicou a
presença de uma mistura complexa de produtos, o que de antemão dificultaria o isolamento de
substâncias com pureza suficiente para elucidação estrutural. Adicionalmente, é de conhecimento
geral a dificuldade de purificação de produtos obtidos por reações mediadas por hidreto de tri-n-
butilestanho (BERGE; ROBERTS, 1979; ROSA et al., 1997; SALOMON et al., 2000).
O resíduo foi submetido à CCS e foram isolados dois produtos em quantidade suficiente para
análise de RMN de 1H e de 13C. Em CCD, um dos produtos apresenta Rf muito próximo daquele
de GAX (mas ligeiramente menor) e o Rf do outro produto é bem menor que o de GAX. As
análises dos espectros de RMN de 1H e de 13C do produto com Rf próximo ao de GAX permitiram
caracterizá-lo como sendo o produto de hidrogenólise, GAXC, que foi isolado como um sólido
branco (faixa de fusão 101,6-105,5 oC; rotação específica +48,2, c 1,05, CHCl3), com 58% de
rendimento. O produto com Rf inferior ao de GAX não foi possível de ser caracterizado, devido à
presença de grande quantidade de derivados de estanho. No entanto, é possível supor que na
mistura não haja os produto de ciclização, GAXA e GAXB, pois nos espectros de RMN encontram-
se sinais típicos de hidrogênios e carbonos olefínicos.
No espectro no infravermelho de GAXC (FIGURA 109) não foram observadas diferenças
significativas em relação ao espectro de GAX (FIGURA 97, p. 106), como se podia esperar.
Observam-se as bandas relativas à deformação axial e angular de ligação N-H (ν 3315 e 1538 cm-1,
respectivamente) e deformação axial de ligação C=O de amida (ν 1639 cm-1).

RESULTADOS E DISCUSSÃO
118
Figura 109 – Espectro no IV (ATR) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-benzoilamino-α-D-
galactopiranosídeo de metila – GAXC.
Os sinais relativos aos hidrogênios aromáticos observados no espectro de RMN de 1H do produto
de Rf próximo ao de GAX (FIGURAS 110 e 111) foram de fundamental importância para identificá-
lo como sendo o produto de hidrogenólise GAXC. O padrão de sinais mais distantes do TMS é
típico de anel aromático monossubstituído com um grupo carbonila, diferentemente dos sinais de
hidrogênios aromáticos observados no espectro de GAX (FIGURAS 98-101, p. 108-109),
compatíveis com anel dissubstituído. As manchas de correlação observadas no mapa de
contornos homonuclear COSY (FIGURA 112), são compatíveis com as atribuições feitas
baseadas na multiplicidade, valores de constantes de acoplamento e deslocamentos químicos.
6
54
3
21
7
8
9
ab
cd
O
OCH3
OBn
BnO
ON
HO
GAXC
b
c
δ 7,73 dd 2 H H-b Jb-c = 6,9 Hz
Jb-d = 1,6 Hz
δ 7,48 tt 1 H H-d Jd-c = 7,3 Hz
Jd-b = 1,6 Hz
δ 7,43 – 7,41 m 2 H H-c -
δ 7,36 – 7,25 m 10 H H aromáticos -
6
54
3
21
7
8
9
ab
cd
O
OCH3
OBn
BnO
ON
HO
GAXC
b
c

RESULTADOS E DISCUSSÃO
119
Figura 110 – Espectro de RMN de 1H (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-
benzoilamino-α-D-galactopiranosídeo de metila – GAXC.
Figura 111 – Seção expandida na região de δ 7,9 – 7,1 do espectro de RMN de 1H (400 MHz, CDCl3) de
4-O-alil-2,3-di-O-benzil-6-desoxi-6-benzoilamino-α-D-galactopiranosídeo de metila – GAXC.
2.01
29
1.07
161.
9966
10.0
72
0.98
80
1.00
00
2.00
05
2.00
41
3.01
75
0.99
76
1.12
151.
0410
3.04
940.
9959
1.00
543.
0264
Inte
gral
7.73
747.
7342
7.72
547.
7204
7.71
667.
5063
7.50
257.
4994
7.49
057.
4836
7.47
857.
4697
7.46
597.
4628
7.42
807.
4085
7.36
307.
3466
7.32
897.
3276
7.32
517.
3074
7.28
857.
2784
7.26
147.
2544
6.66
346.
6559
6.64
646.
6369
5.97
345.
9304
5.91
725.
8742
5.25
935.
2555
5.21
645.
2119
5.19
245.
1879
5.16
655.
1627
4.85
214.
8426
4.82
184.
8135
4.72
074.
6917
4.67
654.
6727
4.66
394.
6462
4.49
154.
4783
4.46
064.
4473
4.14
564.
1279
4.11
404.
0969
3.99
843.
9896
3.97
323.
9644
3.92
143.
9126
3.90
563.
8880
3.88
043.
8065
3.80
083.
4321
3.38
543.
2882
(ppm)
3.03.54.04.55.05.56.06.57.07.58.08.59.09.5
5.93
045.
9172
5.87
42
5.25
935.
2555
5.21
645.
2119
5.19
245.
1879
5.16
655.
1627
4.85
214.
8426
4.82
184.
8135
4.72
074.
6917
4.67
654.
6727
4.66
394.
6462
4.49
154.
4783
4.46
064.
4473
4.14
564.
1279
4.11
404.
0969
3.99
843.
9896
3.97
323.
9644
3.92
143.
9126
3.90
563.
8880
3.88
043.
8065
3.80
08
3.43
213.
3854
(ppm)
3.43.63.84.04.24.44.64.85.05.25.45.65.8
6.66
346.
6559
6.64
646.
6369
(ppm)
H aromáticos
OCH3
H-6’
H açúcar
H-8
H benzílicos e H-1
6
54
3
21
7
8
9
ab
cd
O
OCH3
OBn
BnO
ON
HO
GAXC
b
c
NH
H-9 e H-9’ H-7’ e H-7
H-3, H-6 e H-4 ou H-5
H-7 H-6’
H-2
H-7’
NH
H-8
H-9 e H-9’
H benzílicos e H-1
H-5 ou H-4
2.01
29
1.07
16
1.99
66
10.0
72
Inte
gral
7.73
747.
7342
7.72
547.
7204
7.71
66
7.50
637.
5025
7.49
947.
4905
7.48
367.
4785
7.46
977.
4659
7.46
28
7.42
80
7.40
85
7.36
30
7.34
66
7.32
897.
3276
7.32
517.
3074
7.28
857.
2784
7.26
147.
2544
(ppm)
7.207.257.307.357.407.457.507.557.607.657.707.757.80
H-b
H-d
H-c
6
54
3
21
7
8
9
ab
cd
O
OCHOBn
BnO
ON
HO
b
c

RESULTADOS E DISCUSSÃO
120
Figura 112 – Mapa de contornos COSY (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-
benzoilamino-α-D-galactopiranosídeo de metila – GAXC.
A análise dos espectros de RMN de 13C e DEPT 135 do produto obtido (FIGURA 113) evidencia
que há apenas quatro carbonos não hidrogenados (C=O, dois C ipso dos grupos benzila e C ipso
do grupo benzoíla). Além disso, não há três sinais observados no espectro de GAX (FIGURA 103,
p. 111), característicos da presença de iodo no anel aromático: δ em torno de 90, referente ao
carbono aromático ligado ao iodo, e δ em torno de 140 e 136, relativos a carbonos hidrogenados
desprotegidos pelo iodo em orto. A atribuição dos sinais dos carbonos hidrogenados do grupo
benzoíla (C-b, C-c e C-d) foi possível pela observação das manchas de correlação H-b x C-b, H-c
x C-c e H-d x C-d presentes no mapa de contornos heteronuclear HMQC (FIGURA 114).
6
54
3
21
7
8
9
ab
cd
O
OCH3
OBn
BnO
ON
HO
GAXC
b
c
δ 167,57 C=O
δ 138,53; 138,45 C ipso Bn
δ 134,27 C-a (ipso)
δ 131,49 C-d
δ 128,56 C-c
δ 128,37 – 127,52 C aromáticos
δ 126,76 C-b
H-b x H-d
H-b x H-c
NH x H-6
NH x H-6’
H-8 x H-9 e H-8 x H-9’
H-7 x H-8
OCH2φ x OCH2φ
H-7 x H-7’
H-1 x H-2
H-6 x H-6’
H-7’ x H-8
H-7’ x H-9 e H-7’ x H-9’
6
54
3
21
7
8
9
ab
cd
O
OCH3
OBn
BnO
ON
HO
GAXC
b
c

RESULTADOS E DISCUSSÃO
121
Figura 113 – Espectro de RMN de 13C e DEPT 135 (100 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-
desoxi-6-benzoilamino-α-D-galactopiranosídeo de metila – GAXC.
Figura 114 – Mapa de contornos HMQC (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-
benzoilamino-α-D-galactopiranosídeo de metila – GAXC.
167.
5664
138.
5289
138.
4495
134.
9161
134.
2729
131.
4859
128.
5560
128.
3654
128.
2939
127.
9922
127.
6825
127.
5873
127.
5237
126.
7615
117.
8446
98.8
277
78.7
310
77.3
176
77.0
000
76.6
824
76.3
568
76.2
377
73.9
430
73.6
333
73.3
872
68.5
278
55.2
040
41.0
069
(ppm)
405060708090100110120130140150160170
78.7
310
77.3
176
77.0
000
76.6
824
76.3
568
76.2
377
73.9
430
73.6
333
73.3
872
(ppm)
73747576777879
128.
5560
128.
3654
128.
2939
127.
9922
127.
6825
127.
5873
127.
5237
126.
7615
(ppm)
127.0127.5128.0128.5
C aromáticos
6
54
3
21
7
8
9
ab
cd
O
OCH3
OBn
BnO
ON
HO
GAXC
b
c
C-5 ou C-4 C ipso
C-7
C-1 OCH3
C-6
* C-3, C-4 ou C-5
C benzílicos
C=O
C-8 C-d
C-b C-9
*
C-2
*
C-c
H-b x C-b
H-d x C-d
H-9 + H-9’ x C-9
H-1 x C-1
H-2 x C-2
H-7 x C-7
H-7’ x C-7
OCH2φ x OCH2φ
H-8 x C-8
H-6 x C-6
H-6’ x C-6
OCH3 x OCH3
H-c x C-c
6
54
3
21
7
8
9
ab
cd
O
OCH3
OBn
BnO
ON
HO
GAXC
b
c

RESULTADOS E DISCUSSÃO
122
Nos espectros de massas de GAXC (FIGURA 115) observam-se os picos correspondentes a
[M+H]+ de m/z 518,2805 (calculado: 518,2543), [M+Na]+ de m/z 540,2753 e [M+K]+ de m/z
556,2657.
Assim como no espectro tandem MS/MS da amida inédita GAX (FIGURA 105, p. 113), observam-
se, além do pico correspondente a [M+H]+, dois outros picos de m/z 486,2613 e m/z 378,2063,
relativos aos fragmentos [M+H]+ - MeOH e [M+H]+ - MeOH - BnOH, respectivamente (FIGURA 116).
Figura 115 – Espectros de massas [ESI(+)-MS] e análise seqüencial [ESI(+)-MS/MS] de 4-O-alil-
2,3-di-O-benzil-6-desoxi-6-benzoilamino-α-D-galactopiranosídeo de metila – GAXC.
O
BnO
O
OBn
OCH3
N
O
H
H
+
+ +O
BnO
O
OBn
N
O
H
HO
BnO
O
OBn
N
O
H
O
BnO
O
OBn
N
O
H
H
+
OO
OBn
N
O
H
H+
- CH3OH
m/z 518m/z 486
m/z 378 m/z 486
m/z 486
- BnOH
Figura 116 – Proposta de fragmentação para o íon de m/z 518 após análise seqüencial de 4-O-alil-
2,3-di-O-benzil-6-desoxi-6-benzoilamino-α-D-galactopiranosídeo de metila – GAXC.
[M + H]+
100 200 300 400 500 600 700 800 900 1000 1100 1200 1300 1400m/z0
100
%
gaxc2 16 (0.592) Cm (15:34) TOF MS ES+ 793195.0298
181.0429
79.0797 289.2856518.2805
391.2803 556.2657
[M + H]+ [M +Na]+ [M +K]+
505 510 515 520 525 530 535 540 545 550 555 560 565 570 575 580m/z0
100
%
gaxc2 16 (0.592) Cm (15:34) TOF MS ES+ 91518.2805
513.5342511.3408
540.2753
519.2908
520.3022 533.2702530.3088
556.2657541.2524
542.2579 555.3024557.2573
572.2939570.4800
[M + H]+
50 75 100 125 150 175 200 225 250 275 300 325 350 375 400 425 450 475 500 525 550m/z0
100
%
gaxc_518 37 (1.235) Sm (Mn, 2x3.00); Sb (7,50.00 ); Cm (25:47) TOF MSMS 518.28ES+ 41.2486.2613
378.2063487.2658 518.2805
O
B nO
O
O B n
N
O
H
H+
OO
OB n
N
O
H
H+

RESULTADOS E DISCUSSÃO
123
Porter e colaboradores realizaram diversos estudos que indicaram que a concentração de 3 a 6
mmol/L do substrato favorece a formação do produto de macrociclização (PORTER et al., 1986,
1987). Nandi e colaboradores descreveram ciclização 8-endo de derivados de carboidrato na
concentração de 10 mmol/L e adição da solução de hidreto de tri-n-butilestanho durante 3 horas
para a obtenção de um produto cis, mas para a obtenção do produto trans foi necessário
empregar uma solução do derivado de açúcar na concentração de 4 mmol/L e o tempo de adição
da solução de hidreto de tri-n-butilestanho foi de 12 horas (NANDI et al., 2001). Robertson e
colaboradores obtiveram um macrociclo proveniente de ciclização 13-endo utilizando solução do
substrato a 5 mmol/L e adição da solução de hidreto de tri-n-butilestanho por 6 horas
(ROBERTSON et al., 1997).
Considerando-se estes relatos e o fato de que não se obtiveram macrolactamas nas condições
utilizadas na primeira reação, optou-se por desenvolver a reação de macrociclização radicalar
com a benzamida GAX na concentração final de 2,8 mmol/L e adição da solução de hidreto de tri-
n-butilestanho/AIBN por cerca de 5 h. Outra alteração realizada, em relação à primeira reação, foi
a adição do iniciador radicalar, AIBN, também na solução da amida GAX, como descrito por Nandi
e colaboradores (NANDI et al., 2001).
Da mesma forma que na primeira reação, formou-se uma mistura complexa de produtos,
visualizados por CCD. Cromatografia em coluna de sílica da mistura conduziu a um produto puro
(sólido branco) de Rf igual ao de GAXC, faixa de fusão de 28,1-29,3 oC e rotação específica de
+46,6 (c 1,31, CHCl3). Análise dos espectros de RMN de 1H e de 13C (uni e bidimensionais)
indicou tratar-se de um produto inesperado, 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-fenilbenzoilamino)-
α-D-galactopiranosídeo de metila (GAXD) (FIGURA 117), que foi obtido com rendimento de 35%.
GAXD
O
OCH3
OBn
BnO
ON
HO
Figura 117 – Produto obtido da segunda reação radicalar de GAX: 4-O-alil-2,3-di-O-benzil-6-desoxi-
6-(3-fenilbenzoilamino)-α-D-galactopiranosídeo de metila (GAXD).
Conforme se observa na Figura 117, GAXD apresenta um grupo fenila em substituição ao iodo de
GAX. A formação de GAXD pode ser explicada considerando-se que ocorreu reação
intermolecular entre o radical arila formado a partir de GAX e o solvente usado, o benzeno. A
formação de produtos indesejados, provenientes da reação de radicais com benzeno, quando

RESULTADOS E DISCUSSÃO
124
este solvente foi utilizado em situações de alta diluição, já foi relatada (WALLING, 1985;
BOWMAN et al., 2000; BECKWITH et al., 2004).
O mecanismo proposto para a formação de GAXD envolve substituição aromática por meio de
reação de adição-eliminação radicalar (FIGURA 118) (FOSSEY et al., 1995). O radical arila,
formado por reação da benzamida GAX com o radical tributilestanila, ataca um carbono do
benzeno. Em seguida, o intermediário radicalar não aromático reage com o radical tributilestanila,
levando à eliminação do hidrogênio ligado ao carbono que sofreu a adição do radical arila e à
rearomatização.
Bu3Sn Bu3SnH+NH
O
O
H
+
GAXD
NH
O
O
+NH
O
O
NH
O
O
H
Bu3SnIBu3Sn ++NH
O
O
GAXI
NH
O
O
Figura 118 – Proposta de mecanismo para a formação de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-
fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD.
A possibilidade de que o produto isolado da segunda reação de GAX com Bu3SnH fosse um
macrociclo foi descartada ao se verificar a presença de sinais característico de hidrogênios
olefínicos no espectro de RMN de 1H (FIGURAS 119 e 120). Da mesma forma, descartou-se a
possibilidade de o produto ser o de hidrogenólise (GAXC) ao se verificar que os sinais presentes
na região de hidrogênios aromáticos não eram compatíveis com aqueles de anéis
monossubstituídos. Verificou-se a presença de sinais entre δ 7,98 e 7,25: um tripleto (1 H), dois
tripletos duplos (1 H cada), um dupleto (2 H) e dois multipletos, referentes a três e onze
hidrogênios. A presença do tripleto mais distante do TMS (δ 7,97; J 1,5 Hz) evidenciou que havia
um hidrogênio orto à carbonila, sem hidrogênio vizinho, ou seja, a um dos carbonos meta à
carbonila estaria ligado um substituinte. Pelo número de hidrogênios aromáticos (19), suspeitou-se

RESULTADOS E DISCUSSÃO
125
que o substituinte seria um grupo fenila. No mapa de contornos homonuclear COSY (FIGURA
121), verificaram-se manchas de correlação entre o tripleto, atribuído a H-b, e os dois tripletos
duplos, conseqüentemente atribuídos a H-d e H-f.
6
54
3
21
7
8
9
a
cd
e
f
GAXD
b
O
OCH3
OBn
BnO
ON
HO
c'
b'
d'
a'
b'
c'
Figura 119 – Espectro de RMN de 1H (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-
fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD.
δ 7,97 t 1 H H-b Jb-f = 1,5 Hz
Jb-d = 1,5 Hz
δ 7,71 td 1 H H-d ou H-f Jorto = 7,3 Hz
Jmeta = 1,5 Hz
δ 7,67 td 1 H H-f ou H-d Jorto = 7,7 Hz
Jmeta = 1,5 Hz
δ 7,58 d 2 H H aromáticos Jorto = 7,0 Hz
δ 7,49 – 7,43 m 3 H H aromáticos
δ 7,39 – 7,25 m 11 H H aromáticos
6
54
3
21
7
8
9
a
cd
e
f
GAXD
b
O
OCH3
OBn
BnO
ON
HO
c'
b'
d'
a'
b'
c'
H-d e H-f
H-b’
H-e
H-b
H aromáticos
NH H-8
H-9 e H-9’
H benzílicos e H-1
H-7’ H-7
H açúcar
H-5 ou H-4
OCH3
H-6’
0.96
60
1.13
781.
0228
2.12
47
3.15
33
11.4
75
0.83
81
1.00
00
1.01
770.
9404
1.99
48
3.04
46
0.99
10
1.11
38
4.00
14
1.00
24
1.11
02
2.99
03
Inte
gral
7.46
057.
4486
7.42
947.
3882
7.36
817.
3645
7.34
717.
3068
7.25
016.
6969
6.68
776.
6777
5.97
815.
9653
5.96
165.
9525
5.94
795.
9397
5.93
515.
9223
5.91
775.
9095
5.90
495.
8967
5.89
215.
8793
5.25
745.
2538
5.21
455.
2108
5.17
705.
1514
5.14
864.
8523
4.84
784.
8221
4.81
764.
7243
4.69
414.
6786
4.67
034.
6475
4.49
934.
4865
4.46
824.
4554
4.15
274.
1363
4.12
164.
1052
4.00
283.
9936
3.97
723.
9689
3.95
523.
9461
3.92
963.
9195
3.91
223.
8939
3.88
753.
8171
3.81
073.
4577
3.44
583.
4321
3.42
303.
4184
3.40
923.
3928
3.38
643.
3672
3.30
41
(ppm)
3.43.63.84.04.24.44.64.85.05.25.45.65.86.06.26.46.66.87.07.27.47.67.88.0

RESULTADOS E DISCUSSÃO
126
Figura 120 – Seção expandida na região de δ 8,0 – 7,0 do espectro de RMN de 1H (400 MHz, CDCl3) de 4-O-
alil-2,3-di-O-benzil-6-desoxi-6-(3-fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD.
Figura 121 – Mapa de contornos COSY (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-
fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD.
H-d H-f
H-b’ H-e + 2 H
H-b
H aromáticos
6
54
3
21
7
8
9
a
cd
e
f
GAXD
b
O
OCH3
OBn
BnO
ON
HO
c'
b'
d'
a'
b'
c'
0.96
60
1.13
78
1.02
28
2.12
47
3.15
33
11.4
75
Inte
gral
7.97
817.
9744
7.96
99
7.72
297.
7202
7.71
657.
7037
7.70
107.
6973
7.68
187.
6781
7.66
537.
6617
7.59
587.
5922
7.57
48
7.49
89
7.47
977.
4660
7.46
057.
4486
7.42
94
7.38
82
7.36
817.
3645
7.34
71
7.30
68
7.25
01
(ppm)
7.257.307.357.407.457.507.557.607.657.707.757.807.857.907.958.00
H-b x H-d+H-f
NH x H-6
NH x H-6’
H-8 x H-9 e H-8 x H-9’
H-7 x H-8
H-7’ x H-8
H-7’ x H-9 e H-7’ x H-9’
H-7 x H-7’
H-1 x H-2
H-6 x H-6’
OCH2φ x OCH2φ
6
54
3
21
7
8
9
a
cd
e
f
GAXD
b
O
OCH3
OBn
BnO
ON
HO
c'
b'
d'
a'
b'
c'

RESULTADOS E DISCUSSÃO
127
A análise dos espectros de RMN de 13C e DEPT 135 do produto isolado da segunda reação de
ciclização radicalar (FIGURAS 122 e 123) forneceu mais evidências da obtenção de GAXD.
Observam-se 18 sinais de carbonos aromáticos, entre eles cinco que correspondem a carbonos
não hidrogenados, o que é perfeitamente compatível com a estrutura de GAXD. Com base nos
valores de deslocamentos químicos tabelados atribuíram-se os sinais do carbono carbonílico e
dos carbonos ipso dos grupos benzila. O sinal de C-b e os de C-d ou C-f foram identificados pelas
manchas de correlação C x H observadas no mapa de contornos heteronuclear HMQC (FIGURA
124). Utilizando o mapa de contornos heteronuclear HMBC (FIGURAS 125 e 126), no qual há
manchas de correlação entre hidrogênios e carbonos distanciados por três ligações, foi possível
identificar o sinal do carbono ipso do substituinte fenila (C-a’) ao se observar quatro manchas de
correlação do sinal de H-b. Como três das manchas de correlação são com carbonos previamente
atribuídos (C=O, C-d e C-f), por exclusão a quarta mancha se refere à correlação com C-a’. A
partir da identificação do sinal de C-a’, foi possível diferenciar os sinais de H-d e H-f, pois só H-d
apresenta correlação com C-a’. Além disso, observa-se que o carbono carbonílico só apresenta
correlação com H-b (previamente definido) e H-f, confirmando a atribuição de H-f. Dessa forma,
usando as correlações do HMQC, distinguiram-se os sinais de C-d e C-f. Levando-se em conta
que, de acordo com valores tabelados, carbono ligado a anel fenila é desprotegido de cerca de
12 ppm, considerou-se que o sinal de carbono ipso mais distante do TMS refere-se a C-c (δ 141,71).
Por exclusão, o outro sinal de carbono ipso (δ 129,68) refere-se ao carbono ligado à carbonila (C-a).
As correlações observadas no HMBC para C-c permitem identificar o sinal de H-b’ (dupleto
relativo a dois hidrogênios) e determinar que o sinal de H-e encontra-se no multipleto entre δ 7,49
e 7,43, sobreposto aos sinais de mais dois hidrogênios aromáticos.
6
54
3
21
7
8
9
a
cd
e
f
GAXD
b
O
OCH3
OBn
BnO
ON
HO
c'
b'
d'
a'
b'
c'
δ 167,49 C=O δ 125,74 C-b
δ 141,71 C-c (ipso) δ 125,37 C-f
δ 140,13 C-a’ (ipso) δ 7,71 (td) H-d
δ 138,53;138,44 C ipso Bn δ 7,67 (td) H-f
δ 130,15 C-d δ 7,58 (d, 2 H) H-b’
δ 129,68 C-a (ipso) δ 7,49-7,43 (m, 3 H)
H-e + 2 H aromáticos
δ 128,99- 127,09 C aromáticos

RESULTADOS E DISCUSSÃO
128
Figura 122 – Espectro de RMN de 13C (50 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD.
Figura 123 – Espectro de DEPT 135 (50 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD.
167.4932 141.7120 140.1273 138.5274 138.4360 134.9239 130.1547 129.6823 128.9890 128.8519 128.3719 128.3033 127.9910 127.7320 127.6862 127.5872 127.5263 127.0920 125.7435 125.3702 117.8507 98.8348 78.7370 77.6323 77.0000 76.3676 73.9526 73.6402 73.3964 68.5281 55.2261 41.0328
(ppm) 30405060708090100110120130140150160170(ppm) 126128130132134136138140142 6
54
3
21
7
8
9
a
cd
e
f
GAXD
b
O
OCH3
OBn
BnO
ON
HO
c'
b'
d'
a'
b'
c'
C=O
C ipso
C-d
C-a C-b C-f
C-9 C-1
OCH3
C-6
C-8
C-a’ C-c
C açúcar e C benzílicos
134.9073 130.1664 128.9957 128.8502 128.3776 128.3049 127.9995 127.7377 127.6941 127.5923 127.5268 127.0905 125.7453 125.3672 117.8922 98.8192 78.7210 76.3069 76.2123 73.9582 73.6455 73.3983 68.5119 55.2270 41.0259
(ppm) 35404550556065707580859095100105110115120125130135140*
* C-3, C-4 ou C-5C-4 e
C-1
C benzílicos
C-8
c'
b'
a'
d'
b'
c'
O
OCH3
OBn
BnO
ON
HO
b
GAXD
f
ed
c
a
9
8
7
12
3
4 5
6
C-9
OCH3
C-6
*
C-2 C-5 ou C-4
C-7

RESULTADOS E DISCUSSÃO
129
Figura 124 – Mapa de contornos HMQC (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-
fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD.
Figura 125 – Mapa de contornos HMBC (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-
fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD.
H-6 x C-6
H-6’ x C-6
OCH3 x OCH3
H-b x C-b
H-d x C-d
H-f x C-f H-9 + H-9’ x C-9
H-1 x C-1
H-8 x C-8
OCH2φ x OCH2 φ
H-7 x C-7 H-7’ x C-7
H-2 x C-2 c'
b'
a'
d'
b'
c'
O
OCH3
OBn
BnO
ON
HO
b
GAXD
f
ed
c
a
9
8
7
12
3
4 5
6
H-b x C=O
H-b x C-a’
H-b x C-d H-b x C-f
H-d x C-a’
H-b’ x C-c
H-e x C-c
H-f x C=O
H-6 x C=O H-6’ x C=O
H-7 x C-9
OCH3 x C-1
H-7’ x C-9
H-9 + H-9’ x C-7
H-f x C-d
c'
b'
a'
d'
b'
c'
O
OCH3
OBn
BnO
ON
HO
b
GAXD
f
ed
c
a
9
8
7
12
3
4 5
6

RESULTADOS E DISCUSSÃO
130
Figura 126 – Seção expandida do mapa de contornos HMBC (400 MHz, CDCl3) de 4-O-alil-2,3-di-O-
benzil-6-desoxi-6-(3-fenilbenzoilamino)-α-D-galactopiranosídeo de metila – GAXD.
A obtenção do produto inesperado GAXD pôde ser confirmada pela análise de espectros de
massas (FIGURA 127). Observam-se os picos referentes a [M+H]+ de m/z 594,2786 (calculado:
594,2856), [M+Na]+ de m/z 616,2792 e [M+K]+ de m/z 632,2346.
Assim como nos espectros tandem MS/MS de GAX e GAXC, observam-se, além do pico [M+H]+,
dois outros picos referentes à perda de metanol (m/z 562,2767) e, em seguida, à perda de álcool
benzílico (m/z 454,2053) (FIGURA 128).
H-b x C=O
H-f x C=O
H-b x C-d
H-b x C-f
H-b x C-a’
H-d x C-a’ H-b’ x C-c
H-e x C-c
H-f x C-d
c'
b'
a'
d'
b'
c'
O
OCH3
OBn
BnO
ON
HO
b
GAXD
f
ed
c
a
9
8
7
12
3
4 5
6
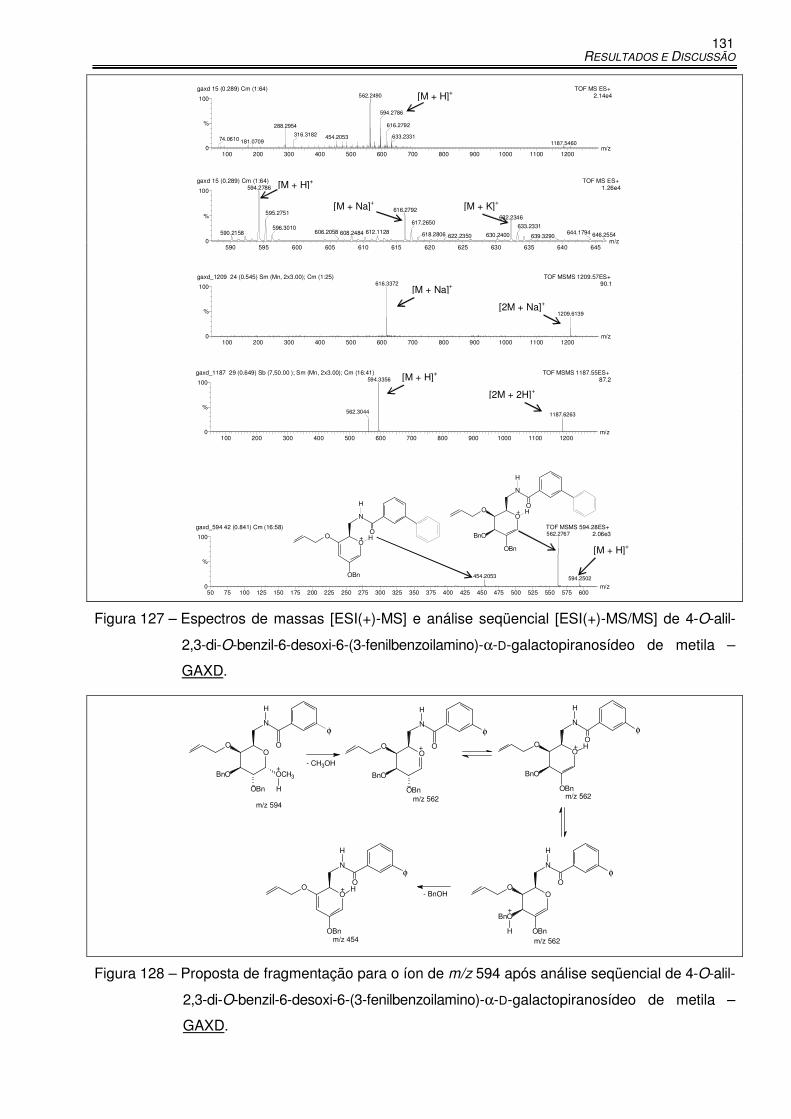
RESULTADOS E DISCUSSÃO
131
Figura 127 – Espectros de massas [ESI(+)-MS] e análise seqüencial [ESI(+)-MS/MS] de 4-O-alil-
2,3-di-O-benzil-6-desoxi-6-(3-fenilbenzoilamino)-α-D-galactopiranosídeo de metila –
GAXD.
O
BnO
O
OBn
OCH3
N
O
H
H
φ
+
+ +O
BnO
O
OBn
N
Oφ
H
HO
BnO
O
OBn
N
O
φ
H
O
BnO
O
OBn
N
Oφ
H
H
+
OO
OBn
N
Oφ
H
H+
- CH3OH
m/z 594m/z 562
m/z 454 m/z 562
m/z 562
- BnOH
Figura 128 – Proposta de fragmentação para o íon de m/z 594 após análise seqüencial de 4-O-alil-
2,3-di-O-benzil-6-desoxi-6-(3-fenilbenzoilamino)-α-D-galactopiranosídeo de metila –
GAXD.
[M + H]+
100 200 300 400 500 600 700 800 900 1000 1100 1200m/z0
100
%
gaxd 15 (0.289) Cm (1:64) TOF MS ES+ 2.14e4562.2490
288.2954
181.070974.0610 454.2053316.3182
594.2786
616.2792
633.23311187.5460
[M + H]+
[M + Na]+ [M + K]+
590 595 600 605 610 615 620 625 630 635 640 645m/z0
100
%
gaxd 15 (0.289) Cm (1:64) TOF MS ES+ 1.26e4594.2786
590.2158
616.2792595.2751
596.3010612.1128608.2484606.2058
632.2346617.2650
618.2806 630.2400622.2350
633.2331
646.2554644.1794639.3290
[M + Na]+
[2M + Na]+
100 200 300 400 500 600 700 800 900 1000 1100 1200m/z0
100
%
gaxd_1209 24 (0.545) Sm (Mn, 2x3.00); Cm (1:25) TOF MSMS 1209.57ES+ 90.1616.3372
1209.6139
[M + H]+
[2M + 2H]+
100 200 300 400 500 600 700 800 900 1000 1100 1200m/z0
100
%
gaxd_1187 29 (0.649) Sb (7,50.00 ); Sm (Mn, 2x3.00); Cm (16:41) TOF MSMS 1187.55ES+ 87.2594.3356
562.3044 1187.6263
[M + H]+
50 75 100 125 150 175 200 225 250 275 300 325 350 375 400 425 450 475 500 525 550 575 600m/z0
100
%
gaxd_594 42 (0.841) Cm (16:58) TOF MSMS 594.28ES+ 2.06e3562.2767
454.2053 594.2502
O
BnO
O
OBn
N
O
H
H+
OO
OBn
N
O
H
H+

RESULTADOS E DISCUSSÃO
132
Considerando-se que não foi possível obter macrociclos nas duas primeiras reações
desenvolvidas (1a - concentração do substrato: 12 mmol/L; tempo de adição da solução de
Bu3SnH/AIBN: 1:15 horas; 2a - concentração do substrato: 2,8 mmol/L; tempo de adição da
solução de Bu3SnH/AIBN: 5:20 horas), tentou-se obtê-los utilizando condições intermediárias
entre a primeira e a segunda reação no que se refere à diluição e tempo de adição (concentração
de GAX: 6 mmol/L; tempo de adição da solução de Bu3SnH/AIBN: 3:20 horas). Condição
semelhante encontra-se descrita na literatura (GHOSH; GHATAK, 1995; RODRIGUEZ et al.,
1999).
Nesta terceira tentativa, também foi obtida uma mistura complexa de produtos visualizada por
CCD, com predominância de dois de Rf muito próximos. Os dois produtos foram isolados
utilizando-se CCS e os espectros de RMN indicaram tratar-se de GAX (16% de recuperação) e
GAXC (38% de rendimento).
Mais uma tentativa foi realizada empregando-se as mesmas condições da segunda reação
radicalar, substituindo-se o benzeno por ciclo-hexano (WALLING, 1985; BOWMAN et al., 2000;
BECKWITH et al., 2004). Tinha-se expectativa de que alta diluição e adição lenta da solução de
Bu3SnH/AIBN minimizasse a formação do produto de hidrogenólise e de que, não ocorrendo a
adição do radical arila ao benzeno, pudesse ocorrer a ciclização. Como tanto GAX quanto o AIBN
não são solúveis em ciclo-hexano, utilizou-se o mínimo de benzeno necessário para a solubilização
da amida GAX e do AIBN. Desta reação, mais uma vez, não foi possível isolar nenhum produto
ciclizado e o produto de hidrogenólise, GAXC, foi isolado com 71% de rendimento.
Portanto, utilizando-se quatro diferentes condições de reação, não foi possível obter os
macrociclos 12-endo GAXA e 11-exo GAXB. Na primeira reação da m-iodobenzamida GAX com
Bu3SnH empregaram-se as mesmas condições que levaram à obtenção das macrolactamas 8 e
19 (FIGURA 54, p. 45) a partir de 7 e 18 (BINATTI et al., 2002; FARACO et al., 2003), isômero de
posição e análogo desprovido de unidade sacarídica de GAX, respectivamente. No entanto, só foi
obtido o produto de hidrogenólise GAXC, assim como ocorreu na terceira reação. Por outro lado,
na segunda tentativa, quando se utilizaram condições para minimizar a ocorrência da reação do
radical arila com o Bu3SnH (maior diluição e maior tempo de adição dos reagentes) não houve
formação de GAXC, mas o radical arila reagiu com o próprio solvente, levando à formação de
GAXD. Alterando-se o solvente na quarta reação para evitar a formação de GAXD, novamente, foi
isolado o produto de hidrogenólise, GAXC.
Algumas hipóteses podem ser aventadas para explicar os diferentes resultados das reações de
ciclização radicalar de GAX, 7 e 18.
Na o-iodobenzamida 7, além da restrição de rotação da ligação C-N, característica das amidas, há
restrição da ligação Ar-CO, atribuída ao efeito estérico do halogênio em orto, e há predominância
do rotâmero no qual o anel aromático e a carbonila não estão coplanares entre si (LEWIN et al.,
1975). Em relação às possíveis conformações correspondentes à ligação C-N, predomina o

RESULTADOS E DISCUSSÃO
133
confôrmero no qual o grupo mais volumoso ligado ao nitrogênio encontra-se syn à carbonila, em
razão da presença do átomo de iodo em orto (SNIECKUS et al., 1990). Portanto, no caso da
benzamida 7 predominaria o confôrmero 7a (FIGURA 129). O tempo de vida de radicais arila não
excede 10-5 s, sendo muito menor que o tempo gasto para interconversão dos rotâmeros syn e
anti de amidas (10-1-10-2 s), o que suporta a hipótese de que não há rotação da ligação C-N
durante a existência dos radicais arila (SNIECKUS et al., 1990). Assumindo que a reatividade dos
rotâmeros syn/anti (7a/7b) seria equivalente frente ao radical tributilestanila, as quantidades relativas
dos radicais formados por abstração de um átomo de iodo seriam determinadas pelas
concentrações de 7a e 7b, portanto seria formado predominantemente o radical 7a’ (FIGURA 129).
Em relação à ligação C-6/C-5, no rotâmero 7a’ há uma conformação alternada (7a’’) estabilizada
por formação de ligação de hidrogênio entre o hidrogênio ligado ao nitrogênio e o oxigênio do grupo
aliloxila, formando um ciclo de seis membros. Nesta conformação (7a’’) o radical arila encontra-se
adequadamente localizado para promover o ataque à ligação dupla, com formação subseqüente do
produto ciclizado 8 (BINATTI et al., 2005).
syn
syn anti
anti
7b'
N
CH2
O
OBn
OBn
H3CO
H
O
O
Bu3Sn. Bu3Sn.
7a
O
OCH3
OBn
OBnO
I
NCH2
H
O
N
CH2
O
OBn
OBn
H3CO
H
OI
O
7b
7a'
4
56
NCH2
O
OCH3
OBn
OBnOH
O
C4O
H6
NH6' H
O
O
H5
Bu3SnH
H5
O C4O
H6
NH6' H
O
7a''
O C4O
H6
NH6' H
O
H5
O
OCH3
OBn
BnO
O NH
O
11-Endo8
X
Figura 129 – Conformações syn e anti de 7 e dos respectivos radicais, conformação 7a’’
estabilizada por ligação de hidrogênio e formação do macrociclo 8.

RESULTADOS E DISCUSSÃO
134
Como 18 também é uma orto-iodobenzamida, pode-se considerar que os mesmos fatores que
conferem restrição de rotação das ligações Ar-CO e N-C=O em 7 o fazem em 18. Assim
predominaria o rotâmero syn (18a), que levaria ao radical arila 18a’ (FIGURA 130). Embora o grau
de liberdade rotacional da cadeia butílica presente em 18 e seus radicais seja maior do que o do
anel piranosídico de 7 e seus radicais, propõem-se que haja uma conformação estabilizada por
ligação de hidrogênio (18a’) na qual o radical arila esteja próximo do grupo alquenila. Esta hipótese
justifica a formação da macrolactama 19 (19%) e o menor rendimento em relação a 8 (33%).
Bu3Sn. Bu3Sn.
18a' 18b'
Bu3SnH
O
N
H
O
I
O
NH
(CH2)4OCH2CH=CH2
syn18a
anti18b
.
O
NH
(CH2)4OCH2CH=CH2
I
O
N(CH2)4OCH2CH=CH2
H
19 12-endo
.
O
N
H
O
X.
O
N
H
O
Figura 130 – Conformações syn e anti de 18 e dos respectivos radicais, conformação de 18a’
estabilizada por ligação de hidrogênio e formação do macrociclo 19.
Em GAX, como o iodo está em meta, a barreira de rotação da ligação Ar-CO é significativamente
menor do que nas orto-iodobenzamidas (LEWIN et al., 1975). Além disso, possivelmente, não haja
predominância do rotâmero syn (GAXa) ou anti (GAXb) (FIGURA 131), uma vez que não há
repulsão estérica entre o halogênio em meta e o substituinte do nitrogênio. A menor restrição
rotacional da ligação Ar-CO e o fato de as conformações das amidas syn e anti coexistirem no
substrato, e portanto nos radicais (GAXa’ e GAXb’), são argumentos que permitem supor que
GAX não conduziu a produtos de ciclização pelo fato de coexistirem muitas conformações, não
havendo predominância de alguma que seja adequada para a ciclização. Ou seja, em GAX não
haveria a pré-organização necessária para a ciclização, requisito essencial para a obtenção de
macrociclos por carbociclização radicalar (BLANKESTEIN; ZHU, 2005). Não ocorrendo a

RESULTADOS E DISCUSSÃO
135
ciclização, os radicais arila reagiram com o Bu3SnH, levando ao produto de hidrogenólise (GAXC),
ou com o próprio solvente (benzeno) para conduzir a GAXD, dependendo das condições de
reação.
Bu3SnH ou Benzeno
R= H GAXC
R= Ph GAXD
R
O
OCH3
OBn
BnO
ON
HO
N
CH2
O
OBn
OBn
H3CO
H
O
I
O
NCH2
H
O
OCH3
OBn
OBnO
I
O
Bu3Sn.
GAXbanti
GAXasyn
Bu3Sn.
X
GAXb'anti
GAXa'syn
NCH2
H
O
OCH3
OBn
OBnO
O
N
CH2
O
OBn
OBn
H3CO
H
O
O
Figura 131 – Conformações syn e anti de GAX e dos respectivos radicais e formação de GAXC e
GAXD.
Outra hipótese pode ser aventada para explicar a razão de não ter sido formado produto ciclizado
na reação de GAX com Bu3SnH: em nenhuma das conformações possíveis para o radical arila
proveniente de GAX haveria a proximidade necessária para que ocorresse o ataque à ligação
dupla e a formação do ciclo de 12 ou 11 membros (GAXA/GAXB).
É importante ressaltar que só foi encontrado um trabalho publicado em que é relatada a obtenção
de benzomacrociclos (14 e 17 membros) por carbociclização radicalar mediada por Bu3SnH a

RESULTADOS E DISCUSSÃO
136
partir de substratos em que o halogênio encontra-se meta em relação ao substituinte envolvido na
ciclização (FIGURA 46, p. 33) (BALRAJU et al., 2005). Em todos os outros trabalhos de obtenção
de macrociclos fundidos a anéis benzênicos utilizaram-se substratos com o halogênio em orto ao
substituinte que contém a ligação dupla (GHOSH; GHATAK, 1995; GIBSON et al., 1997; NANDI et
al., 2001; NANDI; CHATTOPADHYAY, 2002; PRADO et al., 2000; BINATTI et al, 2002; FARACO
et al., 2003; FARACO et al., 2004; DIAS et al., 2006; PIRES et al., 2006; ROCHA; PRADO, 2006).
Esta constatação demonstra a dificuldade de se obter macrociclos utilizando-se precursores meta
halogenados e evidencia a necessidade de estudos experimentais e teóricos para que se possa
explicar este fato.
4.9 TESTES DE ATIVIDADE ANTIBACTERIANA E ANTIFÚNGICA
Todas as soluções de padrões internos (cloranfenicol, gentamicina e anfotericina B) conduziram a
halos de inibição que variaram entre 9,40 e 22,00 mm. Os desvios padrão calculados para todos
os ensaios, realizados em triplicata, foram aceitáveis.
As soluções das substâncias GA6, GA7, GAX, GAXC e GAXD (FIGURA 57, p. 76) não levaram à
formação de halos de inibição nos testes de atividade antimicrobiana realizados.
Portanto, as substâncias testadas não demonstraram atividade antibacteriana e antifúngica contra
os microrganismos usados (TABELA 18, p. 76), nas condições em que os experimentos foram
realizados (item 3.3, p. 75).

CONCLUSÃO
137
5 CONCLUSÃO
Neste trabalho foi sintetizada a aliloxi-m-iodobenzamida inédita 4-O-alil-2,3-di-O-benzil-6-desoxi-6-
(3-iodobenzoilamino)-α-D-galactopiranosídeo de metila (GAX) a partir do α-D-galactopiranosídeo
de metila, por uma rota de síntese de oito etapas. A benzamida foi submetida a reações de
carbociclização radicalar mediadas por hidreto de tri-n-butilestanho.
De nenhuma reação radicalar realizada foi possível isolar macrolactamas. De três reações, nas
quais se variaram as condições da reação (solvente, diluição e tempo de adição dos reagentes),
foi isolado o produto acíclico reduzido 4-O-alil-2,3-di-O-benzil-6-desoxi-6-benzoilamino-α-D-
galactopiranosídeo de metila (GAXC), no qual o átomo de iodo foi substituído por um átomo de
hidrogênio. Em outra tentativa, na qual se empregaram maior diluição e maior tempo de adição
dos reagentes, foi isolado um produto inédito e inesperado, 4-O-alil-2,3-di-O-benzil-6-desoxi-6-(3-
fenilbenzoilamino)-α-D-galactopiranosídeo de metila (GAXD), formado por reação entre o radical
arila proveniente da amida e benzeno, o solvente da reação.
As o-iodobenzamidas 7 e 18, sintetizadas anteriormente em outros trabalhos, isômero de posição
e análogo desprovido de unidade sacarídica de GAX, conduziram às macrolactamas 8 e 19,
respectivamente. Com este trabalho, verificou-se a influência negativa, na ciclização radicalar, do
átomo de iodo em posição meta na benzamida derivada de carboidrato de configuração galacto,
GAX.
A aliloxi-m-iodobenzamida GAX, os produtos isolados das reações radicalares (GAXC e GAXD) e
dois intermediários (GA6 e GA7) foram submetidos a testes de atividade antibacteriana e
antifúngica e não demonstraram atividade.

REFERÊNCIAS BIBLIOGRÁFICAS
138
6 REFERÊNCIAS BIBLIOGRÁFICAS
ALLIN, S. M.; BARTON, W. R. S.; BOWMAN; W. R.; MCINALLY, T. Radical cyclisation onto
pyrazoles: synthesis of withasomnine. Tetrahedron Letters, v. 43, n. 23, p. 4191-4193, 2002.
ANISUZZAMAN, A. K. M.; WHISTLER, R. L. Selective replacement of primary hydroxyl-groups in
carbohydrates - preparation of some carbohydrate-derivatives containing halomethyl groups.
Carbohydrate Research, v. 61, p. 511-518, 1978.
ARAI, T.; SUENAGA, T.; KOYAMA, Y.; HONDA, H. Ascomycin, an antifungal antibiotic. Journal of
Antibiotics, v. 15, n. 6, p. 231-232, 1962.
ARMAREGO, W. L. F.; PERRIN, D. D. Purification of Laboratory Chemicals. 4. ed. Boston:
Butterworth Heinemann, 1996. 529 p.
ASTLEY, M. P.; PATTENDEN, G. Natural furanocembranoids - a synthetic approach to lophotoxin
based on an acyl radical macrocyclization strategy. Synthesis, p. 101-105, 1992.
BAILEY, K. L.; MOLINSKI, T. F. Entropically favorable macrolactamization. Synthesis of
isodityrosine peptide analogues by tandem Erlenmeyer condensation-macrolactamization. Journal
of Organic Chemistry, v. 64, n. 7, p. 2500-2504, 1999.
BALDWIN, J. E. Rules for ring closure. Journal of the Chemical Society, Chemical
Communications, p. 734-736, 1976.
BALDWIN, J. E.; ADLINGTON, R. M.; MITCHELL, M. B.; ROBERTSON, J. Intramolecular SH2'
macrocyclizations. Tetrahedron, v. 47, n. 30, p. 5901-5918, 1991.
BALDWIN, J. E.; ADLINGTON, R. M.; RAMCHARITAR, S. H. Free radical macrocyclisation via
propiolate esters. Tetrahedron, v. 48, n. 16, p. 3413-3428, 1992.
BALRAJU V.; REDDY. D. S.; PERIASAMY, M.; IQBAL, J. Synthesis of small cyclic peptides
constrained with 3-(3-aminomethylphenyl)propionic acid linkers using free radical-mediated
macrocyclization. Tetrahedron Letters, v. 46, n. 31, p. 5207-5210, 2005.
BARREIRO, E. J.; FERREIRA, V. F.; COSTA, P. R. R. Substâncias enantiomericamente puras
(SEP): a questão dos fármacos quirais. Química Nova, v. 20, n. 6, p. 647-656, 1997.
BEAK, P.; MUSICK, T. J.; CHEN, C. Does formal intramolecular transfer of an acidic deuterium to
a site of halogen-lithium exchange show that lithium-halogen exchange is faster than loss of the
acidic deuterium? Evidence in favor of an alternative mechanism. Journal of the American
Chemical Society, v. 110, n. 11, p. 3538-3542, 1988.

REFERÊNCIAS BIBLIOGRÁFICAS
139
BECKWITH, A. L. J.; BOWRY, V. W.; BOWMAN, W. R.; MANN, E.; PARR, J.; STOREY, J. M. D.
The mechanism of Bu3SnH-mediated homolytic aromatic substitution. Angewandte Chemie,
International Edition, v. 43, n. 1, p. 95-98, 2004.
BECKWITH, A. L. J.; DROK, K.; MAILLARD, B.; DEGUEIL-CASTAING, M.; PHILIPPON, A.
Formation of substituted macrocyclic ethers by radical cyclisation. Chemical Communications, p.
499-500, 1997.
BECKWITH, A. L. J.; GARA, W. B. Mechanism of cyclization of aryl radicals containing
unsaturated ortho-substituents. Journal of the Chemical Society, Perkin Transactions 2, n. 7, p.
795-802, 1975.
BEGLEY, M. J.; PATTENDEN, G.; SMITHIES, A.; TAPOLCZAY, D.; WALTER, D. S. Cascade
radical macrocyclisation transannulation approach towards the construction of ring-fused tricycles
and polycycles. Journal of the Chemical Society, Perkin Transactions 1, n.1, p. 21-29, 1996.
BELL, D. J.; LORBER, J. Use of the benzyl radical in syntheses of methylated sugars. Part I: 4:6-
dimethyl glucose. Journal of the Chemical Society, p. 453-455, 1940.
BERGE, J. M.; ROBERTS, S. M. Recommended work-up procedure for reductions employing tri-
normal-butyltin hydride. Synthesis, p. 471-472, 1979.
BERTASSO, M.; HOLZENKAMPFER, M.; ZEECK, A.; STACKEBRANDT, E.; BEIL, W.; FIEDLER,
H. P. Ripromycin and other polycyclic macrolactams from Streptomyces sp. Tu 6239: taxonomy,
fermentation, isolation and biological properties. Journal of Antibiotics, v. 56, n. 4, p. 364-371,
2003.
BINATTI, I. Síntese de benzolactamas por carbociclização radicalar 11-endo de orto-
iodobenzamida derivada da D-galactose. 2001. 70f. (Dissertação) – Programa de Pós-
Graduação em Ciências Farmacêuticas, Faculdade de Farmácia – Universidade Federal de Minas
Gerais, Belo Horizonte, 2001.
BINATTI, I. Síntese de macrolactona derivada de D-galactose e tentativa de síntese de
macrolactona e macrolactamas derivadas de D-glicose, por carbocilização radicalar. 2005.
274f. (tese) – Programa de Pós-Graduação em Química, Departamento de Química – ICEx –
Universidade Federal de Minas Gerais, Belo Horizonte, 2005a.
BINATTI, I.; ALVES, R. B.; PRADO, M. A. F.; ALVES, R. J.; SOUZA-FILHO, J. D.; DIAS, D. F.
Reações de carbociclização de orto-iodoaliloxibenzoato derivados de D-glicose e D-galactose e
comparação com as reações de análogos benzamidas. Química Nova, v. 28, n. 5, p. 1023-1029,
2005b.

REFERÊNCIAS BIBLIOGRÁFICAS
140
BINATTI, I.; PRADO, M. A. F.; ALVES, R. J.; FILHO, J. D. S. Synthesis of benzomacrolactam by
11-endo selective aryl radical cyclization of 2-iodobenzamide derived from D-galactose. Journal of
the Brazilian Chemical Society, v. 13, n. 5, p. 570-575, 2002.
BINKLEY, R. W. Modern Carbohydrate Chemistry. New York: Marcel Dekker, 1988. 343 p.
BLANKENSTEIN, J.; ZHU, J. P. Conformation-directed macrocyclization reactions. European
Journal of Organic Chemistry, n. 10, p. 1949-1964, 2005.
BOGER, D. L.; MATHVINK, R. J. Intramolecular acyl radical alkene addition-reactions -
macrocyclization reactions. Journal of the American Chemical Society, v. 112, n. 10, p. 4008-
4011, 1990.
BORNHOVD, E.; BURGDORF, W. H. C.; WOLLENBERG, A. Macrolactam immunomodulators for
topical treatment of inflammatory skin diseases. Journal of the American Academy of
Dermatology, v. 45, n. 5, p. 736-743, 2001.
BOWMAN, W. R.; FLETCHER, A. J.; POTTS, G. B. S. Synthesis of heterocycles by radical
cyclisation. Journal of the Chemical Society, Perkin Transactions 1, n. 24, p. 2747-2762, 2002.
BOWMAN W. R.; HEANEY, H.; JORDAN, B. M. Synthesis of Oxindoles by radical cyclization.
Tetrahedron Letters, v. 29, n. 50, p. 6657-6660, 1988.
BOWMAN, W. R.; MANN, E.; PARR, J. Bu3SnH mediated oxidative radical cyclisations: synthesis
of 6H-benzo[c]chromen-6-ones. Journal of the Chemical Society, Perkin Transactions 1, n. 17,
p. 2991-2999, 2000.
BULUSU, M. A. R. C.; WALDSTATTEN, P.; TRICOTET, T.; ROCHAIS, C.; STECK, A.; BACHER
M. Synthesis of 6-vinyl and 5-vinylproline analogues of ascomycin. Tetrahedron Letters, v. 45, n.
30, p. 5833-5836, 2004.
CADDICK, S.; HAMZA, D.; WADMAN, S. N.; WILDEN, J. D. Solid-phase intermolecular radical
reactions 2: synthesis of c-glycopeptide mimetics via a novel acrylate acceptor. Organic Letters,
v. 4, n. 10, p. 1775-1777, 2002.
CAREY, J. S.; LAFFAN, D.; THOMSON, C.; WILLIAMS, M. T. Analysis of the reactions used for
the preparation of drug candidate molecules. Organic & Biomolecular Chemistry, v. 4, n. 12, p.
2337-2347, 2006.
CHENG, Y. Q.; TANG, G. L.; SHEN, B. Identification and localization of the gene cluster encoding
biosynthesis of the antitumor macrolactam leinamycin in Streptomyces atroolivaceus S-140.
Journal of Bacteriology, v. 184, n. 24, p. 7013-7024, 2002.

REFERÊNCIAS BIBLIOGRÁFICAS
141
CLYNE, M. A.; ALDABBAGH, F. Photochemical intramolecular aromatic substitutions of the
imidazol-2-yl radical are superior to those mediated by Bu3SnH. Organic & Biomolecular
Chemistry, v. 4, n. 2, p. 268-277, 2006.
COLLINS, P. M.; FERRIER, R. J. Monosacharides: Their Chemistry and Their Roles in
Natural Products. New York: John Wiley & Sons Inc, 1995. 574 p.
COOPER, R.; TRUUMEES, I.; YARBOROUGH, R.; LOEBENBERG, D.; MARQUEZ, J.; HORAN,
A.; PATEL, M.; GULLO, V. Macrolactams - 2 novel homologous series of compounds produced by
Actinomadura sp. SCC-1778. Journal of Antibiotics, v. 45, n. 5, p. 633-638, 1992.
COURI, M. R. C.; EVANGELISTA. E. A.; ALVES, R. B.; PRADO, M. A. F.; GIL, R. P. F.; ALMEIDA,
M. V.; RASLAN, D. S. Microwave-assisted rapid deacetalation of carbohydrates. Synthetic
Communications, v. 35, p. 2025-2031, 2005.
COX, N. J. G.; MILLS, S. D.; PATTENDEN, G. Macrocyclizations using allylic radical intermediates
- a new synthetic approach to natural 14-membered cembranoids. Journal of the Chemical
Society, Perkin Transactions 1, n. 11, p. 1313-1321, 1992.
COX, N. J. G.; PATTENDEN, G.; MILLS, S. D. Radical macrocyclizations in synthesis - a new
approach to mukulol and marine cembranolide lactones. Tetrahedron Letters, v. 30, n. 5, p. 621-
624, 1989.
CULVENOR, C. C. J.; BECK, A. B.; CLARKE, M.; COCKRUM, P. A.; EDGAR, J. A.; FRAHN, J. L.;
JAGO, M. V.; LANIGAN, G. W.; PAYNE, A. L.; PETERSON, J. E.; PETTERSON, D. S.; SMITH, L.
W.; WHITE, R. R. Isolation of toxic metabolites of phomopsis-leptostromiformis responsible for
lupinosis. Australian Journal of Biological Sciences, v. 30, n. 4, p. 269-277, 1977.
CURRAN D. P. The design and application of free-radical chain reactions in organic-synthesis –
Part 1. Synthesis, p. 417-439, 1988.
CURRAN, D. P.; YANG, F. L.; CHEONG, J. H. Relative rates and approximate rate constants for
inter- and intramolecular hydrogen transfer reactions of polymer-bound radicals. Journal of the
American Chemical Society, v. 124, n. 50, p. 14993-15000, 2002.
DE OLIVEIRA, R. B.; DE SOUZA, J. D.; PRADO, M. A. F.; EBERLIN, M. N.; MEURER, E. C.;
SANTOS, L. S.; ALVES, R. J. Synthesis of unexpected six-membered imides by free-radical
carbocyclisation on carbohydrate templates. Tetrahedron, v. 60, n. 44, p. 9901-9908, 2004.
DIAS, D. F.; PRADO, M. A. F.; PINTO, G. D.; ALVES, R. J.; ALVES, R. B.; FILHO, J. D. S. Síntese
de aliloxi-orto-iodobenzamida derivada de D-glicose e reação de ciclização radicalar mediada por
hidreto de tri-n-butilestanho. Quimica Nova, v. 29, n. 3, p. 444-451, 2006.

REFERÊNCIAS BIBLIOGRÁFICAS
142
DUFOUR, M.; GRAMAIN, J. C.; HUSSON, H. P.; SINIBALDI, M. E.; TROIN, Y. Convergent
approach to tetracyclic [ABCE] intermediates in Aspidosperma alkaloids synthesis. Synthetic
Communications, v. 22, n. 2, p. 189-200, 1992.
DUMONT, F. J. FK506, an immunosuppressant targeting calcineurin function. Current Medicinal
Chemistry, v. 7, n. 7, p. 731-748, 2000.
ESCOLANO, C.; JONES, K. Aryl radical cyclisation onto pyrroles: a divergent synthesis of
spiropyrrolidinyloxindoles and pyrroloquinolines. Tetrahedron Letters, v. 41, n. 46, p. 8951-8955,
2000.
FARACO, A. A. G.; PRADO, M. A. F.; ALVES, R. B.; FARACO, R. F. P.; ALVES, R. J.; SOUZA, J.
D.; MEURER, E. C.; EBERLIN, M. N. A new 20-membered macrocyclic dilactam: an unexpected
product of a tri-n-butyltin hydride-mediated radical reaction. Tetrahedron Letters, v. 45, n. 16, p.
3317-3320, 2004.
FARACO, A. A. G.; PRADO, M. A. F.; ALVES, R. J.; SOUZA, J. D.; ALVES, R. B.; FARACO, R. F.
P. Synthesis of benzomacrolactam by 12-endo selective aryl radical cyclization of N-(4-
allyloxybutyl)-2-iodobenzamide. Synthetic Communications, v. 33, n. 3, p. 463-474, 2003.
FARMACOPÉIA brasileira. 4. ed. São Paulo: Atheneu, 1988. Parte I.
FERREIRA, V. F. Carboidratos importantes em síntese orgânica. Química Nova, v. 18, n. 2, p.
267-273, 1995.
FOSSEY, J.; LEFORT, D.; SORBA, J. Free Radicals in Organic Chemistry. Paris: John Wiley &
Sons, 1995, 307 p.
FRÉCHET, J. M. J.; BAER, H. H. Concerning the problem of stereospecific glycosylation.
Synthesis and methanolysis of some 2-O-benzylated D-galactopyranosyl and D-galactofuranosyl
halides. Canadian Journal of Chemistry, v. 53, n. 5, p. 670-679, 1975.
FRYDMAN, B.; BHATTACHARYA, S.; SARKAR, A.; DRANDAROV, K.; CHESNOV, S.;
GUGGISBERG, A.; POPAJ, K.; SERGEYEV, S.; YURDAKUL, A.; HESSE, M.; BASU, H. S.;
MARTON, L. J. Macrocyclic polyamines deplete cellular ATP levels and inhibit cell growth in
human prostate cancer cells. Journal of Medicinal Chemistry, v. 47, n. 4, p. 1051-1059, 2004.
GANGULY, A. K.; WANG, C. H.; DAVID, M.; BARTNER, P.; CHAN, T. M. Synthesis of heterocyclic
compounds using radical reactions. Tetrahedron Letters, v. 43, n. 38, p. 6865-6868, 2002.

REFERÊNCIAS BIBLIOGRÁFICAS
143
GAREGG, J.; SAMUELSSON, B. Novel reagent system for converting a hydroxy-group into a iodo-
group in carbohydrates with inversion of configuration. Part 2. Journal of the Chemical Society,
Perkin Transactions 1, n. 12, p. 2866-2869, 1980.
GENTILE, G.; FATTORI, D.; BOTTA, M.; CORELLI, F.; FUSAR-BASSINI, D.; LAMBA, D.
Synthesis of dimeric and tetrameric macrolactams with cytotoxic activity. Canadian Journal of
Chemistry-Revue Canadienne de Chimie, v. 78, n. 6, p. 925-934, 2000.
GHOSH, K.; GHATAK, U. R. Regioselective 9-endo aryl radical cyclization: a new synthetic route
to decahydro-5h-dibenzo[a,d] and [a,e]-cyclononenols. Tetrahedron Letters, v. 36, n. 27, p. 4897-
4900, 1995.
GIBSON, S. E.; GUILLO, N.; TOZER, M. J. Application of 7-endo, 8-endo and 9-endo radical
cyclisations to the synthesis of conformationally constrained amino acids and comparison with the
corresponding Heck reactions. Chemical Communications, n. 6, p. 637-638, 1997.
GIESE, B. Formation of cc bonds by addition of free-radicals to alkenes. Angewandte Chemie,
International Edition in English, v. 22, n. 10, p. 753-764, 1983.
GIESE, B. Radicals in Organic Synthesis: Formation of carbon-carbon bonds. Oxford:
Pergamon Press, 1986, 294 p.
GRASSBERGER, M.; BAUMRUKER, T.; ENZ, A.; HIESTAND, P.; HULTSCH, T.; KALTHOFF, F.;
SCHULER, W.; SCHULZ, M.; WERNER, F. J.; WINISKI, A.; WOLFF, B.; ZENKE, G. A novel anti-
inflammatory drug, SDZ ASM 981, for the treatment of skin diseases: in vitro pharmacology.
British Journal of Dermatology, v. 141, n. 2, p. 264-273, 1999.
GRIESBECK, A. G.; OELGEMOLLER, M.; LEX, J. Photochemistry of MTM- and MTE-esters of
omega-phthalimido carboxylic acids: macrocyclization versus deprotection. Journal of Organic
Chemistry, v. 65, n. 26, p. 9028-9032, 2000.
HALL, D. M. A practical synthesis of 4-6-O-benzilidene-α- and –β-D-glucopyranoside.
Carbohydrate Research, v. 86, p. 158-160, 1980.
HANDA, S.; PATTENDEN, G. Free radical-mediated macrocyclisations and transannular
cyclisations in synthesis. Contemporary Organic Synthesis, v. 4, n. 3, p. 196-215, 1997.
HARROWVEN D. C.; SUTTON B. J.; COULTON S. Intramolecular radical additions to pyridines.
Organic & Biomolecular Chemistry, v. 1, n. 22, p. 4047-4057, 2003.
HARROWVEN, D. C.; GUY, I. L. KF-silica as a stationary phase for the chromatographic removal
of tin residues from organic compounds. Chemical Communications, n. 17, p. 1968-1969, 2004.

REFERÊNCIAS BIBLIOGRÁFICAS
144
HEGDE, V. R.; PATEL, M. G.; GULLO, V. P.; GANGULY, A. K.; SARRE, O.; PUAR, M. S.
Macrolactams - a new class of antifungal agents. Journal of the American Chemical Society, v.
112, n. 17, p. 6403-6405, 1990.
HEGDE, V. R.; PATEL, M. G.; GULLO, V. P.; PUAR, M. S. Sch-38518 and sch-39185 - 2 novel
macrolactam antifungals. Journal of the Chemical Society, Chemical Communications, n. 12,
p. 810-812, 1991.
HEGDE, V.; PATEL, M.; HORAN, A.; GULLO, V.; MARQUEZ, J.; GUNNARSSON, I.; GENTILE,
F.; LOEBENBERG, D.; KING, A. Macrolactams - a novel class of antifungal antibiotics produced by
Actinomadura spp. SCC-1776 and SCC-1777. Journal of Antibiotics, v. 45, n. 5, p. 624-632,
1992.
HENDRICKSON, J. B.; CRAM, D. J.; HAMMOND, G. S. Organic Chemistry. 3. ed. New York:
McGram-Hill, 1970, 1279 p.
HITCHCOCK, S. A.; PATTENDEN, G. Synthesis of macrocycles via allylic radical intermediates - a
total synthesis of (-)-zearalenone. Tetrahedron Letters, v. 31, n. 25, p. 3641-3644, 1990.
HITCHCOCK, S. A.; PATTENDEN, G. Total synthesis of the mycotoxin (-)-zearalenone based on
macrocyclization using a cinnamyl radical intermediate. Journal of the Chemical Society, Perkin
Transactions 1, n. 11, p. 1323-1328, 1992.
HOULDSWORTH, S. J.; PATTENDEN, G.; PRYDE, D. C.; THOMSON, N. M. Studies towards the
taxane ring system via a cascade macrocyclisation-transannulation strategy. Journal of the
Chemical Society, Perkin Transactions 1, n. 8, p. 1091-1093, 1997.
ISHIBASHI, H.; KATO, I.; TAKEDA, Y.; TAMURA, O. Synthesis of a model compound of mappicine
ketone based on sulfur-directed 5-exo selective aryl radical cyclization onto enamides.
Tetrahedron Letters, v. 42, n. 5, p. 931-933, 2001.
ISHIBASHI, H.; KAWANAMI, H.; NAKAGAWA, H. ; IKEDA, M. Sulfur-controlled 6-exo aryl radical
cyclisation of N-ethenyl-2-(2-bromophenyl)acetamides: Synthesis of (+/-)-tetrahydropalmatine and
saulatine. Journal of the Chemical Society, Perkin Transactions 1, n. 16, p. 2291-2295, 1997.
ISHIBASHI, H.; KOBAYASHI, T.; MACHIDA, N.; TAMURA, O. A new efficient route to (+/-)-
physostigmine and(+/-)-physovenine by means of 5-exo selective aryl radical cyclization of o-
bromo-N-acryloylanilides. Tetrahedron, v. 56, n. 11, p. 1469-1473, 2000.
JEFFERSON, E. A.; SWAYZE, E. E.; OSGOOD, S. A.; MIYAJI, A.; RISEN, L. M.; BLYN, L. B.
Antibacterial activity of quinolone-macrocycle conjugates. Bioorganic & Medicinal Chemistry
Letters, v. 13, n. 10, p. 1635-1638, 2003.

REFERÊNCIAS BIBLIOGRÁFICAS
145
JESSOP, C. N.; PARSONS, A. F.; ROUTLEDGE, A.; IRVINE, D. Cyclisation of dienes using
phosphorus-centred radicals to form organophosphorus adducts. Tetrahedron Letters, v. 44, n. 3,
p. 479-483, 2003.
JONAS, D.; OZLU, Y.; PARSONS, P. J. An intramolecular radical cyclization approach to the
synthesis of periplanone-b. Synlett, n. 3, p. 255-256, 1995.
JONES, K.; WILKINSON, J. A total synthesis of horsfiline via aryl radical cyclization. Journal of
the Chemical Society, Chemical Communications, n. 24, p. 1767-1769, 1992.
KAWAI, M.; GUNAWARDANA, I. W. K.; MOLLISON, K. W.; HSIEH, G. C.; LANE, B. C.; LULY, J.
R. Studies on an immunosuppressive macrolactam, ascomycin: synthesis of a C-33 hydroxyl
derivative. Bioorganic & Medicinal Chemistry Letters, v. 8, n. 8, p. 935-938, 1998.
KISS, J.; BURKHARDT, F. Beta-eliminative decomposition of 4-o-substituted hexopyranoside
uronate derivatives. Helvetica Chimica Acta, v. 53, n. 5, p. 1000-1011, 1970.
KOHYAMA, N.; YAMAMOTO, Y. Total synthesis of stevastelin B, a novel immunosuppressant.
Synlett, n. 5, p. 694-696, 2001.
KOISO, Y.; NATORI, M.; IWASAKI, S.; SATO, S.; SONODA, R.; FUJITA, Y.; YAEGASHI, H.;
SATO, Z. Ustiloxin - a phytotoxin and a mycotoxin from false smut balls on rice panicles.
Tetrahedron Letters, v. 33, n. 29, p. 4157-4160, 1992.
KUPCHAN, S. M.; KOMODA, Y.; THOMAS, G. J.; SMITH, R. M.; BRYAN, R. F.; HALTIWAN, R.
C.; KARIM, A.; COURT, W. A.; GILMORE, C. J. Tumor inhibitors .73. maytansine, a novel
antileukemic ansa macrolide from maytenus-ovatus. Journal of the American Chemical Society,
v. 94, n. 4, p. 1354-1356, 1972.
LAMAS, C.; SAA, C.; CASTEDO, L.; DOMINGUEZ, D. Synthesis of isoquinoline alkaloids through
a 10-membered lactam obtained by radical macrocyclization. Tetrahedron Letters, v. 33, n. 38, p.
5653-5654, 1992.
LANG, E. S.; COMASSETO, J. V. Catálise por transferência de fase. Química Nova, v. 11, n. 2, p.
238-257, 1988.
LEMARCHAND, A.; BACH, T. Synthesis of a para-quinone macrolactam related to geldanamycin
by ring closing metathesis. Tetrahedron, v. 60, n. 43, p. 9659-9673, 2004.
LEWIN, A. H.; FRUCHT, M.; CHEN, K. V. J.; BENEDETTI, E.; DI BLASIO, B. Restricted Rotation
in Amides – VI: Configurations and Conformations of Unsymmetrical Tertiary Benzamides.
Tetrahedron, v. 31, n. 3, p. 207-215, 1975.

REFERÊNCIAS BIBLIOGRÁFICAS
146
LI, P. X.; EVANS, C. D.; FORBECK, E. M.; PARK, H.; BAI, R. L.; HAMEL, E.; JOULLIE, M. M.
Total synthesis and biological evaluation of ustiloxin natural products and two analogs. Bioorganic
& Medicinal Chemistry Letters, v. 16, n. 18, p. 4804-4807, 2006.
LIMA, V. L. E. Os fármacos e a quiralidade: uma breve abordagem. Química Nova, v. 20, n. 6, p.
657-663, 1997.
LUCCHESE, A. M.; MARZORATI, L. Catálise de transferência de fase. Química Nova, v. 23, n. 5,
p. 641-652, 2000.
LUESCH, H.; YOSHIDA, W. Y.; MOORE, R. E.; PAUL, V. J.; MOOBERRY, S. L. Isolation,
structure determination, and biological activity of lyngbyabellin A from the marine cyanobacterium
Lyngbya majuscula. Journal of Natural Products, v. 63, n. 5, p. 611-615, 2000.
MACK, M. M.; MOLINSKI, T. F.; BUCK, E. D.; PESSAH, I. N. Novel modulators of skeletal-muscle
FKBP12/ calcium-channel complex from Ianthella basta: role of FKBP12 in channel gating.
Journal of Biological Chemistry, v. 269, n. 37, p. 23236-23249, 1994.
MARCH, J. Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. New York:
John Wiley & Sons, 1985, 1346 p.
MARCO-CONTELLES, J.; ALHAMBRA, C.; MARTÍNEZ-GRAU, A. Carbocycles from
carbohydrates via free radical cyclizations: synthesis and manipulation of annulated furanoses.
Synlett, p. 693-699, 1998.
MARINOVIC, N. N.; RAMANATHAN, H. The synthesis of fused and bridged ring systems by free
radical carbocyclisation - a general route to masked 1,4-diketones. Tetrahedron Letters, v. 24, n.
18, p. 1871-1874, 1983.
MARTIN, M.; MAS, G.; URPI, F.; VILARRASA, J. High-yielding enantioselective synthesis of the
macrolactam aglycon of Sch 38516 from two units of (2R)-2-ethyl-4-penten-1-ol. Angewandte
Chemie, International Edition, v. 38, n. 20, p. 3086-3089, 1999.
MARTINEZ-GRAU, A.; MARCO-CONTELLES, J. Carbocycles from carbohydrates via free radical
cyclizations: new synthetic approaches to glycomimetics. Chemical Society Reviews, v. 27, n. 2,
p. 155-162, 1998.
MASUNO, M. N.; PESSAH, I. N.; OLMSTEAD, M. M.; MOLINSKI, T. F. Simplified cyclic analogues
of bastadin-5. Structure-activity relationships for modulation of the RyR1/FKBP12 Ca2+ channel
complex. Journal of Medicinal Chemistry, v. 49, n. 15, p. 4497-4511, 2006.

REFERÊNCIAS BIBLIOGRÁFICAS
147
MILLIGAN, K. E.; MARQUEZ, B. L.; WILLIAMSON, R. T.; GERWICK, W. H. Lyngbyabellin B, a
toxic and antifungal secondary metabolite from the marine cyanobacterium Lyngbya majuscula.
Journal of Natural Products, v. 63, n. 10, p. 1440-1443, 2000.
NAKAGAWA, Y.; IRIE, K.; NAKAMURA, Y.; OHIGASHI, H. The amide hydrogen of (-)-indolactam-
V and benzolactam-V8's plays a critical role in protein kinase C binding and tumor-promoting
activities. Bioorganic & Medicinal Chemistry Letters, v. 11, n. 5, p. 723-728, 2001.
NANDI, A.; CHATTOPADHYAY, P. Synthesis of chiral trans-fused pyrano[3,2-c][2]benzoxocines
from D-mannose by regioselective 8-endo-aryl radical cyclization. Tetrahedron Letters, v. 43, n.
34, p. 5977-5980, 2002.
NANDI, A.; MUKHOPADHYAY, R.; CHATTOPADHYAY, P. Synthesis of chiral cis- and trans-
furo[3,2-c][2]benzoxocines from D-glucose by regioselective 8-endo aryl radical cyclisation.
Journal of the Chemical Society, Perkin Transactions 1, n. 24, p. 3346-3351, 2001.
NARUSE, N.; TENMYO, O.; KAWANO, K.; TOMITA, K.; OHGUSA, N.; MIYAKI, T.; KONISHI, M.;
OKI, T. Fluvirucin-A1, fluvirucin-A2, fluvirucin-B1, fluvirucin-B2, fluvirucin-B3, fluvirucin-B4, and
fluvirucin-B5, new antibiotics active against influenza-a virus .1. Production, isolation, chemical-
properties and biological-activities. Journal of Antibiotics, v. 44, n. 7, p. 733-740, 1991.
NEWMAN, D. J.; CRAGG, G. M.; SNADER, K. M. The influence of natural products upon drug
discovery. Natural Product Reports, v. 17, n. 3, p. 215-234, 2000.
NISHIO, M.; KOHNO, J.; SAKURAI, M.; SUZUKI, S.; OKADA, N.; KAWANO, K.; KOMATSUBARA,
S. TMC-135A and B, new triene-ansamycins, produced by Streptomyces sp.. Journal of
Antibiotics, v. 53, n. 7, p. 724-727, 2000.
OJIMA, I.; GENG, X. D.; LIN, S. N.; PERA, P.; BERNACKI, R. J. Design, synthesis and biological
activity of novel C2-C3 ' N-linked macrocyclic taxoids. Bioorganic & Medicinal Chemistry
Letters, v. 12, n. 3, p. 349-352, 2002.
OLIVEIRA, M. T.; PRADO, M. A. F.; ALVES, R. B.; CESAR, A.; ALVES, R. J.; QUEIROGA, C. G.;
SANTOS, L. S.; EBERLIN, M. N. Tri-n-butyltin hydride-mediated radical reaction of a 2-
iodobenzamide: formation of an unexpected carbon-tin bond. Journal of Brazilian Chemical
Society, 2007. (Aceito).
ONISHI, N.; IZAKI, K.; TAKAHASHI, H. A macrocyclic antibiotic m-230b produced by myxococcus-
xanthus: Isolation and characterization. Journal of Antibiotics, v. 37, n. 1, p. 13-19, 1984.

REFERÊNCIAS BIBLIOGRÁFICAS
148
PARK, Y. T.; JUNG, C. H.; KIM, M. S.; SONG, N. W.; KIM, D. Photoreaction of 2-halo-N-
pyridinylbenzamide: Intramolecular cyclization mechanism of phenyl radical assisted with n-
complexation of chlorine radical. Journal of Organic Chemistry, v. 66, n. 7, p. 2197-2206, 2001.
PARK, Y. T.; SONG, N. W.; HWANG, C. G.; KIM, K. W.; KIM, D. Photocyclization mechanism of
halopyridinium salt tethered to arene: flash photolysis observation of a pyridinium sigma,
cyclohexadienyl radicals, and a dihalide radical anion in aqueous solution. Journal of the
American Chemical Society, v. 119, n. 44, p. 10677-10683, 1997.
PATEL, V. F.; ANDIS, S. L.; KENNEDY, J. H.; RAY, J. E.; SCHULTZ, R. M. Novel cryptophycin
antitumor agents: synthesis and cytotoxicity of fragment "B" analogues. Journal of Medicinal
Chemistry, v. 42, n. 14, p. 2588-2603, 1999.
PATTENDEN, G.; THOM, S. M. Polyene macrolactam construction using a stille vinyl-vinyl
coupling protocol: an approach to the antitumor antibiotic substance leinamycin. Synlett, n. 3, p.
215-216, 1993.
PAVIA, D. L.; LAMPMAN, G. M.; KRIZ, G. S. Introduction to Spectroscopy: A Guide for
Students of Organic Chemistry. 3. ed. Stamford: Thomson Learning, 2001, 579 p.
PENG, W. M.; BLAGG, B. S. J. A versatile approach toward the ansamycin antibiotics. Organic
Letters, v. 8, n. 5, p. 975-978, 2006.
PIETRASZKIEWICZ, M.; JURCZAK, J. Synthesis of chiral diaza crow ethers incorporating
carbohydrate units. Tetrahedron, v. 40, n. 15, p. 2967-2970, 1984.
PINTO, G. D.; PRADO, M. A. F. Síntese de orto-iodobenzamida, substrato para a obtenção de
benzomacrolactama. In: XIV Semana de Iniciação Científica – Conhecimento e Cultura – UFMG,
2005, Belo Horizonte. Anais... Belo Horizonte: UFMG, 2005. 1 CD-ROM.
PIRES, M. C.; PRADO, M. A. F.; ALVES, R. J. Reação de carbociclização radicalar para obtenção
de benzomacrolactamas derivadas de carboidratos. In: XX Encontro Regional da Sociedade
Brasileira de Química – MG, 2006, São João Del Rei. Anais... São João Del Rei: UFSJ, 2006. 1
CD-ROM.
PORTER, N. A.; CHANG, V. H. T. Macrolide formation by free-radical cyclization. Journal of the
American Chemical Society, v. 109, n. 16, p. 4976-4981, 1987.
PORTER, N. A.; CHANG, V. H. T.; MAGNIN, D. R.; WRIGHT, B. T. Free-radical macrocyclization-
transannular cyclization. Journal of the American Chemical Society, v. 110, n. 11, p. 3554-3560,
1988.

REFERÊNCIAS BIBLIOGRÁFICAS
149
PORTER, N. A.; MAGNIN, D. R.; WRIGHT, B. T. Free-radical macrocyclization. Journal of the
American Chemical Society, v. 108, n. 10, p. 2787-2788, 1986.
PRADO, M. A. F.; ALVES, R. J.; SOUZA, J. D.; ALVES, R. B.; PEDROSA, M. T. C.; PRADO, R.
F.; FARACO, A. A. G. Synthesis of benzolactams by 11-endo selective aryl radical cyclisation of 2-
iodobenzamides. Journal of the Chemical Society, Perkin Transactions 1, n. 12, p. 1853-1857,
2000.
PUAR, M. S.; GULLO, V.; GUNNARSSON, I.; HEGDE, V.; PATEL, M.; SCHWARTZ, J.
Biosynthesis of macrolactam antifungal agents. Bioorganic & Medicinal Chemistry Letters, v. 2,
n. 6, p. 575-578, 1992.
QUEIROGA, C. G.; PRADO, M. A. F. Síntese e reação mediada por hidreto de tri-n-butilestanho
de orto iodobenzamida derivada de carboidrato de configuração galacto. In: XV Semana de
Iniciação Científica – Conhecimento e Cultura – UFMG, 2006, Belo Horizonte. Anais... Belo
Horizonte: UFMG, 2006. 1 CD-ROM.
RAMASESHAN, M.; ROBITAILLE, M.; ELLINGBOE, J. W.; DORY, Y. L.; DESLONGCHAMPS, P.
Design and synthesis of macro-heterocycles structurally related to tirofiban. Tetrahedron Letters,
v. 41, n. 24, p. 4737-4742, 2000.
ROBERTSON, G. J.; LAMB, R. A. The condensation of α-metilgalactoside with benzaldehyde.
Journal of the Chemical Society, p. 1321-1322, 1934.
ROBERTSON, J.; BURROWS, J. N.; STUPPLE, P. A. Bicyclo[10.2.1]pentadecenone derivatives
by free radical macrocyclisation. Tetrahedron, v. 53, n. 43, p. 14807-14820, 1997.
ROBERTSON, J.; HATLEY, R. J. D. Formal synthesis of roseophilin. Chemical Communications,
n. 16, p. 1455-1456, 1999.
ROCHA, A. P. C.; PRADO, M. A. F.; CORÊA, C. E. M. Reação de carbociclização radicalar
mediada por hidreto de tri-n-butilestanho de substrato derivado de carboidrato. In: XIV Semana de
Iniciação Científica – Conhecimento e Cultura – UFMG, 2005, Belo Horizonte. Anais... Belo
Horizonte: UFMG, 2005. 1 CD-ROM.
ROCHA, A. P. C.; PRADO, M. A. F. Síntese de macrolactama, potencial agente bioativo, por
reação de carbociclização radicalar de substrato derivado de carboidrato. In: XX Encontro
Regional da Sociedade Brasileira de Química – MG, 2006, São João Del Rei. Anais... São João
Del Rei: UFSJ, 2006. 1 CD-ROM.

REFERÊNCIAS BIBLIOGRÁFICAS
150
RODRIGUEZ, G.; CASTEDO, L.; DOMINGUEZ, D., SAA, C.; ADAM, W. Regioselective synthesis
of isoquino[1,2-b][3]benzazepines (homoprotoberberines) through 11-membered-ring stilbene
lactams obtained by radical macrocyclization. Journal of Organic Chemistry, v. 64, n. 13, p.
4830-4833, 1999.
ROSA, A. M.; LOBO, A. M.; BRANCO, P. S.; PRABHAKAR, S. The chemistry and reactivity of aryl
radicals - the c-c bond formation from o-bromobenzylphenylethers with tin hydride and
azobisisobutyronitrile. Tetrahedron, v. 53, n.1, p. 285-298, 1997.
ROXBURGH, C. J. The syntheses of large-ring compounds. Tetrahedron, v. 51, n. 36, p. 9767-
9822, p. 1995.
SALOMON, C. J.; DANELON, G. O.; MASCARETTI, O. A. A practical method for the disposal of
organotin residues from reaction mixtures. Journal of Organic Chemistry, v. 65, n. 26, p. 9220-
9222, 2000.
SHAN, D. D.; CHEN, L.; NJARDARSON, J. T.; GAUL, C.; MA, X. J.; DANISHEFSKY, S. J.;
HUANG, X. Y. Synthetic analogues of migrastatin that inhibit mammary tumor metastasis in mice.
Proceedings of the National Academy of Sciences of The United States of America, v. 102,
n. 10, p. 3772-3776, 2005.
SHEA, K. J.; ODELL, R.; SASAKI, D. Y. Free-radical induced macrocyclizations. Tetrahedron
Letters, v. 33, n. 33, p. 4699-4702, 1992.
SHINDO, K.; KAMISHOHARA, M.; ODAGAWA, A.; MATSUOKA, M.; KAWAI, H. Vicenistatin, a
novel 20-membered macrocyclic lactam antitumor antibiotic. Journal of Antibiotics, v. 46, n. 7, p.
1076-1081, 1993.
SILVEIRA, G. P.; NOME, F.; GESSER, J. C.; SÁ, M. M.; TERENZI, H. Recent achievements to
combat bacterial resistance. Quimica Nova, v. 29, n. 4, p. 844-855, 2006.
SILVERSTEIN, R. M.; BASSLER, G. C.; MORRILL, T. C. Identificação Espectrométrica de
Compostos Orgânicos. 5. ed. Rio de Janeiro: Guanabara Koogan, 1994, 387 p.
SNIECKUS, V.; CUEVAS, J. C.; SLOAN, C. P.; LIU, H.; CURRAN, D. P. Intramolecular alpha-
amidoyl to aryl 1,5-hydrogen atom transfer-reactions - heteroannulation and alpha-nitrogen
functionalization by radical translocation. Journal of the American Chemical Society, v. 112, n.
2, p. 896-898, 1990.
SOLOMONS, T. W. G; FRYHLE, C. B. Organic Chemistry. 7. ed. New York: John Wiley & Sons,
2000, 1258 p.

REFERÊNCIAS BIBLIOGRÁFICAS
151
STACHEL, S. J.; COBURN, C. A.; SANKARANARAYANAN, S.; PRICE, E. A.; PIETRAK, B. L.;
HUANG, Q.; LINEBERGER, J.; ESPESETH, A. S.; JIN, L. X.; ELLIS, J.; HOLLOWAY, M. K.;
MUNSHI, S.; ALLISON, T.; HAZUDA, D.; SIMON, A. J.; GRAHAM, S. L.; VACCA, J. P.
Macrocyclic inhibitors of beta-secretase: functional activity in an animal model. Journal of
Medicinal Chemistry, v. 49, n. 21, p. 6147-6150, 2006.
STEAD, P.; LATIF, S.; BLACKABY, A. P.; SIDEBOTTOM, P. J.; DEAKIN, A.; TAYLOR, N. L.;
LIFE, P.; SPAULL, J.; BURRELL, F.; JONES, R.; LEWIS, J.; DAVIDSON, I.; MANDER, T.
Discovery of novel ansamycins possessing potent inhibitory activity in a cell-based oncostatin M
signalling assay. Journal of Antibiotics, v. 53, n. 7, p. 657-663, 2000.
SZILAGYI, A.; FENYVESI, F.; MAJERCSIK, O.; PELYVAS, I. F.; BACSKAY, I.; FEHER, P.;
VARADI, J.; VECSERNYES, M.; HERCZEGH, P. Synthesis and cytotoxicity of leinamycin
antibiotic analogues. Journal of Medicinal Chemistry, v. 49, n. 18, p. 5626-5630, 2006.
TAKATSU, T.; OHTSUKI, M.; MURAMATSU, A.; ENOKITA, R.; KURAKATA, S. Reblastatin, a
novel benzenoid ansamycin-type cell cycle inhibitor. Journal of Antibiotics, v. 53, n. 11, p. 1310-
1312, 2000.
TROWITZSCH, W.; WRAY, V.; GERTH, K.; HOFLE, G. Structure of myxovirescin-a, a new
macrocyclic antibiotic from gliding bacteria. Journal of the Chemical Society, Chemical
Communications, n. 23, p. 1340-1342, 1982.
UMEZAWA, S.; NISHIMURA, Y.; HATA, Y.; TSUCHIYA, T.; YAGISAWA, M.; UMEZAWA, H.
Synthesis of 4’-deoxykanamycin and its resistance to kanamycin phosphotransferase II. The
Journal of Antibiotics, v. 27, p. 722-725, 1974.
VAULTIER, M., KNOUZI, N.; CARRIE, R. Reduction of azides into primary amines by a general-
method using the Staudinger reaction. Tetrahedron Letters, v. 24, n. 8, p. 763-764, 1983.
VOGEL, A. I. Química Orgânica: Análise Orgânica Qualitativa. 3. ed. Rio de Janeiro: Ao Livro
Técnico, 1981. 3 v.
WAGNER, B.; GONZALEZ, G. I.; DAU, M. E. T. H.; ZHU, J. Total synthesis and conformational
studies of hapalosin, N-desmethylhapalosin and 8-deoxyhapalosin. Bioorganic & Medicinal
Chemistry, v. 7, n. 5, p. 737-747, 1999.
WAGNER, R.; MOLLISON, K. W.; LIU, L. P.; HENRY, C. L.; ROSENBERG, T. A.; BAMAUNG, N.;
TU, N.; WIEDEMAN, P. E.; OR, Y.; LULY, J. R.; LANE, B. C.; TREVILLYAN, J.; CHEN, Y. W.;
FEY, T.; HSIEH, G.; MARSH, K.; NUSS, M.; JACOBSON, P. B.; WILCOX, D.; CARLSON, R. P.;
CARTER, G. W.; DJURIC, S. W. Rapamycin analogs with reduced systemic exposure. Bioorganic
& Medicinal Chemistry Letters, v. 15, n. 23, p. 5340-5343, 2005.

REFERÊNCIAS BIBLIOGRÁFICAS
152
WALLING, C. Some properties of radical reactions important in synthesis. Tetrahedron, v. 41, n.
19, p. 3887-3900, 1985.
WATANABE, Y.; ISHIKAWA, S.; TAKAO, G.; TORU, T. Radical cyclization on solid support:
Synthesis of gamma-butyrolactones. Tetrahedron Letters, v. 40, n. 17, p. 3411-3414, 1999.
WENDEBORN, S.; DE MESMAEKER, A.; BRILL, W. K. D.; BERTEINA, S. Synthesis of diverse
and complex molecules on the solid phase. Accounts of Chemical Research, v. 33, n. 4, p. 215-
224, 2000.
WIDDISON, W. C.; WILHELM, S. D.; CAVANAGH, E. E.; WHITEMAN, K. R.; LEECE, B. A.;
KOVTUN, Y.; GOLDMACHER, V. S.; XIE, H. S.; STEEVES, R. M.; LUTZ, R. J.; ZHAO, R.; WANG,
L. T.; BLATTLER, W. A.; CHARI, R. V. J. Semisynthetic maytansine analogues for the targeted
treatment of cancer. Journal of Medicinal Chemistry, v. 49, n. 14, p. 4392-4408, 2006.
WILLIAMS, D. R.; LI, J. Total synthesis of myxovirescin A1. Tetrahedron Letters, v. 35, n. 29, p.
5113-5116, 1994.
YOKOKAWA, F.; SAMESHIMA, H.; KATAGIRI, D.; AOYAMA, T.; SHIOIRI, T. Total syntheses of
lyngbyabellins A and B, potent cytotoxic lipopeptides from the marine cyanobacterium Lyngbya
majuscula. Tetrahedron, v. 58, n. 46, p. 9445-9458, 2002.
YOON, U. C.; JIN, Y. X.; OH, S. W.; CHO, D. W.; PARK, K. H.; MARIANO, P. S. Comparison of
photomacrocyclization reactions of trimethylsilyl- and tributylstannyl-terminated phthalimido- and
maleimido-polyethers. Journal of Photochemistry and Photobiology A: Chemistry, v. 150, p.
77-84, 2002.
YU, T.; FLOSS, H. G. Ansamitocins (Maytansinoids). In: CRAGG, G. M. L.; KINGSTON. D.;
NEWMAN, D. J. Anticancer Agents from Natural Products. Boca Raton: Taylor & Francis, 2005.
Cap. 17, p.321-333.
ZHANG, W. Recent advances in the synthesis of biologically interesting heterocycles by
intramolecular aryl radical reactions. Current Organic Chemistry, v. 8, n. 9, p. 757-780, 2004.
ZHU, X. W.; GANESAN, A. Intermolecular conjugate addition of alkyl radicals on solid phase.
Journal of Combinatorial Chemistry, v. 1, n. 2, p. 157-162, 1999.





























