Embed Size (px)
Citation preview

. , .UTO Df. e~.,,,!~/ Ullnra!dade de s·o Paulo
.,.,?O.Z&7
UNIVERSIDADE DE SÃO PAULO
INSTITUTO DE QUÍMICA
SÍNTESE ENANTIOSSELETIVA DE
FENILSELENOLACTONAS E FENIL TELUROLACTONAS
ALESSANDRO LEAL NOGUEIRA
DISSERTAÇÃO DE MESTRADO
ORIENTADOR:
PROF. DR. JOÃO VALDIR COMASSETO
J, '
SÃO PAULO
07/06/2002

·/.,
DEDALUS - Acervo - CQ
1111111 ~~ 11111 ~111111111111~ lllll lllll llll l lll Ili 30100005150
Ficha Catalográfica Elaborada pela Divisão de Biblioteca e
Documentação do Conjunto das Químicas da USP.
Nogueira, Alessandro Leal N778s Síntese enantiosseletiva de fenilselenolactonas e
feniltelurolactonas / Alessandro Leal Nogueira. -São Paulo, 2002.
76p.
Dissertação (mestrado) - Instituto de Química da Universidade de São Paulo. Departamento de Químic& Fundamental.
Orientador: Comasseto, João Valdir
l. Síntese : Química orgânica 2. Selênio : Composto : Química orgânica 3. Telúrio: Composto : Química organ1ca 4. Enzima: Química orgânica I. T . II. Comasseto, João Valdir, orientador.
547.2 CDD

"Síntese En1ntiosseletiv1 de Fenilseleno/1ctonu e Fenilteluro/1ctonu"
ALESSANDRO LEAL NOGUEIRA
Dissertação de }'/(estrado submetida ao 9nstituto de Química da Universidade de São Paulo como parte dos requisitos necessários à obtenção do grau de }'l(estre em Oulmica - Area: Química Orgânica.
Aprovado por:
Profa. Ora. LILIANA MARZORATI IQ-USP
(Presidente)
Prof. Dr. CARLOS ROQUE DUARTE CORREIA IQ-UNICAMP
Prof. Dr. HÉLIO ALEXANDRE STEFANI FCF-USP
SÃO PAULO 07 DE JUNHO 2002.

AGRADECIMENTOS
Agradeço a minha família, especialmente a meus pais, por todo
amor, apoio, amizade, respeito e confiança demonstrado ao longo de
toda a minha vida. Obrigado, vocês são maravilhosos.
Agradeço aos colegas de laboratório Ricardo, Maurício, Rodrigo,
Fabiano, Lauri, Priscila, Natália, Eduardo, Carlos Eduardo, Mirian e
Denis pelos momentos divertidos vividos durante o curso de mestrado.
Agradeço ao Professor Comasseto pela orientação.
Agradeço a todos os professores e funcionários do Instituto de
Química, especialmente a Professora Helena Ferraz e os funcionários
Nanci e Alexandre.
Agradeço o meu amigo Tan pelo companheirismo e amizade
desde a nossa graduação.
E finalmente, agradeço a FAPESP pelo apoio financeiro.

índice
ÍNDICE
ÍNDICE
LISTA DE FIGURAS iii
LISTA DE ESQUEMAS iv
LISTA DE TABELAS vi
ABREVIAÇÕES vi
RESUMO vii
ABSTRACT viii
1 INTRODUÇÃO 1
1.1 Biotransformação 1
1. 1. 1 Introdução 1
1.1.2 Estrutura do substrato 5
1.1.3 Reações de hidrólise · 6
1.1.4 Reações de esterificação e transesterificação 7
1.2 Organosselenetos e Organoteluretos 10
1.2.1 Organosselenetos Quirais 1 O
1.2.2 Organoteluretos. 1"2
1.2.3 Síntese de selenolactonas e telurolactonas racêmicas 12
1.2.4 Reações radicalares dos organosselenetos e organotelure-
tos 13

índice ii
2 RESULTADOS E DISCUSSÃO 17
2.1 Introdução 17
2.2 Etapa 1: Preparação do 4-oxipentenoato de propila 19
2.2.1 Reação de alquilação e descarboxilação do dietilmalonato 19
2.2.2 Reção de esterificação do ácido 4-pentenóico 21
2.2.3 Reação de epoxidação do 4-pentenoato de propi/a 23
2.3 Etapa 2: Preparação dos hidroxiésteres racêmicos 25
2.3.1 Introdução dos heteroátomos Se e Te 25
2.4 Etapa 3: Resolução cinética biocatalisada 31
2.5 Determinação do ee 40
3 PARTE EXPERIMENTAL 47
3.1 Preparação de 2-alilmalonato de dietila 47
3.2 Preparação de ácido 4-pentenóico 48
3.3 Preparação de 4-pentenoato de propila 48
3.4 Preparação de 4-oxipentanoato de propila 49
3.5 Preparação de disseleneto de difenila 50
3.6 Preparação de 4-hidroxi-5-fenilselenilpentanoato de propila 51
3.7 Preparação da (R) e (S)-fenilselenolactona 52
3.8 Preparação de ditelureto de difenila 53
3.9 Preparação de 4-hidroxi-5-feniltelurilpentanoato de propila 54
3.1 O Preparação da (R) e (S)-feniltelurolactona 55
3.11 Preparação da valerolactona para medição dos ee 56
4 REFERÊNCIA BIBLIOGRAFICA 59
5 APÊNDICE 61
5.1 Espectros de RMN de hidrogênio 61
5.2 Espectros de RMN de carbono 67
5.3 Espectros no Infra-vermelho 73

índice iii
5.4 Espectro de Massa 75
LISTA DE FIGURAS
Figura 1 Diagrama de energia de uma reação enantiosseletiva catalisada 2
Figura 2 Características dos substratos usados com as lipases 5
Figura 3 Espectro de RMN de H4 do composto 2 na região de 5, 78 ppm 20
Figura 4 Espectro de RMN de H5a e Hsb do composto 2 na região de 5,06 a 20
5, 12 ppm
Figura 5 Espectro de RMN de H2 do composto 3 na região entre 2,45 a 21
2,48 ppm
Figura 6 Espectro de RMN 1 H de H2 e H3 do composto 4 na região de 2,36 22
a 2,43 ppm
Figura 7 Espectro de RMN 1 H de H4 do composto 4 na região de 5, 79 a 23
5,87 ppm
Figura 8 Espectro de RMN 1H de H5a e Hsb do composto 5 24
Figura 9 Espectros de RMN 13C para C4 e C5 para os compostos 4 e 5 nas 25
região de 115, 44 a 136,79 e 46, 81 a 51, 07 ppm respectivamente
Figura 1 O Cromatograma de HPLC da mistura reacional 27
Figura 11 Espectro de RMN 1 H de H3a e H3b do composto 6a na região de 28
1, 75 a 1,94 ppm
Figura 12 Espectro de RMN 1H de H2 do composto 6a na região de 2,39 a 28
2,50 ppm
Figura 13 Espectro de RMN 1 H de H5a e Hsb do composto 6a na região de 29
2,90 a 3, 10 ppm
Figura 14 Cromatograma de HPLC do composto 6b 31
Figura 15 Cromatograma de HPLC do precursor 6a 33

índice iv
Figura 16 Cromatograma de HPLC da reação de ciclização do substrato 33
(6a) com PPL em éter anidro após 22 h
Figura 17 Cromatograma de HPLC da reação de ciclização do substrato 34
(6a) com PPL em éter anidro após 28 h
Figura 18 Cromatogramade HPLC da reação de cic/ização do substrato 34
(6a) com PPL em éter anidro após 47 h
Figura 19 Cromatograma de HPLC da reação de ciclização do substrato 34
(6a) com PPL em éter anidro após 69 h
Figura 20 Cromatograma de HPLC da reação de ciclização do substrato 35
(6a) com PPL em éter anidro após 75 h
Figura 21 Cromatograma de HPLC do precursor 6b 35
Figura 22 Cromatograma de HPLC da reação de ciclização do substrato 36
(6b) com PPL em hexano anidro após 21 h
Figura 23 Cromatograma de HPLC da reação de ciclização do substrato 36
(6b) com PPL em hexano anidro após 23 h
Figura 24 Espectro de RMN 1 H de H2, H3a e H3b do composto 7a na região 39
de 1,92 a 2,61 ppm
Figura 25 Espectro de RMN 1 H de H5a e H5b do composto la na região de 39
2,99 a 3,29 ppm
Figura 26 Espectro de RMN de 125Te da feniltelurolactona 43
LISTA DE ESQUEMAS
Esquema 1 Síntese enantiosseletiva de fenilselenolactonas e v
fenilteluro/ac-tonas.
Scheme 1 Enantioselective synthesis of phenylselenolactones and vi
phenyltelu-rolactones.
Esquema 2 Função biológica das lipases 2
Esquema 3 Mecanismo elucidado para serino hidra/ases 3

índice v
Esquema 4 Formação de intermediários do grupo acila com a enzima e 4
suas reações
Esquema 5 Resolução de álcool secundário 6
Esquema 6 Resolução de ácido carboxílico 6
Esquema 7 Resolução de /actonas 7
Esquema 8 Resolução de ácido carboxílico 8
Esquema 9 Resolução de álcoois secundários 8
Esquema 10 Resolução cinética de um álcool racêmico 8
Esquema 11 Resolução cinética de um diácido enantiotópico 9
Esquema 12 Resolução cinética de hidroxiéster racêmico 9
Esquema 13 Substratos com compostos organometálicos de Sn 1 O
Esquema 14 Reação de ciclofuncionalização estereosseletiva 11
Esquema 15 Abertura estereosseletiva de epóxidos 12
Esquema 16 Síntese diastereoseletiva de selenolactonas 12
Esquema 17 Síntese enantiosse/etiva de organosselenetos quirais imobili- 12
zados
Esquema 18 Exemplos de reações nucleofílica de organoteluretos 13
Esquema 19 Síntese de fenilselenolactona e feniltelurolactona 13
Esquema 20 Mecanismo da reação radica/ar 14
Esquema 21 Reações de redução eliminativa de organosselenetos 14
e organoteluretos
Esquema 22 Exemplos de reações radica/ares com organosselenetos 15
Esquema 23 Exemplo de reação radica/ar com organotelureto 15
Esquema 24 Proposta para a síntese enantiosseletiva da Jasmolactona 16
Esquema 25 Análise retrossintética da selenolactonas e telurolactonas 17
Esquema 26 Esquema da rota sintética da (R) e (S)-fenilselenolactona e 18
(R) e (S)-feniltelurolactona
Esquema 27 Mecanismo de formação da se/enolactona la durante a 26

índice vi
abertura do epóxido 5
Esquema 28 Enantiociclização dos intermediários 6a e 6b 32
Esquema 29 Enantiociclização para obtenção da (S)-hidroxiésteres 38
Esquema 30 Eliminação redutiva para obtenção da y-valero lactona 41
LISTA DE TABELAS
Tabela 1 Porcentagens entre o produto formado, substrato e impureza 35
Tabela 2 Porcentagens entre o produto formado, substrato e impureza 36
Tabela 3 Tempo de reação para consumo de 50 % de 6a catalisada por 37
PPL para dois solventes anidros diferentes
Tabela 4 Tempo de reação para consumo de 50 % de 6b catalisada por 37
PPL para três solventes anidros diferentes
Tabela 5 Relação dos excessos enatioméricos das telurolactonas 42
resolvidas
Tabela 6 Relação dos excessos enatioméricos das selenolactonas 42
resolvidas
Tabela 7 Cromatogramas da valemlactona obtidos por cromatografia 43
gasosa com coluna quiral a partir das (R) e (S)-telurolactonas
Tabela 8 Cromatogramas da valerolactona obtidos por cromatografia 45
gasosa com coluna quiral a partir das (R) e(S)-selenolactonas
PPL
CAL
CAL-B
PSL
CRL
MML
ABREVIAÇÕES USADAS
: Lipase de pancrêas de porco.
: Lipase de Candida antartica.
: Lipase de Candida antartica tipo B.
: Lipase de Pseudomonas sp.
: Lipases de Candida rugosa.
: Lipases de Mucor miehei.
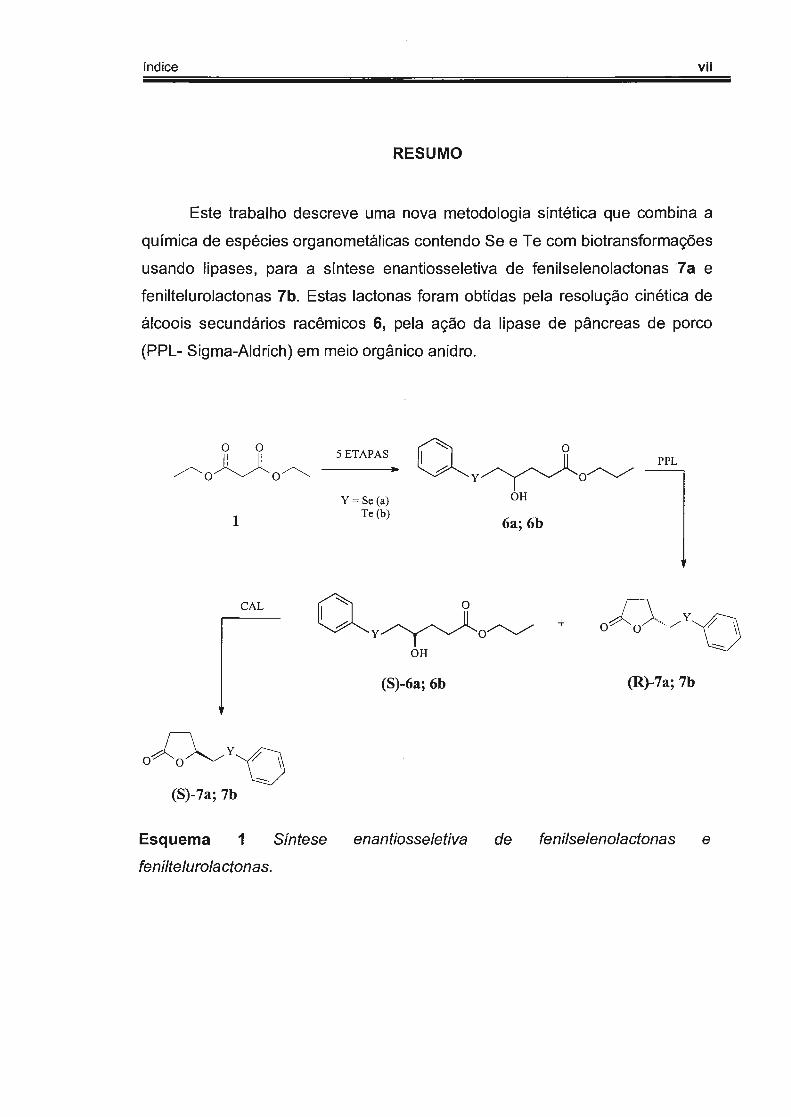
índice vii
RESUMO
Este trabalho descreve uma nova metodologia sintética que combina a
química de espécies organometálicas contendo Se e Te com biotransformações
usando lipases, para a síntese enantiosseletiva de fenilselenolactonas 17a e
feniltelurolactonas 7b. Estas lactonas foram obtidas pela resolução cinética de
álcoois secundários racêmicos 6, pela ação da lipase de pâncreas de porco
(PPL- Sigma-Aldrich) em meio orgânico anidro.
o o
/'-..o~o~
1
CAL
o~YYJ (S)-7a; 7b
5 ETAPAS ~ /'-- /'-- 1 PPL ~v,,, 1 ____,,, 'o/'-....../
Y = Se (a) Te (b)
OH
6a;6b
~ o
~Y~o/'-....../ OH
(S)-6a; 6b
+
(R)-7a; 7b
Esquema 1 Síntese enantiosse/etiva de fenilselenolactonas e
feniltelurolactonas.
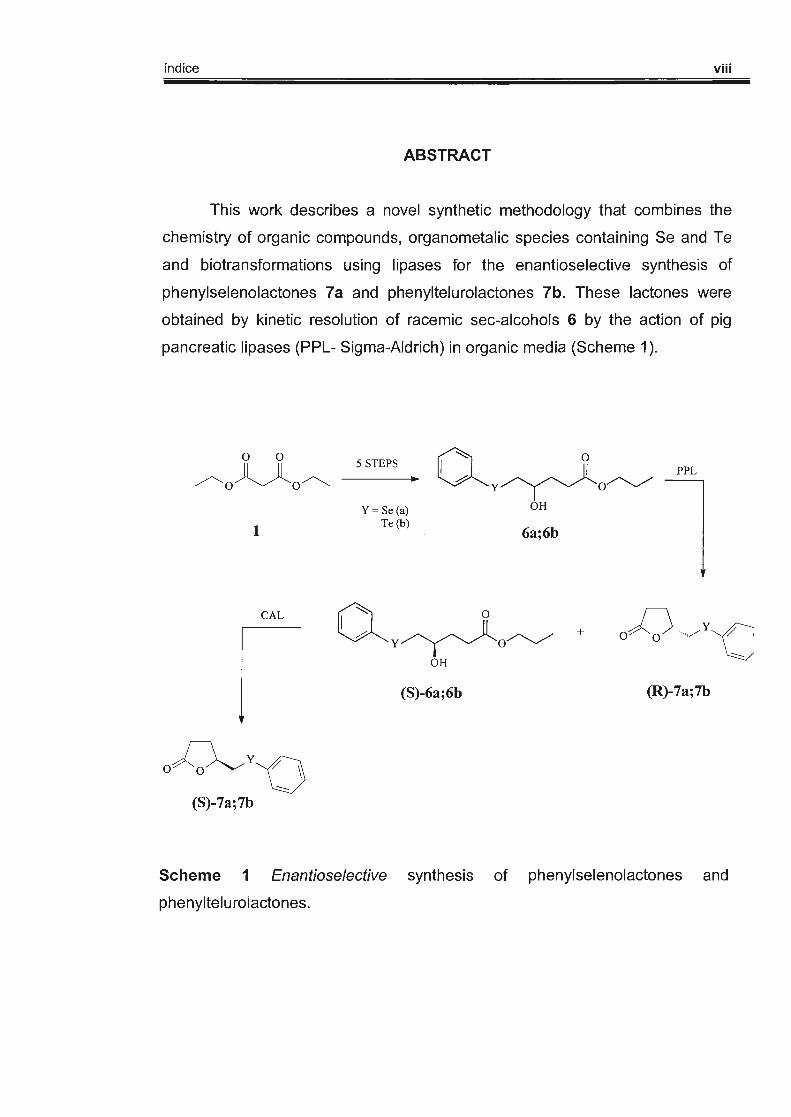
índice viii
ABSTRACT
This work describes a novel synthetic methodology that combines the
chemistry of organic compounds, organometalic species containing Se and Te
and biotransformations using lipases for the enantioselective synthesis of
phenylselenolactones 7a and phenyltelurolactones 7b. These lactones were
obtained by kinetic resolution of racemic sec-alcohols 6 by the action of pig
pancreatic lipases (PPL- Sigma-Aldrich) in organic media (Scheme 1 ).
o o
/"--- o~ o~
1
CAL
o~ yu (S)-7a;7b
5 STEPS
Y = Se (a) Te (b)
6a;6b
~ o
~Y~O~ OH
(S)-6a;6b
+
(R)-7a;7b
Scheme 1 Enantioselective synthesis of phenylselenolactones and
phenyltelurolactones.

Introdução 1 /1
"' INTRODUÇAO

Introdução - Biotransformação 1
1.1 Biotransformação
1.1.1 Introdução
A crescente demanda de moléculas com pureza enantiomérica na
indústria farmacêutica , agroquímica e em laboratórios acadêmicos vem
estimulando o desenvolvimento de novas metodologias sintéticas 1. O uso de
enzima em meio orgânico é uma poderosa ferramenta para a obtenção
destas moléculas devido ao controle régio e estereoquímico proporcionado
pela ação destas proteínas, o que torna a biotransformação uma
metodologia alternativa importante em síntese orgânica enantiosseletiva2.
As enzimas, como os demais catalisadores, atuam reduzindo a
energia do complexo ativado da reação e, conseqüentemente, tornam essas
reações mais velozes. Esse efeito ocorre devido à formação de complexos
intermediários entre o substrato e o biocatalisador com energias de ativação
(L).Gt ) inferiores à dos complexos ativados formados sem a presença do
biocatalisador.
Devido à estrutura tridimensional da enzima, o sítio ativo tem a
capacidade de reconhecer a quiralidade (enatiomérica, diastereotópica e
entre faces) presente no substrato e forma também complexos ativados com
diferentes energias de ativação entre as configurações possíveis do
substrato, levando à formação em maior quantidade do produto que
apresenta menor energia de ativação. Quanto maior essa diferença de
energia (L).L).Gr) entre os complexos formados pelos substratos e o
biocatalisador, maior a seletividade obtida na biotransformação. A Figura 1
ilustra esse comportamento, mostrando a formação de complexos com
diferentes energias de ativação para cada configuração de um substrato
racêmico3.
1. Jones, J. B; Tetrahedron , 42, 3351 , 1986. 2. Loughen, W. A.; Biar. Tech, 74, 49, 2000. 3. Faber, K.; "Biotransformations in Organic Chemistry"; 3ª Ed .; Berlim, Springer Press,
1997

Introdução - Biotransformação 2
As enzimas catalisam quase todos os tipos de reações orgânicas
conhecidas, possibilitando a substituição de reações orgânicas clássicas por
biotransformações equivalentes. As enzimas, de acordo com a "lnternational
Union of Biochemistry", podem ser divididas em seis grupos denominados:
oxidoredutases, transferases, liases, hidrolases, isomerases e ligases 1-3
_
[EA) ···--• E + P .6.G [EA(
[EB] - • E+ O
(\_ __ -------- ].6.AG70
.1 \ [EBt
I \_{\ / E+A orB ,
- ___ _____ __ _____ ____ ___ '\_ E+P E +Q
Reaction coord inate
Figura 1 Diagrama de energia de uma reação enantiosseletiva catalisada.
Dentro do grupo das hidrolases estão as lipases, cuja função biológica
é a hidrólise de triglicerídeos em ácidos graxos e glicero14-5 (Esquema 2). As
lipases apresentam uma particularidade que as tornam diferentes das
demais hidrolases, elas não seguem a equação de Michaelis-Menten, sendo
que somente hidrolizam seus substratos acima da concentração micelar
crítica.
{
OCOR1
R20 CO
OCOR3
lipase --_,.,..,. Ácidos graxos + diglicerideos + monoglicerideos + glicerol
Esquema 2 Função biológica das lipases.
4. Wong, C; Whitesides, G.M .;"Enzymes in Synthetic Organic Chemistry" 12, London, Pergamon Press, 1995
5. Roche Molecular Biochemicals, Industrial Biochemicals; "Biocatalysts for lndustry; 2000

Introdução - Biotransformação 3
Essas proteínas são amplamente estudadas como catalisadores em
reações orgânica (30 % das biotransformações relatadas na literatura)
devido a seu grande potencial para preparação de intermediários
assimétricos. As lipases são em sua maioria serino hidrolases4 (o sítio ativo
da enzima apresenta o resíduo de aminoácido serina que realiza a operação
química. (Esquema 3). O sítio ativo é formado por três resíduos de
aminoácidos, denominado trio catalítico.
No mecanismo elucidado para as serino hidrolases, o grupo hidroxila
do aminoácido serina ataca o grupo carbonila do éster (substrato) formando
o intermediário enzima - substrato e liberando o álcool presente na estrutura
do éster. Em seguida, água (nucleófilo) presente no meio, ataca o
intermediário formando uma nova substância e regenerando o sítio ativo da
enzima para uma nova etapa de catálise.
Etapa 2
Asp His Ser intermediário .l. T T Enz-Substrato
O 0-H ;=\ ~;= >J ~ _N N-H ~ O ""---/ Y, ~ ( Ri
N e u.
Esquema 3 Mecanismo elucidado para serino hidra/ases.
-------•
As reações naturais das lipases são realizadas em água (nucleófilo)
levando os ésteres a ácidos carboxílicos (Esquema 2), porém a
biotransformação de moléculas orgânicas é normalmente realizada em meio
orgânico e os nucleófilos são adicionados de acordo com o interesse
sintético.

Introdução - Biotransformação 4
De uma forma geral, as lipases em meio orgânico catalisam a
transferência de grupos acila para aceptores (nucleófilos) diferentes da
água. Dependendo do tipo de doador do grupo acila e do aceptor, podem ser
realizadas reações de hidrólise, esterificação, transesterificação, amidação,
síntese de peptídeos e preparação de álcoois e ácidos carboxílicos
opticamente ativos6 (Esquema 4 ).
doadores do grupo acila complexo ativado nucleófilos enzima-substrato
HO R'
y o ~ /H20
yºyR' RO R'
'Ir---.. R"OH H, o + ~ CH3 Ser
1 RS R' o NH3
y ~ OAR' o ~NH2R" o
Ço R' O R'/ ~ H202 YY o o
R
Esquema 4 Formação de intermediários do grupo acila e enzima e suas
reações.
Essas biotransformações se processam com substratos de tamanhos
e complexidades enantioméricas muito diferentes dos substratos naturais,
caracterizando a habilidade das lipases em assumir uma grande variedades
de conformações para acomodar · esses diversificados substratos. Diante
desta observação experimental, a proposta mecanística mais aceita é a
"forma induzida", sendo que essa proposta sugere que durante a formação
do complexo ativado enzima-substrato, a enzima altera a sua conformação
sob a influência do substrato.3
6. Chen; C; Sih C.J. ; Angew. Chem . lnt. Ed. Eng/.; 28, 695, 1989.

Introdução - Biotransformação 5
A aplicação experimental das lipases também é bastante
diversificada. As lipases são usadas sólidas ou solubilizadas, sendo
solubilizadas através de modificações covalentes ou por formação de
complexos com surfactantes, polímeros e microemulsões. As sólidas podem
ser liofilizadas, extratos crus, cristalizadas ou imobilizadas em suportes
inorgânicos, poliméricos e em polissacarídeos7.
As reações com lipases também podem ser realizadas em três
sistemas de solventes distintos: sistema água e co-solvente orgânico
miscível, sistema orgânico anidro e sistema água e solvente orgânico
imiscível (sistema bifásico). Os parâmetros pH, temperatura , constante
dielétrica do solvente, estrutura do solvente e umidade também influenciam
na velocidade e estereosseletividade das reações biocatalisadas6.
1.1.2 Estrutura do substrato
A escolha do substrato é o fator mais importante para a obtenção de
moléculas oticamente ativas. As lipases são amplamente usadas na
resolução cinética de misturas racêmicas, sendo que o centro estereogênico
pode estar presente tanto na estrutura que corresponde ao ácido carboxílico,
quanto na estrutura que corresponde ao álcool. (Figura 2).
Figura 2 Características dos substratos usados com as lipases.
Para que a resolução cinética biocatalisada seja orientada pelo álcool,
necessariamente esse álcool deve ser secundário, pois álcoois terciários
normalmente não são aceitos pela enzima. Pelo mesmo motivo, a estrutura
do ácido carboxílico também deve apresentar um átomo de hidrogênio. Esse
fato é interessante quando se deseja proteger e evitar a hidrólise enzimática.
7. Koskinem, A M. P.; Klibanov, A M; "Enzymatic Reactions in Organic Media'; 1ª Ed.; New
York; Chapman & Hall Press, 1996

Introdução - Biotransformação 6
1.1.3 Reações de Hidrólise
As reações de hidrólise biocatalisadas ocorrem quando a água
(nucleófilo) ataca o intermediário formado pelo substrato e o sítio ativo da
enzima. Essas reações são realizadas em meio aquoso ou em sistema água
e solvente orgânico miscível. A hidrólise estereosseletiva apresenta um
grande potencial em síntese orgânica, produzindo moléculas funcionalizadas
como ácido carboxílicos, álcoois, e ésteres com elevadas purezas óticas. O
Esquema 5, ilustra a resolução cinética de um éster racêmico, cujo centro
estereogênico está presente na estrutura do álcool8.
OH OAc
CAL-B ~~ ~~
Esquema 5 Resolução de álcool secundário.
As reações de hidrólise biocatalisada de ésteres, cujo centro
estereogênico está na estrutura do ácido carboxílico , também são bastante
eficientes na produção de ácidos carboxílicos (Esquema 6)9.
Uma das vantagens das lipases, além da estereosseletividade, é a
quimiosseletividade. O anel epóxido não sofre reação de abertura na
hidrólise biocatalisada, fato que seria observado em uma hidrólise química
ácida ou básica.
~ COOCH,
o
rac. trans
CAL-B
Esquema 6 Resolução de ácido carboxílico.
8. Lundell , K.; Raijola,T.;Kanerva, L. ; Enzyme Mícrob. Technol, 22, 86, 1998. 9. Ohtani , K.;Nakatsukasa, H; Kamezawa,M; Tachibana,H; Naoshima,Y. ; J. Mo/. Catalysís
B: Enzymatíc; 4. 53, 1998.

Introdução - Biotransformação 7
Outro exemplo interessante é a função lactona. Essa função
apresenta em sua estrutura as funções álcool e ácido carboxílico. O
Esquema 7 mostra a resolução cinética de três lactonas através da hidrólise
biocatalisada por lipase 10-11
.
~ O O C4H9
D O O CsH11
o
PSL
sol.tampão pH=8 T (ºC) = 5
PSL
sol.tampão pH=8 T (°C) = 5
?H R~COOH
[%ee] = 84
[%ee] = 99
o
Ó-CAL
IPE/ H20 T (ºC) = 36
HO~COOHó .... , [%ee] = 94
Esquema 7 Resolução de lactonas.
1.1.4 Reações de Esterificação e Transesterificação
As reações de transesterificação e esterificação ocorrem quando o
nucléofilo que ataca o intermediário enzima-substrato é um álcool. As
esterificações biocatalisadas transformam ácidos carboxílicos em ésteres
com elevada pureza ótica 12 (Esquema 8).Também podem ser usadas na
resolução de misturas racêmicas de álcoois secundários (Esquema 9)13.
10. Vanderdeen, H; et ai; J. Amer. Chem. Soe, 118, 3801, 1996. 11. Enzelberger, M M; Bornscheuer, U T; Gatfield, I; Schmid, R D; J. Bíotechnology; 56,
129, 1997. 12. Shioji, K.; Matsuo,A; Okuma, K.; Nakamura, K; Ohno, A.; Tetrahedron Lett, 41 , 8799,
2000. 13. Sharma, A. ; Chattopadhyay, S.; J. Mo/. Catalysís B: Enzymatíc; 10. 531, 2000.

Introdução - Biotransformação 8
OH O ~ li CRL
~OH _H_O_C_H-2(_C_H-2)-4C_H.,3
OH O OH O
~O(CH2)sCH3 + ~OH tolueno [%ee] = 96 [%ee] = 75
Esquema 8 Resolução de ácido carboxílico.
Q COOH + HOB _MM_L_
1/ hexano °'rº"8 + HOB Esquema 9 Resolução de álcoois secundários.
Umas das reações clássicas das lipases é a resolução de álcoois
secundários com acetato de vinila. O acetato de vinila é interessante nestas
reações de transesterificação porque o álcool vinílico liberado pelo complexo
enz-substrato se transforma rapidamente em acetaldeído, não permitindo
que reações de equilíbrio acorram (reação biocatalisada irreversível).
(Esquema 1 O) 14-15
_ O Esquema 11 ilustra a transformação estereosseletiva
de um composto enantiotópico2 (composto mesa), caracterizando a
capacidade das lipases em reconhecer a quiralidade nestes tipos de
substratos, embora sejam mais comumente aplicadas a resoluções de
álcoois secundários racêmicos.
OH OAc OH
d' PFL V + d' AcO~
t-BuOMe 99%ee 93% ee
HO\):~O) 1 o
ó o __) vº
CAL
AcO~
l'o~ Í l'o~ ACO O ) HO~-'-:: O )
o + 1 o o __) óo __) vº v º [%ee] = 99 [%ee] = 96
Esquema 10 Resolução cinética de um álcool racêmico
14. Roberts, S M; Turner, N J.; J. Bíotechnol; 22; 227; 1992 15. Kijima, T.; Moriya, T.; Kondoh, E.; lzunii, T; Tetrahedron Lett, 41, 2125, 2000.

Introdução - Biotransformação
PFL
Acü~
EtO\__fOH O
EtO~OAc + ~H
(R)71%ee
Esquema 11 Resolução cinética de um diácido enantiotópico.
9
Outra característica importante das lipases, como mencionado
anteriormente, é a capacidade da enzima em aceitar substratos de
tamanhos e complexidades enantioméricas bastante diferentes. As duas
moléculas do Esquema 1 O sofreram a mesma reação, mesmo com
estruturas químicas tão diferentes.
As reações de equilíbrio não são desejáveis em sínteses
assimétricas, pois reduzem o rendimento e o excesso enantioméricos dos
produtos formados. Para se minimizar problemas com a reversibilidade da
reação biocatalisada, os nucleófilos usados nas reações de
transesterificação são usados em excesso, normalmente como solvente da
reação. Entretanto, em alguns casos não é possível utilizar o nucleófilo em
excesso. A síntese enantiosseletiva de lactona por transesterificação é um
bom exemplo desse comportamento. O substrato hidroxiéster precursor da
lactona apresenta em sua estrutura as funções éster e álcool e à medida que
a reação de ciclização da lactona avança, a concentração do álcool liberado
no éster aumenta e, conseqüentemente, reage com a lactona formada mais
rapidamente, regenerando o substrato inicial e racemizando o produto 16
(Esquema 12).
o
YOEt
YºH CAL
t-BuOMe, 20 ºC
25 h
vZ ylOB lt: + YºH [¾ee] = 11 [¾ee] = 13
Esquema 12 Resolução cinética de hidroxiéster racêmico.
16. Adam,W; Groer, P.; Saha-Moller, G. R. ;Sharma, Tetrahedron Assymm, 11, 2239, 2000.

Introdução - Biotransformação 10
As lipases também catalisam reações com substratos que contém
metais em sua estrutura. O Esquema 13 ilustra substratos que apresentam o
heteroátomo Sn em sua estrutura.
OCOR2
Bu~SnBu3
OCOR2
Ph~S11Bu3
PCL
PCL
OH
Bu~SnBu3
[%ee] = 98
OH
Ph~SnBu3
[%ee] = 98
Esquema 13 Substratos com compostos organometálicos de Sn.
A resolução cinética pela ação de lipases possibilita a obtenção de
diversos intermediários sintéticos enantiomericamente enriquecidos de
acordo com a estratégia sintética. As lipases tem a capacidade de aceitar
uma grande variedades de substratos de tamanhos e complexidades
enantioméricas diferentes, como demostrado nos exemplos anteriores.
Essas características estimulam um estudo mais aprofundado com
substratos bastante diferentes dos substratos naturais e permitem uma
maior interação com metodologias sintéticas que utilizem substratos com os
mais variados heteroátomos, como Se, Te, S, Sn, Si, entre outros.
17. ltoh, T; Takagi, Y; Tsukube, H.; J. Mo/. Catalysís B: Enzymatíc; 3. 259, 1997.

Introdução - Organosselenetos e Organoteluretos 11
1.20rganosselenetos e Organoteluretos
1.2.1 Organosselenetos quirais.
Os organosselenetos quirais são em sua maioria disselenetos, cuja
parte orgânica da molécula apresenta o centro quiral (ferrocenos,
bisnaftalenos, canfora, entre outras moléculas orgânicas). Esses
disselenetos podem ser preparados por diversas rotas, entretanto, as rotas
mais importantes envolvem reações de disselenetos metálicos com haletos
ou a preparação de selenóis por metalação dos precursores e posterior
oxidação levando ao disseleneto correspondente 18. Esses organosselenetos
quirais possuem aplicações interessantes, podendo ser utilizados para
induzir a quiralidade atuando como reagentes eletrofílicos, como por
exemplo em reações de ciclofuncionalização estereosseletivas de álcoois e
ácidos carboxílicos insaturados (Figura 14) 18 e também como reagentes
nucleófilicos, promovendo a abertura estereosseletiva de anéis epóxidos
(Esquema 15 e 16)19-20
.
Outra forma interessante é a síntese de moléculas orgânicas quirais
por indução com organosselenetos quirais imobilizados em polímeros 18.
(Esquema 17).
~OH
o
95 %de
Esquema 14 Reação de ciclofuncionalização estereosseletiva.
18. Wirth, T.; Tetrahedron, 1, 55, 1999. 19. Back, G. T.; Dyck, P. B.; Chem. Commun., 2567, 1996. 20. Wirth , T., Liebiigs Ann, 2189, 1997.
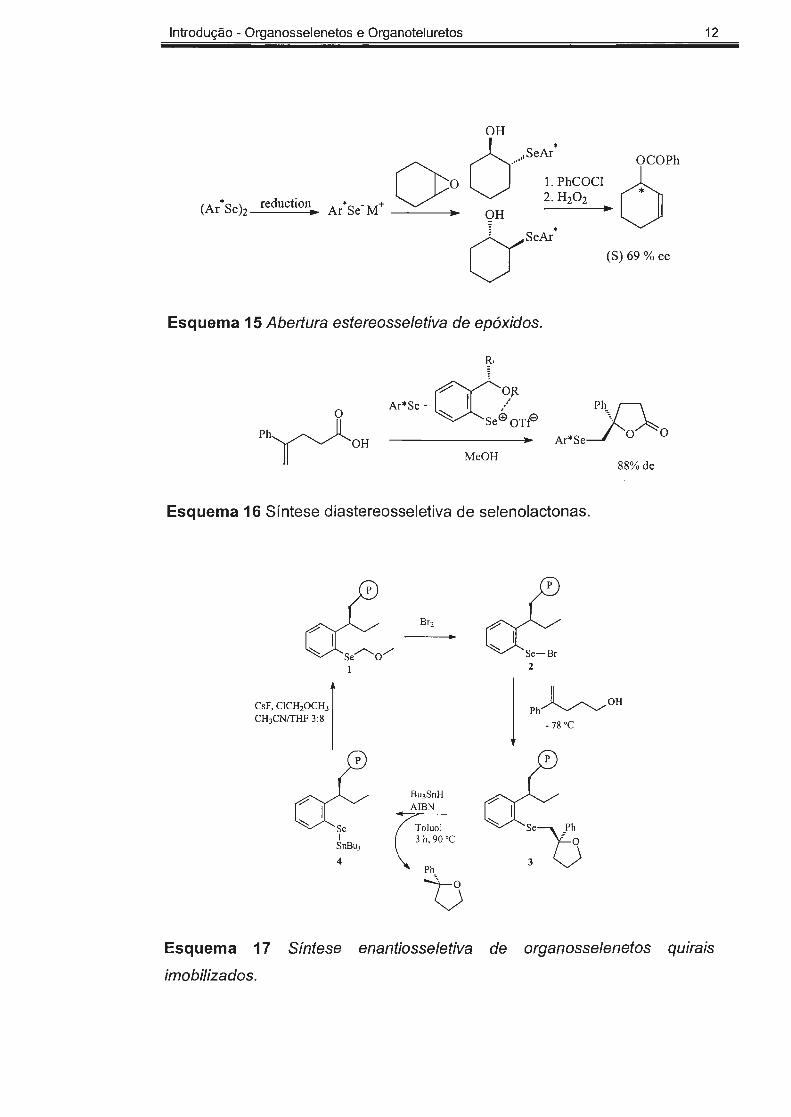
Introdução - Organosselenetos e Organoteluretos 12
(Ar' Se h reduction
Esquema 15 Abertura estereosseletiva de epóxidos.
R,
Ph......._ /"-..... .Jl 1 ~ OH
Ac' So" ex:?" Ph'l\_
______ s_e_<;B_o_T_f2l Ar*Se~ O~ O
MeOH 88% de
Esquema 16 Síntese diastereosseletiva de selenolactonas.
~/
Br2 cl. 2
CsF, CJCH,OCfl , l j ~OH Ph
CH3CN/THF 3: 8 - 78 ºC
ct Bu3SnH AIBN
Toluol 1 3 h, 90 ºC SnBu3
4 Ph
1:)
Esquema 17 Síntese enantiosse/etiva de organosselenetos quirais
imobilizados.

Introdução - Organosselenetos e Organoteluretos 13
1.2.2 Organoteluretos.
Não existem relatos da utilização das técnicas quirais similares as
aplicadas aos organosselenetos em organoteluretos, sendo que os
compostos quirais conhecidos são inorgânicos. Entretanto, os
organoteluretos apresentam reatividade química muito similar aos
organosselenetos, podendo ser preparados a partir do Te metálico21 e
posteriormente realizar reação nucleofílicas e eletrofílicas22-24 (Esquema 18)
O r;hT~ Nf7 ? H ~ _l!'. ___ :J• PhTe~ R
R EtOH
Esquema 18 Exemplos de reações nucleofílica de organoteluretos.
1.2.3 Síntese de selenolactonas e telurolactonas racêmicas
A primeira selenolactona (1) e telurolactona (li) foram sintetizada por
Petragnani23-24 através das reações de adição eletrofílica e posterior
ciclofuncionalização do ácido 4-pentenóico pelos compostos brometo de
arilselênio e tricloreto de ariltelúrio, respectivamente. (Esquema 19).
Entretanto, essa técnica produziu lactonas racêmicas.
ArSeBr ~ SePh o
ct o o I
1. ArTeCl3 ~ TePh
2. [H] o o II
Esquema 19 Síntese de fenilse/eno/actona e fenilteluro/actona .
21. Moura Campos, M; Petragnani , N; Tetrahedron Lett., 11 ; 1959 22. Comasseto J.V, Petragnani , N., Synth. Commun., 13, 889, 1983 23. Haller, S. W.; lrgolic.; J . Organomatel Chem. ; 38, 97, 1972. 24. Petragnani , "Tellurium in Organic Synthesis", Academic Press, London, 1994

Introdução - Organosselenetos e Organoteluretos 14
1.2.4 Reações radicalares dos organosselenetos e organoteluretos.
Os organosselenetos e organoteluretos são compostos muito
versáteis, podendo atuar tanto como nucleófilo quanto como eletrófilos,
como mencionado anteriormente. Podem ser convertidos em alquenos por
oxidação e em hidrocarbonetos por eliminação redutiva e também podem
formar ligações carbono-carbono por mecanismo radicalar24-25
. Entretanto,
essa capacidade dos organosselenetos e organoteluretos em formar
ligações carbono-carbono ainda não foram exploradas em sínteses
assimétricas.
Os heteroátomos Se e Te podem ser eliminados por reações
radicalares promovidas por Ph3SnH, Bu 3SnH e hv na presença do iniciador
de radicais AIBN 26-28 (Esquema 20 e 21 ).
---;>- R· + Ph3Sn-SePh
---e>• RH + Ph3Sn •
Esquema 20 Mecanismo da reação radica/ar.
EtOH ~o
A)=o 1 o SePh
J:>=o o
Esquema 21 Reações de redução eliminativa de organosselenetos e
organoteluretos.
25. Back, T. G.; "Organoseleniun Chemistry" Oxford University Press, Oxford, 1999. 26. Clive , D.L.J., et ali ; J Am. Chem. Soe. , 102:13, 4438, 1980. 27 . Nicolau, K. C.; Seitz, S.P.; Sipio, W.J.;Blount, J.F.; J Am. Chem. Soe.,, 101 :14,3884,
1974. 28. Hu. N. X.; Aso, Y; Otsubo, T.;J. Org. Chem. , 54, 4391, 1989.

Introdução - Organosselenetos e Organoteluretos 15
Entretanto, quando as reações radicalares são promovidas na
presença de captores de radicais, é possível formar ligações carbono
carbono29-31. (Esquema 22 e 23)
Ó S,Ph Bu3SnH ci)+Ó AIBN
o o
Ó(Ph ~ ~ SnBu3
R C6H6 h R
Esquema 22 Exemplos de reações radica/ares com organosselenetos.
o
CH,OPhTe~ o
rOM, Bu3SnH, tolueno
100 ºC. N2
o
Esquema 23 Exemplo de reação radica/ar com organotelureto.
OMe
A geração de organosselenetos e organoteluretos quirais, cria a
possibilidade de desenvolvimento de técnicas aplicadas em sínteses
orgânicas assimétricas. O Esquema 23 ilustra uma proposta sintética que
permite a aplicação das selenolactonas e telurolactonas quirais na síntese
do produtos naturais jasmolactona, molécula presente no jasmim e que é
responsável pela aroma característico da planta.
29. Satoh, T; Chem. Lett, 1949, 1987. 30. Toru , T; et all ; J Am. Chem. Soe.,, 110, 4815, 1990. 31 . Comasseto, J.V. ; Ferraz, H.M.C; Brandt, C.A; Petragnani , N; Tetrahedron Lett,; 28,
561 1, 1987.
32. Missio, J. L. ; Comasseto, J. V.; Tetrahedron asymm, 11 , 4609, 2000.

Introdução - Organosselenetos e Organoteluretos
~YPh o o
Y = Se Te
º~)
16
Jasmolactona
Esquema 24 Proposta para a síntese enantiosseletiva da Jasmolactona

Resultados e Discussão
RESULTADOS E
/.,,,,,;
DISCUSSAO
17/17

Resultados e Discussão 17
2. RESULTADOS E DISCUSSÃO
2.1 Introdução
De acordo com a revisão sobre biotransformação por lipases, os
substratos devem apresentar um centro de descriminação quiral para que a
lipase, através de sua estrutural tridimensional, possa reconhecer essa
quiralidade e promover a biotransformação enantiosseletiva. A estratégia
para obtenção das selenolactonas e telurolactonas em excesso
enantiomérico foi preparar intermediários racêmicos que pudessem sofrer a
resolução cinética enzimática. Esses intermediários, de acordo com a
análise retrossintética da selenolactona (1) e telurolactona (11), poderiam ser
hidroxiésteres racêmicos (Ili) com os heteroátomos Se e Te em suas
estruturas. (Esquema 25). A inserção dos heteroátomos seria através da
abertura do anel epóxido (IV) com selenolato e telurolato de sódio gerados in
situ pela redução de disseleneto e ditelureto de difenila com boroidreto de
sódio, gerando desta forma o intermediário racêmico (111).
/7 YPh
º~º~ I = Se II= Te
o o
==> PhY~O~ ==> n~O~ IGF OH PhY
III IV
Esquema 25 Análise retrossintética da selenolactonas e telurolactonas.
A partir da identificação dos intermediários para a etapa de resolução
cinética biocatalisada, foi estabelecida uma rota sintética para a obtenção
destes intermediários.
O Esquema 26 ilustra as etapas sintéticas para a obtenção dos
enantiômeros da selenolactona (R)-(S)-7a e da telurolactona (R)-(S)-7b
utilizando-se resolução cinética com a lipase de pâncreas de porco (PPL) em
meio orgânico anidro.

Resultados e Discussão 18
1. sol. NaOH 50 %, refluxo - 4 h
o o o o 2. sol. H2S04 3. 178 ºC li li 1. Et&d! / EtOH
~0~0~--2-.-~--B-r--
l 65 %
/"-o o~ 86%
2
o
1. SOCl2
2. PrOH
76%
o
~OH 3
PPL solvente
~º~ o 5
0Y0 Nl
etanol seco
Y= Se (a) 70% Te (b) 53%
A º ~Y~O~
32 ºe
o //
Cº J\'-Y
©J (R)- 7a, 7b
CAL
éter anidro
37%
37% OH
6a,6b (mistura racêmica)
A º ~Y /'-. ·.,, /'-. JL o~ / Y:,o~ ~
+
(R)-6a,6b
(S)-7a, 7b
Esquema 26 Esquema da rota sintética da (R) e (S)-fenilselenolactona e (R)
e (S)-feniltelurolactona,

Resultados e Discussão 19
O trabalho experimental foi divido em três etapas. A primeira etapa foi
a preparação do epóxido (4-oxipentanoato de propila). A definição desta
estrutura foi estratégica, pois de acordo com a análise retrossintética das
lactonas (Esquema 25), seria possível tanto introduzir os orgonosselenetos e
organoteluretos gerando o álcool secundário racêmico.
A segunda etapa foi a preparação do hidroxiésteres racêmico com os
heteroátomos Se e Te. Esses compostos foram preparados pela abertura do
epóxido com selenolato de sódio e telurolato de sódio gerados in situ pela
redução do disseleneto de defenila e do ditelureto de difenila,
respectivamente. E finalmente, a terceira etapa foi a aplicação da lipase PPL
para a resolução do substratos racêmicos e obtenção das lactonas
enriquecidas em suas ambas configurações.
2.2 Etapa 1: Preparação do substrato 4-oxipentanoato de propila.
2.2.1 Reação de alquilação e posterior descarboxi/ação do
dieti/malonato.
O composto 4-oxipentanoato de propila foi preparado em quatro etapas a
partir do dietilmalonato. O trabalho experimental foi iniciado com a alquilação
do malonato de dietila com brometo de alila (1) produzindo 2-alilmalonato de
dietila (2), que posteriormente foi hidrolisado e descarboxilado levando ao
ácido 4-pentenóico. Esses dois primeiros produtos obtidos (2-alilmalonato de
dietila e ácido 4-pentenóico) são compostos conhecidos e já bastante
estudados, não havendo necessidade de maiores discussões.
Entretanto, o espectro de RMN de H do composto 2 apresentou um sinal
interessante para H4. Esse hidrogênio (H4) foi observado com sinal de 1 O
linhas em 5,78 ppm, caracterizando um sinal dddd atribuído a acoplamentos
entre os hidrogênios H5a (17,10 Hz) e H5b (10,17 Hz) e H3 (6,81 Hz)36•
(Figura 3)
33. Hoye,et al; J. Org. Chem., 59, 4096, 1994.

Resultados e Discussão
o o
"' · º
Figura 3 Espectro de RMN de H4 do composto 2 na região de 5, 78 ppm.
20
6 o /"---......í
Para o hidrogênio olefínico terminal H5a , observou-se duplo quarteto
em 5,08 ppm devido a acoplamentos entre os hidrogênios vinílicos (17,15
Hz), geminais (3,01 Hz) e alílicos (1,60 Hz); Hsb apresentou um sinal
semelhante a H5a, sendo observado também um duplo quarteto em 5,03
ppm devido a acoplamentos entre os hidrogênios viníl icos (10,26 Hz),
geminais (3,01 Hz) e alílicos (1,30 Hz). (Figura 4)
o o
Hsa Hsb
Figura 4 Espectro de RMN de Hsa e Hsb do composto 2 na região de 5,06 a
5, 12 ppm.
fOTECA DEQUÍ ir.A
UQfvars!dade de São Paalo

Resultados e Discussão 21
Sinais semelhantes também foram observados para os hidrogênios
vinílicos do composto 3. Entretanto, para H2 foi observado um sinal complexo
de 2ª ordem entre 2,45 e 2,48 ppm. Esse fenômeno ocorreu porque H2 e H3
apresentam ambientes químicos muito semelhantes, relação !iv/J próxima a
1, permitindo desta forma acoplamento spin-spin com H4 (Figura 5).
2 . ,'4
Hsa O
Hsb~OH
H4
3
Figura 5 Espectro de RMN de H2 do composto 3 na região entre 2,45 a
2,48 ppm.
2.2.2 Reção de esterificação do ácido 4-pentenóico.
O ácido 4-pentenóico (3) foi inicialmente transformado em cloreto de
ácido pela reação com cloreto de tionila, sendo em seguida esterificado com
n-propanol, levando ao composto de interesse 4. Esta reação foi de fácil
execução e apresentou bom rendimento.
Optou-se em preparar o produto 4 utilizando-se o álcool n-propílico
porque a atividade das lipases em reações de transesterificação e hidrólise
aumentam de acordo com o tamanho da cadeia carbônica do álcool
presente no estrutura do substrato. Entretanto, cadeias carbônicas muitos
longas (acima de cinco carbonos) criam problemas de emulsificação durante
a etapa de extração do produto e aumentam também a temperatura para
purificações por destilação. Os ésteres com cadeias carbônicas, referentes

Resultados e Discussão 22
ao álcool, inferiores a três carbonos reduzem muito a velocidade da reação
biocatalisada e por esse motivo não são recomendados para resolução de
ésteres com lipases.
O grupo propila apresentou no espectro de RMN 1H sinais dos
hidrogênios dos carbonos 6, 7 e 8 como um tripleto em 4,04 ppm, sexteto
em 1,65 ppm e quarteto em 0,94 ppm, respectivamente. Com a esterificação
do composto 3, os deslocamentos químicos de H2 e H3 passaram a ser
muito próximos, sendo observada a coalescência dos picos. Desta forma,
para H2 e H3, observou-se um multipleto em 2,36-2,43 ppm.(Figura 6). A
coalescência dos picos de H2 e H3 também gerou acoplamentos extras com
os hidrogênios olefínicos, gerando desdobramentos de segunda ordem
muito complicados. Para os hidrogênios olefínicos Hsa e Hsb, observaram-se
multipletos em 5,03-5, 1 O ppm e 4,99-5,02 ppm respectivamente. Para H4
observou-se também a mesma influência, gerando um multipleto em 5,79-
5,87 ppm (Figura 7).
o s 3 11 6 s ~º~ 4 2 7
2 . 45 2.40 2 . 35
Figura 6 Espectro de RMN 1 H de H2 e H3 do composto 4 na região de 2,36 a
2,43 ppm.

Resultados e Discussão 23
, ... !5 .8~ 5,82 '·"
Figura 7 Espectro de RMN 1 H de H4 do composto 4 na região de 5, 79 a 5,87
ppm.
No espectro no infravermelho, observou-se o desaparecimento da
banda de estiramento do grupo hidroxila presente no ácido 4-pentenóico 3 e
o aparecimento da banda de estiramento do grupo carbonila do éster em v
c=o em 1738 cm-1, de acordo com a Figura 41.
2.2.3 Reação de epoxidação do 4-pentenoato de propila
A dupla ligação do composto 4 foi oxidada com ácido m
cloroperbenzóico levando a um anel epóxido. Foram encontrados problemas
para a extração do produto 5 do meio reacional, principalmente quando
usado um grande excesso de AMCPB (superior a 40 %). O melhor método
para a purificação do composto 5 foi por destilação a pressão reduzida.
A formação do epóxido alterou bastante o ambiente químico dos
hidrogênios ligados aos carbonos 2 e 3, sendo observado um deslocamento
em torno de 0,50 ppm entre eles, fato que não se observou nos compostos 3
e 4. O espectro de segunda ordem observado para H2 do composto 4 foi
caracterizado no composto 5 como um tripleto em 2,39 ppm. Para H3a e H3b,
observou-se um multipleto em 1,70-1,47 ppm e 1,87-1,92 ppm.

Resultados e Discussão 24
Os hidrogênios ligados ao carbono 5 apresentaram deslocamentos
químicos e acoplamentos diferentes devido às respectivas posições eis (Hsb)
e trans (Hsb) em relação a H4. Para Hsb e Hsa observou-se um duplo dubleto
em 2,43 e 2,69 ppm respectivamente (Figura 8) e para H4, observou-se um
multipleto em 2,90-2,93 ppm.
tn í"l l.OM -q-a'.)--.q-cnru._.. ..... -q- otcl -m-- oru '<1' 01"':fO> m cn mm ...,-r,("}C\J tO lO lDC.O -q--q--q-v
2. 75 2 .50
Hsa Hsb
Figura 8 Espectro de RMN 1H de Hsa e H5b do composto 5
Em comparação com o espectro de RMN 13C do seu precursor 4, para
C4 e C5 observaram-se picos em 51,07 e 46,81 ppm respectivamente,
caracterizando o desaparecimento da dupla ligação (C4 = 136,79 ppm e Cs = 115,44 ppm) e formação do epóxido, conforme Figura 9.

Resultados e Discussão
o s 3 11 6 s ~ o/"---..../
4 2 7
150
í
125 100
25
11
Hsa H O Hsb~ s Ü 2 il 6 8
lX~r'o~ O 4HH 7
5
50
Figura 9 Espectros de RMN 13C para C4 e C5 para os compostos 4 e 5 nas
região de 115, 44 a 136,79 e 46, 81 a 51, 07 ppm respectivamente.
No espectro no infravermelho foi observado o aparecimento da banda
de estiramento simétrica e assimétrica v c-o-c ass em 1258 cm-1e v e-o-e sim em
1180 cm-1, característica dos epóxidos. As demais bandas permaneceram
iguais ás do precursor 4, conforme Figura 42.
2.3 Etapa 2: Preparação dos hidroxiésteres racêmicos
2.3. 1 Introdução dos heteroátomos Se e Te
Para a abertura do epóxido, os nucleófilos selenolato de sódio e
telurolato de sódio gerados in situ pela redução de disseleneto de difenila e
ditelureto de defenila com boroidreto de sódio, respectivamente, foram
adicionados ao composto 5, produzindo os compostos 4-hidroxi-5-
fenilselenilpentenoato e 4-hidroxi-5-feniltelurilpentenoato de propila.
O acompanhamento da reação de substituição foi inicialmente feito
por cromatografia gasosa. Após várias análises, conclui-se que 6a era
ciclizado durante a anál ise por CG, produzindo a selenolactona 7a. Por este

Resultados e Discussão 26
motivo, o método de acompanhamento da reação por CG foi substituído por
HPLC, permitindo desta forma a analise da reação.
Após definido um método analítico para o acompanhamento da
reação, foi possível observar que o composto de interesse 6a não era o
produto principal formado na reação de substituição, sendo obtido como
produto principal a fenilselenolactona racêmica 7a gerada in situ pela reação
de lactonização (Esquema 27). O produto principal obtido era indesejado,
porém esse método apresentou-se como uma rota muito interessante para a
síntese de fenilselenolactonas racêmicas. Entretanto, para a etapa de
resolução cinética biocatalisada, era necessário que o produto principal
formado nesta reação fosse o hidroxiésteres racêmicos 6a.
Esquema 27 Mecanismo de formação da selenolactona 7a durante a
abertura do epóxido 5.
Para contornar esse problema experimental, foram realizados
diversos testes para se determinar os parâmetros ideais de tempo e
temperatura da reação. A condição experimental encontrada que apresentou
melhor resultado para a síntese do hidroxiéster racêmico foi efetuar a reação
a - 72 ºe (banho de etanol e gelo seco) com adição rápida do epóxido. O
tempo de reação também influenciava na formação de produtos
secundários. Desta forma, após a adição do epóxido, a reação era finalizada
após 1 minuto de reação pela adição de solução aquosa 5% de carbonato
de sódio. Esse procedimento reduziu drasticamente a formação de produtos
secundários e conseqüentemente aumentou o rendimento da reação. A
Figura 1 O ilustra o cromatograma de HPLC dos produtos obtidos na abertura
do anel epóxido nas condições otimizadas com a conversão de 86 % do

Resultados e Discussão 27
composto 5. Essa taxa de conversão foi determinada através de uma análise
por cromatografia gasosa da solução obtida após o término da reação, uma
vez que o epóxido não pode ser detectado pelo detector de UV do HPLC.
Data:AE1901' . D01 Method:ASG0l.MET Ch=2 rnAbs Chrom:AE1901' .C0l Atten : 10 1 000
500
[\ .. ,
)-.__ 0-1---- -----__.L___:,.__ _ _ _,é::,,..._ _ __ L_ _ _,,_ _______ ~
10 15
Tempo de Substância Porcentagem retenção (min) (%)
6,331 Selenolactona 7a 04
9,163 Impureza 02
12,801 selenoester 6a 94
Figura 1 O Cromatograma de HPLC da mistura reacional.
O espectro de RMN 1 H e 13C do composto 6a não apresentou
diferenças entre os sinais referentes ao grupo propila (H5 - Ha e C5- Ca) em
comparação com os espectros do seu precursor 5. (Figuras 30,31,36 e 37)
Para os hidrogênios diastereotópicos H3a e H3b, observou-se um
multipleto em 1,75 - 1,82 e 1,89 - 1,94 ppm, respectivamente. Esses
acoplamentos de segunda ordem ocorreram devido a acoplamentos
geminais entre H3a e H3b pela influência dos hidrogênios H4 do carbono
estereogênico no carbono e demais acoplamentos com H2 e H4 (Figura 11 ).
Para H2, observou-se um multipleto em 2,39-2,51 ppm. Esses
acoplamentos de segunda ordem ocorreram devido a acoplamentos
geminais entre H2a e H2b e acoplamentos entre os hidrogênios
diastereotópicos H3a e H3b. (Figura 12). Para os hidrogênios diastereotópicos
H5a e H5b, observaram-se duplos dubletos em 2,91 e 3,08 ppm. H4. (Figura
13)
min

Resultados e Discussão 28
li 12CT10 o ló~4 3 6s
9 S 10~ 2 7
OH 6a (mistura racêmica)
2.0 1 .9 I.B 1.7
Figura 11 Espectro de RMN 1H de H3a e H3b do composto 6a na região de 1,75
a 1,94 ppm.
li
12CT1º º 1-& ~43 6 8
9 S 10~ 2 7
OH 6a (mistura racêmica)
2.5 2.<I
Figura 12 Espectro de RMN 1H de H2 do composto 6a na região de 2,39 a 2,50
ppm.

Resultados e Discussão
3. t 3 . 0
,-.. N_, lll ,-.. IC) PJ C)
P1 N...., O'I cn cn cn <D nn
2. 9
29
Figura 13 Espectro de RMN 1 H de H5a e H5b do composto 6a na região de 2,90
a 3, 10 ppm.
Para H4, observou-se também um multipleto em 3,69 - 3, 73 ppm devido
a acoplamentos com os hidrogênios diastereoméricos H3a , H3b, H5a e H5b,
(figura 18). Para os hidrogênios aromáticos H11 - H12 e H10. observaram-se
multipletos em 7,23-7,27 e 7,50 - 4,52 ppm, sendo essa separação dos sinais
dos hidrogênios do anel característica do efeito de anisotropia diamagnética do
anel aromático e pelo efeito indutivo causado pelo elemento Se, deslocando os
hidrogênios (H 10) mais próximos do heteroátomo para campo mais baixo.
No espectro de RMN 13C do composto 6a foram observados
deslocamentos químicos característicos dos hidrogênios do anel aromático em
127,24 ppm para os C11 -C13e 129,16 ppm para o C10.
No espectro de 5, C4 e C5 apresentaram deslocamento em 66,01 e 51,07
ppm respectivamente, caracterizando os carbonos do anel epóxido. Com a
abertura do anel epóxido com selenolato de sódio, C4 e C5 do composto 6a

Resultados e Discussão 30
passaram a apresentar deslocamento químico em 69,28 e 36,65 ppm
respectivamente. (Figura 37)
No espectro no infravermelho, foi observado o desaparecimento das
bandas de estiramento simétrica e assimétrica v c-o-c ass em 1258 cm-1e v c-o-c
sim em 1180 cm-1, característica dos epóxidos, e o aparecimento do grupo
hidroxila em v OH em 3400 cm-1. (Figura 43).
O método utilizado para introduzir a espécie organometálica contendo
Te foi similar ao descrito para a espécie organometálica contendo Se. O
nucleófilo telurolato de sódio gerado in situ pela reação de redução de difenil
ditelureto com boroidreto de sódio foi adicionado rapidamente sobre o
composto 5 e imediatamente após 1 minuto, a reação foi finalizada com
solução aquosa 5% de Na2C03.
Foi observado que o composto 6b era ainda mais instável comparado a
seu equivalente com o heteroátomo de Se (6a), sofrendo a reação de
lactonização in situ mais rapidamente, conforme mecanismo comentado
anteriormente. O melhor resultado obtido apresentou o produto principal 6b e
as demais impurezas em uma proporção de 65 e 35% respectivamente, com a
conversão de 75 % do epóxido 5.
As impurezas (produtos secundários) eram de difícil separação e, para
se obter o 6b puro, foram realizadas várias tentativas de separação por
cromatografia. O melhor resultado obtido foi através de dois processos de
separação consecutivos, sendo o primeiro por cromatografia em coluna e o
segundo por cromatografia em placa preparativa. Na primeira etapa de
purificação cromatográfica, foram separados os compostos difenil ditelureto e o
epóxido que não havia reagido. A segunda separação foi realizada por
cromatografia em placa preparativa, sendo separados os compostos 6b e 7b.
O composto 6b era muito instável em contato com o ar e solvente
orgânico, sendo observado, após um período de exposição ao ar, a formação
de precipitado branco, dificultando a manipulação e separação do substrato
para reação enzimática. Para se minimizar essas perdas, após cada etapa de
separação, o solvente era rapidamente retirado e o composto colocado sob
atmosfera de N2.

Resultados e Discussão 31
O procedimento de separação foi satisfatório, sendo obtido o composto
6b puro para a etapa enzimática. A Figura 13 mostra o cromatograma por
HPLC do composto 6b após a purificação por cromatografia em coluna e placa
preparativa.
Data,M ISTll0-1.001 Method:ALE.MET Ch=2 m.i\bs Chmnri\HSTt 104 Cüt Attcll'6
(>Q
40
20
o-1---~ ------_..r--- ---~---------------1
10 15 min
Figura 14 Cromatograma de HPLC do composto 6b
O composto 6b, apresentou espectros de RMN 1H, 13C e de UV
semelhantes ao composto 6a. Foram observadas pequenas diferenças nos
deslocamentos químicos no espectro de RMN de 1H e 13C para os hidrogênios
e carbonos do anel aromático e carbono adjacente ao heteroátomo Te.
Entretanto todos os sinais do espectro apresentaram os mesmos acoplamentos
observados nos espectros de 6a.
2.4. Etapa 3: Resolução cinética biocatalisada
Nesta etapa, os substratos racêmicos 6a e 6b sofreram reação de
transesterificação biocatalisada por lipase (Esquema 28). As lipases
disponíveis comercialmente são provenientes de fonte microbiana, vegetal e
animal e podem ser encontradas imobilizadas, em solução ou como extrato cru.
Cada lipase apresenta uma atividade diferente em contato com substratos não

Resultados e Discussão 32
naturais, sendo que a escolha da lipase mais ativa se faz por análise de
resultados obtidos com substratos parecidos e de forma experimental. Foi
utilizada a lipase PPL (Sigma-Aldrich) porque resultados preliminares obtidos
em trabalho do grupo37 apontou a PPL como a enzima mais seletiva entre
quatro lipases testadas para a fenilselenolactona e feniltelurolactona . (CA -
Candida antartica, MM - Mucor miehei e LPS- Pseudomonas sp.)
PPL O solvente
.,,,,....__ .,,,,....__ Jl /'-.. / 32 ºe Z' j "-/'O'"-/ ___ _
OH
6a;6b (mistura racêmica)
Z: SePh (6a) TePh (6b)
o
z -----. ... __ .,,..,_ Jl o~ ,, ?<o::✓ "
(S)-6a;6b
j CAL solvente
32°c
(S)-7a;7b
Esquema 28 Enantiociclização dos intermediários 6a e 6b.
+H~O
z (R)-7a;7b
A PPL é uma lipase de fonte animal (pâncreas de porco) e é
comercializada como um extrato cru que contém, além da PPL, outras enzimas
como a quimiotripsina e colesterol esterase. A aplicação desta lipase foi
bastante simples devido a insolubilidade do extrato em solvente orgânico,
permitindo desta forma, separar a enzima do substrato e produto por filtração.
A pratica experimental consistiu em misturar a lipase PPL aos
compostos 6a e 6b, previamente dissolvidos em solvente orgânico anidro, e
manter a agitação e temperatura (32 ºC) constantes durante o período da
reação.
34. Missio, L.J .; "Síntese de Butirolactonas substituídas"; Tese de Doutorado; UFSCAR, São Carlos, 2000.

Resultados e Discussão 33
De acordo com a estratégia sintética, era esperado que uma das
configurações (50% do substrato racêmico) 6a e 6b sofresse reação de
transesterificação biocatalisada mais rapidamente que a outra, levando
fenilselenolactona 7a e a feniltelurolactona 7b em uma única configuração. O
acompanhamento da reação foi por HPLC, sendo a reação finalizada pela
retirada da enzima por filtração quando consumido 50% do substrato racêmico.
Esse procedimento experimental foi aplicado com o substrato 6a usando
os solventes anidros: éter e hexano; e para o substrato 6b: éter, hexano e
ciclohexano. A reação de transesterificação foi realizada em solventes com
diferentes constantes dielétricas. É conhecido que a constante dielétrica dos
solventes podem também influenciar na seletividade enzimática, possibilitando
também um estudo sobre a influência dos solventes na reação desses
substratos com a lipase PPL. Os cromatogramas de HPLC, representados nas
Figuras entre 15 - 20 e 21 - 23 mostram o progresso da reação em função do
tempo para os substratos 6a em éter anidro e 6b em hexano anidro,
respectivamente.
Data:AE 1901 ' . D01 Me thod : ASGOl.ME:T Ch =2 mAbs Chrom·AE1901' .C0 l At.t.en· 10 1000
500
,,·, M
À..
,., ·~
10
,,,
/\ Figura 15 Cromatograma de HPLC do precursor 6a .
Data:TOL2.D01 Metho d: ASGO l.MET Ch=2 mAbs Chrom: TOL2 . COl Atten: 1 1 2000
1000
10
1 5 mín
15 min
Figura 16 Cromatograma de HPLC da reação de ciclização do substrato (6a) com PPL em éter anidro após 22 h

Resultados e Discussão
Data : TOL4. D01 Method : ASGOl. MET Ch=2 mAbs Chrorn : TOL4 . CO 1 At ten: 12 4 000
10
34
15 min
Figura 17 Cromatograma de HPLC da reação de ciclização do substrato (6a) com PPL em éter anidro após 28 h
Data :TOLG . D01 Method:ASGOl. MET Ch=2 mAbs Chrom: TOL6 . COl Atten: 11 2000
10 15 min
Figura 18 Cromatogramade HPLC da reação de ciclização do substrato (6a) com PPL em éter anidro após 47 h
Data:TOLS.D01 Method:ASGOl.MET Ch•2 rnAbs Chrom : TOL8.C01 Atten : 10 100
500
,, , .. ,, .,,
O+------------'_ ...,__ __ __,.
l , ~
'P.
10 15 min
Figura 19 Cromatograma de HPLC da reação de ciclização do substrato (6a) com PPL em éter anidro após 69 h

Resultados e Discussão
Data : TOLl O.D01 Method:ASGOl.MET Ch=2 rnAbs Chrom : TOL10 .C01 Atten: 11 2000
1000
·,:, ,.
35
º'+----------~~--~----~-~-----------t
10 15 mi n
Figura 20 Cromatograma de HPLC da reação de ciclização do substrato (6a) com PPL em éter anidro após 75 h
Tabela 1 Porcentagens entre o produto formado, substrato e impureza.
Tempo de reação Produto (7a)
(h)
o 22
28
47
69
75
Da1~:MIST! 104.DOI Mcthod:ALE.l\lET Ch=2 mAb5 Chrom·MIST1104 C0 I Attcn-6
60
40
(%)
04
20
25
32
40
47
Ili
Substrato (6a)
(%)
94
71
66
60
51
47
15
Figura 21 Cromatograma de HPLC do precursor 6b.
Impureza
(%)
02
09
09
08
08
06
min

Resultados e Discussão
D:ua:HE..X \3042001 Mc1hod:ALE.l\1Er 0 1=2 mAhs Oirom+IE.Xt.30-P 011 Attc11"4 -
5
o
~ .,;
s; 5 Ã
~ i;j \. Â R \.
1() 15 min
Figura 22 Cromatograma de HPLC da reação de ciclização do substrato (6b) com PPL em hexano anidro após 21 h.
Data:HE..X \3043.DOl ~lcthod:ALE.l\lET Ch=2 m1\bs Chrom·HEX13043 COl Attcn-4
15
1
~ N
.,;
~
:~
~ :.s \ Ã R \ 1
Ili 15 min
Figura 23 Cromatograma de HPLC da reação de ciclização do substrato (6b) com PPL em hexano anidro após 23 h
Tabela 2 Porcentagens entre o produto formado, substrato e impureza.
Tempo de reação Produto (7b) · Substrato (6b) Impureza
(h) (%) (%) (%)
o 01 98 01
21 41 54 05
23 50 45 05
36

Resultados e Discussão 37
Os cromatogramas anteriormente mencionados ilustram como foi feito a
acompanhamento da reação por HPLC. As reações com os diferentes
solventes apresentaram tempos de reação diferentes, como mostra as Tabelas
3 e4.
Tabela 3 Tempo de reação para consumo de 50 % de 6a catalisada por PPL para dois solventes anidros diferentes.
Solvente Tempo (h)
Eter 75 Hexano 86
Tabela 4 Tempo de reação para consumo de 50 % de 6b catalisada por PPL para três solventes anidros diferentes.
Solvente Tempo (h}
Ciclohexano 12 Éter 21 Hexano 23
Essa grande diferença de velocidade nas reações de transesterificação
biocatalisada entre os substratos 6a e 6b, ocorreu provavelmente devido a
substituição do complexo enzimático PPL usado com o substrato 6a por outro
recentemente adquirido usado com 6b. A PPL antiga havia sido adquirida há 4
anos atrás e provavelmente grande parte da enzima presente no complexo já
havia denaturado, fato que provavelemente reduziu a sua atividade.
Após período descrito nas Tabelas 3 e 4, os compostos (R)-7a e (S)-6a
e (R)-7b e (S)-6b presentes na solução filtrada, foram separados por
cromatografia em placa preparativa.
Os hidroxiésteres (S)-6a e (S)-6b que não reagiram, após a separação
por cromatografia, foram novamente submetidos a transesterificação
biocatalisada pela ação da lipase CAL 37 (lipase da candida antartica) em
condições semelhantes às descritas nas reações com PPL, produzindo os
compostos (S)-7a e (S)-7b (Esquema 29). Optou-se em utilizar a enzima

Resultados e Discussão 38
porque a ciclização em meio ácido não ocorreu com os hidroxiésteres (S)-6a e
(S)-6b. Outra vantagem do uso da enzima foi a facilidade de separação da
enzima e do produto formado, sendo que a enzima CAL imobilizada foi
separada da fenilselenolactona e da feniltelurolactona por filtração em papel.
Y= se (a) (S)-6a,6b Te (b)
o
CAL Cº - é-te-r a-ni-dr-o --- f -_ y H-© (S)- 7a,7b
Esquema 29 Enantiociclização para obtenção da (S)-hidroxiesteres.
O composto 7a foi analisado por RMN 1H e apresentou acoplamentos
semelhantes aos hidrogênios do anel aromático em relação ao seu precursor
6a (Figura 49).
Para os hidrogênios diastereotópicos H3a e H3b observaram-se
multipletos em 2,38 - 2,45 e 1,92 - 2,00 ppm, respectivamente. Esses
espectros de segunda ordem ocorreram devido a acoplamentos geminais entre
H3a e H3b pela influência do centro estereogênico presente no carbono 4 e
demais acoplamentos com H2 e H4 (Figura 29).
Para H2 observou-se um multipleto em 2,48-2,61 ppm. Esses espectros
de segunda ordem ocorreram também devido a acoplamentos geminais entre
H2a e H2b e acoplamentos entre os hidrogênios diatereotópicos H3a e H3b.
(Figura 24) Para os hidrogênios diastereotópico H5a e Hsb, observaram-se
duplos dubletos em 3,29 e 3,00 ppm, respectivamente. Esses hidrogênios são
diastereotópicos devido a influência dó centro estereogênico do composto,
apresentando desta forma, acoplamentos entre os hidrogênios geminais e H4,
de acordo com a Figura 25.
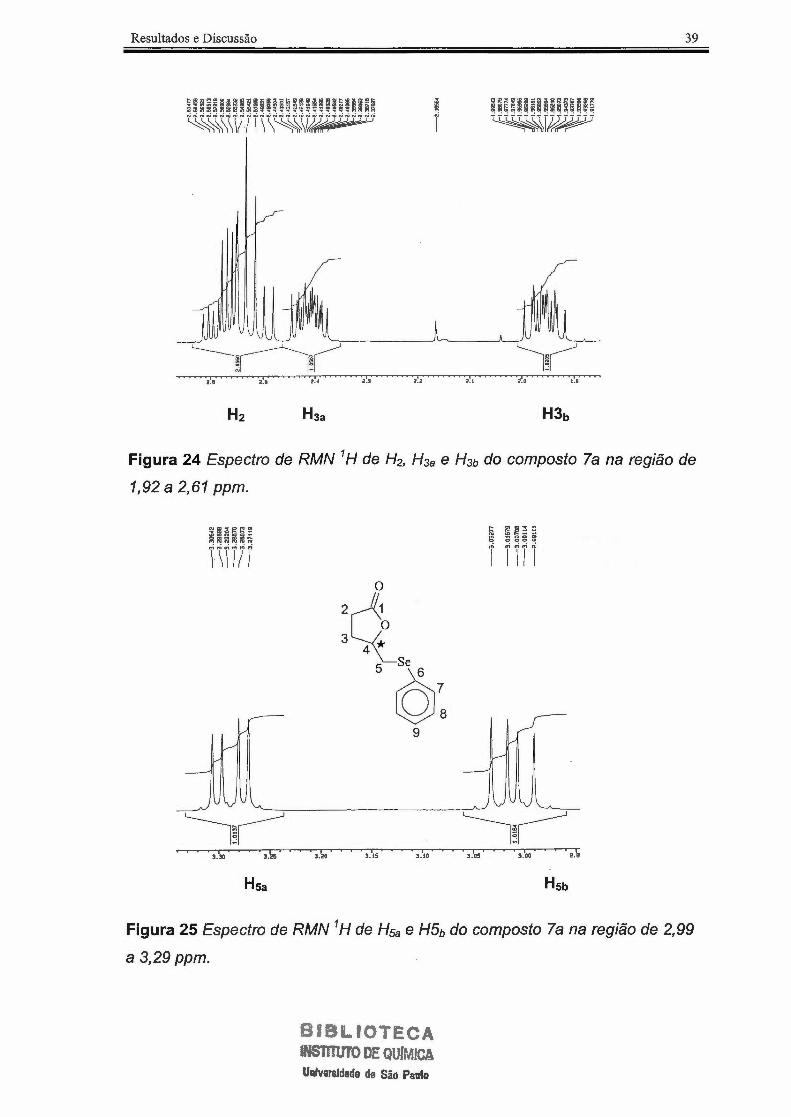
Resultados e Discussão
,., ,., , .. ,., z.z ,.,
~;~~1,~ii~i~ii~~ ~!~
z.o ,.,
39
Figura 24 Espectro de RMN 1 H de H2, H3a e H3b do composto la na região de
1,92 a 2,61 ppm.
3.30 3 .25 3 . 20 3 .15 3 . 10 3 . 05 3 . 00 2 .9
Hsa
Figura 25 Espectro de RMN 1 H de H5a e H5b do composto la na região de 2,99
a 3,29 ppm.
B LIOTIECA DNSTmJ'rO DE QUfMICA UQfv;rsfdade de São Paalo

Resultados e Discussão 40
Para H4 , observou-se também um multipleto em 4,63 - 4,68 ppm devido
a acoplamentos com os hidrogênios diastereoméricos H3a, H3b, Hsa e Hsb
O espectro de RMN 13C do composto 7a apresentou sinais semelhantes
ao espectro do seu precursor 6a, exceto para os carbonos do grupo propila (C6
a C8), eliminado pela reação de hidrólise biocatalisada.
No espectro no infravermelho, foi observado o desaparecimento da
banda de estiramento do grupo hidroxila em v oH 3400 cm-1, caracterizando o
álcool secundário.
O composto 7b apresentou também características analíticas muito
parecidas com as do seu equivalente com o heteroátomo de Se, sendo apenas
diferenciados por pequenos desvios nos deslocamentos químicos e constantes
de acoplamento no espectro de RMN de 1H e 13C no anel aromático e carbono
adjacente ao elemento Te.
2.5. Determinação do ee
Após a resolução cinética biocatalisada, a próxima etapa foi medir os
ees obtidos. Os ees podem ser medidos por cromatografia gasosa ou por
cromatografia líquida utilizando colunas quirais ou por RMN utilizando
reagentes de deslocamento.
Foram muitas as tentativas feitas para medir o excesso enantiomérico
das feniltelurolactonas 7a e feniltelurolactonas 7b por cromatografia gasosa
quiral; entretanto não houve separação dos picos dos enantiômeros. Para
contornar esse problema, os grupos organometálicos PhSe e PhTe foram
eliminados, produzindo desta forma a õ-valerolactona 8. Como descrito na
introdução, os compostos organometálicos de Se e Te podem ser convertidos
em hidrocarbonetos pelo método de redução eliminativa radicalar. Desta forma,
a fenilselenolactona e feniltelurolactona reagiram com Bu3SnH, eliminando os
grupos volumosos e pesados que impediam a separação dos picos dos
enantiômeros na coluna quiral. O produto formado apresentou-se mais
favorável para a análise por cromatográfica quiral (Esquema 30).

Resultados e Discussão
~y
O Y~SeouTe 'O (R), (S)-7a, b
Bu3SnH, AIBN tolueno
refluxo 3 h
~ o o (R), (S)-Sa, b
Esquema 30 Eliminação redutiva para obtenção da y-valero lactona.
41
Os produtos das eliminações redutivas das fenilselenolactonas e
feniltelurolactonas sintetizadas na presença de diferentes solventes foram
analisadas por CG utilizando-se uma coluna quiral de 30 m, com filme (0,25
µm) de p dextrina da marca Supelco. A partir da integração da área dos picos
dos enantiômeros separados na coluna, foram feitos os cálculos de ee
utilizando a Equação 1.
Equação1 Cálculo do ee das áreas dos picos obtidos no cromatograma.
ee (%) = [A(R) - A(S)] / [A(R) + A(S)] x 100
A y-valerolactona (8) obtida pela eliminação redutiva radicalar apresenta
grupos prioritários, de acordo com regra de nomenclatura (regra de Cahn,
lngold e Prelog), diferentes do precursor fenilselenolactona e feniltelurolactona.
Desta forma, a eliminação redutiva da (R)-fenilselenolactona produz a (S)- y
valerolactona.
Os valores dos ees obtidos para as valerolactonas devem estar
diretamente relacionados com os ees dos seus precursores, pois a função
lactona não sofre racemização ou qualquer outra mudança de configuração.
Uma forma interessante para demostrar que a reação radicalar não afeta a
configuração da função lactona seria determinar os ees da fenilselenolactona e
feniltelurolactona por RMN de 77Se e 125Te utilizando reagentes de
deslocamento quirais. Para o experimento de RMN de 77Se e 125Te utilizam-se
soluções de referência externa, normalmente soluções 1,0 M de disseleneto de
difenila e ditelureto de difenila em CDCl3 , respectivamente35 e
35.
35. Duddeck, H; Wagner; BiallaB, A, P; Magnetíc Resonance ín Chemístry, 29,248, 1991 36. Duddeck, BiallaB, A, P; Magnetíc Resonance ín Chemistry, 32, 303, 1994
Kesultados e 1J1scussão 42
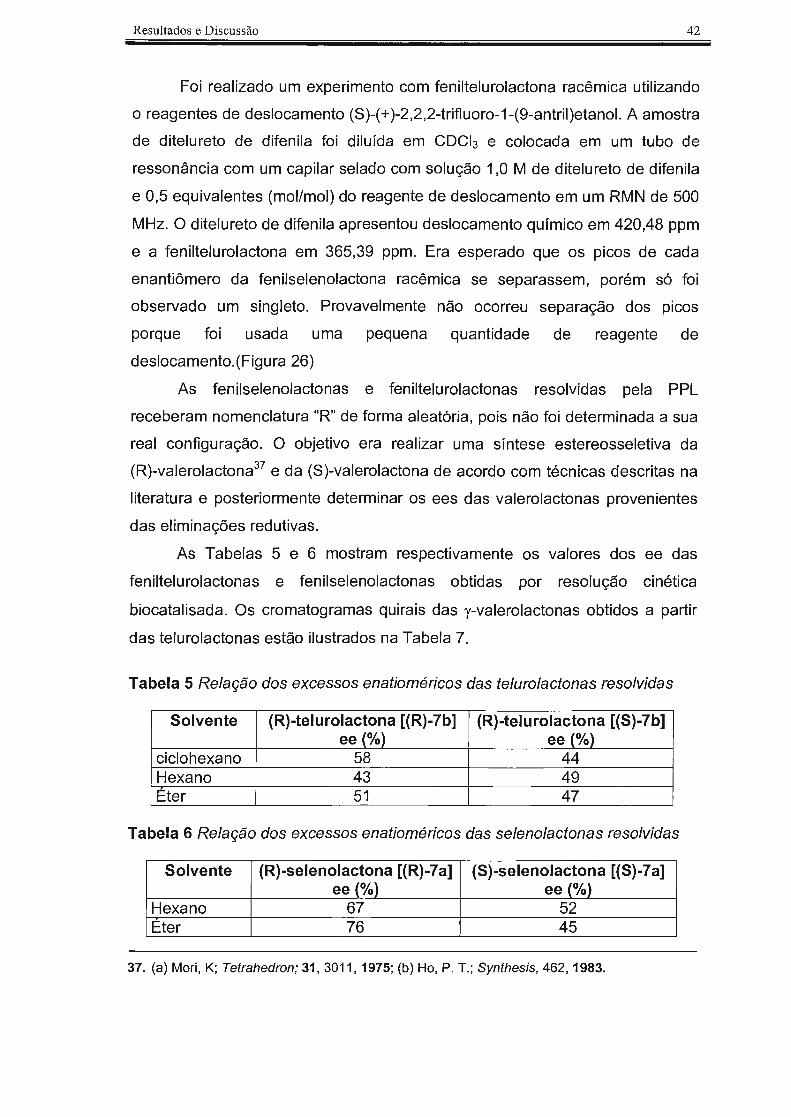
Foi realizado um experimento com feniltelurolactona racêmica utilizando
o reagentes de deslocamento (S)-(+)-2,2,2-trifluoro-1-(9-antril)etanol. A amostra
de ditelureto de difenila foi diluída em CDCl3 e colocada em um tubo de
ressonância com um capilar selado com solução 1,0 M de ditelureto de difenila
e 0,5 equivalentes (mol/mol) do reagente de deslocamento em um RMN de 500
MHz. O ditelureto de difenila apresentou deslocamento químico em 420,48 ppm
e a feniltelurolactona em 365,39 ppm. Era esperado que os picos de cada
enantiômero da fenilselenolactona racêmica se separassem, porém só foi
observado um singleto. Provavelmente não ocorreu separação dos picos
porque foi usada uma pequena quantidade de reagente de
deslocamento.(Figura 26)
As fenilselenolactonas e feniltelurolactonas resolvidas pela PPL
receberam nomenclatura "R" de forma aleatória, pois não foi determinada a sua
real configuração. O objetivo era realizar uma síntese estereosseletiva da
(R)-valerolactona37 e da (S)-valerolactona de acordo com técnicas descritas na
literatura e posteriormente determinar os ees das valerolactonas provenientes
das eliminações redutivas.
As Tabelas 5 e 6 mostram respectivamente os valores dos ee das
feniltelurolactonas e fenilselenolactonas obtidas por resolução cinética
biocatalisada. Os cromatogramas quirais das y-valerolactonas obtidos a partir
das telurolactonas estão ilustrados na Tabela 7.
Tabela 5 Relação dos excessos enatioméricos das telurolactonas resolvidas
Solvente (R)-telurolactona [(R)-7b] (R)-telurolactona [(S)-7b] ee (%) ee {%)
ciclo hexano 58 44 Hexano 43 49 Eter 51 47
Tabela 6 Relação dos excessos enatioméricos das selenolactonas resolvidas
Solvente (R)-selenolactona [(R)-7 a] (S)-selenolactona [(S)-7a] ee (%) ee (%)
Hexano 67 52 Eter 76 45
37. (a) Mori, K; Tetrahedron; 31, 3011, 1975; (b) Ho, P. T.; Synthesis, 462, 1983.

Resultados e Discussão
Figura 26 Espectro de RMN de 125Te da feniltelurolactona.
Tabela 7 Cromatogramas da valerolactona obtidos por cromatografia gasosa com coluna quiral a partir das (R) e (S)-teluro/actonas
Solventes R-valerolactona 5-valerolactona .., ., .,, '.
M m
' o '
~ Ça Mistura -
racêmica (R) (S)
l
43

Resultados e Discussão 44
an an ,:,
,-. .... (• M ,.,
"" ,,
Éter "' N (•J (•J "" "" Ca'J .~, ,-..
N
\ - ..
r-.,,, ,,, r-
"'
•.r,
"' ll'1
r-N
Hexano .... .::, CJ
r -N
., ""
r-,,, ,,,
t \
IJ1 CI ., N .... r• .... N ..,
1"•J
ciclo hexano r-N "" t'•J o
1~ ,-..
r-(,J
·1
\_ - \
A Tabela 8 mostra os cromatogramas das 8-valerolactonas obtidas das
fenilselenolactonas. Os cromatogramas desta análise, embora analisados com

Resultados e Discussão 45
a mesma coluna, apresentaram tempos de retenção diferentes dos
cromatogramas mostrados na Tabela 7. Ambos os cromatogramas
correspondem as mesmas substâncias, porém as amostras foram analisadas
em cromatógrafo gasoso e condições cromatográficas diferentes.
Tabela 8 Cromatogramas da valerolactona obtidos por cromatografia gasosa com coluna quiral a partir das (R) e(S)-selenolactonas
Solventes R-valerolactona 1 S-valerolactona r-a-. N
-·· <r .., a-. r--IJ)
<r : .., . . .
Mistura racêmica
\ \....__
Li) ÇS) a:, N
r{'.1 iD .,..
sr- .... --i
Éter
. r·-O"\ I..O
" --i o s
~ '
_j\__ vL

Resultados e Discussão 46
N .... "' r-- "' e-: .,. .,. .... ....
Hexano
J ) '.::; ~ U)
,,.. " ....
,\
- l_ -
Entre os resultados obtidos na resolução das fenilselenolactonas, os
melhores ee de (R) e (S)-7a foram respectivamente, 76 % em éter e 52 % em
hexano. Para as feniltelurolactonas, os melhores ee de (R) e (S)-7b foram de
58 % em ciclohexano e 49 % em hexano.
Os resultados obtidos demonstram a viabilidade desta metodologia na
síntese enantiosseletiva de fenilselenolactonas e feniltelurolactonas, porém os
resultados precisam ser otimizados.
Os organosselenetos se mostraram mais seletivos do que os respectivos
organoteluretos, embora se esperasse que a enzima PPL utilizada na
resolução dos organoteluretos proporcionasse uma maior seletividade por ser
nova. Provavelmente, o maior volume do substrato contendo o heteroátomo de
Te dificultou uma boa interação com o sítio ativo da enzima e
consequentemente reduziu a seletividade da reação biocatalisada.
Também ficou evidenciado que os solventes interferem na seletividade
na resolução desses substratos racêmicos. O solvente n-hexano proporcionou
a menor seletividade na resolução dos dois substratos para a produção da (R)
telurolactona e (R)-selenolactona; entretanto, produziu os melhores resultados
para a (S)-telulactona e (S)-selenolactona.

Parte Experimental
PARTE EXPERIMENTAL
47/47

Parte Experimental 47
3. PARTE EXPERIMENTAL
3. 1 Síntese do composto 2-alilmalonato de dietila (2).
o o 6
/'--o o~7
C10H1604
pm = 200,23 g 1 s liquido incolor
Em um balão de duas bocas de 2000 ml provido de agitação
magnética, condensador de refluxo e funil de adição de líquido, foram
adicionados 500 ml de etanol seco e pequenos pedaços de sódio metálico
(8,00 g; 350 mmol). Após todo o sódio ter sido consumido, o malonato de
dietila (56,06g; 350 mmol) foi adicionado lentamente (30 minutos) a
temperatura ambiente. Em seguida foi adicionado lentamente (30 minutos)
brometo de alila (46,58g; 385 mmol) e a mistura foi aquecida mantendo-se
refluxo por 16 horas. Após esse período, a mistura reacional foi resfriada e
posteriormente filtrada em funil de Büchner. O solvente foi eliminado por
rotaevaporação e o bruto reacional extraído em (3x 100 ml) acetato de etila.
O produto (2) foi destilado sob pressão reduzida (12 mmHg; 11 O ºe). Rendimento: 45, 55 g (65%).
RMN 1H (500 MHz, CDC'3) : õ (ppm) 1,27 (t, J = 7, 12 Hz, 6 H, H7); 2,65 (qq,
JH3-H2 = 7,60 Hz, JH3-H4 = 6,80 Hz, JH3-H5a = 1,53 Hz, JH3-H5b = 1, 15, 2H, H3);
3,42 (t, JH3-H2 = 7,57 Hz, 1 H, H2); 4, 15 - 4,25 (m, 2 H, H5), 5,06 (dq, JHsb-H4 = 10,20 Hz, JHsa-H5b = 3,01 Hz, JH3-H5b = 1, 15 Hz, 1 H, Hsb); 5, 12 (dq, JHsa-H4 = 17,08 Hz, JHsa-H5b = 3,01 Hz, JH3-H5a = 1,53 Hz, 1 H, Hsa); 5, 78 ( qt, JH5a-H4 = 17,08 Hz, JH5b-H4 = 10,20 Hz, JH3-H4 = 6,80 Hz, 1 H, H4). RMN 13C (500 MHz,
CDCl3): õ (ppm) 14,01 (C1); 32,85 (C3); 51,72 (C2); 61,41 (C5); 117,51 (C5);
134,14 (C4); 168,95 (C1).IV (filme): v c=o 1737 cm-1; v c=c 1643 cm-1.

Parte Experimental
3.2 Síntese do ácido 4-pentenóico(3).
o s 3 li ~OH
4 2 C5HsO2 pm= 100,11 g líquido incolor
48
Em um balão de duas bocas de 250 ml, provido de um condensador
de refluxo e funil de adição de líquido, foi dissolvido NaOH (25,09 g; 627,3
mmol) em 50 ml de água e em seguida o 2-alilmalonato de dietila (2) (49,26
g; 246,01 mmol) foi adicionado lentamente (45 minutos) sobre a solução
alcalina. A mistura foi aquecida mantendo-se refluxo por 4 horas. O etanol
formado na hidrólise básica foi eliminado por rotaevaporação. A mistura
reacional foi resfriada a O ºe.e acidificada com H2SO4 concentrado até pH 3.
O ácido 2-alilmalônico formado foi extraído em acetato de etila (3x150 ml) e
seco com sulfato de magnésio anidro. O solvente foi eliminado por
rotaevaporação obtendo-se o ácido 2-alilmalônico cristalizado. O ácido
formado foi aquecido a 178 ºe, sendo descarboxilado e em seguida
destilado. Rendimento: 21,22g (86%).
RMN 1H (500 MHz, CDCb) : õ (ppm) 2,39 (qq, JH3-H2 = 7,70 Hz, JH3-H4 = 6,36
Hz, JH3-H5a = 1,60 Hz, JH3-H5b = 1,30, 2H, H3); 2,47 (td, JH3-H2 = 7,70, JH4-H2 = 4,60 Hz, 1 H, H2); 5,03 (dq, JHsb-H4 = 10,26 Hz, JHsa-H5b = 3,01 Hz, JH3-H5b =
1,30 Hz, 1 H, Hsb); 5,08 (dq, JHsa-H4 = 17,15 Hz, JHsa-H5b = 3,01 Hz, JH3-H5a =
1,60 Hz, 1 H, Hsa); 5,84 ( qt, JHsa-H4 = 17, 15 Hz, JH5b-H4 = 10,26 Hz, JH3-H4 = 6,36 Hz, 1 H, H4). RMN 13C (500 MHz, CDCl3): õ (ppm) 28,52 (C3); 33,36 (C2);
115,76 (C5); 136,32 (C4); 179,45 (C1); IV (filme): v c=o 1711 cm-1; v e-o 1285
cm-1; õ OH 1422; V C=C 1644 cm-1.
3.3 Síntese do composto 4-pentenoato de propila (4)

Parte Experimental 49
Em um balão de duas bocas de 250 ml, provido de agitação
magnética, condensador de refluxo conectado a um lavador de gás e um
funil de adição de líquido, foi adicionado o cloreto de tionila (49,97 g; 420,06
mmol) e em seguida, lentamente_ (4 minutos), o ácido 4-pentenóico (3)
(33,04 g; 329,64 mmol). Após a adição do composto 3, deixou-se reagir por
60 minutos.
Em um outro balão de duas bocas de 250 ml, também provido de
condensador de refluxo, funil de adição de líquido e agitação magnética, foi
adicionado n-propanol (23,44g; 390,02 mmol) em um banho de gelo. O
cloreto de ácido formado na etapa anterior foi adicionado lentamente (45
minutos) sobre o n-propanol. Após a adição do cloreto de ácido, o banho de
gelo foi retirado e deixou-se reagir a temperatura ambiente por 60 minutos. A
mistura foi lavada com solução 5% de bicarbonato de sódio (3x 15 ml) e
com água (3x 15 ml). O produto foi destilado a 143 ºC. Rendimento: 35,36 g
(76%).
RMN 1H (500 MHz, CDC'3) : 8 (ppm) 0,94 (t, J = 7,42 Hz, 3 H, H8); 1,65 (se, J
= 7,36 Hz, 2H, H1); 2,36-2,43 (m, 4 H, H2 e H3), 4,04 (t, J = 6,72 Hz, 2 H, H6);
4,99 - 5,02 {m, 1 H, Hsb); 5,03 (m, 1 H, H53); 5,79-5,87 (m, 1 H, H4); RMN 13C
(500 MHz, CDCl3): 8 (ppm) 10,40 (Ca);22,05 (C1); 28,97 (C3); 33,65 (C2);
66,01 (C5); 115,44 (Cs); 136,79 (C4); 173, 16 (C1); IV (filme): v c=o 1738 cm-1;
V C=C 1642 cm-1.
3.4 Síntese do composto 4-oxipentanoato de propila (5).
o ~ l ll 6 8 lX ~21 1" º~
O 7
C gH 14Ü3
pm= 158,19 g líquido incolor
Em um balão provido de agitação magnética e funil de adição de
líquido, o ácido 3-cloroperbenzóico (7,00 g; 40,6 mmol) foi dissolvido em 50

Parte Experimental 50
ml de diclorometano seco. Em seguida o 4-pentenoato de proprila (4) (4,00
g; 28, 13 mmol), também dissolvido em 50 ml de diclorometano, foi
adicionado lentamente (30 minutos) sobre a solução de 3-cloroperbenzóico a
O ºC.
A mistura permaneceu sob agitação por 17 horas a temperatura
ambiente. Após esse período, a mistura foi lavada com solução 10% de
bicarbonato de sódio (4 x 15 ml) e seca em sulfato de magnésio anidro. O
solvente foi eliminado por rotaevaporação e o produto (5) destilado a
pressão reduzida (100 ºe; 6 mmHg). Rendimento: 3,40 g (77 %).
RMN 1H (500 MHz, CDCb): 8 (ppm) 0,87 (t, J = 7,45 Hz, 3 H, H8); 1,58 (se, J
= 7,37 Hz, 2H, H7); 1,70 - 1,74 (m, 1 H, H3b); 1,87 - 1,92 (m, 1 H, H38); 2,39
(t, J = 7,87 Hz, 2 H, H2); 2,43 - 2,44 (dd, J5a-5b = 4,96 Hz, J5b-4 = 2,68 Hz, 1 H,
Hsb); 2,68 - 2,70 (dd , J5a-5b = 4,90 Hz, J5a-4 = 4, 1 O Hz, 1 H, Hsa); 2,90 - 2,93
(m, 1 H, H4); 3,98 (t, J = 6,72 Hz, 2 H, H6); RMN 13C (500 MHz, CDC13): 8
(ppm) 10,17 (Cs);21,88 (C1); 27,53 (C3); 30,27 (C2);46,81 (Cs); 51 ,07 (C4);
66,01 (Cs); 173, 16 (C1); IV (filme): v c=o 1730 cm-1; v e-o-e ass 1258 cm-1; v c
o-c sim 1180 cm-1; CG-MS 70 eV [m/z (% rei)]: (M+. 158); 99 (PB); 71 (77); 55
(49).
3.5 Síntese do composto disseleneto de difenila.
2 3
(g--sese-04 C12H10Se2 pm=315,27g sólido amarelado
Em um balão de três bocas de 3000 ml, provido de agitação
mecânica, funil de adição de líquido e condensador de refluxo, foi adicionado
magnésio (6,75 g; 280 mmol) e alguns cristais de iodo em 200 ml de éter
etílico anidro.
Em seguida foi adicionada uma grande quantidade do bromobenzeno
(7,5 g; 40 mmol) ao magnési,>. Após o consumo do bromobenzeno,
observado pela mudança da colo1 ação da mistura, foi iniciada a adição lenta

Parte Experimental 51
de bromobenzeno (33 g; 21 O mmol) dissolvido em 50 ml de éter anidro,
sendo a velocidade de adição controlada pelo refluxo do éter.
Após consumo do magnésio, foi adicionado selênio em pó (20 g; 250
mmol) em pequenas quantidades durante um período de 60 minutos. Ao
término da adição, a mistura reacional permaneceu sob agitação por um
período de 120 minutos e foi finalizada com adição lenta de solução
saturada de cloreto de amônia até a neutralização.
O intermediário formado permaneceu exposto ao ar por 15 horas,
sendo o disseleneto de difenila formado extraído com acetato de etila (4 x
100 ml) e seco com sulfato de magnésio anidro. O solvente foi eliminado
por rotaevaporação e o produto recristalizado em hexano. Rendimento:
32,54g (41%).
3.6 Preparação do composto 4-hidroxi-5-fenilse/enilpentanoato de
propila (6a).
11 1ª10 O lh9~43 6 8
1 3 s 1 o~ 14 2 7
OH
C14H20Se03 pm=315 ,27g líquido amarelado
Em um balão de duas bocas de 250 ml, provido de agitação
magnética e sob atmosfera de N2, disseleneto de difenila (1,72g; 5,5 mmol)
foi dissolvido em etanol absoluto (30ml). Foi adicionado a solução pequenas
quantidades (0,3g) de boroidreto de sódio até a solução permanecer
levemente amarelada, formando selenolato de sódio in situ.
Foi adicionado rapidamente o epóxido 5 (1,58g; 1 O mmol) ao
selenolato em solução alcoólica e deixou-se reagir por 1 minuto em um
banho de gelo seco em etanol. O composto 6a foi extraído em acetato de
etila (150 ml), lavado com solução 10% de NaCO3 (3 x 20 ml) e separado
em coluna cromatográfica. Rendimento: 1,78 g (57 %).

Parte Experimental 52
RMN 1H (500 MHz, CDCl3): õ (ppm) 0,81 (t, J = 7,45 Hz, 3 H, H8); 1,61 (se, J
= 7,37 Hz, 2H, H1); 1,75 - 1,82 (m, 1 H, H3b); 1,89 - 1,94 (m, 1 H, H33 ); 2,39
- 2,51 (m, 2 H, H2); 2,90 - 2,94 (dd, J5a-5b = 12,73 Hz, J5b-4 = 8, 13 Hz, 1 H,
Hsb); 3,07 - 3, 1 O (dd, J5a-5b = 12,73 Hz, J5a-4 = 4, 19 Hz, 1 H, Hsa); 3,69 - 3,73
(m, 1 H, H4); 4,00 (t, J = 6,72 Hz, 2 H, Hs); 7,23 - 7,27 (m, 3H, H11, H12 e H1 3);
7,50 - 7,52 (m, 2H, H10 e H14); RMN 13C (300 MHz, CDCl3): õ (ppm) 10,29
(Ca); 21,88 (C1); 30,71 (C3); 31,39 (C2); 36,65 (C4); 66,09 (C6 ); 69,28 (C5);
127,24 (C11, C12 e C13); 129,16 (C10 e C14); 132,91 (C9); 173,74 (C1); IV
{filme):v c=o 1730 cm-1; v e-o-e ass 1258 cm-1; v e-o-e sim 1180 cm-1
3. 7 Resolução cinética do composto (S)-feni/selenolactona.[(S)-7a] e
(R)-fenilselenolactona [(R)-7a].
(R)-7a
C11 H12Se02 pm=255,17 g liquido amarelo
Em um erlenmeyer de 250 ml, foi dissolvido 4-hidroxi-5-
fenilselenilpentanoato de propila (6a) (1,00 g; 3,20 mmol ) em éter seco (50
ml). Em seguida foi adicionado o complexo enzimático PPL [2,00 g - 1 :2
(M/M)] e deixou-se reagir por um período de 75 horas sob agitação a 32 ºe. Ao término das 75 horas, a mistura foi filtrada sendo obtida uma
solução contendo os compostos (R)-7a e (S)-6a. O solvente foi eliminado por
rotaevaporação e os dois compostos separados por cromatografia em
coluna. (R)-fenilselenolactona [(R)-7a)]: Rendimento: 0,28 g (35 %); (S)-4-
hidroxi-5-fenilselenilpentanoato de propila - [(S)-6a)]. Rendimento: 0,38 g
Tabela 9 Rendimento obtido na resolução do (R)-7a.
Solvente Massa do Massa de Rendimento Tempo de substrato (S)-7a reação
(q) {q) (%) (h) Eter 1,00 0,28 35 75 Hexano* 0,96 0,24 31 86

Parte Experimental 53
*O procedimento experimental descrito anteriormente foi realizado de forma
semelhante para o hexano, respeitando-se a mesma proporção de acordo
com a massa do substrato.
D,2 3
7 . Se 6 º,✓-:::: 1 º 4 ······r v8
11l8J9
(R)-7a 10
C11H12Se02 pm=255,17 g líquido amarelo
O composto (5)-6a (0,38 g; 1,21 mmol) obtido na etapa anterior de
separação foi dissolvido em éter seco (20 ml) e em seguida foi adicionado a
lipase CAL [0,38 g -.1: 1 (M/M)] Deixou-se reagir 12 horas sob agitação a 32
ºe. Após esse período, a lipase foi filtrada e o solvente eliminado por
rotaevaporação, sendo obtido o composto (R)-7a. Rendimento: 0,29 g (72
%).
Tabela 1 O Rendimento obtido na resolução do (S)-7a.
Solvente Massa de (S)-6a Massa de (5)-7a Rendimento (g) (g)
(%) Eter 0,38 0,29 72 Hexano* 0,33 0,27 69
*O procedimento experimental descrito anteriormente foi realizado de forma
semelhante para o hexano, respeitando-se a mesma proporção de acordo
com a massa do substrato.
RMN 1H (500 MHz, CDCI3): õ (ppm) 1,92 - 2,00 (m, 1 H, H3b); 2,38 - 2,45
(m, 1 H, H3a); 2,48 - 2,61 (m, 2 H, H2); 2,99 - 3,02 (dd, Jsa-sb = 12,84 Hz, Jsb-4
= 7,99 Hz, 1H, Hsb); 3,27 - 3,30 (dd, Jsa-Sb = 12,84 Hz, Jsa-4 = 4,77 Hz, 1H,
H5a); 4,63 - 4,68 (m, 1 H, H4); 7,23 - 7,27 (m, 3H, H11, H12 e H13); 7,50 - 7,52
(m, 2H, H10 e H14); RMN 13C (300 MHz, CDC'3): õ (ppm) 30,71 (C3); 31,39
(C2); 36,65 (C4); 69,28 (Cs); 127,24 (C11, C12 e C13); 129, 16 (C10 e C14);

Parte Experimental 54
132,91 (Cg); 173,74 (C1); IV (filme): v c=o 1730 cm-1; v e-o-e ass 1258 cm-1; v
C-O-C sim 1180 Cm-1.
3.8 Síntese do composto ditelureto de difenila.
2 3 O-TeTe-0 4
C12H10Te2 pm = 363,91 g sólido avermelhado
Em um balão de três bocas de 3000 ml, provido de agitação
mecânica, funil de adição e condensador de refluxo, foi adicionado magnésio
(15,5 g; 550 mmol) e alguns cristais de iodo em 500 ml de THF anidro.
Em seguida foi adicionada bromobenzeno (13 g; 80 mmol) ao
magnésio. Após consumo do bromobenzeno, observado pela mudança da
coloração, foi iniciada a adição do bromobenzeno restante (65,5g ; 420
mmol) diluído em 100 ml de THF, sendo a velocidade de adição controlada
pelo refluxo do THF.
Após consumo do magnésio, foi adicionado telúrio (66 g; 500 mmol) e
a mistura resfriada a O ºe. Posteriormente foi borbulhado 5 L de 02 durante
um período de 1 hora. Após etapa de oxidação, o banho de gelo foi retirado
e deixou-se sob agitação vigorosa por 1,5 horas. A mistura reacional foi
colocada em um béquer e permaneceu por 12 horas exposta ao ar. Após
esse período foi neutralizada com solução saturada de cloreto de amônio. O
ditelureto de difenila sintetizado foi extraído com acetato de etila (300 ml),
lavado com solução 10% de NaCI (3 X 50 ml), e recristalizado em etanol 95
%. Rendimento:12,47g (13 %).

Parte Experimental 55
3.9 Preparação do composto 4-hidroxi-5-feniltelurilpentanoato de
propila.
11 12010 13~
14 T OH
o 6 8
1 o~ 7
C14H20Te03 pm= 363,91 g líquido avermelhado
Em um balão de duas bocas de 250 ml, provido de agitação
magnética e sob atmosfera de N2, ditelureto de difenila (1,82g; 5,0 mmol) foi
dissolvido em etanol absoluto (25 ml). Foi adicionada a solução pequenas
quantidades (0,3g) de boroidreto de sódio até a solução permanecer
levemente alaranjada, formando telurolato de sódio in situ.
Foi adicionado rapidamente o epóxido 5 (1,90g; 12 mmol) ao
telurolato em solução alcoólica e deixou-se reagir por 1 minutos em um
banho de gelo seco em etanol. O composto 7a foi extraído com acetato de
etila (150 ml), lavado com solução 10% de NaCO3 (3 x 20 ml), separado em
coluna cromatográfica e purificada em placa preparativa. Rendimento: 1,71 g
(47 %).
RMN 1H (500 MHz, CDCl3): 8 (ppm) 0,81 (t, J = 7,45 Hz, 3 H, H8 ); 1,61 (se, J
= 7,37 Hz, 2H, H7); 1,75 - 1,82 (m, 1 H, H3b); 1,89 - 1,94 (m, 1 H, H33 ); 2,39
- 2,51 (m, 2 H, H2); 2,90 - 2,94 (dd, Jsa-sb = 12,73 Hz, Jsb-4 = 8, 13 Hz, 1 H,
Hsb); 3,07 - 3, 1 O (dd, Jsa-5b = 12,73 Hz, Jsa-4 = 4, 19 Hz, 1 H, Hsa); 3,69 - 3,73
(m, 1 H, H4); 4,00 (t, J = 6,72 Hz, 2 H, H5); 7,23 - 7,27 (m, 3H, H11, H12 e H1 3);
7,50 - 7,52 (m, 2H, H10 e H14); RMN 13C (300 MHz, CDCl3): 8 (ppm) 10,29
(Cs); 21,88 (C1) ; 30,71 (C3); 31,39 (C2); 36,65 (C4); 66,09 (C5); 69,28 (C5);
127,24 (C11, C12 e C13); 129,16 (C10 e C14); 132,91 (Cg); 173,74 (C1); IV
(filme):v C=O 1730 Cm-1; V C-0-C ass 1258 Cm-1; V C-0-C sim 1180 Cm-1

Parte Experimental 56
3.10 Resolução cinética do composto (S)-feni/terulolactona.[(S)-7b] e
(R)-fenilterulolactona [(R)-7b].
C11H12Te0 2 pm = 303,81 g líquido vermelho
Em um erlenmeyer de 250 ml, foi dissolvido o composto 4-hidroxi-5-
feniltelurilpentanoato de propila (6b) (0,72 g; 1,98 mmol ) em éter seco (36
ml). Em seguida foi adicionado o complexo enzimático cru PPL [2,26 g - 1 :3
(M/M)] e deixou-se reagir por 21 horas sob agitação a 32 ºe. Ao término deste período, a mistura foi filtrada sendo obtida uma
solução contendo os compostos (R)-7b e (S)-6b. O solvente foi eliminado
por rotaevaporação e os dois compostos separados por cromatografia em
placa prepartiva. (R)-feniltelurolactona - [(R)-7b]:rendimeto O, 19g (53 %);
(R)-4-hidroxi-5-feniltelurilpentanoato de propila [(5)-6b]: rendimento 0,22 g
(54 %)
Tabela 11 Rendimento obtido na resolução do (R)-7b.
Solvente Massa do Massa de Rendimento Tempo de substrato (R)-7b reação
(q) (a) (%) (h) Eter 0,72 0,19 31 23 Hexano* 0,66 0,12 22 21 Ciclohexano* 0,39 0,10 32 12
*O procedimento experimental descrito anteriormente foi realizado de forma
semelhante para cada um dos solventes, respeitando-se as mesmas
proporções de acordo com as massas dos substratos.
2 3
01:)4 "'( T°'rAo7 8 JJ~ 9
10
C 11 H 12Te0 2 pm = 303,81 g líquido vermelho

Parte Experimental 57
O composto (S)- 6b (0,22 g; 0,60 mmol) obtido na etapa anterior de
separação foi dissolvido em um erlenmeyer de 250 ml com éter anidro (11
ml) e a seguir foi adicionado a enzima CAL [0,43 g -1 :2 (M/M)]. Deixou-se
reagir por um período de 12 horas sob agitação a 32 ºe. Após esse período,
a lipase foi separada por filtração e o solvente eliminado por rotaevaporação,
sendo obtido (R)-7b. Rendimento O, 16g (89 %).
Tabela 12 Rendimento obtido na resolução do (S)-7b.
solvente Massa de Massa de Rendimento (S)-6b (S)-7b
(g) (a) (%) Éter 0,22 0,16 89 Hexano* O, 11 0,08 87 Ciclohexano* 0,14 O, 11 92
*O procedimento experimental descrito anteriormente foi realizado de forma
semelhante para cada um dos solventes, respeitando-se as mesmas
proporções de acordo com as massas dos substratos.
RMN 1H (500 MHz, CDCl3): 8 (ppm) 1,92 - 2,00 (m, 1 H, H3b); 2,38 - 2,45
(m, 1 H, H33 ); 2,48 - 2,61 (m, 2 H, H2); 2,99 - 3,02 (dd, J5a-5b = 12,84 Hz, J5b-4
= 7,99 Hz, 1 H, Hsb); 3,27 - 3,30 (dd, J5a-5b = 12,84 Hz, J5a-4 = 4,77 Hz, 1 H,
Hsa); 4,63 - 4,68 (m, 1 H, H4); 7,23 - 7,27 (m, 3H, H11, H12 e H13); 7,50 - 7,52
(m, 2H, H10 e H14); RMN 13C (300 MHz, CDCl3): 8 (ppm) 30,71 (C3); 31,39
(C2); 36,65 (C4); 69,28 (Cs); 127,24 (C11, C12 e C13); 129, 16 (C10 e C14);
132,91 (Cg); 173,74 (C1); IV (filme) : v c=o 1730 cm·1; v c-o-cass 1258 cm·1; v
e-o-e sim 1180 cm·1.

Parte Experimental 58
3.11 Reação de clivagem das {R) e (5)-seleno. e terulolactona.
Benzeno, refluxo
2 3
o~ 5
(S)-7b (R)-8
Em um balão de 2 bocas de 50 ml, provido de agitação magnética,
condensador de refluxo e sob atmosfera de N2, foi adicionada o composto
(5)-7b (21,6 mg; 0,07 mmol), Bu3SnH (58,21 mg; 020 mmol) e AIBN (49,3
mg; 020 mMol) em 1 O ml de tolueno seco. A mistura foi colocada em banho
de óleo a 120 ºe e deixou-se reagir por 2,5 h. Em seguida a mistura foi
injetada no CG com coluna quiral Beta Dextrina 120™ (30m, 025mm,0,25
µm filem fino Supelco) e mediu-se o ee da valera lactona obtida (8).
Esse mesmo procedimento experimental foi aplicado para todas as
seleno e telurolactona preparadas, respeitando as mesmas proporções entre
os reagentes.

Referência Bibliográfica 59/59
"' REFERENCIA , BIBLIOGRAFICA

Bibliografia 59
4. BIBLIOGRAFIA
1. Jones, J. B; Tetrahedron, 42, 3351, 1986.
2. Loughen, W. A.; Biar. Tech , 74, 49, 2000.
3. Faber, K.; "Biotransformations in Organic Chemistry"; 3ª Ed.; Berlim,
Springer Press, 1997
4. Wong, C; Whitesides, G.M.;"Enzymes in Synthetic Organic Chemistry"
12, London, Pergamon Press, 1995
5. Roche Molecular Biochemicals, Industrial Biochemicals; "Biocatalysts for
lndustry; 2000
6. Chen; C; Sih C.J .; Angew. Chem . lnt. Ed. Eng!.; 28, 695, 1989.
7. Koskinem, A M. P.; Klibanov, A M; "Enzymatic Reactions in Organic
Media'; 1ª Ed. ; New York; Chapman & Hall Press, 1996
8. Lundell, K. ; Raijola,T.;Kanerva, L.; Enzyme Microb. Technol, 22, 86,
1998.
9. Ohtani, K.;Nakatsukasa, H; Kamezawa,M; Tachibana,H; Naoshima,Y.; J.
Mo/. Catalysis B: Enzymatic; 4. 53, 1998.
1 O. Vanderdeen, H; et ai; J. Amer. Chem. Soe, 118, 3801, 1996.
11. Enzelberger, M M; Bornscheuer, U T; Gatfield, I; Schmid, R D; J.
Biotechnology; 56, 129, 1997.
12. Shioji, K.; Matsuo,A; Okuma, K.; Nakamura, K; Ohno, A.; Tetrahedron
Lett, 41, 8799, 2000.
13. Sharma, A.; Chattopadhyay, S.; J. Mo/. Catalysis B: Enzymatic; 1 O. 531,
2000.
14. Roberts, S M; Turner, N J.; J. Biotechnol; 22; 227; 1992
15. Kijima, T.; Moriya, T.; Kondoh, E.; lzumi, T; Tetrahedron Lett, 41, 2125,
2000.
16.Adam,W; Groer, P.; Saha-Moller, G. R.;Sharma, Tetrahedron Assymm,
11, 2239, 2000.
17. ltoh , T; Takagi, Y; Tsukube, H.; J. Mo/. Catalysis B: Enzymatic; 3. 259,
1997.
18.Wirth, T.; Tetrahedron, 1, 55, 1999.

Bibliografia
19. Back, G. T.; Dyck, P. B.; Chem. Commun., 2567, 1996.
20. Wirth, T., Liebiigs Ann , 2189, 1997.
21. Moura Campos, M;Petragnani, N; Tetrahedron Lett., 11 ; 1959
22.ComassetoJ.V, Petragnani, N., Synth. Commun., 13,889, 1983
23. Haller, S. W.; lrgolic.; J. Organomatel Chem.; 38, 97, 1972.
60
24. Petragnani, "Tellurium in Organic Synthesis", Academic Press, London,
1994
25. Back, T. G.; " Organoseleniun Chemistry" Oxford University Press,
Oxford , 1999.
26. Clive, D.L.J. , et all; J Am. Chem. Soe., 102:13, 4438, 1980.
27. Nicolau, K. C.; Seitz, S.P.; Sipio, W.J. ;Blount, J.F.; J Am. Chem. Soe.,,
101:14,3884, 1974.
28. Hu , N. X.; Aso, Y; Otsubo, T.;J. Org. Chem., 54, 4391, 1989.
29.Satoh , T; Chem. Lett, 1949, 1987.
30. Toru , T; et all ; J Am. Chem. Soe.,, 11 O, 4815, 1990.
31. Comasseto, J.V.; Ferraz, H.M.C; Brandt, C.A; Petragnani , N; Tetrahedron
Lett,; 28, 5611, 1987.
32. Missio, J. L.; Comasseto, J. V.; Tetrahedron asymm, 11, 4609, 2000.
33. Hoye,et ai; J. Org. Chem.,59, 4096, 1994.
34. Missio, L.J.; "Síntese de Butirolactonas substituídas"; Tese de Doutorado;
UFSCAR, São Carlos, 2000.
35. Duddeck, H; Wagner; BiallaB, A, P; Magnetie Resonance in Chemistry,
29,248, 1991
36. Duddeck, BiallaB, A, P; Magnetie Resonanee in Chemistry, 32, 303, 1994
37.Mori, K; Tetrahedron; 31, 3011 , 1975; (b) Ho, P. T.; Synthesis, 462, 1983.

Apêndice 61/61
"' APENDICE
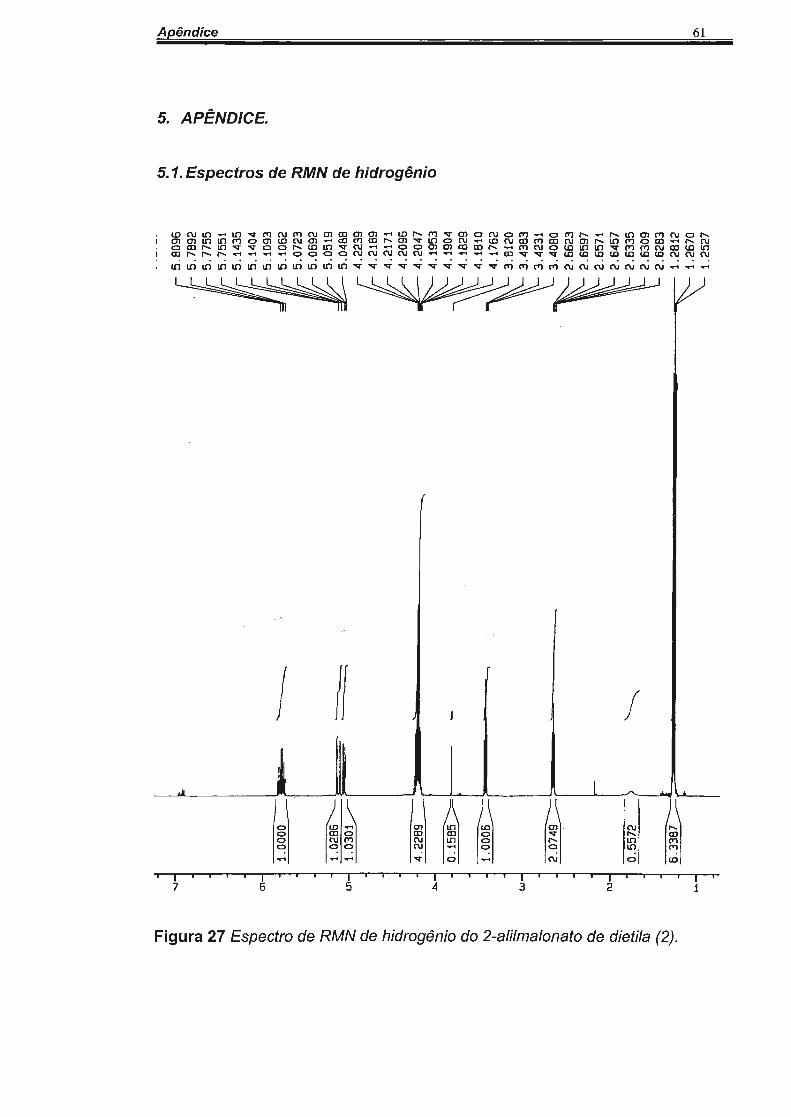
cri
1.00
00
4. 2
289 ~--
-~ ~-
------
---.
..!..:
Q§_
_
w
~>
----
----
'=~
----~
0.55
72
..... ~!
:==-
------
------
------
------
---~
~ 5.80
96
5.78
92
5.7
755
5.7
551
5 .1
435
5 .1
404
5 .1
093
5.10
62
5.0
723
, 5.0
692
5.0
519
5.0
488
4.22
39
4. 2
189
4.2
171
4.2
096
4.2
047
4.1
953
4.19
04
4 .1
829
: : !~!~
3.81
20
3.4
383
3.4
231
3.4
080
2.6
623
2.65
97
2.6
571
2.64
57
2.6
335
2.6
309
2.6
283
1.28
12
1.2
670
1.2
527
(Ih :::i
0.. Õ'
(1)
O\

"T1
)::,,
5.8
628
(1),
e.e
5.84
21
:::i
e:
0..
., 5
.828
8 Õ'
ti
)
(1)
"' 5.
8081
0
0
5.10
41
w
5. 1
011
"O
(1)
01
5.0
979
e,
~
5.0
947
........ c3 Q.
5 .0
668
(1) ::ti
5.06
36
5.0
604
~ ~
5.0
411
<.1l ~
5. 0
388
Q.
(1)
5.0
360
:::r- -. Q. c3
5.0
333
· 5
.020
9 (Q
CD
, 5
.018
3 :J
5.01
54
Õ'
b.
Q.
I]fi
l>=
~
5.01
28
o 3
. 810
5 -i::
:..
2.4
832
-6
(1)
2.4
800
:J
........
(1)
2.4
690
:J
t,J
2.4
665
O, n·
o 2
.453
1 w
~
~~
2.4
515
2.4
115
~
2.40
93
.._
l'l.l
2.4
068
2.39
37
2.3
801
2.3
671
O\
2.3
643
N

.,, (O
5.8
546
)::.
e: 5.
8341
(1
), ..,
::,
Q)
5.8
328
Q., -·
"->
5.82
05
o
(O
(1)
5.80
00
w
5.0
802
1:::)
0
1
5 .0
775
<D
o 0.
3333
5
.076
9 ~
o 5
.046
0
fü-5.
0426
::o ~
~5
01
34
s::
5.0
110
U1
~
<
5.0
099
fü-4.
9929
4
. 989
5 ::J
- ~
§-4.
0530
4
. 039
6 o
~
CQ
.I>
. ~
4.02
62
<D, ~
~
3.8
093
:::i Õ" ~
~
3.60
72
~
--o..
2.
4117
o
2.4
098
~
-6
2.4
011
<D
w
2.39
86
:::i
2.39
66
......
<D
2.3
944
:::i o
1.44
71
2.38
87
Q) ......
2.38
63
o o..
2.38
37
<D
ru
1.67
04
1:::)
~
1.65
70
c3 1:::)
~
1.6
556
~
1.64
36
1.64
22
~
'--'
.....
~
1.62
87
~
1.62
73
0. 9
575
O\
0.94
26
w
0.9
406
0.92
78

"T1
)::.,
(C
e: .., [J
1
C)
(,.)
o
w
1:::1 <D
C
') ~
.....
.Is..
c3 ~ ..
.
CD,
::,
0..
13 99
400
Õ'
CD
3.98
057
3.9
6713
2
.923
60
2.91
446
a..
2.69
995
<D :o
ã(D
~
(1)
-;ã
m
a..
w ~
·-F
<D ~
d_
:::i-
~
~o
õ.:
'"
c3 0.
5117
:P
o
-1
(Q
=
s.m
CD
, ~
3,::
o ~
Õ'
~>
a..
IU
.o
:scm
---o
0.54
52--
---
~
1
~
o x -s·
<D ~ cu
.... ~
~ ~
o Q)
1 L..
. l r
~
....
2.69
123
2.69
015
2.6
8213
!2.4444
' 2
.439
08
2.43
451
2. 4
2914
~2
.408
25
2.3
9252
=
"---
2.3
78
41
t!.8
90
83
1.
7413
9 1.
7276
1 1
.713
54
1.60
648
1.59
280
.....
o 1.
578
07
a..
<D
1.56
442
1:::1
1.54
980
c3 1:::1
o 0
.888
61
~
0.8
7376
- ~ 0
.858
86
°' ~

a.. "
T1
<D
-·
"O 'ê
):
.
a ..,
"O
D)
(!),
::::.:
w
:::,
Q) ~
(D
S: C')
(!)
Õ>
rn ~~
1.98
90
<D
3.1
095
C') .....
. a .....
fü- :::o ~
01
a..
<D
::J--
4. 0
3279
a: a 4
.010
35
(Q
3.9
8794
3
. 708
58
<D,
:::s õ·
... ~
g. 3
.117
14
.!..:.
.._
3 .0
8826
3
.074
66
~
1
1-'·""'
::J--
2. 9
1993
a: w
~:
~~~~
2.
904
87
a j::;;~
><:
,·
2.6
926
2 .4
7075
o,
1
2.4
6683
CD'
"'
2.4
6363
:::s
~
2.4
4651
~
-~
~2
.440
88
<D
~
1::~
ci5'"
:::s
1.8
24
95
-s ~
1.8
02
19
1.
796
20
<D
1''""
:::s cu
1.6
31
67
1
.607
09
:::s
t.5
84
28
o
o 0
.941
00
Q) õ
O. 9
1639
0
.89
15
2
O\
V,
::!!
3 7
.556
82
):,,
(1),
7.55
380
:J
7.5
5099
S:
7
.548
46
e.e
r::
""'I
Q)
(")
(1)
7.5
4574
7
.543
94
w
CD
"' 7
.542
33
T.5
4042
7
.537
82
n, ~
,, 2
9'69
C/)
~
7.2
9155
-o
7.2
8824
(1)
7.2
8531
C") .......
-..J
7.2
8311
c3
7.2
7892
o.
7.26
004
(1) ::o
s:: a
, <
o.
(1
) :::r-
j_' 6652'
4.
6557
0 4.
6504
6
Q
4.64
089
c3 U
l
--j; ::::
(Q
(1),
~
3.2
9688
:::i
3.28
073
o·
3.27
119
fü-
-!!}""'
....
3.0
1679
C/)
~
3.00
708
(1) (D
2.9
9111
:::i
~
2.57
919
o
2.56
900
m ~
C")
w
....... o :::i
a.,
2.5
5984
2
.552
32
2.5
4985
2
.534
21
-'-J ~ "'
2.5
159
9 2.
4985
1
12,M
SB
2.
4194
9 2
.405
82
!. 51
83
1. 9
7774
O
\ 1
, 962
88
O\
1. 9
5210
1.
6007
0

Apêndice 67
5.2. Espectros de RMN de carbono
lD lD CI) .... r-- (TJ (TJ r-- o C\J .... CT") o C\J lD ..... ..... ..... l!1 o ~
.... l!1 CT") o CI) .... r-- CI) .... CI) .... r-- r-- r-- LO .... ..... C\J .... LO CT") .... r-- r-- r-- lD lD (TJ .... ..... .... ....
. 1 1 1 ~ 1 1 1
1 1 1 1
175 150 125 100 75 50 25
Figura 33 Espectro de RMN de carbono do 2-alilmalonato de dietila (2).
P.pena,ce 68
..,. (\J 01 ru ro ...- LC1 (X) LC1 (\J LC1 01 CTl ai LC1 ..... ..,. cri r-- (\J o r-- cri LC1
01 <D lCl r-- r-- <D (T) ai ,-.... ~ ..... r-- ,-.... " (T) (\J - .....
1 1 ~ 1 1
1 1 1 1
'º 175 150 125 100 75 50 25
Figura 34 Espectro de RMN de carbono do 4-pentenóico (3).

·(p) e11do1d ep oJeoueJued-p op ouoq;e~ ep NWcJ ep 01peds3 SE eJn61~
oç; ç;L Çicl
1
1
1
1 1 1
1 1 1 1 (1\ 1 ,-.... ..... ..... .... I\J I\J w CJ') .................. .... w ......
o I\J O) w CJ') CJ') ............ U1 cn w .... o lO cn o O) o w .... ...... fU o w m .t>. I\J ..... cn I\J .... O) o o m W IU .... w ...... -.... fU o
69

o 1
~
1 -o
--..J l"\J
OL
·(ç) e11do1d ep 01eoue1ued1xo-v op ouoq1ea ep NWc:J ep 01peds3 9E eJn61:1
OOJ OSJ ' 1 1 , 1 1
1
wr 1
1 1\ 1 1 1 (1\ /A\ ,_,. ,_,. ,_,. ,_,. ,_,. 1\) I'\) w A U1 Cl'l -..J -..J -..J ru ru I'\) w -..J ..... -..J o m
-Cl'l m -....i -....i -..J lO lO W l"\J
m lJl ru m o o -..J o ru IO lJl lO O -..J w w m
--..J o A O Ul -..J. CD I'\) 01 w
--lD A 1\) CD CD ru CD U1 ru -..J lO .b.

Apêndice
LO ..._ <Jl CD C'1 - líl [T') r-- Q') .... C\I
C'1 C\I Q') ..... ..... [T') C\I C\I - ---1 \ 1/
,
. -· ~ -~
1 1 1 i 1 1 1 1 ' 1
175 150 125 1 1 1
100
CD C'1 ..., r-- <Jl C\IOCDt--<Jl .... oinruo . ... . r-- r-- I.O O') I.O ..... r-- ..... I.O I.O
~
1 75
1/
1
1 50
1
<r1 -,q- .... co lílOl-r--U)[T')r--(I)
C.0.-tQ.-t C'1 C'l [T') C\I
1 \/
1
1 25
1
71
Figura 37 Espectro de RMN de carbono do 4 -hidroxi-5-fenilse/enilpentanoato de propila (6a).
o Q') C\I
o -

·(eL) euope1oue1es ep ouoq1e~ ep NJ/Vc:J ep 01peds3 8f: eJn61:1
!õH • 1
... ,, .. ,., ,. . _,
., • .., 1 .. l
/1 \ \ !!\ \ . 1
-.......... ~ - 1\) 1\) w -.J -.J -.J -.J 1\) 1\) 1\) w -.J -.J CD ,_.. 01 -.J -.J 1D -.J CD 1D W 01
m -.J 1D -.JOI\JW -.JCDWW [J1 CD Ol lJl -.J W CD cn l\lbCDO o __., [J1 __., 1D b -.J -.J 1D W b 1D w
u

Apêndice 73
5.3. Espectros no Infra-vermelho
1P
6
5p
~p ~---.------.-------,----------.------,----~-- ----1 3500 3000 2500 2000 1500 1000 500
Figura 39 Espectro no Infra-Vermelho do 2-alilmalonato de dietila (2).
100
90
80
70
60
50
3500 3000 2500 2000 1500 1000 500
Figura 40 Espectro no Infra-Vermelho do 4-pentenóico (3).
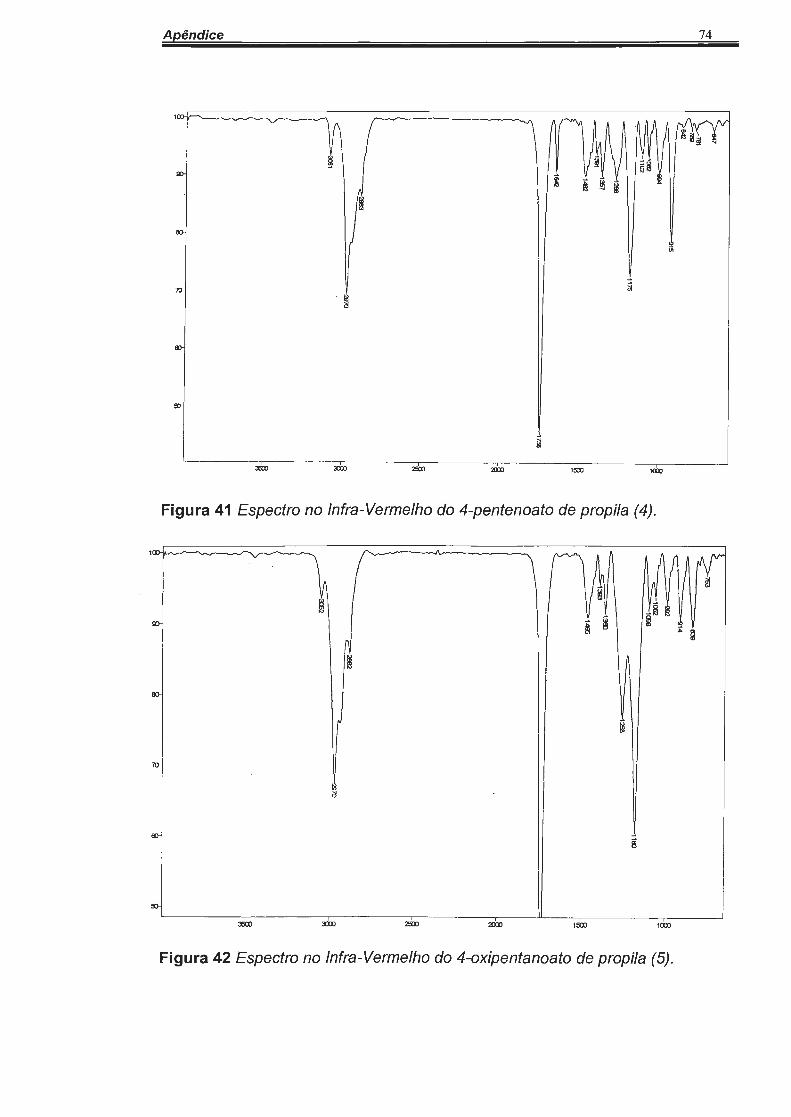
Apêndice 74
2500 1500 100'.l
Figura 41 Espectro no Infra-Vermelho do 4-pentenoato de propila (4).
2500 = 1500 1CXX)
Figura 42 Espectro no Infra-Vermelho do 4-oxipentanoato de propila (5).

Apêndice 75
3500 1500 100J
Figura 43 Espectro no Infra-Vermelho do 4-hidroxi-5-fenilselenilpentanoato de propila (6a) .
5.4. Espectros de Massa
Sca.n 154 (7,297 rnirí ) of DATA : !G?.D 1. 0[5
8 . HE4 il.l 55 0 G.0E4 / e
,-::J ü .:i 4. 0[4 e i 2.0[4. iJ
0 ' llLJL.-.i
' 71
__ ..,...,.-. 9
50 80 10(21 12~1 140 Ma..ss / Charge,
Scan 1 (7 .297 Min of DATA:1 .O
18 0
Figura 44 Espectro de Massa do 4-pentenoato de propila (4).
180 200
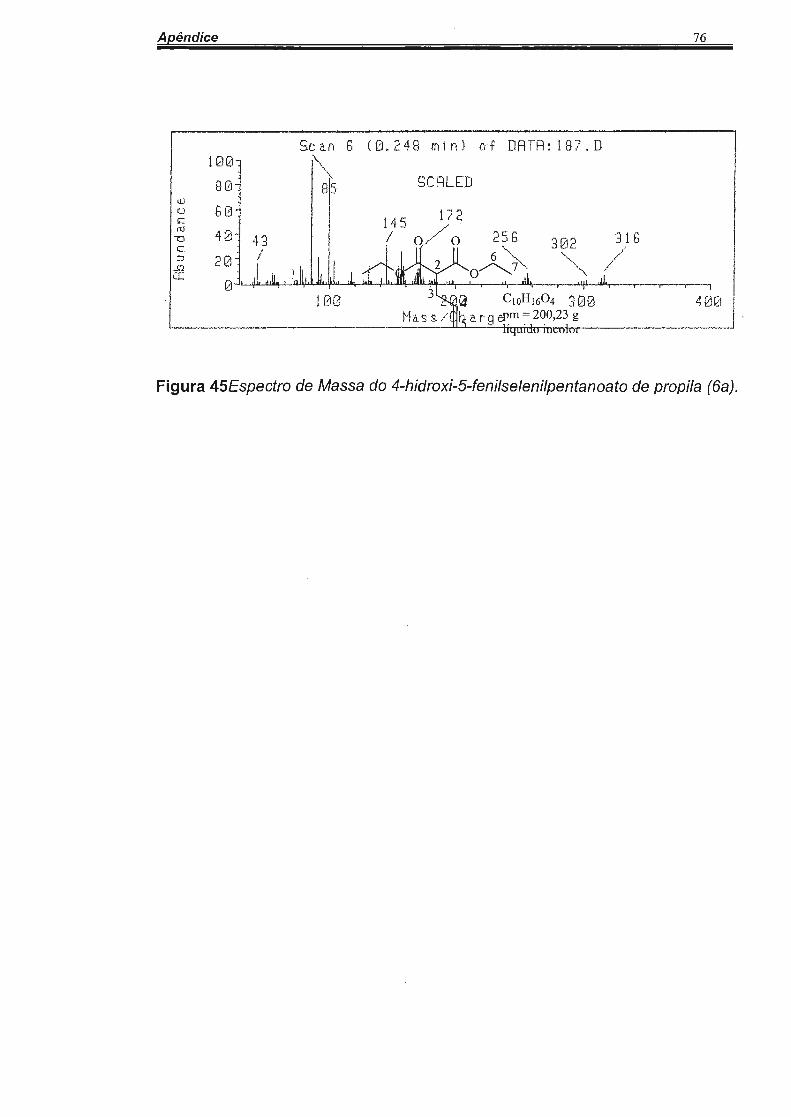
Apêndice 76
Se an 6 (0.248 min) of DRTR:187.D 1 (l0
80 "'-,.,,
85 SCRLED ü)
u 80 e ;rj
40 tl 43 e :::, 20 _i.,
a:: 0 ' 1 1 1 1
3 ~ --:1@ C10H1604 3 00 4DD Ma.ss / t arqepm=200,23g
'-----------------'-~-~d:rnt1· rcotinr---------'
Figura 45Espectro de Massa do 4-hidroxi-5-fenilselenilpentanoato de propila (6a).





























