Embed Size (px)
Citation preview

Química Biológica
Objectivo
Aplicação de uma técnica de separação de moléculas através de uma membrana semi-
permeável.
Introdução
A diálise é utilizada para separar moléculas grandes e pequenas e depende do facto de
uma membrana semi-permeável permitir que as pequenas moléculas passem através dela mas
impedir a passagem de grandes moléculas. Na prática, uma mistura de moléculas grandes e
pequenas é colocada num saco de diálise imerso num grande volume de um solvente aquoso.
As pequenas moléculas passam através da membrana para o fluido externo até que o
equilíbrio seja atingido (ver figura).
A mistura pode ser virtualmente “limpa” de pequenas moléculas por meio de uma
diálise contra água corrente ou por sucessivas trocas do solvente. A taxa de diálise depende de
vários factores como:
A qualidade da membrana.
A permeabilidade da membrana (existem membranas com limites de
permeabilidade bem definidos).
A natureza do solvente (de um modo geral a taxa de diálise é maior em água,
embora seja necessária uma solução aquosa com pH e força iónica definidos
para estabilizar as moléculas em estudo).
Temperatura.
Pressão.
Diálise

Química Biológica
A experiência que irá ser efectuada pretende demonstrar a passagem de moléculas
através de uma membrana de diálise. Nesta experiência uma mistura de uma proteína (por
exemplo, caseína ou albumina), um aminoácido e cloreto de sódio será submetida a diálise. O
aminoácido e o cloreto de sódio atravessarão facilmente a membrana, o mesmo não
acontecendo à proteína.
São três as reacções utilizadas no trabalho experimental para a detecção da albumina, do
aminoácido e dos iões cloreto:
Teste do biureto. Este teste permite detectar a presença de ligações peptídicas, podendo
ser utilizado na determinação da concentração de proteínas em solução. O sulfato de cobre
alcalino reage com compostos com ligações peptídicas originando um complexo violeta.
Teste da ninidrina. É uma reacção característica da função amina primária e é usada na
detecção de aminoácidos. A ninidrina é um reagente oxidante que reage com todos os α-
aminoácidos entre pH 4 e 8, originando um composto de cor púrpura.
Teste do nitrato de prata. Quando a uma solução de cloreto de sódio se adiciona uma
solução de nitrato de prata, observa-se a formação de um precipitado branco de cloreto de
prata.
Material e Reagentes
Manga de diálise (previamente fervida numa solução aquosa de EDTA); solução de
NaCl 5 M; solução de uma proteína (caseína ou albumina) a 1%; solução de um aminoácido
(por exemplo, glicina) a 1%; solução de nitrato de prata 0.5M; reagente do biureto.
Procedimento
1. Prepare 2 sacos de diálise dando um nó numa extremidade de cada um de modo a
ficarem “hermeticamente” fechados. No saco A introduza 10 mL da solução de
albumina e 1 mL da solução de cloreto de sódio. No outro saco (B) introduza 10 mL
da solução de albumina e 5 mL de solução de aminoácido.
2. Expulsar tanto quanto possível o ar dos sacos e atar a outra extremidade de cada um
deles à semelhança.
3. Coloque cada um dos sacos (A e B) em copos de 150 mL, contendo cerca de 80 mL de
água destilada. Os sacos têm que ficar totalmente imersos. O conteúdo dos copos
deverá ser permanentemente agitado.
4. Ao fim de 15 minutos retire duas amostras de 2 mL, do líquido exterior aos sacos A e
também duas amostras de 2mL do líquido exterior ao copo B. Uma das amostras de
cada copo (A e B) vai ser usada para testar a presença de proteína. A outra amostra
deverá ser usada para testar a presença de cloreto de sódio (copos A) e aminoácido
(copo B).
5. Espere mais 30 minutos e repita o passo anterior.
6. Abra os 2 sacos de diálise e retire de cada um deles duas amostras de 2 mL para
verificar do mesmo modo a presença de iões cloreto (saco A), do aminoácido (saco B)
e de proteína (sacos A e B). (NOTA: Antes de retirar as amostras dos sacos de diálise
agite-os bem).

Química Biológica
7. Efectue também os testes para os iões cloreto, para o aminoácido e para a proteína a
amostras de 2 mL de água (controlos negativos), e a três tubos contendo
respectivamente cloreto de sódio (teste aos iões cloreto), solução de aminoácido (teste
da ninidrina) e albumina (teste à proteína) – controlos positivos.
Pesquise a presença de iões cloreto adicionando a cada tubo de ensaio preparado para esse
fim, algumas gotas da solução de nitrato de prata. O aparecimento de uma turvação ou de um
precipitado branco indica a existência de iões cloreto.
Pesquise a presença de aminoácido adicionando a cada tubo de ensaio preparado para o efeito,
algumas gotas da solução de ninidrina. Ferva os tubos de ensaio num banho de água durante
cerca de 2 minutos. O aparecimento de uma cor púrpura é indicativo da presença de
aminoácidos.
Pesquise a presença de albumina realizando a reacção do biureto, que consiste em adicionar a
cada tubo de ensaio preparado para esse fim, 2 mL de reagente do biureto. O aparecimento de
uma cor violeta é indicativo da presença de albumina.
Tratamento de Resultados
1. Registe os resultados obtidos.
2. Que conclusões pode tirar desses resultados?
Bibliografia
D. J. Plummer, An Introduction to Pratical Biochemistry, 2nd ed.; McGraw-Hill: London,
1978; Cap. 3.
Preparação da aula prática
A diálise é utilizada para separar moléculas grandes de moléculas pequenas. Indique
vantagens e desvantagens deste método relativamente a outros que lhe permitam fazer o
mesmo tipo de separação.

Química Biológica
Objectivo
Estudo de reacções coradas de aminoácidos e proteínas. Identificação da composição de
uma solução desconhecida. Estudo de agentes desnaturantes de proteínas: calor e precipitação
por agentes acídicos.
Introdução
As proteínas são polipéptidos constituídos por 50 ou mais resíduos de L- aminoácidos.
Estas macromoléculas são constituintes fundamentais da célula onde desempenham funções
diversas: umas funcionam como enzimas e são particularmente importantes, pois são
responsáveis pela catálise da maior parte das reacções celulares; outras (as hormonas)
controlam os processos metabólicos; outras ainda (os anticorpos) ajudam na protecção do
organismo e algumas (como a hemoglobina e as lipoproteínas) efectuam o transporte de
compostos no sangue.
Uma vez que as proteínas possuem vários grupos ionizáveis, são electrólitos. São
também anfotéricas pois apresentam propriedades de ácido e de base fracas.
A solubilidade da maior parte das proteínas em água deve-se às interacções que se
estabelecem entre os grupos ionizados e/ou polares presentes na superfície das moléculas
proteicas e as moléculas de água. Os agentes que alteram a constante dieléctrica ou a força
iónica de uma solução aquosa influenciam portanto a solubilidade destas moléculas.
Geralmente ocorre precipitação quando as forças atractivas entre as diferentes moléculas de
proteína (forças de coesão) excedem as forças de atracção proteína-água.
Neste trabalho pretende-se observar algumas reacções coradas características de certos
grupos químicos presentes nos aminoácidos e ainda a reacção do biureto, que é típica de
compostos com ligações peptídicas.
Os -aminoácidos (ver Figura) apresentam todas as reacções químicas características de
compostos que contêm grupos amina e carboxilo. De entre estas destaca-se a reacção da
ninidrina, característica da função amina primária e muito usada na detecção de aminoácidos.
A ninidrina é um agente oxidante poderoso que reage com todos os -aminoácidos entre pH 4
e 8, originando um composto de cor púrpura.
H3N COO
R H
Caracterização de Aminoácidos e Proteínas

Química Biológica
Cada tipo de aminoácido pode apresentar ainda reacções típicas de grupos químicos
presentes nas suas cadeias laterais (grupo R). Estas reacções podem ser usadas como testes
qualitativos para detectar especificamente a presença de alguns destes compostos.
Por exemplo, a reacção de Vogel é característica de grupos R tiólicos. Após o
aquecimento dos compostos em meio básico, o enxofre presente na cadeia lateral do
aminoácido origina sulfureto de sódio. Com a adição de acetato de chumbo à solução forma-
se sulfureto de chumbo, que precipita (precipitado escuro).
A reacção xantoproteica é característica de grupos R benzénicos. Os aminoácidos que
contêm um grupo aromático na sua estrutura formam derivados nitrosos amarelos, quando
aquecidos com ácido nítrico concentrado. Os sais destes derivados são côr de laranja.
A reacção do biureto detecta a presença de ligações peptídicas, podendo ser utilizada na
determinação da concentração de proteínas em solução. O sulfato de cobre alcalino reage com
compostos com ligações peptídicas, originando um complexo violeta.
Neste trabalho pretende-se ainda estudar a acção de alguns agentes desnaturantes de
proteínas: calor e adição de reagentes acídicos ao meio.
Quando as proteínas se encontram sob a sua forma natural activa, designam-se por
proteínas nativas. A desnaturação de uma proteína corresponde à perda da sua estrutura nativa
devido à acção de agentes vários (calor, ácidos ou bases, solventes, oxidação, redução,
detergentes, agitação, etc.). Este processo pode ocorrer com uma extensão variável. Como a
função de uma proteína depende das suas características conformacionais específicas, a sua
alteração profunda por desnaturação provoca a perda da sua actividade.
Material e Reagentes
Reacção da ninidrina: Solução de clara de ovo a 5% (v/v); solução de glicina a 1%;
solução de tirosina a 1%; solução de cisteína a 1%; solução de um aminoácido desconhecido a
1%; solução de ninidrina a 1%.
Reacção de Voguel: Solução de clara de ovo a 5% (v/v); solução de glicina a 1%;
solução de tirosina a 1%; solução saturada de cisteína; solução de um aminoácido
desconhecido 1%; solução de NaOH a 40% (p/v); solução saturada de acetato de chumbo.
Reacção xantoproteica: Solução de clara de ovo a 5% (v/v); solução de glicina a 1%;
solução de tirosina a 1%; solução de cisteína a 1%; solução de um aminoácido desconhecido
1%; ácido nítrico concentrado; solução de NaOH a 40% (p/v).
Reacção do biureto: Solução de clara de ovo a 5% (v/v); solução de glicina a 1%;
solução de tirosina a 1%; solução de cisteína a 1%; solução de um aminoácido desconhecido
1%; Reagente do Biureto.
Procedimento
Reacção da ninidrina
1. Prepare 6 tubos de ensaio. Cinco deles irão conter respectivamente 2 mL de solução
de: glicina, tirosina, cisteína, solução desconhecida e água destilada. O sexto tubo
deverá conter 3 mL da solução de clara de ovo.
2. Adicione 4 gotas da solução de ninidrina aos cinco primeiros tubos de ensaio e 3 mL
da mesma solução ao sexto tubo.

Química Biológica
3. Ferva em simultâneo os vários tubos de ensaio, durante cerca de dois minutos.
4. Registe o que observa.
Reacção de Vogel
1. Prepare cinco tubos de ensaio contendo 2 mL de cada uma das soluções a testar
(glicina, tirosina, cisteína, solução desconhecida e clara de ovo) e um de controle (com
2 mL de água).
2. Adicione a cada um 2 mL de NaOH a 40%.
3. Aqueça os tubos de ensaio em banho-maria durante 2 minutos e seguidamente
adicione a todos os tubos algumas gotas da solução de acetato de chumbo.
4. Registe as alterações observadas.
Reacção xantoproteica
1. Prepare cinco tubos de ensaio contendo 2 mL de cada uma das soluções a testar
(glicina, tirosina, cisteína, solução desconhecida e clara de ovo) e um de controle (com
2 mL de água).
2. Adicione a todos os tubos 0.5 mL de ácido nítrico concentrado.
3. Aqueça os tubos de ensaio em banho-maria durante 2 minutos e observe a alteração de
cor.
4. Arrefeça os tubos à torneira e alcalinize com a solução de NaOH a 40%.
5. Registe a cor observada no final em todos os tubos.
Reacção do biureto
1. Prepare cinco tubos de ensaio contendo 2 mL de cada uma das soluções a testar
(glicina, tirosina, cisteína, solução desconhecida e clara de ovo) e um de controle (com
2 mL de água).
2. Num erlenmeyer prepare uma solução contendo 18 gotas de sulfato de cobre e 15 mL
da solução de NaOH a 10% . Esta solução constitui o reagente do Biureto. Se já
estiver preparado, utilize o reagente já fornecido.
3. Junte 2 mL desta solução a cada um dos tubos de ensaio preparados anteriormente.
4. Registe o que observa.
Agentes desnaturantes de proteínas
Desnaturação pelo calor
1. Coloque num tubo de ensaio cerca de 3 mL da solução de clara de ovo.
2. Aqueça o tubo de ensaio num banho fervente durante alguns minutos.
3. Registe o que observa.
Desnaturação por agentes acídicos
1. Coloque 1 mL da solução de clara de ovo em três tubos de ensaio.
2. Ao tubo nº 1 adicione 1 mL de ácido tricloroacético (TCA) a 10%.
3. Ao tubo nº 2, adicione 1 mL de ácido perclórico a 10%.
4. Ao tubo nº 3 adicione 1 mL de ácido metafosfórico a 10%.

Química Biológica
5. Registe o que observa.
Tratamento de Resultados
1. Tire conclusões dos resultados obtidos.
2. Com base nestes resultados e tendo em conta que a solução desconhecida de
aminoácido contém um dos aminoácidos utilizados, identifique esse aminoácido.
Bibliografia
J. M. Clarck, R. L. Switzer, Experimental Biochemistry, 2nd ed.; W.H. Freeman, 1977.
D. T. Plummer, An Introduction to Practical Biochemistry, 2nd ed.; McGraw-Hill, 1978.
R. Scopes, Protein Purification. Principles and Practice, 1st ed.; Springer-Verlag: New York,
1982.
Preparação da aula prática
Apresente os esquemas das reacções referentes aos testes da ninidrina e do biureto.
Apresente as estruturas químicas dos aminoácidos: glicina, alanina, cisteína, ácido glutâmico,
fenilalanina, tirosina e triptofano. Explique o mecanismo da desnaturação das proteínas
provocado pelo calor.

Química Biológica
Objectivo
Doseamento das proteínas da clara do ovo por um método espectrofotométrico.
Introdução
Neste trabalho ião ser quantificadas espectrofotometricamente as proteína da clara do
ovo usando o método do Biureto.
Método do Biureto
A cor púrpura da reacção do biureto é devida à complexação do Cu2+
(em solução
alcalina) pelas ligações peptídicas das proteínas. A reacção ocorre com compostos que
contenham duas ou mais ligações peptídicas. Este método tem uma sensibilidade de 1-10
mg/mL de proteína.
Material e Reagentes
Espectrofotómetro UV-Vis; clara de ovo; solução padrão de albumina de soro bovino
(BSA) a 3.0 mg/mL,
O reagente do biureto foi preparado da seguinte forma: juntar 6.0g de tartarato de sódio
e potássio tetrahidratado em 500 mL de água destilada; adicionar 1.5 g de sulfato de cobre
pentahidratado; adicionar lentamente e com agitação 300 mL de NaOH a 10% (p/v); diluir
para 1 L com água destilada. O reagente é estável, devendo ser guardado em frascos de
polietileno.
Procedimento
Preparação da amostra desconhecida
1. Filtre uma clara de ovo através de uma camada dupla de gaze para um copo de 100
mL. Comprima suavemente a gaze contra a parede do copo sem forçar. O material que
passa através da gaze é o filtrado.
2. Dilua 5 mL de filtrado num balão de 50 mL com água destilada e identifique-o. A
concentração proteica desta solução irá ser doseada pelo método do Biureto.
Método do Biureto
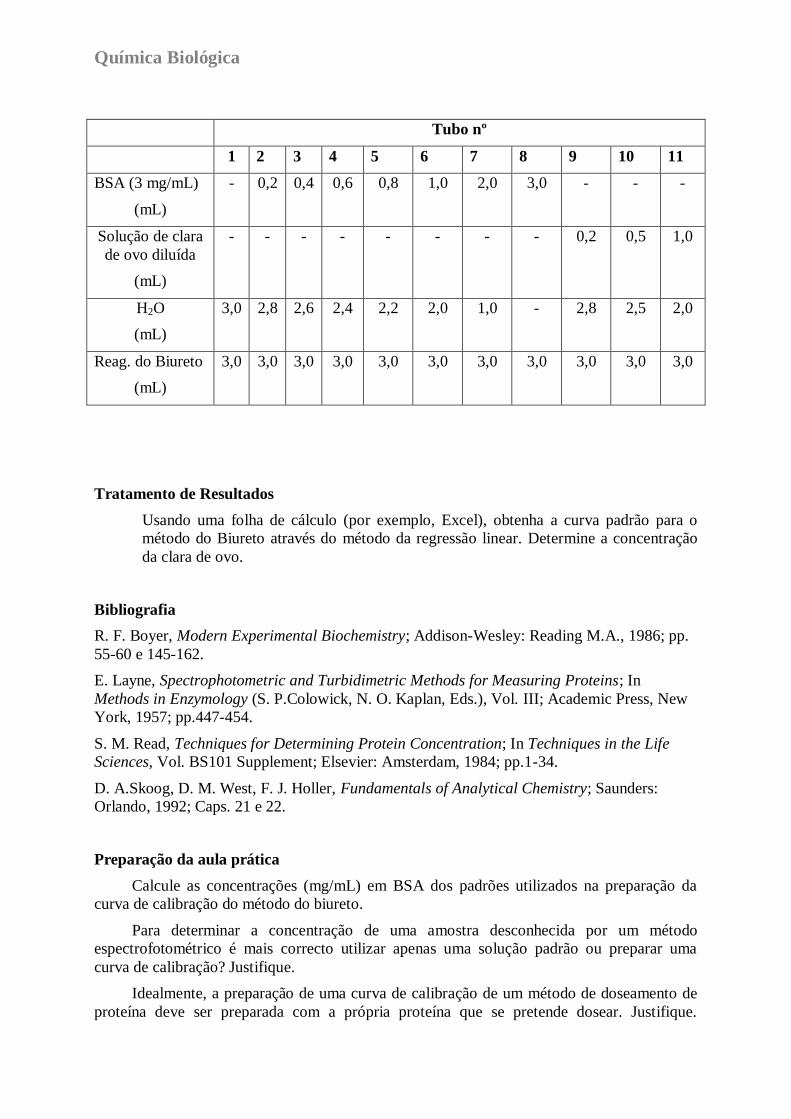
3. Prepare 11 tubos de ensaio de acordo com o quadro que se segue.
4. Misture o conteúdo dos tubos e deixe à temperatura ambiente durante 30 minutos.
5. Leia a absorvência de cada amostra a 540 nm, ajustando o zero do espectrofotómetro
com o conteúdo do tubo 1 (a cor é estável até 1-2 h após a incubação, mas depois
aumenta ao longo do tempo).
Métodos Espectrofotométricos para o Doseamento de Proteínas
Química Biológica
Tubo nº
1 2 3 4 5 6 7 8 9 10 11
BSA (3 mg/mL)
(mL)
- 0,2 0,4 0,6 0,8 1,0 2,0 3,0 - - -
Solução de clara
de ovo diluída
(mL)
- - - - - - - - 0,2 0,5 1,0
H2O
(mL)
3,0 2,8 2,6 2,4 2,2 2,0 1,0 - 2,8 2,5 2,0
Reag. do Biureto
(mL)
3,0 3,0 3,0 3,0 3,0 3,0 3,0 3,0 3,0 3,0 3,0
Tratamento de Resultados
Usando uma folha de cálculo (por exemplo, Excel), obtenha a curva padrão para o
método do Biureto através do método da regressão linear. Determine a concentração
da clara de ovo.
Bibliografia
R. F. Boyer, Modern Experimental Biochemistry; Addison-Wesley: Reading M.A., 1986; pp.
55-60 e 145-162.
E. Layne, Spectrophotometric and Turbidimetric Methods for Measuring Proteins; In
Methods in Enzymology (S. P.Colowick, N. O. Kaplan, Eds.), Vol. III; Academic Press, New
York, 1957; pp.447-454.
S. M. Read, Techniques for Determining Protein Concentration; In Techniques in the Life
Sciences, Vol. BS101 Supplement; Elsevier: Amsterdam, 1984; pp.1-34.
D. A.Skoog, D. M. West, F. J. Holler, Fundamentals of Analytical Chemistry; Saunders:
Orlando, 1992; Caps. 21 e 22.
Preparação da aula prática
Calcule as concentrações (mg/mL) em BSA dos padrões utilizados na preparação da
curva de calibração do método do biureto.
Para determinar a concentração de uma amostra desconhecida por um método
espectrofotométrico é mais correcto utilizar apenas uma solução padrão ou preparar uma
curva de calibração? Justifique.
Idealmente, a preparação de uma curva de calibração de um método de doseamento de
proteína deve ser preparada com a própria proteína que se pretende dosear. Justifique.

Química Biológica
Anexo I
Cuidados gerais a observar durante a realização de medidas espectrofotométricas
Ligue o espectrofotómetro algum tempo antes da realização das medidas, para o aparelho
estabilizar.
Assegure-se que utiliza o “branco” adequado para acertar o zero do espectrofotómetro.
Sempre que realizar várias medidas com a mesma cuvette , mantenha a sua orientação no
porta-células para aumentar a sua precisão.
Evite tocar na superfície óptica das cuvettes. No caso de utilizar cuvettes de plástico tenha
em atenção a existência de faces com características diferentes.
Nunca encha uma cuvette em cima do espectrofotómetro.
É boa prática realizar sempre as medidas espectrofotométricas da solução mais diluída
para a mais concentrada, de modo a minimizar as interferências nas leituras.
Lave sempre as cuvettes imediatamente após a sua utilização com detergente e água. No
caso das cuvettes de vidro ou quartzo e em extrema necessidade pode ser utilizado ácido
nítrico a 50% na lavagem.
Após a realização das medidas, certifique-se que as cuvettes ficam guardadas na
respectiva caixa.

Química Biológica
Separação dos Componentes de uma Mistura por Filtração em Gel
Objectivo
Aplicação de um processo cromatográfico utilizado com frequência em bioquímica, para
separar moléculas de diferentes dimensões.
Introdução
Os processos cromatográficos são largamente utilizados em Bioquímica na separação de
componentes de misturas. A cromatografia é um método de separação de misturas complexas
de moléculas que se baseia na distribuição destas entre uma fase móvel e uma fase
estacionária. A fase móvel pode ser um líquido ou um gás, e a fase estacionária um sólido ou
um sólido impregnado de um líquido. A distribuição das moléculas pelas duas fases é
governada principalmente por um dos quatro processos básicos: adsorção, troca iónica,
partição e filtração em gel. A acção destes processos, acoplada ao movimento da fase móvel,
origina uma migração diferencial das moléculas a separar ao longo da fase estacionária.
Na cromatografia por filtração em gel, a fase estacionária é constituída por redes
porosas tridimensionais, compostas por cadeias lineares de polímeros entrecruzadas e que
formam a matriz de um gel. Os géis mais usados são obtidos por polimerização de cadeias de
polissacáridos (géis de “Sephadex”) ou por polimerização de cadeias de poliacrilamidas (géis
de “Biogel”). As partículas esféricas que formam o gel têm poros e o tamanho dos poros é
função do grau de entrecruzamento das cadeias de polímeros.
A fase móvel é constituída pelo líquido que passa através da coluna. A facilidade de
uma molécula de soluto penetrar na rede do gel presente na coluna é função do seu tamanho e
da sua forma. Quando as moléculas são demasiado grandes para penetrar na matriz do gel,
passam somente através do líquido presente fora das partículas, ou seja, através do volume
morto da coluna V0 (Figuras 1 e 2). Quando as moléculas são suficientemente pequenas,
podem penetrar em quase todo o volume interior das partículas do gel, Vi . Se o volume das
partículas do gel (isto é, o volume da coluna impenetrável ao solvente) for Vg, então o volume
total da coluna, Vt, (Figura 1), será:
gi0t VVVV

Química Biológica
Figura 1 Ilustração da definição de volume morto, V0, e volume total, Vt, de uma
coluna de cromatografia em gel (nota: Vt-V0 inclui o volume do
material sólido que constitui a matriz)
A ordem de eluição de uma mistura de macromoléculas (com formas moleculares
idênticas) introduzida no topo de uma coluna de gel, será por ordem decrescente do seu
tamanho molecular.
Figura 2 Representação esquemática do comportamento de macromoléculas de
diferentes tamanhos ao passarem por um gel (A proteína maior é
excluída dos poros do gel, a proteína de tamanho médio pode penetrar
apenas parcialmente e a proteína de menor tamanho penetra
completamente)
O volume de solvente necessário para a remoção de uma macromolécula da coluna é
denominado volume de eluição, Ve. Este parâmetro por si só não é suficiente para definir o
comportamento de uma substância na coluna pois vai depender do volume total do gel
equilibrado, Vt. Deste modo, a eluição de um soluto é melhor caracterizada por um coeficiente
de distribuição, dK :
i0ed /)( VVVK
onde 0V é o volume de eluição das moléculas que apenas estão distribuídas na fase móvel
porque são maiores do que os poros do gel e iV é o volume de solvente incluído nos poros do
gel e que está disponível para as moléculas pequenas.
No entanto, na prática iV é de difícil determinação pelo que se usa outro coeficiente, o
coeficiente de partição médio, avK . Ignorando a contribuição do volume sólido das partículas
do gel para o volume total da coluna, obtém-se:
)/()( 0t0eav VVVVK
Em condições ideais, o Kav pode variar entre 0 e 1 e os seus valores podem ser
interpretados do seguinte modo:
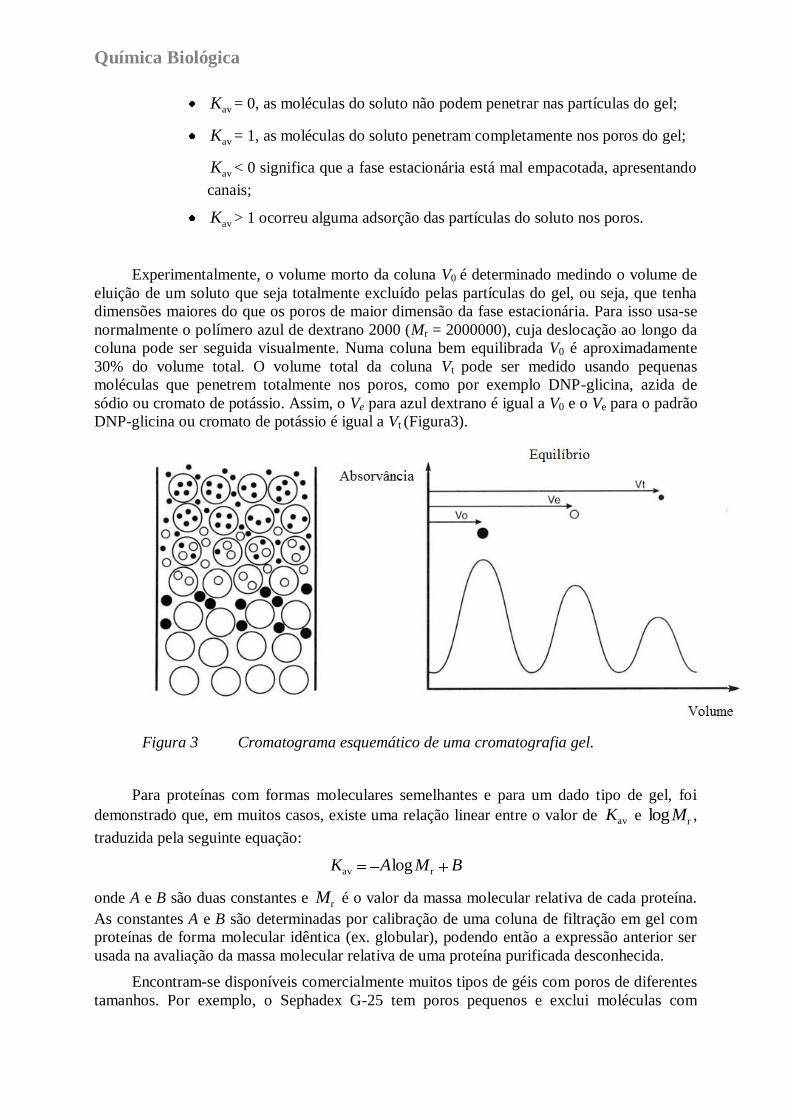
Química Biológica
avK = 0, as moléculas do soluto não podem penetrar nas partículas do gel;
avK = 1, as moléculas do soluto penetram completamente nos poros do gel;
avK < 0 significa que a fase estacionária está mal empacotada, apresentando
canais;
avK > 1 ocorreu alguma adsorção das partículas do soluto nos poros.
Experimentalmente, o volume morto da coluna V0 é determinado medindo o volume de
eluição de um soluto que seja totalmente excluído pelas partículas do gel, ou seja, que tenha
dimensões maiores do que os poros de maior dimensão da fase estacionária. Para isso usa-se
normalmente o polímero azul de dextrano 2000 (Mr = 2000000), cuja deslocação ao longo da
coluna pode ser seguida visualmente. Numa coluna bem equilibrada V0 é aproximadamente
30% do volume total. O volume total da coluna Vt pode ser medido usando pequenas
moléculas que penetrem totalmente nos poros, como por exemplo DNP-glicina, azida de
sódio ou cromato de potássio. Assim, o Ve para azul dextrano é igual a V0 e o Ve para o padrão
DNP-glicina ou cromato de potássio é igual a Vt (Figura3).
Figura 3 Cromatograma esquemático de uma cromatografia gel.
Para proteínas com formas moleculares semelhantes e para um dado tipo de gel, foi
demonstrado que, em muitos casos, existe uma relação linear entre o valor de avK e rlogM ,
traduzida pela seguinte equação:
BMAK rav log
onde A e B são duas constantes e rM é o valor da massa molecular relativa de cada proteína.
As constantes A e B são determinadas por calibração de uma coluna de filtração em gel com
proteínas de forma molecular idêntica (ex. globular), podendo então a expressão anterior ser
usada na avaliação da massa molecular relativa de uma proteína purificada desconhecida.
Encontram-se disponíveis comercialmente muitos tipos de géis com poros de diferentes
tamanhos. Por exemplo, o Sephadex G-25 tem poros pequenos e exclui moléculas com

Química Biológica
massas moleculares relativas ( rM ) superiores a 5000. Este é um tipo de gel útil na separação
de moléculas com valores de rM compreendidos entre 1000 e 5000.
Tipo de Sephadex rM
Limite inferior Limite superior
G-10 700
G-15 1500
G-25 1000 5000
G-50 1500 30000
G-75 3000 70000
G-100 4000 150000
G-150 5000 400000
G-200 5000 800000
Material e Reagentes
Coluna cromatográfica; espectrofotómetro; solução de NaCl a 0.9% (w/v).; Sephadex G-
100 (12 g em 210 mL de solução de NaCl a 0.9%, previamente preparada). Amostra: 4 mg de
azul de dextrano 2000, 4 mg de cromato de potássio e 30 mg de hemoglobina em 2 mL de
solução de NaCl a 0.9% alcalinizada com hidróxido de sódio.
Procedimento
Caso a coluna já se encontre preparada comece por introduzir 10 cm3 da solução de NaCl no
topo e deixe eluir até o nível de líquido estar 1 cm acima do nível do gel. Em seguida inicie o
procedimento no ponto 5.
Preparação da coluna
1. O gel que foi deixado a inchar durante a noite na solução de NaCl, foi previamente
decantado de modo a ficar uma mistura contendo aproximadamente 75% de gel
sedimentado e 25% de solução de NaCl a 0.9%.
2. Monte a coluna na vertical. Encha-a com cerca de 5 mL de solução de NaCl (eluente)
e deixe correr para lavar a coluna e verificar que o líquido flui.
3. Decante a mistura de Sephadex para a coluna com a ajuda de uma vareta ou de um
funil, tendo o cuidado de evitar o aprisionamento de bolhas de ar.

Química Biológica
4. Deixe o gel sedimentar 2 - 3 minutos, abra a saída da coluna e continue a aplicação da
mistura de Sephadex e/ou da solução de NaCl, até a altura da coluna estabilizar em
cerca de 15 cm. Idealmente esta operação deve ser realizada de uma só vez.
Nota: Durante a operação de empacotamento da coluna assim como no resto da experiência
nunca deixe secar a coluna. Esta eventualidade provoca a formação de canais no gel e
impossibilita a separação.
Aplicação e eluição da amostra
5. Numere e marque cerca de 20 tubos de ensaio com o volume de 2 mL.
6. Aplique 1 mL da amostra que lhe é fornecida no topo da coluna. Para isso remova o
excesso da solução de NaCl presente na parte superior, com cuidado, até que o seu
nível fique a cerca de 1 - 2 mm acima do gel. Feche a saída da coluna. Com o auxílio
de uma pipeta de Pasteur ou uma pipeta de 1 mL aplique a misture a cerca de 1 cm do
topo do gel e contra a parede interna da coluna, executando um movimento circular.
Evite danificar a superfície do gel. Coloque nesta altura o tubo de ensaio nº 1 na saída
da coluna.
Nota: Em opção a começar imediatamente a recolher amostras de 2 mL pode recolher o
eluente para uma proveta de 10 ou 20 mL até se aproximar a saída do azul de dextrano.
Nessa altura, tome nota do volume de eluente na proveta e inicie a recolha de amostras de
2 mL.
7. Abra a saída da coluna e deixe penetrar a amostra no gel. Feche novamente a saída da
coluna. Deite cerca de 2 mL de eluente no topo da coluna e volte a abrir a saída da
coluna. Não se esqueça de alternar os tubos de ensaio de recolha das fracções à saída
da coluna.
8. Encha a coluna com eluente até quase ao seu topo e mantenha a pressão hidrostática
adicionando mais eluente à medida que vai recolhendo as várias fracções. Recolha
tantas fracções quantas as necessárias para que a solução emergente da coluna venha
incolor (pode ser necessário utilizar mais do que 20 tubos de ensaio para completar a
eluição).
9. Quando terminar a eluição da amostra da coluna, meça a altura de gel e o diâmetro
interno da coluna como um processo alternativo para determinar o seu volume total.
No final regenere a coluna passando cerca de 1 volume de eluente pela coluna.
10. Adicione a cada fracção 4 mL de água destilada. Misture e meça a sua absorvência a
410 nm e a 620 nm contra um branco de água destilada.
Bibliografia
R. F. Boyer, Modern Experimental Biochemistry; Addison-Wesley, 1986; Cap. 3, p. 61-63 e
87-96.
D. Freifelder, Physical Biochemistry. Applications to Biochemistry and Molecular Biology,
2nd ed.; Freeman: S. Francisco, 1982.
Gel filtration-principles and methods, Handbooks of Amersham Biosciences, Amersham
Biosciences, Uppsala, Suécia;
http://kirschner.med.harvard.edu/files/protocols/GE_gelfiltration.pdf consultado a 23 de
Janeiro de 2013

Química Biológica
Tratamento de Resultados
1. Descreva a evolução da mistura ao longo da sua migração na coluna. Identifique,
justificando, a ordem de eluição dos vários componentes da mistura.
2. Trace um gráfico de eluição da mistura (represente A410 e A620 em função do número
da fracção).
3. Determine os valores de eV para cada componente da mistura.
4. Determine o volume total da coluna ( tV ) e compare este valor com o valor do Ve para
o cromato de potássio.
5. Calcule os valores de avK para cada componente e discuta o seu significado.
Preparação da aula prática
Qual a base do método de separação de filtração em gel?
Quais são os componentes separados na experiência e qual a sua massa molecular
relativa?

Química Biológica
Objectivo
Observar a ação de um enzima sobre um substrato e verificar a influência de vários tipos
de fatores sobre a sua atividade enzimática.
Introdução
As polifenóis oxidases (OPP) são enzimas intracelulares que contêm cobre no seu centro
activo e podem ser encontrados em plantas, animais, bactérias e fungos. Estes enzimas
classificam-se em três principais classes de acordo com o seu substrato específico e o
mecanismo de ação: catecolases, lacases , e cresolases nas plantas e tirosinases nos animais.
As tirosinases, presentes apenas em animais, catalisam a o-hidroxilação de monofenóis a o-
difenóis, e a subsequente oxidação dos o-difenóis a o-quinonas (Figura1).
Figura 1: Mecanismo de acção da tirosinase
As tirosinases estão envolvidas principalmente na biossíntese de melaninas, sendo
responsáveis pela transformação da L-tirosina em L-DOPA e L-DOPA quinona (Figura 2).
Polifenoloxidase

Química Biológica
Figura 2: Primeiros passos na biossíntese de melaninas animais.
Esta sequência de reações está localizada nos melanócitos ou células de pigmentos, nas quais
se faz por este processo a produção das melaninas, que dão a coloração à pele, aos cabelos e
aos olhos.
As cresolases são o equivalente da tirosinases nas plantas mas têm um papel muito menos
importante.
As catecoloxidases, também conhecidas por o-difenoloxidases catalisam a oxidação pelo
oxigénio molecular, de o-difenóis a o-quinonas. Ao contrário das tirosinases estes enzimas
não possuem atividade hidroxilase e portanto os monofenóis não são seus substratos
(Figura2). Estes enzimas são encontrados principalmente nas plantas podendo também existir
em alguns dos outros organismos.
Figura 2: Mecanismo de acção da catecoloxidase
As lacases são enzimas de espectro mais largo capazes de oxidar vários substratos aromáticos
(Figura 3). Estes enzimas são típicos de fungos fitopatogénicos mas também se encontram em
algumas plantas superiores.
Figura 3: Mecanismo de acção da lacase
As polifenoloxidases vegetais são responsáveis pelo escurecimento de certos produtos
vegetais, como as batatas ou as maçãs, ricas em compostos polifenólicos, após serem cortadas
ou descascadas. Este escurecimento é devido à formação de quinonas e à reação que estas
sofrem posteriormente dando origem aos pigmentos castanhos conhecidos por melaninas.
Este escurecimento dos vegetais e frutos, embora na maioria das vezes não corresponda
a uma diminuição do seu valor nutricional, causa imensas perdas económicas no mercado
agrícola, uma vez que diminui a aceitabilidade do produto por parte do consumidor. Um
modo de impedir estas reações é inactivar as polifenoloxidases. Uma vez que possuem cobre

Química Biológica
no seu centro activo, este pode ser complexado com vários quelantes, tornando o enzima
inativo. Outros inibidores do enzima são a feniltioureia, o ditiocarbamato de dietilo, a azida de
sódio, o ião cianeto ou a cisteína.
Neste trabalho vamos observar a ação do enzima polifenoloxidase isolado da batata
sobre o catecol (um difenol) e verificar alguns fatores que influenciam a sua atividade como a
temperatura, o pH, a concentração do substrato e do enzima, e a presença de inibidores.
Material e Reagentes
Homogeneizador; gelo; batatas; Tampão de fosfatos 0,01M pH 6,8; solução de catecol
0.1 M; solução de ácido tricloroacético (TCA) a 10%; feniltioureia; fenol 0.1 M; hidroquinona
0.1 M; solução de HCl 0.1 M; tampão acetato 0.1 M, pH 5; tampão de fosfatos 0.1 M, pH 7;
tampão Tris 0.1 M, pH 9.
Procedimento
Preparação do extrato enzimático
1. Coloque num homogeneizador uma batata média descascada e cortada em bocados.
2. Junte 50 mL do tampão de fosfatos 0,01M pH 6,8 (que deverá estar no frio).
3. Homogenize durante 1 a 2 minutos.
4. Filtre num funil de Büchner. O filtrado obtido é o extrato enzimático.
5. Guarde o extrato enzimático num erlenmeyer coberto com papel de alumínio e
coloque-o num banho de gelo, onde deve ser mantido durante toda a aula.
A. Ação da polifenoloxidase
1. Prepare 3 tubos: B1: 15 gotas de extrato + 15 gotas de solução de catecol
B2: 15 gotas de extrato + 15 gotas de água
B3: 15 gotas de água + 15 gotas de solução de catecol
2. Agite os tubos e coloque-os no termóstato a 37 ºC durante 10 minutos, agitando de vez
em quando. Registe o que observa após a incubação.
B. Natureza química da polifenoloxidase
1. Prepare 3 tubos: C1: 15 gotas de extrato + 15 gotas de solução de catecol.
Agite.
C2: 15 gotas de extrato + 15 gotas de solução de TCA
Agite. Espere 5 minutos. Junte 15 gotas de solução de catecol.
C3: 15 gotas de extrato + alguns cristais de feniltioureia.
Agite durante 5 minutos. Junte 15 gotas de solução de catecol.
Coloque os três tubos durante 10 minutos no termostato a 37oC. Compare C1, C2 e
C3.

Química Biológica
C. Especificidade para o substrato
1. Prepare 3 tubos: D1: 15 gotas de extrato + 15 gotas de solução de catecol
D2: 15 gotas de extrato + 15 gotas de fenol
D3: 15 gotas de extrato + 15 gotas de hidroquinona
2. Agite os tubos e incube 10 minutos a 37 ºC. Observe e registe as alterações que sofrem
os respetivos conteúdos.
D. Efeito da concentração do substrato
1. Prepare 3 tubos: E1: 1 gota de solução de catecol + 3.95 mL de água
E2: 1 mL de solução de catecol + 3 mL de água
E3: 2 mL de solução de catecol + 2 mL de água
2. Adicione 2 mL de extrato a cada tubo. Agite os tubos e incube-os 10 minutos a 37ºC.
Observe e registe as tonalidades nos 3 tubos.
E. Efeito da concentração do enzima
1. Prepare 2 tubos: F1: 15 gotas de solução de catecol + 30 gotas de extrato
F2: 15 gotas de solução de catecol + 5 gotas de extrato + 25
gotas de água
2. Agite e incube a 37 ºC durante 10 minutos. Observe e registe as observações.
F. Efeito da temperatura
1. Prepare 3 tubos: G1: 15 gotas de extrato (0 ºC – gelo)
G2: 15 gotas de extrato (37 ºC – termostato a 37 ºC)
G3: 15 gotas de extrato (70 ºC – termostato a 70 ºC)
2. Incube cada um dos tubos durante 2 minutos à temperatura acima indicada.
3. Junte 15 gotas de catecol a cada tubo, mantendo-os às respetivas temperaturas e deixe-
os ficar mais 5 minutos às mesmas temperaturas. Registe o que observou.
4. Retire os tubos dos locais de incubação às diferentes temperaturas e incube-os todos a
37ºC durante 15 minutos. Registe as alterações.
G. Efeito do pH
1. Prepare 4 tubos: H1: 2 mL de solução de HCl (pH 1)
H2: 2 mL de tampão acetato (pH 5)
H3: 2 mL de tampão de fosfatos (pH 7)
H4: 2 mL de tampão Tris (pH 9)
2. Junte 15 gotas de solução de catecol e 15 gotas de extrato a cada tubo.
3. Agite os tubos e incube-os 15 minutos a 37 ºC. Observe cada tubo e registe as
alterações.

Química Biológica
Bibliografia
M. Dixon, E. C. Webb, Enzymes, 3rd ed.; Longman: London, 1979.
T. Aniszewskl, R. Lieberei, K. Gulewicz, Acta Biol. Crac. Ser. Bot. 2008, 50, 7–18.
Tratamento de Resultados
Interprete e discuta os resultados obtidos relativamente a cada um dos efeitos estudados.
Preparação da aula prática
Esboce as curvas que descrevem a variação da atividade enzimática máxima com a
temperatura e o pH.
Explique por que razão é importante ter em cada experiência um tubo “controlo”.

Química Biológica
Objectivo
Utilizar um processo de separação de moléculas baseado na utilização de um campo
eléctrico.
Introdução
A electroforese consiste na migração de partículas carregadas num campo eléctrico. A
técnica, que tem sido usada pelos bioquímicos há pelo menos 50 anos, é especialmente
aplicável a polímeros e outras moléculas biológicas. De facto, a electroforese é muito usada
em química clínica e os materiais de estudo incluem proteínas séricas, eritrócitos, urina,
líquido cefalo-raquidiano e outros líquidos fisiológicos, assim como proteínas dos tecidos.
As técnicas electroforéticas podem ser classificadas como electroforese de fronteira
móvel (ou electroforese em solução), em que uma solução tamponada das macromoléculas em
estudo é sujeita a um campo eléctrico, e electroforese de zona, em que é utilizada uma matriz
sólida saturada em tampão como suporte. As moléculas possuindo uma carga eléctrica, em
virtude da ionização de um protão, mover-se-ão para o cátodo ou para o ânodo do sistema
electroforético, dependendo da natureza da carga da molécula. A velocidade de migração
depende de vários factores:
carga eléctrica da molécula
tamanho e forma da molécula
força do campo eléctrico
natureza do meio de suporte
temperatura da operação
A mobilidade electroforética – velocidade de migração por unidade de campo eléctrico
– é directamente proporcional à carga e inversamente proporcional ao tamanho da molécula e
à viscosidade do meio.
Uma mistura de proteínas que diferem na sua carga total podem ser separadas em
fracções num campo eléctrico. Podem ser usados dois tipos de meios de suporte. O primeiro
grupo inclui papel, gel de agar e acetato de celulose. Estes meios têm poros suficientemente
largos para que as proteínas se movam livremente. Os métodos de separação das proteínas
séricas que utilizam estes suportes permitem a separação destas proteínas em 4 a 7 fracções. O
segundo grupo de meios de suporte inclui o gel de amido e a acrilamida; os quais têm o
diâmetro dos poros tão pequeno que podem actuar como”peneira” molecular. As técnicas que
utilizam estes materiais fraccionam as proteínas de acordo com o tamanho das suas moléculas
e a sua carga, e são capazes de separar as proteínas séricas em 20 ou mais fracções. O seu
poder de resolução provou ser de grande valor no estudo de proteínas com variantes genéticas
e alguns isoenzimas.
Electroforese das Proteínas do Soro

Química Biológica
No entanto, em muitos trabalhos de rotina muitas bandas originam confusão e, por isso,
em laboratórios clínicos usa-se acetato de celulose ou gel de agar, que têm uma estrutura mais
uniforme e permitem uma boa separação. Estes meios podem ser transparentizados,
permitindo uma melhor vizualização da imagem.
O tampão barbital a pH 8.6 é o tampão padrão para a electroforese de rotina das
proteínas séricas.
O sangue é um tecido que circula num sistema fechado de vasos sanguíneos, constituído
por elementos sólidos (glóbulos vermelhos, glóbulos brancos e plaquetas) em suspensão num
meio líquido (o plasma). Após a coagulação sanguínea, a fase líquida que se obtém constitui o
soro, que não possui os factores de coagulação (incluindo o fibrinogéneo) que estão
normalmente presentes no plasma. O plasma é formado por água, electrólitos, metabolitos,
nutrientes, proteínas e hormonas.
As proteínas do plasma compreendem a maior parte dos sólidos (7-7.5 g/dL) e são uma
mistura complexa que inclui proteínas simples e proteínas conjugadas, tais como
glicoproteínas e vários tipos de lipoproteínas. É costume dividir as proteínas do plasma em
três grupos principais: albumina, globulinas e fibrinogénio.
A albumina é a proteína que existe em maior concentração e é a que tem peso molecular
mais baixo.
As globulinas séricas são uma mistura heterogénea e complexa de proteínas que são
frequentemente designadas por α-, β- e γ-globulinas, com base na sua mobilidade
electroforética. No soro normal estão presentes 15 proteínas em concentração suficiente para
influenciar o modelo electroforético após coloração para proteínas mas, uma vez que várias
delas têm mobilidades semelhantes, originam só seis bandas distintas (albumina, α1, α2, β1,
β2 e γ).
Na figura seguinte representa-se a imagem electroforética dum soro normal em acetato
de celulose e a intensidade relativa das bandas lidas num fotodensitómetro.

Química Biológica
Material e Reagentes
Soro de coelho; tampão barbital sódico 0.04 M, pH 8.6, força iónica 0.05; solução de
coloração (0,5 g Ponceau S em 100 mL de uma solução diluída de ácido tricloroacético a 5%);
solução de ácido acético a 5%; tiras de acetato de celulose; tina de electroforese.
Procedimento
1. Encha a tina de electroforese com tampão. Coloque a ponte.
2. Ensope as tiras de acetato de celulose com tampão durante cerca de 20 minutos num
recipiente à parte; para evitar a adesão de bolhas de ar, deixe primeiro flutuar as tiras
sobre o tampão de forma a mergulharem quando saturadas.
3. Coloque as tiras entre duas folhas de papel de filtro para remover o excesso de
tampão.
4. Coloque as tiras sobre a ponte de modo a ficarem paralelas umas às outras e aos lados
da tina.
5. Coloque os fixadores contra os lados das pontes para assegurar o contacto entre as
tiras e o tampão e para que fiquem esticadas.
6. Aplique cerca de 3 µL de soro em cada tira a aproximadamente 2 cm do bordo
catódico. Desloque o soro contido numa micropipeta atravessando a tira com um
movimento prolongado mas rápido. Em alternativa pode usar um aplicador especial.
7. Coloque a tampa da tina imediatamente após a aplicação dos soros. Uma tampa
abaulada assegura que qualquer mistura que possa ter evaporado durante o processo e
condensado na tampa caia para o lado do tanque e não sobre as tiras.
8. Ligue os fios eléctricos da tina à fonte de alimentação, tendo o cuidado de assegurar a
polaridade correcta.
9. Ligar a corrente, fixar a voltagem em 200V e seguir a variação da intensidade da
corrente.
10. Após 30 minutos, desligar a fonte de alimentação, remover rapidamente as tiras da tina
e colocá-las numa solução corante Ponceau S, durante 5 a 10 minutos. Durante todo o
processo manuseie as tiras com pinças.
11. Coloque as tiras em ácido acético a 5% para remover o excesso de corante. O ácido
tricloroacético terá fixado as proteínas coradas às tiras.
12. Após descoloração da tira coloque sobre uma placa de vidro e proceda à fixação da
imagem da mesma (foto). Separe a tira da placa de vidro, conservando-a entre papéis.
Tratamento de Resultados
1. Faça um esquema da tira de acetato de celulose e do ponto de aplicação da amostra,
referenciando os bordos catódico e anódico.
2. Junte a tira de acetato de celulose após a electroforese ou, no caso de não ser possível,
faça um esquema das bandas observadas.
3. Comparando com o que foi referido na introdução do trabalho, que conclusões pode
tirar sobre as várias bandas, no que respeita à sua identificação, tamanho e carga
eléctrica?

Química Biológica
Bibliografia
J. Stenesh, Experimental Biochemistry, Allyn & Bacon: London, 1984.
D. J. Plummer, An Introduction to Pratical Biochemistry, 2nd ed.; London, 1978.
Preparação da aula prática
Qual o princípio deste método de separação?
Por que razão foi escolhido um tampão com pH 8.6?

Química Biológica
Objectivo
Utilização de um método para o doseamento de glúcidos com base no seu poder redutor.
Introdução
O desenvolvimento de métodos que permitam dosear os glícidos presentes nos
alimentos é extremamente importante, uma vez que estes compostos são um dos constituintes
fundamentais da nossa dieta. Com efeito, os poliósidos glicogénio e amido são importantes
formas de armazenamento de energia, estando presentes em tecidos animais e vegetais
respectivamente.
A capacidade redutora dos glúcidos tem constituído a base química de vários métodos
de análise quantitativa destes compostos. No presente trabalho vai-se determinar o conteúdo
em glúcidos redutores de farinhas alimentares através do método do 3,5-dinitrosalicilato
(DNS). Em meio alcalino, o DNS forma um produto de redução castanho avermelhado, o 3-
amino-5-nitrosalicilato, quando aquecido na presença de glúcidos redutores. Devido ao facto
de os poliósidos não serem redutores, é necessário proceder a uma hidrólise ácida prévia da
amostra de farinha alimentar, de modo a obter as suas oses constituintes.
Material e Reagentes
Almofariz; placa de aquecimento; espectrofotómetro; farinha alimentar; solução de
H2SO4 1.5 M; solução de NaOH a 10%; solução de glucose 3 mg/mL.
Procedimento
1. Reduza a pó fino, com o auxílio de um almofariz, uma pequena quantidade de farinha
alimentar. Pese seguidamente uma amostra de cerca de 50 mg, registando a massa até
ao décimo de mg.
2. Transfira o material pesado para um tubo de ensaio e adicione 10 mL de H2SO4 1.5 M.
Aqueça o tubo de ensaio num banho de água em ebulição durante 20 minutos,
agitando de vez em quando.
3. Arrefeça o tubo à torneira, deite o conteúdo do tubo para dentro de um pequeno
erlenmeyer, e adicione cuidadosamente 12 mL de NaOH a 10%.
4. Aqueça ligeiramente o erlenmeyer contendo o hidrolisado e filtre enquanto quente
para um balão de 50 mL. Se a amostra não estiver aquecida, a filtração far-se-á mais
lentamente. Lave o erlenmeyer e o tubo de ensaio que continha o hidrolisado com
água destilada quente e adicione-a ao filtro, de modo a que o material hidrolisado seja
todo transferido. Perfaça o volume do balão para 50 mL. Agite bem.
5. Prepare a curva padrão para o método do DNS de acordo com o quadro seguinte:
Determinação de Glúcidos Redutores em Produtos Alimentares
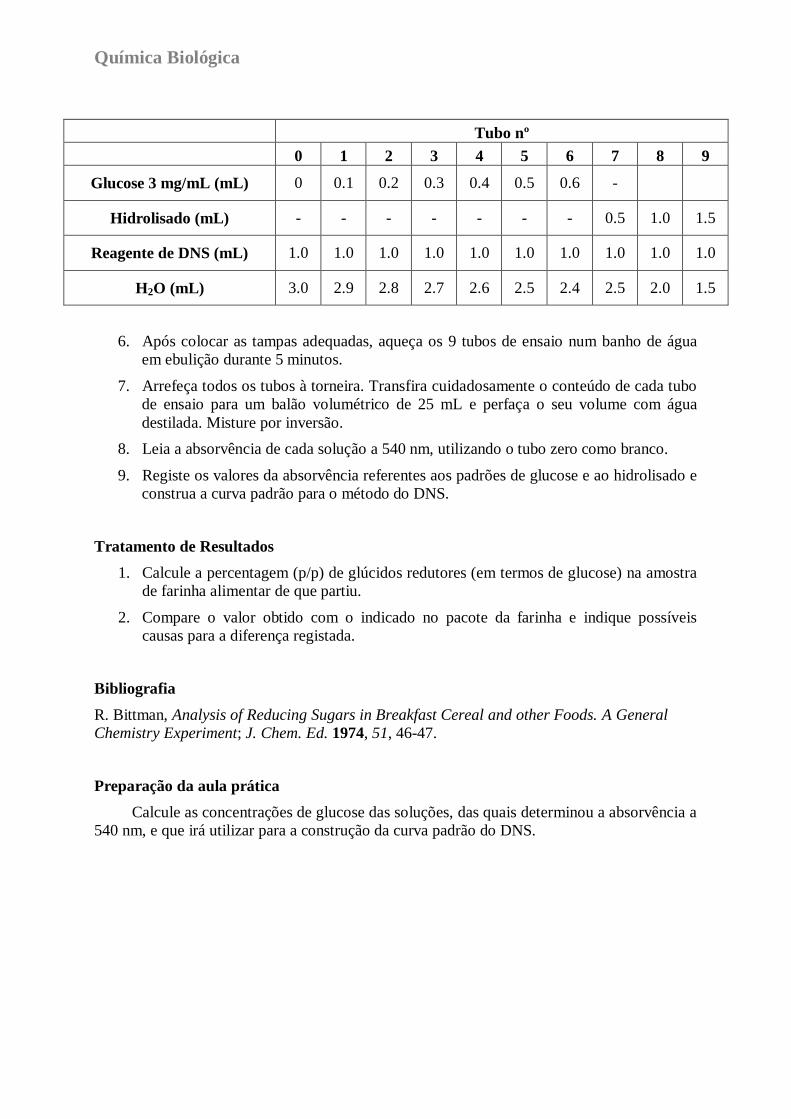
Química Biológica
Tubo nº
0 1 2 3 4 5 6 7 8 9
Glucose 3 mg/mL (mL) 0 0.1 0.2 0.3 0.4 0.5 0.6 -
Hidrolisado (mL) - - - - - - - 0.5 1.0 1.5
Reagente de DNS (mL) 1.0 1.0 1.0 1.0 1.0 1.0 1.0 1.0 1.0 1.0
H2O (mL) 3.0 2.9 2.8 2.7 2.6 2.5 2.4 2.5 2.0 1.5
6. Após colocar as tampas adequadas, aqueça os 9 tubos de ensaio num banho de água
em ebulição durante 5 minutos.
7. Arrefeça todos os tubos à torneira. Transfira cuidadosamente o conteúdo de cada tubo
de ensaio para um balão volumétrico de 25 mL e perfaça o seu volume com água
destilada. Misture por inversão.
8. Leia a absorvência de cada solução a 540 nm, utilizando o tubo zero como branco.
9. Registe os valores da absorvência referentes aos padrões de glucose e ao hidrolisado e
construa a curva padrão para o método do DNS.
Tratamento de Resultados
1. Calcule a percentagem (p/p) de glúcidos redutores (em termos de glucose) na amostra
de farinha alimentar de que partiu.
2. Compare o valor obtido com o indicado no pacote da farinha e indique possíveis
causas para a diferença registada.
Bibliografia
R. Bittman, Analysis of Reducing Sugars in Breakfast Cereal and other Foods. A General
Chemistry Experiment; J. Chem. Ed. 1974, 51, 46-47.
Preparação da aula prática
Calcule as concentrações de glucose das soluções, das quais determinou a absorvência a
540 nm, e que irá utilizar para a construção da curva padrão do DNS.





























