Embed Size (px)
Citation preview

DPOC E TABAGISMO:
UM BINÔMIO PERIGOSO
Marlon Akio da Silva Issobe
Rio de Janeiro
2012

ii
MARLON AKIO DA SILVA ISSOBE
Aluno do curso de Farmácia
Matrícula 0823800119
DPOC E TABAGISMO:
UM BINÔMIO PERIGOSO
Trabalho de Conclusão de Curso, TCC,
apresentado ao Curso de Graduação em
Farmácia, da UEZO como parte dos requisitos
para a obtenção do grau de Bacharel em
Farmácia, sob a orientação do Professor André
Luiz Fonseca de Souza.
Rio de Janeiro
Julho de 2012

iii
DPOC E TABAGISMO:
UM BINÔMIO PERIGOSO
Elaborado por Marlon Akio da Silva Issobe
Aluno do curso de Farmácia da UEZO
Este trabalho de Graduação foi analisado e aprovado com
Grau: ______
Rio de Janeiro, 03 de Julho de 2012.
_______________________________________
Prof. André Luiz Fonseca de Souza, Ph.D., da UEZO – Presidente
______________________________________
Prof.ª Isabele Barbieri dos Santos, Ph.D., da Fiocruz
_______________________________________
Prof.ª Alessandra Micherla Rodrigues do Nascimento, Ph.D., da UEZO
RIO DE JANEIRO, RJ - BRASIL
Julho de 2012

iv
DEDICATÓRIA
Dedico este trabalho...
A toda minha família pelo imenso apoio, suporte, compreensão e
torcida, a mim dedicados durante toda a trajetória de graduação.
Aos meus amigos por todo carinho, companheirismo e atenção,
partilhados ao longo desses 4 longos anos.
A meu sobrinho, Miguel, cuja chegada trouxe muita luz e serviu
de inspiração.

v
AGRADECIMENTOS
A Deus, pela benção, por ter guiado meus passos e pela proteção em
todos os dias de minha vida.
Aos meus pais, Takeshi e Edith, pelo amor, base e dedicação que
sempre me deram e por todos os sacrifícios feitos até hoje por mim e, apesar
de todas as dificuldades passadas nunca pouparam esforços para que eu
pudesse atingir meus objetivos.
A minha irmã, Michele, pelo companheirismo e apoio, não só durante
este período, mas por toda vida.
A minha tia Ercília, pelo enorme carinho e zelo, e por me acolher de
forma tão generosa.
A Paola e Rosilane, pessoas que conheci na graduação e que com
certeza levarei para sempre comigo. Esta jornada não teria sido tão especial,
nem tido a mesma graça sem vocês ao meu lado. Muito obrigado pela
amizade, convívio, companheirismo e por terem dividido angústias e
multiplicado alegrias.
A todos os meus amigos, que de alguma forma me alegraram,
motivaram e impulsionaram durante esta jornada.
A todos os companheiros da 1ª turma de farmácia da UEZO, pelos
momentos divertidos que passamos (que não foram poucos), por todas as
dificuldades e provações que enfrentamos para que pudéssemos concluir este
curso. Sentirei saudades.
Ao Dr. André Luiz Fonseca de Souza, meu orientador, pessoa com um
dos maiores níveis de conhecimento, que tive a honra de conhecer, e um
professor como poucos.
A todos os meus professores pelos ensinamentos, muito uteis, e que
contribuíram para meu crescimento profissional. Cada um dos professores tem
uma participação especial nesta etapa de minha vida.

vi
RESUMO
A doença pulmonar obstrutiva crônica (DPOC) é caracterizada por uma
limitação irreversível no fluxo de ar pulmonar associada com inflamação
crônica e hipersecreção de muco (bronquite crônica) e pela destruição
patológica dos espaços aéreos alveolares que conduzem a enfisema.
Atualmente sabe-se que esta doença envolve uma complexa variedade de
manifestações celulares, anatômicas, funcionais e clínicas. É uma doença
progressiva e está relacionada a uma resposta inflamatória anormal dos
pulmões à inalação de partículas e/ou gases tóxicos. Na prática e na pesquisa
clínica, a definição da DPOC baseia-se na extensão da obstrução ao fluxo
aéreo medido por meio da espirometria. DPOC é considerada uma doença
evitável e tratável que, embora acometa primariamente os pulmões, há
diversas manifestações extrapulmonares significativas que podem também
contribuir para a gravidade da doença em pacientes individuais. DPOC é
causada principalmente pela exposição à fumaça do tabaco, sendo uma das
principais causas de incapacidade e mortalidade, representando a quarta maior
causa de morte no mundo, e sua prevalência só aumentará nos próximos anos.
Apesar do enorme crescimento da prevalência e da mortalidade da DPOC, não
existe nenhuma abordagem terapêutica que consiga controlar a evolução da
doença estabelecida, sendo que os tratamentos são baseados nos alívios dos
sintomas dos pacientes, principalmente com a utilização de broncodilatadores.
Por este motivo a cessação tabágica é a única medida que melhora o
prognóstico e a sobrevida dos doentes com DPOC.
Palavras-chaves: DPOC, Tabagismo, Inflamação, Estresse oxidativo.

vii
ABSTRACT
The chronic obstructive pulmonary disease (COPD) is characterized by
an irreversible limitation in lung airflow associated with chronic inflammation and
mucus hypersecretion (chronic bronchitis) and the pathological destruction of
the alveolar air spaces leading to emphysema. Currently it is known that this
disease involves a complex diversity of cellular, anatomical, functional and
clinical events. It is a progressive disease and is associated with an abnormal
inflammatory response of lungs to inhaled particles and/or toxic gases. In
practice and medical research, the definition of COPD based on the extent of
airflow obstruction measured by spirometry. COPD is considered a treatable
and preventable disease that, although primarily affects the lungs, there are
several significant extrapulmonary manifestations which may also contribute to
the severity of the disease in individual patients. COPD is primarily caused by
exposure to tobacco smoke, being a major cause of disability and mortality,
representing the fourth leading cause of death worldwide, and its prevalence
will only increase in coming years. Despite the enormous increase in
prevalence and mortality of COPD, there is no therapeutic approach that could
control the evolution of established disease, the treatments are based on relief
of symptoms of patients, mainly with the use of bronchodilators. Therefore
smoking cessation is the only action that improves the prognosis and survival of
patients with COPD.
Key Words: COPD, Smoking, Inflammation, Oxidative Stress.

viii
A realidade do seu sucesso frequentemente tem a ver
com quem irá ganhar a batalha que está sendo travada
dentro dos dois de você: o “você” que quer parar,
desistir, relaxar, ou o “você” que se recusa a se
entregar e não irá desistir até conseguir alcançar seus
sonhos.
Chris Widener

ix
LISTA DE FIGURAS
Figura 1. Características histológicas da bronquite crônica. ........................... 20
Figura 2. Foto de histopatologia de tecido pulmonar ....................................... 21
Figura 3. Enfisema representa um desequilíbrio ............................................. 23
Figura 4. Enfisema centrolobular e panacinar ................................................. 24
Figura 5. As células inflamatórias envolvidas na patogênese da DPOC ......... 31
Figura 6. O estresse oxidativo e seus efeitos .................................................. 35
Figura 7. Geração de uma resposta imune adaptativa em pacientes com
DPOC ............................................................................................................... 42
Figura 8. Fisiopatologia das manifestações sistêmicas e locais da DPOC. .... 50
Figura 9. Diagnóstico da DPOC ...................................................................... 52
Figura 10. Mecanismo de ação dos agonistas β-adrenérgicos ....................... 57
Figura 11. Fórmula Estrutural da N-acetilcisteína ............................................ 59

x
LISTA DE TABELAS
Tabela 1. Estágios GOLD de gravidade da DPOC .......................................... 19
Tabela 2. As 10 principais causas de morte no mundo em 2008 ..................... 26
Tabela 3. As 10 principais causas de óbito no Brasil, 2005 ............................. 28
Tabela 4. Fatores de risco para DPOC ............................................................ 29
Tabela 5. Orientações terapêuticas de acordo com os estádios da DPOC ..... 55
xi
LISTA DE ABREVIATURAS E SIGLAS
ALAT Associação Latino-Americana de Tórax
AMP Adenosina monofosfato
AP-1 Ativador de proteína-1
CVF Capacidade vital forçada
DNA Ácido desoxirribonucleico
DPOC Doença Pulmonar Obstrutiva Crônica
ENA Ativador neutrofílico derivado do epitélio
EROs Espécies reativas de oxigênio
GCL Glutamato-cisteína ligase
GM-CSF Fator estimulador de colônias de granulócitos-
macrófagos
GOLD Iniciativa Global para Doença Pulmonar Obstrutiva
Crônica
GPxe Glutationa peroxidase extracelular
GRO Oncogene relacionado ao crescimento
GSTM1 Glutationa S-transferase M1
GSTP1 Glutationa S-transferase P1
IFN-γ Interferon gama
Ig Imunoglobulina
IL Interleucina
IMC Índice de massa corpóreo
LBA Lavado broncoalveolar

xii
LTB4 Leucotrieno B4
MAPK Proteína quinase ativada por mitógenos
MDA Malondialdeído
MMP Metaloproteinase
NF-κB Fator nuclear-κB
NK Células Natural Killer
NO Óxido Nítrico
OMS Organização Mundial de Saúde
PaCO2 Pressão parcial do CO2 no sangue arterial
PaO2 Pressão parcial de oxigênio no sangue arterial
PCO2 Pressão parcial de dióxido de carbono
PDE Fosfodiesterase
PLATINO Projeto Latino-americano de Investigação em
Obstrução Pulmonar
PMNs Leucócitos polimorfonucleares
SIRT1 Proteína sirtuína-1
SOD Superóxido dismutase
SpO2 Saturação periférica de oxigênio
Tc1 Células T citotóxicas do tipo 1
Tc2 Células T citotóxicas do tipo 2
Th1 T helper 1
Th2 T helper 2
TNF-α Fator de necrose tumoral alfa
VEF1 Volume expiratório forçado no primeiro segundo

xiii
SUMÁRIO
Página
RESUMO............................................................................................................ vi
ABSTRACT ....................................................................................................... vii
LISTA DE FIGURAS .......................................................................................... ix
LISTA DE TABELAS ........................................................................................... x
LISTA DE SIGLAS E ABREVIATURAS ............................................................. xi
1. INTRODUÇÃO ............................................................................................. 15
2. OBJETIVOS ................................................................................................. 17
2.1. OBJETIVOS ESPECÍFICOS ............................................................... 17
3. REVISÃO BIBLIOGRÁFICA ........................................................................ 18
3.1. DOENÇA PULMONAR OBSTRUTIVA CRÔNICA .............................. 18
3.1.1. Bronquite Crônica ................................................................ 19
3.1.2. Enfisema Pulmonar .............................................................. 20
3.2. EPIDEMIOLOGIA................................................................................ 24
3.3. FATORES DE RISCO ......................................................................... 27
3.4. PATOGÊNESE ................................................................................... 30
3.4.1. Genética da DPOC ............................................................... 33
3.4.2. O Estresse Oxidativo ........................................................... 34
3.4.3. Inflamação ............................................................................ 38
3.4.3.1. Células inflamatórias e seus mediadores ................... 38
3.4.3.2. Células estruturais ..................................................... 39
3.4.3.3. Macrófagos ................................................................ 40

xiv
3.4.3.4. Linfócitos .................................................................... 41
3.4.3.5. Neutrófilos .................................................................. 45
3.4.3.6. Quimiocinas na patogênese da DPOC ...................... 45
3.4.3. Desequilíbrio Protease – Antiprotease .............................. 47
3.5. PATOLOGIA DA DPOC ...................................................................... 47
3.6. SINTOMATOLOGIA ............................................................................ 49
3.6.1. Comorbidades ...................................................................... 51
3.7. DIAGNÓSTICO ................................................................................... 52
3.7.1. Avaliação Espirométrica ..................................................... 53
3.7.2. Avaliação Radiológica ......................................................... 54
3.7.3. Avaliação Gasométrica e do pH.......................................... 54
3.8. TRATAMENTO ................................................................................... 55
3.8.1. Broncodilatadores ............................................................... 55
3.8.1.1. β2-agonistas ............................................................... 56
3.8.1.2. Anticolinérgicos .......................................................... 56
3.8.1.3. Xantinas ..................................................................... 57
3.8.2. Corticoides ........................................................................... 57
3.8.3. N-acetilcisteína ..................................................................... 58
3.8.4. Oxigenoterapia ..................................................................... 59
3.8.5. Fisioterapia Respiratória ..................................................... 59
3.8.6. Novas Opções Terapêuticas ............................................... 60
3.8.7. Tratamento das Comorbidades .......................................... 60
4. CONCLUSÃO .............................................................................................. 62
5. REFERÊNCIAS BIBLIOGRÁFICAS ............................................................ 63

15
1. INTRODUÇÃO
A doença pulmonar obstrutiva crônica (DPOC) é um termo genérico usado
para descrever um grupo de doenças pulmonares crônicas, caracterizadas pela
limitação por um longo período ao fluxo aéreo nos pulmões que não é totalmente
reversível. O termo inclui “bronquite crônica” e “enfisema pulmonar”, que não
estão mais sendo usados, mas incluídos dentro do diagnóstico da DPOC (WHO,
2011).
Na bronquite crônica a lesão pulmonar se localiza nos brônquios e
bronquíolos, tornando-os cronicamente inflamados, espessos e com constante
produção de muco. O paciente com bronquite crônica apresenta além dos
sintomas de falta de ar e cansaço, um quadro de tosse crônica com expectoração
(FERREIRA, 2010).
O enfisema se caracteriza pela destruição e alargamento dos bronquíolos
terminais e alvéolos, que perdem sua elasticidade e favorecem o aprisionamento
do ar dentro dos pulmões. No enfisema notamos uma hiperinsuflação mantida dos
pulmões devido ao ar que nunca sai por completo (FERREIRA, 2010).
A obstrução no fluxo de ar do pulmão é geralmente progressiva e está
associada a uma resposta inflamatória anormal dos pulmões à inalação de
partículas nocivas ou gases tóxicos e a um processo lesivo do tecido pulmonar,
tendo como uma das principais causas o cigarro (JARDIM; OLIVEIRA;
NASCIMENTO, 2004).
O tabagismo é um fator de risco para seis das oito principais causas de
morte no mundo. E cerca de 85% a 90% de todas as mortes por DPOC são
atribuíveis ao tabagismo, e estudos mostram evidências suficientes para se
chegar à conclusão que existe uma relação causal entre tabagismo e morbidade e
mortalidade por DPOC. A exposição ocupacional a poluentes e a poluição
ambiental também constituem fatores de risco, mas só representam 10 a 15% dos
casos registrados (MEIRELLES, 2009).
O tabagismo leva à DPOC pelo desequilíbrio dos sistemas enzimáticos e
dos sistemas de proteases e antiproteases e pela limitação ao fluxo aéreo
(MARTINELLO, 2009).

16
A DPOC é evitável, mas não curável. O tratamento pode simplesmente
ajudar a retardar a progressão da doença. Por isso é tão importante que fumantes
interrompam o uso do cigarro, para evitar que este propicie o surgimento e/ou a
piora do quadro desta doença, que em 2005 atingiu 65 milhões de pessoas, de
acordo com estimativas recentes divulgadas pela Organização Mundial de Saúde
(OMS) e que vitimou neste mesmo ano mais de 3 milhões de pessoas pelo
mundo (correspondendo a 5% de todas as mortes globais). E o total de óbitos por
DPOC pode aumentar em mais de 30% nos próximos 10 anos, tornando esta
doença a terceira maior causa de morte até 2030 (WHO, 2011).
Por isso a cessação tabágica é considerada um componente fundamental
de ações preventivas, e deveria ser evidente tanto para doentes quanto para os
profissionais de saúde, que a cessação tabágica é o tratamento mais importante
na DPOC. É reconhecido que nenhuma outra intervenção, além desta, melhora a
sobrevida destes doentes independentemente do estado de gravidade da doença
(PAMPLONA; MENDES, 2009).

17
2. OBJETIVOS
O objetivo geral deste trabalho é realizar uma revisão bibliográfica acerca
da relação da doença pulmonar obstrutiva crônica (DPOC) com o uso do tabaco
(tabagismo).
2.1. OBJETIVOS ESPECÍFICOS
Dentre os objetivos específicos estão:
1. A caracterização da DPOC, no que tange a sua patogênese,
epidemiologia, sintomatologia, diagnóstico e tratamento;
2. E a caracterização dos mecanismos pelos quais os agentes nocivos
presentes no cigarro levam ao surgimento da DPOC.

18
3. REVISÃO BIBLIOGRÁFICA
3.1. DOENÇA PULMONAR OBSTRUTIVA CRÔNICA
A doença pulmonar obstrutiva crônica é uma doença heterogênea com
diferentes apresentações clínicas, sendo caracterizada pela presença de
obstrução crônica do fluxo aéreo das vias pulmonares, geralmente progressiva e,
parcialmente reversível. É evitável em sua fase inicial e tratável em todas as
etapas (BRIGGS, 2004). Associa-se a uma resposta inflamatória anormal das vias
aéreas à inalação de gases tóxicos ou partículas nocivas, causada, sobretudo,
pelo tabagismo. A DPOC não é uma doença que compromete somente as vias
aéreas, mas também todo o corpo, através das manifestações sistêmicas da
inflamação da DPOC. Na verdade essas manifestações são responsáveis por
grande parte da morbidade e mortalidade associadas a essa doença (JARDIM;
OLIVEIRA; NASCIMENTO, 2004).
É descrita como uma combinação variada de alterações das vias aéreas
(bronquite crônica) e do parênquima pulmonar (enfisema pulmonar), produzidas
pelo processo inflamatório crônico. Cada componente tem características
singulares e se distingue do outro pela causa da obstrução do fluxo aéreo, pelo
surgimento da doença, localização e reversibilidade da obstrução. A
predominância de um ou outro componente é variável em cada indivíduo, tendo
relação com os sintomas apresentados (FERREIRA, 2010).
Para Bethlen (2001) a DPOC é descrita como um conjunto de alterações
clínicas, radiológicas, funcionais e patológicas do pulmão, que engloba a
bronquite crônica e o enfisema pulmonar.
De acordo com as diretrizes da Iniciativa Global para Doença Pulmonar
Obstrutiva Crônica (GOLD - Global Initiative for Chronic Obstructive Lung
Disease) a gravidade da DPOC pode ser classificada em 4 estágios (Tabela 1),
baseada principalmente em testes de função pulmonar, ou seja, capacidade vital
forçada (CVF) e volume expiratório forçado no primeiro segundo (VEF1)
(MARWICK; CHUNG, 2010; FISCHER; PAVLISKO; VOYNOW, 2011).

19
Tabela 1: Estágios GOLD de gravidade da DPOC
Gravidade da DPOC Características
VEF1/CVF VEF1
Estágio I (leve) < 70%
Estágio II (moderado) < 70% < 50%–70%
Estágio III (grave) < 70% < 30%–50%
Estágio IV (muito grave) < 70% < 30% ou < 30%–50% com insuficiência
respiratória (definida por PaO2, 60 mmHg
e/ou PCO2, 50 mmHg, ao nível do mar).
Abreviações: VEF1, volume expiratório forçado no primeiro segundo; CVF, capacidade vital
forçada; PaO2, pressão parcial de oxigênio no sangue arterial; PCO2, pressão parcial de dióxido de
carbono. Adaptado de: MARWICK; CHUNG, 2010
3.1.1. Bronquite Crônica
A bronquite crônica é uma doença pulmonar caracterizada por excessiva
produção de muco na arvore brônquica, suficiente para causar expectoração
excessiva de escarro, com relato de expectoração na maioria dos dias por mais
de três meses no ano, durante dois ou mais anos consecutivos, sempre que
outras causas estejam descartadas (WEST, 2002; ALAT, 2004). As principais
causas são o consumo de tabaco, a exposição ocupacional por longo período em
ambiente poluído e infecção. Estes fatores desencadeiam: a produção de muco
mais espesso, dificultando o movimento ciliar e gerando limitação ao fluxo aéreo;
o aumento da quantidade de muco intraluminal, produzindo alterações nas
pequenas vias aéreas, características da inflamação crônica (WEST, 2002).
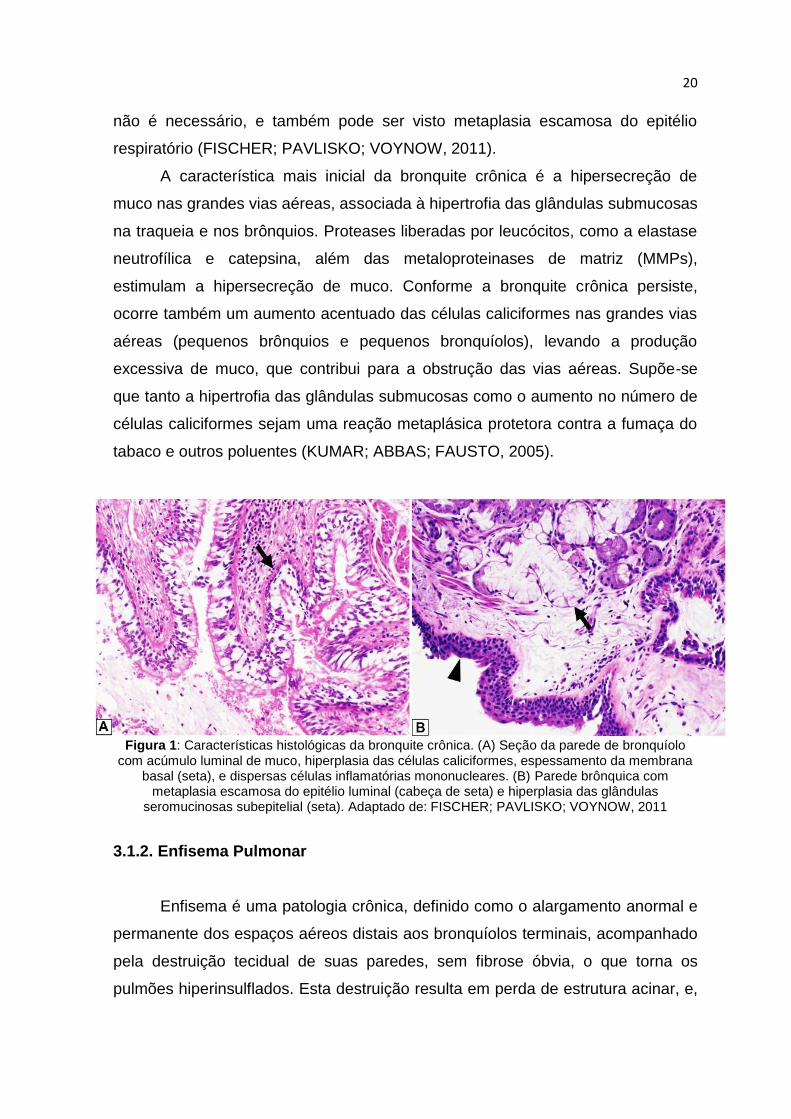
O exame macroscópico do tecido pulmonar em um paciente com bronquite
crônica mostra paredes brônquicas espessas com estreitamento luminal e
obstrução da mucosa ou detritos mucopurulentos nas vias aéreas.
Microscopicamente, estes resultados brutos correspondem à hiperplasia de
células caliciformes, espessamento da membrana basal subepitelial, fibrose da
parede brônquica, e hiperplasia das glândulas seromucinosas subepiteliais
(Figura 1). Um infiltrado inflamatório crônico pode também estar presente, mas
20
não é necessário, e também pode ser visto metaplasia escamosa do epitélio
respiratório (FISCHER; PAVLISKO; VOYNOW, 2011).
A característica mais inicial da bronquite crônica é a hipersecreção de
muco nas grandes vias aéreas, associada à hipertrofia das glândulas submucosas
na traqueia e nos brônquios. Proteases liberadas por leucócitos, como a elastase
neutrofílica e catepsina, além das metaloproteinases de matriz (MMPs),
estimulam a hipersecreção de muco. Conforme a bronquite crônica persiste,
ocorre também um aumento acentuado das células caliciformes nas grandes vias
aéreas (pequenos brônquios e pequenos bronquíolos), levando a produção
excessiva de muco, que contribui para a obstrução das vias aéreas. Supõe-se
que tanto a hipertrofia das glândulas submucosas como o aumento no número de
células caliciformes sejam uma reação metaplásica protetora contra a fumaça do
tabaco e outros poluentes (KUMAR; ABBAS; FAUSTO, 2005).
Figura 1: Características histológicas da bronquite crônica. (A) Seção da parede de bronquíolo
com acúmulo luminal de muco, hiperplasia das células caliciformes, espessamento da membrana basal (seta), e dispersas células inflamatórias mononucleares. (B) Parede brônquica com
metaplasia escamosa do epitélio luminal (cabeça de seta) e hiperplasia das glândulas seromucinosas subepitelial (seta). Adaptado de: FISCHER; PAVLISKO; VOYNOW, 2011
3.1.2. Enfisema Pulmonar
Enfisema é uma patologia crônica, definido como o alargamento anormal e
permanente dos espaços aéreos distais aos bronquíolos terminais, acompanhado
pela destruição tecidual de suas paredes, sem fibrose óbvia, o que torna os
pulmões hiperinsulflados. Esta destruição resulta em perda de estrutura acinar, e,

21
por conseguinte redução da área disponível para troca gasosa (Figura 2). As
pequenas vias aéreas estão estreitadas, tortuosas, e reduzidas em número, além
de possuírem paredes finas e atrofiadas. A perda de tecido elástico leva ao
colapso dessas pequenas vias aéreas e obstrui a saída de ar do interior dos
pulmões, que é frequentemente uma característica proeminente da doença.
Dentre as causas estão o tabagismo (principal fator etiológico), a poluição
atmosférica e a deficiência de α-1 antitripsina (KEMP; POLKEY; SHAH, 2009;
FERREIRA, 2010).
Figura 2: Foto de histopatologia de tecido pulmonar (x40). Painel A mostra o tecido de um pulmão
normal e painel B um pulmão enfisematoso, mostrando a perda das estruturas alveolares e da área de superfície no enfisema, sem fibrose aparente. Adaptado de: KEMP; POLKEY; SHAH,
2009
O fumo do cigarro provoca uma resposta inflamatória no trato respiratório
inferior, caracterizada pela acumulação de macrófagos alveolares carregados de
pigmento em conjunto com o recrutamento de números menores de neutrófilos.
Estas células inflamatórias ativadas liberam uma variedade de mediadores,
incluindo proteases, oxidantes e peptídeos tóxicos, que podem danificar as
estruturas pulmonares e acredita-se ser a causa principal da destruição de
tecidos, através da qual o enfisema é definido (RENNARD; TOGO; HOLZ, 2006).
O enfisema por deficiência de α-1 antitripsina atinge um em cada 2.000-
5.000 indivíduos. É um distúrbio genético de descoberta recente, resultante de
diferentes mutações no gene SERPINA1. A α-1 antitripsina é produzida
principalmente no fígado e atua como uma antiprotease, cuja função principal é
inativar a elastase neutrofílica, impedindo a ocorrência de dano tecidual. A

22
mutação mais frequentemente relacionada à doença clínica é o alelo Z, que
determina polimerização e acúmulo dentro dos hepatócitos. O acúmulo e a
consequente redução dos níveis séricos de α-1 antitripsina determinam,
respectivamente, doença hepática e pulmonar, sendo que esta se manifesta
principalmente sob a forma de enfisema de aparecimento precoce, habitualmente
com predomínio basal (CAMELIER et al., 2008).
Em face da lesão, o pulmão, como a maioria dos tecidos, pode iniciar
mecanismos de reparação. A destruição da rede tecidual que caracteriza
enfisema representa um desequilíbrio entre a destruição dos tecidos e os
processos de reparação de tecidos, análogo ao equilíbrio protease-antiprotease.
Na hipótese protease-antiprotease “clássica” de enfisema, a destruição do
tecido resulta quando a elastase neutrofílica prevalece sobre a proteção
endógena fornecida pela α-1 antitripsina. Este equilíbrio pode ser rompido, quer
por aumento da carga de elastase (por exemplo, com inflamação originada do
fumo) ou através da diminuição da proteção antielastase (por exemplo, pela
deficiência genética de α-1 antitripsina) (Figura 3 A). Na hipótese dos mediadores
de lesão expandidos, o conceito clássico foi expandido para incluir várias classes
de antiproteases, cada uma com muitos membros, bem como outros mediadores
prejudiciais, incluindo oxidantes e peptídeos tóxicos. Existe um conjunto
correspondente de inibidores para cada um destes mediadores, sendo que o
saldo global determina os resultados de lesão tecidual (Figura 3 B). Na hipótese
da reparação da lesão, os tecidos podem reparar lesões, a integridade do tecido
pode ser mantida em face da lesão se a reparação é adequada. Destruição dos
tecidos, por conseguinte, representa um desequilíbrio entre a lesão do tecido e a
reparação (Figura 3 C) (RENNARD; TOGO; HOLZ, 2006).
Embora a deficiência de α-1 antitripsina continue a ser o único distúrbio
genético bem caracterizado associado com DPOC, um número de outros
candidatos têm sido sugeridos. Entre eles estão outras antiproteases,
antioxidantes, e mutações em genes defensina (um tipo de peptídeo tóxico),
consistente com o conceito de um desequilíbrio entre lesão tecidual e proteção do
tecido (RENNARD; TOGO; HOLZ, 2006).
O enfisema pode ser classificado em alguns subtipos: centrolobular (ou
centroacinar), panlobular (ou panacinar) e parasseptal. Enfisema centrolobular é

23
caracterizado pela perda de bronquíolos respiratórios, com um grau escasso dos
alvéolos distais, e afeta predominantemente as porções superiores do pulmão.
Este padrão é aquele que é mais comumente visto em fumantes. O enfisema
panacinar afeta o ácino inteiro de modo uniforme (destruição generalizada do
tecido alveolar); é visto predominantemente nos lobos inferiores, e é o padrão
mais associado com a deficiência de α-1 antitripsina. Enfisema parasseptal
(também conhecido como acinar distal) é localizado em torno do septo e pleura, e
afeta as estruturas acinares distais (KEMP; POLKEY; SHAH, 2009).
Figura 3: Enfisema representa um desequilíbrio. Adaptado de: RENNARD; TOGO; HOLZ, 2006
O enfisema centrolobular geralmente acomete as regiões mais superiores do
pulmão, causando grande alteração na relação ventilação-perfusão, pois o
suprimento de sangue está relativamente preservado para os alvéolos, mas o
oxigênio que chega aos capilares é reduzido devido aos danos nas vias aéreas
proximais. Como o enfisema panacinar afeta mais os lobos inferiores (Figura 4),
seu efeito é menos drástico na relação ventilação-perfusão, pois o suprimento

24
sanguíneo reduz à medida que há a diminuição da ventilação nessas regiões
(LIMA et al., 2007).
Figura 4: Enfisema centrolobular e panacinar. Observar que no enfisema centrolobular, a
destruição é limitada aos bronquíolos terminais e respiratórios (BT e BR). No enfisema panacinar os alvéolos periféricos (A) também são comprometidos. Adaptado de: WEST, 1996
No exame microscópio do tecido pulmonar de um paciente com enfisema
observam-se alvéolos anormalmente grandes, separados por finos septos que
apresentam somente uma fibrose centroacinar focal. Vê-se o desaparecimento
das fixações dos alvéolos à parede externa das vias aéreas. Com a progressão
da doença, observam-se espaços aéreos anormais ainda maiores e
possivelmente bolhas. Geralmente, os bronquíolos respiratórios e a vasculatura
do pulmão estão deformados e comprimidos pela distorção enfisematosa dos
espaços aéreos (KUMAR; ABBAS; FAUSTO, 2005).
As manifestações clínicas do enfisema não aparecem até que pelo menos
um terço do parênquima pulmonar funcionante seja danificado. A dispneia
costuma ser o primeiro sintoma, que começa de forma insidiosa e progride de
forma constante. Em alguns pacientes, a tosse é a principal queixa, facilmente
confundida com asma (KUMAR; ABBAS; FAUSTO, 2005).
3.2. EPIDEMIOLOGIA
A DPOC é uma das doenças mais comuns e é uma das principais causas de
morte em todo o mundo. Além de gerar custos elevados de saúde, a DPOC impõe
uma carga significativa em termos de incapacidade e qualidade de vida
comprometida. Ao contrário de muitas causas de morte e invalidez, a incidência

25
da DPOC é projetada para aumentar em muitas partes do mundo com o
envelhecimento da população e principalmente com a ampla difusão de seu
principal fator etiológico, o fumo do tabaco. O uso crescente de tabaco no mundo
em desenvolvimento permite a previsão de um grande aumento no número de
casos no futuro próximo e distante (PLANTIER; BOCZKOWSKI; CRESTANI,
2007; HALBERT et al., 2006).
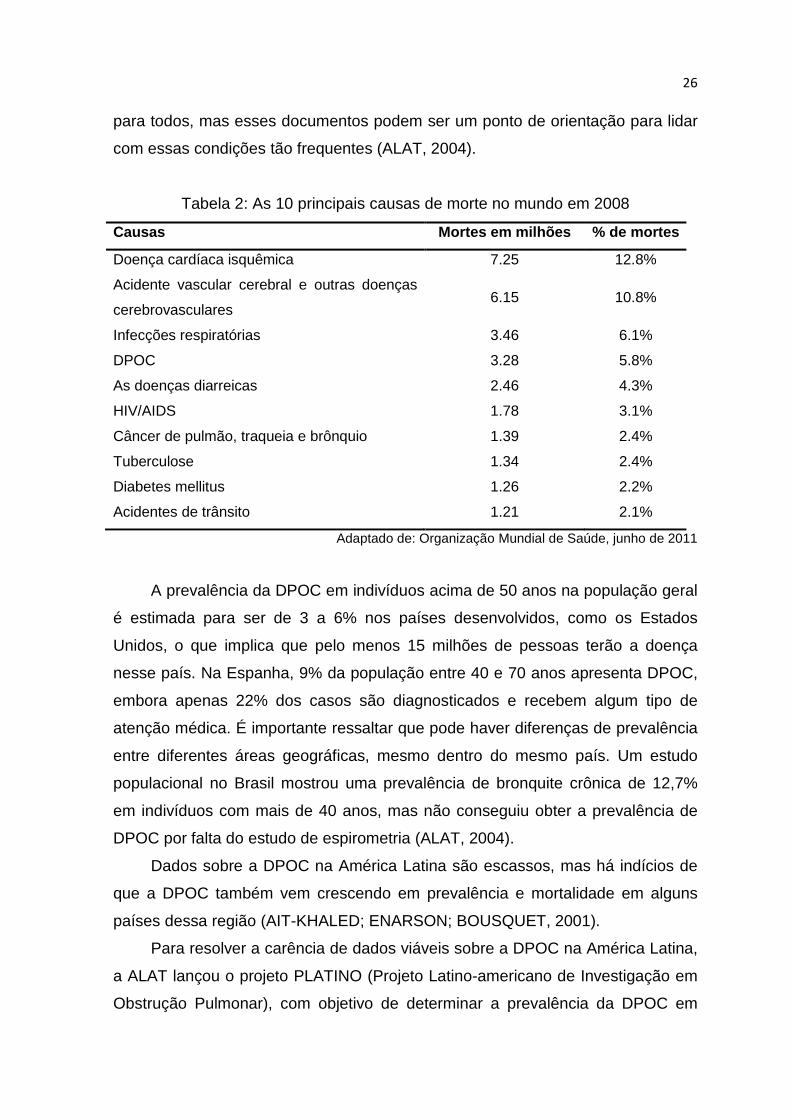
De acordo com a OMS, em 2002, a DPOC foi a quinta maior causa de
morte. E segundo estimativas, em 2007, 210 milhões de pessoas em todo o
mundo tinham DPOC, dos quais 80 milhões se encontravam na faixa de
gravidade moderada à grave. E neste mesmo ano, mais de 3 milhões de pessoas
morreram devido a esta doença, correspondendo a 5% do total de óbitos
registrados no mundo. E conforme boletim divulgado pela OMS em 2011,
referente a dados de 2008, ela já passa a ser a quarta maior causa de morte,
representando 5,8% do total de mortes (Tabela 2).
O total de óbitos por DPOC são projetados para aumentar em mais de 30%
nos próximos 10 anos, a menos que medidas urgentes sejam tomadas para
reduzir os fatores de risco, em especial o uso do cigarro. As estimativas mostram
que a DPOC torna-se em 2030 a terceira maior causa de morte no mundo (WHO,
2011).
Uma meta-análise realizada por Halbert e seus colaboradores sugere que a
prevalência da DPOC é maior entre homens do que mulheres, entre fumantes e
ex-fumantes do que não fumantes, e entre os indivíduos com mais de 40 anos do
que aqueles com menos de 40. No entanto, a prevalência da DPOC pode variar
de forma significativa para uma determinada população, dependendo de qual
definição é usada (HALBERT et al., 2006).
No ano de 2000, a Associação Latino-Americana de Tórax (ALAT), reuniu
um grupo multidisciplinar de especialistas para elaborar as primeiras
recomendações sobre a exacerbação infecciosa da DPOC e pneumonia adquirida
na comunidade. Estas orientações foram publicadas em 2001 e pretendiam ser
uma adaptação à realidade latino-americana de conhecimento sobre a gestão e
tratamento destas infecções respiratórias. Devido ao tamanho e heterogeneidade
da América Latina, é difícil estabelecer um padrão que possa ser igualmente útil
26
para todos, mas esses documentos podem ser um ponto de orientação para lidar
com essas condições tão frequentes (ALAT, 2004).
Tabela 2: As 10 principais causas de morte no mundo em 2008
Causas Mortes em milhões % de mortes
Doença cardíaca isquêmica 7.25 12.8%
Acidente vascular cerebral e outras doenças
cerebrovasculares 6.15 10.8%
Infecções respiratórias 3.46 6.1%
DPOC 3.28 5.8%
As doenças diarreicas 2.46 4.3%
HIV/AIDS 1.78 3.1%
Câncer de pulmão, traqueia e brônquio 1.39 2.4%
Tuberculose 1.34 2.4%
Diabetes mellitus 1.26 2.2%
Acidentes de trânsito 1.21 2.1%
Adaptado de: Organização Mundial de Saúde, junho de 2011
A prevalência da DPOC em indivíduos acima de 50 anos na população geral
é estimada para ser de 3 a 6% nos países desenvolvidos, como os Estados
Unidos, o que implica que pelo menos 15 milhões de pessoas terão a doença
nesse país. Na Espanha, 9% da população entre 40 e 70 anos apresenta DPOC,
embora apenas 22% dos casos são diagnosticados e recebem algum tipo de
atenção médica. É importante ressaltar que pode haver diferenças de prevalência
entre diferentes áreas geográficas, mesmo dentro do mesmo país. Um estudo
populacional no Brasil mostrou uma prevalência de bronquite crônica de 12,7%
em indivíduos com mais de 40 anos, mas não conseguiu obter a prevalência de
DPOC por falta do estudo de espirometria (ALAT, 2004).
Dados sobre a DPOC na América Latina são escassos, mas há indícios de
que a DPOC também vem crescendo em prevalência e mortalidade em alguns
países dessa região (AIT-KHALED; ENARSON; BOUSQUET, 2001).
Para resolver a carência de dados viáveis sobre a DPOC na América Latina,
a ALAT lançou o projeto PLATINO (Projeto Latino-americano de Investigação em
Obstrução Pulmonar), com objetivo de determinar a prevalência da DPOC em

27
diferentes países da área e verificar a associação entre DPOC e alguns fatores de
risco, utilizando para isso um protocolo comum (ALAT, 2004). Cinco grandes
centros foram eleitos para representar as diferentes regiões da América Latina. O
critério de seleção das cidades foi baseado em sua posição geográfica, tamanho
da população, relevância em relação ao país e a disponibilidade de centros de
pesquisa locais de colaboração, sendo eles: São Paulo (Brasil), Cidade do México
(México), Santiago (Chile), Caracas (Venezuela) e Montevidéu (Uruguai)
(PLATINO, 2006).
O estudo PLATINO realizado no Brasil avaliou indivíduos com idade igual ou
superior a 40 anos, residentes na área metropolitana da grande São Paulo. Os
resultados mostraram que a prevalência total de DPOC variou de 6 a 15,8%, de
acordo com a definição utilizada, o que seria equivalente a 2.800.000 a 6.900.000
indivíduos com DPOC (JARDIM; OLIVEIRA; NASCIMENTO, 2004).
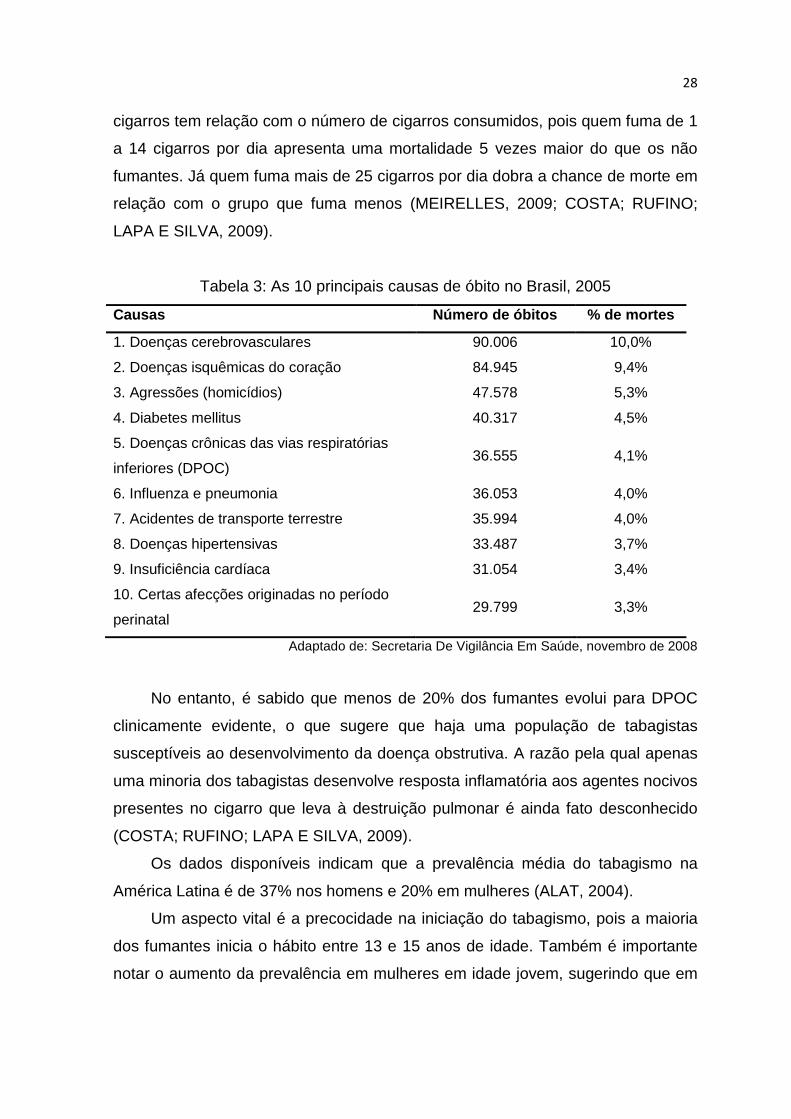
A DPOC, em 2003, foi a quinta maior causa de internamento no sistema
público de saúde do Brasil, em maiores de 40 anos, com 196.698 internações e
gastos aproximados de 72 milhões de reais. E vem ocorrendo um aumento no
número de óbitos por DPOC nos últimos 20 anos, em ambos os sexos, tendo a
taxa de mortalidade passado de 7,88 em cada 100.000 habitantes, na década de
1980, para 19,04 em cada 100.000 habitantes na década de 1990, com um
crescimento de 340%. A DPOC nos últimos anos vem ocupando da 4ª à 7ª
posição entre as principais causas de morte no Brasil (Tabela 3) (JARDIM;
OLIVEIRA; NASCIMENTO; 2004).
3.3. FATORES DE RISCO
A DPOC se desenvolve devido à presença e atuação de vários fatores de
risco. Embora se tenha identificado vários destes fatores, o tabagismo é de longe
o principal fator de risco isolado para a doença (ALAT, 2004). E mais: está
relacionado a um risco aumentado de óbito devido a DPOC, carcinoma pulmonar
e doença coronariana isquêmica. O aumento de risco está diretamente
relacionado à carga tabágica. Fumantes de cigarros apresentam um risco 10 a 14
vezes maior de morte por DPOC, e os de charutos e cachimbos um risco 6 vezes
maior de morte por essa patologia. A mortalidade por DPOC em fumantes de
28
cigarros tem relação com o número de cigarros consumidos, pois quem fuma de 1
a 14 cigarros por dia apresenta uma mortalidade 5 vezes maior do que os não
fumantes. Já quem fuma mais de 25 cigarros por dia dobra a chance de morte em
relação com o grupo que fuma menos (MEIRELLES, 2009; COSTA; RUFINO;
LAPA E SILVA, 2009).
Tabela 3: As 10 principais causas de óbito no Brasil, 2005
Causas Número de óbitos % de mortes
1. Doenças cerebrovasculares 90.006 10,0%
2. Doenças isquêmicas do coração 84.945 9,4%
3. Agressões (homicídios) 47.578 5,3%
4. Diabetes mellitus 40.317 4,5%
5. Doenças crônicas das vias respiratórias
inferiores (DPOC) 36.555 4,1%
6. Influenza e pneumonia 36.053 4,0%
7. Acidentes de transporte terrestre 35.994 4,0%
8. Doenças hipertensivas 33.487 3,7%
9. Insuficiência cardíaca 31.054 3,4%
10. Certas afecções originadas no período
perinatal 29.799 3,3%
Adaptado de: Secretaria De Vigilância Em Saúde, novembro de 2008
No entanto, é sabido que menos de 20% dos fumantes evolui para DPOC
clinicamente evidente, o que sugere que haja uma população de tabagistas
susceptíveis ao desenvolvimento da doença obstrutiva. A razão pela qual apenas
uma minoria dos tabagistas desenvolve resposta inflamatória aos agentes nocivos
presentes no cigarro que leva à destruição pulmonar é ainda fato desconhecido
(COSTA; RUFINO; LAPA E SILVA, 2009).
Os dados disponíveis indicam que a prevalência média do tabagismo na
América Latina é de 37% nos homens e 20% em mulheres (ALAT, 2004).
Um aspecto vital é a precocidade na iniciação do tabagismo, pois a maioria
dos fumantes inicia o hábito entre 13 e 15 anos de idade. Também é importante
notar o aumento da prevalência em mulheres em idade jovem, sugerindo que em

29
poucos anos o número de tabagismo entre homens e mulheres tende a equalizar
(ALAT, 2004).
A prevalência do principal fator de risco para a DPOC - tabagismo - é alta na
América Latina. A média de consumo de cigarros anuais por pessoa, na maioria
dos países latino-americanos varia de 500 a 1499. Como na maioria dos países
de alta renda, há alguma evidência de que a prevalência de tabagismo em
homens é ligeiramente decrescente, enquanto que para as mulheres está
aumentando. Mesmo que as taxas de prevalência do tabagismo caiam
ligeiramente, o número absoluto de fumantes pode aumentar devido ao
crescimento populacional (MENEZES et al., 2005).
Embora de menor relevância, é importante notar outros fatores de risco para
a ocorrência da DPOC (Tabela 4). A poluição atmosférica é um fator que tem sido
associada com aumento da mortalidade, especialmente em pessoas com
doenças pulmonares crônicas. A DPOC pode ser agravada (embora haja menos
evidências) pela exposição à poluição ambiental, especialmente causada por
partículas, e esta pode causar sintomas de exacerbação, com internações e
mortalidade (ALAT, 2004).
Tabela 4: Fatores de risco para DPOC
Fatores externos Fatores individuais
Tabagismo (Principal) Deficiência de alfa-1 antitripsina
Poeira ocupacional Deficiência de glutationa transferase
Irritantes químicos Alfa-1 antiquimotripsina
Fumaça de lenha Hiper-responsividade brônquica
Infecções respiratórias graves na infância Desnutrição
Condição socioeconômica Prematuridade
Adaptado de: JARDIM; OLIVEIRA; NASCIMENTO; 2004
A poluição em domicílio pelo consumo de lenha ou outra biomassa tem sido
implicada na patogênese da DPOC. Em alguns países latino-americanos, fogões
a lenha podem ser encontrados em áreas rurais, emitindo quantidades
significativas de fumaça intradomicílio. Fumaça de madeira tem muitas
substâncias tóxicas e cancerígenas, semelhantes àquelas encontradas no cigarro,

30
e considera-se que 50% da população mundial recorrem a ela como fonte de
energia (ALAT, 2004).
Outros fatores de risco para a incidência da DPOC variam em diferentes
grupos populacionais. A exposição ocupacional a fumaças e poeiras, embora a
sua importância seja pequena se não for associada ao consumo de tabaco.
Também deve ser considerado: graves infecções respiratórias na infância, que
colocam uma maior susceptibilidade a outros agentes que podem ser adicionados
no futuro, como fumaça de tabaco, a exposição à fumaça de lenha ou exposições
ocupacionais (ALAT, 2004).
Finalmente, devemos ter em mente que fatores específicos ao indivíduo, tais
como hiper-reatividade brônquica ou mutações em genes que codificam proteínas
com efeito protetor da região pulmonar, como a α-1 antitripsina, podem influenciar
a suscetibilidade de ter DPOC (ALAT, 2004).
3.4. PATOGÊNESE
A compreensão da patogênese da DPOC tem aumentado ao longo dos
últimos 25 anos. É agora evidente que uma variedade de processos patológicos
está envolvida no desenvolvimento da DPOC. A resposta do hospedeiro a gases
e partículas inaladas desempenha um papel importante no desenvolvimento da
doença (ANDERSON; MACNEE, 2009).
O tabagismo é o principal fator de risco ambiental para a DPOC. Além disso,
outras exposições ambientais (por exemplo, fumaça de madeira) e ocupacionais,
bem como a genética contribuem para a patogênese da DPOC. Por conseguinte,
as alterações patológicas e sintomas clínicos estão ligados à interação dos
fatores do hospedeiro com os fatores ambientais. Essas interações geram a tríade
patológica da DPOC: inflamação persistente, desequilíbrio protease–antiprotease
e estresse oxidativo. Esta tríade resulta em metaplasia e hiperplasia de células
mucosas/caliciformes, hipersecreção de muco, fibrose, alterações do músculo liso
e destruição do tecido pulmonar, que levam aos principais sintomas, que são
tosse crônica, produção de catarro e falta de ar incapacitante. A patologia
subjacente é causada por uma “resposta inflamatória anormal do pulmão a
partículas ou gases nocivos” sendo os neutrófilos, macrófagos e linfócitos T as
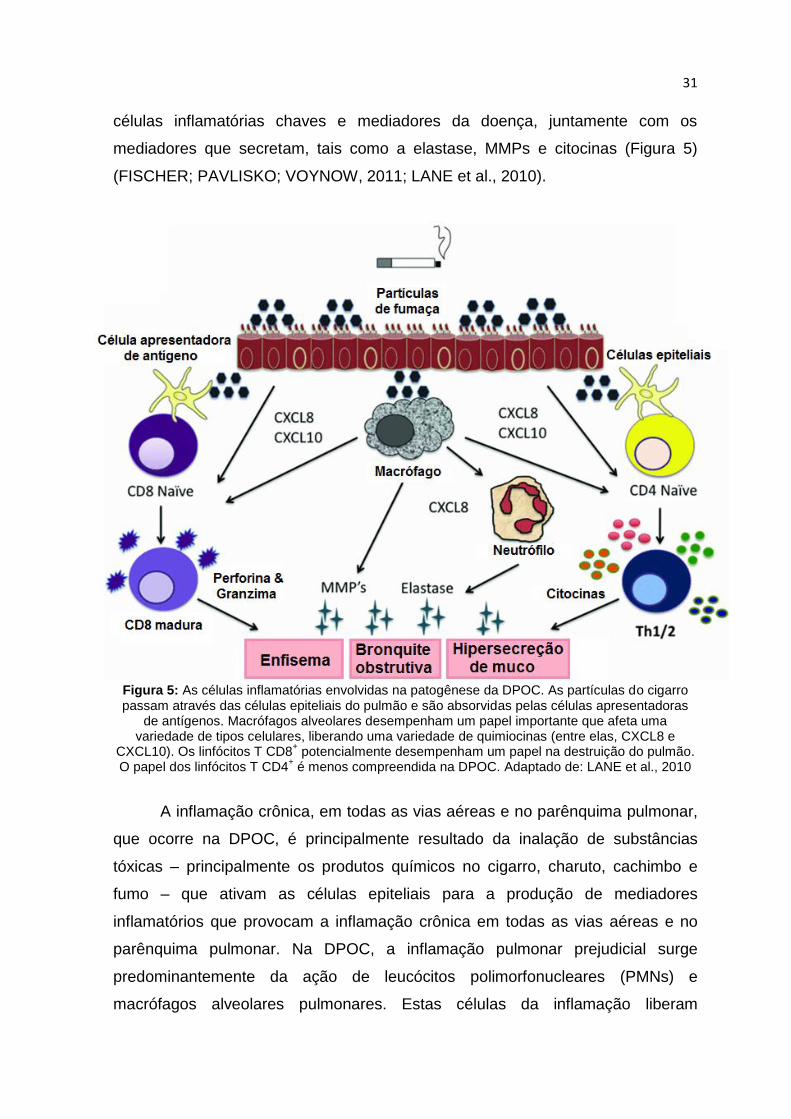
31
células inflamatórias chaves e mediadores da doença, juntamente com os
mediadores que secretam, tais como a elastase, MMPs e citocinas (Figura 5)
(FISCHER; PAVLISKO; VOYNOW, 2011; LANE et al., 2010).
Figura 5: As células inflamatórias envolvidas na patogênese da DPOC. As partículas do cigarro passam através das células epiteliais do pulmão e são absorvidas pelas células apresentadoras
de antígenos. Macrófagos alveolares desempenham um papel importante que afeta uma variedade de tipos celulares, liberando uma variedade de quimiocinas (entre elas, CXCL8 e
CXCL10). Os linfócitos T CD8+ potencialmente desempenham um papel na destruição do pulmão.
O papel dos linfócitos T CD4+ é menos compreendida na DPOC. Adaptado de: LANE et al., 2010
A inflamação crônica, em todas as vias aéreas e no parênquima pulmonar,
que ocorre na DPOC, é principalmente resultado da inalação de substâncias
tóxicas – principalmente os produtos químicos no cigarro, charuto, cachimbo e
fumo – que ativam as células epiteliais para a produção de mediadores
inflamatórios que provocam a inflamação crônica em todas as vias aéreas e no
parênquima pulmonar. Na DPOC, a inflamação pulmonar prejudicial surge
predominantemente da ação de leucócitos polimorfonucleares (PMNs) e
macrófagos alveolares pulmonares. Estas células da inflamação liberam

32
proteases que irritam as vias aéreas, aumentam a secreção de muco, destroem
as paredes alveolares, e perpetuam e agravam a atividade inflamatória em curso.
Além disso, níveis elevados de certas citocinas, principalmente, a interleucina (IL)-
8, leucotrieno B4 (LTB4), e fator de necrose tumoral alfa (TNF-α), ampliam e
perpetuam o processo inflamatório na DPOC, mesmo depois da cessação
tabágica (BRIGGS, 2004).
Como a DPOC é uma doença inflamatória crônica dos pulmões que
progride lentamente, a maioria dos pacientes são, portanto, idosos. Existe uma
evidência crescente para uma relação estreita entre o envelhecimento e doenças
inflamatórias crônicas que conduzem à hipótese de que o envelhecimento
acelerado do pulmão em resposta ao estresse oxidativo pode estar envolvido na
patogênese e progressão da DPOC, particularmente enfisema. Isto resulta de
uma falha dos órgãos em reparar danos no ácido desoxirribonucleico (DNA) pelo
estresse oxidativo (envelhecimento não programado) e do encurtamento dos
telômeros, como resultado da divisão celular (envelhecimento programado).
Comprimento dos telômeros é efetivamente considerado como um marcador para
o envelhecimento biológico, e o encurtamento em excesso dos telômeros é
consistentemente encontrado na DPOC (COSIO; AGUSTÍ, 2010).
O tabagismo é um gatilho para a senescência, o processo natural de
envelhecimento ao nível celular. Uma das sequelas da senescência é o
desenvolvimento de um fenótipo pró-inflamatório. Em tecidos pulmonares de
pacientes com DPOC comparados com fumantes saudáveis e indivíduos-controle
não fumantes, a ativação do fator nuclear (NF)-κB está aumentada em células
senescentes alveolares do tipo II. Além disso, em comparação com controles não
fumantes, há uma redução na expressão da proteína sirtuína-1 (SIRT1), uma
proteína anti-inflamatória e antienvelhecimento que regula negativamente fatores
de transcrição tais como NF-κB, nos pulmões de pacientes com DPOC. Assim, as
células senescentes podem propagar a resposta inflamatória na DPOC
(FISCHER; PAVLISKO; VOYNOW, 2011).
Uma hipótese intrigante sugerindo um papel para a autoimunidade na
patogênese da DPOC tem emergido nos últimos anos e está amadurecendo
rapidamente (COSIO; AGUSTÍ, 2010). A observação de que apenas 20-30% de
fumantes habituais vão desenvolver DPOC sugere um grau de suscetibilidade

33
genética entre os indivíduos. Adicionado a isto, o fato de que a inflamação
pulmonar progressiva permanece, mesmo após a cessação tabágica, levanta a
possibilidade de que houve uma avaria na autotolerância resultante da lesão
tecidual causada pelos estímulos nocivos iniciais. De fato, os avanços recentes na
compreensão da patogênese da doença indicam que pacientes com DPOC
apresentam muitos dos mesmos recursos que pacientes que sofrem de doenças
autoimunes clássicas (STEFANSKA; WALSH, 2009).
Propõe-se que os mecanismos imunológicos que levam a DPOC seguem
três etapas. A primeira é caracterizada por uma resposta imunológica inata a
fumaça do cigarro que se baseia no reconhecimento de danos nos tecidos por
receptores Toll-like e é acionado por células epiteliais, macrófagos e neutrófilos. A
segunda etapa envolve a ativação das células T e proliferação, bem como a
maturação de células dendríticas conhecidas por aumentar nos pulmões de
pacientes com a progressão da DPOC. O terceiro e último passo seria uma
reação imune adaptativa impulsionada por células T citotóxicas CD8+, células T
helper 1 e células B oligoclonais, todas as quais são encontradas em pacientes
com DPOC grave. Estas células expressam o receptor de quimiocina CXCR3 e os
seus ligantes, que contribuem para formação de folículos linfoides. Este último
passo contribuiria para a persistência dos anos da resposta inflamatória após a
cessação tabágica, em resposta a autoantígenos produzidos ao longo de todo
este processo, uma característica de doenças autoimunes. No entanto, devem ser
realizadas mais pesquisas para demonstrar a relação causa-efeito entre
autoimunidade e o desenvolvimento da DPOC (COSIO; AGUSTÍ, 2010).
3.4.1. Genética da DPOC
A DPOC é influenciada por múltiplos determinantes genéticos, mas a grave
deficiência de α-1 antitripsina é o único fator de risco genético comprovado. Dado
o papel claro do tabagismo nessa doença e as diferenças interindividuais na
resposta à fumaça de cigarro, etiologia da DPOC é esperada para incluir
interações gene-ambiente (HIZAWA, 2009).
Numerosos genes candidatos que podem estar ligados à patogênese da
doença têm sido implicados na genética da DPOC. Esses estudos têm fornecido

34
evidências para o possível papel de muitos mediadores inflamatórios e seus
receptores, proteases, antiproteases e antioxidantes na fisiopatologia da DPOC.
Por exemplo, uma revisão sistemática da literatura caracterizou a evidência de
que os genes que codificam enzimas antioxidantes contribuem para a etiologia da
DPOC e traços relacionados; os efeitos mais fortes e mais consistente eram nos
genes que codificam glutamato-cisteína ligase (GCL), glutationa S-transferase M1
(GSTM1), glutationa S-transferase P1 (GSTP1), e superóxido dismutase 3
(SOD3) (HIZAWA, 2009).
3.4.2. O Estresse Oxidativo
Há evidências consideráveis de aumento do estresse oxidativo na DPOC. O
aumento do estresse oxidativo nas vias aéreas de pacientes com DPOC pode
desempenhar um importante papel fisiopatológico na doença, amplificando a
resposta inflamatória na DPOC (Figura 6). As células inflamatórias e estruturais
que são ativadas nas vias aéreas de pacientes com DPOC produzem espécies
reativas de oxigênio (EROs), incluindo, neutrófilos, eosinófilos, macrófagos e
células epiteliais (BARNES; SHAPIRO; PAUWELS, 2003).
O estresse oxidativo ocorre quando EROs são produzidas em excesso dos
mecanismos de defesa antioxidantes e resultam em efeitos nocivos, incluindo
danos aos lipídios, proteínas e DNA (BARNES; SHAPIRO; PAUWELS, 2003).
A inalação da fumaça de cigarro, e demais derivados do tabaco produtores
de fumaça, expõe os pulmões a altas concentrações de agentes oxidantes e
radicais livres que diminuem a capacidade antioxidante que normalmente protege
as células epiteliais (MEIRELLES, 2009). A fumaça do tabaco contém um
conjunto de mais de 4.700 compostos químicos. Na verdade, cada tragada
contém 1015 radicais livres na fase gasosa e 1018 radicais livres por grama de
alcatrão, e inclui oxidantes potentes, tais como peróxido de hidrogênio, ânion
hidroxila e radicais orgânicos (TUDER et al., 2006).
O fumo do cigarro em si contém uma elevada concentração de EROs, e
estas apresentam vários efeitos sobre as vias respiratórias, principalmente
aumento da resposta inflamatória. Estes efeitos podem ser mediados por ações
diretas das EROs nas células alvo das vias aéreas, mas podem também ser

35
mediados indiretamente através da ativação de vias de transdução de sinal e dos
fatores de transcrição e através da formação de mediadores oxidados, tais como
isoprostanos e hidroxi-nonenal (BARNES; SHAPIRO; PAUWELS, 2003).
Figura 6: O estresse oxidativo e seus efeitos. Oxidantes contidos na fumaça do cigarro irritam as células epiteliais (1), liberando citocinas ativadoras que induzem o recrutamento dos neutrófilos e a liberação de oxidantes derivados de células (2) e proteases (3). Antioxidantes inibem os danos mediados por oxidante ao pulmão (4), mas quando surge um desequilíbrio (talvez por causa dos
polimorfismos dos genes) resulta no estresse oxidativo (5). As consequências do estresse oxidativo incluem a ativação de macrófagos (6), que conduz à produção de mais proteases, hipersecreção de muco, apoptose de células epiteliais, inflamação e a inibição da ação das
antiproteases. Adaptado de: HEALTHY LIFESTYLE, 2007
Embora haja no corpo uma bateria de sistemas antioxidantes, os quais
controlam a produção de oxidantes e os seus potenciais efeitos negativos, a
presença da fumaça do tabaco altera o equilíbrio entre oxidantes e antioxidantes
e conduz o sistema a uma situação de estresse oxidativo (PUERTO-NEVADO et
al., 2010).
A produção normal de oxidantes é neutralizada por vários mecanismos
antioxidantes no trato respiratório humano. Os principais antioxidantes
intracelulares nas vias aéreas são catalase, SOD e glutationa, formada pelas
enzimas γ-glutamil-cisteína sintetase e glutationa sintetase. No pulmão,
antioxidantes intracelulares são expressos em níveis relativamente baixos e não

36
são induzidos pelo estresse oxidativo, enquanto que os principais antioxidantes
são extracelulares. Antioxidantes extracelulares, particularmente glutationa
peroxidase, são marcadamente regulados em resposta à fumaça de cigarro e
estresse oxidativo. O sistema da glutationa é o principal mecanismo antioxidante
nas vias aéreas. Existe uma elevada concentração de glutationa reduzida no
fluido de revestimento epitelial do pulmão e as concentrações são aumentadas
nos fumantes de cigarros. Glutationa peroxidase extracelular (GPxe) é um
importante antioxidante nos pulmões e pode ser secretada pelas células epiteliais
e macrófagos, particularmente em resposta à fumaça do cigarro ou ao estresse
oxidativo. GPxe inativa as EROs H2O2 e O2-, mas também pode bloquear as
espécies reativas de nitrogênio. Antioxidantes extracelulares também incluem
ácido úrico, lactoferrina, SOD3 extracelular, e antioxidantes da dieta: vitamina C
(ácido ascórbico) e vitamina E (α-tocoferol) (BARNES; SHAPIRO; PAUWELS,
2003).
No entanto, a exposição à fumaça do tabaco produz alterações importantes
na homeostase da glutationa, produzindo uma diminuição na sua concentração
assim como na atividade das enzimas envolvidas neste ciclo redox (PUERTO-
NEVADO et al., 2010).
O tabagismo expõe a árvore respiratória e os pulmões às EROs, resultando
em estresse oxidativo e lesão. Isso desencadeia a produção de outras EROs e
peroxidação lipídica e inflamação pulmonar subsequente. Por exemplo, o
aumento da expressão de 4-hidroxi-2-nonenal, um produto de peroxidação
lipídica, está presente nas vias aéreas e no epitélio alveolar de pacientes com
DPOC. Do mesmo modo, o malondialdeído (MDA, C3H4O2), um produto final da
peroxidação lipídica também aumenta no sangue de pacientes com DPOC e
aumenta com a gravidade da doença (FISCHER; PAVLISKO; VOYNOW, 2011).
EROs ativam o NF-κB, que liga múltiplos genes inflamatórios, resultando em
amplificação da resposta inflamatória. As vias moleculares pelas quais o estresse
oxidativo ativa NF-κB não foram completamente elucidadas. Muitos dos estímulos
que ativam o NF-κB parecem fazê-lo através da formação de EROs,
particularmente H2O2. EROs ativa NF-κB numa linha celular epitelial e aumenta a
liberação de citocinas pró-inflamatórias. Estresse oxidativo resulta em ativação da
atividade de histona-acetiltransferase o que abre a estrutura da cromatina e está

37
associada com a transcrição aumentada de múltiplos genes inflamatórios. Outro
fator de transcrição que ativa genes inflamatórios é o Ativador de proteína-1 (AP-
1), um heterodímero de proteínas Fos e Jun. O estresse oxidativo pode refletir na
ativação de NF-κB e de AP-1, que em seguida, induzem uma via inflamatória
neutrofílica aumentando a expressão de IL-8 e outras quimiocinas CXC, TNF-α e
MMP-9. NF-κB é ativado nas vias aéreas e macrófagos alveolares de pacientes
com DPOC e está mais ativado durante as exacerbações. É provável que o
estresse oxidativo seja um importante ativador deste fator de transcrição em
pacientes com DPOC (BARNES; SHAPIRO; PAUWELS, 2003).
O fumo do cigarro, e em menor extensão, o dióxido de nitrogênio, impõe um
estresse oxidativo nas vias aéreas, gerando H2O2, um potente ativador da via da
proteína quinase ativada por mitógenos (MAPK) p38 que regula a expressão de
muitos genes inflamatórios e de sobrevivência em certas células, e a propagação
dos macrófagos (BARNES; SHAPIRO; PAUWELS, 2003).
O estresse oxidativo pode também prejudicar a função de antiproteases
como α-1 antitripsina e Inibidor da leucoprotease secretora, e, assim, acelerar a
quebra de elastina no parênquima pulmonar (BARNES; SHAPIRO; PAUWELS,
2003).
Estresse oxidativo pode também induzir a apoptose em células estruturais,
células epiteliais alveolares e células endoteliais. A apoptose de pneumócitos tipo
1 pode contribuir para o desenvolvimento de enfisema e isto pode ser induzido
por linfócitos T citotóxicos ou por inibição dos receptores do fator de crescimento
endotelial vascular. EROs podem induzir a apoptose por ativação da via de NF-
κB, por danos diretos no DNA através da ativação da poli adenosina difosfato
ribose e através da geração de 4-hidroxi-nonenal (um produto de peroxidação
lipídica). O sinal de apoptose regulado pela quinase-1 é realizado em uma
conformação inativa por tiorredoxina e quando oxidados por EROs isso dispara
vias apoptóticas (BARNES; SHAPIRO; PAUWELS, 2003; FISCHER; PAVLISKO;
VOYNOW, 2011)

38
3.4.3. Inflamação
Pacientes com DPOC em comparação com indivíduos controle
experimentam um estado geral elevado de inflamação sistêmica refletida por
biomarcadores, como citocinas e óxido nítrico. Da mesma forma, lavado
broncoalveolar (LBA) e escarro de pacientes também demonstram um aumento
de biomarcadores inflamatórios, como citocinas, proteases, e receptores de
citocinas solúveis. Além disso, os níveis de citocinas podem aumentar com a
gravidade da doença DPOC (FISCHER; PAVLISKO; VOYNOW, 2011).
O cigarro e outros tipos de fumaças inaladas desencadeiam uma resposta
inflamatória nos pulmões, resultando no influxo de diferentes tipos de células
inflamatórias. Os neutrófilos, macrófagos, linfócitos T, bem como eosinófilos e
mastócitos, têm sido associados com bronquite crônica ou DPOC. É interessante
notar que há um forte aspecto imunológico para DPOC que é caracterizado por
ativação de células T, proliferação em células T do tipo efetoras, e em seguida,
quimiotaxia para o pulmão com base no receptor de quimiocina do tecido
específico e expressão do ligante (FISCHER; PAVLISKO; VOYNOW, 2011).
3.4.3.1. Células inflamatórias e seus mediadores
Alguns autores demonstraram a presença de linfócitos T e macrófagos na
parede das vias aéreas de tabagistas, enquanto que neutrófilos são
preferencialmente coletados na luz alveolar. Nesse processo inflamatório existe a
participação não apenas das células inflamatórias, mas de células estruturais
como epiteliais, musculares e fibroblastos. O acúmulo de macrófagos e de
linfócitos na parede das pequenas vias aéreas tem recebido muito interesse por
parte dos pesquisadores da especialidade. Os neutrófilos, através da liberação de
elastases e EROs são pensados para desempenhar um papel significativo na
destruição do tecido enquanto os macrófagos também têm sido implicados na
promoção de alargamento do espaço aéreo alveolar, principalmente através da
liberação de MMPs. Diferente do que ocorre na asma, onde existe proliferação de
linfócitos T com predomínio de CD4, vários achados apontam para o acúmulo
especialmente de linfócitos T CD8. Esta dicotomização do perfil inflamatório, que

39
é particular a cada uma das doenças inflamatórias pulmonares, poderia explicar o
aparecimento de mediadores inflamatórios específicos que seriam determinantes
no desenvolvimento da lesão estrutural que ocorre na DPOC. Entre os
mediadores que causam acúmulo de células no pulmão enfisematoso e que
propiciam a manutenção do processo inflamatório, certamente existe um lugar
para as quimiocinas relacionadas ao fenótipo 1 de inflamação. CXCL9/MIG,
CXCL10/ IP-10, CXCL11/I-TAC e CCL5/RANTES têm sido aventadas como
possíveis quimiocinas implicadas no recrutamento de linfócitos T e monócitos
sanguíneos, facilitando o aumento de macrófagos alveolares e linfócitos T,
especialmente CD8, no parênquima dos pacientes com DPOC. O reconhecimento
do mecanismo pelo qual ocorre o tráfego de células mononucleares em direção
ao pulmão é fundamental não só para um maior entendimento da patogênese da
DPOC, mas para o desenvolvimento de terapêuticas adequadas. O bloqueio de
receptores específicos que reduzisse o recrutamento de células inflamatórias
diretamente relacionadas com a destruição pulmonar pode ser uma proposta
interessante de atuação sobre esta doença (COSTA; RUFINO; LAPA E SILVA,
2009; STEFANSKA; WALSH, 2009).
3.4.3.2. Células estruturais
O processo inflamatório da DPOC envolve uma variedade de células
inflamatórias e seus mediadores. No entanto, tem sido sugerida a participação
das células estruturais na fisiopatologia da doença (COSTA; RUFINO; LAPA E
SILVA, 2009).
As células epiteliais que revestem as grandes e pequenas vias aéreas
representam a primeira linha de defesa na resposta imune inata pulmonar. Bem
como proporcionam uma barreira física contra micro-organismos patogênicos e
toxinas inaladas, tais como as presentes no fumo do tabaco, células epiteliais
respiratórias são agora reconhecidas por desempenhar um papel central na
iniciação e mediação das respostas inflamatórias no pulmão (STEFANSKA;
WALSH, 2009).
As células epiteliais podem ser ativadas diretamente pela fumaça de cigarro
passando a produzir mediadores inflamatórios como IL-8, CCL20 e TNF-α. Após a

40
exposição à fumaça do cigarro, células epiteliais respiratórias são submetidas a
uma gama de alterações estruturais, resultando em perda da função de barreira,
diminuição da depuração mucociliar e metaplasia escamosa. É possível que as
células epiteliais deixem de exercer adequadamente seu papel de defesa, com a
perda da integridade estrutural da monocamada epitelial, facilitando a ocorrência
de infecções respiratórias e a liberação de citocinas (COSTA; RUFINO; LAPA E
SILVA, 2009; STEFANSKA; WALSH, 2009).
3.4.3.3. Macrófagos
Os macrófagos representam a maioria das células inflamatórias recolhidas
das vias aéreas tanto de pessoas saudáveis quanto de pacientes com DPOC. No
entanto, o número de macrófagos está aumentado entre 5 e 10 vezes no LBA de
pacientes com DPOC, quando comparado com voluntários não tabagistas. O
número de macrófagos no LBA parece correlacionar-se com a gravidade da
doença. O acúmulo de macrófagos nas paredes das vias aéreas e no parênquima
pulmonar dos tabagistas que desenvolvem DPOC pode ser explicado tanto pelo
prolongamento do tempo de vida da célula no pulmão como pelo aumento do
recrutamento de monócitos (seu precursor) da circulação. Alguns trabalhos
sugerem que a participação dos macrófagos na patogênese da DPOC tem
relação com a sua capacidade de produzir enzimas MMP como a MMP-1, MMP-9
e MMP-12. Acredita-se que as metaloproteinases sejam capazes de degradar
proteínas de forma semelhante às enzimas neutrofílicas e recrutar células
inflamatórias da circulação, facilitando sua infiltração nos tecidos lesados.
Macrófagos recolhidos do LBA de pacientes com DPOC apresentam aumento da
expressão de receptores para MMP-1 e MMP-9, enquanto que tanto
camundongos com deficiência de MMP-12 quanto o uso de inibidores de MMP em
cobaias apresentaram efeito protetor à exposição à fumaça de cigarro (COSTA;
RUFINO; LAPA E SILVA, 2009).
Essas células são a fonte de um número de mediadores potencialmente
significativos, tais como TNF-α, LTB4, EROs, e várias quimiocinas incluindo IL-8
(que promove a infiltração de neutrófilos, enquanto amplifica a destruição do
tecido através da secreção de protease) e o agonista pró-inflamatório do CCR2,

41
CCL2. A produção deste fator pode ser particularmente crítica porque a produção
de CCL2 fornece uma oportunidade para o recrutamento adicional de macrófagos
do subtipo pró-inflamatório para os pulmões (KIM; ROGERS; CRINER, 2008).
Os macrófagos também têm a capacidade de promover a infiltração de
células T através da expressão de quimiocinas específicas Th1/Tc1, tais como
CXCL9, CXCL10 e CXCL11 (Figura 7). Os níveis aumentados destas quimiocinas
têm sido relatados no escarro de pacientes com DPOC e correlacionada
diretamente com aumento observado na infiltração de macrófagos, bem como
diminuição da função pulmonar (STEFANSKA; WALSH, 2009).
Apesar de não se saber ao certo como os macrófagos atuam na DPOC, está
claro que, além de quantitativamente aumentados, estas células estão
relacionadas com a destruição pulmonar. Avaliando tecido pulmonar biopsiado de
pacientes com DPOC foi verificado que as áreas mais destruídas eram
circundadas por processo macrofágico e linfocitário, fazendo com que alguns
autores sugerissem uma possível interação entre essas células na patogênese da
DPOC (COSTA; RUFINO; LAPA E SILVA, 2009).
3.4.3.4. Linfócitos
Estudos demonstraram que tanto os linfócitos T CD4 (T helper) como os
CD8 (citotóxico) estavam aumentados nas vias aéreas e no parênquima pulmonar
dos pacientes com DPOC (COSTA; RUFINO; LAPA E SILVA, 2009).
Existem dois padrões de resposta das células T CD4: T helper 1 (Th1) e T
helper 2 (Th2). O padrão Th1 está relacionado com a liberação de IL-2, interferon
gama (IFN-γ) e TNF-α; enquanto que o padrão Th2 está associado à secreção de
IL-4, IL-5, IL-10 e IL-13, à produção de imunoglobulina (Ig)-E e IgG4, e se associa
com a ativação de mastócitos e eosinófilos. Este é o padrão, por exemplo, da
asma (FREEMAN; CURTIS; CHENSUE, 2007).
Assim como as células T CD4, as CD8 também apresentam dicotomia de
expressão, neste caso, sendo chamadas de Tc1 (células T citotóxicas do tipo 1) e
Tc2 (células T citotóxicas do tipo 2). No primeiro padrão, observa-se produção de
IFN-γ, mas não de IL-4. No segundo, as células liberam IL-4, mas não INF-γ
(FREEMAN; CURTIS; CHENSUE, 2007).
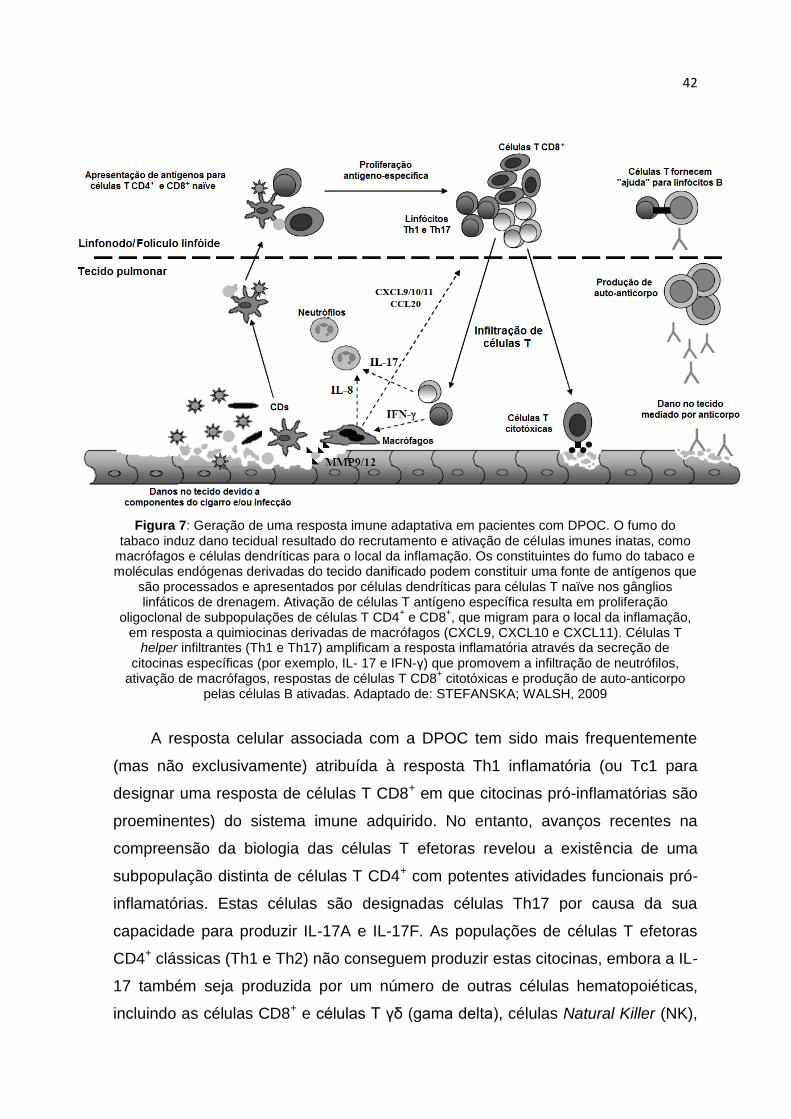
42
Figura 7: Geração de uma resposta imune adaptativa em pacientes com DPOC. O fumo do tabaco induz dano tecidual resultado do recrutamento e ativação de células imunes inatas, como
macrófagos e células dendríticas para o local da inflamação. Os constituintes do fumo do tabaco e moléculas endógenas derivadas do tecido danificado podem constituir uma fonte de antígenos que
são processados e apresentados por células dendríticas para células T naïve nos gânglios linfáticos de drenagem. Ativação de células T antígeno específica resulta em proliferação
oligoclonal de subpopulações de células T CD4+ e CD8
+, que migram para o local da inflamação,
em resposta a quimiocinas derivadas de macrófagos (CXCL9, CXCL10 e CXCL11). Células T helper infiltrantes (Th1 e Th17) amplificam a resposta inflamatória através da secreção de
citocinas específicas (por exemplo, IL- 17 e IFN-γ) que promovem a infiltração de neutrófilos, ativação de macrófagos, respostas de células T CD8
+ citotóxicas e produção de auto-anticorpo
pelas células B ativadas. Adaptado de: STEFANSKA; WALSH, 2009
A resposta celular associada com a DPOC tem sido mais frequentemente
(mas não exclusivamente) atribuída à resposta Th1 inflamatória (ou Tc1 para
designar uma resposta de células T CD8+ em que citocinas pró-inflamatórias são
proeminentes) do sistema imune adquirido. No entanto, avanços recentes na
compreensão da biologia das células T efetoras revelou a existência de uma
subpopulação distinta de células T CD4+ com potentes atividades funcionais pró-
inflamatórias. Estas células são designadas células Th17 por causa da sua
capacidade para produzir IL-17A e IL-17F. As populações de células T efetoras
CD4+ clássicas (Th1 e Th2) não conseguem produzir estas citocinas, embora a IL-
17 também seja produzida por um número de outras células hematopoiéticas,
incluindo as células CD8+ e células T γδ (gama delta), células Natural Killer (NK),

43
e neutrófilos. A atividade inflamatória das células Th17 é aparentemente devido à
produção de um complexo único de citocinas pró-inflamatórias, incluindo IL-6, IL-
17A, a IL-17F, GM-CSF (fator estimulador de colônias de granulócitos-
macrófagos) e TNF-α. Estas células também expressam receptores para IL-18 e
IL-23 (esta serve como um fator de crescimento para promover a atividade da
população Th17), citocinas tipicamente produzidas por monócitos, macrófagos e
células dendríticas. No que diz respeito ao pulmão, IL-17 é um potente indutor da
produção de IL-6 por células epiteliais brônquicas, e IL-6 e IL-17 são fortes
indutores da produção de mucinas Muc5AC e Muc5B por células epiteliais do
pulmão. Além disso, a IL-17 induz as células epiteliais a liberarem CXCL8, uma
quimiocina que é importante para a atração de neutrófilos (KIM; ROGERS;
CRINER, 2008).
Os linfócitos T CD4 e CD8 estão aumentados no parênquima pulmonar e na
parede das vias aéreas de pacientes com DPOC. Em modelo animal de enfisema
induzido pela exposição à fumaça de cigarro foi observado que, além do número
de linfócitos T estarem aumentado, havia correlação direta entre esse número e a
gravidade da doença pulmonar (TAKUBO et al., 2002).
Vários artigos também tem demonstrado o predomínio do padrão Tc1 das
células CD8 no escarro, no LBA e na biópsia dos pacientes, quando comparado
aos tabagistas sem limitação do fluxo aéreo e aos voluntários não tabagistas. No
entanto, os linfócitos T CD4 também estão aumentados no parênquima de
pacientes com DPOC, especialmente naqueles com doença mais grave. É
possível que elas tenham importância na manutenção da memória imunológica
que perpetuaria o processo mesmo após a suspensão do tabagismo (COSTA;
RUFINO; LAPA E SILVA, 2009).
A polarização da inflamação pulmonar da DPOC em favor do tipo 1 resulta
na produção e liberação de um determinado tipo de citocinas, especialmente o
IFN-γ, liberado pelos linfócitos CD4 e CD8 e capaz de ativar os macrófagos, que,
por sua vez, passam a produzir uma série de citocinas, incluindo IL-12, que
mantém a diferenciação de linfócitos desviada para o fenótipo 1 (COSTA;
RUFINO; LAPA E SILVA, 2009).
Linfócitos T CD8 estão aumentados na parede brônquica e no parênquima
pulmonar de pacientes com DPOC quando comparado com tabagistas sem

44
obstrução brônquica. Mais ainda, a intensidade da infiltração linfocitária está
diretamente relacionada com o grau de obstrução brônquica. Dessa forma, a
proliferação de células T CD8 no pulmão de tabagistas estaria relacionada com a
progressão da doença obstrutiva. Uma das funções das células T CD8 é a
apoptose de células-alvo, que no caso do enfisema está relacionada com a
destruição pulmonar. Foi demonstrada a relação tanto da apoptose quanto da
infiltração de células T CD8 na parede brônquica com a carga tabágica e com a
destruição pulmonar em pacientes com DPOC (COSTA; RUFINO; LAPA E
SILVA, 2009).
O mecanismo pelo qual os linfócitos T CD8 se acumulam nas paredes das
vias aéreas e no parênquima pulmonar dos pacientes com DPOC ainda não está
claro. No entanto, o recrutamento de células para o pulmão deve necessitar da
ativação linfocitária, que, por sua vez, provavelmente será seguida por adesão e
quimiotaxia seletiva. Há evidências de que as células T expressam vários
receptores de quimiocinas pró-inflamatórias, incluindo CCR5 e CXCR3,
relacionados às quimiocinas responsáveis pelo recrutamento de linfócitos T, mas
não é de todo certo que estes receptores de quimiocinas sejam responsáveis pela
a acumulação de células T CD8+ nos pulmões de pacientes. As quimiocinas que
atuam no receptor CXCR3 são CXCL9, CXCL10 e CXCL11, enquanto que as
relativas ao receptor CCR5 compreendem CCL3, CCL4, CCL5 e CCL11. Todas
têm atividade quimiotáxica de linfócitos (COSTA; RUFINO; LAPA E SILVA, 2009;
KIM; ROGERS; CRINER, 2008).
Deve-se considerar a possibilidade de que as células T CD8+ no pulmão
doente não representem a população inflamatória mais importante, mesmo que
elas sejam uma população relativamente abundante. No entanto, a simples
presença destas células quase certamente contribui como uma fonte de
mediadores patogênicos, que claramente têm o potencial para participar de danos
no tecido pulmonar. Além disso, estas células possuem o potencial para promover
a destruição do interstício pulmonar através da liberação de proteases, tais como
perforina e granzima, bem como as citocinas citotóxicos tais como o TNF-α (KIM;
ROGERS; CRINER, 2008).

45
3.4.3.5. Neutrófilos
A primeira e mais consistente anormalidade encontrada nas vias aéreas de
pacientes com DPOC é a infiltração celular da parede e luz brônquicas. Estudos
em animais demonstraram que a exposição à fumaça de cigarro promove a
proliferação de neutrófilos nas vias aéreas. Dessa forma vários autores têm
publicado estudos relatando o aumento de neutrófilos no escarro e no LBA de
pacientes com DPOC. Os neutrófilos estão implicados na liberação de citocinas
inflamatórias, mediadores lipídicos e enzimas capazes de promover dano tecidual.
Além disso, podem promover hipersecreção de muco por meio de efeito
secretagogo e pela proliferação de glândulas mucosas (COSTA; RUFINO; LAPA
E SILVA, 2009).
Desde o advento da tese de que a destruição pulmonar estaria relacionada
ao desequilíbrio entre proteases e antiproteases, o neutrófilo foi tido como fator
importante para o desencadeamento da doença, já que esse desequilíbrio seria
mantido através da liberação de enzimas neutrofílicas, o que ocasionaria o
aumento de proteases no pulmão (COSTA, RUFINO, LAPA E SILVA, 2009).
Alguns autores têm relatado que, embora a inflamação neutrofílica esteja
constantemente presente nos fumantes, ela provavelmente representa uma
resposta inespecífica das vias aéreas à agressão causada pelo tabagismo, não
estando, necessariamente, relacionada à progressão para DPOC. E mais: seria
necessária a ocorrência de interação com macrófagos e linfócitos T, os quais
desencadeariam resposta inflamatória ampliada e anormal apenas na parcela de
tabagistas que desenvolveria quadro clínico de DPOC (COSTA; RUFINO; LAPA E
SILVA, 2009).
3.4.3.6. Quimiocinas na patogênese da DPOC
Através da regulação do trânsito de células inflamatórias em direção aos
pulmões, as quimiocinas desempenham um papel fundamental no processo
inflamatório da DPOC. Algumas quimiocinas estão sendo estudadas há mais
tempo e estão mais claramente implicadas ao processo, enquanto outras
desempenham papel ainda incerto (COSTA; RUFINO; LAPA E SILVA, 2009).

46
a) IL-8/CXCL8
É um potente fator quimiotático de neutrófilos e seu nível está aumentado no
escarro induzido de pacientes com DPOC em comparação com os controles não
tabagistas. O nível de IL-8 está relacionado ao número absoluto e relativo de
neutrófilos no escarro induzido e está especialmente aumentado nos pacientes
com deficiência de α-1 antitripsina e naqueles com exacerbação brônquica
(COSTA; RUFINO; LAPA E SILVA, 2009).
b) Oncogene relacionado ao crescimento (GRO)-α/CXCL1
Produzida e liberada por macrófagos alveolares e células epiteliais, GRO-α
tem sido implicada na ativação de neutrófilos, monócitos, basófilos e linfócitos T
por meio de sua ligação com o receptor CXCR2. Sua concentração está
aumentada no escarro, mas não no LBA de pacientes com DPOC em
comparação com tabagistas e não tabagistas. É capaz de recrutar monócitos,
mecanismo pelo qual deve ocorrer o acúmulo de macrófagos alveolares no
pulmão de pacientes com DPOC (COSTA; RUFINO; LAPA E SILVA, 2009).
c) Ativador neutrofílico derivado do epitélio (ENA)-78/CXCL5
Assim como o GRO-α, o ENA-78/CXCL5 atua por meio do receptor CXCR2
e está aumentado no LBA de pacientes com DPOC em relação a controles não
tabagistas, embora em níveis não superiores àqueles encontrados em fumantes.
Foi relatado que a sua expressão nas células epiteliais está aumentada durante
as fases de exacerbação da DPOC (COSTA; RUFINO; LAPA E SILVA, 2009).
d) Quimiocinas dos receptores CXCR1 e CXCR2
Estes receptores estão presentes principalmente em neutrófilos e monócitos.
CXCL6 e CXCL8 são quimiocinas que se ligam aos receptores e estão envolvidas
com processos inflamatórios neutrofílicos. A expressão dos receptores CXCR1 e

47
CXCR2 está aumentada nos pacientes com DPOC (COSTA; RUFINO; LAPA E
SILVA, 2009).
3.4.4. Desequilíbrio Protease – Antiprotease
Num sentido simplista, enfisema é causado pelo desequilíbrio de proteases
e antiproteases que resulta na destruição do parênquima pulmonar. O exemplo
clássico é deficiência de α-1 antitripsina, onde sem oposição da elastase
neutrofílica tem efeitos deletérios sobre os pulmões. No entanto, na patogênese
geral, são as interações entre EROs e proteases ou antiproteases que
desencadeiam este desequilíbrio. Exposição à fumaça de cigarro pode inativar
antiproteases endógenas, bem como desencadear uma resposta pulmonar
aguda, que ativa os macrófagos alveolares residentes e promove o influxo de
neutrófilos para os pulmões. À medida que a exposição à fumaça continua e
torna-se crônica, há acumulação de macrófagos, neutrófilos e células T CD8+ nos
pulmões. Os macrófagos e neutrófilos liberam uma variedade de proteases,
incluindo a elastase neutrofílica, proteinase 3, MMPs e as catepsinas. Essas
proteinases “apoiam” umas as outras, ativando umas as outras ou inibindo seus
inibidores endógenos, tais como elastase neutrofílica inibi inibidores teciduais de
MMPs e MMPs degradam α-1 antitripsina. Estas proteinases clivam componentes
da matriz extracelular, fibras de elastina e colágeno, gerando fragmentos de
elastina ou peptídeos derivados de colágeno, tais como prolina-glicina-prolina,
que tem sido demonstrado ser quimiotáctico para monócitos, a célula precursora
de macrófagos ou neutrófilos. Coletivamente, fragmentos de peptídeos
quimiotáticos perpetuam a acumulação de macrófagos e neutrófilos e destruição
pulmonar (FISCHER; PAVLISKO; VOYNOW, 2011).
3.5. PATOLOGIA DA DPOC
As características patológicas da DPOC são a inflamação das vias aéreas
periféricas e destruição do parênquima pulmonar contribuindo para a
consequência funcional da limitação do fluxo aéreo expiratório (WOUTERS,
2005).

48
As alterações patológicas são detectadas muito cedo nos bronquíolos,
onde fibrose peribronquiolar resulta em obstrução ao fluxo aéreo. Alterações
precoces também podem ser detectadas em todos os brônquios, onde
hipersecreção de muco, células inflamatórias, e a contração da musculatura lisa
brônquica também obstruem o fluxo de ar. A hipersecreção de muco é uma
característica saliente da hipertrofia das glândulas submucosas e hiperplasia de
células caliciformes. Fatores quimiotáticos de neutrófilos também emanam de
macrófagos ativados e de células epiteliais e linfócitos CD8+. Na verdade, os
macrófagos, pela liberação de fatores quimiotáticos de neutrófilos e enzimas
proteolíticas, desempenham um papel proeminente na perpetuação da resposta
inflamatória PMN na DPOC (BRIGGS, 2004).
Na DPOC, a inflamação crônica em todas as vias aéreas e o parênquima
pulmonar produzem alterações patológicas acumulativas ao longo do tempo,
culminando na deterioração progressiva da função pulmonar. Inflamação
constante provoca a hipertrofia das glândulas de muco, hiperplasia de células
caliciformes, fibrose peribronquiolar, e espessamento da vasculatura pulmonar
que conspiram para debilitar o funcionamento pulmonar. Estas características
patológicas da DPOC, juntamente com aumento do tônus vagal, provoca aumento
no tônus do músculo liso, disfunção ciliar, edema e recolhimento elástico
diminuído no tecido pulmonar, juntos obstruem o fluxo de ar. Pacientes já não
podem exalar de forma eficiente e, em resposta, o volume do pulmão aumenta,
especialmente durante o exercício, aprisionando ar, diminuindo a capacidade
inspiratória, e induzindo dispneia. De fato, muitas pessoas que morrem de DPOC
sucumbem a defeitos de trocas gasosas que produzem níveis reduzidos de
oxigênio e níveis aumentados de dióxido de carbono no corpo, ou seja,
insuficiência respiratória. A hipertensão pulmonar da hipoxemia e perda de leito
vascular e cor pulmonale (forma de insuficiência cardíaca, onde há diminuição da
capacidade de funcionamento das câmaras direitas do coração, por doença
pulmonar), são resultados inexoráveis para pacientes com DPOC avançada.
Importantemente, a DPOC também pode causar uma perda de massa magra,
uma manifestação da inflamação sistêmica e do estado catabólico dos pacientes
com doença avançada. Os pacientes com baixo índice de massa corporal (IMC)
desta caquexia pulmonar têm maior mortalidade. Alguns têm notado que, em

49
pacientes com DPOC, o diâmetro transversal do músculo quadríceps prediz a
mortalidade melhor do que a diminuição do VEF1 (BRIGGS, 2004).
3.6. SINTOMATOLOGIA
A DPOC é caracterizada por inflamação crônica que induz o estreitamento
dos brônquios, resultando em uma redução progressiva do fluxo aéreo e
hiperinsuflação dinâmica. Principais sintomas da DPOC podem incluir dispneia,
tosse crônica, produção de expectoração, e às vezes chiado. Entretanto, os
sintomas podem estar ausentes ou negados por pacientes com doença precoce.
Apesar de que já se acreditou ser irreversível e inexoravelmente
progressiva, a sintomatologia da DPOC pode ser detida, ou mesmo revertida, com
o diagnóstico precoce, tratamento broncodilatador adequado, e principalmente
pela interrupção do ato de fumar. No entanto, para gerir eficazmente as sequelas
desta doença, é importante reconhecer a verdadeira natureza da DPOC: uma
doença inflamatória que afeta não somente todas as vias aéreas, mas também
todo o corpo (Figura 8). Na verdade, as manifestações sistêmicas da inflamação
da DPOC, tais como inflamação sistêmica (desequilíbrio entre a formação de
radicais livres de oxigênio e capacidade antioxidante resultando no estresse
oxidativo sistêmico e ativação das células inflamatórias), disfunção músculo
esquelética (perda da massa muscular esquelética e anomalias bioenergéticas) e
principalmente a aterosclerose, são responsáveis por grande parte da sua
morbidade e mortalidade associadas (BRIGGS, 2004; DOURADO et al., 2006;
FERREIRA, 2010).
A presença de sintomas respiratórios crônicos no paciente com hábito
tabágico (cigarro, cigarrilha, cachimbo, charuto) deve levar a suspeita clínica de
DPOC. Quanto maior a intensidade do tabagismo, maior a tendência ao
comprometimento da função pulmonar, embora a relação não seja obrigatória.
Aproximadamente 15% dos fumantes desenvolvem DPOC (JARDIM; OLIVEIRA;
NASCIMENTO, 2004).
A tosse é o sintoma mais encontrado, podendo ser diária ou intermitente e
pode preceder a dispneia ou aparecer simultaneamente a ela. O aparecimento da
tosse no fumante é tão frequente que muitos pacientes não a percebem como
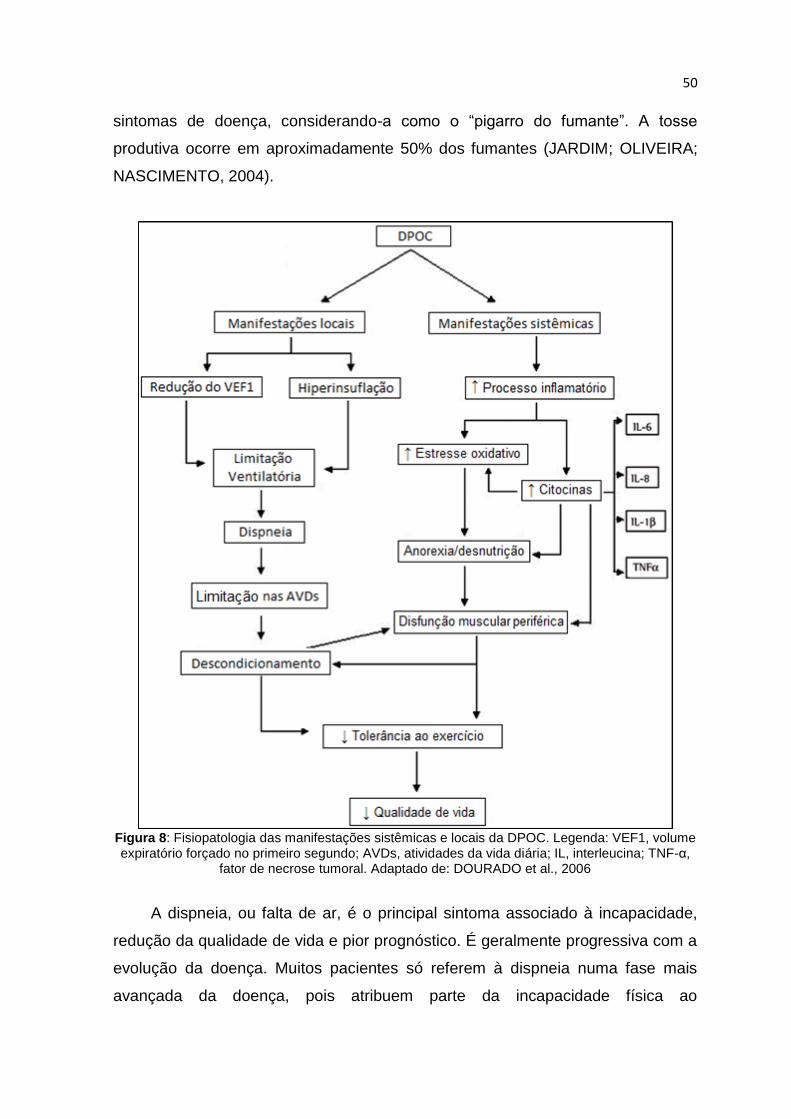
50
sintomas de doença, considerando-a como o “pigarro do fumante”. A tosse
produtiva ocorre em aproximadamente 50% dos fumantes (JARDIM; OLIVEIRA;
NASCIMENTO, 2004).
Figura 8: Fisiopatologia das manifestações sistêmicas e locais da DPOC. Legenda: VEF1, volume expiratório forçado no primeiro segundo; AVDs, atividades da vida diária; IL, interleucina; TNF-α,
fator de necrose tumoral. Adaptado de: DOURADO et al., 2006
A dispneia, ou falta de ar, é o principal sintoma associado à incapacidade,
redução da qualidade de vida e pior prognóstico. É geralmente progressiva com a
evolução da doença. Muitos pacientes só referem à dispneia numa fase mais
avançada da doença, pois atribuem parte da incapacidade física ao

51
envelhecimento e à falta de condicionamento físico (JARDIM; OLIVEIRA;
NASCIMENTO, 2004).
A perda de massa esquelética é um preditor de mortalidade em pacientes
com DPOC e, pode estar associada á acentuação dos sintomas, piora da
qualidade de vida e, intolerância ao exercício (JARDIM; OLIVEIRA;
NASCIMENTO, 2006).
A prevalência da desnutrição é variável, oscilando entre 26% e 47% dos
pacientes portadores de DPOC. Estudos retrospectivos indicam que reduções do
peso do corpo, resultando em valores abaixo de 90% do peso ideal e em valores
baixos de IMC, são fatores prognósticos negativos independentemente da
gravidade da doença. Há relação inversa entre o IMC e a sobrevida em pacientes
com DPOC. Em todos os grupos, a perda de peso está associada com
mortalidade aumentada; além disso, pacientes com DPOC grave e IMC menor
que 25 kg/m2 apresentam aumento da sobrevida quando ocorre ganho de peso
(DOURADO et al., 2006).
Em decorrência da progressão da DPOC o individuo também limita a
execução de suas atividades da vida diária para amenizar os sintomas,
apresentando um quadro de descondicionamento cardiovascular, disfunção
musculo esquelética e transtornos psicológicos, dentre eles a depressão
(FERREIRA, 2010).
3.6.1. Comorbidades
A comorbidade, descrita como a coexistência simultânea de doenças de
natureza distinta da DPOC no paciente portador desta condição constitui um fator
associado ao pior prognóstico da DPOC (LAST, 2001).
Como uma doença multicomponente, a DPOC é frequentemente associada
com disfunção de diferentes órgãos/sistemas. De particular interesse é a sua
associação com aumento da morbidade e mortalidade por doença cardiovascular
(COSIO; AGUSTÍ, 2010).
Entre as comorbidades mais comuns associadas com DPOC estão a
hipertensão, doença cardíaca isquêmica, insuficiência cardíaca, diabetes mellitus
e transtornos de humor (ANCOCHEA; GARCÍA; DÍEZ, 2010).

52
3.7. DIAGNÓSTICO
O diagnóstico precoce é a chave para evitar muitas das consequências
deletérias da DPOC, e isso requer um médico para identificar os pacientes de
risco – principalmente os fumantes. No entanto, os clínicos podem muitas vezes
ignorar os pacientes de risco durante contatos com pacientes de rotina ou mesmo
durante um exame físico, já que a DPOC pode ser assintomática em sua fase
inicial, ou pacientes podem negar os sintomas ou o estilo de vida alterado.
Quando isso ocorre, o diagnóstico só pode ser confirmado tardiamente, quando a
DPOC e as comorbidades avançaram consideravelmente (BRIGGS, 2004).
O diagnóstico de DPOC deve ser considerado em qualquer individuo com
tosse, expectoração, dispneia e/ou história de exposição a fatores de risco (Figura
9) e a espirometria deve ser considerada na definição diagnóstica dos casos da
doença visto que outras condições podem apresentar quadro clinico semelhante a
esta condição (FERREIRA, 2010; PEREIRA, 2002).
Figura 9: Diagnóstico da DPOC. Adaptado de: JARDIM; OLIVEIRA; NASCIMENTO, 2006

53
3.7.1. Avaliação Espirométrica
Simples, barata e confiável, a espirometria é a ferramenta de diagnóstico
essencial para a confirmação da DPOC (BRIGGS, 2004). A espirometria é a
medida do ar que entra e sai dos pulmões, durante respiração lenta ou durante
manobras expiratórias forçadas. É um teste que auxilia na prevenção e permite o
diagnóstico e a quantificação dos distúrbios ventilatórios, exigindo a compreensão
e colaboração do paciente, além de equipamentos exatos e emprego de técnicas
padronizadas aplicadas por pessoal especialmente treinado. Os valores obtidos
devem ser comparados a valores previstos adequados para a população avaliada.
E sua interpretação deve ser feita à luz dos dados clínicos e epidemiológicos
(PEREIRA, 2002).
A espirometria com obtenção da curva expiratória volume-tempo é
obrigatória na suspeita clínica de DPOC, e por meio deste exame avalia-se o grau
de obstrução ao fluxo aéreo expiratório forçado, devendo ser realizada antes e
após administração de broncodilatador, de preferência em fase estável da
doença. A espirometria permite a avaliação de uma multiplicidade de parâmetros,
porém os mais importantes do ponto de vista de aplicação clínica são a CVF, o
VEF1, e a relação VEF1/CVF, pois mostram menor variabilidade inter e intra-
individual. O VEF1 e a CVF são medidas obrigatórias para a identificação e
estadiamento da gravidade da DPOC. A existência de limitação do fluxo aéreo é
definida pela presença da relação VEF1/CVF abaixo de 0,70 pós-broncodilatador
(JARDIM; OLIVEIRA; NASCIMENTO, 2004; BRIGGS, 2004).
Segundo a GOLD os critérios diagnósticos da DPOC pela espirometria
exigem que a relação entre VEF1/CVF pós-broncodilatador seja menor que 0,70.
De acordo com estes critérios estabelecem-se os estadiamentos da DPOC,
dispostos em quatro níveis de gravidade de acordo com o VEF1 pós-
broncodilatador e critérios clínicos. O Estádio I (DPOC leve) apresenta VEF1
maior ou igual a 80% do previsto; o Estádio II (DPOC moderada) VEF1 entre 50%
e 80% do previsto; o Estádio III (DPOC grave) VEF1 entre 30% e 50% do previsto;
o Estádio IV (DPOC muito grave) VEF1 menor do que 30% do previsto ou VEF1
entre 30% e 50% do previsto, com sinais de insuficiência respiratória e/ou

54
insuficiência ventricular direita. O VEF1 diminui de acordo com o aumento da
idade, uma redução de 0,003 L/ano (FERREIRA, 2010).
3.7.2. Avaliação Radiológica
Na DPOC deve-se solicitar, rotineiramente, uma radiografia simples de
tórax nas posições póstero-anterior e perfil, não para definição da doença, mas
para afastar outras doenças pulmonares, principalmente a neoplasia pulmonar. A
radiografia de tórax pode ainda identificar bolhas, com possível indicação
cirúrgica. A tomografia computadorizada de tórax está indicada na DPOC
somente em casos especiais, como suspeita da presença de bronquiectasias ou
bolhas, indicação de correção cirúrgica destas ou programação de cirurgia
redutora de volume (JARDIM; OLIVEIRA; NASCIMENTO, 2004).
3.7.3. Avaliação Gasométrica e do pH
A avaliação da oxigenação pode ser feita, inicialmente, de maneira não
invasiva pela oximetria de pulso. Se for identificada uma saturação periférica de
oxigênio (SpO2) igual ou inferior a 90%, está indicada a realização de gasometria
arterial para avaliação da PaO2 (pressão arterial parcial de O2) e da PaCO2
(pressão parcial do CO2 no sangue arterial) (JARDIM; OLIVEIRA; NASCIMENTO,
2004).
Enfim, para o diagnóstico da DPOC deve-se investigar a sintomatologia
clínica, o resultado da espirometria e os exames complementares (gasometria
radiografias, entre outros), porém deve-se levar em consideração também outros
fatores importantes que influenciam no impacto e na evolução desta doença, tais
como: baixa composição de massa corporal e diminuição na capacidade de
exercício que estão associadas ao alto risco de mortalidade bem como diminuição
da capacidade funcional (FERREIRA, 2010).

55
3.8. TRATAMENTO
3.8.1. Broncodilatadores
Os broncodilatadores são a base do tratamento sintomático das doenças
pulmonares obstrutivas. A via de administração preferencial é a inalatória, pela
ação direta nas vias aéreas e menor incidência de efeitos colaterais (JARDIM;
OLIVEIRA; NASCIMENTO, 2004).
Não existe consenso quanto ao tipo de broncodilatador para iniciar o
tratamento da DPOC. O único acordo na literatura é que as xantinas deveriam ser
consideradas como a última opção terapêutica (Tabela 5).
Tabela 5: Orientações terapêuticas de acordo com os estádios da DPOC
Estádios Fármacos
I β2-agonista de curta duração e/ou ipratrópio, quando necessário.
II
Reabilitação pulmonar
– Sintomas eventuais: β2- agonista de curta duração e/ou ipratrópio
(quando necessário)
– Sintomas persistentes: β2-agonista de longa duração e/ou tiotrópio.
III
Reabilitação Pulmonar
β2-agonista de longa duração e tiotrópio
Acrescentar xantina de longa duração (se persistirem sintomas)
Corticoide inalatório se exacerbações frequentes (> 2 exacerbações
ao ano)
IV
Reabilitação Pulmonar
β2-agonista de longa duração e tiotrópio
Acrescentar xantina de longa duração (se persistirem sintomas)
Corticoide inalatório se exacerbações frequentes (> 2 exacerbações
ao ano)
Oxigenoterapia
Estudar indicações cirúrgicas para o tratamento do enfisema (cirurgia
redutora de volume pulmonar, bulectomia ou transplante pulmonar)
Adaptado de: JARDIM; OLIVEIRA; NASCIMENTO, 2004
Broncodilatadores de curta ação têm sido indicados em estágios iniciais da
doença, podem melhorar o estado de saúde e controle sintomático, mas não a
mortalidade. Broncodilatadores de ação prolongada também foram mostrados

56
para melhorar os sintomas, a capacidade de exercício e as taxas de exacerbação
(ANDERSON; MACNEE, 2009). Outro aspecto a considerar quando se pautar
broncodilatadores de longa duração é o de melhor complacência e adesão à
terapêutica que permite a facilidade de administração (ANCOCHEA; GARCÍA,
DÍEZ, 2010).
3.8.1.1. β2-agonistas
São broncodilatadores potentes e seguros que atuam abrindo os canais de
potássio e aumentando o AMP (adenosina monofosfato) cíclico (Figura 10). Os
β2-agonistas de longa duração, formoterol e salmeterol, quando comparados aos
β2-agonistas de curta ação, fenoterol, salbutamol e terbutalino e ao anticolinérgico
brometo de ipratrópio, são mais eficazes, resultando em redução da dispneia e
melhora funcional mais acentuada e mais duradoura. O único β2-agonista de ação
por 24 horas é o bambuterol, que, no entanto, não apresenta número suficiente de
registros na literatura que apoiem o seu uso sistemático na DPOC (JARDIM;
OLIVEIRA; NASCIMENTO, 2004).
3.8.1.2. Anticolinérgicos
O brometo de ipratrópio é um antagonista inespecífico dos receptores
muscarínicos. O pico de ação do ipratrópio varia de 30 a 90 minutos e a duração
de seu efeito varia entre 4 e 6 horas. O brometo de tiotrópio é um anticolinérgico
de longa duração, mais eficaz que o ipratrópio, com seletividade farmacocinética
para os receptores muscarínicos M1 e M3, permitindo a sua utilização em dose
única diária. O número de efeitos colaterais é pequeno, sendo seu efeito colateral
mais frequente a boca seca, em 16% dos indivíduos que utilizam a medicação. O
brometo de tiotrópio reduz o número de exacerbações e hospitalizações e
melhora a qualidade de vida relacionada ao estado de saúde, comparado com
placebo e ipratrópio e aumenta o VEF1 em pacientes com DPOC estável
(JARDIM; OLIVEIRA; NASCIMENTO, 2004; ANDERSON; MACNEE, 2009).

57
Figura 10: Mecanismo de ação dos agonistas β-adrenérgicos. Onde: GDP (difosfato de
guanosina), GTP (trifosfato de guanosina), Gs (proteína ativa), ATP (trifosfato de adenosina), AMPc (monofosfato cíclico de adenosina). Adaptado de: TELLES FILHO, 2012
3.8.1.3. Xantinas
As xantinas, como a teofilina, estão reservadas como terceira linha de
opção, devido à sua alta carga de efeitos adversos, e só são recomendados para
a doença muito grave. Mesmo assim continuam sendo usadas em larga escala,
apesar de seu efeito broncodilatador ser inferior ao das demais. A bamifilina é
uma xantina de ação de 12 horas e tem a vantagem de provocar menos efeitos
adversos do que a teofilina. São necessários mais estudos para definir seu papel
no tratamento da DPOC (JARDIM; OLIVEIRA; NASCIMENTO, 2004).
3.8.2. Corticoides
Em duas recentes metanálises sobre os benefícios do uso de corticoide
inalatório em DPOC, observou-se a ocorrência de diminuição no número de
exacerbações, porém sem alteração na taxa de mortalidade e com maior índice
de efeitos colaterais do que com o placebo (JARDIM; OLIVEIRA; NASCIMENTO,
2004).
Os corticoides são muito menos eficazes na DPOC e não reduzem a
progressão da doença. Em contraste com pacientes com asma, aqueles com

58
DPOC não demonstram nenhuma reação anti-inflamatória significativa aos
corticoides. Macrófagos alveolares de pacientes com DPOC mostram uma
acentuada redução na capacidade de resposta para os efeitos anti-inflamatórios
de corticoides, em comparação com as células de fumantes normais e não
fumantes. Estudos recentes sugerem que pode haver uma ligação entre estresse
oxidativo e à pobre resposta aos corticoides em pacientes com DPOC (BARNES;
SHAPIRO; PAUWELS, 2003).
Três trabalhos publicados em 2003 estudando corticoides inalatórios e beta-
agonistas de ação prolongada, fluticasona+salmeterol e budesonida+formoterol,
sugerem que esta associação de drogas pode reduzir a mortalidade, reduzir as
exacerbações e melhorar a qualidade de vida em portadores de DPOC (JARDIM;
OLIVEIRA; NASCIMENTO, 2004).
O corticoide sistêmico está indicado apenas nas exacerbações infecciosas e
não infecciosas, pois já foi demonstrado que o seu uso por 14 dias reduz os
sintomas e melhora o VEF1 e a PaO2 em menor tempo. Na fase estável dos
pacientes portadores de DPOC não há indicação do uso de corticoides sistêmicos
de manutenção (oral ou injetável), devido não ocorrer melhora da função
pulmonar, além de levar a efeitos sistêmicos indesejáveis (JARDIM; OLIVEIRA;
NASCIMENTO, 2004).
3.8.3. N-acetilcisteína
Na patogênese da DPOC, é importante a participação do estresse
oxidativo, o qual inicia-se antes mesmo do processo inflamatório, devido à
inalação de radicais livres presentes na fumaça do cigarro, mas que permanece e
se intensifica durante o processo inflamatório. Uma revisão sistemática recente
mostrou diminuição das exacerbações e dias de internação em pacientes com
DPOC que utilizaram N-acetilcisteína (JARDIM; OLIVEIRA; NASCIMENTO,
2004).
N-acetilcisteína tem propriedades antioxidantes diretas e indiretas. O seu
grupo tiol livre (Figura 11) é capaz de interagir com os grupos eletrofílicos das
EROs, tendo o potencial de interagir diretamente com oxidantes como o peróxido
de hidrogênio, o radical hidroxila, e ácido hipoclorito. Durante a absorção, a N-

59
acetilcisteína é rapidamente metabolizada a cisteína, cujo grupo tiol tem
propriedades antioxidantes e que é um precursor direto da glutationa. A glutationa
serve como um fator central na proteção contra agentes tóxicos internos (tais
como respiração aeróbica celular e metabolismo de fagócitos) e agentes externos
(tais como NO, óxido de enxofre, e outros componentes do fumo do cigarro, e
poluição). E devido à sua capacidade para reduzir pontes dissulfeto, N-
acetilcisteína é amplamente utilizada para reduzir a viscosidade e elasticidade do
muco (DEKHUIJZEN; VAN BEURDEN, 2006; SADOWSKA et al., 2006).
Figura 11: Fórmula Estrutural da N-acetilcisteína. Fonte: DEKHUIJZEN, BEURDEN, 2006
3.8.4. Oxigenoterapia
A oxigenoterapia tem demonstrado ser, de forma inequívoca, o principal
tratamento para melhorar a sobrevida e a morbidade de pacientes hipoxêmicos
portadores de DPOC, tendo como objetivo a manutenção da saturação arterial da
oxiemoglobina acima de 90% (SaO2 ≥ 90%), documentada na gasometria arterial
(JARDIM; OLIVEIRA; NASCIMENTO, 2004).
3.8.5. Fisioterapia Respiratória
A fisioterapia respiratória constitui componente de grande valor no
tratamento da DPOC. O plano fisioterápico visa oferecer o melhor comportamento
funcional do paciente, sendo útil o seu início o mais precocemente possível.
Consta de exercícios respiratórios, exercícios de tosse, drenagem postural de
todos os segmentos pulmonares, técnicas de percussão torácica, associadas à
drenagem postural, prática de exercícios destinados a coordenar a atividade física
com a respiração, movimentação ativa e passiva de membros superiores e

60
inferiores, inclusive em pacientes hospitalizados, associação com a terapêutica
inalatória, podendo as sessões de fisioterapia ser realizadas, após nebulizações
de broncodilatadores ou simultaneamente à inalação de oxigênio (CARDOSO;
LEMLE; BETHLEM, 2000).
3.8.6. Novas Opções Terapêuticas
Os inibidores da fosfodiesterase (PDE)-4 promovem o acúmulo intracelular
de AMP cíclico, que deprime a atividade inflamatória dos neutrófilos, aumenta os
níveis de IL-10 e inibe a secreção de IL-8 e LTB4 pelos macrófagos, além de
provocarem o relaxamento da musculatura lisa.
O Roflumilaste é único entre os inibidores de PDE, tendo seletividade para a
PDE-4 e nenhum efeito inibitório aparente sobre a PDE-3, que tem sido associado
com a cardiotoxicidade em outros agentes desta classe. Roflumilaste está
associado com a atividade celular inflamatória reduzida, e os seus efeitos
terapêuticos incluem broncodilatação, a indução da liberação de citocinas e
quimiocinas a partir de células inflamatórias, e inibição do vazamento
microvascular e tráfico celular. Estudos clínicos com monoterapia de roflumilaste
demonstraram melhora da função pulmonar, redução das exacerbações, menor
necessidade de medicamentos anti-inflamatório/anti-infecciosos, e de uma
melhoria da qualidade de vida (RUSSELL; ANZUETO; WEISMAN, 2011).
3.8.7. Tratamento das Comorbidades
Cada uma das comorbidades requer uma abordagem única que deve ser
individualizada em pacientes com DPOC. Por exemplo, no tratamento de doenças
cardiovasculares, a utilização de cardioseletivos beta-bloqueadores, quando
indicado, é segura, embora se recomende tratamento de partida em doses baixas
que aumente gradativamente até a dose necessária. Além disso, o uso de
diuréticos na insuficiência cardíaca deve ser feito com cuidado devido ao risco de
ocorrência de alcalose metabólica, com o consequente risco de aumentar a
PaCO2 (ANCOCHEA; GARCÍA; DÍEZ, 2010).

61
Transtornos de humor, ansiedade e depressão, aparecem em uma alta
porcentagem de pacientes com DPOC. Deve-se ter em mente que os
benzodiazepínicos têm um efeito depressivo sobre o centro respiratório, por isso
devem ser manuseados com cuidado nestes pacientes. Os mais seguros são os
inibidores seletivos de serotonina e norepinefrina, porque não afetam o centro
respiratório. Geralmente o tratamento é iniciado com doses baixas e não se deve
esperar resultados até depois de (pelo menos) 2 a 4 semanas (ANCOCHEA;
GARCÍA; DÍEZ, 2010).

62
4. CONCLUSÃO
A DPOC é, portanto, um importante problema de saúde pública, não só
devido a sua alta morbidade e mortalidade (a DPOC mata uma pessoa em média
a cada 10 segundos), mas também porque é uma doença evitável. Atualmente, é
a quarta maior causa de morte no mundo, e é a única, dentre as principais
doenças do mundo, que vem crescendo em prevalência e em mortalidade,
estando previsto um aumento substancial nas próximas décadas, a menos que
medidas preventivas urgentes sejam tomadas para reduzir os fatores de risco
subjacentes, em especial o uso do tabaco.
O tabagismo é o principal e mais importante fator de risco para a DPOC
(85% a 90% de mortes por DPOC são atribuíveis ao tabaco). Parar de fumar é o
meio mais eficaz de se prevenir e a única forma comprovada de modificar o curso
natural desta doença e poderia prevenir milhões de casos.
Infelizmente, a DPOC é progressiva e irreversível após uma certa
quantidade de danos ocorrerem aos pulmões, caso em que o tratamento
terapêutico pode apenas aliviar os sintomas e melhorar a qualidade de vida. Por
isso é de fundamental importância o diagnóstico precoce e preciso da DPOC,
para impedir o impacto e a progressão da doença. Como a relação entre a dose
de tabaco consumida e a progressão da DPOC é evidente, deveria ser notório
que a cessação tabágica é o tratamento mais importante na DPOC.
A cessação tabágica é a única medida efetiva que pode trazer uma
significativa atenuação do comprometimento da função pulmonar, e quanto mais
cedo ocorrer, menor será este comprometimento, podendo a obstrução das vias
aéreas ser estabilizada com melhora significativa dos sintomas. Parar de fumar
melhora também o prognóstico, e mais importante, melhora a sobrevida dos
pacientes com DPOC, independente da gravidade da doença.

63
5. REFERÊNCIAS BIBLIOGRÁFICAS
AIT-KHALED, N.; ENARSON, D.; BOUSQUET J. Chronic respiratory diseases in developing countries: the burden and strategies for prevention and management. Bulletin of the World Health Organization, v. 79, p. 971-979, 2001. ANCOCHEA, J.; GARCÍA, T. G.; DÍEZ, J. M. Hacia um tratamento individualizado e integrado del paciente con EPOC. Arch Bronconeumol., v. 46, p.14-18, 2010. ANDERSON, D.; MACNEE, W. Targeted treatment in COPD: a multi-system approach for a multi-system disease. International Journal of COPD, v. 4, p. 321–335, 2009. ASOCIACIÓN LATINOAMERICANA DEL TÓRAX (ALAT). Actualización de las recomendaciones ALAT sobre la exacerbación infecciosa de la EPOC. Arch. Bronconeumol., v. 40, n. 7, p. 315-325, 2004.
BARBOSA, L. F. Danos em DNA promovidos pela enzima Cu, Zn-Superóxido Dismutase. Implicações para a apoptose em um modelo celular de Esclerose Lateral Amiotrófica. 2008. 140p. Tese (Doutor em Ciências – Bioquímica), Instituto de Química, Universidade de São Paulo, São Paulo, 2008. Orientador: Marisa Helena Gennari de Medeiros.
BARNES, P. J.; SHAPIRO, S. D.; PAUWELS, R. A. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J., v. 22, p. 672-688, 2003. BETHLEN, N. Pneumologia. 4° ed. São Paulo, Editora Atheneu, 2002. BRIGGS, D.D. Chronic Obstructive Pulmonary Disease Overview: Prevalence, Pathogenesis, and Treatment. Journal of Managed Care Pharmacy, v. 10, n. 4, Jul. 2004. CAMELIER, A. A.; WINTERII, D. H.; JARDIM, J. R.; BARBOZA, C. E. G.; CUKIER, A.; MIRAVITLLES, M. Deficiência de alfa-1 antitripsina: diagnóstico e tratamento. J. bras. pneumol., São Paulo, v. 34, n. 7, p. 514-527, 2008.
CARDOSO, A. P.; LEMLE, A.; BETHLEM, N. Doenças pulmonares obstrutivas crônicas. Pneumologia. 4ª ed. São Paulo: Atheneu, cap. 35, p. 600-621, 2000. COSIO, B. G.; AGUSTÍ, A. Update in Chronic Obstructive Pulmonary Disease 2009. Am J Respir Crit Care Med., v. 181, n. 7, p. 655-660, Abr 2010. COSTA, C. H.; RUFINO, R.; LAPA E SILVA, J. R.. Células inflamatórias e seus mediadores na patogênese da DPOC. Rev. Assoc. Med. Bras., São Paulo, v. 55, n. 3, 2009.

64
DEKHUIJZEN, P. N. R.; VAN BEURDEN, W. J. C. The role for N-acetylcysteine in the management of COPD. Int J Chron Obstruct Pulmon Dis., v. 1, n. 2, p. 99–106, 2006. DOURADO, V. Z.; TANNI, S. E.; VALE, S. A.; FAGANELLO, M. M.; SANCHEZ, F. F.; GODOY, I. Manifestações sistêmicas na doença pulmonar obstrutiva crônica. J. Bras. Pneumol., São Paulo, v. 32, n. 2, p. 161-171, 2006. FERREIRA, V. C. Independência Funcional do Idoso Com Doença Pulmonar Obstrutiva Crônica. 2010. 174p. Dissertação (Mestre em Ciências), Escola de Enfermagem de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, 2010. Orientador: Sueli Marques.
FISCHER, B. M.; PAVLISKO, E.; VOYNOW, J. A. Pathogenic triad in COPD: oxidative stress, protease–antiprotease imbalance, and inflammation. Int J Chron Obstruct Pulmon Dis., v. 6, p. 413-421, 2011. FREEMAN, C. M.; CURTIS, J. L.; CHENSUE, S. W. CC chemokine receptor 5 and CXC chemokine receptor 6 expression by lung CD8 cells correlates with chronic obstructive pulmonary disease severity. Am J Pathol., v. 171, p. 767-776, 2007.
GIEMBYCZ, M. A.; FIELD, S. K. Roflumilast: first phosphodiesterase 4 inhibitor approved for treatment of COPD. Drug Des Devel Ther., v. 4, p. 147-158, 2010. HALBERT, R. J.; NATOLI, J. L.; GANO, A.; BADAMGARAV, E.; BUIST, A. S.; MANNINO, D. M. Global burden of COPD: systematic review and meta-analysis. Eur. Respir. J., v. 28, n. 3, p. 523-532, 2006.
HEALTHY LIFESTYLE. Oxidative Stress And Free Radicals. Outubro de 2007. Disponível em: <http://healthy-lifestyle.most-effective-solution.com/2007/10/01/oxidative-stress-and-free-radicals/>. Acessado em: Jun. de 2012. HIZAWA, NOBUYUKI. Genetic Backgrounds of Asthma and COPD. Allergology International, v. 58, n. 3, p. 315-322, 2009. JARDIM, J.; OLIVEIRA, J.; NASCIMENTO, O. II Consenso Brasileiro de Doença Pulmonar Obstrutiva Crônica (DPOC). J. Pnemol. São Paulo, v. 30, p. S1-S42, 2004. KEMP, S.V.; POLKEY, M.I.; SHAH, P.L. The Epidemiology, Etiology, Clinical Features, and Natural History of Emphysema. Thorac. Surg. Clin., p. 149–158, 2009.
KIM, V.; ROGERS, T. J.; CRINER, G. J. New Concepts in the Pathobiology of Chronic Obstructive Pulmonary Disease. Proc Am Thorac Soc., v. 5, n. 4, p. 478–485, 2008.

65
KUMAR, V.; ABBAS, A. K.; FAUSTO, N. Robbins e Cotran – Patologias - Bases Patológicas das Doenças. 7ª ed. Rio de Janeiro: Elsevier, 2005.
LANE, N.; ROBINS, R. A.; CORNE, J.; FAIRCLOUGH, L. Regulation in chronic obstructive pulmonary disease: the role of regulatory T-cells and Th17 cells. Clinical Science, v. 119, p. 75-86, 2010.
LAST J. Editor, A dictionary of Epidemiology, 4ª ed., Oxford University Press, 2001. LIMA, F. M. R.; DI PACE, A. M.; MEDEIROS, V. M. L.; VIRGÍNIO, F. B. Enfisema pulmonar: aspectos gerais. Maio de 2007. Disponível em: <http://www.wgate.com.br/conteudo/medicinaesaude/fisioterapia/respiratoria/enfisema_fabiola/enfisema_fabiola.htm> Acessado em: abril de 2012. MARTINELLO, M. Avaliação e intervenção fisioterapêutica ambulatorial em paciente portador de doença pulmonar obstrutiva crônica (DPOC) e asma. Revista Digital, Buenos Aires, Ano 14, n. 138, Novembro de 2009.
MARWICK, J. A.; CHUNG, K. F.Glucocorticoid insensitivity as a future target of therapy for chronic obstructive pulmonary disease. International Journal of Chronic Obstructive Pulmonary Disease, v. 5, p. 297–309, 2010. MEIRELLES, R.H.S. Tabagismo e DPOC – dependência e doença – fato consumado. Pulmão RJ - Atualizações Temáticas, v. 1, p. 13-19, 2009. MENEZES, A. M.; PEREZ-PADILLA, R.; JARDIM, J.R.; MUIÑO, A.; LOPEZ, M. V.; VALDIVIA, G.; MONTES DE OCA, M.; TALAMO, C.; HALLAL, P. C.; VICTORA, C. G.; PLATINO TEAM. Chronic obstructive pulmonary disease in five Latin American cities (the PLATINO study): a prevalence study. Lancet., v. 366, p. 1875-1881, 2005. MENEZES, A. N.; et al. Prevalence of chronic obstructive pulmonary disease and associated factors: the PLATINO Study in São Paulo, Brazil. Cad. Saúde Pública, Rio de Janeiro, v. 21, n. 5, Oct. 2005. PAMPLONA, P.; MENDES, B. Estratégia de tratamento do tabagismo na DPOC. Rev Port Pneumol, v. 15, n. 6, p. 1121-1156, Nov. 2009.
PEREIRA, C. A. C. Espirometria. J. Pneumol., Brasília, DF, v. 28, p. 1-82, out. 2002.
PLANTIER, L.; BOCZKOWSKI, J.; CRESTANI, B. Defect of alveolar regeneration in pulmonary emphysema: Role of lung fi broblasts. International Journal of COPD, n. 2, p. 463–469, 2007. PLATINO – PROJETO LATINO-AMERICANO DE INVESTIGAÇÃO EM OBSTRUÇÃO PULMONAR. Associação Latino-Americana de Tórax (ALAT). Coord.: Ana MB Menezes, 2006.

66
PUERTO-NEVADO, L.; PÉREZ-RIAL, S.; GIRÓN-MARTÍNEZ, A.; PECES-BARBA, G. Papel de la inflamación en la etiopatogenia de la EPOC. Arch Bronconeumol., v. 46, n. 11, p. 2-7, 2010. RENNARD, S. I.; TOGO, S.; HOLZ, O. Cigarette Smoke Inhibits Alveolar Repair. A Mechanism for the Development of Emphysema. Proc. Am. Thorac. Soc., v. 3, p. 703–708, 2006.
RUSSELL, R.; ANZUETO, A.; WEISMAN, I. Optimizing management of chronic obstructive pulmonary disease in the upcoming decade. International Journal of COPD, v. 6, p. 47–61, 2011.
SADOWSKA, A. M.; VERBRAECKEN, J.; DARQUENNES, K.; DE BACKER, W. A. Role of N-acetylcysteine in the management of COPD. Int J Chron Obstruct Pulmon Dis., v. 1, n. 4, p. 425–434, 2006. SECRETARIA DE VIGILÂNCIA EM SAÚDE (SVS). Saúde Brasil 2007 – Uma Análise da Situação de Saúde. Ministério da Saúde. Nov. 2008. Disponível em: <http://portal.saude.gov.br/portal/arquivos/pdf/coletiva_saude_061008.pdf>. Acesso em: fev. de 2012.
STEFANSKA, A. M.; WALSH, P. T. Chronic Obstructive Pulmonary Disease: Evidence for an Autoimmune Component. Cellular & Molecular Immunology, v. 6, n. 2, p. 81-86, 2009. TAKUBO, Y.; GUERASSIMOV, A.; GHEZZO, H.; TRIANTAFILLOPOULLOS, A.; BATES, J. H.; HOIDAL, J. R.; et al. Alpha 1-Antitrypsin determines the pattern of emphysema and function in tobacco smoke-exposed mice: parallels with human disease. Am J Respir Crit Care Med., v. 166, n. 12, p. 596-603, 2002.
TELLES FILHO, P. A. Asma brônquica: Tratamento da asma. Disponível em: <http://www.asmabronquica.com.br/medical/tratamento_asma_broncodilatadores.html>. Acessado em: jun de 2012.
TUDER, R. M.; YOSHIDA, T.; ARAP, W.; PASQUALINI, R.; PETRACHE, I. State of the art. Cellular and molecular mechanisms of alveolar destruction in emphysema: an evolutionary perspective. Proc Am Thorac Soc., v. 3, p 503-510, 2006.
WEST, J. B. Doenças obstrutivas. In: ___. Fisiopatologia pulmonar moderna. 4ª ed. São Paulo: Manole, cap.4, p.57-86, 1996. WEST, J. B. Fisiopatologia moderna. 2ª ed. São Paulo: Manole, 2002. WORLD HEALTH ORGANIZATION (WHO). Burden of COPD. Disponível em: <http://www.who.int/respiratory/copd/burden/en/>. Acesso em: 17 de nov. 2011.

67
WORLD HEALTH ORGANIZATION (WHO). Chronic obstructive pulmonary disease (COPD). Disponível em: <http://www.who.int/respiratory/copd/en/>. Acesso em: 17 de nov. 2011. WORLD HEALTH ORGANIZATION (WHO). Enfermedad pulmonar obstructiva crónica (EPOC). Nov. de 2011 Disponível em: <http://www.who.int/mediacentre/factsheets/fs315/es/index.html>. Acesso em: jan. de 2012. WORLD HEALTH ORGANIZATION (WHO). The top 10 causes of death. Jun. 2011. Disponível em: <www.who.int/mediacentre/factsheets/fs310/en/index.html>. Acesso em: fev. de 2012. WOUTERS, E. F. Local and systemic inflammation in chronic obstructive pulmonary disease. Proc Am Thorac Soc., v. 2, n. 1, p. 26-33, 2005.





























