Embed Size (px)
Citation preview

UNIVERSIDADE FEDERAL DE SERGIPE
PRÓ-REITORIA DE PÓS-GRADUAÇÃO E PESQUISA
PROGRAMA DE PÓS-GRADUAÇÃO EM QUÍMICA
ALAN DIEGO DA CONCEIÇÃO SANTOS
DESENVOLVIMENTO E VALIDAÇÃO DE MÉTODOS POR HPLC-DAD-
ELSD PARA CONTROLE DE QUALIDADE QUÍMICO DO LÁTEX DO
CAULE E DO FRUTO DE MANGABA (Hancornia speciosa GOMES)
São Cristóvão – Sergipe
FEVEREIRO/2012

ALAN DIEGO DA CONCEIÇÃO SANTOS
DESENVOLVIMENTO E VALIDAÇÃO DE MÉTODOS POR HPLC-DAD-
ELSD PARA CONTROLE DE QUALIDADE QUÍMICO DO LÁTEX DO
CAULE E DO FRUTO DE MANGABA (Hancornia speciosa GOMES)
Orientador: Prof. Dr. Paulo Cesar de Lima Nogueira
Co-orientadora: Profª. Drª. Valéria Regina de Souza Moraes
São Cristóvão – Sergipe
2012
Dissertação apresentada ao Programa de
Pós-Graduação em Química da
Universidade Federal de Sergipe como
parte dos requisitos para obtenção do
título de Mestre em Química.

FICHA CATALOGRÁFICA ELABORADA PELA BIBLIOTECA CENTRAL UNIVERSIDADE FEDERAL DE SERGIPE
S237d
Santos, Alan Diego da Conceição Desenvolvimento e validação de métodos por HPLC-DAD-
ELSD para controle de qualidade químico do látex do caule e do fruto de mangaba (Hancornia speciosa Gomes) / Alan Diego da Conceição Santos ; orientador Paulo Cesar de Lima Nogueira. – São Cristóvão, 2012.
152 f. : il.
Dissertação (Mestrado em Química) – Universidade Federal de Sergipe, 2012.
1. Química vegetal. 2. Mangaba. 3. Látex. 4. Análise cromatográfica. I. Nogueira, Paulo Cesar de Lima, orient. II. Título.
CDU 547.9:581.192:582.923.5

Com amor e gratidão dedico este trabalho aos
meus pais, Juciara e Azequias, por
consumirem suas vidas em virtude do meu
crescimento e bem estar...

Agradecimentos especiais
A Jesus Sacramentado por todo amor misericordioso e amizade. Sem sua presença
silenciosa nada disso seria possível!
Aos meus pais, Juciara e Azequias, por todo amor incondicional e por me
ensinarem que a vida é um presente de valor inestimável do criador. Aos meus irmãos Wesley
e Taciara por construírem parte do que eu sou.
A minha namorada, Vanessa Ávila, por aceitar dividir comigo o lado árduo da
vida acadêmica. Obrigado pelo companheirismo, pela compreensão, pela paciência com
minha ausência e pensamentos distantes...
Aos meus avós, tios (as), primos (as), padrinhos e madrinhas por todo o incentivo
acompanhado de mimos inestimáveis. Vocês são lugar de repouso.
Aos meus amigos do Grupo de Oração Sementes de Paz (em especial: Ivo e Irone,
Paulo e Regi e Otávio Neto) por toda a acolhida, ensinamentos e oração. Vocês são prova que
o “ser irmão” vai além dos laços sanguíneos. A minha grande amiga "acadêmica” Thalita
Bispo, por abrir os meus olhos para a beleza da pesquisa, minha incentivadora!!! Ao meu
amigo Andryel Vilanova, pela presença dissimulada!

Agradecimentos
Agradeço ao Prof. Dr. Paulo Cesar de Lima Nogueira (amante da ciência) por me
inserir nesse universo intrigante que é a pesquisa, pelos ensinamentos e confiança.
A Universidade Federal de Sergipe e ao Departamento de Química pela
oportunidade de execução deste trabalho.
A todos os professores do Departamento de Química/UFS, em especial aos
professores, Valéria Regina de Souza Moraes, Emmanoel Vilaça Costa, Samísia Maria F.
Machado e Sandro Navickiene pela contribuição na minha formação.
Aos meus “parceiros” durante o curso de mestrado, Givanilton Brito e Vilma
Menezes, pelas horas de estudo e desabafos.
A geração antiga do LABORGANICS Daniele Santos, Sandra Ribeiro e Wesley
Gomes e a Adriano Aquino, hoje doutorandos, pela amizade e incentivo.
Aos meus amigos do LABORGANICS (Aline Menezes, Charlene Souza, Eraldo,
Iara Lisboa, Jemmyson Romário, Leociley Menezes, Lívia Dutra, Marília Sampaio, Pedro
Ernesto, Susy, Thanany Brasil e Valéria Santana) e do LPPN (Darlisson Alexandria,
Givanildo, Hugo Cesar, Paloma Prata e Rafaely Lima). Agradeço a todos pela amizade, apoio,
incentivo, colaboração e momentos descontraídos. Vocês tornaram meu caminho menos
árduo!!
Ao Prof. Dr. Antonio G. Ferreira do Departamento de Química da UFSCar, pela
colaboração e aquisição dos espectros de ressonância magnética nuclear.
A CNPq pela concessão da bolsa de estudo
A FAPITEC/SE, COPES/UFS, PAIRD/UFS e PROCAD/CAPES pelo apoio
financeiro que permitiu a realização desse trabalho.
Enfim, a todos que diretamente ou indiretamente que contribuíram para a
realização deste trabalho.

SUMÁRIO
LISTA DE FIGURAS................................................................................................................I
LISTA DE TABELAS..........................................................................................................VII
LISTA DE ABREVIATURAS E SIGLAS...........................................................................IX
RESUMO...............................................................................................................................XII
ABSTRACT.........................................................................................................................XIII
1. INTRODUÇÃO...................................................................................................................01
1.1 - Látex.................................................................................................................................05
1.2 - Controle de qualidade de amostras vegetais.....................................................................06
1.3 - Arranjo de diodos (DAD) e Espalhamento de luz (ELSD) como detectores para análise
de amostras vegetais por HPLC................................................................................................07
1.4 - Perfil cromatográfico e quantificação de marcadores químicos para controle de qualidade
de amostras vegetais..................................................................................................................10
2. REVISÃO BIBLIOGRÁFICA...........................................................................................11
2.1 - Aspectos botânicos...........................................................................................................12
2.2 - Aspectos químicos............................................................................................................14
2.3 - Aplicações terapêuticas e aspectos farmacológicos.........................................................18
2.4 - Propriedades tecnológicas do látex de H. speciosa Gomes..............................................20
3. OBJETIVOS........................................................................................................................21
3.1 – Objetivos Gerais...............................................................................................................22
3.2 – Objetivos Específicos.......................................................................................................22
4. PARTE EXPERIMENTAL...............................................................................................23
4.1 – Materiais e equipamentos.................................................................................................24
4.2 – Limpeza do material.........................................................................................................25
4.3 – Material vegetal................................................................................................................25
4.3.1 – Obtenção do látex do caule da mangabeira.........................................................25
4.3.2 – Obtenção do látex dos frutos da mangabeira.......................................................26
4.3.3 – Polpa comercial do fruto da mangabeira.............................................................26
4.4 – Preparo das amostras do látex de H. speciosa e dos marcadores
químicos....................................................................................................................................27

4.4.1 – Látex do caule......................................................................................................27
4.4.2 – Látex dos frutos...................................................................................................27
4.4.3 – Marcadores químicos do látex dos frutos............................................................27
4.5 – Condições cromatográficas de análise.............................................................................28
4.5.1 – Método qualitativo para o látex do caule da mangabeira....................................28
4.5.2 – Método qualitativo para o látex dos frutos da mangabeira..................................28
4.6 – Validação de métodos cromatográficos analíticos...........................................................29
4.6.1 – Validação do método analítico para o perfil cromatográfico do látex do caule de
Hancornia speciosa...................................................................................................................29
4.6.1.1 – Teste da precisão de injeção....................................................................29
4.6.1.2 – Teste da repetibilidade............................................................................29
4.6.1.3 – Estabilidade da amostra do látex do caule..............................................30
4.6.1.3.1 – Estabilidade de curta duração.....................................................30
4.6.1.3.2– Estabilidade após ciclos de descongelamento e
congelamento............................................................................................................................30
4.6.1.4 – Robustez..................................................................................................30
4.6.2 – Validação do método cromatográfico para a quantificação de éster diidroxilado
de lupeol em látex do fruto de H. speciosa e polpa
comercial...................................................................................................................................31
4.6.2.1 – Condições analíticas do método quantitativo..........................................31
4.6.2.2 – Preparo da solução estoque, padrões de calibração e amostras controle de
qualidade...................................................................................................................................31
4.6.2.3 – Linearidade..............................................................................................32
4.6.2.4 – Seletividade.............................................................................................32
4.6.2.5 – Limite de detecção (LD) e quantificação (LQ).......................................32
4.6.2.6 – Precisão...................................................................................................33
4.6.2.7 – Exatidão...................................................................................................33
4.6.2.8 – Robustez..................................................................................................34
4.6.2.9 – Estabilidade.............................................................................................34
4.6.2.9.1 – Estabilidade de curta duração.....................................................34
4.6.2.9.2– Estabilidade após ciclos de descongelamento e
congelamento............................................................................................................................34

4.7 – Isolamento dos marcadores químicos do látex do caule de H. speciosa
Gomes.......................................................................................................................................35
4.8 – Identificação dos marcadores químicos...........................................................................35
5. RESULTADOS E DISCUSSÃO........................................................................................36
5.1 – Otimização das condições cromatográficas de análise: látex do caule de H. specisoa
Gomes.......................................................................................................................................37
5.1.1 – Primeira etapa do processo de otimização: uso do gradiente
exploratório...............................................................................................................................38
5.1.2 – Segunda etapa do processo de otimização: avaliação dos parâmetros da eluição
gradiente....................................................................................................................................42
5.1.3 – Preparo da amostra do látex do caule..................................................................45
5.2 – Validação do método analítico para o perfil cromatográfico do látex do caule de H.
speciosa.....................................................................................................................................52
5.3 – Aplicação do método analítico desenvolvido: análise das amostras comerciais do látex
do caule de H. speciosa.............................................................................................................56
5.4 – Isolamento dos marcadores químicos do látex do caule de H. speciosa
Gomes.......................................................................................................................................61
5.5 – Identificação do marcadores químicos.............................................................................66
5.5.1 – Identificação e determinação estrutural HSG 2...................................................66
5.5.2 – Identificação e determinação estrutural HSG 3...................................................75
5.5.3 – Identificação e determinação estrutural HSG 6...................................................80
5.6 – Otimização das condições cromatográficas de análise: látex dos frutos de H.
speciosa.....................................................................................................................................89
5.7 – Validação do método cromatográfico para quantificação do marcador químico éster
diidroxilado 3-β-O-acil lupeol................................................................................................100
5.7.1 – Seletividade.......................................................................................................101
5.7.2 – Linearidade .......................................................................................................103
5.7.3 – Limite de detecção e limite de quantificação....................................................105
5.7.4 – Precisão e exatidão............................................................................................105
5.7.5 – Estabilidade ......................................................................................................107
5.7.6 – Robustez ...........................................................................................................108
5.8 – Aplicação do método.....................................................................................................108

6. CONCLUSÃO...................................................................................................................111
7. REFERÊNCIAS................................................................................................................114

I
LISTA DE FIGURAS
Figura 1: Foto de um espécime (1) e do fruto (2) de Hancornia speciosa Gomes..................13
Figura 2: Extração do látex do caule Harcornia speciosa Gomes...........................................26
Figura 3: Cromatogramas para a seleção de condições para o método em HPLC-DAD. Os
números correspondem às condições apresentadas na Tabela
1.................................................................................................................................................41
Figura 4: Cromatogramas (λ = 220 nm) para a seleção de condições para o método em
HPLC-DAD. Os números correspondem às condições apresentadas na Tabela
2.................................................................................................................................................44
Figura 5: Cromatogramas para a seleção do modo de preparo do látex do caule de H.
speciosa para análise em HPLC-DAD: (preto) látex tratado com ácido acético e secado em
centrifugador; (vermelho) látex sem ácido acético e secado em centrifugador e (azul) látex
secado em liofilizador. Os cromatogramas foram obtidos utilizando a condição 11 descrita na
Tabela 2....................................................................................................................................46
Figura 6: Perfil Cromatográfico do látex do caule de H. speciosa obtido com a eluição
gradiente: 5-50% H2O (A): ACN (B) em 50 min; vazão: 1,0 mL/min; λ= 220nm; Vinj = 20 µL;
coluna analítica Phenomenex LUNA®
5μm C18 (250 x 4,6 mm) adaptada a uma coluna guarda
C18 (4 x 3,0 mm, Phenomenex)...............................................................................................48
Figura 7: Espectros de absorção UV das bandas cromatográficas assinaladas na Figura 6
(apresentados de 1 a 7)..............................................................................................................49
Figura 8: Projeção tridimensional obtido por HPLC-DAD da amostra do látex do caule da H.
speciosa.....................................................................................................................................50

II
Figura 9: Perfil cromatográfico do látex do caule da H. speciosa por HPLC-ELSD obtido
com a eluição gradiente: 5-50% H2O (A): ACN (B) em 50 min; vazão: 1,0 mL/min; ELSD:
temperatura do tubo mantido em 70˚C e fluxo de N2 pressurizado em 353 KPa; Vinj = 20 µL;
coluna analítica Phenomenex LUNA®
5μm C18 (250 x 4,6 mm) adaptada a uma coluna guarda
C18 (4 x 3,0 mm, Phenomenex)...............................................................................................51
Figura 10: Perfil cromatográfico comparativo do látex do caule de H. speciosa por HPLC-
DAD-ELSD. (Preto) Cromatograma monitorado pelo DAD e (Vermelho) cromatograma
monitorado pelo ELSD.............................................................................................................52
Figura 11: Cromatogramas sobrepostos (n = 6) de amostras de látex do caule de H. speciosa
monitorados pelo DAD (220 nm). LCA (látex proveniente de Aracaju), LCP (látex adquirido
em Pontal), LCI (látex obtido em Itabaiana), LCB (látex proveniente de Boquim) e LCU
(látex adquirido em Umbaúba).................................................................................................57
Figura 12: Cromatogramas sobrepostos (n = 6) de amostras de látex do caule de H. speciosa
monitorados pelo ELSD. LCA (látex proveniente de Aracaju), LCP (látex adquirido em
Pontal), LCI (látex obtido em Itabaiana), LCB (látex proveniente de Boquim) e LCU (látex
adquirido em Umbaúba)............................................................................................................58
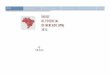
Figura 13: Cromatogramas fingerprint das amostras de látex do caule de H. speciosa (HPLC-
DAD). LCA (látex proveniente de Aracaju), LCP (látex adquirido em Pontal), LCI (látex
obtido em Itabaiana), LCB (látex proveniente de Boquim) e LCU (látex adquirido em
Umbaúba). Obs.: * Picos presentes em todas as amostras do látex do caule............................59
Figura 14: Cromatogramas fingerprint das amostras de látex do caule de H. speciosa (HPLC-
ELSD). LCA (látex proveniente de Aracaju), LCP (látex adquirido em Pontal), LCI (látex
obtido em Itabaiana), LCB (látex proveniente de Boquim) e LCU (látex adquirido em
Umbaúba). Obs.: * Picos presentes em todas as amostras do látex do caule.......................................60
Figura 15: Cromatograma (λ = 220 nm) do látex do caule de H. speciosa em sistema semi-
preparativo. Condições cromatográficas: item 4.7. Os picos coletados estão indicados no
cromatograma............................................................................................................................62
Figura 16: Perfil cromatográfico do látex do caule de H. speciosa acompanhado dos
compostos isolados em escala analítica (λ = 220 nm)..............................................................63

III
Figura 17: Cromatogramas (λ = 220 nm) das bandas cromatográficas HSG 2, HSG 3, HSG 4
e HSG5 purificadas no sistema semi-preparativo, com seus respectivos espectros de absorção
UV no tempo de retenção em que eluem. Condições de análise: item 4.5.1............................64
Figura 18: Espectro de RMN de 1H (400 MHz, CD3OD) da amostra HSG 2.........................66
Figura 19: Ampliação da região entre 6,98-7,06 ppm do espectro de RMN de 1H (400 MHz,
CD3OD) da amostra HSG 2......................................................................................................67
Figura 20: Ampliação da região entre 6,05-6,17 ppm do espectro de RMN de 1H (400 MHz,
CD3OD) da amostra HSG 2......................................................................................................67
Figura 21: Ampliação da região entre 3,93-4,27 ppm do espectro de RMN de 1H (400 MHz,
CD3OD) da amostra HSG 2......................................................................................................68
Figura 22: Ampliação da região entre 3,56-3,91 ppm do espectro de RMN de 1H (400 MHz,
CD3OD) da amostra HSG 2. ....................................................................................................68
Figura 23: Ampliação da região entre 2,012-2,080 ppm do espectro de RMN de 1H (400
MHz, CD3OD) da amostra HSG 2. ..........................................................................................69
Figura 24: Espectro de RMN de 13
C (100 MHz, CD3OD) da amostra HSG 2........................69
Figura 25: Mapa de correlação HSQC (1H: 400 MHz,
13C: 100 MHz, CD3OD) da amostra
HSG 2........................................................................................................................................70
Figura 26: Ampliações do mapa de correlação HSQC (1H: 400 MHz,
13C: 100 MHz,
CD3OD) da amostra HSG 2......................................................................................................71
Figura 27: Mapa de correlação HMQC (1H: 400 MHz,
13C: 100 MHz, CD3OD) da amostra
HSG 2........................................................................................................................................72
Figura 28: Ampliações do mapa de correlação HMQC (1H: 400 MHz,
13C: 100 MHz,
CD3OD) da amostra HSG 2......................................................................................................72
Figura 29: Estrutura química do ciclohexiletanóide glicosilado cornosídeo (HSG 2)............73
Figura 30: Espectro de RMN de 1H (400 MHz, CD3OD) da amostra HSG 3.........................75

IV
Figura 31: Ampliação da região entre 4,08-7,00 ppm do espectro de RMN de 1H (400 MHz,
CD3OD) da amostra HSG 3......................................................................................................76
Figura 32: Ampliação da região entre 2,41-3,94 ppm do espectro de RMN de 1H (400 MHz,
CD3OD) da amostra HSG 3......................................................................................................77
Figura 33: Espectro de RMN de 13
C (100 MHz, CD3OD) da amostra HSG 3........................78
Figura 34: Mapa de correlação HMBC (1H: 400 MHz,
13C: 100 MHz, CD3OD) da amostra
HSG 3........................................................................................................................................80
Figura 35: Estrutura química do ciclohexiletanóide glicosilado dihidrocornosídeo (HSG
3)...............................................................................................................................................80
Figura 36: Espectro de RMN de 1H (400 MHz, CD3OD) da amostra HSG 6.........................83
Figura 37: Ampliação da região entre 6,65-7,12 ppm do espectro de RMN de 1H (400 MHz,
CD3OD) da amostra HSG 6......................................................................................................83
Figura 38: Ampliação da região entre 4,38-5,08 ppm do espectro de RMN de 1H (400 MHz,
CD3OD) da amostra HSG 6......................................................................................................84
Figura 39: Ampliação da região entre 3,10-3,90 ppm do espectro de RMN de 1H (400 MHz,
CD3OD) da amostra HSG 6......................................................................................................84
Figura 40: Espectro de RMN de 13
C (100 MHz, CD3OD) da amostra HSG 6........................85
Figura 41: Ampliação da região entre 56,0 e 87,0 ppm do espectro de RMN de 13
C (100
MHz, CD3OD) da amostra HSG 6............................................................................................85
Figura 42: Mapa de correlação HSQC (1H: 400 MHz,
13C: 100 MHz, CD3OD) da amostra
HSG 6........................................................................................................................................86
Figura 43: Ampliações do mapa de correlação HSQC (1H: 400 MHz,
13C: 100 MHz,
CD3OD) da amostra HSG 6......................................................................................................87
Figura 44: Mapa de correlação HMBC (1H: 400 MHz,
13C: 100 MHz, CD3OD) da amostra
HSG 6........................................................................................................................................87

V
Figura 45: Ampliações do mapa de correlação HMBC (1H: 400 MHz,
13C: 100 MHz,
CD3OD) da amostra HSG 6......................................................................................................88
Figura 46: Estrutura química da (7,8)-treo-4,7,9,9’-tetrahidroxi-3,3’-dimetoxi- 8-O-4’-
neolignana-7-O-β-D-glicopiranosídeo (HSG 6).......................................................................89
Figura 47: Cromatograma obtido a partir das seguintes condições: coluna Microsorb MV100-
5C18, modo isocrático 100% de ACN por 30 min em um fluxo de 0,8 mL/min, volume de
injeção 20µL.............................................................................................................................90
Figura 48: Cromatogramas (λ = 200 nm) obtidos a partir das seguintes condições: coluna
Microsorb MV100-5C18, fluxo de 0.8 mL/min, volume de injeção 20µL: (Análise 1) modo
isocrático 85% de ACN:H2O, (Análise 2) modo isocrático 80% de ACN:H2O ambos por 30
min............................................................................................................................................92
Figura 49: Cromatogramas (λ = 200 nm) obtidos a partir das seguintes condições: coluna
hexil-fenil LUNA, fluxo de 0.8mL/min, volume de injeção 20µL: (Análise 3) modo isocrático
100% de ACN:H2O, (Análise 4) modo isocrático 90% de ACN:H2O ambos por 30
min............................................................................................................................................93
Figura 50: Cromatogramas (λ = 200 nm) obtidos a partir das seguintes condições: coluna
hexil-fenil LUNA®, modo isocrático 90% de ACN:H2O, fluxo de 0,8 mL/min, volume de
injeção 20µL: (A) lupeol; (B) mistura de ésteres 3- β-O-acil lupeol; (C) éster diidroxilado 3-
β-O-acil lupeol e (D) extrato diclorometano do látex dos frutos de H.
speciosa.....................................................................................................................................94
Figura 51: Cromatogramas (λ = 200 nm) para a seleção de condições para análise do látex
dos frutos de H. speciosa Gomes em HPLC-DAD obtidos a partir das condições de análise (5-
7) descritas na Tabela 11...........................................................................................................95
Figura 52: Projeção tridimensional obtido por HPLC-DAD da amostra do látex dos frutos de
H. speciosa................................................................................................................................96
Figura 53: (A) Perfis cromatográficos monitorado com o DAD (200nm): extrato
diclorometano do látex dos frutos (preto); marcadores químicos: éster diidroxilado 3-β-O-acil
lupeol (vermelho), mistura de ésteres 3- β-O-acil lupeol (azul), e lupeol (magenta); (B) Perfil

VI
cromatográfico do látex dos frutos de H. speciosa monitorado pelo DAD (preto) e pelo ELSD
(vermelho).................................................................................................................................97
Figura 54: (A) Cromatogramas obtidos por DAD (200 nm): extrato diclorometano do látex
dos frutos (preto), látex dos frutos liofilizado (vermelho) e polpa comercial (azul); (B)
Cromatogramas obtidos por ELSD: extrato diclorometano do látex dos frutos (magenta), látex
dos frutos liofilizado (verde) e polpa comercial (azul).............................................................98
Figura 55: Estrutura química do éster diidroxilado 3-β-O-acil lupeol...................................100
Figura 56: Cromatograma típico: polpa da mangaba (vermelho), éster diidroxilado 3-β-O-acil
lupeol (azul) e polpa da mangaba fortificada (preto); (A) obtido pelo DAD; (B) obtido pelo
ELSD. As condições experimentais para obtenção desses cromatogramas são descritos no
item 4.6.2.1..............................................................................................................................102
Figura 57: Curvas analíticas obtidas para a quantificação de éster diidroxilado 3-β-O-acil
lupeol pelo DAD: padronização externa (A) e padronização por adição de padrão (B); e pelo
ELSD: padronização externa (C)............................................................................................104
Figura 58: Cromatograma representativo da polpa comercial de mangaba, obtido pelo DAD
(vermelho) e pelo ELSD (preto). Picos selecionados para quantificação: (1) éster diidroxilado
3-β-O-acil lupeol e (2-4) mistura de ésteres de lupeol............................................................109

VII
LISTA DE TABELAS
Tabela 1: Condições cromatográficas avaliadas na primeira etapa da otimização do método
para análise do látex do caule de H. speciosa, com detecção em 220 nm................................40
Tabela 2: Condições cromatográficas avaliadas na segunda etapa da otimização do método
para análise do látex do caule de H. speciosa, com detecção em 220 nm................................43
Tabela 3: Valores de desvio padrão relativo entre os tempos de retenção, área e altura das
bandas cromatográficas avaliadas nas análises da amostra do látex do caule de H. speciosa
(precisão de injeção).................................................................................................................53
Tabela 4: Valores de desvio padrão relativo entre os tempos de retenção, área e altura das
bandas cromatográficas avaliadas nas análises de amostra de H. speciosa
(repetibilidade)..........................................................................................................................53
Tabela 5: Valores de desvio padrão relativo entre os tempos de retenção, área e altura das
bandas cromatográficas avaliadas nas análises de amostra de H. speciosa Gomes
(estabilidade).............................................................................................................................54
Tabela 6: Valores de desvio padrão relativo entre os tempos de retenção, área e altura das
bandas cromatográficas avaliadas nas análises de amostra de H. speciosa
(robustez)..................................................................................................................................55
Tabela 7: Valores de massa e de rendimento obtido no isolamento dos marcadores
químicos....................................................................................................................................62
Tabela 8: Dados de RMN de 1H (400 MHz) e de
13C (100 MHz) em CD3OD da HSG-2
(cornosídeo) em comparação com dados da literatura..............................................................74
Tabela 9: Dados de RMN de 1H (400 MHz) e de
13C (100 MHz) em CD3OD da HSG-3
(dihidrocornosídeo) em comparação com dados da literatura..................................................79
Tabela 10: Dados de RMN de 1H (400 MHz) e de
13C (100 MHz) em CD3OD da HSG 6 em
comparação com dados da literatura.........................................................................................82
Tabela 11: Condições cromatográficas avaliadas na otimização do método para análise do
látex dos frutos da H. speciosa, com detecção em 220 nm, volume de injeção de 20 µL e
vazão de 0,8 mL/min.................................................................................................................91

VIII
Tabela 12: Parâmetros de linearidade usando as soluções padrões.......................................105
Tabela 13: Dados do estudo de precisão, intra- e inter-dias, do método para quantificação de
éster diidroxilado 3-β-O-acil lupeol........................................................................................106
Tabela 14: Dados do estudo de exatidão, intra- e inter-dias, do método para quantificação de
éster diidroxilado 3-β-O-acil lupeol........................................................................................107
Tabela 15: Teor de éster diidroxilado 3-β-O-acil lupeol de polpa e látex liofilizado utilizando
DAD e ELSD..........................................................................................................................109
Tabela 16: Teor dos ésteres de lupeol (indicados na figura 11) em polpa comercial de
mangaba por HPLC-DAD-ELSD...........................................................................................110

IX
LISTA DE ABREVIATURAS E SIGLAS
1D Unidimensional
2D Bidimensional
ACE Angiotensin Converting Enzyme (Enzima Conversora de Angiotensina)
ACN Acetonitrila
ANVISA Agência Nacional de Vigilância Sanitária
C18 Fase sólida à base de sílica modificada com grupos octadecil
C8 Fase sólida à base de sílica modificada com grupos octil
CQA Controle de Qualidade de Alta Concentração
CQB Controle de Qualidade de Baixa Concentração
CQM Controle de Qualidade de Média Concentração
d Dupleto
DAD Diode array detector (Detector de arranjo de diodos)
dd Dupleto duplo
ddd Duplo dupleto duplo
DPR Desvio padrão relativo
ELSD Evaporative light scattering detector (Detector evaporativo por espalhamento de luz)
F Fluxo
UFSCar Universidade Federal de São Carlos
GC Gas Chromatography (Cromatografia em fase gasosa)
HCOOH Ácido fórmico
HMBC Heteronuclear multiple bond correlation
HPLC High Performance Liquid Chromatography (Cromatografia Líquida de Alta
Eficiência)
HPLC-DAD-ELSD High Performance Liquid Chromatography coupled with diode array
detection and evaporative light scattering detector (Cromatografia Líquida de Alta Eficiência
com detectores de arranjo de diodos e evaporativo por espalhamento de luz)

X
HSQC Heteronuclear single quantum coherence
Hz Hertz
IBGE Instituto Brasileiro de Geografia e Estatística
J Constante de acoplamento escalar
LA Comprimento da coluna analítica
LABORGANICS Laboratório de Pesquisa em Química Orgânica de Sergipe
LCA Látex do caule de Aracaju
LCB Látex do caule de Boquim
LCI Látex do caule de Itabaiana
LCP Látex do caule de Pontal
LCU Látex do caule de Umbaúba
LD Limite de Detecção
LP Comprimento da coluna semi-preparativa
LQ Limite de Quantificação
m Mutipleto
MeOH Metanol
MHz Mega Hertz
NO Óxido nítrico
OMS Organização Mundial da Saúde
PF Porcentagem final
PI Porcentagem inicial
ppm Partes por milhão
q Quadrupleto
quintl Quintupleto largo
RA Diâmetro da coluna analítica
RMN 13
C Ressonância Magnética Nuclear de Carbono - 13
RMN 1H Ressonância Magnética Nuclear de Hidrogênio

XI
RMN Ressonância Magnética Nuclear
RP Diâmetro da coluna semi-preparativa
S Fator de escalonamento
s Simpleto
t Tripleto
THF Tetrahidrofurano
tl Tripleto largo
TPA Tissue Plasminogen Activator (Ativador de Plasminogênio)
tR Tempo de retenção
UFS Universidade Federal de Sergipe
UV Ultravioleta
Vinj. Volume de injeção
WHA World Health Assembly (Assembleia Mundial da Saúde)
δ Deslocamento Químico
λ Comprimento de onda

XII
RESUMO
O presente trabalho apresenta o desenvolvimento e aplicação de métodos analíticos para o
controle de qualidade do látex do fruto e do caule de H. speciosa Gomes utilizando
cromatografia líquida de alta eficiência com os detectores de arranjo de diodos e evaporativo
por espalhamento de luz (HPLC-DAD-ELSD). No primeiro momento, um método analítico
para a obtenção do perfil cromatográfico foi desenvolvido e validado para a análise de uma
amostra autêntica do látex do caule de H. speciosa; em seguida foi utilizado o método
otimizado para a análise de amostras do látex do caule comercializadas em feiras livres do
Estado de Sergipe. A comparação visual dos perfis cromatográficos das diferentes amostras
do látex do caule permitiu averiguar a autenticidade e as dissimilaridades dos perfis químicos
dessas amostras. Utilizando uma coluna semi-preparativa, sete marcadores químicos foram
purificados a partir do látex do caule de H. speciosa; a caracterização por RMN 1H,
13C
possibilitou a identificação das seguintes substâncias: ciclohexiletanóide glicosilado
cornosídeo, ciclohexiletanóide glicosilado dihidrocornosídeo e (7,8)-treo-4,7,9,9’-
tetrahidroxi-3,3’-dimetoxi- 8-O-4’-neolignana-7-O-β-D-glicopiranosídeo. No segundo
momento, um método cromatográfico para análise qualitativa dos marcadores químicos
lupeol, α-amirina, β-amirina e ésteres 3- β-O-acil lupeol no látex dos frutos de H. speciosa e
em polpa comercial de mangaba foi desenvolvido. Por fim, um método analítico para a
quantificação do teor de ésteres de lupeol em látex dos frutos de H. speciosa e em polpa
comercial foi desenvolvido e validado utilizando HPLC-DAD-ELSD. No estudo da validação
foram avaliadas as figuras de mérito seletividade, linearidade, limite de quantificação e
detecção, precisão, exatidão, estabilidade e robustez conforme as normas descritas na RE nº
899/03 (ANVISA). A quantificação do teor de ésteres de lupeol por ambos os detectores se
mostraram significativamente similares (259,44 µg/mg para o DAD e 269,58 µg/mg para o
ELSD) com coeficiente de variação de 2,7 %. Este trabalho apresenta uma contribuição ao
controle de qualidade de amostras de H. speciosa.
Palavras-chave: Hancornia speciosa Gomes, látex, perfil cromatográfico, marcadores
químicos, quantificação, HPLC-DAD-ELSD, mangaba.

XIII
ABSTRACT
This work describes the development and application of analytical methods to establish
parameters to the quality control of fruit and trunk latex of H. speciosa using High
Performance Liquid Chromatography (HPLC) with diode array detect and evaporative light
scattering detector (ELSD). As a first step chromatographic profile analytical method was
developed and validated for the analysis of authentic sample of trunk latex of H. speciosa, and
then the optimized method was used for the analysis of commercial samples of trunk latex
sold in open markets from Sergipe, Brazil. The visual comparison of the chromatographic
profiles of different samples of trunk latex allowed checking the authenticity and
dissimilarities of chemical profiles of these samples. Seven chemical markers were purified
by semi-preparative HPLC-DAD from the trunk latex of H. speciosa, the characterization by
1H,
13C NMR allowed the identification of the following substances cyclohexylethanoid
glucoside cornoside, cyclohexylethanoid glucoside dihydrocornoside and (7,8)-treo-4,7,9,9’-
tetrahydroxy-3,3’-dimethoxy-8-O-4’-neolignan-7-O-β-D-glucopyranoside. As a second step
of our work chromatographic method for qualitative analysis of chemical markers lupeol, α-
amyrin, β-amyrin e 3- β-O-acyl lupeol esters in the fruit latex of H. speciosa and in the
mangaba commercial pulp was developed, the optimized method showed itself appropriate to
the identification of such substances with adequate separation. In the end one analytical
method for the quantification of the lupeol ester content in fruit latex and commercial pulp
was developed and validated using the HPLC-DAD-ELSD. In the validation study were
evaluated the figures of merit selectivity, linearity, limit of quantification and detection,
precision, accuracy, stability and robustness according to the standards described in RE nº
899/03 (ANVISA). The quantification of lupeol ester by both detectors was significantly
similar (259.44 µg/mg DAD and 269.58 µg/mg ELSD) with one coefficient of variation of
2.7%. This paper presents a contribution to the quality control of H. speciosa samples.
Keywords: Hancornia speciosa Gomes, latex, chromatographic profile, chemical markers,
quantification, HPLC-DAD-ELSD, mangaba.

1
Introdução

2
1 – INTRODUÇÃO
O consumo mundial de frutas tem aumentado em decorrência das evidências
científicas sobre os seus efeitos benéficos para a saúde humana. [1-3]
Juntamente com
outras partes dos vegetais, as frutas são uma excelente fonte de nutrientes vitais e de
compostos que são biologicamente ativos, tais como aqueles que são capazes de agir
como antioxidante celular ou como agente anti-inflamatório.[3-6]
Dentro desse contexto,
as plantas em geral podem ser consideradas um laboratório biossintético não só ao que
se refere aos compostos químicos utilizados como alimentos pelos seres humanos e
pelos animais, mas também no que se refere a uma infinidade de compostos que
exercem os mais diversos efeitos fisiológicos.[7,8]
Os compostos bioativos que não estão relacionados às funções básicas das
plantas são chamados de metabólitos secundários, os quais ocorrem com fantástica
variedade e complexidade e são biossintetizadas como mecanismo de defesa dos
vegetais às condições ambientes.[8,9]
Alguns metabólitos secundários são responsáveis pelos efeitos benéficos e
toxicológicos encontrados em plantas medicinais. Portanto, o uso seguro destas espécies
requer estudos que avaliem os seus aspectos químicos, farmacológico e toxicológico. [10]
A utilização da maioria das plantas medicinais cultivadas em nosso país está
fundamentada somente no seu uso popular sem nenhuma comprovação pré-clínica nem
clínica.[11]
As razões para falta de pesquisa nesta área são, em parte, pela escassez de
políticas de saúde pública, mas também pela ausência de metodologias de pesquisas
adequadas ou aceitas para a avaliação do uso da medicina tradicional. Mediante a
importância do tema, um processo coordenado de todos os atores (fitoquímicos,
químicos sintéticos, farmacólogos, farmacêuticos, médicos etc.) precisa ser realizado a
fim de descobrir metodologias efetivas para o controle de qualidade. [12,13]

3
Entre as metodologias disponíveis para o controle de qualidade de amostras
vegetais, o uso de perfis cromatográficos (fingerprint) tem ganhado bastante atenção.
Como uma ferramenta analítica, o fingerprint cromatográfico representa as
características químicas da planta. Em uma abordagem tradicional deste conceito, o
cromatograma padrão é construído a partir da relação entre os múltiplos compostos
presentes na amostra da planta. Nesse contexto, o perfil cromatográfico tem potencial
para determinar a identidade, autenticidade das plantas medicinais partindo-se do
princípio de que amostras com fingerprint similares têm propriedades similares.[14-15]
A cromatografia, com destaque para a cromatografia líquida de alta
eficiência (sigla HPLC, do inglês, high performance liquid chromatography) e
cromatografia a gás (sigla GC, do inglês, gás chromatography), é a técnica mais
utilizada na obtenção de fingerprints. Com rápida separação, o HPLC apresenta
eficiência e sensibilidade similar a GC. No entanto, sua aplicação é mais abrangente
visto que é capaz de analisar a maior parte dos compostos presentes em plantas, uma
vez que não está limitada a volatilidade ou estabilidade térmica das substâncias na
amostra.[13,16]
Normalmente, a análise química de amostras de origem vegetal é realizada
por meio da investigação qualitativa e quantitativa dos padrões de referência
(marcadores químicos). A quantificação de substâncias marcadoras em plantas
medicinais comumente apresenta os seguintes desafios: teor desconhecido das
moléculas de interesse na amostra, alta variabilidade do teor o que demanda métodos
que cubram um grande faixa e pequeno número de substâncias padrão disponível no
mercado. Faz-se necessário considerar que dificilmente os compostos quantificados
sejam, isoladamente, responsáveis pela atividade biológica ou pela eficácia terapêutica,
uma vez que plantas constituem matrizes complexas onde a sua totalidade química pode
ser considerada o “princípio ativo”.[13,17-19]
No Brasil pode ser encontrada uma grande variedade de fruteiras com
potencial para o mercado agroindustrial e que podem tornar-se uma fonte de renda para
a população local.[7]
Nesse contexto, a biodiversidade brasileira constitui um fascinante
tema de pesquisa acadêmica, visto que muitas espécies utilizadas na medicina
tradicional (entre elas as fruteiras) não têm sido devidamente estudadas quanto a suas

4
propriedades e atividades benéficas à saúde. Neste cenário, encontra-se a mangabeira
(Hancornia speciosa Gomes), uma fruteira de expressão regional e o objeto de estudo
deste trabalho.
A mangabeira é uma planta tipicamente tropical, nativa do Brasil, que
possui grande potencial para exploração econômica. O mercado para esta fruta
encontra-se, principalmente, nas regiões Norte e Nordeste do Brasil. Em Sergipe, a
mangaba é uma das frutas mais abundantes e procuradas nas feiras livres, atingindo um
preço superior ao da uva e de outras frutas nobres. Além do consumo in natura, o maior
aproveitamento da mangaba se dá na forma de sucos, doces e sorvetes, sendo esse
processamento feito, na maioria das vezes, por pequenas empresas produtoras de
sorvete da região Nordeste. Segundo dados do Instituto Brasileiro de Geografia e
Estatística (IBGE) referentes ao ano de 2009 o valor da produção sergipana da
mangaba, que corresponde a 55,3% da produção nacional, foi de R$ 705 mil, quase
65% do R$ 1,09 milhão produzidos nacionalmente pela extração do fruto.[20-22]
A mangabeira, como outras espécies pertencentes à família Apocynaceae é
produtora de látex. Quimicamente, o látex em geral é uma mistura complexa de
proteínas, carboidratos, óleo, metabólitos secundários e borracha tendo a função de
proteger a planta contra herbivoria, microorganismos e também selar ferimentos. Na H.
speciosa, o látex pode ser encontrado em diversas partes principalmente no tronco e nos
frutos. Apesar de seu potencial comercial devido algumas propriedades similares ao
látex da seringueira (Hevea brasiliensis), o látex da mangabeira encontra maior
utilização na medicina popular contra tuberculose, úlcera, fungos e certos tipos de
inflamações.[23-27]
Uma das principais preocupações dos produtores de frutas da região
Nordeste do Brasil é a agregação de valor às fruteiras regionais, as quais são geralmente
produzidas em regime extrativista. Dessa forma, o conhecimento químico da H.
speciosa e seu emprego como insumo para as indústrias farmacêutica, de alimentos e/ou
cosméticos poderia ser uma alternativa para agregar valor ao fruto, melhorar a renda dos
produtores da região e contribuir para diminuição da devastação desta espécie.

5
1.1 – Látex
O látex ocorre no reino das plantas em mais de 12.000 espécies pertencentes
a 900 gêneros.[28]
As estruturas vegetais denominadas de laticíferos são responsáveis
pela produção do “leite vegetal” podendo ser localizados em todos os tecidos epiteliais
da planta, sendo a ocorrência mais frequente na casca do tronco. Quando seccionados,
os vegetais laticíferos deixam fluir o látex, que coagula e veda o corte feito na planta
por reações enzimáticas.[ 29,30]
Do ponto de vista funcional, não há conhecimento de
qualquer influência do látex no metabolismo primário, por outro lado, sua função tem
sido frequentemente relacionada a defesa contra herbívoros.[31]
De forma geral, o látex natural apresenta-se com um sistema bifásico, no
qual a fase polimérica está na forma de emulsão. O látex é normalmente composto por
uma complexa mistura de diferentes componentes, incluindo macromoléculas. O
poliisopreno está presente em todas as espécies laticíferas e é um dos componentes
majoritários podendo ser encontrado na forma cis e/ou trans. Outros constituintes
presentes em látex e relatados em estudos fitoquímicos são: polissacarídeos,
flavonóides, lipídeos, fosfolipídios, proteínas, alcanos, triterpenóides, alcalóides,
taninos.[29,30,32]
Alguns destes compostos fornecem resistência contra herbívoros por
meio de efeitos antinutritivos e tóxicos, enquanto que outros estão envolvidos na
viscosidade que podem capturar diferentes insetos. Em geral os compostos encontrados
no látex estão diretamente relacionados com o metabolismo, defesa e interações
ecológicas das plantas.[32]
No contexto humano, o látex pode ser ingerido a partir do consumo das
frutas frescas (e seus derivados) ou como bebida.[33]
A ingestão de látex deve ser bem
avaliada, uma vez que propriedades corrosivas e queratolíticas são conhecidas nesse
tipo de matriz.[31-32]
Por outro lado, o reconhecimento do efeito benéfico das substâncias
presentes em látex na saúde humana tem sido relatado.[34-36]
Nesse sentido, a caracterização química de látices de diferentes espécies
vegetais tem sido realizada permitindo o conhecimento de diferentes substâncias que
por sua vez viabiliza o entendimento da funcionalidade do látex para plantas, bem como
assegura o emprego do mesmo na medicina popular. A cromatografia líquida de alta

6
eficiência é uma das técnicas empregadas para o isolamento e identificação de
compostos nesse tipo de matriz.[37-39]
1.2 – Controle de qualidade de amostras vegetais
A Organização Mundial da Saúde (OMS) através das resoluções WHA
31.33 (1978) e 40.33 (1987) afirmou a importância do uso de plantas medicinais nos
cuidados da saúde e a necessidade da criação de programas globais para a identificação,
validação, preparação, cultivo e conservação das plantas medicinais utilizadas na
medicina tradicional, bem como certificar o controle de qualidade delas.[40]
Assegurar a qualidade do material vegetal é um dos fatores que contribui
para o uso eficaz e seguro do produto. A eficácia é dada pela comprovação, por meio de
ensaios farmacológicos pré-clínicos e clínicos, dos efeitos biológicos preconizados para
esses recursos terapêuticos, e a segurança é determinada pelos ensaios que comprovam
a ausência de efeitos tóxicos. [40-41]
A avaliação da qualidade dos produtos de origem
vegetal pode ser feita através de procedimentos de análises químicas, físicas, físico-
químicas e microbiológicas.[42]
O controle de qualidade químico de plantas torna-se uma tarefa difícil
devido a variabilidade e a complexidade dos compostos presente nos vegetais. Em
muitos casos os princípios ativos não estão suficientemente estabelecidos ou ainda a
atividade biológica é atribuída aos efeitos sinérgicos das substâncias. Esses fatores
tornam as metodologias para o controle de qualidade laboriosas e limitadas.[13,43]
Convencionalmente, um dos primeiros e imprescindíveis passos para o
controle de qualidade é a verificação da autenticidade da matéria-prima vegetal, que
pode ser feita através da identificação botânica (análise macro e microscópica) e através
de métodos químicos, baseados na presença de substâncias que, preferencialmente,
possuam relação com a atividade terapêutica e/ou com a identificação das espécies, os
chamados marcadores químicos e/ou quimiotaxonômicos.[17]

7
Marcadores químicos são substâncias de referência que podem ser
considerados a base para estabelecer a qualidade de medidas analíticas e,
consequentemente, do material vegetal. Qualquer substância química com estrutura
conhecida pode ser usada como um padrão para teste de identidade (análise
qualitativa).[44]
Com o avanço das técnicas cromatográficas, os métodos químicos clássicos
de caracterização baseados na presença de grupos funcionais têm caído em desuso, uma
vez que são pouco sensíveis e inespecíficos. Paralelo a esse fato, a cromatografia líquida
de alta eficiência tem possibilitado a caracterização de amostras vegetais a partir de
análises qualitativas de padrões conhecidos na matriz e do isolamento em escala semi
ou preparativa de substâncias, de forma rápida, eficaz e economicamente viável.[45-47]
1.3 – Arranjo de diodos (DAD) e Evaporativo por
espalhamento de luz (ELSD) como detectores para análise de amostras
vegetais por HPLC
Para a detecção dos compostos presente em extratos brutos de planta,
eficientes métodos de separação são requeridos. Nesse sentido, a cromatografia líquida
de alta eficiência tem sido reconhecida como a mais versátil técnica, dispensando, em
muitos casos, métodos complexos de preparação da amostra. O HPLC tem se
desenvolvido ao longo dos anos em termos de conveniência, velocidade, opções de fase
estacionária, alta sensibilidade, aplicabilidade e habilidade para hifenação com
detectores espectrométricos e espectroscópicos.[48]
A diversidade estrutural das substâncias presentes nos produtos naturais
resulta na alta variabilidade das propriedades físico-químicas destes compostos, o que
torna a detecção universal um desafio. Nenhum dos detectores disponíveis para o HPLC
é capaz de detectar todas as substâncias, em um dado extrato, dentro de uma única
análise. Nesse contexto, a escolha apropriada do detector é de grande relevância e
conduz para a obtenção dos dados químicos apropriados, tendo em vista a classe de
metabólitos secundários que será analisada.[17,48]

8
Na tentativa de se ter uma visão mais ampla da composição química das
plantas, métodos cromatográficos têm sido desenvolvidos com o uso de diferentes
detectores o que possibilita a determinação simultânea de compostos com propriedades
diferentes. Por exemplo, o detector de arranjo de diodos (DAD – do inglês, diode array
detector) juntamente com o detector evaporativo por espalhamento de luz (ELSD – do
inglês, evaporative light scattering detector) podem ser empregados para determinar
simultaneamente compostos que apresentam pouca ou nenhum absorção na região do
ultravioleta (UV).[16,49-51]
Entre os detectores utilizados no HPLC o mais simples e largamente
empregado é o UV. Três tipos de detectores de UV estão disponíveis: os fotômetros de
comprimento de onda fixo, os espectrofotômetros e os detectores por arranjo de
fotodiodos. O detector de comprimento de onda fixo é o mais barato e apresenta alta
sensibilidade visto que a luz é emitida em um específico comprimento de onda através
de uma dada lâmpada. No entanto, o detector de múltiplos comprimentos de onda é
mais versátil, através da escolha de um comprimento de onda adequado para o analito, a
baixa sensibilidade do mesmo pode ser compensada. Já o detector de arranjo de diodos
oferece completo espectro de UV o que permite verificar a pureza dos picos resultantes
da separação.[48,52]
Embora tenha algumas limitações, particularmente para compostos que não
possuem grupos cromóforos, a detecção por UV possui a melhor combinação de
sensibilidade, linearidade, versatilidade e confiabilidade. A maior parte dos compostos
encontrados em plantas absorve no UV (200-400) devido à presença de substâncias com
uma ou mais ligações duplas ou elétrons desemparelhados. Esse fato torna o DAD o
detector espectrofotométrico mais frequentemente usado para análise de vegetais. [48,52]
O ELSD é considerado um detector quasi-universal, com ele pode ser
detectado qualquer analito menos volátil que a fase móvel, independente de qualquer
outro tipo de propriedade (ótica, eletroquímica etc.). Uma vantagem particular desse
tipo de detecção é a possibilidade de se usar a eluição no modo isocrático ou gradiente.
Estima-se que 1,5 % dos trabalhos que envolvem o HPLC na análise de produtos
naturais empregam o detector evaporativo por espalhamento de luz.[48,52]

9
O detector evaporativo por espalhamento de luz tem como princípio a
nebulização da fase móvel com um gás inerte e evaporação do solvente para produzir
pequenas partículas que serão detectadas em uma cela de espalhamento de luz. A
intensidade da luz espalhada depende do tamanho, da forma e das propriedades
superficiais das partículas formadas durante o processo de nebulização. Os principais
parâmetros que afetam a resposta do ELSD são o fluxo do gás nebulizador e a
temperatura utilizada no tubo.[48]
Diferente do DAD, a resposta gerada pelo ELSD depende da massa do
analito, ou seja, não tem nenhuma relação com as propriedades espectrais ou físico-
químicas da substância de interesse. Em teoria, isso significa que o ELSD gera uma
resposta similar para quantidades iguais de massa gerando assim um fator de resposta
universal. Contudo, a resposta do ELSD depende também da volatilidade do composto e
da composição da fase móvel o que faz com que tais procedimentos de quantificação
sejam experimentalmente inacessíveis. A soma desses fatores leva a uma resposta não
linear do ELSD, em outras palavras, a relação entre a concentração do analito e o sinal
produzido pelo detector de espalhamento de luz não obedece a Lei de Beer, mas sim
uma equação exponencial, I = kmb, onde I é a intensidade da luz, m a massa da partícula
espalhada, k e b são constantes determinadas principalmente pela natureza da fase
móvel e parâmetros do detector. Para facilitar o uso do detector, a equação torna-se
linear na forma logarítmica: log I = log k + b x log m.[48,53-55]
Várias são as aplicações, incluindo a determinação de resinas, polímeros,
polietilenoglicóis, carboidratos, lipídios, triglicerídeos, fosfolipídeos, esteróides,
adoçantes, aditivos de petróleo, agentes surfactantes, entre outras com o uso deste.[56]

10
1.4 – Perfil Cromatográfico e Quantificação de marcadores
químicos para controle de qualidade de amostras vegetais
As abordagens qualitativas (perfis cromatográficos) e quantitativas
(quantificação de marcadores químicos) têm sido utilizadas para estabelecer parâmetros
de autenticidade e qualidade de amostras vegetais.[17,57,58]
Os perfis cromatográficos (ou fingerprints) possibilitam a formação de um
padrão de reconhecimento específico dos múltiplos compostos presentes na amostra,
permitindo o reconhecimento de semelhanças e diferenças entre extratos submetidos às
mesmas condições de análise. Dessa forma, a qualidade química é avaliada baseada na
complexidade característica das plantas, uma vez que considera a ausência e presença
de marcadores químicos e a proporcionalidade existente entre os analitos detectados. O
conceito de fingerprint tem sido empregado em diferentes aplicações: (1) determinação
de autenticidade e identificação de espécies, (2) avaliação da qualidade química e da
fitoequivalência, (3) análise de estabilidade entre diferentes extratos, (4) análise de
consistência (variação lote-a-lote) de matérias-primas e de fitoterápicos e (5) estudos de
variabilidade sazonal e acompanhamento de cultivares.[15-17]
Embora a quantificação de marcadores químicos não seja suficiente para
assegurar a qualidade de uma amostra vegetal devido à presença metabólitos
secundários comuns a diferentes espécies, esta abordagem é bastante utilizada pela
pesquisa acadêmica e pela indústria.[17]
Na literatura, estão disponíveis diversos
exemplos de métodos cromatográficos por HPLC para a quantificação de substâncias de
referência em amostras vegetais.[59-61]
Nesse contexto, o presente trabalho apresenta o desenvolvimento, validação
e aplicação de métodos analíticos para estabelecer parâmetros para o controle de
qualidade do látex do fruto e do caule de H. speciosa Gomes utilizando cromatografia
líquida de alta eficiência com os detectores de arranjo de diodos e evaporativo por
espalhamento de luz (HPLC-DAD-ELSD).

11
Revisão
Bibliográfica

12
2 – REVISÃO BIBLIOGRÁFICA
2.1 – Aspectos botânicos
Apocynaceae é uma das maiores e mais representativas famílias de
Angiospermas, contendo em seus limites cerca de 400-480 gêneros e 4.300-4.800
espécies com variados hábitos, como árvores, arbustos, subarbustos, lianas e ervas.
Dentre estas espécies algumas se destacam pelo grande potencial econômico (como por
exemplo, a Funtumia elastica Stapf. usada para produção de borracha); outras
apresentam grande importância medicinal (a citar: Catharantus roseus L. e
Vincetoxicum officinale Moench); outras ainda são cultivadas como ornamentais devido
ao seu valor paisagístico, como Mandevilla Lindl. e Nerium oleander L. Além destas
existem diversas espécies como o amapazeiro (Parahancornia amapa (Huber) Ducke),
a sorva (Couma utilis (Mart.) Müll. Arg.) e a mangaba (Hancornia speciosa Gomes),
cujos frutos são comestíveis e usados na fabricação de sucos, compotas e licores.[62]
Hancornia speciosa Gomes, a mangabeira, pertence à classe Equisetopsida
C. Agardh, subclasse Magnoliidae Novák ex Takht, superordem Asteranea Takht.,
ordem Gentianales Juss. Ex Bercht. & J. Presl., família Apocynaceae Juss., subfamília
Rauvolfioideae, tribo Willughbeieae. Hancornia é um gênero de única espécie
possuindo 11 sinônimos, compreendendo duas variedades que se diferenciam por
algumas características morfológicas, principalmente da folha e da flor: H. speciosa var.
speciosa e H. speciosa var. pubescens (Nees & Mart.) Mull. Arg.[63]
A mangabeira é uma planta arbórea de porte médio, possuindo de 2 a 10 m
de altura, podendo chegar a até 15 m, dotada de copa irregular, tronco tortuoso, bastante
ramificado e áspero; ramos lisos e avermelhados, exsudando látex em toda a sua
extensão (Figura 1). A casca da árvore é escura e fendilhada. Suas folhas são elípticas
de 5 a 6 cm de comprimento e 2 cm de largura. Sua inflorescência possui de 1 a 7 flores
perfumadas e de coloração branca. O fruto é uma baga ovóide e globosa, verde
amarelada ou verde rosada. Alguns autores classificam os frutos como baga piriforme,
que apresenta na sua superfície manchas encarnadas e encerram sementes comprimidas
e de forma triangular. As sementes são do tipo recalcitrante, isto é, não suportam
ressecamento, perdendo rapidamente o poder germinativo assim que são retiradas do
fruto.[64-66]

13
(1) (2)
Figura 1: Foto de um espécime (1) e do fruto (2) de Hancornia speciosa Gomes.
Fonte: (1) NOGUEIRA, P. C. L. (2009) e (2) SANTOS, A. D. C. (2011).
Embora a mangabeira também seja produtora de látex, o fruto, denominado
“mangaba” é o seu principal produto; este nome tem origem na língua tupi-guarani e
significa “coisa boa de comer”. A mangaba apresenta ótimo sabor e aroma o que leva a
ter uma excelente aceitação no mercado; com teor de proteína de 1,3 a 3,0%, podendo
ser indicada para alimentação de pessoas doentes e convalescentes, em função da sua
alta digestibilidade, valor nutricional e propriedades medicinais.[66,67]
No cenário atual, tem-se verificado uma crescente valorização da mangaba
pelos consumidores, o que tem estimulado a demanda, porém, paralelamente, tem-se
observado uma grande ameaça de diminuição da oferta, tendo em vista a crescente
devastação da vegetação nativa na qual a espécie está inserida, motivada pelo
crescimento das zonas urbanas, pela implantação da monocultura e pela falta de
conhecimento dos agricultores dos poucos saberes técnico disponível a respeito do
cultivo de mangaba. [66,68]

14
Neste sentido, há estudos promissores visando gerar conhecimentos e
tecnologias sobre a cultura da mangabeira, bem como o desenvolvimento dessa fruteira
em bases sustentáveis e competitivas, tornando a sua exploração sistemática e
superando as barreiras do extrativismo. Entre os aspectos agronômicos pesquisados
podemos citar: influência de substrato[69]
, organogênese[70]
, germinação in vitro[71]
,
influência da profundidade de semeadura e luminosidade[72]
, irrigação [73,74]
, avaliação
dos frutos pós-colheita[75,76]
e parâmetros genéticos em progênies.[77]
.
2.2 – Aspectos químicos
Embora possam ser encontrados diversos trabalhos de isolamento e
identificação de metabólitos secundários em outros gêneros de Apocynaceae, existem
poucos trabalhos a respeito dos aspectos químicos (isolamento e identificação de
substâncias) da H. speciosa.[78]
O nosso grupo de pesquisa (LABORGANICS) tem sido o pioneiro no
isolamento e identificação das substâncias presentes na mangabeira. O estudo
fitoquímico do látex dos frutos realizado por Sampaio (2008) permitiu o isolamento de
doze substâncias, sendo elas sete ésteres 3-β-O-acil lupeol : 3-β-O-hexadecanoato de
lupeoíla (2), 3-β-O-9-octadecenoato de lupeoíla (3), 3-β-O-octadecanoato de lupeoíla
(4), 3-β-O-3’-hidroxihexadecanoato de lupeoíla (8), 3-β-O-3’-hidroxioctadecanoato de
lupeoíla (9), 3-β-O-3’-hidroxiicosanoato de lupeoíla (10), 3-β-O-3’- hidroxidocosanoato
de lupeoíla (11), uma mistura contendo os triterpenos α-amirina (12), β-amirina (13),
lupeol (14), um 3-β-O-3’,5’-diidroxiicosanoato de lupeolíla (15), além da sacarose na
sua forma peracetilada (16).[86]
Esses triterpenos pentacíclicos são uma classe de
substâncias caracterizada por apresentar uma grande variedade de atividade
biológica.[79-82]

15
+
9'( )
7
1
3 45
10
25
9
O
O
( )5
26
827
1417
18
1928
20
29
30
2423
1'
3'
1'
2423
30
29
2028
19
18
1714
278
26
n O
OR1
)(
925
10
54
3
1
17
20
30 29
3 27
28
23
26
HO
1
4 5
8
9
10
1213
14
18
25
24
(13)
23
261
4 5
8
9
10
13
14
18
25
24
HO
273
28
29
192030
17
(14)
29
14
5'1' O
OOHOH
3'
3
20
4 5
30 19
28
1718
13
14
27
26
8
9
10
251
23 24
30
3
23
17
20
28
18
19
24
27
26
HO
1
4 5
8
9
10
1213
14
29
25
(12)
1'
2'O
CH2OAc
AcO
OAcCH2OAc
3'4'
5'6'
1
O
23
45
6
OCH2OAc
AcOAcO
AcO
(3)
(15)
(16)
R1= H, n= 10 (1), 12 (2), 14 (4), 16 (5), 18 (6), 20 (7)
R1=OH, n= 12 (8), 14 (9), 16 (10), 18 (11)

16
n = 22 - docosano (17)
23 - tricosano (18)
24 - tetracosano (19)
25 - pentacosano (20)
26 - hexacosano (21)
27 - heptacosano (22)
Já o estudo fitoquímico das folhas e ramos desta espécie proporcionou a
identificação de 22 substâncias. Novamente a mistura de triterpenos α-amirina, β-
amirina e lupeol e uma mistura de ésteres 3- β-O acil lupeol (1, 2, 4, 5, 6, 7, 8-11), além
de uma mistura de n-alcanos C22-C33 (17-28). Estes triterpenos pentacíclicos isolados da
H. speciosa são semelhantes aos que foram isolados por Carvalho et al. (2001) da casca,
látex e raiz da Parahancornia amapa, gênero quimiotaxinomicamente mais próximo da
Hancornia.[83,84]
A avaliação fitoquímica do extrato etanólico das folhas realizados por
Endringer et al. (2010) permitiu o isolamento do ácido quínico (29), do L-(+)-
bornesitol (30) e da rutina (31).[85]
CH3n
n
n = 28 - octacosano (23)
29 - nonacosano (24)
30 - triacontano (25)
31 - hentriacontano (26)
32 - dotriacontano (27)
33 - titriacontano (28)
OH OH
HO
HO
O
OH
(29)
HOHO
HO
O
OH
OH
(30)

17
Quanto à composição dos voláteis, há dois trabalhos na literatura: um que
trata da identificação dos voláteis da mangaba nos três estádios de maturação (verde,
“de vez” e maduro)[86]
e o outro que estuda os voláteis presentes nas folhas da
mangabeira variando-se o tempo de secagem e a época de colheita.[83]
Em ambos os
trabalhos, foi possível verificar a ocorrência da variação quantitativa e qualitativa dos
compostos.
Honda et al. (1990) apud Moraes et al. (2008), relataram a presença de
flavonóides e taninos na casca da mangabeira, bem como esteróides, triterpenos e
taninos nas folhas.[87]
A proantocianidina do tipo B epicatequina-(4β → 8) catequina
(32) e sua forma C-glicosilada (33) foram determinadas na infusão da casca da H.
speciosa por meio de injeção em fluxo direto em espectrômetro de massas seqüencial,
equipado com sistema de ionização por eletronebulização e analisador por armadilha de
íons. A atividade anti-úlcera atribuída à mangabeira pode estar relacionada à alta
concentração de proantocianidinas, uma vez que esses compostos são reconhecidos
como antioxidantes.[88]
O
OH
OHHO
OH O
O
O
OOOH
OH OH OH
OH
OH
(31)

18
2.3 – Aplicações terapêuticas e aspectos farmacológicos
Além da exploração econômica dos frutos e do potencial agroindustrial do
látex, as diversas partes da mangabeira encontram apreço na medicina popular. Em
algumas regiões, por exemplo, a casca possui propriedades adstringentes, e são
comumente utilizadas para o tratamento de dermatites, diabetes, doenças hepáticas,
como também usadas como anti-inflamatório. Já o látex é empregado contra a
tuberculose, úlceras, herpes, pancadas, inflamações, dermatoses e verrugas. O chá das
folhas é usado para cólica menstrual, reumatismo e hipertensão sendo as duas últimas
aplicações também atribuídas ao decocto da raiz.[64,65,89,90]
Recentemente, há trabalhos publicados ressaltando os aspectos
farmacológicos desta espécie. Serra et al. (2005) demonstraram que o extrato etanólico
das folhas de H. speciosa inibi a enzima conversora de angiotensina I (ACE); Ferreira et
al. (2007b) mostraram que estes extratos induzem uma potente vasodilatação em
artéria de ratos através de um mecanismo dependente da produção de NO; além desses
resultados, estes autores demonstraram também que o flavonóide rutina contribui para o
efeito vasodilatador da H. speciosa. Os resultados obtidos por Ferreira et al. (2007a)
corroboraram o uso popular desta planta para o tratamento de hipertensão. A atividade
antimicrobiana do extrato etanólico das folhas foi avaliado por Costa et al. (2008) que
observou o efeito inibitório para os microorganismos gram-positivos e gram-negativos.
[90-93]
O
OH
HO
OH
OH
OH
OHO
OH
OH
OH
OH
(32)
OHO
OH
OH
OH
OH
O
OH
HO
OH
OH
OH
oHO
HO
OH
OH
(33)

19
A avaliação de atividade antiinflamatória e quimiopreventiva do extrato
etanólico bruto das folhas de H. speciosa e sua fração metanólica foi realizada por
Endringer et al. (2009), neste estudo ambas atividades foram confirmadas pelo ensaio
de inibição NF-kB induzido por TPA em células HepG2-Luc ( que avalia a
quimioprevenção) e ensaio de inibição da enzima cicloxigenase-2, que avalia a ação
inflamatória.[85]
Moraes et al. (2008) reportaram que a casca da H. speciosa tem eficácia no
combate a úlcera e sugeriram que esta eficácia está baseada na habilidade do extrato
hidroalcólico de H. speciosa em estimular a síntese de muco e de produzir efeito anti-
secretor. Associado com esse efeito, o extrato hidroalcólico também tem propriedade
anti-Helicobacter pylori com ausência de efeito toxicológico. Segundo os autores essas
ações estariam relacionadas à presença de ácidos fenólicos e proantocianidinas,
previamente identificados nessa espécie. [87]
Em contraposição ao uso popular, o látex da mangabeira não apresentou
atividade microbiana frente às condições de análise realizada por Santos et al. (2007).[94]
Por outro lado, Marinho et al. (2011) demonstrou que o látex da H. speciosa apresentou
atividade anti-inflamatória confirmando o seu uso popular.[95]
Ainda no que se diz
respeito ao látex da mangabeira, Sampaio (2008) realizou testes bioquímicos para
avaliação da atividade antioxidante do látex dos frutos verdes, que apresentou efeito
hepatoprotetor significativo.[86]
Foi possível encontrar na literatura dois estudos relacionados à polpa da
mangaba. Trindade et al. (2002) investigaram a biodiversidade de leveduras em polpas
de frutas maduras e congeladas[96]
, enquanto Araújo et al. (2004) averiguaram a
atividade biológica das proteínas provenientes de frutas tropicais, sendo a mangaba uma
das frutas que apresentou atividade inibitória elevada.[97]
Um registro de patente feito por Endringer et al. (2010) descreveu a
obtenção de um extrato padronizado de folhas de H. speciosa e de uma fração
padronizada com atividade inibidora da enzima conversora de angiotensina (ACE),
vasodilatadora, anti-hipertensiva e antioxidante. O registro compreende ainda a
obtenção de composições farmacêuticas que contêm o extrato ou frações derivadas do

20
extrato de folhas da espécie H. speciosa ricas em ciclitóis e flavonóides, bem como sua
utilização para o tratamento de distúrbios cardiovasculares como hipertensão arterial,
aterosclerose, restenose, isquemia cardíaca ou cerebral não limitantes.[98]
2.4 – Propriedades tecnológicas do látex de H. speciosa
Gomes
Devido ao aumento das pesquisas em busca de fontes alternativas de
borracha natural com propriedades similares ao da Hevea brasiliensis, diversas fontes
têm sido estudadas nos últimos anos entre elas a mangabeira. Recentemente, Malmonge
et al. (2008) avaliaram as propriedades tecnológicas do látex da mangabeira, as quais se
mostraram bastante similar a da H. brasiliensis, tais resultados mostram que o látex da
H. speciosa é adequado para o uso em aplicações industriais. O baixo teor de proteína
da Hancornia, quando comparado com o látex da Hevea, sugere que o látex pode ter
importante aplicação em situações que necessitem do uso de borracha antialérgica.[99]
Já o comportamento térmico do látex da mangabeira foi avaliado por
Medeiros et al. (2010). O estudo indicou que a borracha extraída da H. brasiliensis é um
pouco mais estável termicamente que aquela extraída da H. speciosa. Ambos os
trabalhos mostram o potencial da Hancornia como fonte alternativa para a produção da
borracha natural.[100]

21
Objetivos

22
3 – OBJETIVOS
3.1 – Geral
Desenvolver e validar métodos analíticos em cromatografia líquida de alta
eficiência com os detectores de arranjo de diodos e evaporativo por
espalhamento de luz (HPLC-DAD-ELSD) para controle de qualidade do látex
do caule e dos frutos de Hancornia speciosa Gomes.
3.2 – Específicos
Desenvolver e validar métodos analíticos utilizando HPLC-DAD-ELSD para a
caracterização química do látex do caule e dos frutos de H. speciosa;
Aplicar o método analítico para determinar a autenticidade de amostras
comerciais do látex do caule da mangabeira;
Isolar marcadores químicos a partir do látex do caule de H. speciosa e
caracterizar quimicamente sua estrutura por RMN de 1H e de
13C em uma e duas
dimensões;
Desenvolver e validar método por HPLC-DAD-ELSD para a quantificação de
marcadores químicos em amostras de látex dos frutos de H. speciosa Gomes.

23
Parte Experimental

24
4 – PARTE EXPERIMENTAL
4.1 – Materiais e equipamentos
As análises por cromatografia líquida foram realizadas em equipamento da
marca SHIMADZU (Kyoto, Japão), modelo Prominence, constituídos pelos seguintes
módulos: duas bombas LC-6A, desgaseificador DGU-20A3, auto-injetor SIL-10A,
detector de UV com arranjo de diodos SPD-M20Avp, detector evaporativo de
espalhamento de luz (ELSD-LT II); ambos os detectores conectado a uma interface
CBM 20A. O equipamento foi gerenciado pelo software LC solution. Diferentes
colunas analíticas foram utilizadas na otimização das condições cromatográficas, sendo
elas: coluna C18 (5µm; 25,0 x 1,0 cm, Varian), coluna Hexil-fenil LUNA®
(10 µm, 15,0
x 0,46 cm, Phenomenex) e coluna C18 LUNA® (5 µm, 25,0 x 0,46 cm, Phenomenex).
Os solventes grau HPLC utilizados nas análises cromatográficas foram
adquiridos da JT BAKER (Phillipsburg, NJ, USA) e TEDIA (Fairfield, OH, USA).
Antes do uso, os solventes foram filtrados através de uma membrana filtrante de Nylon
de 47 mm (diâmetro da membrana) e 0,45 μm (diâmetro dos poros) da marca MFS.
A água desionizada utilizada na composição da fase móvel e no preparo das
amostras foi obtida em um sistema MILLI-Q (Millipore, São Paulo, Brasil). A retirada
do solvente das diferentes amostras foi realizada em evaporador rotatório da marca
HEIDOLPH®.
Os extratos comerciais do látex do caule da mangabeira foram secos em um
liofilizador LIOTOP® modelo L101 (Liobrás, São Carlos-SP, Brasil) e em concentrador
e centrifugador CENTRIVAP (LABCONCO®). As pesagens foram realizadas em
balança analítica (DENVER INSTRUMENT®) com precisão de 0,1 mg. As amostras
foram homogeneizadas em agitador do tipo Vortex (QL-901) da BIOMIXER®. No
preparo das soluções, usaram-se micropipetas de marca LAB MATE - HIGH TECH
LAB®.

25
4.2 – Limpeza do material
As vidrarias utilizadas durante o trabalho foram enxaguadas com água
corrente por várias vezes, ensaboadas com detergente, enxaguadas novamente,
sonicadas por 30 min em uma solução de água destilada e isopropanol (50:50 v/v) e por
fim levadas a estufa a 120 ˚C. As vidrarias volumétricas foram secas a temperatura
ambiente . Em seguida, o material foi coberto com papel filme e guardado em armário
fechado.
4.3 – Material Vegetal
4.3.1 – Obtenção do látex do caule da mangabeira
Uma amostra do látex do caule da H. speciosa foi adquirida no povoado de
Pontal, município de Indiaroba-SE (LCP). A extração foi realizada no dia 13 de janeiro
de 2011 às 5h e 45min com auxílio da Sra. Ednalva Tavares dos Santos, uma das
participantes do projeto “Catadoras de Mangaba”. Neste processo, pequenas incisões
foram feitas no caule da mangabeira e a seiva branca e leitosa oriunda de vários táxons
foi coletada em recipientes de plástico (Figura 2). O látex obtido (~100 mL) foi diluído
com 700 mL de água destilada e filtrado com auxílio de um pano fino. O volume
utilizado para a diluição está de acordo com aquele empregado no uso popular daquela
região. Um frasco âmbar revestido com papel alumínio foi utilizado para armazenar
esse material vegetal (“leite de mangabeira”), o qual foi mantido sob refrigeração
durante o transporte e em freezer até as análises por cromatografia líquida. Para a
identificação taxonômica da espécie, uma exsicata (#20.585) do material botânico foi
depositada no Herbário da Universidade Federal de Sergipe (ASE/UFS).
O estudo do látex do caule consistiu também na análise de amostras
comercializadas em feiras livres de diferentes cidades do Estado de Sergipe: Aracaju
(LCA), Itabaiana (LCI), Umbaúba (LCU) e Boquim (LCB). Tais amostras foram
adquiridas ao longo do desenvolvimento da pesquisa.

26
Figura 2: Extração do látex do caule Harcornia speciosa Gomes
Fonte: SANTOS, A. D. C. (2011)
4.3.2 – Obtenção do látex dos frutos da mangabeira
O látex da mangaba foi obtido a partir dos frutos verdes adquiridos na feira
livre de Pedrinhas-SE. A extração do látex foi feita por meio de diversos cortes nos
frutos (229 g), seguida da coleta do líquido branco em um béquer, o qual foi diluído em
água e, em seguida, extraído com diclorometano (6 x 50 mL). Após a remoção do
solvente em evaporador rotatório, obteve-se 480 mg do extrato diclorometânico.[86]
Um segundo extrato do látex dos frutos verdes foi preparado usando frutos
da mangabeira coletados no Bairro Augusto Franco, Aracaju-SE. Para a obtenção desse
extrato foram feitos diversos cortes nos frutos coletando-se o suco leitoso, o qual em
seguida foi liofilizado até completa remoção da água.
4.3.3 – Polpa comercial do fruto da mangabeira
As polpas de mangaba da marca VIP® (100 g) foram adquiridas em
supermercado da cidade de Aracaju-SE e posteriormente liofilizadas para a remoção da
água.

27
4.4 – Preparo das amostras do látex de Hancornia speciosa e dos
marcadores químicos
4.4.1 – Látex do caule
Para a análise cromatográfica, 3 mL das amostras do látex do caule foram
liofilizados. Após a completa retirada da água, foram adicionados 3 mL de metanol e
sonicados por 20 min. Em seguida, a parte insolúvel (borracha) foi retirada, a solução
resultante foi seca e a massa residual determinada. Sete miligramas desta amostra foi
pesada e dissolvida em 1 mL da solução aquosa de acetonitrila a 5% em volume; a qual
foi filtrada em uma membrana de nylon 0,45µm antes da análise por HPLC.
4.4.2 – Látex dos frutos
O látex dos frutos e a polpa comercial ambos liofilizados foram
solubilizados em uma mistura de hexano:metanol (7:1 v/v). Após filtração simples, o
sobrenadante foi secado e diluído em uma solução de 10% tetrahidrofurano:isopropanol.
Já para a análise do extrato diclorometano do látex dos frutos foi realizada a dissolução
direta em isopropanol. As amostras foram filtradas em uma membrana de nylon 0,45
µm e então analisadas por cromatografia líquida.
4.4.3 – Marcadores químicos do látex dos frutos
Os padrões utilizados nesse trabalho foram dissolvidos em isopropanol,
filtrados em membrana de nylon 0,45 µm e analisados por cromatografia líquida. Uma
amostra do triterpeno lupeol foi gentilmente cedida pela Profa. Dra. Valéria Regina de
Souza Moraes (DQI/UFS). Os demais marcadores químicos: uma mistura contendo os
triterpenos α-amirina, β-amirina e lupeol; uma mistura contendo sete (07) ésteres 3-β-O-
acil lupeol; e um éster diidroxilado 3-β-O-acil lupeol foram isolados do extrato
clorofórmio do látex dos frutos previamente pela MSc. Taís Santos Sampaio.[86]

28
4.5 – Condições cromatográficas de análise
4.5.1 – Método qualitativo para o látex do caule da mangabeira
Para as análises cromatográficas do látex do caule foi empregada uma coluna
analítica C18 LUNA®
(5 μm, 250 x 4,6 mm, Phenomenex) adaptada a uma coluna de guarda
C18 (4,0 x 3,0 mm, Phenomenex). A separação dos compostos se deu por meio da eluição
gradiente no modo reverso, com fase móvel constituída por água (A) e acetonitrila (B) na
seguinte proporção: 5-40% de (B) em 50 min; 40-100% de (B) em 5 min; 100% de (B) por 5
min. Ao término da corrida empregou-se um gradiente de retorno de 100-5% por 5 min com
tempo de espera para o recondicionamento da coluna de 10 min. Nos experimentos, utilizou-
se vazão de 1,0 mL/min e volume de injeção de 20 µL. Os cromatogramas foram monitorados
pelo DAD (λ= 220 nm) e pelo ELSD (temperatura do tubo mantido em 70˚C e fluxo de N2
pressurizado em 353 KPa).
4.5.2 – Método qualitativo para o látex dos frutos da mangabeira
As amostras obtidas conforme os itens 4.3.2 e 4.3.3 e preparadas conforme
procedimentos descritos nos itens 4.4.2 e 4.4.3 foram analisadas empregando-se uma
coluna analítica Hexil-fenil LUNA® (10 μm, 150 x 4,6 mm, Phenomenex). Uma vazão
de 0,8 mL/min foi utilizada e volume de injeção foi de 20 µL.
As análises cromatográficas foram realizadas em eluição gradiente no modo
reverso, com fase móvel constituída por água (A) e acetonitrila (B), seguindo a
programação: 86-90% (B) por 10 min; 90-100% (B) por 30 min; 100-86% (B) por 5
min para o retorno do gradiente, finalizando com 86% (B) por 15 min.
Os cromatogramas foram registrados com detector de arranjo de diodos em
varredura de 190-450 nm, sendo 200 nm o comprimento de onda escolhido para a
obtenção dos cromatogramas.
Depois do DAD, o efluente da coluna cromatográfica foi submetido ao
detector ELSD. A nebulização foi proporcionada por um fluxo de N2 pressurizado (351
KPa) e o efluente nebulizado foi evaporado a 50˚C.

29
4.6 – Validação de métodos cromatográficos analíticos
4.6.1 – Validação do método analítico para o perfil cromatográfico do
látex do caule de Hancornia speciosa
Para a validação do método analítico utilizou-se o látex do caule coletado
em Indiaroba. O tempo de retenção juntamente com a área e a altura foram utilizados
como parâmetros no processo de validação.
4.6.1.1 – Teste da precisão de injeção
A precisão na injeção dos extratos foi determinada pela análise da amostra
do látex ao longo de um dia de trabalho. As amostras foram preparadas
independentemente em triplicata e analisadas em quintuplicata. A precisão foi avaliada
através do desvio padrão relativo (DPR), utilizando a Equação 1.
Onde:
S = Desvio padrão experimental
X = Média das medições
4.6.1.2 – Teste da repetibilidade
A repetibilidade foi avaliada pela análise das amostras do látex do caule
preparadas por três vezes independentemente em três dias diferentes e analisadas em
quintuplicata. O DPR foi tomado como parâmetro para a avaliação da repetibilidade.
Equação 1:

30
4.6.1.3 – Estabilidade da amostra do látex do caule
A estabilidade foi avaliada em duas formas: após ciclos de congelamento e
descongelamento e a estabilidade de curta duração.
4.6.1.3.1 – Estabilidade de curta duração
As amostras do látex do caule permaneceram no autoinjetor do
cromatógrafo um período total de 48h, sendo analisadas ao completar períodos de 24 e
48 h. Os resultados foram comparados com aqueles obtidos nas análises das amostras
recém-preparadas.
4.6.1.3.2– Estabilidade após ciclos de descongelamento e congelamento
As amostras foram congeladas por 24h em freezer, sendo então submetidas
ao descongelamento à temperatura ambiente. Quando completamente descongeladas, as
amostras foram congeladas novamente por 12h e assim sucessivamente até completar os
três ciclos. Os resultados foram comparados com aqueles obtidos da análise das
amostras recém-preparadas.
4.6.1.4 – Robustez
Para avaliar a robustez do método foram feitas as seguintes alterações:
Fluxo da fase móvel: 0,8 - 1,2 mL/min;
Proporção da fase móvel inicial: 3 - 7% de ACN:H2O;
Proporção da fase móvel final: 38 - 42% de ACN:H2O;
Lote da coluna C18 (Phenomenex) : 423579-1 e 416537-1.
A avaliação das diferentes condições se deu em triplicata e separadamente,
ou seja, para averiguar um parâmetro, mantiveram-se os demais fixos.

31
4.6.2 – Validação do método cromatográfico para a quantificação do
éster diidroxilado 3-β-O-acil lupeol em látex do fruto de H. speciosa e na polpa
comercial
4.6.2.1 – Condições analíticas do método quantitativo
A separação cromatográfica foi realizada utilizando coluna analítica Hexil-
fenil LUNA® (10 μm, 150 x 4,6 mm, Phenomenex) e fase móvel constituída de água
(A) e acetonitrila (B). As análises foram feitas em eluição isocrática no modo reverso
utilizando 95% de B por 40 min. Uma vazão de 0,8 mL/min foi utilizado e volume de
injeção de 20 µL. Os cromatogramas foram registrados com detector de arranjo de
diodos em 200 nm e com ELSD usando as mesmas condições descritas em 4.5.2. O pico
cromatográfico referente ao éster diidroxilado 3-β-O-acil lupeol foi determinado na
amostra real pela comparação do tempo de retenção do pico gerado pela análise da
solução padrão. A pureza do éster diidroxilado foi verificada por normalização
utilizando o ELSD o que resultou em uma pureza de 97,13%.
4.6.2.2 – Preparo da solução estoque, padrões de calibração e amostras
controle de qualidade
A solução estoque do éster diidroxilado 3-β-O-acil lupeol foi preparada em
isopropanol na concentração de 2 mg/mL. A partir desta foram preparadas soluções de
trabalho nas concentrações: 19,4; 38,8; 77,7; 155,4; 310,8 e 466,2 µg/mL.
Da solução estoque foram preparadas soluções de controle de qualidade nas
seguintes concentrações: CQB - Controle de Qualidade Baixo: 43,7 µg/mL; CQM -
Controle de Qualidade Médio: 207,9 µg/mL e CQA – Controle de Qualidade Alta:
372,9 µg/mL. Tais soluções foram utilizadas nos experimentos de precisão (item
4.6.2.6) e exatidão (item 4.6.2.7). Os valores de concentração dos controles de
qualidade do método foram definidos baseados na ANVISA. Segundo esta agência o
controle de qualidade de baixa concentração deve ser menor ou igual três vezes o limite
inferior de quantificação, já o controle de qualidade de média concentração deve ser
aproximadamente a média entre CQB e CQA, enquanto que o controle de qualidade de
alta concentração deve corresponder de 75 a 90% da maior concentração usada na curva

32
de calibração.As soluções estoque, de calibração e de controle de qualidade foram
mantidas em refrigerador a zero graus Celsius.
4.6.2.3 – Linearidade
A faixa de linearidade foi avaliada de acordo com os níveis de éster
diidroxilado esperados nas matrizes (polpa comercial e látex liofilizado). O estudo da
linearidade se deu pelo método do padrão externo e por adição de padrão.
No método do padrão externo os seis níveis de concentração (mostrados no
item 4.6.2.2) foram avaliados em triplicata. Para o método da adição de padrão duzentos
microlitros das soluções de calibração foram pipetados. Após a completa evaporação do
isopropanol, duzentos microlitros da polpa (preparado conforme o item 4.4.2) foi
adicionado e misturados por 30 segundos usando agitador tipo vortex. Da mesma forma
que no método do padrão externo, as soluções de calibração da adição de padrão foram
avaliadas em triplicata. As curvas de calibração foram obtidas por regressão linear, a
partir da relação entre as áreas dos picos e as concentrações das amostras para ambos os
métodos.
4.6.2.4 – Seletividade
A seletividade do método foi avaliada de duas formas: adicionando o éster
diidroxilado 3-β-O-acil lupeol na polpa comercial de mangaba e comparando-se o
espectro de UV do padrão com o pico correspondente obtido na análise do extrato da
matriz. Além disso, a pureza do pico cromatográfico referente ao analito foi avaliada
usando o DAD.
4.6.2.5 – Limite de detecção (LD) e limite de quantificação (LQ)
O limite de detecção (LD) e o limite de quantificação (LQ) foram estimados
com base na relação de três vezes o ruído da linha de base e dez vezes a relação sinal-
ruído, respectivamente.

33
4.6.2.6 – Precisão
A repetibilidade e precisão intermediária foram determinadas pela análise
das soluções de controle de qualidade em três diferentes dias. Para cada uma das três
concentrações, as amostras foram preparadas independentemente por três vezes e
analisadas em quintuplicata. O DPR foi tomado como medida de precisão.
4.6.2.7 – Exatidão
A exatidão foi determinada pelo teste de recuperação. A amostra da polpa
comercial de mangaba preparada conforme o item 4.4.2 foi fortificada com o composto
de referência em três níveis de concentração (as mesmas usadas no estudo da precisão).
Duzentos microlitros da amostra da polpa foram pipetados e colocados em tubos de
ensaio; após a retirada do solvente foram adicionados 200 µL da solução de controle de
qualidade, por fim a amostra foi homogeneizada usando agitador tipo vortex por 30
segundos. Para cada concentração, as amostras foram independentemente preparadas e
fortificadas em triplicata. As amostras foram analisadas em quintuplicata em três dias
diferentes. A exatidão foi determinada através da concentração média experimental em
relação à concentração teórica, em valores percentuais (Equação 2).
Equação 2:

34
4.6.2.8 – Robustez
A robustez do método foi avaliada por meio das seguintes alterações:
Fluxo da fase móvel: 1,0 – 0,6 mL/min.
Proporção da fase móvel: 93 – 97% ACN:H2O.
Lote da coluna Hexil-fenil LUNA®
: 423579-1 e 416537-1.
A análise cromatográfica das diferentes condições analíticas foi analisada
em triplicata e separadamente, ou seja, para avaliação de um parâmetro, mantiveram-se
os demais fixos.
4.6.2.9 – Estabilidade
A estabilidade foi avaliada de duas formas: após ciclos de congelamento e
descongelamento e a estabilidade de curta duração.
4.6.2.9.1 – Estabilidade de curta duração
Os resultados das amostras de controle de qualidade recém-preparadas
foram comparados com aqueles obtidos na análise de tais amostras após permanecerem
no autoinjetor por 24h.
4.6.2.9.2– Estabilidade após ciclos de descongelamento e congelamento
As amostras do controle de qualidade foram congeladas por um período de
24 h no freezer, sendo então submetida ao descongelamento à temperatura ambiente.
Quando completamente descongeladas, as amostras foram congeladas novamente por
12h e assim sucessivamente até completar os três ciclos. Os resultados foram
comparados com aqueles obtidos na análise das amostras recém-preparadas.

35
4.7 – Isolamento dos marcadores químicos do látex do caule de
Hancornia speciosa Gomes
O látex do caule, preparado conforme o item 4.4.1, foi utilizado para o
isolamento dos marcadores químicos por HPLC em escala semi-preparativa. Para tanto
empregou-se uma coluna Hexil-fenil LUNA®
(250 x 10 mm; Phenomenex), vazão de
4,5 mL/min e detecção em 220 nm. A fase móvel constituída por H2O (A) e ACN (B)
variou na seguinte proporção: 5-40% de (B) em 50 min. Antes de uma nova injeção, foi
feita a limpeza da coluna com 100% de ACN seguida do recondicionamento em 5% de
(B) por 10 min. As bandas cromatográficas coletadas em erlenmeyer foram
centrifugadas. Em seguida, os compostos isolados foram transferidos para frascos
âmbar, identificados e pesados.
4.8 – Identificação dos marcadores químicos por RMN de 1H e de
13C
Os espectros de ressonância magnética nuclear (RMN) foram adquiridos
utilizando aparelho Bruker Avance DRX-400 (9,4 Tesla) operando a 400 MHz para
RMN 1H e 100 MHz para RMN
13C. As amostras foram solubilizadas em metanol
deuterado (CD3OD) e os deslocamentos químicos estão expressos em ppm (δ) e as
multiplicidades dos sinais estão indicadas conforme convenção: s (simpleto), sl
(simpleto largo), d (dupleto), dd (duplo dupleto), ddd (duplo duplo dupleto), ddl (duplo
dupleto largo), t (tripleto), tl (tripleto largo), dt (duplo tripleto), dtd (duplo tripleto
duplo) e td (tripleto duplo), q (quadrupleto), quintl (quintupleto largo). As constantes de
acoplamento (J) foram registradas em Hertz (Hz). Estes experimentos foram realizados
no Laboratório de Ressonância Magnética Nuclear do Departamento de Química da
Universidade Federal de São Carlos (DQ/UFSCar) sob a responsabilidade do Prof. Dr.
Antonio Gilberto Ferreira.

36
Resultados e
discussão

37
5 – RESULTADOS E DISCUSSÃO
5.1 – Otimização das condições cromatográficas de análise: látex do
caule de H. speciosa Gomes
O objetivo do processo de otimização é definir as melhores condições
cromatográficas para resultar em alta qualidade da separação dos compostos no menor
tempo possível. Não existe uma metodologia ou conjunto de regras que definam
exatamente o caminho a se percorrer durante o estudo de otimização, o que exige do
analista bom senso e criatividade. Na literatura são poucos os artigos que discutem
plausivelmente as estratégias de otimização, normalmente os autores se restringem a
citar as melhores condições cromatográficas sem nenhum tipo de discussão sobre os
parâmetros avaliados.[101]
Neste trabalho, o processo de otimização do método consistiu na avaliação
da influência de diferentes fatores na separação cromatográfica, entre eles: fase
estacionária, natureza do modificador orgânico (solvente B) e proporção da fase móvel.
A condição cromatográfica escolhida foi estabelecida por comparação da resolução,
linha de base, tempo de eluição e número de picos no cromatograma.
A avaliação dos parâmetros em busca da melhor separação cromatográfica
se deu em duas etapas. Na primeira, foi possível selecionar a fase estacionária e o
modificador orgânico por meio de experimentos utilizando um gradiente exploratório.
Na segunda fase, os parâmetros da eluição gradiente foram devidamente ajustados,
baseado nos dados adquiridos na etapa anterior.[101]

38
5.1.1 – Primeira etapa do processo de otimização: uso do gradiente
exploratório
O gradiente exploratório, proposto por Snyder e Dolan (1996), consiste na
variação do modificador orgânico de 5 a 100% por 60 min a um fluxo de 2 mL/min. Os
autores sugerem ainda que as análises sejam feitas inicialmente em uma coluna C8 ou
C18 de 15 x 0,46 cm.[102]
O uso do gradiente exploratório permite conhecer a
complexidade da amostra fornecendo um importante indicativo de qual fase móvel e
fase estacionária podem resultar em separação mais adequada.
Durante esta etapa, optou-se por fazer adaptações no método sugerido por
Snyder e Dolan (1997). Variando-se amplamente a força do solvente orgânico e usando
diferentes fases estacionárias foram testadas diversas combinações modificando os
seguintes fatores:
Fase estacionária: hexil-fenil LUNA® (10 µm, 150 x 4,6 mm,
Phenomenex) e C18 LUNA® (5 µm, 250 x 4,6 mm, Phenomenex);
Solvente (A): H2O e solução aquosa de ácido fórmico (HCOOH) 0,5%
(v/v);
Solvente (B): metanol (MeOH) e acetonitrila (ACN).
A cromatografia em fase reversa é a mais utilizada em HPLC, uma vez que
permite a separação de uma grande variedade de solutos e o uso de fase móvel aquosa.
A fase estacionária quimicamente ligada ao grupo octadecilsilano (C18) é, em geral, a
primeira opção de escolha em análises realizadas no modo reverso. Esse fato pode ser
justificado pela baixa polaridade e pelas interações hidrofóbicas que ocorrem entre a
parte não polar das substâncias a serem separadas e as cadeias hidrocarbônicas da fase
estacionária.[103]
Além da coluna C18 utilizou-se a fase estacionária hexil-fenil, pois além de
interações hidrofóbicas, inerentes ao modo reverso, nesta coluna ocorrem interações do
tipo π-π, o que diferencia sua seletividade na análise de substâncias com anéis
benzênicos e duplas ligações em sistema conjugados como taninos, flavonóides etc.[104]

39
A escolha do tipo de solvente no modo reverso é comumente o mais
significativo procedimento para alterar a seletividade e alcançar a separação dos
múltiplos compostos presente na amostra. Usualmente três solventes são recomendados
para a investigação da seletividade, são eles: metanol (MeOH), acetonitrila (ACN) e
tetrahidrofurano (THF). Obviamente que são as propriedades desses solventes, como
miscibilidade em água, compatibilidade com absorção no UV e viscosidade, que
permitem o amplo uso em cromatografia de fase reversa.[105]
Um importante ponto a ser observado na otimização da separação
cromatográfica é o efeito causado pela diminuição do pH da fase móvel pela adição de
ácidos fracos.[106]
Essa medida possibilita a supressão da ionização de algumas
substâncias que em solução aquosa podem estar parcial ou totalmente ionizadas, como
por exemplo, compostos fenólicos e ácidos carboxílicos. A supressão da ionização é
uma condição muitas vezes desejável, pois torna as substâncias mais hidrofóbicas,
aumentando sua retenção.[107]
As condições cromatográficas de análise testadas na primeira etapa da
otimização são demonstradas, com seus respectivos perfis cromatográficos, na Tabela 1
e na Figura 3.

40
Tabela 1: Condições cromatográficas avaliadas na primeira etapa da
otimização do método para análise do látex do caule de H.speciosa com detecção em
220 nm.
Fase Estacionária Análise Condição Cromatográfica
C18 Luna
(5 µm, 250 x 4,6 mm)
1
Fase Móvel: ACN: H2O
Gradiente: 5-100% ACN por 60 min
Vazão: 1,0 mL/min
Volume de injeção: 20 µL
2
Fase Móvel: ACN: solução aquosa de HCOOH a
0,5%
Gradiente: 5-100% ACN por 60 min
Vazão: 1,0 mL/min
Volume de injeção: 20 µL
3
Fase Móvel: MeOH: H2O
Gradiente: 5-100% MeOH por 60 min
Vazão: 1,0 mL/min
Volume de injeção: 20 µL
4
Fase Móvel: MeOH: solução aquosa de HCOOH a
0,5%
Gradiente: 5-100% MeOH por 60 min
Vazão: 1,0 mL/min
Volume de injeção: 20 µL
Hexil-fenil Luna
(10 µm, 150 x 4,6
mm)
5
Fase Móvel: ACN: H2O
Gradiente: 5-100% ACN por 60 min
Vazão: 1,0 mL/min
Volume de injeção: 20 µL
6
Fase Móvel: MeOH: H2O
Gradiente: 5-100% MeOH por 60 min
Vazão: 1,0 mL/min
Volume de injeção: 20 µL

41
0 10 20 30 40 50 60 70
-2,0x106
-1,5x106
-1,0x106
-5,0x105
0,0
5,0x105
Absorb
ância
(m
Abs)
Tempo (min)
0 10 20 30 40 50 60 70
0,0
5,0x104
1,0x105
1,5x105
2,0x105
2,5x105
Absorb
ância
(m
Abs)
Tempo (min)
0 10 20 30 40 50 60 70
-2,0x106
-1,5x106
-1,0x106
-5,0x105
0,0
Absorb
ância
(m
Abs)
Tempo (min)
0 10 20 30 40 50 60
0,0
5,0x104
1,0x105
1,5x105
Absorb
ância
(m
Abs)
Tempo (min)
0 10 20 30 40 50 60 70
0,0
5,0x104
1,0x105
1,5x105
Absorb
ância
(m
Abs)
Tempo (min)
0 10 20 30 40 50 60
0
1x105
2x105
3x105
Absorb
ância
(m
Abs)
Tempo (min)
Figura 3: Cromatogramas para a seleção de condições para o método em HPLC-DAD. Os
números correspondem às condições apresentadas na Tabela 1.
1 2
3
4
5 6

42
Observando os cromatogramas da Figura 3 algumas considerações podem ser
feitas. A solução aquosa de ácido fórmico pode ser retirada do método, uma vez que
desestabiliza a linha de base. A interferência do ácido fórmico na detecção se dá por
apresentar valores de cut-off (210 nm) próximos ao utilizado nos registros dos
cromatogramas, 220 nm.[108]
Com a percepção deste fenômeno tornou-se desnecessário fazer
a combinação da solução aquosa de ácido fórmico com metanol.
Dentre as condições de análise testadas, a condição 6, a priori, foi a mais
adequada; uma vez que resultou no aparecimento de um maior número de picos e melhor
separação. Dessa forma, a fase estacionária hexil-fenil e o metanol foram selecionados para os
experimentos da segunda etapa.
5.1.2 – Segunda etapa do processo de otimização: avaliação dos
parâmetros da eluição gradiente
O objetivo dessa etapa foi aperfeiçoar a seletividade do método, ou seja, melhorar
a separação entre os picos. Para tanto, o procedimento mais eficaz e conveniente é a mudança
da proporção e da composição da fase móvel.
Embora haja ferramentas computacionais que determinam a porcentagem de B
mais precisamente e com o mínimo de experimentos, o gradiente foi definido através de
experimentos de tentativa e erro. [109]
O início e o fim da porcentagem do solvente orgânico no método cromatográfico
foram ajustados a partir do perfil cromatográfico 6. Devido à ampla faixa de retenção em que
os compostos foram registrados no cromatograma 6, a separação mais adequada deve ser
alcançada por meio da eluição gradiente. As condições de análise testadas nesta fase são
mostradas na Tabela 2 e os respectivos cromatogramas na Figura 4.

43
Tabela 2: Condições cromatográficas avaliadas na segunda etapa da otimização
do método para análise do látex do caule de H.speciosa com detecção em 220 nm.
Análise Gradiente de eluição Fase móvel Fase estacionária
7
5% (B) por 10 min; 5-30% (B) por 5 min;
30-70% (B) por 30 min; 70-100% (B) por
10 min; isocrático em 100% de B por 5
min.
MeOH:H2O
Hexil-fenil Luna
(10 µm, 150 x 4,6 mm)
8
5-35% (B) por 10 min; 35-70% (B) por
40 min; 70-100% (B); isocrático em
100% (B) por 5 min.
MeOH:H2O
Hexil-fenil Luna
(10 µm, 150 x 4,6 mm)
9
5-25% (B) por 15 min; 25-45% (B) por
25 min; 45-100% (B) por 10 min;
isocrático em 100% (B) por 5 min.
ACN: H2O
Hexil-fenil Luna
(10 µm, 150 x 4,6 mm)
10
5-40% (B) por 50 min; 40-100% (B) por
5 min; isocrático 100% (B) por 5 min.
ACN: H2O
Hexil-fenil Luna
(10 µm, 150 x 4,6 mm)
11
5-40% (B) por 50 min; 40-100% (B) por
5 min; isocrático 100% (B) por 5 min.
ACN: H2O
C18 Luna
(5 µm, 250 x 4,6 mm)
12
5-40% (B) por 70 min; 40-100% (B) por
5 min; isocrático 100% (B) por 5 min.
ACN: H2O
C18 Luna
(5 µm, 250 x 4,6 mm)

44
0 10 20 30 40 50 60
0,0
5,0x104
1,0x105
1,5x105
2,0x105
Ab
so
rbâ
ncia
(m
Ab
s)
Tempo (min)
0 10 20 30 40 50 60
0,0
5,0x104
1,0x105
1,5x105
2,0x105
Ab
so
rbâ
ncia
(m
Ab
s)
Tempo (min)
0 10 20 30 40 50
0
1x105
2x105
3x105
4x105
Ab
so
rbâ
ncia
(m
Ab
s)
Tempo (min)
0 5 10 15 20 25 30 35 40 45 50
0
1x105
2x105
3x105
4x105
Ab
so
rbâ
ncia
(m
Ab
s)
Tempo (min)
0 5 10 15 20 25 30 35 40 45 50
0
1x105
2x105
3x105
Ab
so
rbâ
ncia
(m
Ab
s)
Tempo (min)
0 10 20 30 40 50 60 70
0
1x105
2x105
3x105
Ab
so
rbâ
ncia
(m
Ab
s)
Tempo (min)
Figura 4: Cromatogramas (λ = 220 nm) para a seleção de condições para o método em
HPLC-DAD. Os números correspondem às condições apresentadas na Tabela 2.
7 8
9 10
11 12

45
Foi verificado que não houve melhora significativa nas condições de análise 7 e 8.
Nesses experimentos foram utilizadas diferentes rampas de eluição gradiente. Em vista dos
resultados insatisfatórios foi alterado o modificador orgânico de metanol para acetonitrila e
foi testado a condição 9. Os picos permaneceram sem a devida resolução apresentando o
mesmo perfil observado nas tentativas anteriores. Dessa forma, a condição 9 apresentou uma
separação inadequada dos picos. Na condição 10, novas rampas de eluição gradiente foram
empregadas sem ocasionar nenhuma alteração satisfatória na resolução.
A estratégia seguinte consistiu em utilizar o melhor gradiente observado
utilizando uma fase estacionária C18. O resultado, apresentado no cromatograma 11, mostra
que a décima primeira condição permitiu uma melhor resolução dos picos iniciais (com
tempos de retenção entre 0 e 10 minutos) e uma significativa abertura na segunda parte do
cromatograma (no intervalo de 20 a 45 minutos).
Por fim, testou-se a condição 12, na qual foi alterada a inclinação do gradiente
mudando o tempo de análise de 50 para 70 min. Com essa mudança a inclinação do gradiente
variou de 0,7 (condição 11) para 0,5 %B/min. No entanto, nenhuma alteração significativa
pôde ser notada. Dessa forma, a condição 11 foi a mais adequada. A descrição completa está
descrita no item 4.5.1.
5.1.3 – Preparo da amostra do látex do caule
Até o presente momento nenhuma averiguação da influência do preparo da
amostra tinha sido realizada neste trabalho. Nos experimentos acima descritos foi feita a
retirada da água por centrifugação e para facilitar a coagulação da borracha foi utilizado 150
µL de ácido acético no preparo da amostra de 3 mL de látex.[110]
Sendo assim, avaliar
pequenas alterações no modo de preparo tornou-se indispensável.
O látex do caule da H. speciosa apresentou algumas características que torna o seu
manuseio trabalhoso, como por exemplo, fotossensibilidade e fácil polimerização.
Normalmente, a extração do látex da mangabeira acontece nas primeiras horas do dia e este é
logo diluído em água, permitindo assim a sua conservação.

46
Nesse caso, o preparo da amostra do látex do caule para a análise em HPLC
consistiu na retirada da água e da borracha, bem como na seleção do solvente (ou mistura de
solventes) mais compatível com a fase móvel. Sendo assim, a necessidade do uso do ácido
acético[120]
no processo de coagulação da borracha e o processo de retirada de água da amostra
é mais eficiente (centrifugação ou liofilização) foram avaliados.
Assim experimentos foram realizados, sendo um por liofilização sem o uso de
ácido acético e outros dois fazendo-se uso de uma centrifuga à vácuo (CENTRIVAP),
avaliando-se o uso do ácido acético. O resultado são apresentados na Figura 5.
Figura 5: Cromatogramas para a seleção do modo de preparo do látex do caule de H.
speciosa para análise em HPLC-DAD: (preto) látex tratado com ácido acético e seco em
centrifugador; (vermelho) látex sem ácido acético e seco em centrifugador e (azul) látex seco
em liofilizador. Os cromatogramas foram obtidos utilizando a condição 11 descrita na Tabela
2.
0 20 40
0
1x101
Ab
sorb
ân
cia
(m
Ab
s)
Tempo (min)

47
Nos perfis cromatográficos apresentados na Figura 5 não se observou mudança
significativa nas condições testadas. O processo de liofilização foi escolhido pela
simplicidade e por possibilitar o menor manuseio da amostra, o que consequentemente
permitiu preservar os compostos na matriz. Uma vez definida a porcentagem inicial da fase
móvel da corrida cromatográfica (discutido no item 5.1.2), uma solução aquosa a 5% de ACN
foi preparada para dissolver o material resultante do processo da retirada da água. As
condições finais podem ser vistas no procedimento experimental item 4.4.1.
Aplicando o preparo da amostra otimizado, foi obtido o perfil cromatográfico
ilustrado na Figura 6, o qual apresentou bons valores de tempo de retenção e adequada
separação entre os picos. A avaliação efetuada com o DAD mostrou um alto grau de pureza
dos picos, acima de 0,99999. Os espectros de UV dos picos assinalados na Figura 6
(apresentados de 1 a 7) podem ser vistos na Figura 7.

48
Figura 6: Perfil Cromatográfico do látex do caule de H. speciosa obtido com a eluição gradiente: 5-50% H2O (A): ACN (B) em 50 min; vazão: 1,0
mL/min; λ= 220nm; Vinj = 20 µL; coluna analítica C18 LUNA®
(5μm ,250 x 4,6 mm, Phenomenex) adaptada a uma coluna guarda C18 (4 x 3,0 mm,
Phenomenex). Condição cromatográfica 11.
0 10 20 30 40 50
0
1x105
2x105
3x105
4x105
5x105
6x105
7x105
Ab
so
rbâ
ncia
(m
Ab
s)
Tempo (min)
1
2
3
4
5
6
7

49
Figura 7: Espectros de absorção UV das bandas cromatográficas dos picos assinaladas de 1 à 7 na Figura 6.
200 300 400 500 600 700 nm
0
500
1000
1500
2000mAU
195
323
492
393
200 300 400 500 600 700 nm
0
100
200
mAU
230
400
200 300 400 500 600 700 nm
0
250
500
mAU
220
317
200 300 400 500 600 700 nm
0
250
mAU
198
279
395
648
200 300 400 500 600 700 nm
0
250
500
750
mAU
197
279
343
494
616
200 300 400 500 600 700 nm
0
250
500mAU
199
228
279
616
726
200 300 400 500 600 700 nm
0
50
100
150
mAU
279
494
547
617
647
(1) (2) (3)
(4) (5) (6)
(7)

50
Com auxílio da projeção tridimensional da amostra do látex do caule de H.
speciosa obtido por HPLC-DAD (Figura 8), o comprimento de onda em 220 nm foi o
selecionado, pois neste valor se observou a máxima absorção dos compostos.
Figura 8: Projeção tridimensional obtido por HPLC-DAD da amostra do látex do
caule da H. speciosa.
Após a conclusão dos trabalhos com o uso do DAD, novas análises foram
realizadas utilizando o detector ELSD. A análise da amostra do látex do caule por HPLC-
ELSD (Figura 9) permitiu verificar a presença de um pico não detectado pelo DAD (tR= 6,7
min). A comparação entre os perfis cromatográficos obtidos por ambos os detectores pode ser
visto na Figura 10. Portanto, o uso dos detectores DAD-ELSD em série permitiu uma visão
mais completa da amostra tornando o método mais seguro e confiável.

51
Figura 9: Perfil cromatográfico do látex do caule da H. speciosa por HPLC-ELSD obtido com a eluição gradiente: 5-50% H2O (A): ACN (B) em 50
min; vazão: 1,0 mL/min; ELSD: temperatura do tubo mantido em 70˚C e fluxo de N2 pressurizado em 353 KPa; Vinj = 20 µL; coluna analítica C18
LUNA®
(5μm ,250 x 4,6 mm, Phenomenex) adaptada a uma coluna guarda C18 (4 x 3,0 mm, Phenomenex). Condição cromatográfica 11.
. (Alan inclua as condições do ELSD aqui e indique na figura o numero dos picos como feito anteriormente...
1
3
0 10 20 30 40 50
0,0
2,0x105
4,0x105
6,0x105
8,0x105
1,0x106
1,2x106
mV
Tempo (min)
Pico registrado somente pelo ELSD
2
4
5
6

52
0.0 5.0 10.0 15.0 20.0 25.0 30.0 35.0 40.0 45.0 min
0.00
0.25
0.50
0.75
1.00
uV(x1,000,000)
Figura 10: Perfil cromatográfico comparativo do látex do caule de H. speciosa por HPLC-
DAD-ELSD. (Preto) Cromatograma monitorado pelo DAD e (Vermelho) cromatograma
monitorado pelo ELSD.
5.2 – Validação do método analítico para o perfil cromatográfico do látex
do caule de H. speciosa
Após a otimização das condições cromatográficas, o método qualitativo foi
validado por meio da averiguação das seguintes figuras de mérito: precisão de injeção,
repetibilidade, estabilidade e robustez.[111,112]
A avaliação destes parâmetros foram baseadas
nos valores de tempo de retenção (tR), área e altura de bandas cromatográficas que
apresentaram boa resolução e intensidade. Nos experimentos do estudo da validação foi
utilizada a amostra do látex do caule (LCP) adquirida em Pontal município de Indiaroba-SE, a
qual foi preparada conforme o item 4.4.1. A Figura 6 mostra as bandas cromatográficas
selecionadas para a validação (bandas cromatográficas de 1 a 7).
Em todos os parâmetros um valor de desvio padrão relativo (DPR) menor ou igual
a 15 % foi aceito como preconiza o Guia da ANVISA[113]
para quantificação em métodos
bioanalíticos. A Tabela 3 mostra os valores de DPR da precisão instrumental que variou de
0,01 – 0,42 %; 5,2 – 7,4 % e 5,1 – 7,1 % para os valores de tempo de retenção, área e altura,
respectivamente.

53
Tabela 3: Valores de desvio padrão relativo entre os tempos de retenção, área e
altura das bandas cromatográficas avaliadas nas análises da amostra do látex do caule de H.
speciosa (precisão de injeção).
De acordo com a Tabela 4, os valores de DPR médios entre as análises do estudo
de repetibilidade foram de 0,04 a 0,28 % para o tempo de retenção; de 5,1 a 7,0 % para os
valores de área e por fim de 3,4 a 6,5 % para os valores de altura. Os valores obtidos na
avaliação da repetibilidade e precisão de injeção demonstraram que a variabilidade
instrumental e preparo de amostra apresentaram boa reprodutibilidade.
Tabela 4: Valores de desvio padrão relativo entre os tempos de retenção, área e
altura das bandas cromatográficas avaliadas nas análises de amostra de H. speciosa
(repetibilidade).
Banda 1º dia 2º dia 3º dia Média
DRP (%)
Tr Área Altura Tr Área Altura Tr Área Altura Tr Área Altura
1 0,38 2,8 1,8 0,28 8,8 7,8 0,19 6,3 5,5 0,28 5,9 5,0
2 0,27 2,7 2,2 0,19 10,7 10,1 0,14 7,7 7,2 0,2 7,0 6,5
3 0,24 2,8 2,4 0,17 9,3 8,9 0,12 6,6 6,3 0,18 6,2 5,9
4 0,14 2,9 2,6 0,16 8,1 3,8 0,12 5,7 3,8 0,14 5,6 3,4
5 0,14 2,7 2,9 0,17 8,5 7,8 0,12 6,0 5,5 0,14 5,7 5,4
6 0,06 2,7 2,6 0,10 8,9 9,1 0,07 6,4 6,5 0,08 6,0 6,1
7 0,01 2,4 2,0 0,06 7,6 9,5 0,04 5,4 6,7 0,04 5,1 6,1
Banda Precisão da injeção
DPR (%)
Tr Área Altura
1 0,42 6,0 7,1
2 0,19 7,4 6,9
3 0,21 5,8 5,7
4 0,13 5,2 5,2
5 0,13 5,8 5,1
6 0,04 5,7 5,6
7 0,01 5,6 6,2

54
Para verificar a estabilidade das amostras do látex em termos de tempo (período
de permanência no auto-injetor) e temperatura (após ciclos de congelamento e
descongelamento), os resultados das soluções recém-preparadas foram comparados com os
resultados adquiridos após 24 e 48 h de permanência no auto-injetor bem como após três
ciclos de congelamento e descongelamento. A Tabela 5 sumariza os valores de DPR
encontrados. Como pode ser observado, somente as amostras que passaram por ciclos de
congelamento e descongelamento apresentaram valores de DPR maior que 15 % para os
valores de área e altura. Sendo assim, as soluções das amostras do látex do caule não foram
estocadas no congelador, sempre que preparadas eram colocadas no auto-injetor do
equipamento por um tempo de permanência máximo de 48 horas.
Tabela 5: Valores de desvio padrão relativo entre os tempos de retenção, área e
altura das bandas cromatográficas avaliadas nas análises de amostra de H. speciosa Gomes
(estabilidade).
Através dos experimentos de robustez, foram avaliados a influência da variação
do fluxo (0,8 e 1,2 mL/min), da porcentagem inicial do método cromatográfico (3 e 7 % de
acetonitrila), da porcentagem final do método cromatográfico (38 e 42 % de acetonitrila) e do
lote da fase estacionária, cujo resultados estão mostrados na Tabela 6. Admitiu-se que o
método se mantinha robusto para variação de DRP igual ou menor a 15% para cada parâmetro
avaliado. Dentro desse contexto o método se mostrou robusto frente à variação do fluxo de
1,2 mL/min, à variação na porcentagem final do método cromatográfico e à variação do lote
da coluna. Sendo assim, os demais parâmetros devem ser mantidos para o bom desempenho
do método desenvolvido.
Banda Estabilidade (24 h) Estabilidade (48 h) Estabilidade (Congelamento)
DPR (%)
Tr Área Altura Tr Área Altura Tr Área Altura
1 0,0 3,7 5,5 0,7 5,8 0,5 0,5 27,5 34,8
2 0,2 7,4 5,2 1,0 9,8 8,9 1,1 29,9 32,1
3 0,2 5,1 8,0 0,8 7,3 8,2 0,8 28,3 27,1
4 0,1 5,5 4,7 0,5 6,0 5,6 0,6 29,1 29,0
5 0,1 6,4 5,9 0,5 9,1 7,7 0,6 25,6 27,2
6 0,0 5,9 5,0 0,1 7,1 6,4 0,1 29,0 29,6
7 0,0 13,0 8,3 0,2 6,8 8,7 0,3 28,7 27,7

55
Tabela 6: Valores de desvio padrão relativo entre os tempos de retenção, área e altura das bandas cromatográficas avaliadas nas
análises de amostra de H. speciosa (robustez).
* F = Fluxo, ** PI = Porcentagem inicial e *** PF = Porcentagem final
Banda *F (0,8 mL/min) *F (1,2 mL/min) **PI (3%) **PI (7%) ***PF (38%) ***PF(42%) LOTE
DPR (%)
Tr Área Altura Tr Área Altura Tr Área Altura Tr Área Altura Tr Área Altura Tr Área Altura Tr Área Altura
1 15,7 23,9 11,0 13,3 4,4 10,0 22,8 8,5 13,8 16,5 8,3 14,3 0,17 8,3 10,5 0,4 10,2 10,2 2,2 8,3 4,4
2 15,1 26,3 18,5 12,3 1,8 0,1 19,9 12,7 14,7 18,5 3,4 8,9 0,17 11,8 9,9 0,0 12,2 13,8 1,5 2,3 14,1
3 14,8 23,6 15,1 12,1 4,6 2,4 19,7 8,8 7,4 18,7 4,1 4,3 0,27 8,3 6,6 0,1 8,5 8,1 1,6 5,4 12,1
4 8,8 18,2 20,0 6,8 3,6 3,3 9,7 7,4 8,9 11,5 5,1 6,2 1,9 8,2 5,4 1,6 6,3 9,5 1,0 0,3 15,0
5 8,5 23,4 21,8 6,6 6,4 5,4 9,3 7,6 9,0 11,1 8,5 7,2 1,9 7,2 5,0 1,7 7,6 10,2 0,9 1,6 14,8
6 5,6 22,8 20,3 4,3 2,3 1,6 5,4 10,0 10,3 6,6 10,8 7,1 9,8 9,8 6,3 2,5 9,4 10,2 0,5 7,3 12,7
7 3,9 25,0 21,3 3,0 2,7 3,9 2,8 8,7 10,6 3,4 7,4 5,4 8,9 8,9 4,9 2,9 10,3 10,9 0,3 8,0 6,3

56
5.3 – Aplicação do método analítico desenvolvido: análise das amostras
comerciais do látex do caule de H. speciosa
O método desenvolvido e validado foi utilizado para a obtenção de perfis
cromatográficos das amostras de látex de caule comercializados em feiras livres do estado de
Sergipe. As amostras foram denominadas com os seguintes códigos: LCA (látex proveniente
de Aracaju), LCI (látex obtido em Itabaiana), LCB (látex proveniente de Boquim) e LCU
(látex adquirido em Umbaúba). Nas Figuras 11 e 12 estão apresentados os cromatogramas do
látex do caule da H. speciosa realizados em sextuplicata por DAD e pelo ELSD,
respectivamente. As sobreposições dos cromatogramas (n = 6), para cada amostra do látex do
caule, mostraram a adequada separação e a boa reprodutibilidade do método o que corrobora a
sua eficiência para a obtenção de fingerprint cromatográfico.
O látex do caule da mangabeira (LCP) adquirido em Pontal município de
Indiaroba-SE foi considerado padrão, uma vez que se tem convicção da sua originalidade. A
comparação visual do látex LCP com as amostras de látex comercializados (LCA, LCI, LCB
e LCU) permitiu a identificação de picos comuns em todas as amostras. No entanto, com
diferentes intensidades (Figura 13 e Figura 14). As diferenças entre os perfis
cromatográficos das amostras de látex do caule ficam mais acentuadas quando se observa pelo
DAD do que quando analisada pelo ELSD.
Embora haja diferença entre os perfis cromatográficos dos látex comercializados
e do látex considerado padrão, pode-se concluir que tais amostras são de fato provenientes da
mangabeira uma vez que possuem os picos majoritários encontrados no látex LCP.

57
LCI LCU
0 10 20 30 40 50
0,0
5,0x104
1,0x105
1,5x105
2,0x105
2,5x105
Ab
so
rbâ
ncia
(m
Ab
s)
Tempo (min)0 10 20 30 40 50
0
1x105
2x105
3x105
4x105
5x105
6x105
7x105
Ab
so
rbâ
ncia
(m
Ab
s)
Tempo (min)
0 10 20 30 40 50
0
1x105
2x105
3x105
4x105
5x105
Ab
so
rbâ
ncia
(m
Ab
s)
Tempo (min)
0 10 20 30 40 50
0
1x105
2x105
3x105
4x105
5x105
6x105
7x105
Ab
so
rbâ
ncia
(m
Ab
s)
Tempo (min)
0 10 20 30 40 50
0
1x105
2x105
3x105
4x105
5x105
6x105
7x105
Ab
so
rbâ
ncia
(m
Ab
s)
Tempo (min)
Figura 11: Cromatogramas sobrepostos (n = 6) de amostras de látex do caule de H. speciosa monitorados
pelo DAD (220 nm). LCA (látex proveniente de Aracaju), LCP (látex adquirido em Pontal), LCI (látex
obtido em Itabaiana), LCB (látex proveniente de Boquim) e LCU (látex adquirido em Umbaúba).
LCA LCB
LCP
57

58
0 10 20 30 40 50
0,0
2,0x105
mV
Tempo (min)
0 10 20 30 40 50
0,0
2,0x105
4,0x105
6,0x105
8,0x105
1,0x106
1,2x106
mV
Tempo (min)
0 10 20 30 40 50
0,0
2,0x105
4,0x105
6,0x105
8,0x105
1,0x106
1,2x106
mV
Tempo (min)
0 10 20 30 40 50
0,0
2,0x105
4,0x105
6,0x105
8,0x105
1,0x106
1,2x106
mV
Tempo (min)
0 10 20 30 40 50
0,0
2,0x105
4,0x105
6,0x105
8,0x105
1,0x106
1,2x106
mV
Tempo (min)
Figura 12: Cromatogramas sobrepostos (n = 6) de amostras de látex do caule de H. speciosa
monitorados pelo ELSD. LCA (látex proveniente de Aracaju), LCP (látex adquirido em Pontal), LCI
(látex obtido em Itabaiana), LCB (látex proveniente de Boquim) e LCU (látex adquirido em Umbaúba).
LCA LCB
LCI LCU
LCP
58
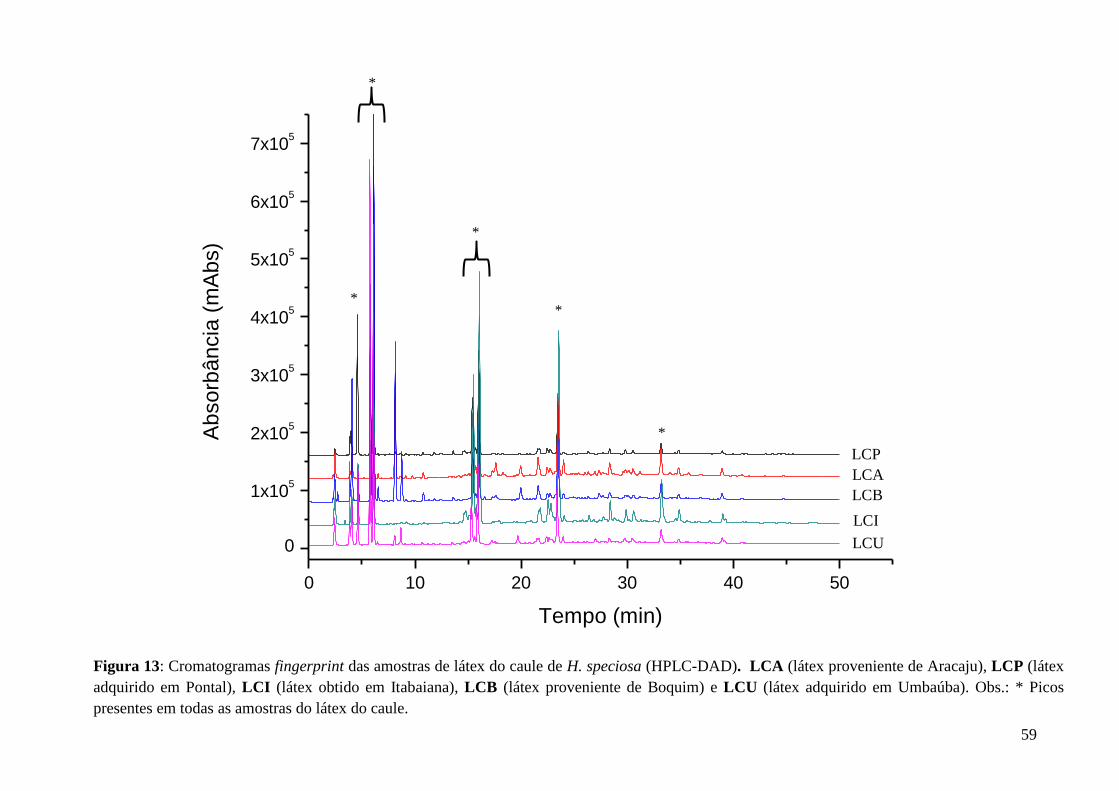
59
LCB
0 10 20 30 40 50
0
1x105
2x105
3x105
4x105
5x105
6x105
7x105
Ab
so
rbâ
ncia
(m
Ab
s)
Tempo (min)
LCP
LCI
LCU
LCA
Figura 13: Cromatogramas fingerprint das amostras de látex do caule de H. speciosa (HPLC-DAD). LCA (látex proveniente de Aracaju), LCP (látex
adquirido em Pontal), LCI (látex obtido em Itabaiana), LCB (látex proveniente de Boquim) e LCU (látex adquirido em Umbaúba). Obs.: * Picos
presentes em todas as amostras do látex do caule.
*
*
*
*
*
59

60
LCB
Figura 14: Cromatogramas fingerprint das amostras de látex do caule de H. speciosa (HPLC-ELSD). LCA (látex proveniente de Aracaju), LCP (látex
adquirido em Pontal), LCI (látex obtido em Itabaiana), LCB (látex proveniente de Boquim) e LCU (látex adquirido em Umbaúba). Obs.: * Picos
presentes em todas as amostras do látex do caule.
0 10 20 30 40 50
0,0
2,0x105
4,0x105
6,0x105
8,0x105
1,0x106
1,2x106
mV
Tempo (min)
LCP
LCI
LCU
LCA
*
*
* *

61
5.4 – Isolamento dos marcadores químicos do látex do caule de H. speciosa
Gomes
Uma vez obtido os cromatogramas fingerprint foi feito o isolamento dos
marcadores químicos do látex do caule da H. speciosa. Até o presente momento não há relatos
na literatura a respeito do isolamento de substâncias no látex do caule da mangabeira.
Para o isolamento empregou-se as condições cromatográficas baseadas naquelas
usadas para os cromatogramas fingerprint em escala semi-preparativa. A transferência do
método analítico para o semi-preparativo requereu o ajuste de alguns parâmetros. Esta
adaptação que tem por finalidade reproduzir em escala preparativa ou semi-preparativa os
resultados obtidos em escala analítica é chamada de escalonamento.[58]
Para o isolamento das bandas cromatográficas a amostra do látex do caule obtida
do município de Pontal foi selecionada. Nas análises em escala semi-preparativa utilizou-se
uma coluna com fase estacionária hexil-fenil LUNA®
(250 x 10 mm; Phenomenex) diferente
daquela empregada nas análises em escala analítica. A vazão utilizada foi 4,5 mL/min; este
valor foi encontrado aplicando a Equação 3. A Figura 15 mostra o cromatograma do látex
do caule em sistema semi-preparativo.
Equação 3:
Onde: S = fator de escalonamento, RP e RA são os diâmetros e LP e LA são os
comprimentos das colunas preparativa e analítica, respectivamente.

62
0 5 10 15 20 25 30 35 40 45 50
0,0
2,0x105
4,0x105
6,0x105
Ab
so
rbâ
ncia
(m
Ab
s)
Tempo (min)
Figura 15: Cromatograma (λ = 220 nm) do látex do caule de H. speciosa em
sistema semi-preparativo. Condições cromatográficas: item 4.7. Os picos coletados estão
indicados no cromatograma com as siglas HSG2 a HSG8.
As bandas cromatográficas coletadas durante o isolamento foram denominadas
HSG2 a HSG8 (conforme pode ser vista na Figura 15). No isolamento foi coletada a parte
central das bandas cromatográficas para aumentar o grau de pureza dos compostos isolados.
Os rendimentos para cada banda, após 11 injeções (1,120 g da amostra de látex), pode ser
visualizado na Tabela 7.
Tabela 7: Valores de massa e de rendimento obtido no isolamento dos
marcadores químicos usando 1,120 g do látex de caule.
Banda Cromatográfica Massa obtida (mg) Rendimento (% m/m)
HSG 2 8,0 0,7
HSG 3 17,6 1,6
HSG 4 3,6 0,32
HSG 5 8,9 0,8
HSG 6 5,3 0,5
HSG 7 0,7 0,06
HSG 8 0,7 0,06
HSG 2
HSG 3
HSG 4
HSG 5
HSG 6 HSG 7 HSG 8

63
Os compostos HSG 2 a HSG 8 foram reanalisados em escala analítica com o
objetivo de verificar o grau de pureza. Na Figura 16 pode-se observar a comparação entre os
tempos de retenção dos compostos isolados e do perfil cromatográfico do látex do caule de H.
speciosa enquanto que na Figura 17 os compostos purificados estão acompanhados com o
espectro de UV. Verificou-se um grau de pureza superior a 0,9999 para todos os compostos
isolados. Devido a baixa quantidade das amostras HSG 7 e HSG 8 somente as amostras HSG
2, HSG 3, HSG 4, HSG 5 e HSG 6 foram enviadas para análise por ressonância magnética
nuclear.
Figura 16: Perfil cromatográfico do látex do caule de H. speciosa acompanhado
dos compostos isolados em escala analítica (λ = 220 nm).
0 10 20 30 40 min0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
uV(x1,000,000)
Látex do caule
HSG 2
HSG 3
HSG 4
HSG 5
HSG 6
HSG 7
HSG 8

64
0 5 10 15 20 25 30 35 40 45 50
0,0
5,0x105
1,0x106
1,5x106
Abso
rbânci
a (
mA
bs)
Tempo (min)
200 300 400 500 600 700 nm
0
500
1000
1500
2000mAU
230
324
623
0 5 10 15 20 25 30 35 40 45 50
0,0
5,0x105
1,0x106
1,5x106
2,0x106
Abso
rbânci
a (
mA
bs)
Tempo (min)
0 5 10 15 20 25 30 35 40 45
0,0
2,0x105
4,0x105
6,0x105
8,0x105
Abso
rbânci
a (
mA
bs)
Tempo (min)
200 300 400 500 600 700 nm
0
1000
2000
mAU
194
279
657
617
0 5 10 15 20 25 30 35 40 45 50
0,0
2,0x105
4,0x105
6,0x105
8,0x105
1,0x106
Abso
rbânci
a (
mA
bs)
Tempo (min)
200 300 400 500 600 700 nm
0
500
1000
1500
2000mAU
217
315
492
718
200 300 400 500 600 700 nm
0
1000
2000
mAU
195
279
636
680
727
HSG 2 HSG 3
HSG 4 HSG 5
Figura 17: Cromatogramas (λ = 220 nm) das bandas cromatográficas HSG 2, HSG 3, HSG 4 e HSG5 purificadas no sistema semi-preparativo,
com seus respectivos espectros de absorção UV no tempo de retenção em que eluem. Condições de análise: item 4.5.1.
64

65
0 5 10 15 20 25 30 35 40 45 50
0,0
5,0x104
1,0x105
1,5x105
Abso
rbânci
a (
mA
bs)
Tempo (min)
200 300 400 500 600 700 nm
0
50
100
150
200
mAU
206
281
512
617
727
200 300 400 500 600 700 nm
0
100
200
300
mAU
202
280
616
726
0 5 10 15 20 25 30 35 40 45
0,0
2,0x105
4,0x105
6,0x105
Abso
rbânci
a (
mA
bs)
Tempo (min)
0 5 10 15 20 25 30 35 40 45 50
0,0
2,0x104
4,0x104
6,0x104
Abso
rbânci
a (
mA
bs)
Tempo (min)
200 300 400 500 600 700 nm
0
500
1000
1500
mAU
196
228
279
366
476
HSG 6 HSG 7
HSG 8
Continuação da Figura 17
Figura 17: Cromatogramas (λ = 220 nm) das bandas cromatográficas HSG 6, HSG 7 e HSG 8 purificadas no sistema semi-preparativo, com
seus respectivos espectros de absorção UV no tempo de retenção em que eluem. Condições de análise: item 4.5.1.
65

66 ppm (t1)
0.01.73.35.06.78.310.0
7.0
37
2
7.0
32
8
7.0
28
1
7.0
12
2
7.0
07
4
7.0
02
8
6.1
36
1
6.1
33
8
6.1
29
0
6.1
23
9
6.1
22
6
6.1
13
7
6.1
12
3
6.1
08
4
6.1
03
5
6.1
01
4
6.0
98
7
4.9
05
5
4.2
22
6
4.2
03
1
4.0
23
7
4.0
07
3
3.9
98
5
3.9
90
8
3.9
82
0
3.9
65
5
3.8
63
5
3.8
58
9
3.8
32
1
3.8
29
8
3.6
65
7
3.6
53
0
3.6
37
4
3.6
28
5
3.6
22
0
3.6
12
3
3.5
96
1
3.3
25
5
3.3
12
9
3.3
08
9
3.3
04
8
3.3
00
8
3.2
96
7
3.2
80
1
3.2
56
6
3.2
50
7
3.2
40
3
3.2
38
0
3.2
34
5
3.1
54
4
3.1
34
7
3.1
32
03
.11
23
2.0
60
92
.04
46
2.0
28
30
.00
00
2.0
0
2.1
9
1.0
1
1.0
51
.36
2.8
3
1.5
62
.87
2.3
7
5.5 – Identificação dos marcadores químicos
A partir das análises por HPLC-DAD semi-preparativo foi possível o isolamento
de 5 bandas cromatográficas denominadas (HSG 2, HSG 3, HSG 4, HSG 5 e HSG 6). Até o
momento, foram identificados dois ciclohexiletanóides glicosilados cornosídeo (HSG 2) e
dihidrocornosídeo (HSG 3) e uma 8-O-4’-neolignana glicosilada (HSG 6). A determinação
estrutural das substâncias foram baseadas nos dados espectrais de RMN 1D (1H e
13C ) e 2D
(HMBC e HSQC).
5.5.1 – Identificação e determinação estrutural HSG 2
A banda cromatográfica HSG 2 (tR = 6,8 min ; 8,0 mg) apresentou-se como óleo
marrom. Pela análise do espectro de RMN de 1H (400 MHz, CD3OD) (Figuras 18-24),
verificou-se a presença de sinais de olefina em δ 7,01 (2H, d) e δ 6,11 (2H, d). A porção
glicose foi confirmada pelo sinal relativo à posição anomérica em δ 4,21 (1H, d), além de
outros sinais relativos aos grupos metinos e metilenos oxigenados em δ 3,92- δ 3,53. Além
destes, um sinal de grupo metileno oxigenado em δ 3,99 (1H, dt) e δ 3,63 (1H, dt) e um sinal
de grupo metileno em δ 2,04 (2H, t) também foram observados indicando a presença de um
grupo etila oxigenado.
ppm (f1)
6.9907.0007.0107.0207.0307.0407.0507.060
7.0
37
2
7.0
32
8
7.0
28
1
7.0
12
2
7.0
07
4
7.0
02
8
2.0
0
O
HO OO
HO
OH
OH
OH
1 2
34
7
8
1' 2'3'
4'5'
6'
5
6
Figura 18: Espectro de RMN de 1H (400 MHz, CD3OD) da amostra HSG 2.

67
ppm (f1)
6.1006.150
6.1
36
1
6.1
33
8
6.1
29
0
6.1
23
9
6.1
22
6
6.1
13
7
6.1
12
3
6.1
08
4
6.1
03
5
6.1
01
4
6.0
98
7
2.1
9
Figura 20: Ampliação da região entre 6,05-6,17 ppm do espectro de RMN de 1H (400
MHz, CD3OD) da amostra HSG 2.
O
HO OO
HO
OH
OH
OH
1 2
34
7
8
1' 2'3'
4'5'
6'
5
6
ppm (f1)
6.9907.0007.0107.0207.0307.0407.0507.060
7.0
37
2
7.0
32
8
7.0
28
1
7.0
12
2
7.0
07
4
7.0
02
8
2.0
0
Figura 19: Ampliação da região entre 6,98-7,06 ppm do espectro de RMN de 1H (400
MHz, CD3OD) da amostra HSG 2.
O
HO OO
HO
OH
OH
OH
1 2
34
7
8
1' 2'3'
4'5'
6'
5
6

68
ppm (f1)
3.9504.0004.0504.1004.1504.2004.250
4.2
22
6
4.2
03
1
4.0
23
7
4.0
07
3
3.9
98
5
3.9
90
8
3.9
82
0
3.9
65
5
1.0
1
1.0
5
ppm (f1)
3.6003.6503.7003.7503.8003.8503.900
3.8
63
5
3.8
58
9
3.8
32
1
3.8
29
8
3.6
65
7
3.6
53
0
3.6
37
4
3.6
28
5
3.6
22
0
3.6
12
3
3.5
96
1
1.3
6
2.8
3
Figura 21: Ampliação da região entre 3,93-4,27 ppm do espectro de RMN de 1H (400
MHz, CD3OD) da amostra HSG 2.
Figura 22: Ampliação da região entre 3,56-3,91 ppm do espectro de RMN de 1H (400 MHz,
CD3OD) da amostra HSG 2.
O
HO OO
HO
OH
OH
OH
1 2
34
7
8
1' 2'3'
4'5'
6'
5
6
O
HO OO
HO
OH
OH
OH
1 2
34
7
8
1' 2'3'
4'5'
6'
5
6

69
A análise do espectro de RMN 13
C (100 MHz, CD3OD) (Figura 24) mostrou a
presença de 14 sinais, consistindo de um sinal em δ 186,6 típico de cetona α, β -insaturada
confirmado pelos sinais das duas olefinas em δ 153,1 e δ 153,0 e δ 126,6 e δ 126,5, além de
seis sinais característicos de unidade de glicose (δ 104,33, 75,09, 78,00, 71,64, 78,12 e 62,77).
ppm (t1)
2.0202.0302.0402.0502.0602.070
2.0
60
9
2.0
44
6
2.0
28
3
2.3
7
O
HO OO
HO
OH
OH
OH
1 2
34
7
8
1' 2'3'
4'5'
6'
5
6
Figura 23: Ampliação da região entre 2,012-2,080 ppm do espectro de RMN de 1H
(400 MHz, CD3OD) da amostra HSG 2.

70
As corretas atribuições dos sinais relativos à unidade glicose e a posição da ligação
glicosídica foram feitas através dos mapas de correlação HMBC e HSQC (Figuras 25-28),
sendo que a posição anomérica δ 4,21 (H-1’) correlaciona a 3J com o sinal do carbono em δ
65,75 e a 2J com o sinal do carbono em δ 75,09 (C-2’).
ppm (f1)
0255075100125150175200
18
8.0
28
15
4.5
73
15
4.4
76
12
8.0
03
12
7.9
54
10
4.3
31
78
.12
3
77
.99
9
75
.09
3
71
.64
0
69
.32
7
65
.75
1
62
.77
5
49
.47
6
49
.26
3
49
.05
0
48
.83
7
48
.62
4
41
.06
9
O
HO OO
HO
OH
OH
OH
1 2
34
7
8
1' 2'3'
4'5'
6'
5
6
Figura 24: Espectro de RMN de 13
C (100 MHz, CD3OD) da amostra HSG 2.
Figura 25: Mapa de correlação HSQC (1H: 400 MHz, 13C: 100 MHz, CD3OD) da amostra HSG 2.
O
HO OO
HO
OH
OH
OH
1 2
34
7
8
1' 2'3'
4'5'
6'
5
6
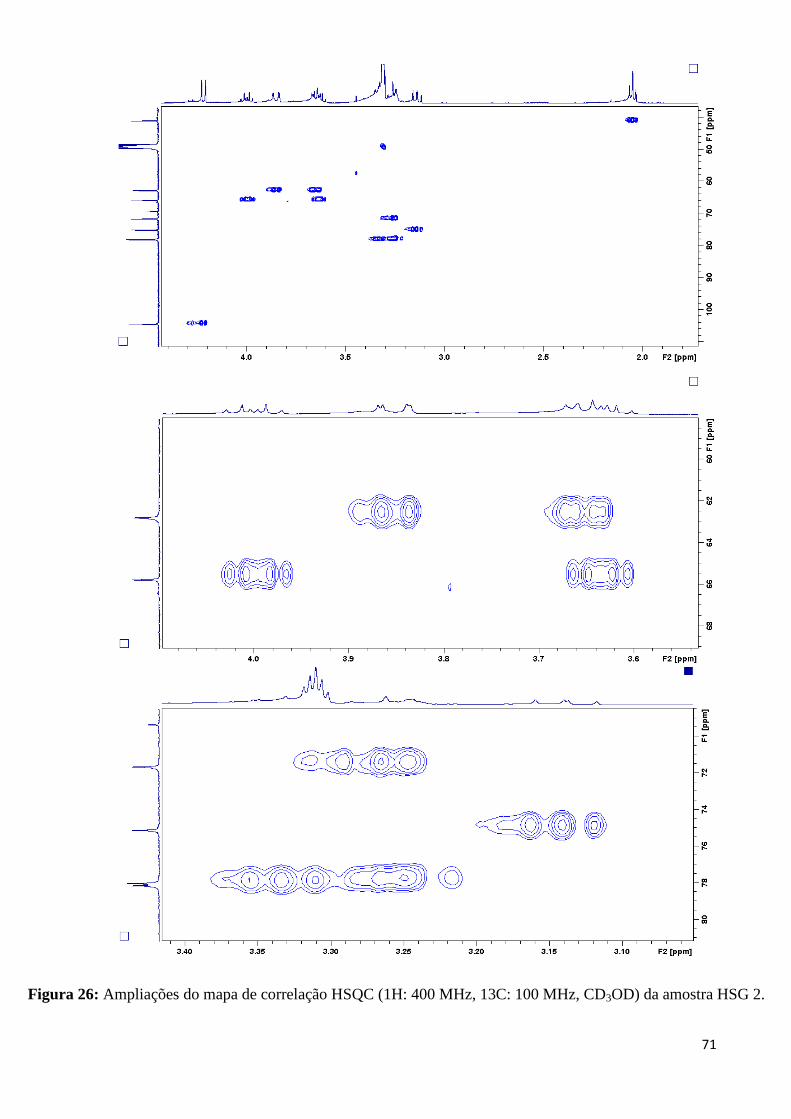
71
Figura 26: Ampliações do mapa de correlação HSQC (1H: 400 MHz, 13C: 100 MHz, CD3OD) da amostra HSG 2.

72
Figura 27: Mapa de correlação HMQC (1H: 400 MHz, 13C: 100 MHz, CD3OD) da amostra HSG 2.
O
HO OO
HO
OH
OH
OH
1 2
34
7
8
1' 2'3'
4'5'
6'
5
6
Figura 28: Ampliações do mapa de correlação HMQC (1H: 400 MHz, 13C: 100 MHz, CD3OD)
da amostra HSG 2.
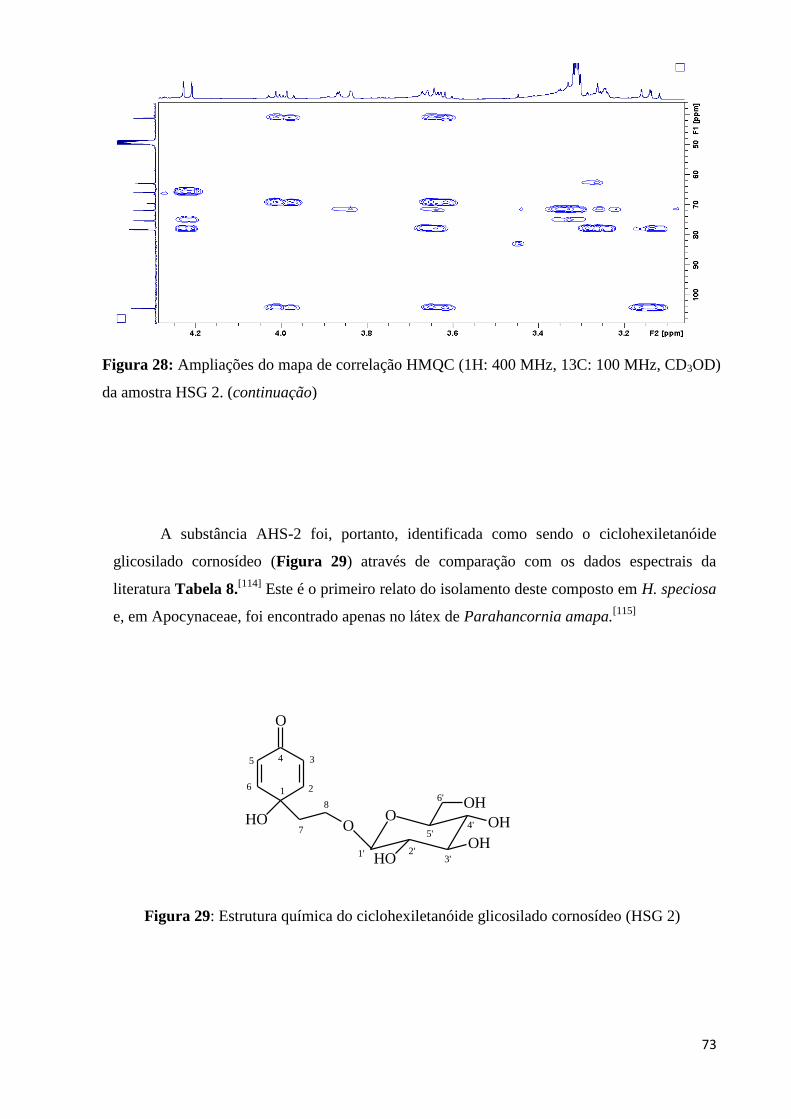
73
A substância AHS-2 foi, portanto, identificada como sendo o ciclohexiletanóide
glicosilado cornosídeo (Figura 29) através de comparação com os dados espectrais da
literatura Tabela 8.[114]
Este é o primeiro relato do isolamento deste composto em H. speciosa
e, em Apocynaceae, foi encontrado apenas no látex de Parahancornia amapa.[115]
Figura 28: Ampliações do mapa de correlação HMQC (1H: 400 MHz, 13C: 100 MHz, CD3OD)
da amostra HSG 2. (continuação)
O
HO OO
HO
OH
OH
OH
1 2
34
7
8
1' 2'3'
4'5'
6'
5
6
Figura 29: Estrutura química do ciclohexiletanóide glicosilado cornosídeo (HSG 2)

74
Tabela 8: Dados de RMN de 1H (400 MHz) e de
13C (100 MHz) em CD3OD da HSG-2
(cornosídeo) em comparação com dados da literatura[114]
.
Posição AHS-2
Cornosídeo114
1H δ (mult., J em Hz)
13C δ
1H δ (mult., J em Hz)
13C δ
1 - 69,33 (C0) - 69,2
2 7,02 (1H, dt, 10,2 e 1,8) 154,57 (CH) 7,01 (1H, d, 9,6Hz) 154,4
3 6,12 (1H, dtl, 8,2 e 1,9) 128,00 (CH) 6,11 (1H, d, 9,6Hz) 127,8
4 - 188, 03 (C0) - 187,8
5 6,12 (1H, dtl, 8,2) 127,95 (CH) 6,11 (1H, d, 9,6Hz) 127,8
6 7,02 (1H, dt, 10,2 e 1,8) 154,48 (CH) 7,01 (1H, d, 9,6 Hz) 154,3
7 2,04 (2H, t, 6,5) 41,07 (CH2) 2,04 (2H, t, 6,4 Hz) 41,0
8 3,99 (1H, dt, 10,1 e 6,4) e
3,60-3,67 (1H, m)
65,75 (CH2) 3,99 (1H, dt, 10,0 e 6,4 Hz) e
3,63 (1H, dt, 10,0 e 6,4Hz)
65,7
1’ 4,21 (1H, d, 7,8) 104,33 (CH) 4,21 (1H, d, 7,6 Hz) 104,2
2’ 3,13 (1H, dd, 9,0 e 7,9) 75,09 (CH) - 75,0
3’
3,24-3,33 (3H, m)
78,00 (CH) - 77,9
4’ 71,64 (CH) - 71,6
5’ 78,12 (CH) - 78,0
6’ 3,85 (1H, ddl, 12,1 e 1,4)
e 3,64 (1H, m)
62,77 (CH2) - 62,7
* sinais em sobreposição com sinal do solvente (CD3OD).

75
5.5.2 – Identificação e determinação estrutural HSG-3
A banda cromatográfica HSG 3 (tR= 7,5 min ; 17,6 mg) apresentou-se como óleo marrom. A
análise do espectro de RMN 13
C (100 MHz, CD3OD) mostrou 14 sinais, consistindo de um sinal em
δ 188,03 típico de cetona α, β-insaturada confirmado pelos sinais da olefina em δ 154,57 e δ 128,0
ppm.
A comparação dos dados de RMN com aqueles de HSG 2 (Figuras 30-33, Tabela 9) sugere
que HSG-3 possui estrutura similar, com a principal diferença sendo a observação de sinais de dois
CH2 a mais (δ C 35, 43 (CH2-5) e 36,21 ppm (CH2-6). Além disso, pela análise HMBC (Figura 34)
foi visível as correlações entre H-1” com C-8 e entre C-4 com H-7 confirmando a posição dos
substituintes no ciclohexiletanóide.
ppm (f1)
0.01.73.35.06.78.310.0
6.9
80
96
.97
90
6.9
55
36
.95
34
5.8
74
25
.84
87
4.2
89
24
.26
97
4.1
77
74
.16
13
4.1
52
6
4.1
45
44
.13
62
4.1
20
3
3.8
86
9
3.8
83
63
.85
80
3.8
53
7
3.8
01
03
.79
33
3.7
85
1
3.7
75
9
3.7
68
9
3.7
60
0
3.7
43
8
3.6
72
8
3.6
66
0
3.6
58
9
3.6
43
1
3.6
34
1
3.6
29
4
3.3
77
4
3.3
68
9
3.3
65
3
3.3
46
6
3.3
44
5
3.3
24
0
3.3
13
6
3.3
09
5
3.3
05
4
3.3
01
3
3.2
97
3
3.2
72
7
3.2
67
9
3.2
61
4
3.2
56
4
3.2
53
0
3.1
75
7
3.1
56
1
3.1
53
0
3.1
33
4
2.5
89
4
2.5
75
3
2.5
59
5
2.5
46
1
2.5
31
2
2.5
16
5
2.5
04
2
2.4
92
5
2.4
80
0
2.4
73
6
2.4
60
7
2.4
49
1
2.4
36
7
2.2
66
1
2.2
63
5
2.2
53
92
.25
06
2.2
47
22
.23
33
2.2
30
12
.22
01
2.2
17
12
.20
42
2.2
01
52
.10
37
2.0
87
82
.07
32
2.0
67
42
.06
43
2.0
51
52
.04
10
2.0
35
82
.03
07
2.0
16
31
.99
99
1.9
83
61
.98
06
1.9
63
81
.94
76
1.9
21
81
.90
53
1.8
88
80
.00
00
1.0
0
0.9
5
1.2
71
.37
1.5
61
.63
1.5
9
8.6
71
.60
2.7
8
1.5
0
5.2
7
Figura 30: Espectro de RMN de 1H (400 MHz, CD3OD) da amostra HSG 3.
1 2
345
6
7
8
1'2'
3'
4'5'
6'
O
HO OO
HO
OH
OH
OH

76
ppm (f1)
6.9406.9506.9606.9706.9806.990
6.9
80
9
6.9
79
0
6.9
55
3
6.9
53
4
1.0
0
ppm (f1)
5.8505.900
5.8
74
2
5.8
48
7
0.9
5
ppm (f1)
4.1004.1504.2004.2504.300
4.2
89
2
4.2
69
7
4.1
77
7
4.1
61
3
4.1
52
6
4.1
45
4
4.1
36
2
4.1
20
3
1.2
7
1.3
7
Figura 31: Ampliação da região entre 4,08-7,00 ppm do espectro de RMN de 1H (400
MHz, CD3OD) da amostra HSG 3.

77
ppm (f1)
3.6003.6503.7003.7503.8003.8503.900
3.8
86
9
3.8
83
6
3.8
58
0
3.8
53
7
3.8
01
0
3.7
93
3
3.7
85
1
3.7
75
9
3.7
68
9
3.7
60
0
3.7
43
8
3.6
72
8
3.6
66
0
3.6
58
9
3.6
43
1
3.6
34
1
3.6
29
4
1.5
6
1.6
3
1.5
9
ppm (f1)
3.1003.1503.2003.2503.3003.3503.400
3.3
77
4
3.3
68
9
3.3
65
3
3.3
46
6
3.3
44
5
3.3
24
0
3.3
13
6
3.3
09
5
3.3
05
4
3.3
01
3
3.2
97
3
3.2
72
7
3.2
67
9
3.2
61
4
3.2
56
4
3.2
53
0
3.1
75
7
3.1
56
1
3.1
53
0
3.1
33
4
8.6
7
1.6
0
ppm (f1)
2.4502.5002.5502.600
2.5
89
4
2.5
75
3
2.5
59
5
2.5
46
1
2.5
31
2
2.5
16
5
2.5
04
2
2.4
92
5
2.4
80
0
2.4
73
6
2.4
60
7
2.4
49
1
2.4
36
7
2.7
8
Figura 32: Ampliação da região entre 2,41-3,94 ppm do espectro de RMN de 1H (400
MHz, CD3OD) da amostra HSG 3.

78
ppm (t1)
050100150200
20
1.9
8
15
7.1
4
12
8.5
0
10
4.3
6
78
.15
78
.02
75
.10
71
.69
70
.36
66
.38
62
.81
49
.69
49
.48
49
.26
49
.05
48
.84
48
.62
48
.41
40
.09
36
.21
35
.43
Figura 33: Espectro de RMN de 13
C (100 MHz, CD3OD) da amostra HSG 3.
1 2
345
6
7
8
1'2'
3'
4'5'
6'
O
HO OO
HO
OH
OH
OH
1 2
345
6
7
8
1'2'
3'
4'5'
6'
O
HO OO
HO
OH
OH
OH
Figura 34: Mapa de correlação HMBC (1H a 400 MHz;
13C a 100 MHz, CD3OD) da amostra
HSG 3.

79
Tabela 9: Dados de RMN de 1H (400 MHz) e de
13C (100 MHz) em CD3OD da HSG-3
(dihidrocornosídeo) em comparação com dados da literatura.[116,117]
Posição AHS-3
Dihidrocornosídeo116,117
1H δ (mult., J em Hz)
13C δ
1H δ (mult., J em Hz)
13C δ
1 - 70,36 (C0) - 68,9
2 6,97 (1H, dd, 9,5 e 0,8) 157,14 (CH) 6,96 (1H, dd, 10,0 e 1,0) 155,9
3 5,86 (1H, d, 10,2) 128,50 (CH) 5,86 (1H, d, 10,0) 127,6
4 - 201,98 (C0) - 198,8
5 2,52 (2H, m) 35,43 (CH2) 2,47 (1H, ddd, 17,0, 11,5 e 5,0) e
2,55 (1H, ddd, 17,0 , 6,5 e 5,0)
35,1
6 2,20-2,27 (1H, m) e 2,03-
2,10 (1H, m)
36,21 (CH2) 2,19-2,26 (1H, m) e 1,95-2,05
(1H, m)
36,2
7 1,89-2,10 (2H, m) 40,09 (CH2) 2,025-2,103 (2H, m) 40,0
8 4,15 (1H, dt, 10,0 e 6,6) e
3,77 (1H, dt, 10,0 e 6,6)
66,38 (CH2) 4,14 (1H, dt, 10,0 e 6,0) e 3,77
(1H, dt, 10,0 e 6,0)
65,9
1’ 4,28 (1H, d, 7,8) 104,36 (CH) 4,28 (1H, d, 7,5) 104,6
2’ 3,15 (1H, dd, 9,1 e 7,8) 75,10 (CH) 3,15 (1H, dd, 9,0 e 7,5) 75,0
3’ 3,25-3,38 (3H, m) 78,15 (CH) - 78,5
4’ 71,69 (CH) - 71,6
5’ 78,02 (CH) 3,64 (3H, m, H-5’ e H-6’a) 78,4
6’ 3,87 (1H, dd, 11,8 e 1,5) e
3,65 (1H, m)
62,81 (CH2) 3,86 (2H, ddl, 12,0 e ca. 1,0) e
3,64 (3H, m)
62,6
* sinais em sobreposição com sinais do solvente (CD3OD).

80
A substância HSG-3 foi, portanto, identificada como sendo o ciclohexiletanóide
glicosilado dihidrocornosídeo (Figura 35) através de comparação com os dados espectrais da
literatura.[116,117]
Este é o primeiro relato do isolamento deste composto em Apocynaceae.
Figura 35: Estrutura química do ciclohexiletanóide glicosilado dihidrocornosídeo (HSG 3)
5.5.3 – Identificação e determinação estrutural HSG 6
A banda cromatográfica HSG 6 (tR = 30,2 min, 5,3 mg) apresentou-se como óleo marrom. A
análise do espectro de RMN de 1H (CD3OD) de HSG 6 ( Figuras 36-39) mostrou a presença de seis
hidrogênios aromáticos [δ 7,08 (1H, d, 1,7), 6,89 (1H, d, 8,1), 6,74 (1H, d, 8,1), 6,84 (1H, d, 1,7),
6,90 (1H, d, 8,2), 6,69 (1H, dd, 8,2 e 1,8)] revelando dois sistemas de hidrogênios 1,3,4-
trissubstituídos de dois anéis aromáticos. Além destes, foi possível verificar a presença de duas
metoxilas em δ 3,85 (s) e 3,82 (s), dois hidrogênios metilênicos [δ 1,80 (quintl, 7,1 Hz), 2,61 (tl,
7,7Hz, H-7’)], dois hidrogênios metínicos [δ 5,05 (1H, d, 5,4), 4,42 (1H, q, 5,0)], além de um
hidrogênio anomérico referente à glicose em δ 4,55 (d, 7,7 Hz, H-1”).
1 2
345
6
7
8
1'2'
3'
4'5'
6'
O
HO OO
HO
OH
OH
OH

81
ppm (f1)
0.01.73.35.06.78.310.0
7.0
80
57
.07
63
6.9
10
7
6.9
04
4
6.8
90
2
6.8
84
2
6.8
40
9
6.8
36
6
6.7
50
9
6.7
30
6
6.7
00
7
6.6
96
3
6.6
80
2
6.6
75
9
5.0
55
7
5.0
42
2
4.5
64
0
4.5
44
8
4.4
43
4
4.4
30
9
4.4
18
3
4.4
05
8
3.8
48
6
3.8
24
2
3.7
99
8
3.7
88
7
3.7
70
3
3.7
59
1
3.7
23
5
3.7
17
6
3.6
93
7
3.6
88
0
3.5
96
8
3.5
83
8
3.5
66
9
3.5
50
7
3.5
34
4
3.4
46
9
3.4
33
9
3.4
17
2
3.4
04
3
3.3
85
1
3.3
62
9
3.3
45
5
3.3
40
7
3.3
14
7
3.3
10
9
3.3
06
8
3.3
02
8
3.2
98
8
3.2
90
5
3.2
77
7
3.2
68
0
3.2
55
1
3.1
81
2
3.1
75
4
3.1
68
33
.16
22
3.1
58
13
.15
17
3.1
44
53
.13
86
2.6
31
32
.61
26
2.5
93
01
.83
74
1.8
21
21
.80
13
1.7
83
11
.76
67
0.0
00
0
1.0
0
2.0
10
.97
1.0
20
.97
0.8
1
0.9
10
.94
6.1
71
.25
1.1
13
.19
2.4
91
.03
0.9
7
2.0
3
2.0
7
Figura 36: Espectro de RMN de 1H (400 MHz, CD3OD) da amostra HSG 6
ppm (f1)
6.7006.7506.8006.8506.9006.9507.0007.0507.100
7.0
80
5
7.0
76
3
6.9
10
7
6.9
04
4
6.8
90
2
6.8
84
2
6.8
40
9
6.8
36
6
6.7
50
9
6.7
30
6
6.7
00
7
6.6
96
3
6.6
80
2
6.6
75
9
1.0
0
2.0
1
0.9
7
1.0
2
0.9
7
Figura 37: Ampliação da região entre 6,65-7,12 ppm do espectro de RMN de 1H (400
MHz, CD3OD) da amostra HSG 6.
O OH
O
OH
OCH3
CH3O
OH
O
OH
HOHO
OH
1
2
34
5
6
78
9
1'
2'
3'
4'
5'
6'
7'
8'
9'
1"
2"
3"
4" 5"
6"
O OH
O
OH
OCH3
CH3O
OH
O
OH
HOHO
OH
1
2
34
5
6
78
9
1'
2'
3'
4'
5'
6'
7'
8'
9'
1"
2"
3"
4" 5"
6"

82
ppm (f1)
4.404.504.604.704.804.905.00
5.0
55
7
5.0
42
2
4.5
64
0
4.5
44
8
4.4
43
4
4.4
30
9
4.4
18
3
4.4
05
8
0.8
1
0.9
1
0.9
4
Figura 38: Ampliação da região entre 4,38-5,08 ppm do espectro de RMN de 1H (400
MHz, CD3OD) da amostra HSG 6
ppm (f1)
3.203.303.403.503.603.703.80
3.8
48
6
3.8
24
2
3.7
99
8
3.7
88
7
3.7
70
3
3.7
59
1
3.7
23
5
3.7
17
6
3.6
93
7
3.6
88
0
3.5
96
8
3.5
83
8
3.5
66
9
3.5
50
7
3.5
34
4
3.4
46
9
3.4
33
9
3.4
17
2
3.4
04
3
3.3
85
1
3.3
62
9
3.3
45
5
3.3
40
7
3.3
14
7
3.3
10
9
3.3
06
8
3.3
02
8
3.2
98
8
3.2
90
5
3.2
77
7
3.2
68
0
3.2
55
1
3.1
81
2
3.1
75
4
3.1
68
3
3.1
62
2
3.1
58
1
3.1
51
7
3.1
44
53
.13
86
6.1
7
1.2
5
1.1
1
3.1
9
2.4
9
1.0
3
0.9
7
Figura 39: Ampliação da região entre 3,10-3,90 ppm do espectro de RMN de 1H (400
MHz, CD3OD) da amostra HSG 6
O OH
O
OH
OCH3
CH3O
OH
O
OH
HOHO
OH
1
2
34
5
6
78
9
1'
2'
3'
4'
5'
6'
7'
8'
9'
1"
2"
3"
4" 5"
6"
O OH
O
OH
OCH3
CH3O
OH
O
OH
HOHO
OH
1
2
34
5
6
78
9
1'
2'
3'
4'
5'
6'
7'
8'
9'
1"
2"
3"
4" 5"
6"

83
Com a observação do espectro de RMN de 13
C (CD3OD) de HSG 6 (Figuras 40 e
41) foi possível verificar 26 sinais de carbono, incluindo dois carbonos de metoxila em δ
56,48 e 56,44, 18 carbonos das duas unidades fenilpropanóide (um grupo guaiacil e outro
dihidroconiferil) formando a neolignana, além de seis carbonos da porção O-glicose (δ
104,80, 77,75, 77,87, 71,47, 78,02 e 62,59).
ppm (f1)
050100150200
15
1.4
8
14
8.6
5
14
7.2
5
14
7.2
3
13
7.9
4
13
1.9
0
12
1.8
9
12
1.3
8
11
8.6
9
11
5.6
5
11
3.9
1
11
2.6
3
10
4.8
0
85
.55
81
.46
78
.01
77
.87
75
.75
71
.47
62
.59
62
.23
61
.46
56
.48
56
.44
49
.69
49
.48
49
.26
49
.05
48
.84
48
.62
48
.41
35
.58
32
.72
Figura 40: Espectro de RMN de 13
C (100 MHz, CD3OD) da amostra HSG 6.
O OH
O
OH
OCH3
CH3O
OH
O
OH
HOHO
OH
1
2
34
5
6
78
9
1'
2'
3'
4'
5'
6'
7'
8'
9'
1"
2"
3"
4" 5"
6"

84
A partir do mapa de correlação HSQC (Figuras 42 e 43) obtido foi possível atribuir os
sinais de carbono ao sinal dos hidrogênios correspondentes. Posteriormente, a estrutura parcial de
HSG 6 foi confirmada a partir do mapa de correlação HMBC (Figuras 44 e 45). As correlações no
mapa de correlação HMBC de H-7 com C-1, C-2, C-6, C-8, C-9 e C-1”; de H-2’/6’ com C-1’, C-
3’/5’, C-4’ e C-7’; de OMe-3/3’ com C-3/3’, junto com seus deslocamentos, confirmaram a posição
dos substituintes e a ligação 8-O-4’em AHS 6. As correlações do hidrogênio anomérico H-1” com
C-7 em δ 81,46 ppm e H-7 com C-1” em δ 104.80 pelo HMBC, indicando a posição da ligação com
a unidade glicose em C-7. A ligação glicosídica foi determinada ser do tipo δ devido ao maior valor
da constante de acoplamento do dupleto referente H-1” em δ 4,55 (J=7,7 Hz).[118]
ppm (f1)
60.065.070.075.080.085.0
85
.55
81
.46
78
.01
77
.87
75
.75
71
.47
62
.59
62
.23
61
.46
56
.48
56
.44
Figura 41: Ampliação da região entre 56,0 e 87,0 ppm do espectro de RMN de 13
C (100
MHz, CD3OD) da amostra HSG 6.
O OH
O
OH
OCH3
CH3O
OH
O
OH
HOHO
OH
1
2
34
5
6
78
9
1'
2'
3'
4'
5'
6'
7'
8'
9'
1"
2"
3"
4" 5"
6"

85
Figura 42: Mapa de correlação HSQC (1H: 400 MHz, 13C: 100 MHz, CD3OD) da amostra HSG 6.
Figura 43: Ampliações do mapa de correlação HSQC (1H: 400 MHz,
13C: 100 MHz,
CD3OD) da amostra HSG 6.
O OH
O
OH
OCH3
CH3O
OH
O
OH
HOHO
OH
1
2
34
5
6
78
9
1'
2'
3'
4'
5'
6'
7'
8'
9'
1"
2"
3"
4" 5"
6"

86
Figura 44: Mapa de correlação HMBC (1H: 400 MHz, 13C: 100 MHz, CD3OD) da amostra HSG 6.
Figura 45: Ampliações do mapa de correlação HMBC (1H: 400 MHz,
13C: 100 MHz,
CD3OD) da amostra HSG 6.
O OH
O
OH
OCH3
CH3O
OH
O
OH
HOHO
OH
1
2
34
5
6
78
9
1'
2'
3'
4'
5'
6'
7'
8'
9'
1"
2"
3"
4" 5"
6"

87
A substância AHS 6 foi, portanto, identificada como sendo uma 8,4’-O-neolignana
(Figura 46) através de comparação com os dados espectrais da literatura (Tabela 10).[118,119]
Este é
o primeiro relato do isolamento deste composto em Apocynaceae.
O OH
O
OH
OCH3
CH3O
OH
O
OH
HOHO
OH
1
2
34
5
6
78
9
1'
2'
3'
4'
5'
6'
7'
8'
9'
1"
2"
3"
4" 5"
6"
Figura 46: Estrutura química da (7,8)-treo-4,7,9,9’-tetrahidroxi-3,3’-dimetoxi- 8-O-4’-
neolignana-7-O-β-D-glicopiranosídeo (HSG 6)

88
Tabela 10: Dados de RMN de 1H (400 MHz) e de
13C (100 MHz) em CD3OD da HSG 6 em
comparação com dados da literatura.[118]
Posição AHS 6
(7,8)-treo-4,7,9,9’-tetrahidroxi-3,3’-
dimetoxi- 8-O-4’-neolignana-7-O-β-D-
glicopiranosídeo 118,119
1H δ (mult., J em Hz)
13C δ
1H δ (mult., J em Hz)
13C δ
1 - 131,90 (C0) - 131,9 (C-1)
2 7,08 (1H, d, 1,7) 112,63 (CH) 7,08 (1H, d, 1,8) 112,6 (C-2)
3 - 148,65 (C0) - 148,6 (C-3)
4 - 147,23 (C0) - 147,3 (C-4)
5 6,74 (1H, d, 8,1) 115,65(CH) 6,74 (1H, d, 8,0) 115,6 (C-5)
6 6,89 (1H, d, 8,1) 121,38 (CH) 6,89 (1H, dd, 8,0 e 1,8) 121,4 (C-6)
7 5,05 (1H, d, 5,4) 81,46 (CH) 5,05 (1H, d, 5,1) 81,5 (C-7)
8 4,42 (1H, q, 5,0) 85,55(CH) 4,42 (1H, m) 85,5 (C-8)
9 3,78 (1H, dd, 11,8 e 4,4)
e 3,43 (1H, dd, 11,9 e
5,2)
61,46(CH2) 3,87 (1H, dd, 13 e 6) e
3,55 (1H, dd, 13 e 6)[119]
61,5 (C-9)
1’ - 137,94 (C0) - 137,9 (C-1’)
2’ 6,84 (1H, d, 1,7) 113,91(CH) 6,84 (1H, d, 1,8) 113,9 (C-2’)
3’ - 151,48 (C0) - 151,5 (C-3’)
4’ - 147,25 (C0) - 147,3 (C-4’)
5’ 6,90 (1H, d, 8,2) 118,69 (CH) 6,90 (1H, d, 8,0) 118,7 (C-5’)
6’ 6,69 (1H, dd, 8,2 e 1,8) 121,89 (CH) 6,69 (1H, dd, 8,0 e 1,8) 121,9 (C-6’)
7’ 2,61 (2H, tl, 7,7) 32,72(CH2) 2,61 (2H, t) 32,7 (C-7’)
8’ 1,80 (2H, quintl, 7,1) 35,58 (CH2) 1,80 (2H, m) 35,6 (C-8’)
9’ 3,55 (2H, t, 6,5) 62,23 (CH2) 3,55 (2H, t) 62,2 (C-9’)
1” 4,55 (1H, d, 7,7) 104,80 (CH) 4,55 (1H, d, 7,6) 104,8 (C-1’’)
2” 3,26-3,32 (1H, m)* 75,75(CH) 3,21 (1H, dd, 9 e 8) [119]
75,7 (C-2”)
3” 3,26-3,39 (3H, m)* e
3,14-3,18 (1H, m)
77,87(CH)
3,35-3,10 (3H, m) [119]
77,9 (C-3”)
4” 3,26-3,39 (3H, m)* 71,47(CH) 71,5 (C-4”)
5” 3,26-3,39 (3H, m)* e
3,14-3,18 (1H, m)
78,02 (CH) 78.0 (C-5”)
6” 3,71 (1H, dd, 11,9 e 2,3)
e 3,59 (1H, d, 5,2)
62,59(CH2) 3,83 (1H, dd, 12 e 2) e
3,68(1H, dd, 12 e 6) [119]
62,6 (C-6”)
OCH3-3’ 3,85 (3H, s) 56,48 3,85 (3H, s) 56,5
OCH3-3 3,82 (3H, s) 56,44 3,82 (3H, s) 56,4
* sinais em sobreposição com sinais do solvente (CD3OD).

89
5.6 – Otimização das condições cromatográficas de análise: látex dos frutos
de H. speciosa
As substâncias isoladas do extrato clorofórmio do látex dos frutos da mangabeira
(mistura de ésteres 3-β-O-acil lupeol (2, 4, 8-11), lupeol (14) e éster diidroxilado 3-β-O-acil
lupeol (15)) previamente isolados pela MSc. Taís Sampaio[86]
foram utilizadas como
marcadores químicos do látex dos frutos de H. speciosa. Um método cromatográfico foi
desenvolvido para a identificação destas substâncias nas diferentes amostras do látex desta
espécie.
A análise de triterpenos pentacíclicos por HPLC utilizando UV, DAD, ELSD e
MS como detector é descrito na literatura.[120-125]
Entre os métodos disponíveis foi
selecionado a metodologia proposta por Martelanc e colaboradores como base para o
desenvolvimento do método. No trabalho em questão, os pesquisadores avaliaram a presença
de lupeol, α-amirina e β-amirina por HPLC-UV. As condições utilizadas foram: coluna
Hypersil BDS C18 (250 mm x 3 mm, 3µm ), temperatura ambiente, 100% de acetonitrila
como fase móvel, volume de injeção: 20 µL, fluxo de 0,8 mL/min e o tempo da corrida de 30
min. O comprimento de onda escolhido para obtenção dos cromatogramas foi 200 nm. As
amostras foram dissolvidas em n-propanol.[121]

90
Baseado nas condições citadas acima foi feita análise da mistura de lupeol, α-
amirina e β-amirina. Como não se tinha disponível no laboratório a coluna Hypersil BDS
C18, foi realizada a análise com a coluna Microsorb MV100-5C18 (250 mm x 4,6 mm, 5 µm,
(VARIAN); sendo que as demais condições foram mantidas. O cromatograma pode ser
visualizado na Figura 47.
Figura 47: Cromatograma obtido a partir das seguintes condições: coluna Microsorb MV100-
5C18, modo isocrático 100% de ACN por 30 min em um fluxo de 0,8mL/min, volume de
injeção 20µL, λ = 200 nm.
Com pode ser observado não houve reprodução do método[121]
, fato esse que pode
ser explicado em virtude das alterações realizadas no método. As substâncias apresentaram
bastante afinidade com a fase móvel eluindo na parte inicial do cromatograma. Para diminuir
tal afinidade a porcentagem do solvente orgânico foi reduzida, inicialmente, para 85%
(Análise 1 da Tabela 11) e em seguida para 80% (Análise 2 da Tabela 11). O resultado pode
ser visto na Figura 48.

91
Tabela 11: Condições cromatográficas avaliadas na otimização do método para análise do
látex dos frutos da H. speciosa, com detecção em 220 nm, volume de injeção de 20 µL e
vazão de 0,8 mL/min.
Análise Gradiente de eluição Fase móvel Fase estacionária
1
85% (B) por 30 min
H2O (A):ACN (B)
Microsorb MV100-5C18
(5 µm, 250 mm x 4,6 mm)
2
80 % (B) por 30 min
H2O (A):ACN (B)
Microsorb MV100-5C18
(5 µm, 250 mm x 4,6 mm)
3
100% (B) por 30 min
H2O (A):ACN (B)
Hexil-fenil Luna
(10 µm, 150 x 4,6 mm)
4
90% (B) por 30 min
H2O (A):ACN (B)
Hexil-fenil Luna
(10 µm, 150 x 4,6 mm)
5
86-90% (B) por 10 min; 90-100% (B)
por 30 min; 100-86% (B) por 5 min e
86% (B) por 15 min.
H2O (A):ACN (B)
Hexil-fenil Luna
(10 µm, 150 x 4,6 mm)
6
80-90% (B) por 30 min; 90-100% (B)
por 30 min; 100-86% (B) por 5 min;
86% (B) por 15 min; 86-80% (B) por 5
min e 80% (B) por 5 min.
H2O (A):ACN (B)
Hexil-fenil Luna
(10 µm, 150 x 4,6 mm)
7
70-90% (B) por 30 min; 90-100% (B)
por 30 min; 100-86% (B) por 15 min;
86-70% (B) por 10 min e 70% (B) por 5
min.
H2O (A):ACN (B)
Hexil-fenil Luna
(10 µm, 150 x 4,6 mm)

92
Figura 48: Cromatogramas (λ = 200 nm) obtidos a partir das seguintes condições: coluna
Microsorb MV100-5 C18, fluxo de 0,8 mL/min, volume de injeção 20 µL: (Análise 1) modo
isocrático 85% de ACN:H2O, (Análise 2) modo isocrático 80% de ACN:H2O ambos por 30
min.
A baixa seletividade e retenção observadas nas análises cromatográficas empregando a
coluna Microsorb MV100-5C18 nos levou a testar a fase estacionária hexil-fenil LUNA ®
(150
mm x 4,6 mm, 10 µm), mantendo em um primeiro momento a fase móvel 100 % de ACN
(Análise 3) e depois modificando para 90% de ACN:H2O (Análise 4). Os respectivos
cromatogramas podem ser vistos na Figura 49.
(A)
(B)
Análise 1
Análise 2

93
Figura 49: Cromatogramas (λ = 200 nm) obtidos a partir das seguintes condições: coluna
hexil-fenil LUNA, fluxo de 0,8 mL/min, volume de injeção 20µL: (Análise 3) modo
isocrático 100% de ACN:H2O, (Análise 4) modo isocrático 90 % de ACN:H2O ambos por 30
min.
Mediante a aparente separação da mistura de lupeol, α-amirina e β-amirina,
usando a fase estacionária hexil-fenil e fase móvel 90% de ACN:H2O, foi feita a injeção do
extrato diclorometano do látex dos frutos bem como dos demais marcadores químicos: lupeol,
éster diidroxilado 3-β-O-acil lupeol e mistura de ésteres 3- β-O-acil lupeol. Os cromatogramas
podem ser visualizados na Figura 50.
Análise 3
Análise 4

94
O método se mostrou eficiente para a detecção do lupeol (Figura 50A). No
entanto, para os ésteres e o extrato do látex dos frutos, foi observado o alargamento dos picos
possivelmente em consequência da interação excessiva com a fase estacionária (Figura 50B-
D).
Figura 50: Cromatogramas (λ = 200 nm) obtidos a partir das seguintes condições: coluna
hexil-fenil LUNA®, modo isocrático 90 % de ACN:H2O, fluxo de 0,8mL/min, volume de
injeção 20µL: (A) lupeol; (B) mistura de ésteres 3- β-O-acil lupeol; (C) éster diidroxilado 3-
β-O-acil lupeol e (D) extrato diclorometano do látex dos frutos de H. speciosa.
(A) (B)
(C) (D)

95
A partir dos resultados discutidos acima foi feita a opção pelo método gradiente
ao invés do isocrático. Utilizando o extrato diclorometano do látex dos frutos foram testados
três sistemas (Análises 5 a 7) em gradiente conforme a descrição da Tabela 11. Os
cromatogramas destes experimentos são apresentados na Figura 51.
Figura 51: Cromatogramas (λ = 200 nm) para a seleção de condições para análise do látex
dos frutos de H. speciosa Gomes em HPLC-DAD obtidos a partir das condições de análise (5-
7) descritas na Tabela 11.
Análise 5 Análise 6
Análise 7

96
Tendo em vista os objetivos do presente trabalho, a condição de análise 5
proporcionou a melhor resposta com uma boa separação entre os picos de interesse em menor
tempo. O comprimento de onda foi escolhido baseado na projeção tridimensional obtido por
HPLC-DAD da amostra do látex dos frutos (Figura 52). A análise do cromatograma 3D
mostrou que 200 nm proporcionou a detecção adequada da maioria dos picos de interesse.
Figura 52: Projeção tridimensional obtido por HPLC-DAD da amostra do látex dos frutos de
H. speciosa.
Para avaliar a presença das substâncias de interesse no látex dos frutos, os
marcadores químicos foram analisados na condição de análise 5. Como pode ser visto na
Figura 53A, os tempos de retenção dos triterpenos são compatíveis com os picos do
cromatograma do extrato do látex dos frutos da mangabeira. Além das análises por DAD, o
extrato do látex dos frutos verdes de H. speciosa também foi analisado por ELSD. Ao se
comparar os cromatogramas obtidos com os dois detectores, foi observado um perfil
cromatográfico similar, destacando-se a maior sensibilidade do detector ELSD para a amostra
analisada (Figura 53B).

97
0 10 20 30 40 50 min0
250000
500000
750000
1000000
1250000
uV
Figura 53: (A) Perfis cromatográficos monitorado com o DAD (λ = 200 nm): extrato
diclorometano do látex dos frutos (preto); marcadores químicos: éster diidroxilado 3-β-O-acil
lupeol (vermelho), mistura de ésteres 3- β-O-acil lupeol (azul), e lupeol (magenta); (B) Perfil
cromatográfico do látex dos frutos de H. speciosa monitorado pelo DAD (preto) e pelo ELSD
(vermelho)
0.0 5.0 10.0 15.0 20.0 25.0 30.0 35.0 40.0 45.0 50.0 55.0 min
0
50000
100000
150000
200000
250000
300000
350000
400000
450000
500000
550000
600000
uV
(A)
(B)

98
0.0 5.0 10.0 15.0 20.0 25.0 30.0 35.0 40.0 45.0 50.0 55.0 min0
25000
50000
75000
100000
125000
150000
175000
200000
225000
250000
uV
0.0 5.0 10.0 15.0 20.0 25.0 30.0 35.0 40.0 45.0 50.0 55.0 min0
25000
50000
75000
100000
125000
uV
Além da análise do extrato diclorometano, foram investigadas as amostras do látex
liofilizado e de uma polpa comercial. Os cromatogramas monitorados pelo DAD e pelo ELSD
são mostrados na Figura 54. Os picos característicos dos marcadores químicos podem ser
notados tanto na polpa quanto no látex liofilizado.
Figura 54: Cromatogramas obtidos por DAD (λ = 200 nm): (A) extrato diclorometano do
látex dos frutos de mangaba (preto), látex dos frutos de mangaba liofilizado (vermelho) e
polpa de mangaba comercial (azul); (B) Cromatogramas obtidos por ELSD: extrato
diclorometano do látex dos frutos (magenta), látex dos frutos liofilizado (verde) e polpa
comercial (azul).
(A)
(B)

99
A identificação dos marcadores químicos nas amostras de H. speciosa através das
condições otimizadas corrobora a seletividade do método para estes compostos. Dessa forma,
as melhores condições foram: coluna hexil-fenil LUNA® (10 µm, 150 mm x 4,6 mm,
Phenomenex), fase móvel ACN:H2O em eluição gradiente 86-90% (B) por 10 min; 90-100%
(B) por 30 min; 100-86% (B) por 5 min e 86% (B) por 15 min, volume de injeção de 20 µL e
vazão de 0,8 mL/min. O uso da ACN como modificador orgânico possibilitou a seleção de
200 nm como comprimento de onda para o registro dos cromatogramas.
No entanto, a presença de compostos de baixa polaridade nas amostras de H.
speciosa torna pouco apropriado o uso de ACN como solvente de diluição. Com o intuito de
aumentar a compatibilidade entre o solvente usado no preparo das amostras de látex dos
frutos e a fase móvel, foi utilizado a metodologia mostrada no item 4.4.2. Os marcadores
químicos, por sua vez, foram dissolvidos em isopropanol[121]
. A diferença entre a fase móvel e
o solvente utilizado na solubilização das amostras ocasiona uma distorção (pico do solvente)
que pode ser observado no início do cromatograma (54 A).

100
5.7 – Validação do método cromatográfico para quantificação do marcador
químico éster diidroxilado 3-β-O-acil lupeol
A confiabilidade do método foi avaliada pelo estudo da seletividade, linearidade,
sensibilidade, precisão, exatidão, estabilidade e robustez. Todos os experimentos realizados
no estudo de validação seguiram as normas estabelecidas na RE 899/03 (ANVISA).[113]
O objetivo dessa etapa do trabalho consistiu em quantificar o éster diidroxilado 3-
β-O-acil lupeol na polpa comercial de mangaba e no látex liofilizado bem como estimar a
quantidade de ésteres de lupeol na polpa comercial de mangaba usando o éster diidroxilado
como padrão. A estrutura do éster diidroxilado 3-β-O-acil lupeol pode ser vista na Figura 55.
A validação foi acompanhada pelo DAD em série ao ELSD, ou seja, depois do DAD, o
efluente da coluna cromatográfico foi submetido à detecção por ELSD. O grau de pureza do
padrão de éster diidroxilado foi verificado por normalização, este se mostrou igual a 97,13%.
Figura 55: Estrutura química do éster diidroxilado 3-β-O-acil lupeol.
Para a quantificação dos triterpenos pentacíclicos um novo método, baseado nas
condições anteriores (item 5.2), foi desenvolvido. As condições do método quantitativo estão
descritas no item 4.6.2.1. O uso da eluição isocrática permitiu a diminuição do tempo de
análise sem comprometer a separação dos picos referentes aos ésteres de lupeol; tais
características fizeram o novo método mais conveniente para a quantificação, uma vez que
diminuiu drasticamente o tempo de análise.
O
OOHOH
14
(15)

101
5.7.1 - Seletividade:
No processo de validação de um método a seletividade é o primeiro passo a ser
avaliado. A possível existência de substâncias que podem sofrer degradação requer a
freqüente reavaliação deste parâmetro durante a validação, uma vez que a seletividade não
esteja assegurada, outros parâmetros como a linearidade, exatidão e precisão, estarão
seriamente comprometidos. [126,127]
Nesse trabalho a seletividade foi avaliada pelo grau de pureza do pico de interesse
e pela adição do padrão na amostra. A Figura 56 mostra um típico cromatograma do éster
diidroxilado 3-β-O-acil lupeol, da polpa da mangaba e da polpa da mangaba fortificada
monitorado pelo DAD e pelo ELSD. Os cromatogramas mostram que o método desenvolvido
separou o éster diidroxilado 3-β-O-acil lupeol na amostra de tal forma que nenhum pico
interferente eluiu na região de interesse. Este fato foi confirmado pelas medidas da pureza do
pico adquirido pelo DAD que mostrou alta pureza espectral. Essas análises evidenciaram a
seletividade do método.[126]

102
5.0 10.0 15.0 20.0 25.0 30.0 35.0 min0.0
1.0
2.0
3.0
4.0
5.0uV(x100,000)
0.0 5.0 10.0 15.0 20.0 25.0 30.0 35.0 min
1.0
2.0
3.0
4.0
5.0
6.0
7.0
uV(x100,000)
Figura 56: Cromatograma típico: polpa da mangaba (vermelho), éster diidroxilado 3-β-O-acil
lupeol (azul) e polpa da mangaba fortificada (preto); (A) obtido pelo DAD; (B) obtido pelo
ELSD. As condições experimentais para obtenção desses cromatogramas são descritos no
item 4.6.2.1.
(A)
(B)
éster diidroxilado 3-β-O-acil lupeol
éster diidroxilado 3-β-O-acil lupeol

103
5.7.2 - Linearidade:
A linearidade foi avaliada por dois métodos de calibração: padronização externa e
padronização por adição de padrão. Embora não exista uma regra específica para escolha do
método de quantificação este deve ser selecionado para fornecer a melhor exatidão possível e
um alto nível de precisão. Normalmente, a padronização externa é utilizada para amostras que
não precisam de pré-tratamento extenso enquanto que a padronização por adição de padrão é
empregada nas seguintes situações: quando não é possível obter a matriz isenta do analito; em
matrizes muito complexas; quando há fortes interações entre o analito e a matriz e quando é
difícil encontrar um padrão interno adequado.[126,127]
O uso das curvas analíticas obtidas pelos métodos descritos acima resulta em uma
importante informação sobre a seletividade do método. Uma vez que as curvas de calibração,
quando comparadas, apresentem-se paralelas a seletividade do método é garantida e não há
interferência da matriz na quantificação. Dessa forma, a quantificação do analito em
diferentes amostras pode ser obtida usando a regressão oriunda da padronização externa.[17]
A Figura 57 mostra as curvas analíticas resultantes do nosso estudo de
linearidade. Para a construção destas curvas seis concentrações conhecidas do éster
diidroxilado 3-β-O-acil lupeol foram preparadas e analisadas em triplicata. O preparo das
soluções estoque e padrões de calibração estão descrito no item 4.5.1. Para a detecção por
DAD, a equação de regressão foi calculada na forma de Y = A.X + B, onde Y e X são área do
pico e a correspondente concentração, respectivamente. Por outro lado, a curva de calibração
do ELSD foi construída usando a conversão logarítmica tanto da área do pico (Y) quanto da
concentração (X). Este específico tratamento é devido ao comportamento não linear do ELSD
que tem sido descrito pelo modelo empírico Y=amb onde b é o slope, m é a massa do
composto injetado e a o intercepto da regressão linear.[53, 128]
A similaridade entre os coeficientes angulares das curvas de calibração adquiridas
pela padronização externa e padronização por adição de padrão monitorados pelo DAD
(coeficiente de variação de 0,2%) corroboram a seletividade do método e a não influência da
matriz na quantificação. Dessa forma, a quantificação dos ésteres nas amostras de H. speciosa
pode ser realizada pela padronização externa.

104
Y= 7367,95179X - 22369,23508
r2 = 0,996
-0,6 -0,4 -0,2 0,0 0,2 0,4 0,6 0,8 1,0
4,5
5,0
5,5
6,0
6,5
7,0
7,5
Are
a
Concentraçao ( g/mL)
Figura 57: Curvas analíticas obtidas para a quantificação de éster diidroxilado 3-β-O-acil
lupeol pelo DAD: padronização externa (A) e padronização por adição de padrão (B); e pelo
ELSD: padronização externa (C).
As equações de regressão linear e os valores de coeficientes de correlação, obtidas
pela padronização externa com o DAD e o ELSD são mostradas na Tabela 12. Ambos os
detectores apresentaram coeficientes de relação acima do valor mínimo preconizado pela
ANVISA (r2= 0,98). A partir destes resultados é possível concluir que dentro da faixa
estudada há uma boa correlação entre a concentração do composto de interesse e a área
obtida. O coeficiente de variação referente às replicatas (n = 3) das concentrações das duas
curvas não ultrapassou 5%.
(A)
Y= 7387,16845X + 770488,94322
r2 = 0,998
(B)
0 100 200 300 400 500
0,0
5,0x106
1,0x107
1,5x107
2,0x107
2,5x107
3,0x107
3,5x107
Áre
a
Concentração ( g/mL)
0 100 200 300 400 500
0,0
5,0x106
1,0x107
1,5x107
2,0x107
2,5x107
3,0x107
3,5x107
4,0x107
4,5x107
Áre
a
Concentração ( g/mL)
(C)
Y= 1,85731x+ 5,45295
r2 = 0,999

105
Tabela 12: Parâmetros de linearidade usando as soluções padrões.
Detecção
Faixa de Linearidade
(µg/mL)
Equação da reta
r2
LD
(µg/mL)
LQ
(µg/mL)
DAD 19,4 - 466,2 y= 7367,95x – 22369,23 0,996 14,5 41,2
ELSD 19,4 - 466,2 y= 1,85x + 5,47 0,999 8,3 15,8
5.7.3 – Limite de detecção e limite de quantificação:
A habilidade de um método em quantificar e detectar baixas concentrações é
comprovada em termos de limite de detecção (LD) e limite de quantificação (LQ). O LD é
definido como a menor concentração de um analito que o método é capaz de diferenciar,
confiavelmente, do ruído de fundo. O LQ, por sua vez, representa a menor concentração do
composto de interesse em uma amostra, que pode ser quantitativamente determinado com
valores aceitáveis de precisão e exatidão.[113,126]
Os valores de LD e LQ são reportados na
Tabela 12 para ambos os detectores. O ELSD apresentou valores de LD e LQ mais baixos
que o DAD.
5.7.4 – Precisão e exatidão:
A precisão juntamente com a exatidão determinam erros de uma medida analítica e
são os principais critérios usados para julgar a qualidade de um método. Enquanto a precisão
avalia os resultados obtidos em uma série de medidas de uma amostragem múltipla de uma
mesma amostra, a exatidão pondera a proximidade dos resultados obtidos pelo método em
estudo em relação ao valor verdadeiro.[125,126]
A avaliação da precisão e da exatidão foi realizada em três dias diferentes a partir
de três concentrações denominadas soluções de controle de qualidade. Tais concentrações
contemplam a faixa de linearidade do método e são designadas pelas siglas CQB (controle de
qualidade de baixa concentração, CQM (controle de qualidade de média concentração) e CQA
(controle de qualidade de alta concentração).

106
O DPR foi tomado como medida de precisão. Os resultados do estudo de precisão
são mostrados na Tabela 13. Como pode ser notado, o valor médio de DPR variou entre 0,8 e
5,5% quando o experimento foi avaliado pelo DAD, enquanto que para a detecção por ELSD,
o valor de DPR variou entre 1,1 e 10,0%.
Tabela 13: Dados do estudo de precisão, intra- e inter-dias, do método para quantificação de
éster diidroxilado 3-β-O-acil lupeol.
Concentração
1º Dia
2º Dia
3ºDia
Média
DPR(%)a
DPR(%)a
ELSD DAD ELSD DAD ELSD DAD ELSD DAD
CQB* 8,7 5,3 10,0 5,5 5,5 3,0 8,0 4,6
CQM** 4,8 3,7 5,3 4,1 2,9 1,5 4,3 3,1
CQA*** 1,1 2,4 5,5 2,2 2,9 0,8 3,1 1,8
a Desvio padrão relativo (DPR%), para n =5
* CQB (43,7 µg/mL),
** CQM (207,9 µg/mL) e
*** CQA (372,9 µg/mL)
A exatidão do método foi calculada como percentagem de recuperação de uma
quantidade conhecida do analito adicionada na amostra. A recuperação média foi estimada
pela razão entre a concentração experimental e a quantidade adicionada na matriz. Os
resultados mostraram que as amostras fortificadas variaram entre 97,1 e 109,7% quando
detectados pelo DAD; e entre 81,5 e 112,5% quando os experimentos foram monitorados pelo
ELSD. A Tabela 14 sumariza os resultados obtidos no estudo da exatidão.
A comparação dos valores de DPR e de recuperação percentual obtido pelos
detectores mostrou melhores resultados com o uso de DAD. No entanto, os valores do ELSD
podem ser melhorados pelo ajuste de condições experimentais como, por exemplo, o fluxo do
gás nebulizante.[55]

107
Tabela 14: Dados do estudo de exatidão, intra- e inter-dias do método para quantificação de
éster diidroxilado 3-β-O-acil lupeol.
Concentração
1º Dia
2º Dia
3º Dia
Média
Recuperação (%)a
Recuperação (%)
ELSD DAD ELSD DAD ELSD DAD ELSD DAD
CQB* 97.4 97.1 81.5 104.6 93.8 100.8 90.9 100.8
CQM** 112.5 98.5 107.4 107.6 110.9 101.7 110.2 102.6
CQA*** 90.3 108.7 100.4 109.7 98.3 98.0 96.3 105.5
a Percentagem de recuperação, para n =5
* CQB (43,7 µg/mL),
** CQM (207,9 µg/mL) e
*** CQA (372,9 µg/mL)
Segundo a ANVISA, são aceitos desvios de até 15% para a precisão e exatidão nas
concentrações acima do limite de quantificação (LQ). Uma vez que os valores médios obtidos
na avaliação da precisão e exatidão estão dentro dos limites aceitáveis pela ANVISA. Os
resultados de precisão e exatidão foram considerados satisfatórios para subseqüente
determinação dos ésteres em amostras de mangaba.
5.7.5 – Estabilidade:
O grande número de experimentos a serem realizados durante o processo de
validação exige, na maioria das vezes, a realização de análises durante a noite por meio de
equipamentos automatizados. Nesse sentido, a estabilidade das amostras e dos padrões é um
importante ponto a ser averiguado. Aqui, a estabilidade foi verificada em termos de
temperatura (estabilidade após ciclos de congelamento e descongelamento) e tempo
(estabilidade de curta duração).
A comparação entre as áreas do pico do analito nas soluções recém-preparadas
com soluções depois de 24 h (em temperatura ambiente) e após três ciclos de congelamentos e
descongelamento resultou em um desvio menor que 12%. Esse fato mostrou que o éster
diidroxilado 3-β-O-acil lupeol foi estável em ambas as condições.

108
5.7.6 – Robustez:
A robustez de um método mede a sensibilidade que este apresenta em relação a
pequenas variações, ou seja, o método se mostra robusto quando não há variações
significativas na resposta do equipamento por uma pequena modificação nos parâmetros. Os
testes de robustez são de fundamental importância para que o analista conheça quais fatores
devem ser controlados na execução de uma metodologia.[113,126,129]
O método desenvolvido se mostrou robusto com relação à variação na
porcentagem do modificador orgânico da fase móvel, bem como a diferentes lotes da coluna
analítica. Contudo, a mudança no fluxo da fase móvel causou uma significativa alteração.
Este fato mostra que o controle do fluxo da fase móvel é um ponto crítico na execução do
método.
5.8 – Aplicação do método
Embora significativas diferenças entre as respostas dos dois detectores possam ser
percebidas, ambos podem ser usados para quantificar o éster diidroxilado 3-β-O-acil lupeol
em amostras de látex de mangaba, uma vez que os valores obtidos no processo da validação
estão dentro do limite aceitável pela ANVISA.
Dessa forma, o método desenvolvido foi empregado para a quantificação do éster
diidroxilado 3-β-O-acil lupeol em polpa de mangaba e no látex dos frutos liofilizado
(preparados conforme o item 4.4.2). A Tabela 15 mostra os teores do éster diidroxilado 3-β-
O-acil lupeol nas amostras de H. speciosa . Como pode ser notado, os detectores apresentaram
diferentes teores de éster diidroxilado, com variação de 1,5% na polpa e 2,4% no látex
liofilizado.

109
Tabela 15: Teor de éster diidroxilado 3-β-O-acil lupeol de polpa e látex liofilizado utilizando
DAD e ELSD.
É possível quantificar os demais ésteres de lupeol presente nas amostras de H.
speciosa utilizando o éster diidroxilado como padrão devido à similaridade estrutural entre
estes compostos.[18]
Logo, foi viável estimar a quantidade de ésteres de lupeol, até o momento
conhecidos, presente na polpa comercial de mangaba por meio da soma do teor de cada éster.
Na Figura 58 pode-se verificar os picos referentes ao éster diidroxilado (1) e a mistura de
ésteres (2-4) em um típico cromatograma da polpa de mangaba. A Tabela 16 mostra a
contribuição de cada substância para o teor total do éster quando monitorado pelo DAD e pelo
ELSD. A comparação do teor dos ésteres de lupeol em polpa comercial de mangaba entre os
dois detectores resultou na variação de 2,7%.
Figura 58: Cromatograma representativo da polpa comercial de mangaba, obtido pelo DAD
(vermelho) e pelo ELSD (preto). Picos selecionados para quantificação: (1) éster diidroxilado
3-β-O-acil lupeol e (2-4) mistura de ésteres de lupeol.
Detector Polpa
(µg/mg)
Látex liofilizado
(µg/mg)
DAD 99,3 295,00
ELSD 101,49 284,81

110
Tabela 16: Teor dos ésteres de lupeol (indicados na figura 11) em polpa comercial de
mangaba por HPLC-DAD-ELSD.
Composto Concentração (µg/mg)a
Detector
DAD ELSD
1 99,34 101,49
2 99,93 99,62
3 20,39 31,17
4 39,78 37,30
Total 259,44 269,58 a
Média da triplicata

111
Conclusão

112
6 – CONCLUSÃO
Por meio das análises por cromatografia líquida de alta eficiência acoplada ao
detector de arranjo de diodos e evaporativo por espalhamento de luz (HPLC-DAD-ELSD), foi
possível obter pela primeira vez os cromatogramas fingerprint inéditas do látex do caule de H.
speciosa. O método desenvolvido mostrou-se representativo para as amostras submetidas ao
estudo, com separação de bandas cromatográficas capazes de caracterizar quimicamente o
látex do caule da mangabeira. O procedimento de validação do método apresentou boa
precisão e excelente desempenho analítico; o mesmo permitiu também garantir a estabilidade
das soluções das amostras de látex por 48 h a temperatura ambiente, além de demonstrar os
parâmetros que precisam ser mantidos para o bom funcionamento do método.
O presente trabalho demonstrou que é possível averiguar a autenticidade das
amostras de látex do caule comercializados pela inspeção visual das características químicas
peculiares apresentadas nos cromatogramas fingerprint. Foi possível também perceber as
diferenças entre os perfis químicos das amostras analisadas provenientes dos diferentes
localidades.
A utilização da cromatografia líquida em escala semi-preparativa permitiu o
isolamento de sete compostos (HSG 2-8) com elevado grau de pureza a partir do látex do
caule. Três destes compostos tiveram suas estruturas elucidadas: ciclohexiletanóide
glicosilado cornosídeo (HSG 2), ciclohexiletanóide glicosilado dihidrocornosídeo (HSG 3) e
(7,8)-treo-4,7,9,9’-tetrahidroxi-3,3’-dimetoxi- 8-O-4’-neolignana-7-O-β-D-glicopiranosídeo
(HSG 6).
O método cromatográfico desenvolvido para a análise qualitativa dos marcadores
químicos lupeol, α-amirina, β-amirina e ésteres 3- β-O-acil lupeol no látex dos frutos de H.
speciosa e em polpa comercial de mangaba apresentou-se apropriado com adequada
separação.
Um método analítico baseado na detecção por DAD e ELSD em série foi
desenvolvido e validado para a quantificação de ésteres de lupeol em látex dos frutos e em
polpa comercial de H. speciosa utilizando o éster diidroxilado 3-β-O-acil lupeol como padrão.
Embora, uma diferença no teor de ésteres de lupeol tenha sido verificada, os resultados se
mostraram dentro da faixa aceita pela ANVISA para ambos os detectores.

113
Dessa forma, os métodos cromatográficos qualitativos e quantitativos
desenvolvidos neste trabalho podem ser usados como uma ferramenta de controle de
qualidade para amostras de H. speciosa.

114
Referências

115
7 – REFERÊNCIAS
[1] HEVERT-HERNÁNDEZ, D.; GARCÍA, O. P.; ROSADO, J. L.; GOÑI, I. The
contribuiton of fruits and vegetables to dietary intake of polyphenols and antioxidant capacity
in a Mexican rural diet: Importance of fruit and vegetable variety. Food Research
International, v. 44, n. 5, p. 1182-1189, 2011.
[2] SANTIAGO-SILVA, P.; LABANCA, R. A.; GLORIA, M. B. A. Functional potential of
tropical fruits with respect to free bioactive amines. Food Research International, v. 44, n.
5, p. 1264-1268, 2011.
[3] LIMA, V. L. A. G.; MELO, E. A.; LIMA, D. E. S. Fenólicos e carotenóides totais em
pitanga. Scientia Agricola, v. 59, n. 3, p. 447-450, 2002.
[4] BOWEN-FORBES, C.; MULABAGAL, V.; LIU, Y.; NAIR, M. G. Ursolic acid
analogues: non-phenolic functional food components in Jamaican raspberry fruits. Food
Chemistry, v. 116, n. 3, p. 633-637, 2009.
[5] DEIGHTON, N.; BRENNAN, R.; FINN, C.; DAVIES, H. V. Antioxidant properties of
domesticated and wild Rubus species. Journal of the Science of Food and Agriculture, v.
80, n. 9, p. 1307-1313, 2000.
[6] SEERAM, N. P.; MOMIN, R. A.; NAIR, M. G.; BOURQUIN, L. D. Cyclooxygenase
inhibitory and antioxidant cyaniding glycosides in cherries and berries. Phytomedicine:
International Journal of Phytotherapy and Phytopharmacology, v. 8, n. 5, p. 362-369,
2001.
[7] RUFINO, M. S. M.; ALVES, R. E.; BRITO, E. S.; PÉREZ-JIMÉNEZ, J.; SAURA-
CALIXTO, F.; MANCINI-FILHO, J. Bioactive compounds and antioxidant capacities of 18
non-traditional tropical fruits from Brazil. Food Chemistry, v.121, n. 4, p. 996-1002, 2010.

116
[8] ROBBERS, J. E.; SPEEDIE, M. K.; TYLER, V. E. Farmacognosia e
farmacobiotecnologia. Editora Editorial Premier, Araraquara-SP, p. 1-16, 1997.
[9] WAKSMUNDZKA-HAJNOS, M.; SHERMA, J. Phytochemistry, phytopharmacology,
and the biological role of plant metabolites. In: High performance liquid chromatography in
phytochemical analysis. Editora CRC Press, Taylor e Francis Group, New York, p. 90-104,
2011.
[10] YUNES, R. A.; CALIXTO, J. B. Plantas medicinais sob a ótica da química medicinal.
Editora Argos, Santa Catarina, p. 29-43, 2001.
[11] MONTANARI, C. A.; BOLZANI, V. S. Planejamento racional de fármacos baseados em
produtos naturais. Química Nova, v. 24, n. 2, p. 105-111, 2001.
[12] YUNES, R. A.; PEDROSA, R. C.; FILHO, V. C. Fármacos e fitoterápicos: a necessidade
do desenvolvimento da indústria de fitoterápicos e fitofármacos no Brasil. Química Nova, v.
24, n. 1, p. 147-152, 2001.
[13] LIANG, Y.; XIEP, P.; CHAN, K. Quality control of herbal medicines. Journal of
Chromatography B, v. 812, n. 1-2, p. 53-70, 2004.
[14] DING, Y.; WU, E. Q.; LIANG, C.; CHEN, J.; TRAN, M. N.; HONG, C. H.; JANG, Y.;
PARK, K. L.; BAE, K.; KIM, Y. H.; KANG, J. S. Discrimination of cinnamon bark and
cinnamon twig samples sourced from various countries using HPLC-based fingerprint
analysis. Food Chemistry, v. 127, n. 2, p. 755-760, 2011.
[15] FAN, X.; CHENG, Y.; YE, Z.; LIN, R.; QIAN, Z. Multiple chromatographic fingerprint
and its application to the quality control of herbal medicines. Analytica Chimica Acta, v.
555, n. 2, p. 217-224, 2006.

117
[16] JIANLIANG, Z.; LIANWEN, Q.; PING, L. Quality control of Chinese herbal medicines
with chromatographic fingerprint. Chinese Journal of Chromatography, v. 26, n. 2, p. 153-
159, 2008.
[17] MARTINS, L. R. R. Perfil cromatográfico e análise multivariada para o controle de
qualidade de amostras comerciais do gênero Phyllanthus (quebra-pedra). Tese
(Doutorado em Química) – Universidade Federal de São Carlos, 2008.
[18] THEUNIS, M. H. B. L.; FOUBERT, K.; POLLIER, J.; GONZALEZ-GUZMAN, M.;
GOOSSENS, A.; VLIENTICK, A. J.; PIETERS, L. A. C.; APERS, S. Determination of
saponins in Maesa lanceolata by LC-UV: Development and validation. Phytochemistry, v.
68, n. 22-24, p. 2825-2830, 2007.
[19] XIE, P.; CHEN, S.; LIANG, Y.; WANG, X.; TIAN, R.; UPTOM, R. Chromatographic
fingerprint analysis: a rational approach for quality assessment of traditional chinese herbal
medicine. Journal of Chromatography A, v. 1112, n. 1-2, p. 171-180, 2006.
[20] LEMOS, R. P. Caracterização fenológica e teores de nutrientes da mangabeira
(Hancornia speciosa Gomes). Areia: UFPB/CCA, 1988. 44p. Monografia (Graduação em
Agronomia) - Universidade Federal da Paraíba.
[21] LEDERMAN, I. E.; SILVA JÚNIOR, J. F.; BEZERRA, J. E. F.; ESPÍNDOLA, A.C.M.
Mangaba (Hancornia speciosa Gomes). Jaboticabal: São Paulo, 2000. 35p. (Série Frutas
Nativas, 2).
[22] INSTITUTO BRASILEIRO DE GEOGRAFIA E ESTATÍSTICA. Extração vegetal e
silvicultura, IBGE, 2009. Disponível em:
<http://www.ibge.gov.br/estadosat/temas.php?sigla=df&tema=extracaovegetal2009>
Acessado em: 05 Out. de 2011.

118
[23] MINORSKY, P. V. Overexpression of talin alters auxin-mediated responses. Plant
Physiology, v. 15, n. 1, p. 1-2, 2009. Disponível em:
<http://www.plantphysiol.org/content/151/1/1> Acessado em: 19 Ago. de 2011.
[24] TEIXEIRA, R. D.; RIBEIRO, H. A. L.; GOMES, M. R.; LOPES, M. T. P.; SALAS, C.
E. The proteolytic activities in latex from Carica candamarcensis. Plant Physiology and
Biochemistry, v. 46, n. 956, p. 956-961, 2008.
[25] SANTOS, P. O.; BARBOSA JUNIOR, A. M.; MÉLO, D. L. F. M.; TRINDADE, R. .C.
Investigação da atividade antimicrobiana do látex da mangabeira (Hancornia speciosa
Gomes). Revista Brasileira de Planta Medicinal, v. 9, n. 2, p. 108-111, 2007.
[26] SAMPAIO, T. S.; NOGUEIRA, P. C. L. Volatile components of mangaba fruit
(Hancornia speciosa Gomes) at three stages of maturity. Food Chemistry, v. 95, n. 4, p.
606-610, 2006.
[27] MATHIAS, J. A produção da fruta requer regiões quentes e com alta luminosidade, mas
vai bem mesmo em solos pobres e com carência de água. Globo Rural, Como plantar.
Disponível em: < http://revistagloborural.globo.com/GloboRural/0,6993,EEC1674241-
4529,00.html> . Acessado em: 19 Ago. de 2011.
[28] HONORATO, S. B. Efeito antioxidante de componentes do látex da seringueira e
mangabeira sobre a degradação termo-oxidativa do Poli (1,4- cis-isopreno) sintético.
Dissertação (Mestrado em Química) – Universidade Federal do Ceará, 2005.
[29] NASCIMENTO, A. M. R. Estudo do látex e fruto da sorva pela espectroscopia de
ressonância magnética nuclear. Tese (Doutorado em Ciências) – Universidade Federal do
Rio de Janeiro, 2006.
[30] CARDINAL, Á. B. B. Influência da relação enxerto vs. Porta-enxerto no aumento do
vigor e produção de clones superiores de seringueira. Dissertação (Mestrado em
Tecnologia da Produção Agrícola) – Universidade Estadual de Campinas, 2006

119
[31] AGRAWAL, A. A.; LAJEUNESSE, M. J.; FISHBEIN, M. Evolution of latex and its
constituent defensive chemistry in milkweeds (Asclepias): a phylogenetic test of plant defense
escalation. Entomologia Experimentalis et Applicata, v. 128, n. 1, p. 126-138, 2008.
[32] OLIVEIRA, A. P.; SILVA, L. R.; ANDRADE, P. B.; VALENTÃO, P.; SILVA, B. M.
GONÇALVES, R. F.; PEREIRA, J. A.; PINHO, P. G. Further Insight into the látex
metabolite profile of Ficus carica. Journal of Agricultural and Food Chemistry, v. 58, n.
20, p. 10855-10863, 2010.
[33] AMBIENTE BRASIL. Látex-Borracha vegetal. Disponível em: <
http://ambientes.ambientebrasil.com.br/florestal/artigos/latex_-_borracha_vegetal.html >
Acesso em 03/01/2011.
[34] LAGARDA, M. J.; GARCÍA-LLATAS, G.; FARRÉ ,R. Analysis of phytosterols in
foods. Journal of Pharmaceutical and Biomedical Analysis, v. 41, n. 5, p. 1486-1496,
2006.
[35] AMORIN, A.; BORBA, H. R.; CARAUTA, J. P. P.; LOPES, D.; KAPLAN, M. A. C.
Anthelmintic activity of the latex of Ficus species. Journal of Ethnopharmacology, v. 64, n.
3, p. 255-258,1999.
[36] FERNANDEZ-ARCHE, A.; SAENZ, M. T.; ARROYO, M.; DE LA PUERTA, R.;
GARCIA, M. D. Topical anti-inflammaoty effect of tirucallol, a triterpene isolated from
Euphorbia lactea látex. Phytomedicine, v. 17, n. 2, p. 146-148, 2010.
[37] MARTI, G.; APARVIER, V.; MORETTI, C.; SUSPLUGAS, S.; PRADO, S.;
GRELLIER, P.; RETAILLEAU, P.; GUÉRITTE, F.; LITAUDON, M. Antiplasmodial
benzophenones from the trunk latex of Moronobea coccinea (Clusiaceae). Phytochemistry, v.
70, n. 1, p. 75-85, 2009.

120
[38] CALLAHAN, D. L.; ROESSNER, U.; DUMONTET, V.; PERRIER, N.; WEDD, A. G.;
O’HAIR, R. A. J.; BAKER, A. J. M.; KOLEV, S. D. LC-MS and GC-MS metabolite profiling
of nickel (II) complexes in the latex of the nickel-hyperaccumulating tree Sebertia acuminate
and identification of methylated aldaric acid as a new nickel (II) ligand. Phytochemistry, v.
69, n. 1, p. 240-251, 2008.
[39] SHOYAMA, Y.; KAWACHI, F.; TANAKA, H.; NAKAI, R.; SHIBATA, T.; NISHI, K.
Genetic and alkaloid analysis of Papaver species and their F1 hybrid by RAPD, HPLC and
ELISA. Forensic Science International, v. 91, n. 3, p. 207-217, 1998.
[40] MIGUEL, M. D.; MIGUEL, O. G. Desenvolvimentos de fitoterápicos. Editora Robe
Editorial, São Paulo, 1999.
[41] MELO, J. G.; MARTINS, J. D. G. R.; AMORIM, E. L. C.; ALBUQUERQUE, U. P.
Qualidade de produtos a base de plantas medicinais comercializados no Brasil: castanha-da-
índia (Aesculus hippocastanum L.), capim-limão (Cymbopogon citratus (DC.) Stapf) e centela
(Centella asiática (L.) Urban). Acta Botanica Brasilica, v. 21, n. 1, p. 27-36, 2007.
[42] BARA, M. T. F.; RIBEIRO, P. A. M.; ARANTES, M. C.; AMORIM, L. L. S. S.;
PAULA, J. R. Determinação do teor de princípios ativos em matérias-primas vegetais.
Revista Brasileira de Farmacognosia, v. 16, n. 2, p. 211-215, 2006.
[43] CALIXTO, J. B. Efficacy, safety, quality control, marketing and regulatory guidelines
for herbal medicines (phytotherapeutic agents). Brazilian Journal of Medical and
Biological Research, v. 33, n. 2, p.1 79-189, 2000.
[44] REIF, K.; SIEVERS, H.; STEFFEN, J. P. O papel dos padrões químicos de referência
como instrumentos analíticos no estabelecimento da qualidade de materiais botânicos – Uma
perspectiva Européia. HelbalGram, v. 63, p. 38-43, 2004. Disponível em: <
http://www.abfit.org.br/p&d.shtml > Acessado em: 03/01/2012.

121
[45] MOUCO, G.; BERNARDINO, M. J.; CORNÉLIO, M. L. Controle de qualidade de ervas
medicinais. Revista Biotecnologia, Ciência e Desenvolvimento, v. 31, p. 68-73, 2003.
Disponível em: < http://www.biotecnologia.com.br/revista/bio31/bio31.pdf> Acessado em
03/01/2012.
[46] NYIREDY, S. Separation strategies of plant constituents-current status. Journal of
Chromatography B, v. 812, n. 1-2, p. 35-51, 2004.
[47] WEI, H.; SUN, L.; TAI, Z.; GAO, S.; XU, W.; CHEN, W. A simple and sensitive HPLC
method for the simultaneous determination of eight bioactive components and fingerprint
analysis of Schisandra sphenanthera. Analytica Chimica Acta, v. 662, n. 1, p. 97-104, 2010.
[48] WOLFENDER, J. L. HPLC in natural product analysis: the detection issue. Plant
Medica, v. 75, n. 7, p.719-734, 2009.
[49] LI, W.; CHEN, S.; FABRICANT, D.; ANGERHOFER, C. K.; FRONG, H. H. S.;
FARNSWORTH, N. R.; FITZLOFF, J. F. High-performance liquid chromatographic analysis
of Black Cohosh (Cimicifuga racemosa) constituents with in-line evaporative light scattering
and photodiode array detection. Analytica Chimica Acta, v. 471, n. 1, p. 61-75, 2002.
[50] YU, Q.; QI, L.; LI, P.; YI, L.; ZHAO, J.; BI, Z. Determination of seventeen main
flavonoids and saponins in the medicinal plant Huang-qi (Radix Astragali) by HPLC-DAD-
ELSD. Journal of Separation Science, v. 30, n. 9, p.1 292-1299, 2007.
[51] YE, J.; ZHANG, X.; DAI, W.; YAN, S.; HUANG, H.; LIANG, X.; LI, Y.; ZHANG, W.
Chemical fingerprint of Liuwei Dihuang Pill and simultaneous determination of its major
bioactive constituents by HPLC coupled with multiple detections of DAD, ELSD and ESI-
MS. Journal of Pharmaceutical and Biomedical Analysis, v. 49, n. 3, p. 638-645, 2009.

122
[52] WAKSMUNDZKA-HAJNOS, M.; SHERMA, J. Photodiode Array (PDA) and other
detection methods in HPLC of plant metabolites. In: High performance liquid
chromatography in phytochemical analysis. Editora CRC Press, Taylor e Francis Group, New
York, p. 331-346, 2011.
[53] MEGOULAS, N. C.; KOUPPARIS M. A. Twenty years of evaporative light scattering
detection. Critical Reviews in Analytical Chemistry, v. 35, n. 4, p. 301-316, 2005.
[54] CHAI, K.; LI, S.; Li, P. Quality evaluation of Flos Lonicerae through a simultaneous
determination of seven saponins by HPLC with ELSD. Journal of Chromatography A, v.
1070, n. 1-2, p. 43-48, 2005.
[55] Li, W.; FITZLOFF, J. F. Determinatio of 24(R)- pseudoginsenoside F11 in North
American ginseng using high performance liquid chromatography with evaporative light
scattering detection. Journal of Pharmaceutical and Biomedical Analysis, v. 25, n. 2, p.
257-265, 2001.
[56] PIETRO, A. C. Desenvolvimento de métodos cromatográficos para o controle de
qualidade e para a análise de resíduos em leite bovino de medicamentos veterinários.
Tese (Doutorado em Química) – Universidade Federal de São Carlos, 2008.
[57] RIBEIRO, S. S. Estudo fitoquímico e atividades biológicas de jatropha curcas L.
Dissertação (Mestrado em Química) – Universidade Federal de Sergipe, 2010.
[58] GOMES, S. V. F. Desenvolvimento de método por Cromatografia Líquida de Alta
Eficiência para diferenciação de genótipos de Lippia Gracilis Schauer. Dissertação
(Mestrado em Química) – Universidade Federal de Sergipe, São Cristóvão, 2009.
[59] XU, G.; YE, X.; LIU, D.; MA, Y.; CHEN, J. Composition and distribution of phenolic
acids in Pokan (Citrus poonensis Hort. Ex Tanaka) and Huyou (Citrus paradise Macf.
Changshanhuyou) during maturity. Journal of Food Composition and Analysis, v. 21, n. 5,
p. 382-389, 2008.

123
[60] OLSZEWSKA, M. A.; GUDEJ, J. Quality evaluation of golden saxifrage
(Chrysosplenium alternifolium L.) through simultaneous determination of four bioactive
flavonoids by high-performance liquid chromatography with PDA detection. Journal of
Pharmaceutical and Biomedical Analysis, v. 50, n. 5, p. 771-777, 2009.
[61] LIANG, X.; ZHANG, X.; DAÍ, W.; LV. Y.; YAN, S.; ZHANG, W. A combined HPLC-
PDA and HPLC-MS method for quantitative and qualitative analysis of 10 major constituents
in the traditional Chinese medicine. Journal of Pharmaceutical and Biomedical Analysis,
v. 49, n. 4, p. 931-936, 2009.
[62] AGUIAR, S. Morfoanatomia de frutos e sementes em Apocynaceae. Tese (Doutorado
em Biologia) – Universidade Estadual de Campinas, 2009.
[63] RAPINI, A.; KOCH, I.; KINOSHITA, L. S.; SIMÕES, A. O.; SPINA, A. P.
Apocynaceae in Lista de Espécies da Flora do Brasil. Jardim Botânico do Rio de Janeiro,
2010. Disponível em: < http://floradobrasil.jbrj.gov.br/2010/FB015557> Acessado em 01 set.
2011.
[64] JUNIOR, J. F. S. A Cultura da Mangaba. Revista Brasileira de Fruticultura, v. 26, n.
1, p. 1-192, 2004.
[65] SOUSA, C. S.; SILVA, S. A.; COSTA, M. A. P. C.; DANTAS, A. C. V. L.; FONSECA,
A. A.; COSTA, C. A. L. C.; De ALMEIDA, W. A. B.; PEIXOTO, C. P. Mangaba:
perspectivas e potencialidades. Bahia Agrícola, v. 7, n. 1, p. 29-31, 2005.
[66] FERREIRA, E. G.; MARINHO, S. J. O. Produção de frutas da mangaba para o consumo
in natura e industrialização. Tecnologia e Ciência Agropecuária, v. 1, n. 1, p. 9-14, 2007.
[67] VIEIRA NETO, R. D; CINTRA, F. L. D.; SILVA, A. L. da; SILVA JUNIOR, J. F.;
COSTA, J. L. da S.; SILVA, A. A. G. da; CUENCA, M. A. G. Sistema de Produção de
mangaba para os tabuleiros costeiros e baixadas litorânea. Aracaju: Embrapa Tabuleiros
Costeiros, 2002. Disponível em: < http//www.cpatc.embrapa.br.>. Acessado em: 18 jan. 2010.

124
[68] VIEIRA NETO, R. D.; MELO, V. e S. Caracterização dos sistemas produtivo da
mangabeira no município de Itaporanga D` Ajuda, Sergipe. In: XX CONGRESSO
BRASILEIRO DE FRUTICULTURA, 54TH
ANNUAL MEETING OF THE
INTERAMERICAN SOCIETY FOR TROPICAL HORTICULTURE, 12 a 17 de outubro de
2008 – Centro de Convenções – Vitória/ES. Disponível em: <
http://200.137.78.15/cd_XXCBF/paginas/DesenvolvimentoRegional/20080730_154145.pdf>.
Acessado em: 18 jan. 2010
[69] ROSA, M. E. C.; NAVESS, R. V.; JÚNIOR, J. P. de O. Produção e Crescimento de
Mudas de Mangabeira (Hancornia speciosa Gomes) em diferentes substratos. Pesquisa
Agropecuária Tropical, v. 35, n. 2, p. 65-70, 2005. Disponível em: <
www.revistas.ufg.br/index.php/pat/article/download/2252/2211>. Acessado em: 18 jan. 2010.
[70] SOARES, F. P.; PAIVA, R.; ALVARENGAS, A. A.; NOGUEIRA, R. C., EMRICH, E.
B.; MARTINOTTO,C. Organogênese direta em explantes caulinares de mangabeira
(Hancornia speciosa Gomes). Ciência e Agrotecnologia, v. 31, n. 4, p. 1049-1053, 2007.
Disponível em: <http://www.scielo.br/pdf/cagro/v31n4/16.pdf>. Acessado em: 18 jan 2010.
[71] LÉDO, A. da S.; SECA, G. S. V.; BARBOZA, S. B. S. C.; JUNIOR, J. F. S. Crescimento
inicial de mangabeira (Hancornia speciosa Gomes) em diferentes meios de germinação in
vitro. Ciência e Agrotecnologia, v. 31, n. 4, p. 989-993, 2007. Disponível em:
<http://www.scielo.br/pdf/cagro/v31n4/07.pdf.>. Acessado em: 18 jan. 2010.
[72] FONSECA, E. L.; CONDÉ, R. C. C.; SILVA, J. A. Influência da profundidade de
semeadura e da luminosidade na germinação de sementes de mangaba (Hancornia speciosa
Gomes). Pesquisa Agropecuária Brasileira, v. 29, n. 4, p. 661-666, 1994.
[73] SOUZA, L. A. C.; SILVA, S. M. C.; NASCIMENTO, J. L. Desenvolvimento vegetativo
e produção de mangaba (Hancornia speciosa Gomes), sob as condições de irrigação e
adubação. In: 1º SEMANA DE INICIAÇÃO CIENTÍFICA DA EMBRAPA ARROZ E
FEIJÃO E XII SEMANA DE INICIAÇÃO CIENTÍFICA DA UNIVERSIDADE FEDERAL
DE GÓIAS, 2004, Góias. Anais... Goiás: UFG, 2004 p. 102-105.

125
[74] LOBO, F. A.; JUNIOR, J. H. C.; RODRÍGUEZ-ORTÍZ, C. E.; LUCENA, I. C.;
VOURLITIS, G. L. Leaf and fruiting phenology and gás Exchange of Mangabeira in response
to irrigation. Brazilian Journal of Plant Physiology, v. 20, n. 1, p. 01-10, 2008.
[75] SOUZA, F. G.; FIGUEIREDO, R. W.; ALVES, R. E.; MAIA, G. A.; ARAÚJO, I. A.
Qualidade pós-colheita de frutos de diferentes clones de mangabeira (Hancornia speciosa
Gomes). Ciência e Agrotecnologia, v. 31, n. 5, p. 1449-1454, 2007.
[76] SOUZA, F. G.; VIROLL, S. L. M. Qualidade pós-colheita de um clone de
mangabeira (Hancornia speciosa Gomes). In: IV CONGRESSO CIENTÍFICO DO
CEULP/ULBRA. Disponível em: <
http://paraiso.etfto.gov.br/docente/admin/upload/docs_upload/material_6eb1d56ff8.pdf>.
Acessado em: 18 jan. 2010.
[77] GANGA, R. M. D.; CHAVES, L. J.; NAVES, R. V. Parâmetros genéticos em progênies
de Hancornia speciosa Gomes do cerrado. Scientia Forestalis, v. 37, n. 84, p. 395-404, 2009.
[78] PEREIRA, M. M.; JÁCOME, R. L. R. P.; ALCÂNTARA, A. F. C.; ALVES, R. B.;
RASLAN, D. S. Alcalóide indólicos isolados de espécies do gênero Aspidosperma
(Apocynaceae). Química Nova, v. 30, n. 4, p. 970-983, 2007.
[79] THIBEAULT, D.; GAUTHIER, C.; LEGAULT, J.; BOUCHARD, J.; DUFOUR, P.;
PICHETTE, A. Synthesis and structure-activity relationship study of cytotoxic germanicane-
and lupine-type 3β-O-monodesmosidic saponins starting from betulin. Bioorganic &
Medicinal Chemistry, v. 15, n. 18, p. 6144-6157, 2007.
[80] OTUKI, M. F.; FERREIRA, J.; LIMA, F. V.; MEYRE-SILVA, C.; MALHEIROS, Â.;
MULLER, L. A.; CANI, G. S.; SANTOS, A. R. S.; YUNES, R. A.; CALIXTO, J. B.
Antinociceptive properties of mixture of α-amyrin and β-amyrin triterpenes: evidence for
participation of protein kinase C and protein kinase A pathways. The Journal of
Pharmacology and Experimental Therapeutics, v. 313, p. 310-318, 2005.

126
[81] CHATURVEDI, P. K.; BHUI, K.; SHUKLA, Y. Lupeol: Connotations for
chemoprevention. Cancer Letters, v. 263, n.1, p. 1-13, 2008.
[82] SUDHARSAN, P. T.; MYTHILI, Y.; SELVAKUMAR, E.; VARALAKSHMI, P.
Lupeol and its ester ameliorate the cyclophosphamide provoked cardiac lysosomal damage
studied in rat. Molecular and Cellular Biochemistry, v. 282, n. 1-2, p. 23-29, 2006.
[83] GOMES, W. F. Estudo químico de recursos naturais renováveis do estado de
Sergipe: Hancornia speciosa Gomes. Dissertação (Mestrado em Química)- Departamento de
Química, Universidade Federal de Sergipe, São Cristóvão, 2008.
[84] CARVALHO, M. G.; VELLOSO, C. R. X.; BRAZ-FILHO, R.; COSTA, W. F. Acyl-
lupeol esters from Parahancornia amapa (Apocynaceae). Journal of the Brazilian
Chemical Society, v. 12, n. 4, p. 556-559, 2001.
[85] ENDRINGER, D. C.; PEZZUTO, J. M.; BRAGA, F. C. NF-kB inhibitory activity of
cyclitols isolated from Hancornia speciosa. Phytomedicine, v. 16, n. 11, p. 1064-1069, 2009.
[86] SAMPAIO, T. S. Estudo fitoquímico de Hancornia speciosa Gomes: isolamento,
determinação estrutural e atividade biológica. Dissertação (Mestrado em Química) –
Universidade Federal de Sergipe, São Cristóvão, 2008.
[87] MORAES, T. M.; RODRIGUES, C. M.; KUSHIMA, H.; BAUAB, T. M.; VILLEGAS,
W.; PELLIZZON, C. H.; BRITO, A. R. M. S.; HIRUMA-LIMA, C. A. Hancornia speciosa:
Indications of gastroprotective, healing and anti-Helicobacter pylori actions. Journal of
Ethnopharmacology, v. 120, n. 2, p. 161-168, 2008.
[88] RODRIGUES, C. M.; RINALDO, D.; SANTOS, L. C.; MONTORO, P.; PIACENTE, S.;
PIZZA, C.; HIRUMA-LIMA, C. A.; BRITO, A. R. M.; VILEGAS, W.Metabolic fingerprint using
direct flow injection electrospray ionization tandem mass spectrometry for the characterization of
proanthocyanidins from the barks of Hancornia speciosa. Rapid Communications in Mass
Spectrometry, v. 21, n. 12, p. 1907-1914, 2007.

127
[89] SOARES, F. P. Cultura da Mangabeira (Hancornia speciosa Gomes). Boletim
Agropecuário. Lavras/MG, n. 67, p. 1-12. Disponível em:
<http://www.editora.ufla.br/BolTecnico/pdf/bol_67.pdf>. Acessado em: 18 jan. 2009.
[90] FERREIRA, H. C.; SERRA, C. P.; ENDRINGER, D. C.; LEMOS, V. S.; BRAGA, F. C.;
CORTES, S. F. Endothelium-dependent vasodilation induced by Hancornia speciosa in rat
superior mesenteric artery. Phytomedicine, v. 14, n. 7-8, p. 473-478, 2007 a.
[91] SERRA, C.P.; CÔRTES, S. F.; LOMBARDI, J. A.; OLIVEIRA, A. B.; BRAGA, F. C.
Validation of a colorimetric assay for the in vitro screening of inhibitors of angiotensin-
converting enzyme (ACE) from plant extracts. Phytomedicine, v. 12, n. 6-7, p. 424-432,
2005.
[92] FERREIRA, H.C.; SERRA, C.P.; LEMOS, V.S.; BRAGA, F.C.; CORTES, S.F. Nitric
oxide-dependent vasodilatation by ethanolic extract of Hancornia speciosa via phosphatidyl-
inositol 3-kinase. Journal of Ethnopharmacology, v. 109, n. 1, p. 161-164, 2007 b.
[93] COSTA, E. S.; HIRUMA-LIMA, C. A.; LIMA, E. O.; SUCUPIRA, G. C.; BERTOLIN,
A. O.; LOLIS, S. F.; ANDRADE, F. D. P.; VILEGAS, W.; SOUZA-BRITO, A. R. M.
Antimicrobial activity of some medicinal plants of the cerrado, Brazil. Phytotherapy
Research, v. 22, n. 5, p. 705-707, 2008.
[94] SANTOS, P. O.; BARBOSA JUNIOR, A. M.; MELO, D. L. F. M.; TRINDADE, R. C.
Investigação da atividade antimicrobiana do látex da mangabeira (Hancornia speciosa
Gomes). Revista Brasileira de Planta Médica, v. 9, n. 2, p. 108-111, 2007. Disponível em: <
http://www.ibb.unesp.br/servicos/publicacoes/rbpm/pdf_v9_n2_2007/artigo14_v9n2_108-
111.pdf> Acessado em 23 out. 2011
[95] MARINHO, D. G.; ALVIANO, D. S.; MATHEUS, M. E.; ALVIANO, C. S.;
FERNANDES, P. D. The látex from Hancornia speciosa Gomes possesses anti-inflammatory
activity. Journal of Ethnopharmacology, v. 153, n. 2, p. 530-537, 2011.

128
[96] TRINDADE, R. C.; RESENDE, M. A.; SILVA, C. M.; ROSA, C. A. Yeasts associated
with fresh and frozen pulps of Brazilian tropical fruits. Systematic and Applied
Microbiology, v. 25, p. 294-300, 2002.
[97] ARAÚJO, C. L.; BEZERRA, I. W. L.; DANTAS, I. C.; LIMA, T. V. S.; OLIVEIRA, A.
S.; MIRANDA, M. R. A.; LEITE, E. L.; SALES, M. P. Biological activity of proteins of
pulps of tropical fruits. Food Chemistry, v. 85, n. 1, p. 107-110, 2004.
[98] UFMG. Universidade Federal de Minas Gerais (MG). Denise Coutinho Endringer,
Fernão Castro Braga, Grazielle Caroline da Silva, Steyner de França Côrtes. Extrato e fração
padronizados de folhas de Hancornia speciosa e sua composição farmacêutica. BR n.
PI0802004-3 A2, 19 maio 2008, 12 jan. 2010.
[99] MALMONGE, J. A.; CAMILO, E. C.; MORENO, R. M. B.; MCMAHAN, C. M.;
MATTOSO, L. H. C. Comparative study on the technological properties of látex and natural
rubber from Hancornia speciosa Gomes and Hevea brasiliensis. Journal of Applied
Polymer Science, v. 111, n. 6, p. 2986-2991, 2008.
[100] MEDEIROS, E. S.; GALIANI, P. D.; MORENO, R. M. B.; MATTOSO, L. H. C.;
MALMONGE, J. A. A comparative study of the non-isothermal degradation of natural rubber
from Mangabeira (Hancornia speciosa Gomes) and Seringueira (Hevea brasiliensis).Journal
of Thermal Analysis Calorimetry, v. 100, n. 1, p. 1045-1050, 2010.
[101] ALAERTS, G.; MATTHIIS, N.; SEMEYERS-VERBEKE, J.; HEYDEN, Y. V.
Chromatographic fingerprint development for herbal extracts: A screening and optimization
methodology on monolithic columns. Journal of Chromatography A, v. 1172, n. 1, p. 1-8,
2007.
[102] SNYDER, L. R.; DOLAN, J. W. Initial experiments in high-performance liquid
chromatographic method development – use of a starting gradient run. Journal of
Chromatography A, v. 721, n. 1, p.3-14, 1996.
[103] SNYDER, L. R.; KIRKLAND, J. J.; GLAJCH, J. L. The Colunm. In: Pratical HPLC
Method Development. New York: John Wiley e Sons, 2. Ed.; p. 175-230, 1997.

129
[104] NEUE, U. D.; O`GARA, J. E.; MÉNDEZ, A. Selectivity in reversed-phase separations
– influence of the stationary phase. Journal of Chromatography A, v. 1127, n. 1-2, p. 161-
174, 2006.
[105] SNYDER, L. R.; KIRKLAND, J. J.; GLAJCH, J. L. Non-ionic samples: reversed- and
normal-phase HPLC. In: Pratical HPLC Method Development. New York: John Wiley e
Sons, 2. Ed.; p. 233-291, 1997.
[106] ZHENG, Y.; XIN, Y.; GUO, Y. Study on the fingerprint profile of Monascus products
with HPLC-FD, PDA and MS. Food chemistry, v. 113, n. 2, p. 705-711, 2009.
[107] SNYDER, L. R.; KIRKLAND, J. J.; GLAJCH, J. L. Ionic Samples: reversed-phase,
ion-pair and ion-exchange HPLC. In: Pratical HPLC Method Development. New York: John
Wiley e Sons, 2. Ed.; p.292-316, 1997.
[108] SNYDER, L. R.; KIRKLAND, J. J.; GLAJCH, J. L. Ionic Samples: reversed-phase,
ion-pair and ion-exchange HPLC. In: Pratical HPLC Method Development. New York: John
Wiley e Sons, 2. Ed.; p. 299, 1997.
[109] SNYDER, L. R.; KIRKLAND, J. J.; GLAJCH, J. L. Computer-assited method
development. In: Pratical HPLC Method Development. New York: John Wiley e Sons, 2. Ed.;
p.439-478, 1997.
[110] BURANOV, A. U.; ELMURADOV, B. J. Extraction and Characterization of latex and
natural rubber from rubber-bearing plants. Journal of Agricultural and Food Chemistry, v.
58, n. 2, p. 734-743, 2010
[111] MARTINS, L. R. R.; PEREIRA-FILHO, E. R.; CASS, Q. B. Chromatographic profiles
of Phyllanthus aqueous extracts samples: a proposition of classification using chemometric
models. Analytical and Bioanalytical Chemistry, v. 400 , n.2 , p. 469-481, 2011.

130
[112] CHEN, Y.; YAN, Y. XIE, M. Y; NIE, S. P.; LIU, W.; GONG, X. F.; WANG, Y. X.
Development of a chromatographic fingerprint for the chloroform extracts of Ganoderma
lucidum by HPLC and LC-MS. Journal of Pharmaceutical and Biomedical Analysis, v. 47,
n. 3 , p. 469-477, 2008.
[113] BRASIL. Ministério da Saúde. Agência Nacional de Vigilância Sanitária. Resolução
RE nº 899. Guia para Validação de Métodos Analíticos e Bioanalíticos. 29 de maio de 2003.
[114] Kim, D.-H.; Oh, Y.-J.; Han, K.-M.; Chung, I.-S..; Kim, D.-K.; Kim, S.-H.; Kwon, B.-M.;
Park, M.-H.; Baek, N.-I. Development of biologically active compounds from edible plant sources
XIV. Cyclohexylethanoids from the flower of Campsis grandiflora K. Schum. Agricultural
Chemistry and Biotechnology, v.48, n.1, p.35-37, 2005. (1H a 400 MHz,
13C a 100 MHz,
CD3OD). Deslocamentos químicos (δ) em ppm.
[115] De Carvalho, M.G.; de Albuquerque, L.A.; Alves, C.C.F.; Cascon, V. Cornoside and other
constituents from the latex of Parahancornia amapa (Hub.) Ducke (Apocynaceae) a medicinal
plant in Northern Brazil. Revista Brasileira de Farmacognosia, v.18, supl., p.667-669, 2008.
[116] Hase, T.; Kawamoto, Y.; Ohtani, K.; Kasai, R.; Yamasaki, K.; Picheansoonthon, C.
Cyclohexylethanoids and related glucosides from Millingtonia hortensis. Phytochemistry, v. 39,
n.1, p.235-241, 1995.
[117] Kuwajima, H.; Takai, Y.; Takaishi, K.; Inoue, K. Synthesis of 13C-labeled possible
intermediates in the biosynthesis of phenylethanoid derivatives, cornoside and rengyosides.
Chemical and Pharmaceutical Bulletin, v.46, n.4, p.581-586, 1998.
[118] Matsuda, N.; Kikuchi, M. Studies on the constituents of Lonicera species. X. Neolignan
glycosides from the leaves of Lonicera gracilipes var. glandulosa Maxim. Chemical and
Pharmaceutical Bulletin, v.44, n.9, p.1676-1679, 1996.
[119] Cai, W.-H.; Matsunami, K.; Otsuka, H.; Shinzato, T.; Takeda, Y. Lignan and neolignan
glucosides, and tachioside 2’-O-4”-O-methylgallate from the leaves of Glochidion rubrum. Journal
of Natural Medicines, v.63, p. 408-414, 2009.

131
[120] CHEN, X.; ZAN, K.; YANG, J.; LIU, X.; MAO, Q.; ZHANG, L.; LAI, M.; WANG, Q.
Quantitative analysis of triterpenoids in different parts of Ilex hainanensis, Ilex stewardii and
Ilex pubescens using HPLC-ELSD and HPLC-MS and antibacterial activity. Food
Chemistry, v. 126, n. 3, p. 1454-1459, 2011.
[121] MARTELANC, M.; VOVK, I. SIMONOVSKA, B. Determination of three major
triterpenoids in epicuticular wax of cabbage (Brassica oleracea L.) by high-performance
liquid chromatography with UV and mass spectrometric detection. Journal of
Chromatography A, v. 1164, n. 1-2, p. 145-152, 2007.
[122] MARTELANC, M.; VOVK, I. SIMONOVSKA, B. Separation and identification of
some common isomeric plant triterpenoids by thin-layer chromatography and high-
performance liquid chromatography. Journal of Chromatography A, v. 1216, n. 38, p.
6662-6670, 2009.
[123] MORITA, M.; SHIBUYA, M.; KUSHIRO, T.; MASUDA, K.; EBIZUKA,Y. Molecular
cloning and functional expression of triterpene synthases from pea (Pisum sativum).
European Journal of Biochemistry, v. 267, n. 12, p. 3453-3460, 2000.
[124] LI, G.; ZHANG, X.; YOU, J.; SONG, C.; SUN, Z.; XIA, L.; SUO, Y. Highly sensitive
and selective pre-column derivatization HPLC approach for rapid determination of triterpenes
oleanolic and ursolic acids and application to Swertia species: Optimization of triterpenic
acids extraction and pre-column derivatization using response surface methodology.
Analytica Chimica Acta, v. 668, n. 2, p. 208-218, 2011.
[125] WANG, H.; WANG, Z.; GUO, W. Comparative determination of ursolic acid and
oleanolic acid of Macrocarpium officinalis (Sieb. et Zucc.) Nakai by RP-HPLC. Industrial
Crops and Products, v. 28, n. 3, p. 328-332, 2008.
[126] RIBANI, M.; BOTTOLI, C. B. G.; COLLINS, C. H.; JARDIM, I. C. S. F.; MELO, L.
F. C. Validação em métodos cromatográficos e eletroforéticos. Química Nova, v. 27, n. 5, p.
771-780, 2004.

132
[127] CASSIANO, N. M.; BARREIRO, J. M.; MARTINS, L. R. R.; OLIVEIRA, R. V.;
CASS, Q. B. Validação em métodos cromatográficos para análises de pequenas moléculas em
matrizes biológicas. Química Nova, v.32, n.4, p. 1021-1030, 2009.
[128] BABU, M. K. M. Simultaneous separation and quantitation of four antiepileptic drugs
– a study with potential for use in patient drug level monitoring. Journal of Pharmaceutical
and Biomedical Analysis, v. 34, n. 2, p. 315-324, 2004.
[129] BRASIL. Instituto Nacional de Metrologia, Normalização e Qualidade Industrial
(INMETRO). Orientações sobre validação de métodos de ensaios químicos. DOQ-CGCRE-
008, 2003.