Embed Size (px)
Citation preview

Registro de medicamentos novos,
similares e genéricos
RDC 60/201422/08/2015
1

2
Vanessa Rodrigues Lopes
Farmacêutica, graduada pela Faculdade de Ciências Farmacêuticas da Universidade de São Paulo
– USP, São Paulo com especialização em fármacos e medicamentos. Possui vários cursos de
especialização nas áreas de Validação Analítica, Estabilidade, Controle de Qualidade e Assuntos
Regulatórios por associações independentes. Experiência de 10 anos adquirida nas empresas
Bristol-Myers Squibb nas áreas de controle e garantia de qualidade e Eurofarma na área de
assuntos regulatórios onde atua como Especialista em assuntos regulatórios, como link entre as
áreas técnicas e regulatória na submissão de novos projetos, resposta à exigências e
treinamentos técnicos internos. Responsável pelos contatos técnicos com a ANVISA para desenho
de projetos e participação ativa nas discussões de entidades para revisão da legislação técnica
atual.

Contextualização Histórica
Brasileira de registro3

Contexto Brasileiro4
1996: Retorno do reconhecimento brasileiro do direito de patentes
Até 1999: Medicamentos inovadores e medicamentos similares
Fabricantes escolhiam o medicamento referência para suas cópias;
Não era necessário comprovação de equivalência terapêutica;
Era permitido registro de formas farmacêuticas e dosagens diferentes
em relação ao medicamento referência escolhido;
Similares identificados por marca ou denominação geral;

Contexto Brasileiro5
1999:
Janeiro: Criação da Agência Nacional de Vigilância Sanitária (Anvisa)
- Lei n° 9782, de 26/01/1999
Fevereiro: Publicação da Lei dos Genéricos - Lei n° 9787, de
10/02/1999
– Introduz conceitos de equivalência farmacêutica,
bioequivalência e intercambialidade;
– Estabelece um novo padrão para o desenvolvimento e o registro
de medicamentos no Brasil
– Estabelece preço para produtos genéricos (mínimo 35% menores
que os medicamentos de referência);

Evolução regulatória
e
Pilares do medicamento6

7
Lei 6360/76
Lei 9787/99
RDC 135/03(Genéricos)
RDC 134/03(Adequações)
RDC 133/03(Similar)
RDC 136/03(Novo)
RDC 16/07(Genéricos)
RDC 17/07(Similar)
RDC 60/14(Registro)
Registro de
medicamentos sintéticos
e semi-sintéticos

Qualidade
Segurança
Eficácia
8Novo/Inovação Genérico/Similar
Desenvolvimento, BPF, Controle de qualidade, validação analítica, qualificações, treinamentos
Desenvolvimento, BPF, Controle de qualidade, validação analítica, qualificações, treinamentos
Estudos toxicológicos, préclínicos e clínicos fase I e II(BDR se inovador incremental)
Equivalência Farmacêutica eBE comparativa ao medicamento de referência
Estudos clínicos de fase III Equivalência Farmacêutica eBE comparativa ao medicamento de referência

Organograma ANVISA9
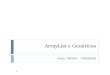
Medicamento novo (radical)10

Genéricos/Similar - Fluxo11
Consultar lista medicamento
referência
Desenvolvimento do produto
Estudos de estabilidade acelerada
RE 01/2005
Avaliação da equivalência in vitro
ao referência
RDC 31/2010
Estudos de Bioequivalência
RE 1170/2006
Estudos de estabilidade de longa
duração
RE 01/2005
Montagem do dossiê
RDC 60/2014Submissão regulatória
Exigências Aprovação

Ciclo de vida – novo/genérico/similar
Submissão• RDC 60/2014
Exigência
Publicação
HMP• RDC 48/2009 – CP 18/2015 (anualmente)
Alterações pós registro
• RDC 48/2009 – CP 18/2015 (sempre que alteração)
Renovação
• RDC 60/2014 (a cada 5 anos)
12

RDC 60/2014
Registro de medicamentos
13

RDC 60/2014 – 13/10/2014
Dispõe sobre os critérios para a concessão e renovação do registro de
medicamentos com princípios ativos sintéticos e semissintéticos,
classificados como novos, genéricos e similares, e dá outras
providências.
Vigência: 90 dias após sua publicação em DOU - 11/01/2015
Substitui integralmente as seguintes normas:
1. RDC 136/2003 – Registro e renovação de medicamentos novos
2. RDC 16/2007 – Registro e renovação de medicamentos genéricos
3. RDC 17/2003 – Registro e renovação de medicamentos similares
14

Organização da norma
Capítulo I – Disposições Iniciais: Objetivos, abrangência e definições
Capítulo II – Disposições Gerais
Capítulo III – Requisitos Gerais para o Registro
1. Das medidas Antecedentes ao registro de Medicamento novo
2. Das medidas Antecedentes ao Registro de Medicamento Genérico e Similar
3. Da documentação administrativa
4. Da documentação técnica da qualidade
Sobre o IFA
Sobre o desenvolvimento da formulação
Sobre o produto terminado
Sobre a produção do produto terminado
Sobre o CQ das matérias primas
Sobre o CQ do produto terminado
Sobre embalagem (primaria, secundária e funcional)
Sobre o envoltório intermediário

Organização da norma
Capítulo IV - dos requisitos específicos para o registro de medicamento novo
1. Do registro de medicamento novo
2. Do registro de nova associação
3. Do registro de nova associação em dose fixa
4. Do registro de nova forma farmacêutica
5. Do registro de nova concentração
6. Do registro de nova via de administração
7. Do registro de nova indicação terapêutica
8. Do Registro de Medicamento com Mesmo(s) IFA(s) de Medicamento Novo já Registrado;
9. Dos Estudos de Biodisponibilidade Relativa
Capítulo V – Dos requisitos específicos para o registro de medicamento
genérico e similar
1. Dos Estudos de Equivalência Farmacêutica e Perfil de Dissolução;
2. Dos Estudos de Bioequivalência;
Capítulo VI – Da renovação de registro
Capítulo VII – Das disposições finais e transitórias

Divisão da normaMedidas
antecedentes
Documentação administrativa
Documentação da qualidade
Requisitos específicos
Submissão do registro
17

Capítulo I
Disposições Iniciais: Objetivos,
abrangência e definições18

Conceitos
MEDICAMENTO NOVO:
Medicamento com Insumo Farmacêutico Ativo (IFA) não registrado no
país, seus novos sais, isômeros ou mistura de isômeros, ésteres, éteres,complexos ou demais derivados igualmente não registrados;
Inovação radical - desenvolvimento de nova molécula não registrada
no país;
Inovação incremental - desenvolvimento de melhorias em relação a
um medicamento já registrado no país;
19

Conceitos
INOVAÇÕES INCREMENTAIS DA RDC 60/2014:
Nova associação (kit)
Nova associação em dose fixa
Nova forma farmacêutica
Nova concentração
Nova via de administração
Nova indicação terapêutica
Medicamento com Mesmo(s) IFA(s) de Medicamento
Novo já Registrado;
20

Conceitos
MEDICAMENTO DE REFERÊNCIA (Inovador ou incremental):
Produto inovador registrado no órgão federal responsável pela
vigilância sanitária e comercializado no País, cuja eficácia,
segurança e qualidade foram comprovadas cientificamente junto
ao órgão federal competente, por ocasião do registro.” (Lei nº.
9.787, de 10/2/1999 e RDC 60/2014 de 13/10/2014)
21
Eleito pela ANVISA

Conceitos
MEDICAMENTO GENÉRICO:
medicamento similar a um produto de referência ou inovador, que
se pretende ser com este intercambiável, geralmente produzido
após a expiração ou renúncia da proteção patentária ou de
outros direitos de exclusividade, comprovada a sua eficácia,
segurança e qualidade, e designado pela DCB ou, na sua
ausência, pela DCI.” (Lei nº. 9.787, de 10/2/1999 e RDC 60/2014 de
13/10/2014)
22

Conceitos
MEDICAMENTO SIMILAR:
Aquele que contém o mesmo ou os mesmos princípios ativos,
apresenta a mesma concentração, forma farmacêutica, via de
administração, posologia e indicação terapêutica, e que é
equivalente ao medicamento registrado no órgão federal responsável
pela vigilância sanitária, podendo diferir somente em características
relativas ao tamanho e forma do produto, prazo de validade,
embalagem, rotulagem, excipientes e veículos, devendo sempre ser
identificado por nome comercial ou marca; (Medida Provisória nº
2.190-34, de 2001 e RDC 60/2014 de 13/10/2014);
23

Conceitos
PRODUTO FARMACÊUTICO INTERCAMBIÁVEL:
equivalente terapêutico de um medicamento de referência,
comprovados, essencialmente, os mesmos efeitos de eficácia e
segurança (Lei nº. 9.787, de 10/2/1999)
Dois medicamentos são considerados terapeuticamente equivalentes se
eles são equivalentes farmacêuticos e, após administração na mesma
dose molar, seus efeitos em relação à eficácia e segurança são
essencialmente os mesmos, o que se avalia por meio de estudos de
bioequivalência apropriados
24

ConceitosEQUIVALENTES FARMACÊUTICOS:
Medicamentos que possuem mesma forma farmacêutica, mesma via de
administração e mesma quantidade da mesma substância ativa, isto é,
mesmo sal ou éster da molécula terapêutica
Devem cumprir com os mesmos requisitos da monografia individual da
Farmacopeia Brasileira, preferencialmente, ou com os de outros
compêndios oficiais, normas ou regulamentos específicos
aprovados/referendados pela Anvisa ou, na ausência desses, com outros
padrões de qualidade e desempenho.
Formas farmacêuticas de liberação modificada que requerem reservatório
ou excesso podem conter ou não a mesma quantidade da substância
ativa, desde que liberem quantidades idênticas da mesma substância
ativa em um mesmo intervalo posológico (RDC 31/2010 e RDC 60/2014)
25

Conceitos
BIOEQUIVALÊNCIA:
Demonstração de biodisponibilidade equivalente entre
produtos, quando estudados sob um mesmo desenho
experimental
BIODISPONIBILIDADE:
Velocidade e extensão de absorção de um princípio ativo
proveniente de uma forma farmacêutica, a partir de sua curva
concentração/tempo na circulação sistêmica ou sua excreção
na urina, medida com base no pico de exposição e na
magnitude de exposição ou exposição parcial (RDC nº 60/2014)
26

Medicamento
RDC 60/14
Princípio AtivoRDC 45/2012 Estabilidade
Excipientes
Embalagem
RDC 71/09 Rotulagem
RDC 47/09 Bula
Estudos in vitro
RDC 31/10 EQFAR
RE 01/2005 Estabilidade
RDC 899/03 Validação
RDC 58/13 Impurezas
NT 03/13 Dissolução
Estudos in vivo
RE 09/15 DDCM
RE 1170 BE
BPF
RDC 17/10
27

Capítulo II
Disposições Gerais28

Informações gerais
Todos os documentos encaminhados em via impressa, numerados e rubricados;
Documentos em português, inglês ou espanhol;
Documentos oficiais (BPF, CPP): Tradução juramentada
Apresentados na ordem descrita na regulamentação
Check list de documentos por petição:
https://www9.anvisa.gov.br/peticionamento/sat/Consultas/ConsultaAssunto
Persistir.asp
Documento em pdf com busca textual (OCR) e marcadores
Inovação radical: peticionamento eletrônico
Apresentações em conformidade com regime posológico/indicação
Dados de estudo clínico: Enviar todas as referências (favoráveis ou não)
29

Informações gerais
Tamanho de lote = utilizado nos estudos de eficácia (biolote)
Genéricos/Similares: Tamanho do lote de bioequivalência
Novo/Inovadores: Tamanho do lote dos estudos clínicos
Permitido registrar faixa de tamanho de lote:
Tamanho de lote “até dez vezes” (biolote: 100.000 unidades)
Perfil de dissolução comparativo com o biolote
Protocolo do estudo de estabilidade
Histórico de alterações (novos/inovadores)
Permitido inclusão de local de fabricação do ativo ou do acabado:
Perfil de dissolução comparativo com o biolote
Relatório do estudo de estabilidade
30

Farmacovigilância
Dados de farmacovigilância protocolados direto à área
Medicamento não comercializado em outros países: Plano de
farmacovigilância/Minimização de riscos
Medicamento já comercializado em outros países: relatório de
farmacovigilância atualizado
31

ANVISA - poderes
Art. 12. A Anvisa poderá, a seu critério e mediante justificativa
técnica, exigir provas adicionais de qualidade de
medicamentos e requerer novos estudos para comprovação de
qualidade, segurança e eficácia.
§ 1º A Anvisa poderá solicitar à empresa os dados brutos dos
ensaios clínicos e não clínicos, assim como os dados de qualidade
do medicamento.
§ 2º A exigência de provas adicionais poderá ocorrer mesmo após
a concessão do registro.
32

Capítulo III
Dos requisitos gerais para o registro
1. Medidas antecedentes ao registro
2. Documentação administrativa
3. Documentação técnica da qualidade
33

1. Medidas Antecedentes ao
registro de medicamento novo
34

Medicamento novo (radical)35

Fases da pesquisa clínica36
http://www.bndes.gov.br/SiteBNDES/export/sites/default/bndes_pt/Galerias/Arquivos/conhecimento/bnset/set3602.pdf
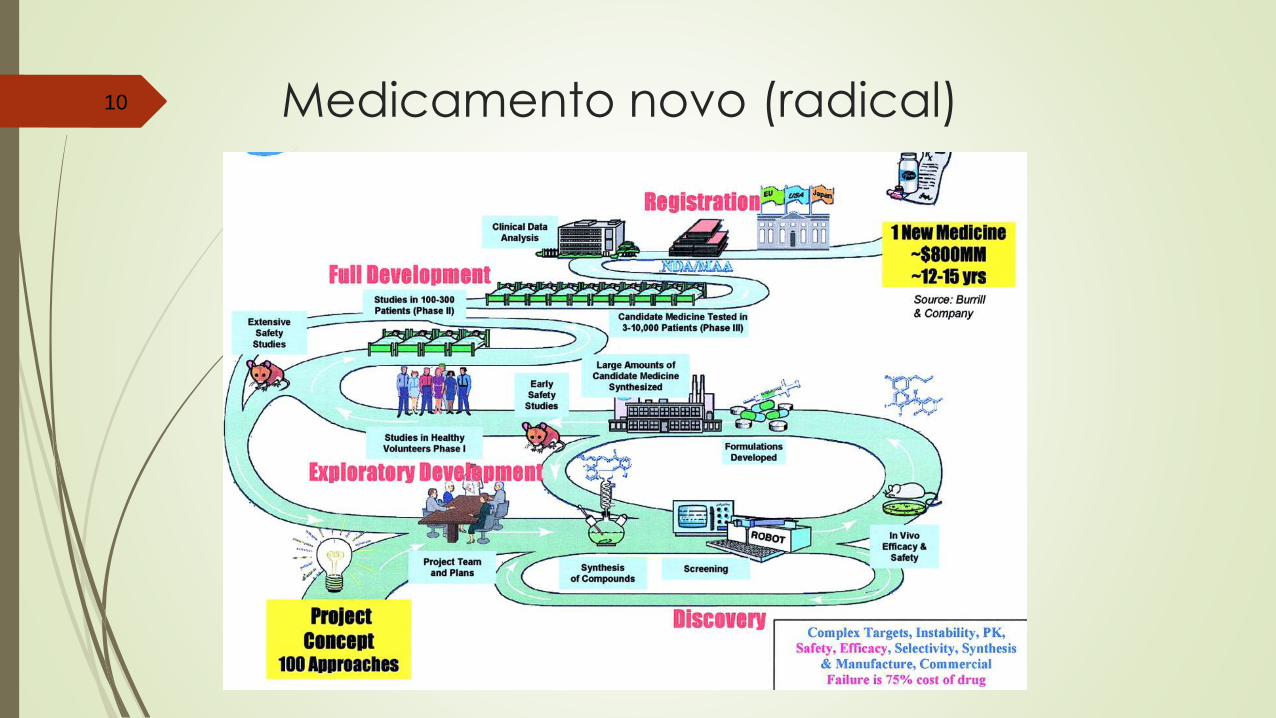
ANVISA – fluxo estudos clínicos37
CEP: Comitê de ética em pesquisa
CONEP: Comitê Nacional de ética em pesquisa
DDCM: Dossiê de desenvolvimento clínico de medicamento
DDCM
18
0 d
ias
(B
ras
il)
90
dia
s (
mu
lti)

Estrutura DDCM e norma anterior38

DDCM – IFA
1. Características físico-químicas e organolépticas;
2. Método geral de obtenção
a. Informações gerais
b. Processo de fabricação
c. Metodologia analítica validade
d. Especificações/limites aceitáveis
e. Resultados de estudo de estabilidade
‒ Completos para EC de fase III
‒ Suficientes para EC de fase I e II
39

DDCM – Medicamento Experimental
1. Lista de todos os componentes e composição quantitativa;
2. Descrição geral do processo de fabricação e embalagem;
a. Informações gerais
b. Informações sobre as etapas de fabricação
c. Informações sobre os excipientes
‒ Excipientes farmacopeicos
‒ Excipientes novos
d. Embalagem
3. Metodologia analítica e limites aceitáveis
‒ Fase I – verificação de métodos
‒ Fase II e III – validação completa
4. Resultados do estudo de estabilidade
40

DDCM – Placebo
1. Composição;
2. Características organolépticas;
3. Processo de fabricação;
4. Controles analíticos;
41

DDCM – Outros documentos
1. Documento de EET;
2. Análise crítica de estudos farmacológicos e toxicológicos não
clínicos;
3. Análise crítica de estudos clínicos já realizados (se aplicável);
4. Autorização do detentor do DDCM (se aplicável);
5. Informação sobre o processo de registro do medicamento
experimental (se aplicável);
6. Toda documentação em CD com busca textual;
42

Genéricos/Similar - Fluxo43
Consultar lista medicamento
referência
Desenvolvimento do produto
Avaliação da equivalência in vitro
ao referência
RDC 31/2010
Estudos de estabilidade acelerada
RE 01/2005
Estudos de Bioequivalência
RE 1170/2006
Estudos de estabilidade de longa
duração
RE 01/2005
Montagem do dossiê
RDC 60/2014Submissão regulatória
Exigências Aprovação

Lista de referência ANVISA44
Para solicitar inclusão de referência:
1. Justificativa técnica;
2. Formulário de solicitação da RDC 35/2012;
Se não disponível no comércio, enviar provas da indisponibilidade;
Não é possível solicitar comparador internacional que não seja de uso hospitalar
ou de programas do governo;

Formulário de solicitação de indicação de
medicamento referência45

Medicamentos que não podem ser genéricos/similares
I. Produtos biológicos, imunoterápicos, derivados do plasma e sangue humano;
II. Medicamentos fitoterápicos;
III. Medicamentos específicos;
IV. Medicamentos dinamizados;
V. Medicamentos de notificação simplificada;
VI. Antissépticos de uso hospitalar;
VII. Produtos com fins diagnósticos e contrastes radiológicos;
VIII. Radiofármacos;
IX. Gases medicinais; e
X. Outras classes de medicamentos que venham a possuir legislação específica para
seu registro.
46

2. Documentação administrativa
de registro Peticionamento do protocolo
Embalagem – RDC 71/2009
Certificado de boas práticas de fabricação
47

Aspectos Gerais
Usar papel formato A4
Apresentar documentação na sequencia dos check lists;
Utilizar folhas separadoras de cores diferentes para separar os documentos
de cada item do check list;
Separar em volumes contendo 200 folhas cada;
Usar colchetes para agrupar as folhas de cada volume do processo;
A primeira folha do volume deverá informar o número do volume e as
páginas que estão contidas;
Não encadernar ou plastificar;
Mídias (CD) devem estar em embalagem colada à folha A4;
48

Aspectos Gerais
Petição individual por forma farmacêutica, mesmo que
diferentes medicamentos de referência;
Todas as concentrações na mesma petição;
49
SildenafilaInjetável
Viagra
•25 mg
•50 mg
•100 mg
Revatio
•20 mg
Sildenafilacomprimido
Viagra
•25 mg
•50 mg
•100 mg
Revatio
•20 mg

2. Documentação administrativa
de registroPeticionamento do processo
50
http://www.anvisa.gov.br/hotsite/protocolo/passoapasso.html

Peticionamento do registro
1. Acesso ao sistema da ANVISA;
2. Petição eletrônica e pagamento de taxa
a. Entrega de documentação em papel
b. Entrega documentação online (não aplicável para novos,
similares e genéricos);
3. Petição manual e pagamento de taxa
a. Entrega de documentação em papel
51

Acesso ao sistema ANVISA52

Acesso ao sistema ANVISA53

Acesso ao sistema ANVISA54

Acesso ao sistema ANVISA55

Petição Eletrônica56

Petição Eletrônica57

Petição Eletrônica58

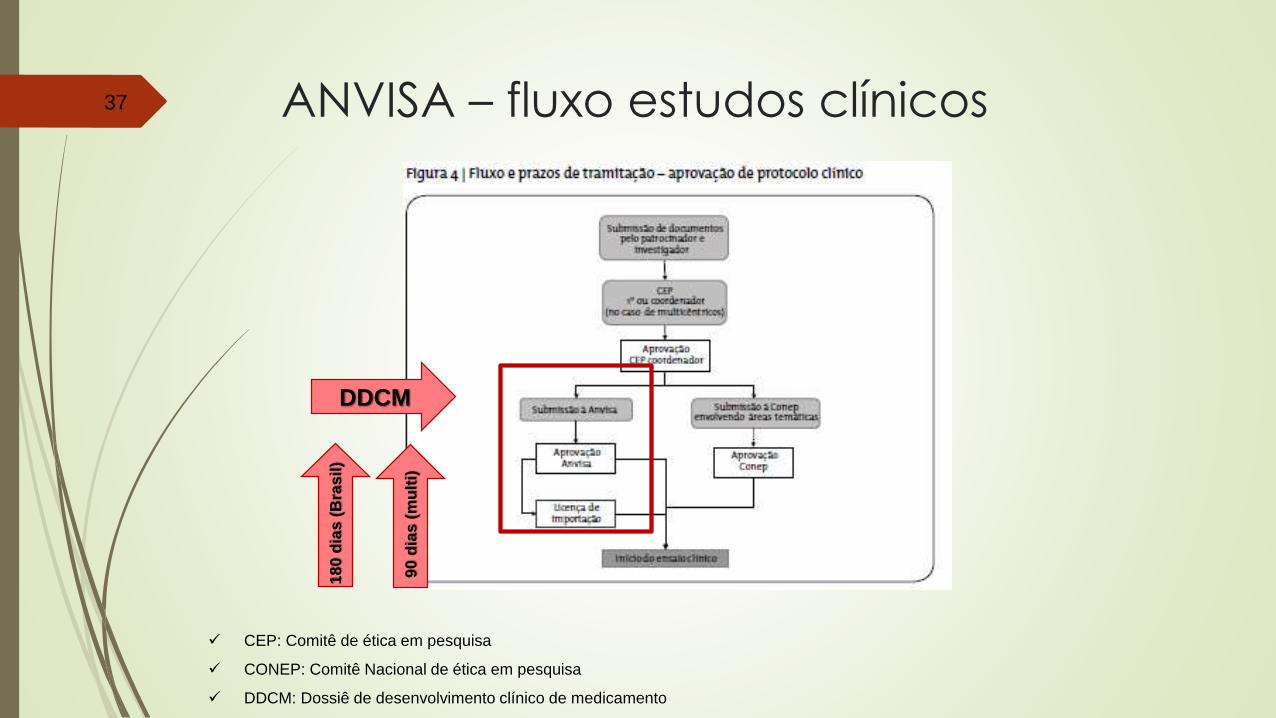
Petição Manual59

Petição Manual60

Petição Manual61

Petição Manual62

Petição Manual63
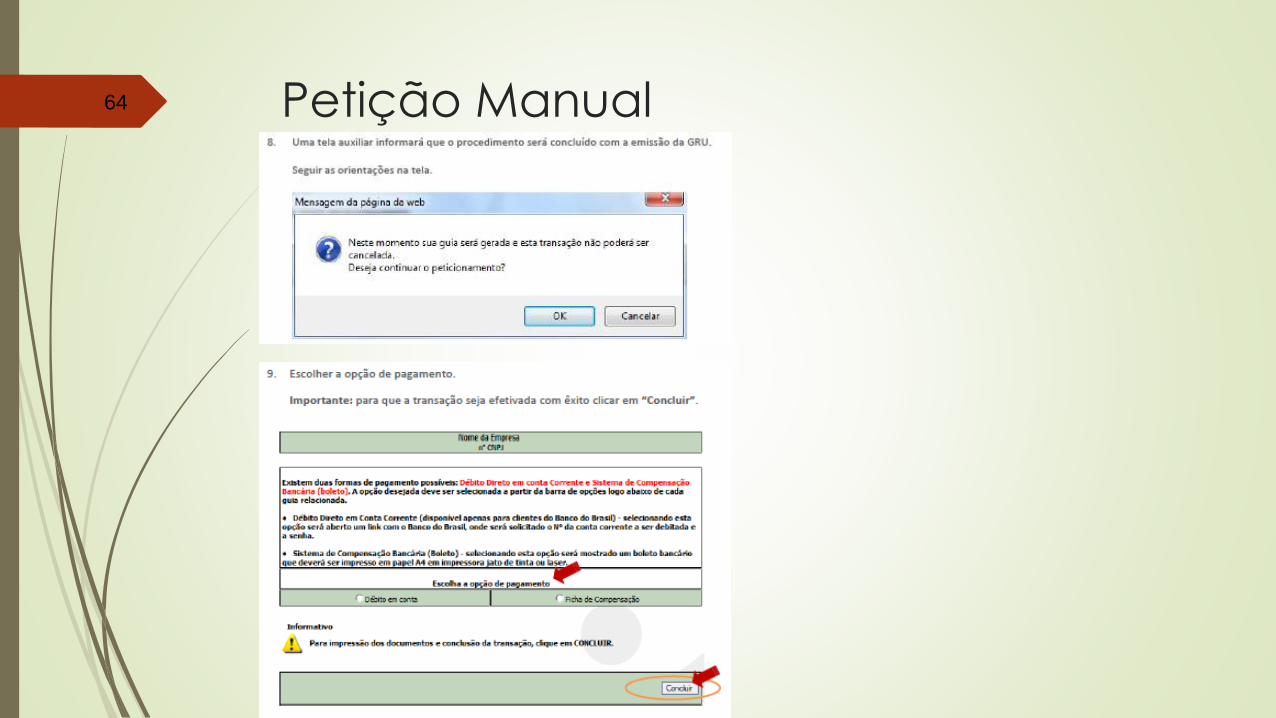
Petição Manual64

Petição Manual65

FP1 - preenchimento66
Classe terapêutica
Lista referência
Consulta produto
Forma física
D.C.B

FP1 -
exemplo67

FP2 - preenchimento68
Assunto petição
Restrição de uso
Cuidados de
conservação
Embalagens

FP2 – Apresentação do produto69

FP2 -
exemplo70

Registro eletrônico de
medicamentos novos
Inovação radical71
http://portal.anvisa.gov.br/wps/wcm/connect/b69d2900474581508da0dd3fbc4c6735/MANUAL+DO+REGISTRO+ELETRONICO_SUBST%C3%82NCIA_PRODUTo+4.pdf?MOD=AJPERES

Divisões para o registro72

2. Documentação administrativa
de registroEmbalagem – RDC 71/2009
73

Embalagens
Embalagem – invólucro, recipiente ou qualquer forma de
acondicionamento, removível ou não, destinada a cobrir, empacotar,
envasar, proteger ou manter, especificamente ou não, medicamentos (Lei
nº6.360, de 23/09/1976, e Resolução-RDC nº 71, de 22/12/2009);
Embalagem primária – embalagem que mantém contato direto com o medicamento;
Embalagem secundária – embalagem externa do produto, que está em contato com a
embalagem primária ou envoltório intermediário, podendo conter uma ou mais embalagens
primárias;
Embalagem secundária funcional – aquela que oferece proteção adicional ou serve para
liberar a dose do produto;
Envoltório intermediário – embalagem opcional que está em contato com a embalagem
primária e constitui um envoltório ou qualquer outra forma de proteção removível, podendo
conter uma ou mais embalagens primárias, conforme aprovação da Anvisa
74

Necesidade PrimáriaSecundári
a
Nome comercial do medicamento; X X
DCB em letras minúsculas; X X
Concentração de cada princípio ativo, por unidade posológica; X X
Via de administração; X X
Número do lote X X
Data de fabricação X X
Data de validade X X
Quantidade total de peso líquido, volume e unidades farmacotécnicas
X
Quantidade total de acessórios dosadores que acompanha as apresentações (se aplicável);
X
Forma farmacêutica; X
Composição qualitativa, conforme DCB, e quantitativa de cada princípio ativo, incluindo, quando aplicável, a equivalência sal base;
X
Código de barras X
75

Necesidade Primária Secundária
Restrição de uso por faixa etária, indicando a idade mínima, em meses ou anos, para qual foi aprovada no registro o uso do medicamento, ou "USO ADULTO e PEDIÁTRICO“ se não houver restrição registrada;
X
Cuidados de conservação, indicando a faixa de temperatura e condições de armazenamento, conforme estudo de estabilidade do medicamento;
X
Nome e endereço da empresa titular do registro no Brasil (registrado por);
X X
Nome e endereço da empresa fabricante e local de fabricação (fabricado por);
X
Nome e endereço da empresa fabricante, quando o medicamento
for importado, citando a cidade e o país precedidos pela frase "Fabricado por" e inserindo a frase "Importado por:" antes dos dados da empresa titular do registro;
X
CNPJ do titular do registro; X
Expressão "Indústria Brasileira", quando aplicável; X
Nome do responsável técnico, CRF e sigla do estado; X
Sigla "MS" adicionada ao número de registro no Ministério da Saúde X
Frases de advertência conforme RDC 60/2010 X
76

Necesidade Primária Secundária
Informações ao paciente, indicação e contra-indicação – vide bula com letra maior que 1,5 mm
X
Frase “TODO MEDICAMENTO DEVE SER MANTIDO FORA DO ALCANCE DE CRIANÇAS”
X
Presença de tinta reativa na lateral da embalagem e lacre ou selo de segurança sob ele a palavra “qualidade”
X
Nome comercial e denominação genérica com letra maior que 1,5 mm e obedecendo a proporcionalidade;
X
Em mesmo destaque e no mesmo campo de impressão, em tamanho de 50% do nome comercial, a denominação genérica seguindo DCB;
X
Telefone do SAC da empresa titular do registro ou de sua responsabilidade
X X
77

2. Documentação administrativa
de registroCertificado de boas práticas de fabricação - CBPF
78

Importância das Boas Práticas de
Fabricação
BPF de medicamentos é a parte da garantia da
qualidade que assegura que os produtos sejam
fabricados em conformidade e controlados em
relação aos padrões de qualidade solicitados pelo
registro sanitário do produto. As BPF de
medicamentos estão relacionadas com os
procedimentos de fabricação e de controle da
qualidade.
Auditoria CBPF: Bi-anualhttp://www.anvisa.gov.br/certificadoBo
asPraticas/principal/index.asp

Reclamações e Recolhimento do Produto
Inspeção e auditoria
Qualificação e Validação
Controle de Qualidade
Documentação e Rastreabilidade
Treinamentos
Controle de Riscos
Pilares da RDC 17/2010

Medicamento importado
Poderá ser aceito CBPF do país se houver equivalência das
medidas de controle de boas práticas com a ANVISA;
Intermediários: Poderá ser apresentado documento de
comprovação de boas práticas de fabricação emitido pelo
órgão responsável pela Vigilância Sanitária do país fabricante.
Cópia do protocolo de solicitação de inspeção + certificado
CBPF do país de origem (impede a aprovação)
Documentos de compromissos assumidos com outras agências;
81

3. Documentação técnica da
qualidade Insumo farmacêutico ativo
Desenvolvimento da formulação
Produção do produto terminado
CQ das matérias primas
CQ do produto terminado
Embalagem primária e secundária funcional
Involtório intermediário
Acessórios
Estudos de estabilidade do produto terminado
82

3. Documentação administrativa
de registro
Insumo farmacêutico ativo
Estereoquímica, isomeria e quiralidade
Solubilidade - Conceito de classificação BCS
Tamanho de partícula
Polimorfismo
Estabilidade – RDC 45/2012
Especificações do IFA
83

Informações para o IFA
a. Nomenclatura: Denominação Comum Brasileira (DCB);
b. Estrutura: fórmula estrutural, estereoquímica relativa e absoluta, fórmula
molecular, e massa molecular relativa;
c. Propriedades físico-químicas: forma física, relação sal/base, ponto de fusão,
solubilidade, tamanho de partícula e pKa;
d. Nome do(s) fabricante(s) do(s) IFA(s) com os respectivo(s) endereço(s) e
documento do órgão oficial sanitário do país de origem comprovando
autorização para a atividade de fabricar IFA;
e. Descrição do processo de síntese: fluxograma do processo de síntese, incluindo
fórmula molecular, estruturas químicas dos materiais de partida, intermediários e
respectivas nomenclaturas, solventes, catalisadores, reagentes e o IFA,
contemplando a estereoquímica;
84

Informações para o IFA
f. Elucidação da estrutura e outras características e impurezas: confirmação da
estrutura e informação sobre potencial isomerismo estrutural e geométrico,
rotação óptica específica, índice de refração, quiralidade, potencial de formar
polimorfos, discriminando as suas características e de outros polimorfos
relacionados ao IFA, e informações sobre impurezas;
g. Controle de qualidade: especificações, justificativa das especificações para IFA
não farmacopeico, métodos analíticos utilizados e validação e laudo de análise
de um lote emitido pelo fabricante do IFA;
h. Estabilidade: um resumo sobre os tipos de estudos conduzidos e os resultados,
conforme legislação específica vigente, incluindo os resultados de estudos de
degradação forçada e condições de stress e respectivos procedimentos
analíticos, bem como as conclusões sobre o prazo de validade ou data de
reteste e material de embalagem.
85

Estereoquimica, isomeria e
quiralidade86

Estereoquímica
Envolve o estudo do arranjo espacial relative
dos átomos dentro das moléculas
É sinônimo da atividade optica que uma
molécula quiral possui;
Também conhecida como quimica “3D” pois
estuda a conformação especial de uma
molécula;
87

Estereoquímica88

Isômeros constitucionais
São moléculas com a mesma formula química
mas diferentes estruturas de átomos e ligações:
89

Estereoisômeros – Isômeros espaciais
São moléculas distintas com a mesma sequencia de ligação de átomos
mas com uma orientação espacial diferente se dividem em:
Enântiomeros: Imagens espelhadas
Diasteroisômeros: Não espelhadas
90

91

Solubilidade
Conceito de classificação
BCS92

Conceitos – BCS ou SCB
SOLUBILIDADE:
Solubilidade de um fármaco (BCS): disolução da dosagem mais alta (em
tomada única) de um medicamento em 250 mL de uma solução tampão de pH
entre 1,0 e 8,0.
Fármaco altamente solúvel: relação dose/solubilidade é menor ou igual a 250;
PERMEABILIDADE:
Um fármaco de alta permeabilidade: biodisponibilidade absoluta é maior que
90% na ausência de instabilidade no trato gastrintestinal ou quando este
parâmetro é determinado experimentalmente.
93

Classificação BCS e exemplos
94
http://www.contractpharma.com/contents/displayImage/5605/

Tamanho de partícula95

Tamanho de partícula, biodisponibilidade e dissolução
96
Tamanho de partícula importante para
fármacos de baixa solubilidade – BCS
classe II ou classe IV.
Deve ser definido antes do estudo de BE e
mantido para o biolote e lotes produtivos.

Polimorfismo97

Definição de polimorfos
É a habilidade de uma substância existir como duas ou mais fases
cristalinas que tem diferentes arranjos ou conformações das moléculas
em sua estrutura cristalina.
Mesma entidade quimica diferentes formas;
Associados a diferentes propriedades físicas do
fármaco
Polimorfismo pode interferir na qualidade, eficácia e segurança do
medicamento
98

Tipos de polimorfismo
Formas cristalinas com diferentes arranjos ou
conformações das moléculas na estrutura cristalina;
Formas amorfas consistindo de arranjos desordenados
das moléculas que não possuem uma estrutura
cristalina distinguivel;
Solvatos consistindo de formas contendo quantidades
estequimétricas ou não de solventes incorporados.
Podem ser hidratos (água) ou solvatos (outros solventes)
99

Importância do polimorfismo Diferentes formas polimórficas
tem diferentes propriedades
quimicas e físicas:
Ponto de fusão, Reatividade
quimica, Solubilidade aparente,
Taxa de dissolução, Propriedades
opticas e mecânicas, Pressão de
vapor, Densidade
Estas propriedades tem um
efeito direto na habilidade de
processar e/ou fabricar o
fármaco ou o produto
acabado:
Estabilidade do produto acabado,
Dissolução, Bioequivalência
100
5-Methyl-2-[(2-nitrophenyl)amino]-3-
thiophenecarbonitrile has been crystallized in ten
polymorphs: The structure of themolecule is shown in
the figure. The systemhas been named ROY for its
red,orange, andyellowcrystal colors.
Thedifferentpolymorphs arenamedas, yellowprisms (Y),
red prisms (R), orange needles (ON), orange plates
(OP), yellowneedles (YN), orange-red plates (ORP), and
red plates (RPL).

Estabilidade – RDC
45/2012 Estudo de estress/degradação forçada
Fotoestabilidade
Estabilidade
101

Estabilidade – RDC
45/2012 Estudo de estress/degradação forçada
102

Material de partida
Intermediário
Fármaco
Produto acabado
Reagentes
Solventes
Catalisadores
Reagentes
Solventes
Catalisadores
Solventes?
Impurezas do
material de partida
Derivados
Derivados
Degradação
Interação com
excipientes
Interação com
excipientes
Impurezas/Produtos de degradação
Potenciais Impurezas
1. Resíduo do material de partida
2. Resíduo dos intermediários
3. Impurezas no material de partida
4. Reagentes
5. Solventes
6. Catalisadores
7. Derivados da reação
8. Produtos de degradação
9. Reações fármaco-excipiente
10. Reações formulação-embalagem

Possíveis
Realização de estudos de stress:
Stress ácido
Stress básico
Stress por calor
Stress por umidade
Stress por oxidação
Fotoestabilidade
Íons metálicos
Garantir degradação de no mínimo 10% do ativo (evitar degradação secundária);
Em cada fabricante do fármaco
Para cada forma farmacêutica/dose/associação do medicamento
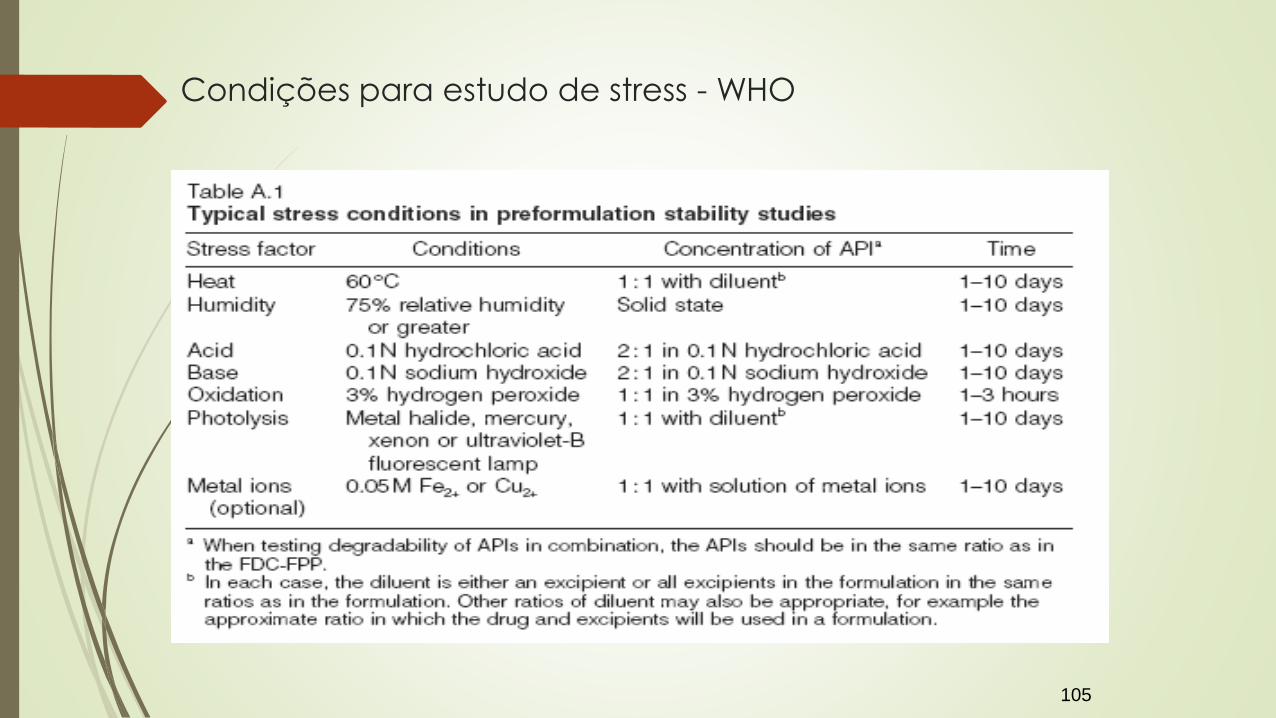
Condições para estudo de stress - WHO
105

Estabilidade – RDC
45/2012 Fotoestabilidade
106

Fotoestabilidade - IFA
Utilizando um lote do IFA (+2 se não conclusivo);
Composto de duas partes
Degradação forçada
Estudo confirmatório:
Luz padrão de emissão D65 (luz do dia)/ID65 (luz indireta de interiores), como uma
lâmpada fluorescente artificial combinando emissão visível e UV;
Usar filtro para eliminar radiação abaixo de 320 nm
Uma proporção significativa da luz UV deve estar entre 320 e 360 nm e entre 360 e
400 nm;
Expostas a no mínimo 1,2 milhões de lux horas, integrados a uma energia UV
próxima de no mínimo 200 watt horas/m2
Amostras expostas e amostras controle: Propriedades físicas, teor e produtos de
degradação (mínimo)
107

Estabilidade – RDC
45/2012 Estabilidade
108

Estabilidade - IFACondição de
armazenamentoEstabilidade longa duração Estabilidade acelerada
até 30 ºC 30 ºC ± 2 ºC / 75% UR ± 5% UR 40 ºC ± 2 ºC /75% UR ± 5% UR
2 ºC a 8 ºC 5ºC ± 3 ºC 25 ºC ± 2 ºC /60% UR ± 5% UR
-15 ºC a -25 ºC -20 ºC ± 5 ºC Não aplicável
Abaixo de -20 ºC Caso a caso Não aplicável
109
1. Alteração significativa no estudo acelerado: Validade conforme longa duração;
2. IFA refrigerado com resultado OOS até 3 meses (acelerado): um lote em tempo inferior a 3
meses, fora da condição de armazenamento recomendada;
3. Para IFA com armazenamento inferior a -15 ºC o prazo de validade é baseado no estudo de
longa duração;
a. Um lote deve ser avaliado em temperaturas mais altas para verificar efeito de excursões;
4. Empresas em diferentes zonas climáticas: realizar o pior caso;
5. Acelerado + longa duração: prazo provisório de 24 meses
6. Um lote por ano em estabilidade de acompanhamento

Especificações do IFA110

Especificações comuns - IFA
Aparência
Solubilidade
Tamanho de partícula
Polimorfismo
Identificação: IV, HPLC
Doseamento
Perda por secagem
Umidade
Impurezas: Resíduos por ignição, Metais pesados, Produtos de
degradação, Solventes residuais
111

Especificação – tamanho de
partícula
112

Impurezas/Produtos de degradação – Especificações
As especificações devem incluir:
Impurezas/produtos de degradação:
Especificação para cada impureza/PD identificado;
Especificação para cada impureza/PD não identificado (≤ identification threshold);
Total de impurezas/PD.
Solventes residuais: Para todos os fármacos e controle no produto
acabado somente se a dose do mesmo for maior que 10 g por dia ou
se os limites no fármaco estiverem acima dos descritos nos guias
internacionais
Impurezas inorgânicas: Somente nos fármacos e utiliza-se monografias e
especificações de capítulos gerais das farmacopéias.

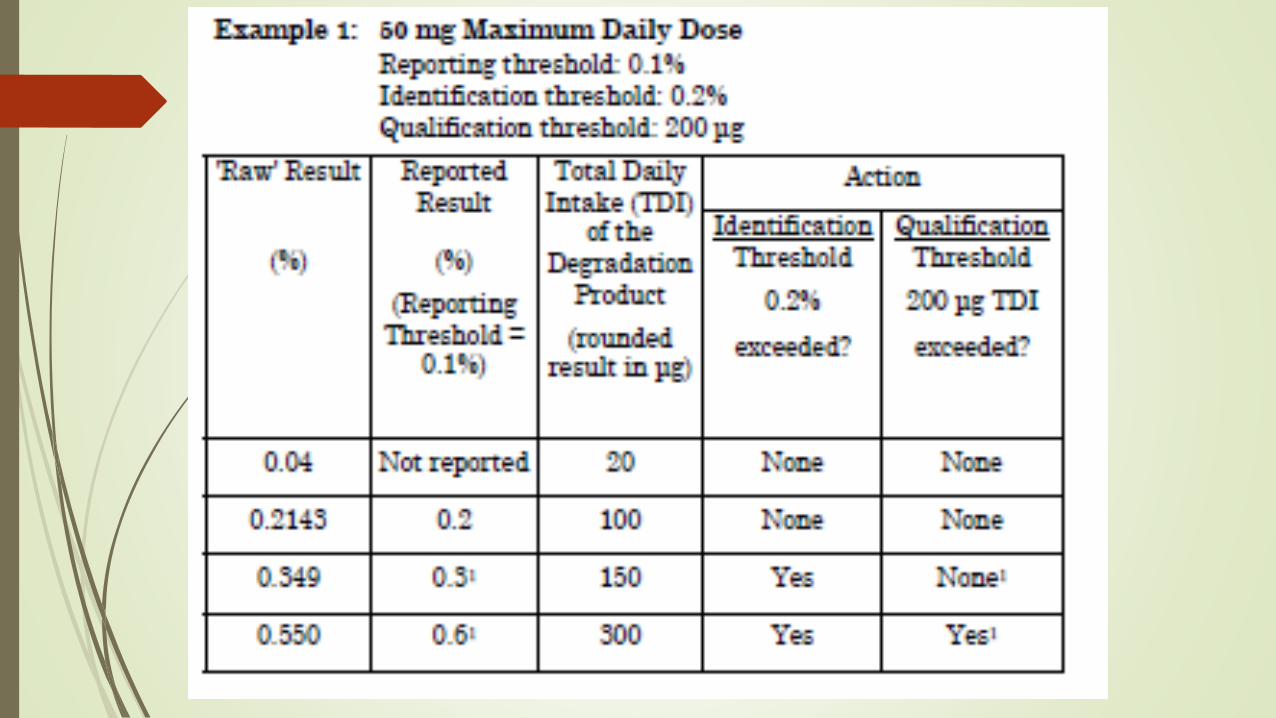
Especificações de impurezas
Qualificação
≤ 2g/dia: 0,15% ou 1 mg/dia ≥ 2g/dia: 0,05%
Identificação
≤ 2g/dia: 0,10% ou 1 mg/dia ≥ 2g/dia: 0,05%
Reporte
≤ 2g/dia: 0,05% ≥ 2g/dia: 0,03%
114

IFA registrado na ANVISA Aciclovir
Ampicilina
Carbamazepina
Carbonato de lítio
Ciclofosfamida
Ciclosporina
Ciprofloxacino
Cl. de clindamicina
Clozapina
Efavirenz
Fenitoína
Fenitoína sódica
Lamivudina
Metotrexato
Claritromicina
Ácido pamidrônico
Bromoprida
Cilostozol
Desogestrel
Gestodeno
Sinvastatina
Nevirapina
D-penicilamina
Rifampicina
Ritonavir
Tiabendazol
Zidovudina
Azitromicina di-hidratada
Benzilpenicilina
Carbegolina
Carboplatina
Cefalexina
Cefalotina
Ceftazidima
Ceftriaxona
Cisplatina
Amoxicilina
Cl. de amiodarona
Cl de metiformina
Cl de paroxetina
Mazindol
Tibolona
115

3. Documentação administrativa
de registro
Desenvolvimento da formulação
Desenvolvimento da formulação
Desenvolvimento do método de dissolução116

Desenvolvimento da formulação
117

Desenvolvimento da formulação
a. Resumo sobre o desenvolvimento da formulação;
b. Estudos de compatibilidade IFA-excipientes e das características
do IFA que influenciam a performance do medicamento;
c. Detalhes de fabricação, caracterização, e controles com
referência bibliográfica para suportar os dados de segurança
para excipientes usados pela primeira vez em um medicamento
ou em uma nova via de administração;
d. Dados e discussão sobre a avaliação de eficácia do sistema
conservante utilizado(s) na formulação; e
e. Justificativa no caso de excesso de ativo
118
Difícil aceitação ANVISA

Etapas do desenvolvimento desenhado por qualidade
Perfil do produto
Atributos críticos de qualidade
Desenho do espaço
Estratégia de controle
Melhoria contínua
119
O que é o produto?
Quais as especificações?
O que se conhece?
Como se mantém?
Posso fazer melhor?

Pharmaceutical Developement - ICH Q8
Desenhar um produto e processo de fabricação de qualidade
que entreguem a performance do produto;
Conhecer o produto clinicamente, toxicologicamente,
quimicamente…
Estabelecer desenho de espaço (design space), especificações
e controles de fabricação;
Novo paradigma: Qualidade não deve ser testada no produto,
ela deve ser inserida no produto pelo desenho e entendimento
dos processos;
120

Novo paradigma – ICH Q8
121

Atividades de desenvolvimento
Desenvolvimento da molécula ou
avaliação do referência
Avaliação das características do
IFA
Avaliação das características dos excipientes
Avaliação da compatibilidade de excipientes
(estudos binários)
Desenvolvimento do processo de
fabricação
Estudos de formulação
Avaliação do perfil de
dissolução piloto
Desenvolvimento do método de
dissolução
Fabricação de lote para piloto
de BEPiloto de BE
Refinamento do método de dissolução
Refinamento do processo fabril
Avaliação do lote para BE
Estudo de estabilidade acelerado
Estudo de BEEstudo de
estabilidade longa duração
122

Estudos de compatibilidade IFA-excipientes123
Stress

Excipientes - segurança
Novos excipientes:
Nunca utilizados em nenhuma forma farmacêutica
Nunca utilizados na via de administração
Utilizados em menor quantidade do que a proposta;
Referências para sustentar uso de excipientes:
Handbook de excipientes:
https://www.medicinescomplete.com/about/publications.htm
Banco de dados de ingredientes inativos do FDA:
http://www.accessdata.fda.gov/scripts/cder/iig/index.Cfm
124

Eficácia do sistema conservante
Substâncias adicionadas à produtos de base aquosa, não estéreis
a fim de proteger a formulação do crescimento microbiano
inadvertidamente introduzido durante ou após o processo de
fabricação ou no caso de repetidas retiradas de dose de
embalagens multi-doses de produtos estéreis;
Não devem ser adicionados para substituição de boas práticas de manipulação;
Deve ser incluído na menor quantidade que se prove eficaz e abaixo do limite
tóxico para humanos;
Avaliação no estudo de desenvolvimento: quantidades decrescentes em
matrizes propositalmente contaminadas
Avaliação na estabilidade do acabado: Quantificação do conservante (em
todos os pontos de estabilidade) e eficácia do Sistema conservante inicial e final
- conside
125

Desenvolvimento do método de
dissolução
126

O que é o teste de dissolução?Teste padronizado que mede a porção do fármaco:
1. Liberada da matriz da forma farmacêutica e
2. Dissolvida no meio de dissolução em condições controladas
durante um período de tempo definido;
Em termos simples:
1. A forma farmacêutica se desintegra;
2. O fármaco então se dissolve no meio;
Quanto mais lenta a desintegração da forma farmacêutica, mais lenta a dissolução.
127

Dissolução e co-relação sistêmica128

Equipamento e aparatos
Aparato 1 (cesta)
Aparato 2 (pás)
Aparato 3 (cilindro reciprocador)
Aparato 4 (célula flow-through)
Aparato 5 (pá sobre disco)
Aparato 6 (cilindro)
Aparato 7 (holder reciprocador)
Escolha depende da forma farmacêutica e da finalidade do teste.
129

Etapas do desenvolvimento do
método de dissolução
Verificação do método de solubilidade
• Especificidade/avaliação de perfil cromatográfico de tampões que serão utilizados no estudo
• Demonstrar que não há pico eluindo onde se espera quantificar o IFA
Solubilidade do IFA
• Usar incubadora com agitação orbital e temperatura de 37⁰C±1⁰C
• Solubilidade em equilíbrio
• Tampões em pH fisiológico (pH 1,2 a 6,8) – 3 meios
• Triplicata – calcular desvio %RSD
• Justificar quantidade de tensoativo utilizada (sink condition)
Desenvolvimento do método
• 3 curvas de dissolução em pH fisiológico;
• Testar aparatos, velocidade e volume dos meios testados
• Confirmar quantidade de tensoativo utilizado
• Testar filtros x centrifugação, testar necessidade de deaeração
• Justificar a escolha de especificação – mostrar discriminatoriedade do método
130

Importância do teste de dissolução
Prever o desempenho in vivo da formulação;
Garantir a qualidade lote a lote do medicamento;
Orientar o desenvolvimento de novas formulações;
Perfil comparativo:
Genérico/similar: Mostrar comparação ao referência
Todos: Assegurar a uniformidade da qualidade e do
desempenho do medicamento depois de alterações;
Reflexo da qualidade da formulação!!!!
131

Fatores que afetam a dissolução
Características do fármaco:
Solubilidade: Alta ou baixa
Características da forma farmacêutica:
Liberação muito rápida;
Liberação rápida;
Liberação prolongada;
Liberação retardada.
132

Dissolução: Características
do fármaco133

Sistema de classificação
biofarmacêutica - BCS Introduzido por Amidon et al. em 1995;
Classifica os fármacos em 4 grupos, levando em
consideração:
Solubilidade aquosa na maior dosagem (de uma tomadaúnica) – alta se dissolver em 250 mL dos meios
Permeabilidade intestinal
134
PermeabilidadeSolubilidadeClasse
AltaAlta1
AltaBaixa2
BaixaAlta3
BaixaBaixa4

Dissolução: Características
da forma farmacêutica135

Sólidos de liberação imediata
Forma farmacêutica em que a dose total da substância ativa é disponibilizada
rapidamente após sua administração. Em ensaios in vitro apresenta, em geral,
dissolução média de no mínimo 75% da substância ativa em até 45 minutos. Tal
forma farmacêutica pode ainda apresentar tipos de dissoluções diferenciadas
em rápida e muito rápida:
Liberação muito rápida: Dissolução média de no mínimo 85% da substância
ativa em até 15 minutos
Liberação rápida: Dissolução média de no mínimo 85% da substância ativa
em até 30 minutos
RDC 31/2010 – Equivalência Farmacêutica
136

Sólidos de liberação modificada
Liberação Prolongada: forma farmacêutica que apresenta liberação
modificada em que a substância ativa é disponibilizada gradualmente da
forma farmacêutica por um período de tempo prolongado;
Liberação Retardada: forma farmacêutica que apresenta liberação
modificada em que a substância ativa é liberada em um tempo diferente
daquele imediatamente após a sua administração. As preparações gastro-
resistentes são consideradas forma de liberação retardada, pois são
destinadas a resistir ao fluido gástrico e liberar a substância ativa no fluido
intestinal;
RDC 31/2010 – Equivalência Farmacêutica
137

Quando os perfis são considerados
similares? Ausência do cálculo de F2 - Fármaco de alta solubilidade e formulação for de liberação imediata com
dissolução muito rápida para ambos os medicamentos:
Fator F2 perde o seu poder discriminativo e, portanto, não é necessário calculá-lo.
O coeficiente de variação no ponto de 15 minutos que não pode exceder 10%.
Calcular fator F2 (entre 50 e 100):
Utilizar, no mínimo, os três primeiros pontos, excluindo o tempo zero;
Incluir apenas um ponto da curva após ambos os medicamentos atingirem a média de 85% de
dissolução – evitar falsos positivos
O RSD para os primeiros pontos de coleta (40% do total de pontos coletados) não podem exceder
20%. Para os demais pontos considera-se o máximo de 10%.

Especificação representativa?
139

3. Documentação administrativa
de registroProduto terminado
140

Produto terminado
a. Descrição detalhada sobre a fórmula completa, conforme a
Denominação Comum Brasileira (DCB);
b. Quantidade de cada componente da fórmula e suas funções,
incluindo os componentes da cápsula, e indicação das
respectivas referências de especificações de qualidade;
c. Descrição detalhada sobre a proporção qualitativa e
quantitativa dos produtos intermediários utilizados na fórmula
do produto terminado; e
d. Justificativa quanto à presença de sulco no comprimido com
os devidos testes.
141

Descrição da formulação – Anexo I142

Vinco/Sulco em comprimidos
Não há guia local para condução do estudo:
FAQ ANVISA tecnologia farmacêutica:
143
http://portal.anvisa.gov.br/wps/wcm/connect/918cda8048c24f5c8623be0a466faa84/FAQ+GRMED.pdf?MOD=AJPERES
1. Genérico/Similar:
Permitido só se tiver
no referência
2. Controle de qualidade
e estabilidade do
comprimido partido
(justificado pela
cronicidade e
posologia);
3. Unif. Dose unitária
4. Perda de massa na
faixa de dureza
5. Friabilidade
6. Dissolução

Guia FDA144
http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm269921.pdf

Documentação administrativa de
registroProdução do Produto terminado
145

Produção do Produto terminado
a. Dossiê de produção referente a 1 (um) lote;
Nos casos em que a solicitação de registro se referir a mais de uma concentração, deverá ser apresentado para a
maior e menor concentração, desde que as formulações sejam qualitativamente iguais, sejam proporcionais e sejam
fabricadas no mesmo local e com mesmo processo produtivo.
b. Nome e responsabilidade de cada envolvido na produção e no controle de qualidade e
estabilidade, incluindo terceirizados;
c. Fluxograma com as etapas do processo de fabricação mostrando onde os materiais entram no
processo, identificando os pontos críticos do processo e os pontos de controle, testes
intermediários;
d. Tamanhos de lotes do produto terminado;
e. Lista dos equipamentos envolvidos na produção, identificados por princípio de funcionamento
(classe) e desenho (subclasse) com suas respectivas capacidades;
f. Controle das etapas críticas com a informação sobre os testes e critérios de aceitação realizados
nos pontos críticos identificados no processo de fabricação, além dos controles em processo; e
g. Relatório sumário da validação do processo de fabricação, incluindo lotes, definição das etapas
críticas de fabricação com as respectivas justificativas, parâmetros avaliados, e indicação dos
resultados obtidos e conclusão.
146

Conceitos de proporcionalidade
1. Todos os componentes exatamente na mesma proporção (% do peso
médio) em todas as dosagens;
2. Proporção (% do peso médio) esteja dentro do que é classificado
como alteração moderada de excipientes:
147

Fluxograma do processo
de fabricação148

Fluxo do processo de fabricação149

Descrição/Detalhamento
dos Equipamentos150

Descrição dos equipamentos151

Classificação de equipamentos152
http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm346049.pdf
SUPAC:
Scale-up and post approval changes

Sumário de validação de
processo153

Informações no sumário de validação
de processoRecomenda-se que o relatório sumário inclua pelo menos os seguintes itens:
Breve descrição do processo produtivo incluindo parâmetros de operação, limites do processo e entrada
de materiais;
Identificação de lotes avaliados;
Resumo das etapas críticas e variáveis presentes nas atividades e procedimentos do processo a ser
investigado e as respectivas justificativas;
Lista dos equipamentos/instalações a serem utilizados durante o desenvolvimento do produto e
escalonamento da produção;
Justificativa dos controles em processo propostos, dos respectivos critérios de aceitação e frequência dos
testes a serem aplicados durante e após a validação;
Plano de amostragem com justificativa para a escolha dos pontos;
Métodos para registro e avaliação dos resultados;
Avaliação do processo em condições extremas (testes desafio/“pior caso”) nos quais se pode determinar a
robustez do processo;
Cronograma de atividades de validação;
Resultados obtidos, considerando os desvios e conformidades observados e a variabilidade intra e inter
lotes;
Conclusão, incluindo a avaliação sobre a possibilidade de reprodução em escala comercial.
154

Documentação administrativa de
registroControle de qualidade das matérias primas
155

CQ das matérias primas
a. Especificações, métodos analíticos e laudo analítico para
os excipientes, acompanhados de referência bibliográfica,
feitos pelo fabricante do medicamento;
b. Informações adicionais para os excipientes de origem
animal de acordo com a legislação específica vigente
sobre controle da Encefalopatia Espongiforme Transmissível;
e
c. Especificações, métodos analíticos e laudo analítico para o
insumo farmacêutico ativo, acompanhados de referência
bibliográfica, realizados pelo fabricante do medicamento.
156

Controle de qualidadeRDC 17/2010
157

Controle de qualidade – RDC 17/2010
Art. 281. O Controle de Qualidade é responsável
pelas atividades referentes à amostragem, às
especificações e aos ensaios, bem como à
organização, à documentação e aos
procedimentos de liberação que garantam que os
ensaios sejam executados e que os materiais e os
produtos terminados não sejam aprovados até que
a sua qualidade tenha sido julgada satisfatória.
158

Monografias compendiais
Se houver monografia nos compêndios reconhecidos pela
ANVISA, deve obrigatoriamente seguir as especificações
definidas;
Os métodos podem diferir desde que sejam validados conforme
RDC 899/2003;
Farmacopeias reconhecidas (RDC 37/2009): Farmacopéia
Alemã, Americana, Argentina, Britânica, Européia, Francesa,
Internacional (OMS), Japonesa, Mexicana, Portuguesa
159

160

Testes gerais para CQ de matérias primas
Identificação
Doseamento
Produtos de degradação
Impurezas orgânicas
Impurezas inorgânicas
Solventes residuais
Metais pesados
Arsênico
Perda por secagem
Água/Umidade
Tamanho de partícula
Solubilidade
Testes específicos para performance – em monografia ou a definir pelo usuário
161

Documentação administrativa de
registroControle de qualidade do produto terminado
162

CQ do produto terminado
a. Especificações, métodos analíticos e laudo
de análise, acompanhados de referência
bibliográfica, incluindo relatórios de
validação de método analítico; e
b. Gráfico do perfil de dissolução, quando
aplicável.
163

Testes convencionais por forma farmacêutica
Testes Sólidos orais Suspensões Semi sólidos Líquidos
Aparência x x x x
Teor do ativo x x x x
Uniformidade das doses unitárias x --- --- ---
Quantificação de produtos de degradação x x x x
Limites microbianos/patógenos/esterilidade x x x x
Dissolução x x --- ---
Friabilidade x --- --- ---
Dureza x --- --- ---
pH --- x x x
Sedimentação pós agitação --- x --- ---
Perda de peso (base aquosa) --- x x x
Separação de fases (emulsões e cremes) --- --- x ---
Claridade da solução --- --- --- x
Tamanho de partícula e permeabilidade (em pós registro somente)
--- x x ---
164

Produtos de degradação
RDC 58/2013
CP 68/2014165

Perfil de degradação166

Decisão para realizar o estudo167
Estudo de degraçãoforçada
Parte bibliográfica
Parte experimental
Ativos
Placebo
Produto acabado
1. Condição controle
2. Ácida
3. Alcalina
4. Oxidante
5. Térmica seca
6. Fotolítica
7. Íons metálicos
Buscar entre 10 a 30% de degradação

Resultados do estudo168
• Perfil de degradação potencial ou possível
• O perfil de degradação real é avaliado na estabilidade do produto
acabado;
• Produtos de degradação devem seguir especificações da RDC
58/2013 – ICH Q1B

Impurezas no produto acabado
1. Impurezas do fármaco: não aumentam durante o tempo, não são controladas,
exceto se também forem produtos de degradação;
2. Produtos de degradação exclusivamente dos excipientes: verificados em
estudos de stress e estabilidade do placebo – não são controlados;
3. Produtos de degradação provenientes de interação com o fármaco –
controlados:
Interação fármaco-excipiente: Verificado em estudos de stress e nos estudos
de estabilidade
Interação fármaco-material de embalagem: Verificado nos estudos de
estabilidade
4. Extraíveis e lixiviáveis: Não cobertos pelo guia ICH, especialmente importantes
para soluções – capítulo geral da USP

Produtos de degradação no produto acabado

Tamanho de partícula e
permeabilidade172

Semi-solidos/Suspensões - Performance
Apresentar resultados comparativos entre
distribuição do tamanho de partícula/gotícula
da condição anteriormente registrada e da nova
condição;
Incluir discussão relativa ao impacto de eventuais
alterações da distribuição do tamanho de
partícula/gotícula;
Apresentar resultados comparativos entre a taxa
de permeação cutânea da condição
anteriormente registrada e da nova condição
(ainda não é solicitado pois não tem guia local)
173

Limites microbianos174

Limites FB 5º edição
175

Limites FB 5º edição
176

Validação analítica177

O que é validação?
• Ato documentado que atesta que qualquer procedimento,
processo, equipamento, material, atividade ou sistema
realmente e consistentemente leva aos resultados
esperados;
– RDC 17/2010 – Boas Práticas de Fabricação
• Process of defining an analytical requirement, and confirming
that the method under consideration has performance
capabilities consistent with what the application requires.
– Eurachem: Fitness for purpose of analytical methods

Porque validar?
• Demonstrar que o método é apropriado para a finalidade
pretendida, ou seja, a determinação qualitativa, semi-quantitativa
e/ou quantitativa de fármacos e outras substâncias em produtos
farmacêuticos.
– RDC 899/2003 – Validação de métodos analíticos
• A validação é uma parte essencial de Boas Práticas de Fabricação
(BPF), sendo um elemento da garantia da qualidade associado a
um produto ou processo em particular.
– RDC 17/2010 – Boas Práticas de Fabricação

Quando validar?
• Os métodos de controle de qualidade devem ser validados antes de serem
adotados na rotina, levando-se em consideração as instalações e os
equipamentos disponíveis.
– Parágrafo único. Os métodos analíticos compendiais não requerem
validação, entretanto antes de sua implementação, devem existir
evidências documentadas de sua adequabilidade nas condições
operacionais do laboratório.
– RDC 17/2010 – Boas Práticas de Fabricação
• No caso de metodologia analítica descrita em farmacopéias ou formulários
oficiais, devidamente reconhecidos pela ANVISA, a metodologia será
considerada validada.
– RDC 899/2003 – Validação analítica


Quando revalidar?
• A metodologia analítica deverá ser revalidada nas seguintes circunstâncias:
– mudanças na síntese da substância ativa,
– mudanças na composição do produto acabado,
– mudanças no procedimento analítico.
– Outras mudanças podem requerer validação dependendo da sua natureza.
– RDC 899/2003 – Validação de métodos analíticos
• Alterações de formulação (excipiente, sabor, cor), alteração de especificações e métodos,
inclusão de novo fabricante do fármaco ou alterações na sua rota de síntese, inclusão de
nova concentração ou forma farmacêutica do medicamento,
– RDC 48/2009 – Alterações pós registro

Requisitos prévios para validação
Padrões de referência:
1.4. Deve-se utilizar substâncias de referência oficializadas pela Farmacopéia Brasileira ou, na ausência
destas, por outros códigos autorizados pela legislação vigente. No caso da inexistência dessas
substâncias, será admitido o uso de padrões de trabalho, desde que a identidade e o teor sejam
devidamente comprovados (RDC 899/2003)
• padrão secundário (padrão de trabalho): padrão utilizado na rotina laboratorial, cujo valor é
estabelecido por comparação a um padrão de referência (RDC 17/2010) – NÃO ACEITOS EM
VALIDAÇÃO!!!
• padrão de referência: são exemplares de fármacos, impurezas, produtos de degradação,
reagentes, dentre outros, altamente caracterizados e da mais elevada pureza, cujo valor é aceito
sem referência a outros padrões (RDC 17/2010);
– Farmacopeico: Adquirido de um compêndio oficial reconhecido pela ANVISA (RDC 37/2009);
– Caracterizado (primario): Análises para determinação absoluta da pureza e identidade;

http://www.pharmtech.com/pharmtech/Peer-Reviewed+Research/Reference-Standard-Material-
Qualification/ArticleStandard/Article/detail/591372

Processos/Parâmetros da validação• No caso de metodologia analítica não descrita em farmacopéias ou
formulários oficiais, devidamente reconhecidos pela ANVISA, a
metodologia será considerada validada, desde que sejam avaliados
os parâmetros relacionados a seguir,
– Especificidade e Seletividade
– Linearidade
– Intervalo
– Precisão
– Limite de detecção (sensibilidade)
– Limite de quantificação
– Exatidão
– Robustez
– RDC 899/2003 – Validação de métodos analíticos

Categorias de testes
Categoria Finalidade
ITestes quantitativos para a determinação do princípio
ativo em produtos farmacêuticos ou matérias–primas
II
Testes quantitativos ou ensaio limite para a determinação de impurezas e produtos de
degradação em produtos farmacêuticos e matérias-
primas
IIITestes de performance (por exemplo: dissolução,
liberação do ativo, etc)
IV Testes de identificação

Ensaios necessários por categoria
ParâmetroCategoria
I
Categoria IICategori
a III
Categori
a IVQuantitativoEnsaio
Limite
Especificidade Sim Sim Sim * Sim
Linearidade Sim Sim Não * Não
Intervalo Sim Sim * * Não
PrecisãoRepe Sim Sim Não Sim Não
Repro ** ** Não ** Não
Limite de detecção Não Não Sim * Não
Limite de
quantificaçãoNão Sim Não * Não
Exatidão Sim Sim * * Não
Robustez Sim Sim Sim Não Não
* pode ser necessário, dependendo da natureza do teste específico.
** se houver comprovação da reprodutibilidade não é necessária a comprovação da Precisão Intermediária.

Especificidade
É a capacidade que o método possui de medir exatamente um composto
em presença de outros componentes tais como impurezas, produtos de
degradação e componentes da matriz
Contém compostos
semelhantes
Contém fármaco
Quantitativamente: Seleção entre o alvo e compostos semelhantes;
Qualitativamente: Ausência de interferência na presença de compostos
semelhantes (comprovar pureza cromatográfica do pico alvo)
• Impurezas “disponíveis”: Contaminar amostra
• Impurezas “indisponíveis”: Estudo de stress ou comparação de métodos

Limite de detecção – LD ou LoD
• Limite de detecção é a menor quantidade do analito presente em uma
amostra que pode ser detectado, porém não necessariamente
quantificado, sob as condições experimentais estabelecidas (RDC
899/2003);
– O limite de detecção é estabelecido por meio da análise de soluções de
concentrações conhecidas e decrescentes do analito, até o menor nível
detectável;
– Métodos não instrumentais: Pode ser feita visualmente, onde o limite de
detecção é o menor valor de concentração capaz de produzir o efeito
esperado (mudança de cor, turvação, etc);
– Métodos instrumentais: estimativa com base na relação de 3 vezes o ruído da
linha de base. Pode ser determinado analisando 3 curvas contendo analito
próximo ao limite de detecção ou de análise de amostras do branco;

Linearidade
É a capacidade de uma metodologia analítica de demonstrar
que os resultados obtidos são diretamente proporcionais à
concentração do analito na amostra, dentro de um intervalo
especificado (RDC 899/2003).
Análise de no mínimo 5 concentrações diferentes, conforme tipo de
validação – usualmente 3 replicatas de cada concentração;
Coeficiente de correlação (mínimo 0,99), intersecção com o eixo Y,
coeficiente angular, soma residual dos quadrados mínimos da
regressão linear e desvio padrão relativo.
Se não houver relação linear, realizar transformação matemática.

Precisão
A precisão é a avaliação da proximidade dos resultados obtidos
em uma série de medidas de uma amostragem múltipla de uma
mesma amostra.
– Repetibilidade (precisão intra-corrida): concordância entre os
resultados dentro de um curto período de tempo com o mesmo
analista e mesma instrumentação = no mínimo, 9 determinações no
linear do método, ou seja, 3 concentrações na baixa, média e alta,
com 3 réplicas cada ou mínimo de 6 determinações a 100% da
concentração do teste (usualmente até 2%)
– Precisão do sistema – uma preparação avaliada 6 vezes;
– Precisão do método – 6 preparações avaliadas 1 vez;

Precisão
Precisão intermediária (precisão inter-corridas): concordância entre os
resultados do mesmo laboratório, mas obtidos em dias diferentes, com
analistas diferentes e/ou equipamentos diferentes. Para a
determinação da precisão intermediária recomenda-se um mínimo de
2 dias diferentes com analistas diferentes (usualmente até 5%)
Reprodutibilidade (precisão inter-laboratorial): concordância entre os
resultados obtidos em laboratórios diferentes. Estes dados não precisam
ser apresentados para a concessão de registro (usualmente até 5%)
A precisão pode ser expressa como desvio padrão relativo (DPR) ou
coeficiente de variação (CV%), não podendo ser superior a 5%

Exatidão A exatidão de um método analítico é a proximidade dos resultados obtidos
pelo método em estudo em relação ao valor verdadeiro.
Fármaco
Uso de metodologia analítica proposta na análise de uma substância de pureza
conhecida (padrão de referência);
Comparação dos resultados entre metodologias;
Forma Farmacêutica
Placebo contaminado com quantidade conhecida de fármaco ou da
substância que se quer avaliar (no caso da indisponibilidade de amostras de
certas impurezas e/ou produtos de degradação = comparação entre
metodologias

Exatidão - Procedimento
• 9 determinações contemplando o intervalo linear do
procedimento:
– 3 concentrações na baixa – 3 replicatas;
– 3 concentrações na média – 3 replicatas;
– 3 concentrações na alta – 3 replicatas;
• Determinar a relação entre a concentração média
determinada experimentalmente e a concentração
teórica correspondente


Robustez
A robustez de um método analítico é a medida de sua capacidade em resistir a
pequenas e deliberadas variações dos parâmetros analíticos. Indica sua
confiança durante o uso normal (RDC 899/2003)
Durante o desenvolvimento da metodologia, deve-se considerar a
avaliação da robustez. Constatando-se a susceptibilidade do método à
variações nas condições analíticas, estas deverão ser controladas e
precauções devem ser incluídas no procedimento.

Intervalo
O intervalo especificado é a faixa entre os limites de
quantificação superior e inferior de um método analítico.
Normalmente é derivado do estudo de linearidade e
depende da aplicação pretendida do método.
É estabelecido pela confirmação de que o método
apresenta exatidão, precisão e linearidade adequados
quando aplicados a amostras contendo quantidades de
substâncias dentro do intervalo especificado.

Estabilidade das soluções
Não detalhada na RDC 899/2003;
Necessária para garantir que o analito tem
estabilidade no mínimo no tempo de análise;
Importante para garantir as condições nas quais
o analito é estável (refrigerado, ambiente) para
estabelecer por exemplo,
soluções-mãe de compostos pouco disponíveis ou caros,
Tempo no qual as soluções podem ser mantidas para uma eventual investigação
de resultados não conforme;

Relatório de validação
• Art. 490. Os relatórios devem refletir os protocolos seguidos
e contemplar, no mínimo, o título, o objetivo do estudo,
bem como fazer referência ao protocolo, detalhes de
materiais, equipamentos, programas e ciclos utilizados e
ainda, os procedimentos e métodos que foram utilizados.
– Art. 491. Os resultados devem ser avaliados, analisados e
comparados com os critérios de aceitação previamente
estabelecidos.
– Art. 492. Os Departamentos responsáveis pelos trabalhos de
qualificação e validação devem aprovar o relatório completo.

Exemplos de
parâmetros para
verificação de
métodos
Não há guia da ANVISA,
cada laboratório deve
fornecer racional para
escolha dos parâmetros de
verificação

Documentação administrativa de
registroEmbalagem primária e secundária funcional
201

Embalagem primária e secundária
funcional
a. Descrição do material de embalagem; e
b. Relatório com especificações, método
analítico e resultados do controle de
qualidade de embalagem.
202

Materiais de embalagem
203

Documentação administrativa de
registroInvoltório intermediário
204

Documentação administrativa de
registroAcessórios
205

Documentação administrativa de
registroEstudo de estabilidade do produto terminado
206

Estabilidade do produto acabado
a. Relatório com os resultados dos estudos de estabilidade
acelerada e de longa duração conduzidos com 3 (três)
lotes, protocolos usados, incluindo conclusões com relação
aos cuidados de conservação e prazo de validade;
b. Resultados de estudos de estabilidade para medicamentos
que, após abertos ou preparados, possam sofrer alteração
no seu prazo de validade original ou cuidado de
conservação original; e
c. Resultados do estudo de fotoestabilidade ou justificativa
técnica para a isenção do estudo;
207

RE 01/2005 – condições de estabilidade208
Tipo do estudo Pontos de análise
Estabilidade acelerada 0, 3, 6 meses
Estabilidade longa duração 0, 3, 6, 9, 12, 18, 24, 36, 48, 60 meses
Estabilidade acompanhamento Anualmente até prazo de validade
Estudo acelerado + longa duração (6 meses) = prazo provisório de 24 meses

Testes obrigatórios por forma farmacêutica
TestesSólidos
orais
Suspensõ
es
Semi
sólidosLíquidos
Aparência x x x x
Teor do ativo x x x x
Quantificação de produtos de
degradaçãox x x x
Limites microbianos x x x x
Dissolução x x --- ---
Dureza x --- --- ---
pH --- x x x
Sedimentação pós agitação --- x --- ---
Perda de peso (base aquosa) --- x x x
Separação de fases (emulsões e cremes) --- --- x ---
Claridade da solução --- --- --- x
Tamanho de partícula e permeabilidade
(em pós registro somente)--- X X ---
209

Requisitos específicos para o registro
de medicamentos novos, genéricos e
similares210

Requisitos específicos211

Equivalência Farmacêutica
RDC 31/2010212

Equivalência Farmacêutica
Seguir orientações da RDC 31/2010;
Usar método e padrão compendial se disponível
Usar método de dissolução desenvolvido conforme NT 03/2013
Precede obrigatoriamente o estudo de BE
Compra de medicamento referência em território Brasileiro
Deve ser realizado em centro autorizado pela ANVISA
http://portal.anvisa.gov.br/wps/content/Anvisa+Portal/Anvisa/Inicio/Medicamentos/Assunto+de+Interesse/Equi
valencia+farmaceutica/Habilitados+Nacionais
Conjunto de ensaios físico-químicos e, quando aplicáveis, microbiológicos e biológicos, que comprovam que dois
medicamentos são Equivalentes (registro de genéricos/similares e alterações pós registro de medicamentos):
Etapa físico química
Etapa microbiológica
Perfil de dissolução comparativo
Seguir templates disponibilizados pela ANVISA: ttp://s.anvisa.gov.br/wps/s/r/Lk
213

214
1. Perfil de dissolução com mesmo
comportamento
2. Desvios conforme tipo de perfil:
a. Muito rápido: 15 minutos ≤ 10%
b. Demais:
40% pontos iniciais: ≤ 20%
Demais pontos: ≤ 10%

Bioequivalência
RE 1170/2006215

Bioequivalência/BDR
Deve seguir orientações RE 1170/2006;
Deve ser realizado em centro autorizado pela ANVISA
• Realizado somente após EQFAR se teor entre teste e referência tiver diferência
inferior a 5%;
Quantificação conforme ANVISA:
Lista de analito para quantificação
Lista de forma de administração
Escolha da dose depende da farmacocinética da molécula – RDC 37/2012;
Utilizando indivíduos saudáveis, acima de 18 anos, peso normal, que assinam
termo de consentimento livre e esclarecido
Exceções para medicamento contendo fármacos tóxicos;
216

Quando não precisa fazer BE?
I - soluções aquosas que contenham o mesmo fármaco, na mesma concentração em relação
ao medicamento de referência e excipientes de mesma função que aqueles presentes no
medicamento comparador;
II - pós para reconstituição que resultem em soluções aquosas orais ou parenterais, desde que
cumpram os requisitos descritos no inciso I;
III - gases;
IV - soluções oleosas parenterais que contenham o mesmo fármaco, na mesma concentração
em relação ao medicamento de referência e qualitativamente o mesmo veículo oleoso
presente no medicamento de referência, em concentrações compatíveis com a função
pretendida;
V - medicamentos de uso oral que contenham fármacos destinados a ação local no trato
gastrintestinal descritos na Lista 3 - Fármacos de ação local no trato gastrintestinal que não
necessitam de estudos de biodisponibilidade relativa / bioequivalência
217

Quando não precisa fazer BE?
VI - medicamentos de aplicação tópica, não destinados a efeitos sistêmicos, que
contenham o mesmo fármaco, na mesma concentração em relação ao medicamento
de referência e excipientes de mesma função que aqueles presentes no medicamento
comparador.
VII - Demais doses de um medicamento submetido à BE (se formulação proporcional);
VIII - Medicamentos de classe 1 no sistema SCB (enviar dados in vitro comprobatórios),
conforme lista da ANVISA
Acido acetilsalicílico, cloridrato de propranolol, cloridrato de doxiciclina, dipirona,
estavudina, fluconazol, hemitartarato de rivastigmina, isoniazida, levofloxacino,
metoprolol, metronidazol, paracetamol, sotalol, ou temozolomida.
218

219 Escolha da dose para estudo de BE

220 Escolha da dose para estudo de BE

221
http://www.uspharmacist.com/content/s/253/c/41306/
Parâmetros plasmáticos para BE
f) dois medicamentos serão
considerados bioequivalentes se
os valores extremos do intervalo
de confiança de 90% da razão
das médias geométricas (ASC0-t
teste/ASC0-t referência e
Cmaxteste/Cmaxreferência) forem
maiores que 0,8 e menores que
1,25.
Outros limites de IC de 90%
para Cmax, previamente
estabelecidos no protocolo,
poderão ser aceitos mediante
justificativas científicas. Quando
clinicamente relevante, Tmax
deverá também ser considerado;

Aditamento dos estudos de BE/BDR
Aditar diretamente à CETER – assuntos:
10416 - GENÉRICO - Aditamento de estudo de biodisponibilidade relativa
10415 - SIMILAR - Aditamento de estudo de biodisponibilidade relativa
557 - NOVO - Aditamento de estudo de biodisponibilidade relativa
222
Documento Centro local Centro internacional
1. Folha de rosto conforme modelo Sim Sim
2. Justificativa para o aditamento Sim Sim
3. Comprovante de isenção de taxa Sim Sim
4. Identificação do estudo que consta na petição com base no modelo de declaração
Sim Sim
5. Lista de outros estudos conforme modelo de comunicação de outros estudos realizados com o mesmo medicamento Teste
Não Sim
6. Cópia do Certificado de Equivalência Farmacêutica; Relatório completo de BD/BE em formato digital PDF, gravado em CD;
Não Sim
7. Planilhas em MS-Excel dos resultados dos parâmetros farmacocinéticos ASC0-t, ASC0-∞, CMAX e TMAX, calculados individualmente e valores individuais das concentrações plasmáticas do fármaco, separados por produto, para todas as fases do estudo
Não (Sineb) Sim
Enviar na forma impressa o relatório final + protocolo e aprovação do comitê de ética para todas as petições que requerem BE/BDR

Estudos Clínicos223

224 Fases dos Estudos Clínicos
https://cern-foundation.org/?page_id=292

Renovação de registro225

Documentação de renovação
Quando? Primeiro semestre do último ano do quinquênio de validade do
registro já concedido, deverão apresentar:
I.Formulários de petição, FP1 e FP2, preenchidos e assinados;
II.Comprovante de pagamento da Taxa de Fiscalização de Vigilância
Sanitária-TFVS e respectiva Guia de Recolhimento da União-GRU, ou
isenção, quando for o caso;
III.Sumário executivo em português referente ao período de cinco anos do
Relatório Periódico de Farmacovigilância do mesmo período; e
IV.Documento comprobatório de venda no último quinquênio de vigência
do registro, contendo os números das notas fiscais emitidas no Brasil e a
relação de estabelecimentos compradores em um mínimo de 1 (uma)
nota fiscal emitida no País, por forma farmacêutica e concentração.
226

Disposições finais e
transitórias227

Enfim... A empresa detentora ou fabricante do medicamento poderá ser
inspecionada para verificação in loco de dados e informações da
petição de concessão e renovação do registro, a critério da Anvisa.
(RDC 20/2015)
Será divulgada informação na página eletrônica da Anvisa com a
decisão final da análise técnica da solicitação do registro do
medicamento. (RDC 20/2015)
228

Após submissão na ANVISA
e antes da aprovação229

Genéricos/Similar - Fluxo230
Escolha da molécula
Prospecção de fabricantes da
molécula
Desenvolvimento inicial da formulação
Escolha dos excipientes
Avaliação de possíveis
processos produtivos
Refinamento do
desenvolvimento da
formulação
Desenvolvimento e validação
de metodologia
analítica
Produção
Lotes pilotos
Avaliação da equivalência in
vitro ao referência
Estudos de estabilidade acelerada
Estudos de Bioequivalênci
a
Estudos de estabilidade de longa duração
Montagem do dossiê
Submissão regulatória
Aprovação

Filas de análise – 15/08/2015
Tipo de registro Tamanho da fila Processo mais antigo
Generico/Similar 1066 processos 09/01/2009
Novo (incremental) 134 processos 22/12/2008
Novo (radical – eletrônico) 5 processos 05/06/2015
231

Medicamentos novos232
http://www.anvisa.gov.br/listadepeticoes/fila_tipo_produto.asp?nomeCombo=MEDICAMENTOS

Medicamentos novos233

Medicamentos Inovadores234
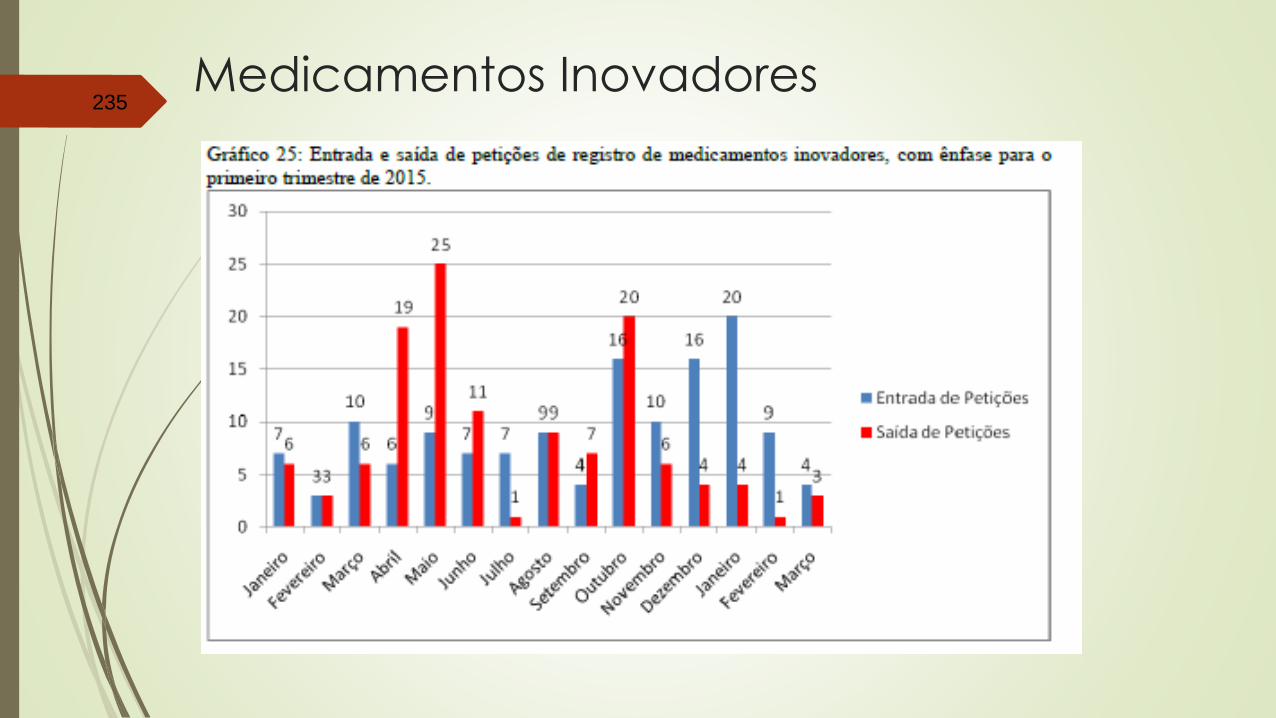
Medicamentos Inovadores235

Medicamentos Genérico/Similar236

Medicamentos Genérico/Similar237

239
Exigência
Resposta
OK?
Sim Não
ExigênciaAprovação
Publicação Resposta
OK?
Não
Indeferimento Recurso Julgamento
OK?
Indeferimento Publicação
120 dias
15 a 60 dias
30 dias
30 dias
120 dias
15 a 60 dias
30 dias
10 dias 2 anos
30 dias

Medicamentos novos240

Medicamentos Inovadores241

Medicamentos Genérico/Similar242

Motivos de indeferimento243

Referências e links1. ANVISA – hotsite medicamentos -
http://portal.anvisa.gov.br/wps/content/Anvisa+Portal/Anvisa/Inicio/Medicamentos
2. Perguntas e Repostas Registro de medicamentos genéricos, novos e similares: Tecnologia
Farmacêutica -
http://portal.anvisa.gov.br/wps/wcm/connect/918cda8048c24f5c8623be0a466faa84/FAQ+
GRMED.pdf?MOD=AJPERES
3. Bases técnicas e cinetíficas para aprovação de registro -
http://www.anvisa.gov.br/datavisa/Fila_de_analise/frmResultado.asp
4. Consulta medicamento referência ANVISA -
http://portal.anvisa.gov.br/wps/content/Anvisa+Portal/Anvisa/Inicio/Medicamentos/Assunto
+de+Interesse/Medicamentos+de+referencia
5. Passo a passo de peticionamento eletrônico -
http://portal.anvisa.gov.br/wps/wcm/connect/6a87918045041a649e95feef4ebdb6ee/Peticio
namento+eletr%C3%B4nico.pdf?MOD=AJPERES
244

Referências e links6. Lista de medicamento referência em avaliação –
http://portal.anvisa.gov.br/wps/wcm/connect/101a3e00460302feb136b57ffa9843d8/Lista+A+em+av
alia%C3%A7%C3%A3o+29-10-2014.pdf?MOD=AJPERES
7. RDC 35/2012 – inclusão e exclusão de medicamento referência -
http://portal.anvisa.gov.br/wps/wcm/connect/84fce9804d642e10b823f9c116238c3b/RESOLU%C3%8
7%C3%83O+rdc+35+de+2012.pdf?MOD=AJPERES
8. NT 46/2013 -
http://portal.anvisa.gov.br/wps/wcm/connect/640e56804eca7e149b239b8a610f4177/nota+tecnica
+n+46_2013.pdf?MOD=AJPERES
9. ANVISA – hotsite medicamentos genéricos -
http://portal.anvisa.gov.br/wps/content/Anvisa+Portal/Anvisa/Inicio/Medicamentos/Assunto+de+Int
eresse/Medicamentos+genericos
10. Lista I da NT 46/2013 - http://s.anvisa.gov.br/wps/s/r/bxnZ
11. ANVISA – hotsite medicamentos similares - http://s.anvisa.gov.br/wps/s/r/ft
12. ANVISA – hotsite medicamento similar único - http://s.anvisa.gov.br/wps/s/r/b0T2
245

Referências e links13. ANVISA – lista de similar e seus medicamentos de referência -
http://portal.anvisa.gov.br/wps/wcm/connect/f611970048af1f74ac42bc0a466faa84/Lista+sit
e+01-06-15.pdf?MOD=AJPERES
14. ANVISA – hotsite Equivalência Farmacêutica -
http://portal.anvisa.gov.br/wps/content/Anvisa+Portal/Anvisa/Inicio/Medicamentos/Assunto
+de+Interesse/Equivalencia+farmaceutica
15. RDC 31/2010 – Equivalência Farmacêutica -
http://portal.anvisa.gov.br/wps/wcm/connect/89d63480474597439fb9df3fbc4c6735/RDC_31
_2010_Disp%C3%B5e+sobre+a+realiza%C3%A7%C3%A3o+dos+Estudos+de+Equival%C3%AAn
cia+Farmac%C3%AAutica+e+de+Perfil+de+Dissolu%C3%A7%C3%A3o+Comparativo.pdf?MO
D=AJPERES
16. RDC 899/2003 – Validação analítica -
http://portal.anvisa.gov.br/wps/wcm/connect/4983b0004745975da005f43fbc4c6735/RE_899_
2003_Determina+a+publica%C3%A7%C3%A3o+do+Guia+para+valida%C3%A7%C3%A3o+de
+m%C3%A9todos+anal%C3%ADticos+e+bioanal%C3%ADticos.pdf?MOD=AJPERES
246

Referências e links17. RDC 37/2009 – Admissibilidade de farmacopeias -
http://portal.anvisa.gov.br/wps/wcm/connect/e9d6f98048e524df8f859f466b74119d/RDC_37_2009_Tr
ata%2Bda%2Badmissibilidade%2Bdas%2BFarmacop%C3%A9ias%2Bestrangeiras..pdf?MOD=AJPERES
18. ANVISA – hotsite Bioequivalência -
http://portal.anvisa.gov.br/wps/content/Anvisa+Portal/Anvisa/Inicio/Medicamentos/Assunto+de+Int
eresse/Bioequivalencia+e+Biodisponibilidade
19. ANVISA – forma de administração para estudos de BE -
http://portal.anvisa.gov.br/wps/wcm/connect/b0be460049584ec18332dbb32cf0f1c1/Lista+1+03+08
+15.pdf?MOD=AJPERES
20. ANVISA – analito para determinação de BE -
http://portal.anvisa.gov.br/wps/wcm/connect/41b0228049584f91833bdbb32cf0f1c1/Lista+2+03+08+
2015.pdf?MOD=AJPERES
21. ANVISA – lista de fármacos com ação no TGI – isentos de BE -
http://portal.anvisa.gov.br/wps/wcm/connect/5bcf8700486e643abdb7bf734e60b39c/02+Lista+3_13
_06_2012.pdf?MOD=AJPERES
247

Referências e links22. ANVISA – hotsite bulário eletrônico - http://s.anvisa.gov.br/wps/s/r/f4
23. RDC 137/2003 – frase de alerta - http://www.cff.org.br/userfiles/file/resolucao_sanitaria/137.pdf
24. ANVISA – hotsite bulas -
http://portal.anvisa.gov.br/wps/content/Anvisa+Portal/Anvisa/Inicio/Medicamentos/Assunto+de+Int
eresse/Bulas+e+Rotulos+de+medicamentos/Bulas
25. RDC 47/2009 – bulas -
http://portal.anvisa.gov.br/wps/wcm/connect/2e6206804745886991f3d53fbc4c6735/Microsoft+Wor
d+-+RDC+n+47-09_bulas_vers%C3%A3o+republicada.pdf?MOD=AJPERES
26. ANVISA – hotsite rotulagem -
http://portal.anvisa.gov.br/wps/content/Anvisa+Portal/Anvisa/Inicio/Medicamentos/Assunto+de+Int
eresse/Bulas+e+Rotulos+de+medicamentos/Rotulos
27. ANVISA – manual de identidade visual de rotulagem -
http://portal.anvisa.gov.br/wps/wcm/connect/fb84e68045c83f28a0bde2d10ee53f37/Manual+identi
dade+visual+medicamentos+SUS_DAF_11_09_14_atualizad.pdf?MOD=AJPERES
248

Referências e links28. ANVISA – hotsite clones -
http://portal.anvisa.gov.br/wps/content/Anvisa+Portal/Anvisa/Inicio/Medicamentos/Assunto+de+Int
eresse/Medicamentos+clones
29. RDC 31/2014 – clones -
http://portal.anvisa.gov.br/wps/wcm/connect/58c5ce80456c04e4a0f7a40bb3a02a58/RDC+31_2014
_CLONES.pdf?MOD=AJPERES
30. OS 06/2013 – indeferimento sumário genéricos e similares -
http://portal.anvisa.gov.br/wps/wcm/connect/a916590040388956b333f3dc5a12ff52/OS+N%C2%BA+
06-2013.pdf?MOD=AJPERES
31. ANVISA – FAQ RDC 09/2015 -
http://portal.anvisa.gov.br/wps/wcm/connect/d450ca8048c807258b6abf0a466faa84/Perguntas+e+
Respostas+SAT+-+RDC+09-2015.pdf?MOD=AJPERES
32. ANVISA – manual de submissão de DDCM -
http://portal.anvisa.gov.br/wps/wcm/connect/df82470047488da08cb0ae2e90f0f661/Microsoft+Wor
d+-+Guia+de+Submiss%C3%A3o+do+DDCM+e+Protocolos+13-02-
2015+Vers%C3%A3o+Final_corrigida.pdf?MOD=AJPERES
249

Referências e links33. ANVISA – manual alteração DDCM -
http://portal.anvisa.gov.br/wps/wcm/connect/901cfa804782f534b323fbfe096a5d32/Manual+Para+S
ubmiss%C3%A3o+de+Emendas+Modifica%C3%A7%C3%B5es+Cancelamentos+e+Suspens%C3%B5es
.pdf?MOD=AJPERES
34. ANVISA – manual qualidade DDCM sintéticos -
http://portal.anvisa.gov.br/wps/wcm/connect/69072a004782f6e8b335fbfe096a5d32/Manual+de+su
bmiss%C3%A3o+dos+requisitos+de+qualidade_sint%C3%A9ticos+%285%29.pdf?MOD=AJPERES
35. ANVISA – hotsite relatórios gerenciais - http://s.anvisa.gov.br/wps/s/r/0a
36. ANVISA – hotsite resultado de priorização de análise - http://s.anvisa.gov.br/wps/s/r/YSf
37. ANVISA – hotsite workshop validação – dez/2013 -
http://portal.anvisa.gov.br/wps/content/Anvisa+Portal/Anvisa/Inicio/Medicamentos/Assunto+de+Int
eresse/Workshop
38. ANVISA – bulário eletrônico - http://www.anvisa.gov.br/datavisa/fila_bula/index.asp
39. ANVISA – manual de vocabulário controlado -
http://portal.anvisa.gov.br/wps/wcm/connect/497d908047458b5f952bd53fbc4c6735/vocabulario_c
ontrolado_medicamentos_Anvisa.pdf?MOD=AJPERES
250

Referências e links41. ANVISA – avaliação de medicamentos novos - http://s.anvisa.gov.br/wps/s/r/kf
42. ANVISA – uso de consultores ad hoc para avaliação de eficácia - http://s.anvisa.gov.br/wps/s/r/cwp
43. ANVISA – fluxo de encaminhamento para consultor ad hoc - http://s.anvisa.gov.br/wps/s/r/QI
44. ANVISA guia ADF para HAS -
http://portal.anvisa.gov.br/wps/wcm/connect/600d4a0047458ec797d6d73fbc4c6735/Guia+para+Registro+de+Associa%C3%A7%C3%B5es+e
m+Dose+Fixa+para+o+Tratamento+da+Hipertens%C3%A3o+Arterial.pdf?MOD=AJPERES
45. ANVISA – guia para ADF -
http://portal.anvisa.gov.br/wps/wcm/connect/979397004745767a8465d43fbc4c6735/Guia+para+Registro+de+Novas+Associa%C3%A7%C3%B
5es+em+Dose+Fixa.pdf?MOD=AJPERES
46. ANVISA – hotsite consultas bancos de dados - http://s.anvisa.gov.br/wps/s/r/bSOe
47. ANVISA – lista de preços de medicamentos - http://s.anvisa.gov.br/wps/s/r/UfT
48. ANVISA – consulta CBPF - http://www.anvisa.gov.br/certificadoBoasPraticas/principal/index.asp
49. ANVISA – consulta medicamentos - http://www7.anvisa.gov.br/datavisa/Consulta_Produto/consulta_medicamento.asp
50. ANVISA – lista preço máximo medicamentos – julho/2015 -
http://portal.anvisa.gov.br/wps/wcm/connect/db3c07804943443db0e8f9b32cf0f1c1/LISTA+CONFORMIDADE_2015-07-20.pdf?MOD=AJPERES
51. ANVISA – hotsite descontinuação medicamento - http://s.anvisa.gov.br/wps/s/r/cWw8
251