Embed Size (px)
Citation preview

Diana Raquel Martins Fernandes Rompante Ferreira
ATAXIA DE FRIEDREICH
REVISÃO BIBLIOGRÁFICA E CASO CLÍNICO
Dissertação de Mestrado Integrado em Medicina
Artigo de Revisão Bibliográfica
Orientador: José Fernando da Rocha Barros, Assistente Graduado de Neurologia e
Professor Auxiliar Convidado de Neurologia
Instituto de Ciências Biomédicas Abel Salazar, Largo Prof. Abel Salazar, 2. 4099-003
Porto
PORTO, 2011

2
Resumo
A ataxia de Friedreich (FRDA) é uma doença neurológica, genética,
degenerativa. Tem uma distribuição epidemiológica universal, sendo a mais frequente
das ataxias autossómicas recessivas, com uma prevalência de 2 a 4/100000 e uma
frequência do estado de portador estimada em 1/120. Pretendemos sintetizar o
conhecimento sobre esta patologia em diferentes perspectivas, complementando o
trabalho com a observação de uma doente, descrição e discussão do seu caso clínico.
Clinicamente a FRDA é caracterizada por início dos sintomas antes dos 25 anos,
ataxia mista progressiva, ausência de reflexos osteotendinosos, perda de sensibilidades
profundas e sinal de Babinski bilateral. Outras alterações são a disfagia, alterações
oculomotoras, cardiomiopatia, deformidades ortopédicas e diabetes mellitus. Podem
ocorrer quadros de apresentação atípica.
A causa é uma mutação no gene FXN que se localiza no cromossoma 9, que
codifica para a frataxina, proteína constituinte da matriz mitocondrial e envolvida no
processo de homeostase do ferro celular. Ela deve-se a uma repetição de tripletos GAA
no primeiro intrão do gene. A maioria dos pacientes é homozigoto para esta mutação.
O diagnóstico é feito pela clínica e por genética molecular. Impõe-se à distinção
clínica com a ataxia por deficiência de vitamina E, ataxia-telangiectasia e ataxia com
apraxia oculomotora. O diagnóstico definitivo é feito por detecção molecular da
mutação no gene FXN.
A FRDA não tem cura ou tratamento etiológico e as tentativas têm sido pouco
robustas. A terapêutica sintomática pode ajudar os doentes e famílias a garantir
dignidade e qualidade de vida.
Concluindo, a FRDA é uma patologia com sintomatologia progressiva e
debilitante e para a qual ainda não existe cura, sendo imperativo prosseguir com a
investigação terapêutica. A genética molecular revolucionou o diagnóstico e alargou o
espectro clínico desta doença.
Palavras-chave: Ataxia de Friedreich; ataxia recessiva; consanguinidade; arreflexia;
síndrome cordonal posterior; cardiopatia hipertófica; escoliose; pés cavus; gene FXN;
frataxina; homeostase do ferro.

3
Abstract
Friedreich‟s ataxia (FRDA) is a neurological, genetic, degenerative disease. It
has an universal epidemic distribution, being the most frequent autossomal recessive
ataxia, with a prevalence of 2 to 4/100000 and a frequency of carrier estimated at 1/120.
We intent to synthesize the knowledge about this pathology in different perspectives,
completing the work with the observation of a patient, description and discussion of his
case history.
Clinically, FRDA is characterized by onset of symptoms before age 25,
progressive mixed ataxia, absence of deep tendon reflexes, loss of deep sensitivity and
bilateral Babinski sign. Others symptons are dysphagia, oculomotor changes,
cardiomiopathy, orthopedic deformities and diabetes mellitus. Clinical presentation can
be atypical.
Its cause is a mutation on FXN gene which is located on chromosome 9,
responsible for encoding frataxin, a protein constituent of the mitochondrial matrix and
involved in cellular iron homeostasis. This is due to a GAA repeat in the first intron of
the gene. Most patients are homozygous for this mutation.
The diagnosis is made by clinical presentation and molecular genetics. The
differential diagnosis must be established with ataxia with vitamin E deficiency, ataxia-
telangiectasia and ataxia with oculomotor apraxia. The definitive diagnosis is made by
molecular detection of gene mutation in FXN.
The FRDA has no cure or etiologic treatment and previous attempts have been
less than robust. The symptomatic therapy can help patients and families to ensure
dignity and quality of life.
In conclusion, FRDA is a disease with progressive and debilitating symptoms,
for which there is still no cure, making it imperative to proceed with therapeutic
research. Molecular genetics has revolutionized the diagnosis and broadened the clinical
spectrum of this disease.
Key-words: Friedreich ataxia; recessive ataxia; consanguinity; areflexia; syndrome of
posterior columns; hypertrophic cardiomiopathy; scoliosis; pes cavus; FXN gene;
frataxin; iron homeostasis.

4
1 - Introdução
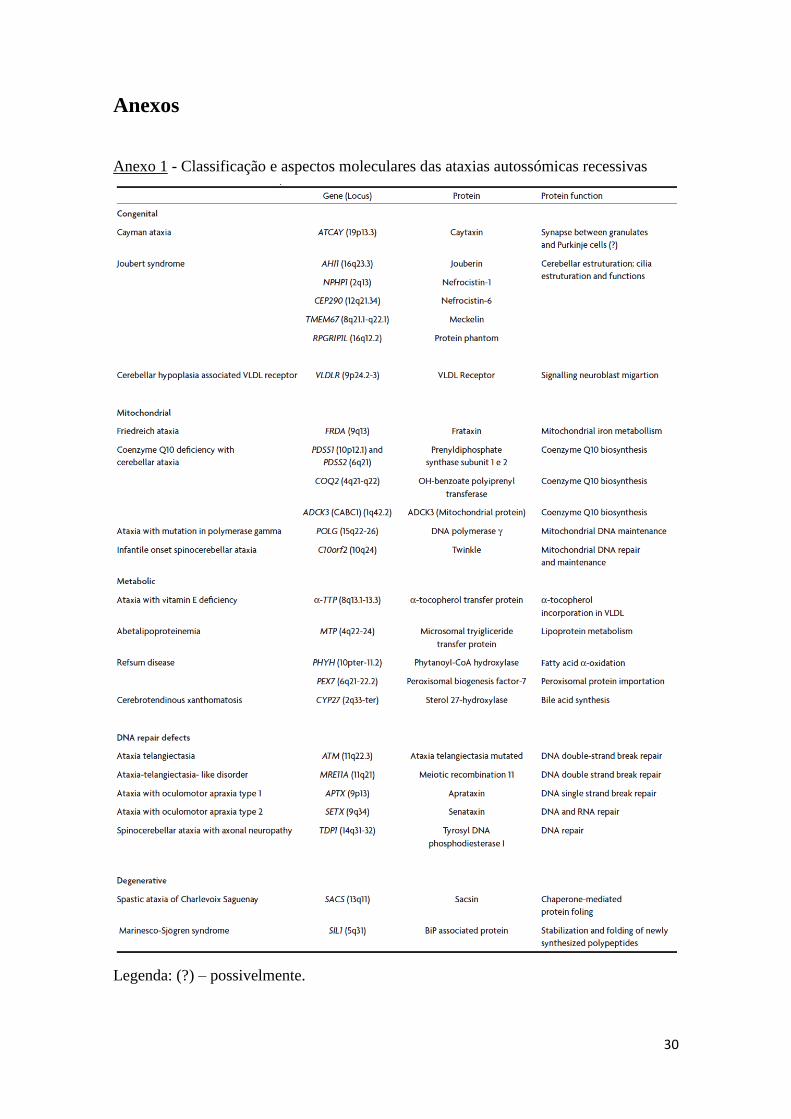
Estão reconhecidos mais de 20 tipos clínicos de ataxias autossómicas recessivas
(AR). Elas são caracterizadas, essencialmente, por desequilíbrio, incoordenação motora,
tremor cinético ou postural e disartria (1). A ataxia de Friedreich (FRDA) pertence a
este grupo, estando na subcategoria das AR mitocondriais (Anexo 1) (2).
A ataxia de Friedreich é uma doença neurológica, genética e degenerativa, que
afecta, principalmente, o sistema nervoso e o coração (3). Foi descrita pela primeira vez
em 1863 pelo Dr. Nicholaus Friedreich como uma “atrofia degenerativa das colunas
posteriores da espinhal medula” (4). A FRDA é a mais frequente das AR, com uma
prevalência de cerca de 2 a 4/100000, uma frequência do estado de portador estimada
em 1/120 e uma distribuição epidemiológica universal (5), embora mais frequente em
humanos de origem europeia. A idade média de morte nestes doentes é
aproximadamente aos 38 anos, podendo variar entre os 5 e os 70 anos (1).

5
2 - Características Clínicas
A FRDA caracteriza-se por início dos sintomas antes dos 25 anos, ataxia
progressiva, ausência de reflexos osteotendinosos, perda de sensibilidades profundas e
sinal de Babinski bilateral. A idade de início mais precoce pode aproximar-se dos 2
anos (6). A ataxia mista (cerebelosa e sensitiva profunda) é o sintoma cardinal. Os
doentes sentem desequilíbrio e têm quedas, além de dificuldades crescentes nas
actividades que requerem destreza (escrita, asseio pessoal). A dismetria, tremor de
intenção e marcha com base alargada com mudança permanente de posição e
titubeação, levam a perda da autonomia em 10 a 15 anos (1,3,7). A disartria
caracterizada por discurso lento, imprecisão na articulação e monotonia surge após 2 a 5
anos de evolução da doença e pode progridir para anartria (1). A disfagia para líquidos é
um sintoma da doença avançada e progride até ser necessária entubação nasogástrica ou
gastrostomia percutânea (4). As funções cognitivas estão preservadas (1,3,4).
As perturbações oculares são tardias, sendo o nistagmo e a instabilidade de
fixação as alterações mais comuns (8). Há outras características clínicas como a
cardiopatia hipertrófica (mais de 50% dos casos), sendo uma importante causa de morte
(9); a escoliose, presente em aproximadamente 100% dos casos, apresentando
curvaturas ligeira a moderada (<40º) ou severa (>60º) (10,11), necessitando de
correcção cirúrgica; pé cavo (96%) (12); atrofias distais (50%); diabetes mellitus (10 –
20%) e surdez neurossensorial (6,13). A escoliose está frequentemente associada a
hipertonia dos músculos para-espinais, sugerindo estas alterações como causa da
deformidade anatómica em alguns casos desta patologia (14).
No entanto existem quadros de apresentação atípica da FRDA, sendo os mais
conhecidos a LOFA (Late-Onset Friedreich Ataxia), caracterizada por uma idade de
início tardia (6) e a FARR (Friedreich Ataxia with Retained Reflexes) caracterizada pela
presença de reflexos osteotendinosos. Os doentes com LOFA não apresentam,
habitualmente, atrofia óptica, surdez neurossensorial, diabetes mellitus ou evidência de
cardiomiopatia (15). Um outro quadro clínico (algumas das mutações missense)
manifesta-se por uma forma menos severa com marcha espástica no início da doença,
progressão lenta, ausência de disartria, hiper/normorreflexia e ausência de ataxia
cerebelosa (3,16). Outras apresentações raras caracterizam-se por alterações do tempo

6
de evolução da doença (casos de progressão rápida com perda de autonomia locomotora
em menos de 10 anos e pacientes que preservam a marcha, precisando apenas de apoio
passados mais de 15 anos de evolução), indivíduos com idade de início tardia e
preservação dos reflexos osteotendinosos, apresentações atípicas das alterações oculares
e movimentos hipercinéticos como tremores (sendo que este surge mais frequentemente
em doentes com quadro clínico típico de FRDA), distonia, mioclonia e coreia (15,17).

7
3 - Patofisiologia
Relativamente à patofisiologia, o sistema somatossensorial que conduz a
sensibilidade proprioceptiva é afectado no início da doença e de forma severa (3). Há
evidências de desmielinização e diminuição do número de neurónios dos cordões
posteriores e feixes espinocerebelosos devido ao atrofiamento e morte dos neurónios
localizados nos gânglios dorsais e no nucleus thoracicus posterior, respectivamente. A
nível do sistema locomotor, observa-se degenerescência progressiva dos feixes
piramidais, sendo esta mais evidente distalmente, sugerindo um processo de dying-back
(3,4,18). A nível do cerebelo observa-se envolvimento do vérmis e núcleo dentado,
sendo esta degenerescência de apresentação tardia (3). As lesões anteriormente referidas
explicam a origem mista da ataxia nesta doença.
Estudos de potenciais evocados demonstraram alterações severas e simétricas
dos SSEPs (Somatosensory Evoked Potencial) e condução motora central anormal na
fase inicial da doença. Também foram detectadas alterações ao nível dos VERs (Visual
Evoked Response) e BAERs (Brainstem Auditory Evoked Response) (4). As
velocidades de condução nervosa (VCNs) confirmam a perda prematura dos SNAPs
(Sensory Nerve Action Potentials), mas com redução apenas ligeira a moderada das
VCNs (4,19).
Ao nível do músculo cardíaco, o ECG demonstra inversão das ondas T,
hipertrofia ventricular, alterações da condução cardíaca (10%), extra-sístoles
supraventriculares e, ocasionalmente, fibrilação auricular. Ecocardiograficamente
observa-se hipertrofia concêntrica ventricular e hipertrofia assimétrica septal (2).
Histologicamente, o coração apresenta perda de fibras nervosas, fibrose difusa, necrose
miocárdica focal e hipertrofia celular (9,13).

8
4 - Patogénese
4.1 - O gene FXN
A causa da FRDA é uma mutação no gene FXN que contém 7 exões e se localiza
no braço longo do cromossoma 9, no locus 9q13-21.1, tendo sido descoberto em 1988 e
associada a esta patologia em 1996 (20,21). O gene FXN é expresso em todas as células,
mas em níveis variáveis nos diferentes tecidos e estes variam durante o
desenvolvimento, sendo a sua expressão elevada naqueles que são mais afectados pela
doença, como é o caso dos tecidos nervoso e cardíaco (também é expresso a nível do
fígado, músculo esquelético e pâncreas) (21,22,23).
A FRDA é uma doença de repetição de tripletos GAA no primeiro intrão do
gene superior há observada nos indivíduos sem patologia (<40 repetições) (4). A
maioria dos pacientes é homozigoto para este mutação (95-98%), sendo que apenas uma
pequena percentagem é heterozigoto, contendo a expansão GAA num alelo e uma
mutação pontual no outro, sendo estas do tipo missense, nonsense, mutações no splice
site e deleções (3,4,24,25). Das mutações missense, algumas são responsáveis por um
fenótipo de evolução atípica e mais benigna (D122Y, R165P, G130V), enquanto outras
agravam a evolução do quadro clínico (W173G, devido a total ausência de actividade)
(3,25,26). O tamanho da expansão GAA (entre 70 a 1000), no alelo mais pequeno
(relação inversa com a expressão genética residual), está inversamente relacionado com
a idade de início da doença e directamente relacionado com a perda da autonomia e o
desenvolvimento de cardiomiopatia (3,4). Outros factores influenciam o fenótipo, entre
eles genes modificadores, variação na sequência de DNA mitocondrial e mosaicismo
celular (4). A transmissão hereditária da expansão GAA é instável, sendo que expansões
ou contracções são igualmente comuns após transmissão materna, enquanto estas
últimas são mais frequentes por transmissão paterna (3,4,27).
O mecanismo molecular através do qual as expansões GAA causam FRDA está
relacionado com a formação de estruturas de DNA anormais (estrutura não-B DNA) que
interferem com a transcrição (3,28,29,30). Duas destas formam uma extensão de DNA
que contém apenas purinas (R) numa fita de DNA ou apenas pirimidinas (Y) na outra
fita, sendo que estas sequências R-Y podem dobrar-se sobre si próprias e formar um
tripleto R-R-Y intramolecular adoptando, posteriormente (através da conjugação de

9
duas regiões R-R-Y), uma estrutura em forma de haltere e que apenas pode sofrer
ruptura através de elevadas temperaturas (>80ºC) e com a adição de EDTA para
remover iões metálicos divalentes (28,31,32,33). Estas estruturas são denominadas de
sticky DNA, impedindo virtualmente todos os processos biológicos incluindo
transcrição (sequestro da RNA polimerase por ligação directa com o sticky DNA),
replicação, reparação e recombinação (4,28).
As expansões GAA apresentam, ainda, um outro mecanismo molecular para
silenciar o gene FXN através da remodelação e condensação da cromatina no núcleo
celular. Sequências repetitivas de DNA têm a propensão para embrulhar regiões
genómicas em estruturas de heterocromatina inacessíveis, levando ao silenciamento
genético. Este fenómeno denomina-se position effect varigation (PEV) e é mimetizado
pelas expansões GAA (28,34,35,36).
A repetição dos tripletos GAA leva, também, a instabilidade da molécula de
DNA, processos de mutagénese e a uma taxa de recombinação elevada (15 vezes o
normal, levando a expansões e contracções) (28,37).
4.2 - A Frataxina e suas funções
O gene FXN codifica para a frataxina, uma proteína altamente conservada de
210 aminoácidos e localização mitocondrial. A sua estrutura consiste numa proteína
globular contendo uma hélice-α N-terminal, uma região em folha-β média, uma segunda
hélice-α e uma cadeia C-terminal em conformação extendida (38,39). Trata-se de uma
proteína constituinte da matriz mitocondrial e está envolvida no processo de homeostase
do ferro celular. Pensa-se que esta participa em pelo menos cinco processos celulares:
envolvimento na biogénese do heme e clusters de ferro-enxofre (ISCs); proteína de
armazenamento de ferro durante excesso celular deste; ajudante na reparação das
aconitase ISCs oxidativamente danificadas; factor de controlo do stress oxidativo
celular ao diminuir as espécies reactivas de oxigénio (ROS); e como participante activo
em cadeias que envolvem conversão energética e fosforilação oxidativa. Fenótipos
associados a deficiência de frataxina incluem acumulação de ferro mitocondrial,
perturbações na biossíntese de heme e ISCs e agravamento progressivo da homeostase
celular do ferro (40). Outras características incluem alterações do funcionamento da
cadeia transportadora de electrões, diminuição da produção de ATP, alterações da

10
homeostase do cálcio e da permeabilidade mitocondrial e, eventualmente, degeneração e
morte celular (41,42).
O motivo pelo qual o sistema nervoso é o mais afectado devido às alterações na
síntese de frataxina é porque este é o sistema que necessita de maior quantidade de
energia para o seu funcionamento, podendo o número de mitocondrias por célula variar
entre centenas a milhares. Como cada um destes organelos possui 2 a 10 cópias de
mtDNA, tem-se vários milhares de cópias deste por cada neurónio. Este tipo de DNA é
de herança quase exclusivamente materna (a hereditariedade paterna é muito rara), no
entanto, o risco de mutação é 10 vezes superior ao do DNA nuclear devido à ausência
de proteínas protectoras (como as histonas) e de um sistema de reparação altamente
eficiente, ocorrendo a presença de mtDNA mutado e normal na mesma célula –
heteroplasmia. Sendo que o mtDNA é o responsável pelo controlo funcional dos
sistemas mitocondriais, quanto maior for a proporção de mtDNA mutado relativamente
ao normal, maior é o nível de disfunção deste organelo. Isso traduz-se num quadro
clínico neurológico mais exuberante e com mais rápida evolução (43).
O papel da frataxina na homeostese do ferro pode ser observado através da
acumulação deste quando ocorre produção deficiente desta proteína (21) e pela
diminuição da função dos ISCs, complexos que servem de grupos prostéticos para
variadas enzimas com diferentes funções incluindo metabolismo energético (aconitase;
complexos I, II e III da cadeia respiratória), metabolismo do ferro (ferroquelatase;
proteína responsiva do ferro I), síntese de purinas e reparação do DNA. A deficiente
produção de ISCs, devido ao insuficiente transporte e fornecimento de ferro pela
frataxina, leva à acumulação deste no espaço intracelular (40,44,45).
Uma outra forma de registar a função dos ISCs é pela actividade da cadeia
respiratória mitocondrial, que é expressa pela razão entre as enzimas citrato sintetase e
aconitase nos diferentes tecidos. Num estudo de 10 doentes com FRDA confirmada
geneticamente, foram observadas reduções significativas da actividade do complexo I,
complexo II/III e da aconitase no tecido cardíaco. Diminuição da actividade destas
enzimas também foi observada no músculo esquelético, o que não ocorreu ao nível do
cerebelo (redução do complexo I) e dos gânglios dorsais (redução da aconitase) (21).
Uma outra função da frataxina é o seu envolvimento na biossíntese de ISCs,
processo mediado pelas proteínas estruturantes do ferro mitocondrial IscU‟s,

11
especialmente a IscU2 (nas leveduras) que, ao receber o ferro da frataxina, envolve-se
nos passos iniciais da síntese de ISCs (36,46).
Ambas as formas monomérica e oligomérica da frataxina podem ligar e
transportar Fe2+
. No entanto, esta proteína pode actuar de uma forma ferritina-like
(proteína oligomerizada, formando agregados), podendo ligar-se, preferencialmente, a
átomos de Fe3+
de forma a destoxificá-lo (evitando a formação de ROS, transformando
o Fe3+
em Fe2+
através da ferroxidase) e funcionando como uma proteína de
armazenagem do ferro (46,47,48). Foram, também, encontradas evidências da presença
de uma forma truncada (~14kDa) da molécula de frataxina (normal é ~17kDa,
encontrada no espaço mitocrondrial após clivagem da sequência-alvo pela MPP –
Mitocondrial Processing Peptidade) devido à actividade da enzima proteolítica DLD
(Dihydrolipoamide Dehydrogenase). Esta forma truncada é incapaz de agregar e
desempenhar as funções de destoxificação do Fe3+
anteriormente referidas in vitro
(49,50).
A deficiência de frataxina também afecta o metabolismo oxidativo (relacionados
com os distúrbios do metabolismo do ferro), podendo isto ser observado pela
sensibilidade celular ao peróxido de hidrogénio (H2O2) (44,51). Este, ao reagir com o
Fe2+
, gera o radical hidroxilo, altamente tóxico, responsável por danificar ácidos
nucleicos, proteínas e lípidos da membrana celular, levando a diminuição da biossíntese
mitocondrial, alterações do funcionamento dos sistemas energéticos celulares e
diminuição da fluidez e aumento da permeabilidade da membrana interna (diminuição
da produção de um fosfolipídeo ácido, a cardiolipina), respectivamente (43,44). O
défice desta proteína leva ao aumento de ambos os compostos e, consequentemente, ao
aumento de ROS. O H2O2 é eliminado pela glutationa peroxidase, através do consumo
da molécula antioxidativa glutationa, cujos níveis estão diminuídos devido ao consumo
excessivo desta (43,52). Um outro ROS é o superóxido (O2-) que, ao reagir com o óxido
nítrico (NO), um neurotransmissor importante ao nível do sistema nervoso central
(SNC) e periferia, produz um agente oxidante potente, o peroxinitrito (53). A
velocidade desta reacção é 3 vezes superior à reacção de destoxificação do O2- pela
enzima superóxido dismutase (SOD), tornando os neurónios vulneráveis a esta molécula
tóxica (43,53).
Nos doentes com FRDA o stress oxidativo é demonstrado por aumento dos
níveis plasmáticos de malondialdeído (MDA, um produto da peroxidação lipídica),

12
aumento do 8-hidroxi-2‟-deoxiguanosina (8-OHdG) urinário (marcador de dano
oxidativo do DNA), diminuição da glutationa livre e aumento da actividade da
glutationa-S-tranferase plasmática (54,55,56).
A função da frataxina na biossíntese do grupo heme é mediada pela
ferroquelatase, enzima que catalisa o último passo da síntese deste produto, sendo que é
necessária a presença de ferro para que estas interajam. A frataxina actua através da
doação de uma molécula de Fe2+
para a inserção no anel de porfirina (40,57,58).
Também foram observadas alterações ao nível de órgãos-alvo como o tecido
cardíaco onde foi observada diminuição da actividade de genes codificadores de
proteínas contrácteis, contribuindo para a cardiomiopatia. O músculo esquelético
revelou alterações em genes envolvidos no metabolismo lipídico e oxidação de ácidos-
gordos e o tecido hepático diminuição da actividade dos genes glicolíticos; ambas estas
alterações podem contribuir para a resistência à insulina e desenvolvimento de diabetes
(36).

13
5 - Diagnóstico
O diagnóstico de FRDA é feito pela clínica típica e por genética molecular,
sendo a síndrome completa altamente sugestiva para médicos informados.
Este impõe-se à distinção clínica com outras AR, sendo as mais relevantes a
ataxia por deficiência de vitamina E (AVED), a ataxia-telangiectasia (AT) e a ataxia
com apraxia oculomotora (AOA) (3).
A AVED apresenta uma idade de início entre os 4 e 20 anos (59), sendo
caracterizada clinicamente por ataxia progressiva do tronco e membros, disartria,
perturbação das sensibilidades postural e vibratória nos membros inferiores, arreflexia
osteotendinosa e sinal de Babinski bilateral (60). Outros sinais frequentes são escoliose
e pé cavo, distonia (13%), titubeação da cabeça (28%) e cardiomiopatia ou um evento
cardíaco agudo, associados a morte prematura nestes doentes (59,61). No entanto, a
idade de início, as manifestações clínicas e a velocidade de progressão são muito
variáveis. Esta patologia é causada por uma mutação no gene TTPA (α-tocopherol
transfer protein gene), provocando uma diminuição dos níveis de vitamina E circulante
(61,62).
A AT tem uma prevalência estimada de 1/40000 (63), apresentando como
manifestações clínicas principais ataxia progressiva com início antes dos 3 anos de
idade, telangiectasias em mais de 90% dos casos com início 2 a 8 anos após os
primeiros sintomas (63,64). Como sintomas associados observam-se variadas alterações
oftalmológicas (nistagmos optocinético – 81%, movimentos sacádicos – 76%,
estrabismo – 38% e apráxia oculomotora – 30%), disartria, disfagia, hipomimia facial,
neuropatia periférica e perturbações dos movimentos como tremor ou coreoatetose
(64,65). A perda da autonomia locomotora ocorre após 10 anos de evolução clínica
(63,64). Imunodeficiência provoca susceptibilidade aumentada para infecção
sinopulmonar crónica e posterior desenvolvimento de doença maligna. A causa da AT é
uma mutação no gene ATM que codifica para a proteína ATM serina/treonina cinase,
pertencente ao complexo fosfatidil-inositol-3-cinase (PI3K), responsável pela reparação
de erros no DNA (63,66).
Por fim, temos a AOA, podendo esta ser de tipo 1 ou tipo 2. A AOA tipo 1
caracteriza-se por idade de início entre os 1 e 20 anos, alterações oculomotoras
(nistagmo - 100% -, apraxia oculomotora, movimentos sacádicos, instabilidade da

14
fixação e pestanejar excessivo) e movimentos involuntários coreicos ou distónicos e/ou
ataxia global progressiva com disartria e tremor das mãos e cabeça e atraso do
desenvolvimento (incapacidade cognitiva variável). Com a progressão da doença
surgem outras manifestações clínicas como neuropatia periférica (com atenuação das
alterações dos movimentos), atrofia distal, pé cavo e hipo/arreaflexia osteotendinosa
(67,68). Esta é provocada por uma mutação no gene APTX, envolvida na reparação
ssDNA (69). Relativamente à AOA tipo 2, esta manifesta-se por idade de início entre os
2 e 25 anos, ataxia global progressiva, disartria, neuropatia axonal sensitivo-motora e
alterações oculomotoras (apraxia oculomotora em menos de 50% dos casos,
movimentos sacádicos em 100% dos casos, nistagmo e estrabismo com abdução
limitada dos olhos bilateralmente). Distonia, tremor postural, disfagia, pé cavo e
escoliose são observados ocasionalmente e a função cognitiva está preservada (70,71).
Uma mutação no gene SETX que codifica para a senataxina (proteína com actividade
DNA e RNA helicase) é a responsável por esta patologia (72).
O diagnóstico definitivo é feito por detecção molecular da mutação no gene
FXN. Após a implementação dos testes genéticos começaram a surgir casos atípicos,
diferentes do paradigma clássico, fenótipos incompletos e idades de início tardias, que
no passado não seriam diagnosticados como FRDA.

15
6 - Tratamento
A FRDA não tem cura ou tratamento etiológico, tendo as tentativas terapêuticas
sido pouco robustas. No entanto, a ausência de cura não significa demissão de
tratamento. A terapêutica sintomática pode ajudar os doentes e famílias a garantir
dignidade e mesmo qualidade de vida. Esta pode ser médica ou cirúrgica, indo desde
agentes farmacológicos como agonistas PPARγ (Peroxisome Proliferator-Activated
Receptor Gamma), quelantes de ferro, agentes antioxidantes, inibidores HDAC (Histone
Deacetylase), eritropoetina e pentamidina (73); vacinas, prevenção de complicações
cardíacas, tratamento da diabetes mellitus, fisioterapia, cinesioterapia respiratória,
natação, ajudas técnicas e correcção de deformidades ortopédicas (escoliose e pé cavo)
(13).
Provavelmente o desenvolvimento passará por terapias precoces susceptíveis de
atrasar a progressão da doença (74). Com base nisto, são necessários instrumentos de
medida da progressão da doença para o desenvolvimento de novos fármacos. Outras
medidas que os clínicos podem ter em conta como sendo úteis são as PRO (Patient-
Reported Outcome) que relatam o impacto da doença na perspectiva do doente (13,75).
Uma vez que o sistema afectado em primeiro lugar e que apresenta mais
alterações patológicas é o SNC, medidas de morbilidade neurológica foram
desenvolvidas estando entre elas as escalas ICARS (Internacional Cooperative Ataxia
Rating Scale) (Anexo 2), a FARS (Friedreich Ataxia Rating Scale) (Anexo 3),
especificamente desenvolvida para avaliar os danos neurológicos desta patologia e a
SARA (Scale for the Assessment and Rating of Ataxia), tendo esta menor importância
que as precedentes (75).
A ICARS é uma escala semi-quantitativa de 100 pontos que consiste de 19 itens
divididos em 4 subescalas: posição postural (34 pontos), ataxia dos membros (52
pontos), disartria (8 pontos) e alterações oculomotoras (6 pontos) (76). Apesar deste
instrumentos ser apenas parcialmente objectivo, isso não diminui a sua eficácia, uma
vez que este apresenta uma boa reprodutibilidade e boa correlação entre resultados total
e subescalas e idade e duração da doença, respectivamente. No entanto apresenta um
efeito tecto significativo, a partir do qual as variações das variáveis já não podem ser
medidas (75,77,78).

16
A FARS tem uma pontuação máxima de 159 ou 167, sendo a mais alta associada
a doença com maior severidade. Estas variações nas pontuações máximas ocorrem
devido a esta escala ter duas versões, uma que inclui e outra que exclui dois itens de 4
pontos na subescala do exame neurológico. Assim sendo, a FARS consiste de 3
subescalas: ataxia (6 pontos), actividades da vida diárias (AVD: 36 pontos) e exame
neurológico (117 ou 125 pontos) (79). Como esta escala ainda não foi submetida a
análise psicométrica, ela pode conter itens redundantes (75).
A SARA foi desenvolvida para medir a severidade da ataxia e foi validada
através de testes em indivíduos com ataxia espinocerebelar autossómica dominante
(80,81). Ela consiste em 8 itens que avaliam a marcha, postura, posição sentada e
disartria e por 4 itens que avaliam a função cinética dos membros. Tem uma pontuação
máxima de 40, directamente proporcional com a severidade da ataxia. Esta apresenta
uma boa correlação inter-observador e boa reprodutibilidade, no entanto, apenas avalia
os factores relativos à ataxia, não sendo, por isso, tão abrangem como as escalas
anteriormente referida para a avaliação da FRDA (75,80).
As medidas PRO consistem em escalas que avaliam a qualidade de vida (QoLS),
desenhadas para averiguar a perspectiva do doente acerca do seu bem-estar e como a
sua vida e a da sua família é afectada pela doença; satisfação do tratamento e medidas
gerais que avaliam o impacto da doença no indivíduo (82,83). A QoLS mais
generalizada é a SF36 (Medical Outcomes Study 36-Item Short Form Health Survey)
(84), sendo que os doentes com FRDA apresentam piores resultados na subescala PCS
(Physical Component Summary) (75). Contudo, uma escala específica para avaliar o
impacto da FRDA na perspectiva do doente ainda não foi desenvolvida, mas é altamente
pertinente.
Relativamente a agentes farmacológicos, os agonistas PPARγ como a
pioglitazona são úteis através da restauração da acção da enzima PGC-1a (Peroxisome
Proliferator-Activated Receptor Gamma Coactivator 1-Alpha), que é um regulador
transcripcional importante dos sistemas de síntese energética e antioxidante
mitocondriais, aumenta a β-oxidação de ácidos gordos e diminui as respostas
inflamatórias (73,85).
Os quelantes de ferro como a deferiprona e desferroxamina actuam através da
diminuição das concentrações de ferro e, consequentemente, dos seus efeitos tóxicos ao
nível das alterações do funcionamento dos sistemas energéticos celulares, diminuição da

17
fluidez e aumento da permeabilidade da membrana mitocondrial interna e lesão do
mtDNA com activação dos mecanismos de apoptose. Relatos sobre melhoria clínica dos
sintomas neurológicos foram obtidos com a administração de deferiprona (36,86,87,88).
Agentes antioxidantes como a ibedenona e coenzima Q10 protegem a
mitocôndria da lesão pelas ROS. Esta pertence à classe das ubiquinonas que actuam
através do seu papel na cadeia transportadora de electrões, recebendo os pares de
electrões do NADH do Complexo I e do FADH2 do Complexo II, sendo que estes
podem ser fornecidos às ROS para as estabilizar e evitar a sua acção lesiva. A
ibedenona, em doses reduzidas (5mg/Kg/dia) reduz a progressão da cardiomiopatia com
consequente melhoria do estado geral e diminuição da fatigabilidade, sem influência na
progressão clínica da doença neurológica (89,90,91). A coenzima Q10, em doses de
400mg/dia melhora a bioenergética do tecido cardíaco e do músculo esquelético,
principalmente quando combinado com vitamina E, um outro antioxidante (36,91).
Em relação aos inibidores HDAC (por exemplo, o BML-210), a sua acção é
demonstrada através da abertura da cromatina, impedindo os mecanismos que
promovem a formação de heterocromatina (dificulta a formação de estruturas não-B
DNA e sticky DNA), uma vez que o silenciamento genético nos alelos FXN expandidos
é acompanhado de hipoacetilação das histonas H3 e H4 e trimetilação de H3, processo
compatível com um mecanismo de repressão mediado por heterocromatina (92,93).
A eritropoetina actua pelo aumento da produção de frataxina ao nível pós-
transcripcional (activação de factor transcripcional específico) oferecendo um efeito
neuroprotector e cardioprotector, estimulando a neurogénese, diferenciação neuronal e
activação dos mecanismos antiapoptóticos, antioxidantes e anti-inflamatórios (36,94).
Um outro grupo de agentes farmacológicos são os compostos que se ligam ao
DNA ao qual pertence a pentamidina que aumentam a transcripção através da sua
ligação preferencial ao DNA com estrutura B, aumentando assim a síntese de frataxina
(92).
A FRDA é uma patologia devido a perda de função, sendo que novas estratégias
baseadas em alvos genéticos desenhadas para aumentar a síntese de frataxina podem vir
a ser uma terapia ideal. Já foi descrita produção desta proteína por vectores víricos em
fibroblastos com alterações por FRDA, ocorrendo correcção parcial da sensibilidade ao
stress oxidativo. No entanto ainda existem várias limitações para a aplicação clínica
destas (73,92,95,96).

18
Em relação às restantes intervenções, estas incidem, maioritariamente, na
prevenção, correcção cirúrgica de potenciais complicações e na promoção da qualidade
de vida. Também a informação fornecida aos doentes sobre a sua patologia desempenha
um papel crucial na forma como estes lidam com esta e influenciam positivamente as
medidas PRO (13).
Há excepção dos antioxidantes e quelantes de ferro, as restantes terapêuticas
farmacológicas ainda se encontram em fase de investigação, sendo que o seu uso para o
tratamento da FRDA ainda não foi aprovado. Por outro lado, as estratégias
farmacológicas actualmente em uso têm um efeito muito limitado ao nível da
progressão e sintomatologia desta doença. As restantes terapêuticas são utilizadas como
forma de correcção de alterações debilitantes ou como forma de prevenção para outras
patologias que possam surgir em associação a esta.

19
7 - Caso Clínico
R.B.M.D.; 29 anos; sexo feminino. É natural de uma aldeia do concelho de
Amarante. Tem o 12º ano de escolaridade. Desempregada.
Desde a infância que apresenta dificuldades na marcha, sendo estas mais
notórias em actividades desportivas (corrida, ginástica e jogos). Ocorreu agravamento
progressivo sendo que, actualmente, é necessário apoio para subir e descer escadas e é
incapaz de manter deambulação em linha recta.
Posteriormente, aos 14 ou 15 anos iniciou alterações da caligrafia e disartria com
agravamento progressivo, dificuldades nas actividades de vida diárias com lentificação
dos movimentos para uso de utensílios de cozinha, vestir-se e fazer a sua higiene
pessoal e perturbações auditivas. Mais tardiamente surgiu disfagia, mais acentuada para
líquidos, e tremor de acção generalizado, com agravamento progressivo.
Ausência de consanguinidade parental ou de história familiar de patologia
neurológica idêntica. Há outras pessoas com a mesma doença na aldeia.
Exame neurológico:
Vigil, orientada no espaço e no tempo e com discurso coerente. Disartria
cerebelosa e disfagia. Força e tónus muscular normais. Arreflexia osteo-tendinosa. Sinal
de Babinski bilateral. Dismetria nas provas dedo-nariz e calcanhar-joelho, com
decomposição do movimento, procura permanente do centro de gravidade axial, marcha
atáxica com base alargada e funânbulo impossível, . Diminuição das sensibilidades
vibratória e postural
Outras alterações são a presença de escoliose discreta e pés cavus.
O diagnóstico de FRDA foi confirmado por teste genético com resultado de
homozigotia para expansão GAA no gene FXN, sendo a expansão dos alelos de 290 e
807 GAA‟s, respectivamente.
Último despiste para patologia cardíaca apresenta ECG e Holter sem alterações e
ecocardiograma com discreta insuficiência tricúspide com PSAP de 26mmHg e despiste
para diabetes mellitus negativo. Foram pedidas novas consultas de Oftalmologia para
despiste de alterações oculares e de Otorrinolaringologia para reavaliação das alterações
previamente detectadas.

20
8 - Conclusões
A Ataxia de Friedreich é uma patologia com sintomatologia progressiva e
debilitante, avançando naturalmente para a morte do doente e para a qual ainda não
existe cura. A importância da sua abordagem reside não só nestes factores, como
também na sua distribuição epidemiológica universal e no facto de ser a mais frequente
das ataxias autossómicas recessivas, com uma prevalência e uma frequência do estado
de portador elevadas.
A identificação de mutações genéticas específicas nomeadamente ao nível do
gene FXN nos pacientes com FRDA revolucionou o diagnóstico desta patologia. Muitos
casos como início tardio dos sintomas, hiperreflexia ou outras apresentações “atípicas”,
que anteriormente não eram possíveis de enquadrar nesta ataxia, podem agora ser
incluídos devido à análise genética. Desta forma, o espectro clínico desta doença é,
agora, muito mais diversificado relativamente às descrições clássicas.
Actualmente a terapêutica é limitada, uma vez que, há excepção dos
antioxidantes e quelantes de ferro, as restantes terapêuticas farmacológicas ainda se
encontram em fase de investigação, sendo que, mesmo essas têm um efeito muito
limitado no controlo da progressão e sintomatologia desta doença. Desta forma, será
imperativo prosseguir com a investigação para a descoberta e melhoria de agentes
farmacológicos para o tratamento desta patologia. Mesmo um pequeno avanço a nível
da terapêutica sintomática pode ajudar os doentes e famílias a garantir dignidade e
qualidade de vida.

21
Bibliografia
1. Fogel BL, Perlman S. Clinical features and molecular genetics of autosomal
recessive cerebellar ataxias. Lancet Neurol 2007;6:245-257. 2. Embiruçu EK, Martyn ML, Schlesinger D. Autossomal Recessive Ataxias – 20
types, and counting. Arq Neuropsiquiatr 2009;67(4):1143-1156.
3. Pandolfo M. Friedreich Ataxia. Arch Neurol 2008;65(10):1296-1303.
4. Pandolfo M. Friedreich ataxia: The clinical picture. J Neurol 2009;256(Suppl
1):3-8.
5. Harding AE, Zilkha KJ. „Pseudo-dominant‟ inheritance in Friedreich‟s ataxia. J
Med Genet. 1981;18:285-287.
6. De Michele G, Filla A, Cavalcanti F, et al. Late onset Friedreich´s disease:
clinical features and mapping of the FRDA locus. J Neurol Neurosurg
Psychiatry 1994;57(8):977-979.
7. Harding AE. Friedreich‟s ataxia: a clinical and genetic study of 90 families with
an analysis of early diagnostic criteria and intrafamilial clustering of clinical
features. Brain 1981;104:589-620.
8. Moschner C, Perlman S, Baloh RW. Comparison of oculomotor findings in the
progressive ataxia syndromes. Brain 1994;117(pt1):15-25.
9. Harding AE, Hewer RL. The heart disease of Friedreich‟s ataxia: a clinical and
electrocardiographic study of 115 patients, with an analysis of serial
electrocardiographic changes in 30 cases. Q J Med. 1983;52:489–502.
10. Cady RB, Bobechko WP. Incidence, natural history, and treatment of scoliosis
in Friedreich‟s ataxia. J Pediatr Orthop. 1984;4:673–676.
11. Shapiro F, Specht L. The diagnosis and orthopaedic treatment of childhood
spinal muscular atrophy, peripheral neuropathy, Friedreich ataxia, and
arthrogryposis. J Bone Joint Surg Am. 1993;75:1699–1714.
12. Campanella G, Filla A, DeFalco F, et al. Friedreich‟s ataxia in the south of
Italy: a clinical and biochemical survey of 23 patients. Can J Neurol Sci.
1980;7:351–357.
13. Maring JR, Croarkin E. Presentation and progression of Friedreich ataxia and
implications for physical therapist examination. Phys Ther. 2007;87:1687–
1696.

22
14. Hensinger RN, MacEwen GD. Spinal deformity associated with heritable
neurological conditions: spinal muscular atrophy, Friedreich's ataxia, familial
dysautonomia, and Charcot–Marie–Tooth disease. J. Bone Jt. Surg., Am.
Vol. 1976;58:13–24.
15. MacCabe JDH, Ryan F, Moore DP, McQuaid S, King MD, Kelly A. Typical
Friedreich‟s ataxia without GAA expansions and GAA expansions without
typical Friedreich‟s ataxia. J Neurol 2000;247:346–355.
16. Cossée M, Dürr A, Schmitt M, et al. Frataxin point mutations and clinical
presentation of compound heterozygous Friedreich ataxia patients. Ann Neurol.
1999;43:200-206.
17. Hou JGG, Jankovic J. Movement disorders in Friedreich‟s ataxia. Journal of the
Neurological Sciences 2003;206:59-64.
18. Koeppen A. The neuropathology of inherited ataxias. In: Manto M, Pandolfo
M, eds. The Cerebellum and its Disorders. New York, NY: Cambridge
University Press 2002:387-405.
19. Santoro L, De Michele G, Perretti A, Crisci C, Cocozza S, Cavalcanti F, et al.
Relation between trinucleotide GAA repeat length and sensory neuropathy in
Friedreich‟s ataxia. J Neurol Neurosurg Psychiatry 1999;66:93–96.
20. Campuzano V, Montermini L, Molto MD, Pianese L, Cossée M, Cavalcanti F,
et al. Friedreich‟s ataxia: autossomal recessive disease caused by an intronic
GAA triplet expansion. Science 1996;42:265-269.
21. Bradley JL, Blake JC, Chamberlain C, Thomas PK, Cooper JM, Schapira AHV.
Clinical, biochemical and molecular genetic correlations in Friedreich‟s ataxia.
Human Molecular Genetics 2000;9(2):275-282.
22. Jiralerspong S, Liu Y, Montermini L, Stifani S, Pandolfo M. Frataxin shows
developmentally regulated tissue-specific expression in the mouse embryo.
Neurobiol Dis. 1997;4(2):103-113.
23. Koutnikova H, Campuzano V, Foury F, Dolle´ P, Cazzalini O, Koenig M.
Studies of human, mouse and yeast homologues indicate a mitochondrial
function for frataxin. Nat Genet. 1997;16(4):345-351.
24. Beauchamp M, Labelle H, Duhaime M, Joncas J. Natural history of muscle
weakness in Friedreich‟s ataxia and its relation to loss of ambulation. Clin
Orthop 1995;311:270–275.

23
25. Cavadini P, Gellera C, Patel PI, Isaya G. Human frataxin maintains
mitochondrial iron homeostasis in Saccharomyces cerevisiae. Hum Mol Genet.
2000;9:2523–2530.
26. Cossée M, Dürr A, Schmitt M, et al. Frataxin point mutations and clinical
presentation of compound heterozygous Friedreich ataxia patients. Ann Neurol.
1999;43:200-206.
27. Pianese L, Cavalcanti F, De Michele G, et al. The effect of parental gender on
the GAA dynamic mutation in the FRDA gene. Am J Hum Genet.
1997;60(2):460-463.
28. Wells RD. DNA triplexes and Friedreich ataxia. The FASEB Journal
2008;22:1625-1634.
29. Jain A, Rajeswari MR, Ahmed F. Formation and thermodynamic stability of
intermolecular (R·R·Y) DNA triplex in GAA/TTC repeats associated with
Friedreich‟s ataxia. J BiomolStruct Dyn. 2002;19:691–699.
30. Bidichandani SI, Ashizawa T, Patel PI. The GAA triplet-repeat expansion in
Friedreich ataxia interferes with transcription and may be associated with an
unusual DNA structure. Am J Hum Genet. 1998;62:111–121.
31. Sakamoto N, Ohshima K, Montermini L, Pandolfo M. Wells R.D. Sticky DNA,
a self-associated complex formed at long GAA_TTC repeats in intron 1 of the
frataxin gene, inhibits transcription. J. Biol. Chem. 2001;276:27171–27177.
32. Napierala M, Dere R, Vetcher A, Wells RD. Structure-dependent recombination
hot spot activity of GAA_TTC sequences from intron 1 of the Friedreich‟s
ataxia gene. J Biol Chem. 2004;279:6444–6454.
33. Sakamoto N, Larson JE, Iyer RR, Montermini L, Pandolfo M, Wells RD.
GGA_TCC interrupted triplets in long GAA_TTC repeats inhibit the formation
of triplex and sticky DNA structures, alleviate transcription inhibition, and
reduce genetic instabilities. J Biol Chem. 2001;276:27178–27187.
34. Saveliev A, Everett C, Sharpe T, Webster Z, Festenstein R. DNA triplet repeats
mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature
2003;422:909–913.
35. Hiragami K, Festenstein R. Heterochromatin protein 1: a pervasive controlling
influence. Cell Mol. Life Sci. 2005;62:2711–2726.

24
36. Babady NE, Carelle N, Wells RD, Rouault TA, Hirano M, Lynch DR, et al.
Advancements in the pathophysiology of Friedreich‟s Ataxia and new prospects
for treatments. Molecular Genetics and Metabolism 2007;92:23-35.
37. Wells RD. Non-B conformations, mutagenesis, and diseases. Trends Biochem.
Sci. 2007;32:271–278.
38. Musco G, Stier G, Kolmerer B, et al. Towards a structural understanding of
Friedreich‟s ataxia: the solution structure of frataxin. Structure 2000;8(7):695-
707.
39. Gibson TJ, Koonin EV, Musco G, Pastore A, Bork P. Friedreich‟s ataxia
protein: phylogenetic evidence for mitochondrial dysfunction. Trends Neurosci
1996;19:465–468.
40. Benze KZ, Kondapalli KC, Cook JD, McMahon S, Millán-Pacheco C, Pastor N,
et al. The Structure and Function of Frataxin. Crit Rev Biochem Mol Biol
2006;41(5):269-291.
41. Stewart VC, Sharpe MA, Clark JB, Heales SJ. Astrocyte-derived nitric oxide
causes both reversible and irreversible damage to the neuronal mitochondrial
respiratory chain. J Neurochem 2000;75:694–700.
42. Halliwell B. Antioxidant defence mechanisms: from the beginning to the end
(of the beginning). Free Radic Res 1999;31:261– 72.
43. Calabrese V, Lodi R, Tonon C, D‟Agata V, Sapienza M, Scapagnini G et al.
Oxidative stress, mitocondrial dysfunction and cellular stress response in
Friedreich‟s ataxia. Journal of Neurological Sciences 2005;233:145-162.
44. Pandolfo M, Pastore A. The pathogenesis of Friedreich ataxia and the structure
and function of frataxin. J Neurol 2009;256(Suppl 1):9-17.
45. Chen OS, Crisp RJ, Valachovic M, Bard M, Winge DR, Kaplan J. Transcription
of the yeast iron regulon does not respond directly to iron but rather to iron-
sulfur cluster biosynthesis. J Biol Chem 2004;279:29513-29518.
46. Yoon T, Cowan JA. Iron–sulfur cluster biosynthesis. Characterization of
frataxin as an iron donor for assembly of [2Fe–2S] clusters in ISU-type
proteins, J. Am. Chem. Soc. 2003;125:6078–6084.
47. Yoon T, Dizin E, Cowan JA. N-terminal iron-mediated selfcleavage of human
frataxin: regulation of iron binding and complex formation with target proteins.
J Biol Inorg Chem 2007;12:535–542.

25
48. Gakh O, Adamec J, Gacy AM, Twesten RD, Owen WG, Isaya G. Physical
evidence that yeast frataxin is an iron storage protein. Biochemistry
2002;41:6798-6804.
49. Cavadini P, Adamec J, Taroni F, Gakh O, Isaya G. Two-step processing of
human frataxin by mitochondrial processing peptidase. Precursor and
intermediate forms are cleaved at different rates, J. Biol. Chem.
2000;275:41469–41475.
50. O‟Neill HA, Gakh O, Isaya G. Supramolecular assemblies of human frataxin
are formed via subunit-subunit interactions mediated by a non-conserved
amino-terminal region, J. Mol. Biol. 2005;345:433–439.
51. Wong A, Yang J, Cavadini P, Gellera C, Lonnerdal B, Taroni F, et al. The
Friedreich‟s ataxia mutation confers cellular sensitivity to oxidant stress which
is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum
Mol Genet 1999;8:425–430.
52. Drake J, Sultana R, Aksenova M, Calabrese V, Butterfield DA. Elevation of
mitochondrial glutathione by gamma-glutamylcysteine ethyl ester protects
mitochondria against peroxynitrite-induced oxidative stress. J Neurosci Res
2003;74:917– 27.
53. Radi R, Peluffo G, Alvarez MN, Naviliat M, Cayota A. Unraveling
peroxynitrite formation in biological systems. Free Radic Biol Med
2001;30:463– 88.
54. Tozzi G, Nuccetelli M, Lo Bello M, Bernardini S, Bellincampi L, Ballerini S. et
al. Antioxidant enzymes in blood of patients with Friedreich‟s ataxia. Arch Dis
Child 2002;86:376–379.
55. Emond M, Lepage G, Vanasse M, Pandolfo M. Increased levels of plasma
malondialdehyde in Friedreich ataxia. Neurology 2000;55:1752–1753.
56. Schulz JB, Dehmer T, Schöls L, Mende H, Hardt C, Vorgerd M. Oxidative
stress in patients with Friedreich ataxia. Neurology 2000;55:1719–1721.
57. Taketani S. Acquisition, mobilization and utilization of cellular iron and heme:
endless findings and growing evidence of tight regulation. Tohoku J Exp Med
2005;205:297–318.
58. Foury F, Cazzalini O. Deletion of the yeast homologue of the human gene
associated with Friedreich‟s ataxia elicits iron accumulation in mitochondria.
FEBS Lett 1997;411:373–377.

26
59. Cavalier L, Ouahchi K, Kayden HJ, et al. Ataxia with isolated vitamin E
deficiency: heterogeneity of mutations and phenotypic variability in a large
number of families. Am J Hum Genet 1998;62:301-310.
60. Marzouki N, Benomar A, Yahyaoui M, et al. Vitamin E deficiency ataxia with
(744del A) mutation on a-TTP gene: genetic and clinical peculiarities in
Moroccan patients. Eur J Med Genet 2005;48:21-28.
61. Mariotti C, Gellera C, Rimoldi M, et al. Ataxia with isolated vitamin E
deficiency: neurological phenotype, clinical follow-up and novel mutations in
TTPA gene in Italian families. Neurol Sci 2004;25:130-137.
62. Arita M, Sato Y, Mayata A, et al. Human a-tocoferol transfer protein: cDNA
cloning, expression and chromosomal localization. Biochem J 1995;306:437-
443.
63. Chun HH, Gatti RA. Ataxia-telangiectasia, an evolving phenotype. DNA Repair
2004;3:1187-1196.
64. Perlman S, Becker-Catania S, Gatti RA. Ataxia-telangiectasia: diagnosis and
treatment. Semin Pediatr Neurol 2003;10:173-182.
65. Farr AK, Shalev B, Crawford TO, Lederman HM, Winkelstein JA, Repka MX.
Ocular manifestations of ataxia-telangiectasia. Am J Ophthalmol 2002;134:891-
896.
66. Taylor AMR, Byrd PJ. Molecular pathology of ataxia telangiectasia. J Clin
Pathol 2005;58:1009-1015.
67. Barbot C, Coutinho P, Chorão R, et al. Recessive ataxia with ocular apraxia:
review of 22 portuguese patients. Arch Neurl 2001;58:201-205
68. Le Ber I, Moreira M-C, Rivaud-Péchoux S, et al. Cerebellar ataxia with
oculomotor apraxia type 1: clinical and genetic studies. Brain 2003;126: 2761-
2772.
69. Ferrarini M, Squintani G, Cavallaro T, Ferrari S, Rizzuto N, Fabrizi GM. A
novel mutation of aprataxin associated with ataxia ocular apraxia type 1:
Phenotypical ang genotypical characterization. J Neurol Sci 2007;260:219-224.
70. Le Ber I, Bouslam N, Rivaud-Péchoux S, et al. Frequency and phenotypic
spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic stydy in 18
patients. Brain 2004;127:759-767.

27
71. Tazir M, Ali-Pacha L, M‟Zahem A, et al. Ataxia with ocolumotor apraxia type
2: a clinical and genetic study of 19 patients. J Neurol Sci 2009;278:77-81.
72. Duquette A, Roddier K, McNabb-Baltar J, et al. Muattions in senataxin
responsible for Quebec cluster of ataxia with neuropathy. Ann Neurol
2005;57:408-414.
73. Mancuso M, Orsucci D, Choub A, Siciliano G. Current and emerging treatment
options in the management of Friedreich ataxia. Neuropsychiatric Disease and
Treatment 2010;6:491-499.
74. Voncken M, Ioannou P, Delatycki MB. Friedreich ataxia – update on
pathogenesis and possible therapies. Neurogenetics 2004;5:1–8.
75. Delatycki MB. Evaluating the progression of Friedreich ataxia and its treatment.
J Neurol 2009;256(Suppl 1):36-41.
76. Storey E, Tuck K, Hester R, Hughes A, Churchyard A. Inter-rater reliability of
the International Cooperative Ataxia Rating Scale (ICARS). Mov Disord
2004;19:190–192.
77. Cano SJ, Hobart JC, Hart PE, Korlipara LV, Schapira AH, Cooper JM.
International Cooperative Ataxia Rating Scale (ICARS): appropriate for studies
of Friedreich‟s ataxia? Mov Disord 2005;20:1585–1591.
78. Ribai P, Pousset F, Tanguy ML, Rivaud- Pechoux S, Le Ber I, Gasparini F, et
al. Neurological, cardiological, and oculomotor progression in 104 patients with
Friedreich ataxia during long-term follow-up. Arch Neurol 2007;64:558–564.
79. Subramony SH, May W, Lynch D, Gomez C, Fischbeck K, Hallett M, et al.
Measuring Friedreich ataxia: interrater reliability of a neurologic rating scale.
Neurology 2005;64:1261–1262.
80. Schmitz-Hubsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C,
et al. Scale for the assessment and rating of ataxia: development of a new
clinical scale. Neurology 2006;66:1717–1720.
81. Weyer A, Abele M, Schmitz-Hubsch T, Schoch B, Frings M, Timmann D, et al.
Reliability and validity of the scale for the assessment and rating of ataxia: a
study in 64 ataxia patients. Mov Disord 2007;22:1633–1637.
82. Atkinson MJ, Lennox RD. Extending basic principles of measurement models
to the design and validation of Patient Reported Outcomes. Health Qual Life
Outcomes 2006;4:65.

28
83. Meyers AR, Gage H, Hendricks A. Health-related quality of life in neurology.
Arch Neurol 2000;57:1224–1227.
84. Ware JE Jr, Sherbourne CD. The MOS 36-item short-form health survey (SF-
36) I. Conceptual framework and item selection. Medical Care 1992;30:473–
483.
85. Marmolino D, Manto M, Acquaviva F, et al. PGC-1alpha downregulation
affects the antioxidant response in Friedreich‟s ataxia. PLoS One.
2010;5(4):e10025.
86. Kontoghiorghes GJ, Efstathiou A, Kleanthous M, Michaelides Y, Kolnagou A.
Risk/benefit assessment, advantages over other drugs and targeting methods in
the use of deferiprone as a pharmaceutical antioxidant in iron loading and non
iron loading conditions. Hemoglobin. 2009;33(5):386–397.
87. Richardson DR. Friedreich‟s ataxia: iron chelators that target the mitochondrion
as a therapeutic strategy? Expert Opin Investig Drugs. 2003;12(2):235–245.
88. Kakhlon O, Manning H, Breuer W, et al. Cell functions impaired by frataxin
deficiency are restored by drug-mediated iron relocation. Blood.
2008;112(13):5219–5227.
89. Rustin P, Rotig A, Munnich A, Sidi D. Heart hypertrophy and function are
improved by idebenone in Friedreich‟s ataxia. Free Radic Res. 2002;36(4):467–
469.
90. Hausse AO, Aggoun Y, Bonnet D, et al. Idebenone and reduced cardiac
hypertrophy in Friedreich‟s ataxia. Heart. 2002;87(4):346–349.
91. Cooper JM, Schapira AHV. Friedreich‟s ataxia: Coenzyme Q10 and vitamin E
therapy. Mitochondrion 2007;7:127-135.
92. Hebert MD. Targeting the gene in Friedreich ataxia. Biochimie 2008;90:1131-
1139.
93. Herman D, Jenssen K, Burnett R, Soragni E, Perlman SL, Gottesfeld JM.
Histone deacetylase inhibitors reverse gene silencing in Friedreich„s ataxia. Nat
Chem Biol. 2006;2(10):551–558.
94. Sturm B, Stupphann D, Kaun C, Boesch S, Schranzhofer M, Wojta J, et al.
Recombinant human erythropoietin: effects on frataxin expression in vitro. Eur
J Clin Invest. 2005;35:711-717.
95. Fleming J, Spinoulas A, Zheng M, et al. Partial correction of sensitivity to
oxidant stress in Friedreich ataxia patient fibroblasts by frataxinencoding

29
adeno-associated virus and lentivirus vectors. Hum Gene Ther. 2005;16(8):947–
956.
96. Gomez-Sebastian S, Gimenez-Cassina A, Diaz-Nido J, Lim F, Wade- Martins
R. Infectious delivery and expression of a 135 kb human FRDA genomic DNA
locus complements Friedreich‟s ataxia deficiency in human cells. Mol Ther.
2007;15(2):248–254.
30
Anexos
Anexo 1 - Classificação e aspectos moleculares das ataxias autossómicas recessivas
Legenda: (?) – possivelmente.

31
Anexo 2 – Principais componentes da escala ICARS
Legenda: KF – kinetic function (função cinética); OD – oculomotor disorders
(distúrbios oculomotores); SD – speech disorders (distúrbios da fala).

32
Anexo 3 – Sumário dos principais componentes da escala FARS
Legenda: ADL - activities of daily living (actividades de vida diárias); DTR - deep
tendon reflexes (reflexos osteotendinosos); FARS - Friedreich Ataxia Rating Scale.





























