Embed Size (px)
Citation preview

UNIVERSIDADE ESTADUAL DE CAMPINAS
FACULDADE DE CIÊNCIAS MÉDICAS
THIAGO MAZZO PELUZZO
ESTUDOS MOLECULARES EM ATAXIA DE FRIEDREICH
CAMPINAS
2017

THIAGO MAZZO PELUZZO
ESTUDOS MOLECULARES EM ATAXIA DE FRIEDREICH
Dissertação apresentada à Faculdade de Ciências Médicas da Universidade
Estadual de Campinas como parte dos requisitos exigidos para a obtenção do
título de Mestre em Ciências.
ORIENTADOR: PROF. DR. MARCONDES CAVALCANTE FRANÇA JUNIOR.
ESTE EXEMPLAR CORRESPONDE À VERSÃO
FINAL DA DISSERTAÇÃO DEFENDIDA PELO
ALUNO THIAGO MAZZO PELUZZO, E ORIENTADO PELO
PROF. DR. MARCONDES CAVALCANTE FRANÇA JUNIOR.
CAMPINAS
2017


BANCA EXAMINADORA DA DEFESA DE MESTRADO
THIAGO MAZZO PELUZZO
ORIENTADOR: PROF. DR. MARCONDES CAVALCANTE FRANÇA JUNIOR
MEMBROS:
1. PROF. DR. MARCONDES CAVALCANTE FRANÇA JUNIOR
2. PROF. DR. JONAS ALEX MORALES SAUTE
3. PROFA. DRA. CLÁUDIA VIANNA MAURER MORELLI
4. PROF. DR. JOSÉ LUIZ PEDROSO
5. PROF. DR. ANDRÉ SCHWAMBACH VIEIRA
Programa de Pós-Graduação em Fisiopatologia Médica da Faculdade de Ciências
Médicas da Universidade Estadual de Campinas.
A ata de defesa com as respectivas assinaturas dos membros da banca examinadora
encontra-se no processo de vida acadêmica do aluno.
Data: 05 de julho de 2017

Dedico aos que identificam na Ciência uma
resposta plausível aos acontecimentos dessa
vida e a todos que de alguma forma
contribuíram para esse estudo.

EPÍGRAFE
“Algumas regras para a vida:
- Questione a autoridade.
- Nenhuma ideia é verdadeira só porque alguém diz que é, incluindo eu.
- Pense por si próprio, questione a si próprio.
- Não acredite em algo só porque quer acreditar, acreditar em algo não o torna verdadeiro.
- Teste ideias pelas evidências adquiridas, pela observação e experimentação.
- Se uma ideia prevalecente falhar num teste bem desenvolvido, está errada. Supere.
- Siga as evidências onde quer que elas levem, se não houver evidências, evite julgamento.
E talvez a regra mais importante de todas:
- Lembre-se, você pode estar errado.”
(Neil deGrasse Tyson)

AGRADECIMENTOS
Consigo me lembrar do dia em que fui aprovado no vestibular da UNICAMP. Todo
um mundo novo. Novas pessoas, novas amizades, novos lugares... Mas minha cabeça
continuava na genética. Eu amo a biologia como um todo. Amo as suas interações – todas elas
– e, ainda assim, meu fascínio pela genética se manteve. Com o primeiro estágio na área, tive
meu primeiro contato com a genética médica, com os pacientes e com a clínica. Nas palavras
de Fernando Pessoa: “Primeiro, estranha-se, depois entranha-se”. Nesse local me vi trilhando
um novo caminho - para mim, pelo menos - e assim se segue até o presente momento em que
entrego essa dissertação.
Não há como dizer que a realização de todo esse trabalho se fez somente com meus
méritos pessoais, seria prepotência – e, no fim das contas – uma mentira. Meus eternos
agradecimentos vão para todas as pessoas que de alguma forma contribuíram para que eu
chegasse até aqui e, por isso, gostaria de citar algumas delas:
Primeiramente, aos meus pais pelo suporte emocional, financeiro e todos os outros que
puderem se encaixar aqui. Rosângela Maria Mazzo Peluzzo e Djalma Roberto Peluzzo, vocês
são o meu norte e sempre serão. Espero ter vocês ao meu lado por mais todo um sempre.
Aos meus irmãos que, mesmo distantes geograficamente, aqui no meu coração, a
distorção do espaço-tempo nos mantém sempre próximos.
À Fernanda Mendes por estar presente ao meu lado, dando apoio profissional e
pessoal. Por topar me acompanhar nessa caminhada e sempre me ajudar, fazendo com que ela
fosse, na maioria das vezes, mais leve e fácil de ser digerida.
Ao meu orientador Prof. Dr. Marcondes Cavalcante França Jr. pela oportunidade de
trabalhar juntos nesse projeto tão interessante. Por todo conhecimento e instrução ao longo
dessa jornada.
Ao Dr.Wilson Marques Junior da USP-RP e a Dra. Laura Bannach Jardim da UFRGS
por cederem as amostras e os dados clínicos para que a realização desse projeto fosse
possível.
À Dra. Luciana Cardoso Bonadia, por ser um exemplo íntegro de profissional que
quero seguir pelo resto de minha vida. Por todas as conversas sobre assuntos diversos,
regados a altas doses de café e risadas. Por todas as dúvidas tiradas e todas os conselhos
proporcionados. Por ser também, com o passar dos anos, um exemplo de mãe a quem já me

espelho quando penso na criação do meu filho. Poderia me estender nesses pontos
infinitamente, pois não há palavras que descreverão toda minha gratidão a você.
À Dra. Simoni Avansini por todo o conhecimento, por todas discussões
metodológicas, todas as pesquisas e papers indicados. Pelos sorrisos mesmo nas horas de
desespero. Pelo companheirismo a todo momento.
À Dra. Tânia Kawasaki de Araujo, por ter me ouvido sempre. Ter esclarecido todas as
minhas dúvidas. Ter respondido meus chamados inúmeras vezes pelo laboratório e sempre ter
me atendido de forma pronta, prestativa e com um sorriso no rosto, independente do dia e
horário. Por ser uma profissional exemplar na qual também me espelho.
Ao Me. Alexandre Hilário Berenguer de Matos pela força tanto teórica quanto
experimental. Certamente, esse trabalho não seria o mesmo se não fosse por toda sua ajuda.
À banca examinadora da qualificação por todas as sugestões e contribuições para a
melhoria tanto da escrita como da apresentação dessa dissertação.
À República Rapina, incluindo todos os seus moradores e ex-moradores que, junto
comigo, em dezembro de 2009 fundaram essa fraternidade com o objetivo de, além de dividir
o mesmo teto, formar uma família. Vida longa à Rapina.
A todos os colegas do laboratório que, de alguma forma, participaram tanto desse
projeto como da minha vida, no dia-a-dia, no cafezinho no espaço gourmet, nos quitutes nas
tardes quentes pela FCM, pelas conversas sobre assuntos diversos. Beijos a todos vocês:
Cynthia, Taty, Karina, Stéphanie, Luciana, Marcella, Marina, Saul, Paty, Danny, Fábio,
André e Maria.
Aos demais familiares, amigos, funcionários e professores da FCM, que não citarei um
por um, para que ninguém se zangue, mas que tiveram seus papéis essenciais na minha vida,
na minha história e na minha formação.
À CAPES e a FAPESP por todo o fomento dessa pesquisa.

RESUMO
A ataxia de Friedreich (AF) é a ataxia hereditária autossômica recessiva mais comum
em populações caucasianas, causada por expansões (GAA) em homozigose no 1º íntron do
gene frataxin (FXN) (OMIM: 606829) em aproximadamente 96% dos pacientes afetados. Os
outros 4% dos pacientes são heterozigotos compostos para a expansão num dos alelos e
mutações pontuais no gene FXN no outro. Em ambos os casos há uma expressão diminuída da
proteína frataxina. A AF trata-se de uma doença neurodegenerativa, de início precoce e
evolução progressiva. A principal característica patológica é a morte dos neurônios dos
gânglios dorsais, seguida da degeneração da coluna posterior da medula espinhal e dos tratos
espinocerebelares, culminando com atrofia medular, especialmente na região cervical e
torácica. Dentre os sintomas da AF estão a instabilidade da marcha e a incoordenação de
movimentos como os primeiros sinais. Porém, também são evidentes: a perda da sensibilidade
vibratória e proprioceptiva, a fraqueza muscular, geralmente associada à escoliose, arreflexia
difusa com sinal de Babinski, disfunções cognitivas sutis, tremores de extremidades, disfagia,
disartria, pés cavos, cardiomiopatia e diabetes. Estudos de identificação das mutações
pontuais em heterozigotos compostos estão se mostrando uma abordagem promissora para
elucidar as vias metabólicas de ação da proteína frataxina. Além disso, há implicações para o
diagnóstico molecular e aconselhamento genético. Neste contexto, o objetivo deste estudo foi
investigar a frequência, perfil mutacional e fenotípico de uma amostra de pacientes brasileiros
heterozigotos compostos para a AF. Para tanto, recrutamos pacientes de três centros de
referências no país (Universidade Estadual de Campinas-UNICAMP, Universidade de São
Paulo Campus Ribeirão Preto-USP-RP e Universidade Federal do Rio Grande do Sul-
UFRGS). Aqueles com apenas uma expansão identificada foram submetidos ao
sequenciamento dos 5 exons e transições exon-intron do gene FXN (técnica de Sanger). Foi
identificada uma nova variante (c.482+1G>T) em um paciente heterozigoto composto para
AF, considerada patogênica de acordo com os critérios da American College of Medical
Genetics and Genomics (ACMG). Também encontramos uma mutação já descrita
previamente na literatura em outros dois pacientes não-relacionados (c.157delC). A
frequência observada de heterozigose composta foi de 2,87% (5/174); quando considerados
somente os casos em que mutações foram encontradas, a taxa diminui para 1,72% (3/174).
Estes são dados inéditos na população brasileira, com relevância para a escolha de técnicas
adequadas de diagnóstico molecular e orientação sobre aconselhamento genético em nossa
população.

Palavras-chave: Ataxia de Friedreich; Análise de Sequência de DNA; Biologia Molecular.

ABSTRACT
Friedreich Ataxia (FRDA) is the most common autosomal recessive ataxia in
Caucasian populations. It is caused by a homozygous GAA expansion in the first intron of the
frataxin gene (FXN) (OMIM: 606829) in 96% of the affected individuals. The remaining
patients are compound heterozygous: they have a GAA expansion in one allele and a point
mutation in the other. As a result, there is a dramatic reduction of the frataxin expression in
both situations. FRDA is a neurodegenerative disease characterized by early onset and
progressive ataxia. Death of the dorsal ganglia neurons, followed by degeneration of the
posterior spinal cord and spinocerebellar tracts, culminating in spinal cord atrophy, especially
in the cervical and thoracic regions, are the most common pathological features for this
disease. The first symptoms of FRDA are gait instability and limb incoordination. However,
other manifestations are also often found: loss of vibratory and proprioceptive sensation,
muscle weakness, usually associated with scoliosis, diffuse areflexia with Babinski sign,
subtle cognitive dysfunction, tremor, dysphagia, dysarthria, pes cavus, cardiomyopathy and
diabetes. Looking at FXN point mutations is a promising strategy to uncover the metabolic
pathways related to frataxin. In addition, it has obvious implications for clinical diagnosis and
genetic counseling. We therefore designed this study to determine the frequency, phenotypic
and mutational profile of Brazilian patients that presented compound heterozygosity for FXN.
To accomplish that, we recruited patients from 3 national reference centers (State University
of Campinas-UNICAMP, São Paulo University at Ribeirão Preto-USP-RP and Federal
University of Rio Grande do Sul-UFRGS). Those patients with a single identified expansion
underwent sequencing of all 5 exons and exon-intron boundaries at FXN (Sanger technique).
We identified a novel variant (c.482+1G>T) considered pathogenic following American
College of Medical Genetics and Genomics (ACMG) guidelines. In addition, another
pathogenic variant previously described in the literature (c.157delC) was found in 2 unrelated
subjects. Compound heterozygosity accounted for 2.87% (5/174) of all patients; however,
when considered only cases in which point mutations were found, the rate decreases to 1,72%
(3/174). These are novel data for the Brazilian population. From a clinical perspective, they
will help clinicians to choose the adequate techniques for FRDA diagnosis and proper genetic
counseling in our country.
Key-words: Friedreich Ataxia; Sequence Analysis, DNA; Molecular Biology

LISTA DE FIGURAS
Figura 1: Estrutura 3D da proteína frataxina, retirada do Protein Data Bank (PDB) com
código de entrada 1EKG (mature human frataxin)... ............................................................... 18
Figura 2: Gel em poliacrilamida mostrando ensaios de PCR para a amplificação das
expansões em pacientes com AF. As setas vermelhas apontam as expansões dos pacientes
testados, com tamanhos que variam entre 2500pb até mais de 3500pb ................................... 20
Figura 3: As figuras mostram os resultados de um teste de TP-PCR para um controle e um
paciente com Ataxia de Friedreich. Os picos dos eletroferogramas mostram a presença do
tripleto GAA. A presença de vários picos significa a presença de repetições dos tripletos. (a)
Teste de TP-PCR para uma amostra controle, nele, a presença de poucos picos revela que a
expansão GAA existente é pequena. (b) Teste de TP-PCR para um paciente de AF em que é
possível verificar várias expansões GAA no gene FXN ........................................................... 21
Figura 4: (a) Eletroferograma do exon 4 do paciente 94, em que a mutação c.482+1G>T em
heterozigose está evidenciada pela seta em vermelho. (b) Eletroferograma do exon 4 de um
controle. A base nitrogenada situada na posição equivalente à alteração descrita acima está
evidenciada pela seta em vermelho .......................................................................................... 32
Figura 5: Eletroferograma do exon 1 do paciente 431 em que é possível verificar a deleção de
um C em heterozigose na posição 157, mudando todo o frame de leitura a partir do ponto
evidenciado pela seta vermelha ................................................................................................ 32
Figura 6: Gel de eletroforese: no primeiro poço à esquerda está o ladder utilizado e os
tamanhos das bandas em pb. No poço 1 está presente a amostra do paciente 94 após a digestão
com a enzima de restrição MvaI, onde é possível observar a presença de duas bandas, uma de
279pb – referente ao amplicon do alelo em que está presente a mutação c.482+1G>T – e uma
banda de 218pb – referente ao alelo selvagem do paciente e que com a presença do sítio de
restrição da enzima, há o corte. No poço 2 observa-se a digestão do amplicon de um paciente
que não tem a mutação c.482+1G>T e por isso só é visualizada uma banda, pois a enzima
MvaI corta toda a amostra. Para fim de comparação, no poço 3 é possível observar um
amplicon (279pb) da PCR do exon 4 do gene FXN sem o tratamento com a enzima .............. 33

Figura 7: Resultados obtidos através da utilização do algoritmo ESE Finder: na figura 7a, é
possível observar, destacado em vermelho, o sítio em que a mutação c.482+1G>T foi
encontrada. Nesse caso, foi inserida no algoritmo uma sequência de até 5000 nucleotídeos
(limitação do algoritmo) na qual foram selecionados excertos anteriores e posteriores ao exon
4 da sequência selvagem do gene FXN. Como resultado, foram obtidos 10 pontos de
prováveis sítios de splicing e foi atribuída uma pontuação para cada um deles (score) com o
intuito de classifica-los por ordem de importância. O sítio em que a mutação c.482+1G>T foi
encontrada obteve a maior pontuação dentre eles. Na figura 7b, em que a sequência de 5000
nucleotídeos inserida no algoritmo foi a mutada (com a inserção da variante c.482+1G>T), os
resultados obtidos foram nove prováveis sítios de splicing e suas pontuações. A perda do sítio
de splicing, onde a nova variante foi encontrada, está evidenciado pelas setas vermelhas.
Note-se que, em ambas as figuras, são apresentados os mesmos prováveis sítios de splicing
para humanos e para camundongos, respectivamente, confirmando a preservação evolutiva da
sequência entre essas espécies .................................................................................................. 35

LISTA DE TABELAS
Tabela 1: Resumo das mutações pontuais/inserções/deleções descritas no HGMD em
heterozigotos compostos para o gene FXN até maio de 2017 com a adição das duas novas
variantes descritas no trabalho de Galea e colaboradores em 2016. Adaptado de: Gellera C,
Castellotti B, Mariotti C, Mineri R, Seveso V, DiDonato S, et al. Frataxin gene point
mutations in Italian Friedreich ataxia patients. Neurogenetics. 2007;8(4):289–99. ................ 22
Tabela 2: Sequencias dos primers utilizados para a realização da PCR e suas temperaturas
ótimas de ancoramento. ........................................................................................................... 27
Tabela 3: Número de pacientes, famílias, homozigotos e heterozigotos compostos estudados
para AF e o centro de origem da coleta dos dados obtidos (com a exclusão do paciente
UNICAMP02 do número total de pacientes, de famílias e de heterozigotos compostos)........ 29
Tabela 4: Pacientes heterozigotos compostos estudados nos quais as mutações de ponto
foram encontradas, juntamente com seus dados clínicos. O paciente 431 é proveniente do HC-
UNICAMP; o paciente POA1, do Hospital de Clínicas da UFRGS; e o paciente 94, do
Hospital de Clínicas da USP-Ribeirão Preto. ........................................................................... 30
Tabela 5: Dados clínicos dos pacientes heterozigotos compostos nos quais não foram
encontradas mutações de ponto no gene FXN, juntamente com os dados do paciente
UNICAMP02, que serviram de base para sua exclusão como possível heterozigoto composto
.................................................................................................................................................. 31
Tabela 6: Algoritmos utilizados para prever a provável patogenicidade da mutação
c.482+1G>T e seus resultados. ................................................................................................. 34

LISTA DE ABREVIATURAS, SIGLAS E SÍMBOLOS
ACMG American College of Medical Genetics
AF Ataxia de Friedreich
A Base nitrogenada Adenina
C Base nitrogenada Citosina
G Base nitrogenada Guanina
T Base nitrogenada Timina
CEP Comitê de Ética em Pesquisa
CGH Comparative genomic hybridization
SDS Dodecilsulfato de Sódio
(GAA)>36 Expansão GAA no 1º íntron do gene FXN com mais de 36 repetições
FRDA Friedreich’s Ataxia
FXN Gene que codifica a proteína Frataxina
°C Graus Celsius
μg Microgramas
μL Microlitros
mL Mililitro
MLPA Multiplex ligation-dependent probe amplification
ng Nanogramas
PVS1 Null variant – Very Strong Pathogenic (by ACMG)
pb Pares de base
pmoles Picomoles
% Porcentagem
PCR Reação em cadeia da Polimerase
RNAm RNA mensageiro
s Segundos
CDS Sequência codificante do DNA
TA Temperatura de Ancoramento
HGMD The Human Gene Mutation Database
UNICAMP Universidade Estadual de Campinas
USP-RP Universidade Estadual de São Paulo – Ribeirão Preto
UFRGS Universidade Federal do Rio Grande do Sul

SUMÁRIO
1.INTRODUÇÃO ................................................................................................................... 17
1.1 Histórico .......................................................................................................................... 17
1.2 Aspectos Genéticos ......................................................................................................... 17
1.3 A Proteína Frataxina ....................................................................................................... 17
1.4 Aspectos Clínicos ............................................................................................................ 18
1.5 O Diagnóstico Molecular da AF ..................................................................................... 20
1.6 Os Heterozigotos Compostos .......................................................................................... 21
1.7 Aconselhamento Genético .............................................................................................. 23
1.8 Justificativa ..................................................................................................................... 24
2. OBJETIVOS ....................................................................................................................... 25
2.1 Geral ................................................................................................................................ 25
2.2 Específicos ...................................................................................................................... 25
3. MATERIAL E MÉTODOS ............................................................................................... 26
3.1 Casuística e Coleta de Amostras ..................................................................................... 26
3.2 Extração do DNA Genômico .......................................................................................... 26
3.3 Amplificação por PCR .................................................................................................... 27
3.4 Detecção de Mutações: Sequenciamento dos Produtos de Amplificação ...................... 28
3.5 Análise de Dados ............................................................................................................ 28
4. RESULTADOS ................................................................................................................... 29
5. DISCUSSÃO ....................................................................................................................... 37
6. CONCLUSÃO ..................................................................................................................... 41
7. REFERÊNCIAS ................................................................................................................. 42
8. ANEXO I ............................................................................................................................ 48

17
1. INTRODUÇÃO
1.1 Histórico
A Ataxia de Friedreich (AF) foi descrita pela primeira vez em 1863 por Nikolaus
Friedreich em Heidelberg, Alemanha, e foi aceita como uma nova doença em 1877 após suas
publicações1. Friedreich descreveu as características clínicas e patológicas fundamentais da
doença, a saber: idade de início dos sintomas na puberdade; atrofia degenerativa da coluna
posterior da medula espinhal, que leva à ataxia progressiva, perda de sensibilidade e fraqueza
muscular; escoliose; deformidades nos pés e sintomas cardíacos1. Ao longo dos anos, estudos
foram surgindo dando mais aprofundamento na clínica e na patologia da doença, porém foi
em 1988 que a doença foi associada a alterações no braço longo do cromossomo 91,2. Quase
uma década depois, em 1996, a sequência do gene frataxin (FXN) foi primeiramente descrita
por Campuzano e colaboradores, sendo a mutação característica da doença identificada1,3. A
partir disso, inúmeros estudos moleculares foram realizados e foi possível tanto conhecer
melhor o substrato molecular da doença, suas vias metabólicas e de ação da proteína
frataxina, quanto correlacionar a presença/ausência da sua expressão com o quadro clínico e
patológico apresentado pelos pacientes.
1.2 Aspectos Genéticos
A AF é a ataxia autossômica recessiva hereditária mais comum na maioria das
populações, possuindo uma prevalência estimada entre 1:50000 e 1:29000 em populações
caucasianas4. É causada por uma mutação que consiste em uma expansão instável e em
homozigose (em aproximadamente 96% dos casos)4–6 da repetição de trinucleotídeos GAA no
primeiro íntron do gene FXN que está localizado no cromossomo 9q13-213. Esse gene
codifica uma proteína de 210 aminoácidos chamada frataxina e essa mutação resulta na sua
expressão reduzida3. Os outros 4% dos pacientes são heterozigotos compostos para uma
mutação pontual, deleção ou inserção, no gene FXN em um dos alelos e uma expansão
(GAA)>36 no outro5,7.
Na literatura, indivíduos normais carregam alelos que variam de 5 até 33 repetições de
GAA. Alelos com expansões entre 34 e 65 repetições são considerados pré-mutados e alelos
com mais de 66 repetições são considerados expandidos, causadores da doença, com
penetrância completa. As expansões podem passar de 1.300 repetições em alguns pacientes5.
1.3 A Proteína Frataxina

18
A frataxina (Figura 1) é uma proteína da matriz mitocondrial, relacionada ao
transporte de ferro e metabolismo de sulfeto ferroso I (FeS)3,4. Assim, pacientes com AF, por
possuírem níveis reduzidos desta proteína, apresentam prejuízo na atividade de enzimas
dependentes de FeS, como as da cadeia respiratória, e acúmulo de ferro intramitocondrial
observado no coração, fígado, núcleo denteado e fibroblastos8. O ferro acumulado, por sua
vez, ao reagir com oxigênio, causa oxidação de elementos celulares; combinado aos defeitos
na cadeia respiratória, há produção de radicais livres, degeneração e morte celular4.
A proteína está expressa em altos níveis no coração do adulto e na medula espinhal,
assim como no gânglio da raiz dorsal e na camada granulosa do cerebelo e também em
tecidos com alta taxa metabólica1,7. A expressão reduzida da frataxina nos lugares onde ela é
altamente expressa explica os achados anátomo-patológicos da doença.
Figura 1: Estrutura 3D da proteína frataxina, retirada do Protein Data Bank (PDB) com
código de entrada 1EKG (mature human frataxin).
1.4 Aspectos Clínicos
Os efeitos sobre o sistema neuromuscular são os mais evidentes na AF. A principal
consequência da alteração genética é a morte dos neurônios dos gânglios dorsais, seguida por

19
degeneração da coluna posterior da medula espinhal e dos tratos espinocerebelares. Assim, o
achado anátomo-patológico mais característico da doença é a atrofia medular, especialmente
na região cervical e torácica. Os núcleos profundos do cerebelo e suas vias eferentes, assim
como os tratos córtico-espinais também sofrem degeneração em estágios mais avançados da
doença9.
Os sintomas de AF geralmente se iniciam de forma precoce e tem caráter
progressivo10. Na maioria dos casos, a instabilidade da marcha e a incoordenação de
movimentos são os primeiros sinais. Porém, também são evidentes a perda da sensibilidade
vibratória e proprioceptiva; a fraqueza muscular, geralmente associada à escoliose; arreflexia
com presença do sinal de Babinski; disfunção cognitiva sutil; tremores de extremidades;
disfagia; disartria; pés cavos; cardiomiopatia e diabetes 8,10.
A disfunção cardíaca pode causar a morte prematura a uma minoria significante de
pacientes, particularmente aqueles em que a AF se manifesta de forma precoce. Os
ecocardiogramas dos pacientes mostram uma hipertrofia concêntrica dos ventrículos ou uma
hipertrofia assimétrica do septo cardíaco. Em muitos casos, essa cardiomiopatia progride para
um estágio dilatado com insuficiência cardíaca às vezes grave 10. A causa mortis mais comum
nos pacientes com AF se dá pelo desenvolvimento desse fenótipo cardíaco11. A gravidade da
doença e a idade de início dos sintomas estão relacionadas com o comprimento da repetição
do tripleto GAA, especialmente no menor alelo12.

20
1.5 O Diagnóstico Molecular da AF
Testes moleculares para a detecção das expansões em AF são realizados a partir de
quatro técnicas: (i) reação em cadeia da polimerase (“Polymerase chain reaction” – PCR)
(Figura 2); (ii) PCR de longo alcance (“long-range PCR” - LR-PCR); (iii) “triplet repeat
primed PCR” (TP-PCR) (Figura 3); e/ou (iv) “Southern blotting”13. Todas essas técnicas têm
como objetivo primário verificar a presença dos alelos expandidos para o gene FXN. Como
objetivo secundário, elas tentam quantificar o número exato de expansões que cada alelo
contém.
Figura 2: Gel em poliacrilamida mostrando ensaios de PCR para a amplificação das
expansões em pacientes com AF. As setas vermelhas apontam as expansões dos pacientes
testados, com tamanhos que variam entre 2500pb até mais de 3500pb.

21
Figura 3: As figuras mostram os resultados de um teste de TP-PCR para um controle e
um paciente com Ataxia de Friedreich. Os picos dos eletroferogramas mostram a
presença do tripleto GAA. A presença de vários picos significa a presença de repetições
dos tripletos. (a) Teste de TP-PCR para uma amostra controle, nele, a presença de
poucos picos revela que a expansão GAA existente é pequena. (b) Teste de TP-PCR para
um paciente de AF em que é possível verificar várias expansões GAA no gene FXN.
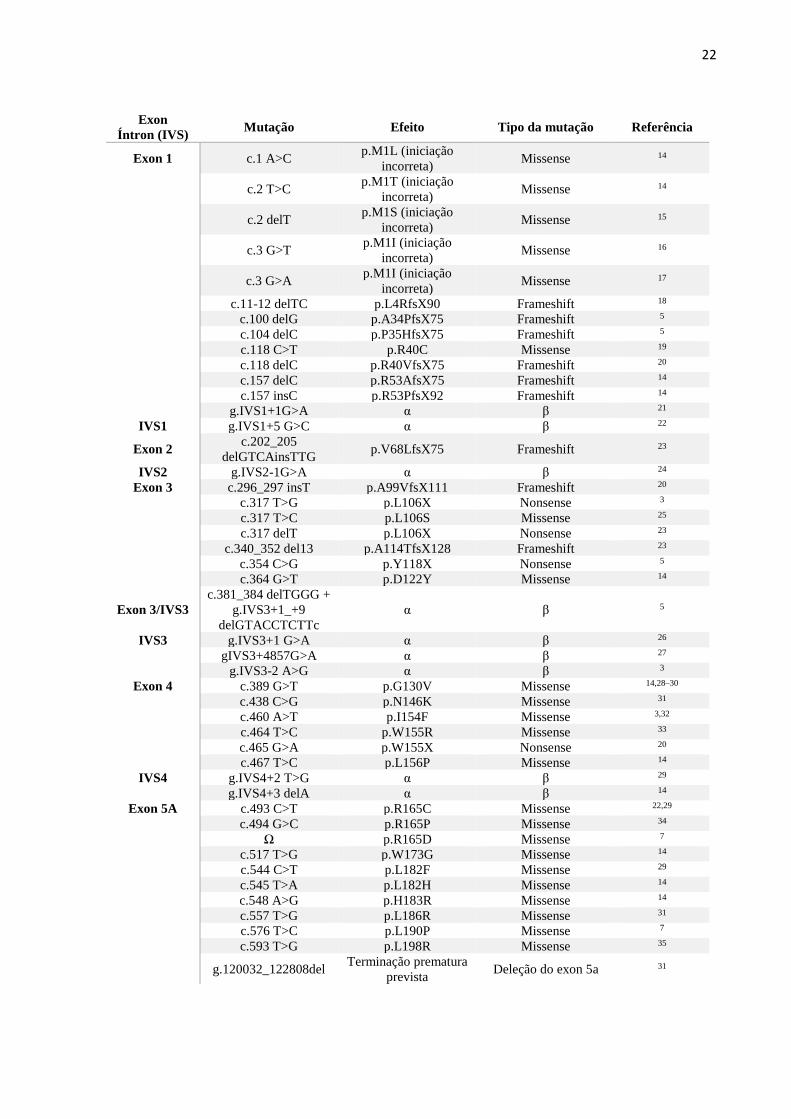
1.6 Heterozigotos Compostos
Nos indivíduos heterozigotos compostos para a AF, 51 mutações
pontuais/inserções/deleções no gene FXN foram descritas no The Human Gene Mutation
Database (HGMD) até o momento5,7. Um resumo dessas alterações genéticas segue abaixo,
apresentado na Tabela 1.
Tabela 1: Resumo das mutações pontuais/inserções/deleções descritas no HGMD em
heterozigotos compostos para o gene FXN até maio de 2017 com a adição das duas novas
variantes descritas no trabalho de Galea e colaboradores em 2016. Adaptado de: Gellera
C, Castellotti B, Mariotti C, Mineri R, Seveso V, DiDonato S, et al. Frataxin gene point
mutations in Italian Friedreich ataxia patients. Neurogenetics. 2007;8(4):289–99.
α: Predição de splicing anormal
β: Mutação em sítio doador de splicing
Ω: Dado não fornecido pela literatura
22
Exon
Íntron (IVS) Mutação Efeito Tipo da mutação Referência
Exon 1 c.1 A>C p.M1L (iniciação
incorreta) Missense 14
c.2 T>C p.M1T (iniciação
incorreta) Missense 14
c.2 delT p.M1S (iniciação
incorreta) Missense 15
c.3 G>T p.M1I (iniciação
incorreta) Missense 16
c.3 G>A p.M1I (iniciação
incorreta) Missense 17
c.11-12 delTC p.L4RfsX90 Frameshift 18
c.100 delG p.A34PfsX75 Frameshift 5
c.104 delC p.P35HfsX75 Frameshift 5
c.118 C>T p.R40C Missense 19
c.118 delC p.R40VfsX75 Frameshift 20
c.157 delC p.R53AfsX75 Frameshift 14
c.157 insC p.R53PfsX92 Frameshift 14
g.IVS1+1G>A α β 21
IVS1 g.IVS1+5 G>C α β 22
Exon 2 c.202_205
delGTCAinsTTG p.V68LfsX75 Frameshift 23
IVS2 g.IVS2-1G>A α β 24
Exon 3 c.296_297 insT p.A99VfsX111 Frameshift 20
c.317 T>G p.L106X Nonsense 3
c.317 T>C p.L106S Missense 25
c.317 delT p.L106X Nonsense 23
c.340_352 del13 p.A114TfsX128 Frameshift 23
c.354 C>G p.Y118X Nonsense 5
c.364 G>T p.D122Y Missense 14
Exon 3/IVS3
c.381_384 delTGGG +
g.IVS3+1_+9
delGTACCTCTTc
α β 5
IVS3 g.IVS3+1 G>A α β 26
gIVS3+4857G>A α β 27
g.IVS3-2 A>G α β 3
Exon 4 c.389 G>T p.G130V Missense 14,28–30
c.438 C>G p.N146K Missense 31
c.460 A>T p.I154F Missense 3,32
c.464 T>C p.W155R Missense 33
c.465 G>A p.W155X Nonsense 20
c.467 T>C p.L156P Missense 14
IVS4 g.IVS4+2 T>G α β 29
g.IVS4+3 delA α β 14
Exon 5A c.493 C>T p.R165C Missense 22,29
c.494 G>C p.R165P Missense 34
Ω p.R165D Missense 7
c.517 T>G p.W173G Missense 14
c.544 C>T p.L182F Missense 29
c.545 T>A p.L182H Missense 14
c.548 A>G p.H183R Missense 14
c.557 T>G p.L186R Missense 31
c.576 T>C p.L190P Missense 7
c.593 T>G p.L198R Missense 35
g.120032_122808del Terminação prematura
prevista Deleção do exon 5a 31

23
As diferenças clínicas que os pacientes com AF homozigotos teriam em relação aos
heterozigotos compostos são bem discutidas na literatura. Nela é possível verificar que as
mutações pontuais no gene FXN levam, em geral, a fenótipos brandos, ou relacionados
diretamente ao tamanho da expansão GAA, no caso de mutações missense; Já no caso de
mutações nonsense ou de frameshift, as alterações levam a quadros clínicos mais graves que
os relacionados diretamente às expansões GAA, ou seja, apresentam, por exemplo, uma data
de início dos sintomas da doença mais precoce5,6. Também foi demonstrado que pacientes
homozigotos são mais propensos a desenvolver cardiomiopatia e que grupos de pacientes
heterozigotos compostos, com mutações que levam a não produção da frataxina, tem 4,5
vezes mais disposição para desenvolver diabetes mellitus7.
Pacientes com mutações pontuais no gene FXN em homozigose ainda não foram
observados na literatura. Isso pode ser explicado, por exemplo, pela raridade da ocorrência
dessas mutações, e também pelo fato de que a inativação do gene homólogo ao FXN em
camundongos leva a um quadro de letalidade embrionária1,36, sugerindo que “null mutations”
– aquelas que resultam na completa ausência da proteína – podem levar a um quadro clínico
muito grave ou a um fenótipo letal1.
1.7 Aconselhamento Genético
O aconselhamento genético é uma etapa importante do cuidado clínico de pacientes
acometidos por doenças hereditárias. Esse processo inclui, por exemplo, o aconselhamento de
casais sobre o risco e a probabilidade da recorrência da doença hereditária em questão na sua
prole. No caso da AF, nos deparamos ocasionalmente com indivíduos que já tem a
confirmação genética da doença (homozigoto para a expansão GAA no gene FXN) e querem
orientação acerca do risco para sua prole.
Considerando que a frequência estimada de portadores (indivíduos que carregam a
expansão GAA no gene FXN em um de seus alelos) em populações caucasianas é de 1 em
9037, o procedimento que pode ser realizado para que seja possível um aconselhamento
genético preciso, além da análise em cima das probabilidades (que consideram todas as
variáveis existentes), é um teste para saber se o cônjuge é portador ou não da expansão. Esse
teste somente pode ser realizado com a autorização do cônjuge.
O teste geralmente é efetuado utilizando-se a técnica de LR-PCR ou a de TP-PCR,
pois são ambas de baixo custo e precisas na verificação da presença ou não da expansão. Elas
podem, entretanto, não detectar outros tipos de variantes patogênicas no gene FXN.

24
Desta forma, nosso estudo de heterozigotos compostos tem uma implicação direta no
aconselhamento genético dos pacientes com AF e essa questão será melhor discutida na seção
que diz respeito à discussão dessa dissertação.
1.8 Justificativa
Não existem tratamentos efetivos para a AF. Contudo, o melhor conhecimento
fisiopatológico está possibilitando o desenvolvimento de intervenções terapêuticas10.
A identificação de mutações pontuais no gene FXN vem se mostrando uma forma de
elucidar a importância metabólica da proteína, a correlação estrutura-função e sua associação
com o quadro clínico patológico da doença.
Não dispomos de dados no Brasil sobre a frequência e o perfil mutacional de pacientes
com heterozigose composta. Portanto, um estudo com este foco pode contribuir para o melhor
entendimento da doença em nosso meio, com implicações para a testagem molecular
diagnóstica e para o aconselhamento genético.

25
2. OBJETIVOS
2.1 Geral
• Caracterização da nossa casuística de pacientes brasileiros heterozigotos compostos
para o gene FXN.
2.2 Específicos
• Determinar a frequência de heterozigotos compostos na nossa casuística e compará-la
em relação à literatura;
• Descrever as novas mutações de ponto encontradas e analisar in silico sua
patogenicidade;
• Caracterizar o fenótipo dos pacientes confirmados heterozigotos.

26
3. MATERIAIS E MÉTODOS
3.1 Casuística e coleta de amostras
Todos os indivíduos participantes deste projeto foram selecionados no ambulatório de
doenças neuromusculares e neurogenética do HC-UNICAMP, no ambulatório de
neurogenética do Hospital de Clínicas da USP-Ribeirão Preto e no ambulatório de
neurogenética do Hospital de Clínicas de Porto Alegre (HCPA ligado à Universidade Federal
do Rio Grande do Sul). Todos assinaram previamente um TCLE nos centros em que suas
amostras foram coletadas.
Essas coletas foram realizadas ao longo de 10 anos, entre 2007 e 2017. Na UNICAMP
essas amostras estão armazenadas em biorrepositório aprovado pelo CEP com CAAE número
12112913.3.0000.5404 (Anexo I). Os dados clínicos desses pacientes foram revisados e
discutidos pelo Dr. Marcondes Cavalcante França Jr. da UNICAMP, Dr.Wilson Marques
Junior da USP-RP e Dra. Laura Bannach Jardim do HCPA-UFRGS.
Os critérios de inclusão para a participação nesse estudo foram:
• Pacientes com AF e expansão GAA confirmada em um ou dois alelos;
• Confirmação do fenótipo clínico do paciente no caso do estudo de heterozigoto
composto.
3.2 Extração do DNA genômico
A extração do DNA genômico foi feita a partir de amostras de sangue periférico
coletadas dos pacientes e familiares em tubos com EDTA para evitar a coagulação. O
protocolo de extração com fenol-clorofórmio segue descrito abaixo38:
Aproximadamente 6 a 12 mL de sangue venoso foram colhidos de cada indivíduo
recrutado para o estudo. As amostras foram centrifugadas por 10 minutos a 2500 rpm e a fase
intermediária, contendo os leucócitos, foi transferida para um tubo de propileno com fundo
cônico. O passo seguinte foi a adição das soluções de RSB 1X até completar o volume de
11mL, e de 60µL de Nonidet®. Após homogeneização em agitador durante 10 minutos, a
solução foi centrifugada a 2500 rpm por mais 10 minutos e o sobrenadante foi descartado. Em
seguida, foram adicionados 3mL de solução SDS a 10% e 60µL de proteinase K (100mg/mL).

27
Posteriormente, as amostras foram incubadas a 37° por 24h. Após a incubação, foram
acrescentados 3mL de fenol saturado (pH 7,8), seguido de agitação dos tubos até
homogeneização e centrifugação por 10 minutos a 2500 rpm. Descartada a fração inferior do
tubo, o processo foi repetido com 1,5 mL de fenol saturado (pH7,8) e 1,5 mL de solução de
clorofórmio-álcool isoamílico (proporção 24:1). Após descartar desta a vez a porção superior,
foi adicionado 3 mL de solução clorofórmio-álcool isoamílico (proporção 24:1). Descartada
novamente a parte superior, o DNA genômico foi precipitado com a adição de 6 mL de etanol
absoluto ao tubo. A “nuvem” de DNA foi transferida para tubos criogênicos de 1.5 mL e com
aproximadamente 200-250µL de TE 1X. Esse protocolo permitiu a obtenção de DNA em
altas concentrações, aproximadamente 700μg de DNA genômico a partir de aproximadamente
12 mL de sangue venoso.
3.3 Amplificação por PCR
As reações de PCR foram realizadas para um volume final de 25μl, contendo 10 pmoles de
cada primer específico (Tabela 2), 2,5 nmoles de cada nucleotídeo (dGTP, dCTP, dTTP e
dATP), 1,0U de Taq DNA polimerase (Fermentas), 50 nmoles de MgCl2 e 50ng de DNA. Os
produtos de amplificação foram visualizados em gel de agarose 1% corado com SYBR Safe
(Invitrogen). As condições para a realização da PCR foram: desnaturação inicial a 95ºC por 4
minutos, seguida por 35 ciclos de 30 segundos a 95ºC, 30 segundos à temperatura de
ancoramento específica dos pares de primers e 1 minuto a 72ºC; e, depois dos ciclos, extensão
final a 72ºC por 10 minutos. Para determinar a temperatura de ancoramento ideal dos pares de
primers, foram realizados PCRs em gradientes de temperatura.
Tabela 2: Sequências dos primers utilizados para a realização da PCR e suas
temperaturas ótimas de ancoramento
Primer Sequência Forward Sequencia Reverse TA
Exon 1 5’-CCAGCGCTGGAGGGCG-3’ 5’-CCGCGGCTGTTCCCGG-3’ 56,7ºC
Exon 2 5’-GGCACTCGAATGTAGAAGTAGC-3’ 5’-AGAGGAAGATACCTATGACGTG-3’ 51,5ºC
Exon 3 5’-AAAATGGAAGCATTTGGTAATCA-3’ 5’-AGTGAACTAAAATTCTTAGAGGG-3’ 51,5ºC
Exon 4 5’-GGTGTATTTTGTGTACTTG-3’ 5’-GTCACATTTCGGAAGTC-3’ 51,5ºC
Exon 5 5’-CTGAAGGGCTGTGCTGTGGA-3’ 5’-TGTCCTTAAAACGGGGCT-3’ 51,5ºC

28
3.4 Detecção de mutações: Sequenciamento dos produtos de amplificação
Os produtos de amplificação foram purificados com a utilização da enzima illustra
ExoProStar (GE Healthcare), de acordo com as instruções do fabricante e, em seguida,
sequenciados em ambas as direções em sequenciador automático ABI 3500 (Applied
Biosystems).
A reação de sequenciamento seguiu as recomendações do fabricante. O kit utilizado
foi o BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). Além do pré-
mix já fornecido no kit, foram adicionados 5 pmoles de primer e 200ng do produto de
amplificação. As condições para a realização do sequenciamento foram: 35 ciclos de 95ºC por
20s, 50ºC por 15s, 60ºC por 1 min. A purificação da reação de sequenciamento foi realizada
com etanol e acetato de amônio 7,5mM, como sugerido pelo fabricante. Após esses passos, as
amostras foram ressuspendidas em formamida para a realização da corrida na plataforma.
3.5 Análise dos dados
Os eletroferogramas gerados pela plataforma de sequenciamento foram analisados
utilizando dois softwares: o FinchTV 1.4.039 e o CodonCode Aligner40.
As mutações encontradas tiveram suas entradas pesquisadas em databases de mutações
nacionais e internacionais para saber se as variantes já haviam sido descritas na literatura.
Aquelas utilizadas nesse estudo foram: 1000 Genomes41, EVS42, BIPMed, ExAC e HGMD43.
As variantes não encontradas na literatura foram submetidas a algoritmos de avaliação
in silico da provável patogenicidade da variante. Aqueles utilizados nesse estudo foram:
NNSplice44, Mutation Taster45, ESE Finder 3.046,47 e NetGene248,49. Ao final, as variantes
foram classificadas de acordo com os critérios do American College of Medical Genetics50.
Novas variantes também foram confirmadas através de reação de digestão enzimática.
Essa confirmação se fez necessária pelo fato das fitas forward e reverse do sequenciamento
automático, em alguns casos, não abrangerem todas as bases dos amplicons, principalmente
aquelas que se encontram próximas à sequência dos primers. Para utilizar uma enzima com
um sítio de clivagem que incluísse as novas variantes deste estudo, utilizamos o algoritmo
Web Cutter 2.051.

29
4. RESULTADOS
Fizeram parte dessa pesquisa 183 pacientes, sendo que 68 pacientes (61 famílias)
foram acompanhados no HC-UNICAMP, 75 no Hospital de Clínicas da UFRGS e 40
pacientes (38 famílias) na USP-Ribeirão Preto.
Dos 183 pacientes estudados, 174 são de famílias não relacionadas e seus diagnósticos
moleculares de AF foram confirmados nos centros em que suas amostras foram coletadas.
Os pacientes considerados heterozigotos compostos tiveram seus quadros clínicos
revisados para a confirmação do fenótipo relacionado à doença. Um desses pacientes – o
UNICAMP02 –, proveniente do HC-UNICAMP, apesar de ter confirmada a presença da
expansão GAA em um de seus alelos, foi excluído como acometido pela AF. Isso se deve ao
fato de que, uma vez obtidos os dados de sua família, foi constatado em seu heredograma que
se tratava de herança autossômica dominante (sua mãe e seu irmão apresentam quadro clínico
semelhante). Outras características clínicas observadas nesse paciente também não condizem
com o esperado para casos de AF, como a presença de atrofia cerebelar e bulbopontina,
sensibilidade inteiramente normal (incluindo eletroneuromiografia) e reflexos exaltados
(Tabela 5). Assim, pelos motivos citados, esse paciente foi excluído do cálculo final da
frequência de heterozigotos compostos. Um resumo da casuística contendo os dados
utilizados para a realização desse cálculo estão apresentados na Tabela 3 abaixo.
Tabela 3: Número de pacientes, famílias, homozigotos e heterozigotos compostos
estudados para AF e o centro de origem da coleta dos dados obtidos (com a exclusão do
paciente UNICAMP02 do número total de pacientes, de famílias e de heterozigotos
compostos).
Centro de
Coleta Pacientes Famílias Homozigotos
Heterozigotos
Compostos
UNICAMP 68 61 66 2
UFRGS 75 75 73 2
USP-RP 40 38 39 1
TOTAL
183 174 178 5
Sendo assim, o cálculo da frequência de heterozigotos compostos na nossa amostra da
população brasileira é de 2,87% (5/174). Se considerarmos somente os pacientes em que
foram encontradas mutações de ponto/inserção/deleção, a taxa é de 1,72% (3/174). As

30
frequências são inferiores àquelas observadas na literatura para populações caucasianas, a
saber, 4%7,14.
Foi realizado o sequenciamento dos cinco exons do gene FXN para os cinco pacientes
heterozigotos compostos que participaram desse estudo, juntamente com o paciente
UNICAMP02, porém mutações (pontuais/inserções/deleções) foram encontradas somente em
três deles. Os resultados podem ser observados na Tabela 4 abaixo.
Tabela 4: Pacientes heterozigotos compostos estudados nos quais as mutações de ponto
foram encontradas, juntamente com seus dados clínicos. O paciente 431 é proveniente
do HC-UNICAMP; o paciente POA1, do Hospital de Clínicas da UFRGS; e o paciente
94, do Hospital de Clínicas da USP-Ribeirão Preto.
Paciente Mutação Início Fenótipo Cardiopatia Diabetes
431 c.157 delC 27 anos Ataxia mista; Reflexos (+);
Sensibilidade alterada - -
POA1 c.157 delC 12 anos Ataxia mista; Reflexos(+/-);
Sensibilidade alterada - +
94 c.482+1G>T 10 anos Ataxia mista; Reflexos (-);
Sensibilidade Alterada + +
Os dados clínicos dos pacientes em que não foram encontradas mutações de ponto
estão descritos na Tabela 5 abaixo. Esses dados serão melhor explorados na seção que
concerne à discussão dessa dissertação.

31
Tabela 5: Dados clínicos dos pacientes heterozigotos compostos nos quais não foram
encontradas mutações de ponto no gene FXN, juntamente com os dados do paciente
UNICAMP02, que serviram de base para sua exclusão como possível heterozigoto
composto.
Paciente Início Padrão de
Herança Fenótipo Cardiopatia Diabetes
UNICAMP01 48 anos Caso Isolado
Ataxia presente;
Reflexos abolidos;
Sensibilidade profunda
alterada
- +
UNICAMP02 32 anos Dominante
Ataxia presente;
Reflexos exaltados;
Sensibilidade normal
- -
POA2 15 anos Caso Isolado
Ataxia presente;
Reflexos preservados;
Sensibilidade reduzida
- -
No paciente 94, foi encontrada uma nova mutação no gene FXN. Essa mutação está
localizada na primeira base nitrogenada logo após o término do exon 4 do gene.
Prioritariamente, as primeiras bases logo antes do começo de um exon, ou logo após o
término dele, são sítios canônicos de splicing e tendem a ser altamente conservados. Assim a
nomenclatura correta para a alteração encontrada é “c.482+1G>T”, onde o “c” significa que a
nomenclatura está tratando do CDS do gene, o “482+1” é referente à posição da alteração no
CDS, em que o exon 4 termina na posição “482” e “+1” refere-se à primeira base após o
término dele e o “G>T” mostra que a mudança se dá pela substituição de uma guanina por
uma timina.
No eletroferograma mostrado na Figura 4a, é possível observar a presença da mutação
em heterozigose. Na Figura 4b é possível observar o eletroferograma de um paciente sem a
mutação, para fim de comparação.

32
Figura 4: (a) Eletroferograma do exon 4 do paciente 94, em que a mutação c.482+1G>T
em heterozigose está evidenciada pela seta em vermelho. (b) Eletroferograma do exon 4
de um controle. A base nitrogenada situada na posição equivalente à alteração descrita
acima está evidenciada pela seta em vermelho.
Os pacientes 431 e POA1 apresentaram a mutação c.157delC, que já foi descrita
previamente na literatura e será melhor discutida na seção de discussão dessa dissertação. Na
figura 5, é possível observar o eletroferograma do paciente 431 e verificar a deleção que muda
o frame de leitura.
Figura 5: Eletroferograma do exon 1 do paciente 431 em que é possível verificar a
deleção de um C em heterozigose na posição 157, mudando todo o frame de leitura a
partir do ponto evidenciado pela seta vermelha.

33
Para a nova variante c.482+1G>T, foram feitas pesquisas em bases de dados nacionais
e internacionais como o 1000 Genomes, ExAC, EVS, HGMD e BIPMed, com o intuito de
saber se essa mutação já havia sido descrita anteriormente e, até o presente momento, essa
variante não se encontra depositada nessas bases de dados.
Para confirmar a presença dessa mutação, conforme descrito na seção de materiais e
métodos dessa dissertação, também foi realizada uma digestão enzimática do produto da PCR
do exon 4 do paciente 94. A enzima de restrição utilizada para esse procedimento foi a MvaI,
selecionada a partir do algoritmo WebCutter 2.0. A sequência do exon 4 foi submetida ao
algoritmo e inúmeras enzimas de restrição com sítios de corte na sequência do nosso
amplicon foram retornadas pelo algoritmo. A enzima MvaI foi selecionada, pois tem o sítio de
restrição que flanqueia as bases onde a nova mutação foi encontrada e também por já existir
alíquota dela no laboratório onde os experimentos foram realizados.
O sítio de restrição da enzima MvaI é CCWGG para a fita forward e é GGWCC para o
seu reverso complementar. W pode ser um A ou um T. No caso da nossa mutação, temos a
sequência CCAGG para a fita selvagem e a sequência CCAGT para a fita mutada, assim,
quando há a presença da mutação na amostra, a enzima de restrição perde seu sítio de ligação
e não realiza o corte do amplicon.
Na Figura 6 é possível visualizar o gel de agarose em que foi testada a enzima MvaI e
a confirmação da presença da mutação.
Figura 6: Gel de eletroforese: no primeiro poço à esquerda está o ladder utilizado e os
tamanhos das bandas em pb. No poço 1 está presente a amostra do paciente 94 após a

34
digestão com a enzima de restrição MvaI, onde é possível observar a presença de duas
bandas, uma de 279pb – referente ao amplicon do alelo em que está presente a mutação
c.482+1G>T – e uma banda de 218pb – referente ao alelo selvagem do paciente e que
com a presença do sítio de restrição da enzima, há o corte. No poço 2 observa-se a
digestão do amplicon de um paciente que não tem a mutação c.482+1G>T e por isso só é
visualizada uma banda, pois a enzima MvaI corta toda a amostra. Para fim de
comparação, no poço 3 é possível observar um amplicon (279pb) da PCR do exon 4 do
gene FXN sem o tratamento com a enzima.
Para analisar a provável patogenicidade da variante encontrada, foram utilizados os
algoritmos Mutation Taster45, NNSplice (Berkeley Drosophila Genome Project)44, ESE
Finder 3.046,47 e NetGene248,49, sendo que os últimos três são específicos para predição de
variações em sítios doadores de splicing. A imagem do resultado obtido através do algoritmo
ESE Finder pode ser observado na Figura 7.
A Tabela 6, que resume os resultados obtidos através da utilização dos algoritmos
selecionados nesse trabalho e que predizem a mutação como altamente patogênica, pode ser
observada abaixo.
Tabela 6: Algoritmos utilizados para prever a provável patogenicidade da mutação
c.482+1G>T e seus resultados.
Algoritmo Mutation Taster NNSplice ESE Finder NetGene2
Alteração
patogênica + + + +
Perda de sítio
doador de Splicing + + + +

35
Figura 7: Resultados obtidos através da utilização do algoritmo ESE Finder: na figura
7a, é possível observar, destacado em vermelho, o sítio em que a mutação c.482+1G>T
foi encontrada. Nesse caso, foi inserida no algoritmo uma sequência de até 5000
nucleotídeos (limitação do algoritmo) na qual foram selecionados excertos anteriores e
posteriores ao exon 4 da sequência selvagem do gene FXN. Como resultado, foram
obtidos 10 pontos de prováveis sítios de splicing e foi atribuída uma pontuação para
cada um deles (score) com o intuito de classifica-los por ordem de importância. O sítio
em que a mutação c.482+1G>T foi encontrada obteve a maior pontuação dentre eles. Na
figura 7b, em que a sequência de 5000 nucleotídeos inserida no algoritmo foi a mutada

36
(com a inserção da variante c.482+1G>T), os resultados obtidos foram nove prováveis
sítios de splicing e suas pontuações. A perda do sítio de splicing, onde a nova variante foi
encontrada, está evidenciado pelas setas vermelhas. Note-se que, em ambas as figuras,
são apresentados os mesmos prováveis sítios de splicing para humanos e para
camundongos, respectivamente, confirmando a preservação evolutiva da sequência
entre essas espécies.
Não foi possível utilizar algoritmos que se baseassem na mudança da sequência de
aminoácidos da proteína, uma vez que essa mutação se encontra num sítio doador de splicing,
e a maioria dos algoritmos são limitados a avaliar a substituição de aminoácidos para prever a
patogenicidade na provável perda ou não da função da proteína.
De acordo com a ACMG, as variantes que resultam na perda de sítios doadores e/ou
receptores de splicing (situadas 1 ou 2 bases depois do fim de cada exon), são classificadas
como PVS1, uma “null variant”. Além disso, esta variante atende um critério moderado de
patogenicidade – ausência em controles nas bases de dados ExAC e 1000 Genomes, e um
critério de suporte – múltiplas linhas de evidência computacional indicam efeito deletério da
variante (vide tabela 6). Portanto, de acordo com os critérios da ACMG, a alteração
c.482+1G>T foi classificada como patogênica.

37
5. DISCUSSÃO
Essa dissertação se constitui como o primeiro estudo de heterozigotos compostos para
AF no Brasil. Nossa casuística foi composta por 183 pacientes, sendo 174 deles pertencentes
a famílias não relacionadas.
Na literatura é possível encontrar uma pequena variação na porcentagem de
heterozigotos compostos nas populações estudadas, Cossée e colaboradores em 199914 e
Galea e colaboradores em 20167 encontraram uma taxa de 4%, enquanto o trabalho de Gellera
e colaboradores em 20075 traz um número em torno de 4,3%. A frequência da nossa amostra
da população brasileira ficou em 2,87% (5/174). Se considerarmos somente os pacientes em
que foram encontradas mutações de ponto/inserção/deleção, a taxa é de 1,72% (3/174).
A frequência encontrada neste estudo ficar abaixo daquela verificada na literatura e
podemos inferir que isso é causado, provavelmente, pela: (i) cobertura das regiões brasileiras
presentes no nosso estudo (Sul e Sudeste), pois, mesmo considerando que muitos pacientes
participantes se locomoveram de outras regiões para comparecer aos nossos centros de coleta,
não podemos garantir que a cobertura do estudo é nacional; e (ii) heterogeneidade de etnias
presentes no nosso país.
Na África e na Ásia, a AF é uma doença incomum e não existem estudos da
frequência de portadores nessas regiões52,53. É esperado que essa frequência seja menor que a
apresentada por populações europeias (aproximadamente 1:90)37, o que reflete diretamente na
frequência de heterozigotos compostos desses continentes, que também é esperado que seja
menor.
Assim, é possível inferir que o resultado de uma frequência de heterozigotos
compostos menor do que aquela encontrada na população europeia e presente nos estudos
citados nessa dissertação é devido à miscigenação da população brasileira, com a mistura das
populações europeias, africanas e asiáticas, por exemplo.
Dos seis pacientes classificados como possíveis heterozigotos compostos que
participaram desse estudo (três coletados no HC-UNICAMP, dois na UFRGS e um na USP-
RP), cinco foram confirmados para essa condição através do teste genético que verificou a
presença de uma expansão GAA no gene FXN em um de seus alelos, juntamente com a
clínica que sugere a AF.
O paciente não confirmado (UNICAMP02) acabou sendo excluído dos cálculos de
frequência e não foi considerado como heterozigoto composto – apesar de também apresentar
a expansão GAA –, uma vez que seus dados clínicos não sugerem quadro de AF pois

38
apresentam: (i) padrão de herança dominante tendo a mãe e um irmão com quadro
semelhante; (ii) sensibilidade normal; (iii) atrofia cerebelar e bulbopontina e (iv) reflexos
exaltados. Esse caso em específico se caracteriza com um achado incidental que, além de se
tratar de um paciente com doença neuromuscular de genética ainda não confirmada, também é
um portador da expansão que, em homozigose, causa a AF.
Dentre os cinco pacientes confirmados, pudemos verificar a presença de mutações
pontuais/inserção/deleção em apenas três deles. Acreditamos que a não verificação de
alterações nos outros dois pacientes é justificada pela limitação da técnica escolhida para a
realização do estudo.
O método de Sanger para sequenciamento de fragmentos de PCR não tem cobertura
sobre grandes inserções ou deleções, por exemplo. E essa técnica tampouco cobre as
alterações do tipo deep intronic (que necessitam da total cobertura intrônica), uma vez que a
nossa cobertura é limitada aos exons do gene estudado.
Vários autores já descreveram deleções no gene FXN através da utilização da técnica
de MLPA31,54–58. Um dos mais recentes estudos desse tipo encontrado na literatura é o de
Dorota Hoffman-Zacharska e colaboradores em 2016 que mostraram uma deleção inteira do
gene FXN em um paciente heterozigoto composto52. Assim, estudos abordando outras
técnicas que não as selecionadas nos nossos experimentos, como o MLPA e/ou CGH Array,
se mostram necessárias para elucidar quais são as alterações no gene FXN desses dois
pacientes nos quais não foram verificadas mutações de ponto.
As três variantes encontradas nesse estudo, uma em cada um dos pacientes
heterozigotos compostos estudados nos quais foram verificadas mutações de ponto, estão
descritas na Tabela 4 que se localiza na seção destinada aos resultados.
A variante c.157delC não teve sua patogenicidade avaliada nos algoritmos utilizados
nesse estudo, pois já havia sido descrita por Cossée e colaboradores em 199914 e também
aparece em outro caso não relacionado no estudo de Gellera e colaboradores em 20075. Ela é
uma variação do tipo frameshift de efeito p.R53AfsX75 que muda todo o read de leitura do
CDS, substituindo uma arginina por uma alanina na posição 53 e terminando num stop-codon
75 posições à frente dessa substituição.
Também no trabalho de Gellera e colaboradores em 20075, há uma sugestão de que
essa variante em questão se encontre situada em um “hot spot” mutacional. Na nossa
casuística isso também pode ser inferido, pois se considerarmos somente os pacientes em que
mutações foram encontradas, notamos uma porcentagem de 66,6% (2/3). Por outro lado, se

39
considerarmos todos os pacientes heterozigotos compostos estudados, essa porcentagem
diminui para 40% (2/5).
Ponderando que há 51 mutações diferentes para o gene FXN descritas na base de
dados HGMD, confirmando uma heterogeneidade de genótipos, verificar a mesma variante
em dois pacientes de famílias não relacionadas na nossa amostra da população brasileira,
também nos permite sugerir que a variante que encontramos se encontra em um “hot spot”
mutacional.
A variante c.482+1G>T encontrada no paciente 94 é uma nova mutação, não tendo
sido descrita anteriormente na literatura até o presente momento. Se trata de uma substituição
de uma guanina por uma timina na posição 482+1, ou seja, na primeira base nitrogenada após
o término do éxon 4 do gene FXN. As primeiras bases, tanto antes do começo de um éxon,
quanto ao seu final, são sítios doadores de splicing, assim mutações nessas posições são
consideradas altamente patogênicas. Para confirmar nossa previsão sobre a variante,
utilizamos algoritmos de predição de patogenicidade. Os resultados podem ser visualizados na
Tabela 6 localizada na seção concernente aos resultados. Segundo os critérios da ACMG, esta
nova variante é classificada como patogênica.
Sendo essa nova mutação encontrada de provável alta patogenicidade, um quadro
clínico agravado dos sintomas seria esperado para esse paciente. Isso foi confirmado no
paciente 94, como observado na tabela 4, que mostra o início dos sintomas aos 10 anos,
fenótipo clássico de AF com presença de diabetes e de cardiomiopatia. Esse paciente chegou a
óbito aos 20 anos.
Comparando o quadro clínico dos três heterozigotos compostos em que pudemos
verificar a presença de mutações no gene FXN (vide Tabela 4 acima), juntamente com aqueles
pertencentes aos pacientes UNICAMP01 e POA2 (vide Tabela 5 acima), apenas um teve
início precoce dos sintomas da doença, mostrando que a nossa casuística não segue o padrão
mostrado pela literatura5,6. Somente um apresentou quadro clínico de cardiomiopatia,
confirmando a tendência descrita na literatura de que a frequência dessa disfunção seria
menor em heterozigotos compostos7.
A presença do quadro clínico de diabetes mellitus é de 9% no trabalho de Cossée e
colaboradores em 199914 e de 8,3% no trabalho de Gellera e colaboradores em 20075. Na
nossa casuística ela se apresenta numa porcentagem de 66,6% (2/3), se considerarmos
somente os pacientes nos quais mutações foram encontradas, e de 60% (3/5) se considerarmos
todos os heterozigotos compostos estudados. Essa porcentagem, em ambos os casos, é alta em

40
comparação com o descrito previamente, mostrando que nossa coorte não segue o padrão
apresentado na literatura.
Ainda que o esperado fosse uma apresentação mais agravada dos quadros clínicos da
doença como resultado das mutações pontuais/inserção/deleção no gene FXN, de modo geral,
não é o que se observa na literatura5,14. Galea e colaboradores em 20167 propuseram que a
presença de um quadro clínico mais grave bem como o aumento da probabilidade de
desenvolvimento da diabete mellitus estaria associada ao impacto do perfil das mutações na
função e estrutura da frataxina, que ocorre quando há presença de mutações que eles
classificam como null mutations (quando elas resultam na não produção da proteína).
Outro ponto importante a ser colocado é que a gravidade do quadro clínico da doença
está associada ao tamanho da expansão GAA no gene FXN, sendo que expansões menores
levam a quadros clínicos mais leves e expansões maiores a quadros clínicos mais graves12.
Pela falta do dado de quantificação da expansão GAA nos nossos pacientes, podemos
apenas inferir que, provavelmente, as expansões presentes nos heterozigotos compostos desse
estudo são menores para os casos de início tardio e maiores para os casos apresentados como
AF clássica. Muito provavelmente, a maioria dos pacientes desse estudo não apresenta uma
expansão enorme, que pudesse levar a um quadro mais grave da doença, como, por exemplo,
o início precoce dos sintomas, como sugere a literatura. No entanto, a quantificação precisa
das expansões GAA no gene FXN desses pacientes se faz necessária em estudos futuros para
melhor avaliação comparativa entre os dados.
O aconselhamento genético para famílias com a AF é feito pela análise das
probabilidades de cada caso analisado nos centros em que os pacientes são acompanhados. Na
maioria dos casos, quando os cônjuges aceitam a realização do teste genético, é feita a busca
pela expansão GAA no gene FXN do cônjuge saudável, com o intuito de classificá-lo como
portador ou não. Mesmo que o cônjuge não seja portador, esse estudo mostra que, ainda
assim, para um aconselhamento genético preciso, é necessária uma busca mais aprofundada
considerando o sequenciamento de seu gene FXN para poder estabelecer se a prole desse
paciente tem chances de herdar sua condição, uma vez que nos casos dos heterozigotos
compostos há uma alta probabilidade de que a mutação pontual tenha sido herdada de seus
parentais.

41
6. CONCLUSÃO
A frequência de heterozigotos compostos para a AF numa amostra da população
brasileira é de 2,87% (5/174). Considerando somente os pacientes em que mutações foram
encontradas, a taxa é de 1,72% (3/174).
Foram encontradas duas mutações: uma deleção já descrita na literatura: c.157delC em
dois pacientes e uma nova mutação c.482+1G>T em um paciente.
Com relação ao fenótipo dos pacientes: dois pacientes apresentaram quadros clássicos,
um apresentou quadro tardio; a cardiomiopatia esteve presente em um paciente e diabetes
esteve presente em dois pacientes.

42
7. REFERÊNCIAS
1. Santos R, Lefevre S, Sliwa D, Seguin A, Camadro J-M, Lesuisse E. Friedreich ataxia:
molecular mechanisms, redox considerations, and therapeutic opportunities. Antioxid
Redox Signal. 2010;13(5):651–90.
2. Chamberlain S, Shaw J, Rowland A, Wallis J, South S, Nakamura Y, et al. Mapping of
mutation causing Friedreich’s ataxia to human chromosome 9. Nature.
1988;334(6179):248–50.
3. Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, et al.
riedreich’s Ataxia: Autosomal Recessive Disease Caused by an Intronic GAA Triplet
Repeat Expansion. Science. 1996;271(5254):1423–7.
4. Alper G, Narayanan V. Friedreich’s ataxia. Pediatr Neurol. 2003;28(5):335–41.
5. Gellera C, Castellotti B, Mariotti C, Mineri R, Seveso V, DiDonato S, et al. Frataxin
gene point mutations in Italian Friedreich ataxia patients. Neurogenetics.
2007;8(4):289–99.
6. Cossée M, Schmitt M, Campuzano V, Reutenauer L, Moutou C, Mandel J-L, et al.
Evolution of the Friedreich’s ataxia trinucleotide repeat expansion: Founder effect and
premutations. Proc Natl Acad Sci U S A. 1997;94(14):7452–7.
7. Galea CA, Huq A, Lockhart PJ, Tai G, Corben LA, Yiu EM, et al. Compound
heterozygous FXN mutations and clinical outcome in friedreich ataxia. Ann Neurol.
2016;79(3):485–95.
8. Ribai P, Pousset F, Tanguy M-L, Rivaud-Pechoux S, Le Ber I, Gasparini F, et al.
Neurological, cardiological, and oculomotor progression in 104 patients with
Friedreich ataxia during long-term follow-up. Arch Neurol. 2007;64(4):558–64.
9. Morral JA, Davis AN, Qian J, Gelman BB, Koeppen AH. Pathology and pathogenesis
of sensory neuropathy in Friedreich’s ataxia. Acta Neuropathol. 2010;120(1):97–108.
10. Pandolfo M. Friedreich ataxia. Arch Neurol. 2008;65(10):1296–303.
11. Kelly M, Bagnall RD, Peverill RE, Donelan L, Corben L, Delatycki MB, et al. A
polymorphic miR-155 binding site in AGTR1 is associated with cardiac hypertrophy in
Friedreich ataxia. J Mol Cell Cardiol. 2011;51(5):848–54.

43
12. Mateo I, Llorca J, Volpini V, Corral J, Berciano J, Combarros O. GAA expansion size
and age at onset of Friedreich’s ataxia. Neurology. 2003;61(2):274–5.
13. Xunclà M, Rodríguez-Revenga L, Madrigal I, Jiménez D, Milà M, Badenas C. Protocol
proposal for Friedreich ataxia molecular diagnosis using fluorescent and triplet repeat
primed polymerase chain reaction. Transl Res. 2010;156(5):309–14.
14. Cossee M, Durr A, Schmitt M, Dahl N, Trouillas P, Allinson P, et al. Friedreich’s
ataxia: point mutations and clinical presentation of compound heterozygotes. Ann
Neurol. 1999;45(2):200–6.
15. Zhu D, Burke C, Leslie A, Nicholson GA. Friedreich’s ataxia with chorea and
myoclonus caused by a compound heterozygosity for a novel deletion and the
trinucleotide GAA expansion. Mov Disord. 2002;17(3):585–9.
16. Zuhlke C, Laccone F, Cossee M, Kohlschutter A, Koenig M, Schwinger E. Mutation of
the start codon in the FRDA1 gene: linkage analysis of three pedigrees with the ATG
to ATT transversion points to a unique common ancestor. Hum Genet.
1998;103(1):102–5.
17. Potter NT, Miller CA, Anderson IJ. Mutation detection in an equivocal case of
Friedreich’s ataxia. Pediatr Neurol. 2000;22(5):413–5.
18. Spacey SD, Szczygielski BI, Young SP, Hukin J, Selby K, Snutch TP. Malaysian
siblings with friedreich ataxia and chorea: a novel deletion in the frataxin gene. Can J
Neurol Sci. 2004;31(3):383–6.
19. Van Driest SL, Gakh O, Ommen SR, Isaya G, Ackerman MJ. Molecular and functional
characterization of a human frataxin mutation found in hypertrophic cardiomyopathy.
Mol Genet Metab. 2005;85(4):280–5.
20. De Castro M, Garcia-Planells J, Monros E, Canizares J, Vazquez-Manrique R, Vilchez
JJ, et al. Genotype and phenotype analysis of Friedreich’s ataxia compound
heterozygous patients. Hum Genet. 2000;106(1):86–92.
21. Lamba LD, Ciotti P, Giribaldi G, Di Maria E, Varese A, Di Stadio M, et al.
Friedreich’s ataxia: A new mutation in two compound heterozygous siblings with
unusual clinical onset. Eur Neurol. 2009;61(4):240–3.
22. McCormack ML, Guttmann RP, Schumann M, Farmer JM, Stolle CA, Campuzano V,

44
et al. Frataxin point mutations in two patients with Friedreich’s ataxia and unusual
clinical features. J Neurol Neurosurg Psychiatry. 2000;68(5):661–4.
23. Pook MA, Al-Mahdawi SA, Thomas NH, Appleton R, Norman A, Mountford R, et al.
Identification of three novel frameshift mutations in patients with Friedreich’s ataxia. J
Med Genet. 2000;37(11):E38.
24. Hoischen A, Gilissen C, Arts P, Wieskamp N, Van Vliet W Der, Vermeer S, et al.
Massively parallel sequencing of ataxia genes after array-based enrichment. Hum
Mutat. 2010;31(4):492–9.
25. Bartolo C, Mendell JR, Prior TW. Identification of a missense mutation in a
Friedreich’s ataxia patient: implications for diagnosis and carrier studies. Am J Med
Genet. 1998;79(5):396–9.
26. Kit Doudney, Mark Pook, Sahar Al-Mahdawi, Jaime Carvajal, Renate Hillermann and
SC. A novel splice site mutation (384+1G-A) in the Friedreich’s ataxia gene. Hum
Mutat. 1997;11(5):415.
27. Shiffman D, Kane JP, Louie JZ, Arellano AR, Ross DA, Catanese JJ, et al. Analysis of
17,576 potentially functional SNPs in three case-control studies of myocardial
infarction. PLoS One. 2008;3(8):1–5.
28. Bidichandani SI, Ashizawa T, Patel PI. Atypical Friedreich ataxia caused by compound
heterozygosity for a novel missense mutation and the GAA triplet-repeat expansion.
Am J Hum Genet. 1997;60(5):1251–6.
29. Forrest SM, Knight M, Delatycki MB, Paris D, Williamson R, King J, et al. The
correlation of clinical phenotype in Friedreich ataxia with the site of point mutations in
the FRDA gene. Neurogenetics. 1998;1(4):253–7.
30. McCabe DJH, Wood NW, Ryan F, Hanna MG, Connolly S, Moore DP, et al.
Intrafamilial phenotypic variability in Friedreich ataxia associated with a G130V
mutation in the FRDA gene. Arch Neurol. 2002;59(2):296–300.
31. Zuhlke CH, Dalski A, Habeck M, Straube K, Hedrich K, Hoeltzenbein M, et al.
Extension of the mutation spectrum in Friedreich’s ataxia: detection of an exon
deletion and novel missense mutations. Eur J Hum Genet. 2004;12(11):979–82.
32. Filla A, De Michele G, Cavalcanti F, Pianese L, Monticelli A, Campanella G, et al. The

45
relationship between trinucleotide (GAA) repeat length and clinical features in
Friedreich ataxia. Am J Hum Genet. 1996;59(3):554–60.
33. Malgorzata Labuda, Josee Poirier MP. A Missense Mutation (W155R) in an American
Patient with Friedreich Ataxia. Hum Mutat. 1999;13(6):506.
34. De Michele G, Filla A, Cavalcanti F, Tammaro A, Monticelli A, Pianese L, et al.
Atypical Friedreich ataxia phenotype associated with a novel missense mutation in the
X25 gene. Neurology. 2000;54(2):496–9.
35. Al-Mahdawi S, Pook M, Chamberlain S. A novel missense mutation (L198R) in the
Friedreich’s ataxia gene. Hum Mutat. 2000;16(1):95.
36. Cossée M, Puccio H, Gansmuller a, Koutnikova H, Dierich a, LeMeur M, et al.
Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality
without iron accumulation. Hum Mol Genet. 2000;9(8):1219–26.
37. Zamba-Papanicolaou E, Koutsou P, Daiou C, Gaglia E, Georghiou A, Christodoulou
K. High frequency of Friedreich’s ataxia carriers in the Paphos district of Cyprus. Acta
Myol. 2009;28(1):24–6.
38. Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd ed.
New York: Cold Spring Harbor Laboratory Press; 1989. 3-4 p.
39. Geospiza, Inc.; Seattle, WA, USA [Internet]. [cited 2017 May 19]. Available from:
http://www.geospiza.com
40. CodonCode Corporation [Internet]. [cited 2017 May 19]. Available from:
www.codoncode.com
41. Aken BL, Achuthan P, Akanni W, Amode MR, Bernsdorff F, Bhai J, et al. Ensembl
2017. Nucleic Acids Res. 2017;45(D1):D635–42.
42. Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP) [Internet]. [cited
2017 May 19]. Available from: http://evs.gs.washington.edu/EVS/
43. Stenson PD, Ball E V, Mort M, Phillips AD, Shiel JA, Thomas NST, et al. Human
Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21(6):577–81.
44. Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie.
J Comput Biol. 1997;4(3):311–23.

46
45. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation
prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–2.
46. Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR. An increased
specificity score matrix for the prediction of SF2/ASF-specific exonic splicing
enhancers. Hum Mol Genet. 2006;15(16):2490–508.
47. Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: A web resource to
identify exonic splicing enhancers. Nucleic Acids Res. 2003;31(13):3568–71.
48. Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor
sites from the DNA sequence. J Mol Biol. 1991;220(1):49–65.
49. Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouze P, Brunak S. Splice site
prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence
information. Nucleic Acids Res. 1996;24(17):3439–52.
50. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and
guidelines for the interpretation of sequence variants: a joint consensus
recommendation of the American College of Medical Genetics and Genomics and the
Association for Molecular Pathology. Genet Med. 2015;17(5):405–23.
51. Heiman M. Webcutter 2.0 [Internet]. 1997 [cited 2017 May 19]. Available from:
rna.lundberg.gu.se/cutter2/
52. Hoffman-Zacharska D, Mazurczak T, Zajkowski T, Tataj R, Górka-Skoczylas P,
Połatyńska K, et al. Friedreich ataxia is not only a GAA repeats expansion disorder:
implications for molecular testing and counselling. J Appl Genet. 2016;57(3):349–55.
53. Ryan F. Best practice guidelines for molecular analysis for Friedreich Ataxia. Eur Mol
Genet Qual Netw. 2001;1–5.
54. Sacca F, Marsili A, Puorro G, Antenora A, Pane C, Tessa A, et al. Clinical use of
frataxin measurement in a patient with a novel deletion in the FXN gene. J Neurol.
2013;260(4):1116–21.
55. Deutsch EC, Santani AB, Perlman SL, Farmer JM, Stolle CA, Marusich MF, et al. A
rapid, noninvasive immunoassay for frataxin: utility in assessment of Friedreich ataxia.
Mol Genet Metab. 2010;101(2–3):238–45.

47
56. Brigatti KW, Deutsch EC, Lynch DR, Farmer JM. Novel diagnostic paradigms for
Friedreich ataxia. J Child Neurol. 2012;27(9):1146–51.
57. Anheim M, Mariani L-L, Calvas P, Cheuret E, Zagnoli F, Odent S, et al. Exonic
deletions of FXN and early-onset Friedreich ataxia. Arch Neurol. 2012;69(7):912–6.
58. van den Ouweland AMW, van Minkelen R, Bolman GM, Wouters CH, Becht-
Noordermeer C, Deelen WH, et al. Complete FXN Deletion in a Patient with
Friedreich’s Ataxia. Genet Test Mol Biomarkers. 2012;16(9):1015–8.

48
8. ANEXO I

49

50





























