Embed Size (px)
Citation preview

IV Seminário de Iniciação Científica
836
www.ueg.br www.prp.ueg.br
GRUPO DE PACIENTES COM DISTROFIA MUSCULAR INICIA
TRATAMENTO FISOTERÁPICO TARDIAMENTE
Carolina Albernaz Toledo1. 4, João Alírio Teixeira Jr 2,4,5, Luciana Caetano Fernandes3,4
1 Bolsista PBIC/UEG 2 Professor Colaborador
3. Pesquisadora - Orientadora 4 Curso de Fisioterapia, Eseffego, UEG
5. CRER
RESUMO
As distrofinopatias são doenças genética sendo a distrofia de Duchenne a mais comum e severa. Trata-se de uma herança recessiva ligada ao sexo, com uma incidência maior entre os meninos (1: 3500). O gene afetado é o da distrofina, proteína importante na estabilidade da fibra muscular esquelética. A ausência de distrofina permite que a fibra muscular degenere mais rapidamente. Há comprometimento do sistema locomotor e do cardio-respiratório, sendo a fisioterapia muito importante para melhorar a qualidade de vida dos distróficos. Mas será que esses pacientes recebem tratamento fisioterápico assim que são diagnosticados? Nesse trabalho foram analisados 3283 prontuários de diferentes clínicas com o objetivo de avaliar o perfil do tratamento dos portadores de distrofia em Goiânia.. Ao todo foram detectados 45 distróficos, todos com comprometimento motor. Mais da metade dos pacientes teve seu diagnóstico correto depois dos 12 anos de idade. A maio ria iniciou tratamento fisioterápico após 2 anos de diagnóstico. Esses dados indicam que o diagnóstico e o tratamento fisioterápico são iniciados tardiamente, o que prejudica a qualidade de vida dos pacientes, visto que a distrofia não tem um tratamento medicamentoso que leve a cura.
Palavras-chave: Distrofia, Duchenne, Distrofina
INTRODUÇÃO
As distrofinopatias representam um grupo heterogêneo de desordens musculares, onde vários genes, que codificam componentes do complexo glicoproteínas – distrofina, podem estar envolvidos (Barton , 2006., Ségalat, 2001 e Shim,2003 ) Esse complexo da distrofina normalmente liga o citoesqueleto à matriz extracelular. Portanto, alterações nesses genes resultam em perda da integridade do sarcolema e torna as fibras musculares mais susceptíveis a danos celulares. A alteração mais bem conhecida e documentada é uma mutação no gene codante para distrofina, localizado no Xp21. Essa proteína participa do arranjo citoesquelético do miócito, ligando este ao sarcolema da matriz extracelular (Mc NALLY, 2004). Os músculos esqueléticos e cardíacos normalmente contêm grande quantidade desta proteína. A

IV Seminário de Iniciação Científica
837
www.ueg.br www.prp.ueg.br
deficiência de distrofina está associada com rupturas do sarcolema e afeta canais de cátions ativados por alongamento. A sua falta possibilita um influxo de cálcio e outras moléculas provenientes do líquido extracelular para o interior. Isso causa um acúmulo de cálcio intracelular, comprometendo o funcionamento das fibras rápidas, culminando muitas vezes com a necrose dessas fibras (Ségalat, 2001), levando a um comprometimento diversos músculos do corpo, inclusive da musculatura respiratória.
Os principais tipos de Distrofia Muscular (DM) são: Distrofia Muscular do tipo Duchenne (DMD), Distrofia Muscular do tipo Becker (DMB), Distrofia Muscular do tipo Cinturas (DMC), e Distrofia Muscular Facio-Escápulo-Umeral (FSH), sendo que a mais grave e mais comum é a Distrofia Muscular Duchenne (DMD).
As distrofias de Duchenne e de Becker são distrofias ligadas ao sexo, onde há deleção total (Duchenne) ou parcial (Becker) da distrofina. A distrofia de Duchenne é a mais severa e mais conhecida, com uma incidência de um a cada 3500 nascidos vivos do sexo masculino (Boland,1996 e Grain, 2001). Os primeiros sintomas são: fraqueza muscular, quedas freqüentes, alterações na postura e no modo de andar, dificuldade de subir escada ou sentar, até chegar a total incapacidade motora. Alguns pacientes podem apresentar comprometimento mental.
Segundo a Associação Brasileira de Distrofia Muscular- ABDIM - no Brasil existem cerca de 80 mil pessoas com distrofia de Duchenne ou portadora de um outro tipo de distrofia muscular. Essas doenças, de origem genética, não têm cura na atualidade O acompanhamento da patologia baseia-se apenas em melhoras sintomáticas ocasionais, uma vez que não há cura e a evolução significa invariavelmente a invalidez total e o óbito. Para se proporcionar uma melhor qualidade de vida ao paciente, assim como diminuir a morbidade inerente à doença, deve-se lançar mão de fisioterapia, cirurgias ortopédicas corretivas, órtoses que possam ajudar a prevenir escoliose em pacientes que não são capazes de deambular, além de um forte apoio psicológico (e financeiro se necessário), sem o qual muitas famílias correm o risco de se desestabilizar emocionalmente.
Existem vários relatos de que a fisioterapia ajuda e muito a manter por mais tempo a qualidade de vida dos pacientes distróficos, retardando, por exemplo, o aumento de peso (substituição das fibras musculares por tecido adiposo), o uso precoce de cadeiras de rodas, ou até mesmo complicações mais sérias do aparelho respiratório ou cardíaco (Alvarez,1994, Araujo, 2004). Porém, a maioria dos pacientes inicia tardiamente o tratamento fisioterápico, principalmente devido a um diagnóstico tardio da doença pelo médico (Bushby, 1999, De Los Angeles, 1999 e Essex,2001): inicialmente suspeita-se de problemas neurológicos e geralmente somente com a perda da locomoção é que o diagnóstico de uma distrofinopatia é sugerido (Alvarez, 1994, Van Essen,1997). Portanto a maioria dos casos, o diagnóstico é baseado apenas na apresentação clínica, história familiar e análise do portador. A análise do DNA é o procedimento ideal para detectar portadores de distrofia muscular, o que infelizmente não acontece na maioria dos casos. Grande parte dos portadores não tem acesso a exames diagnósticos avançados, que lhes permita conhecer ou afastar um tipo específico da enfermidade que sofrem e, com isso, poderem saber o que esperar do futuro e o risco de transmiti- la a seus filhos (Campbell, 2000, Flanigan,2003; Smith,1989 e VAN Essen,1997). O diagnóstico tardio e o alto custo do tratamento fazem com que pacientes de baixa renda evoluam rapidamente para estágios graves da doença (Araujo, 2004, Bushby,1999 e Essex, 2001).

IV Seminário de Iniciação Científica
838
www.ueg.br www.prp.ueg.br
Por estes motivos, nosso grupo iniciou um estudo sobre as distrofinopatias, avaliando se em Goiânia, o diagnóstico da distrofia é precoce ou não, se há diagnóstico molecular e se os pacientes distróficos recebem algum tipo de tratamento fisioterápico, logo após o diagnóstico médico. Essas informações básicas podem dar suporte para os portadores exigirem das autoridades governamentais da área da saúde os seus direitos básicos de saúde.
MATERIAL E MÉTODOS
A pesquisa foi realizada, em quatro instituições de Goiânia, sendo elas a Clínica Escola de Fisioterapia da Escola Superior de Educação Física e Fisioterapia do Estado de Goiás (ESEFFEGO), o Centro de Orientação Reabilitação e Assistência ao Encefalopata (CORAE), o Centro de Reabilitação e Readaptação Dr. Henrique Santillo (CRER) e o Centro Estadual de Apoio ao Deficiente (CEAD). Essa pesquisa teve a aprovação do Comitê de Ética em Pesquisa Humana e Animal (CEPHA) do Hospital Geral de Goiânia (HGG).
Foi aplicado um questionário que foi respondido com base em dados colhidos nos prontuários dos pacientes encontrados. Os dados incompletos foram pesquisados com as famílias via telefone ou contato pessoal e estão aqui, alguns, classificados como dado ignorado se a família, ou o próprio paciente, não souberam informar. Nas quatro instituições foram pesquisado um total de 3.283 prontuários. Desses encontramos 45 pacientes registrados com Distrofia Muscular Progressiva (DMP). Não foi delimitado o tipo de Distrofia a ser pesquisado.
3. RESULTADOS E DISCUSSÃO
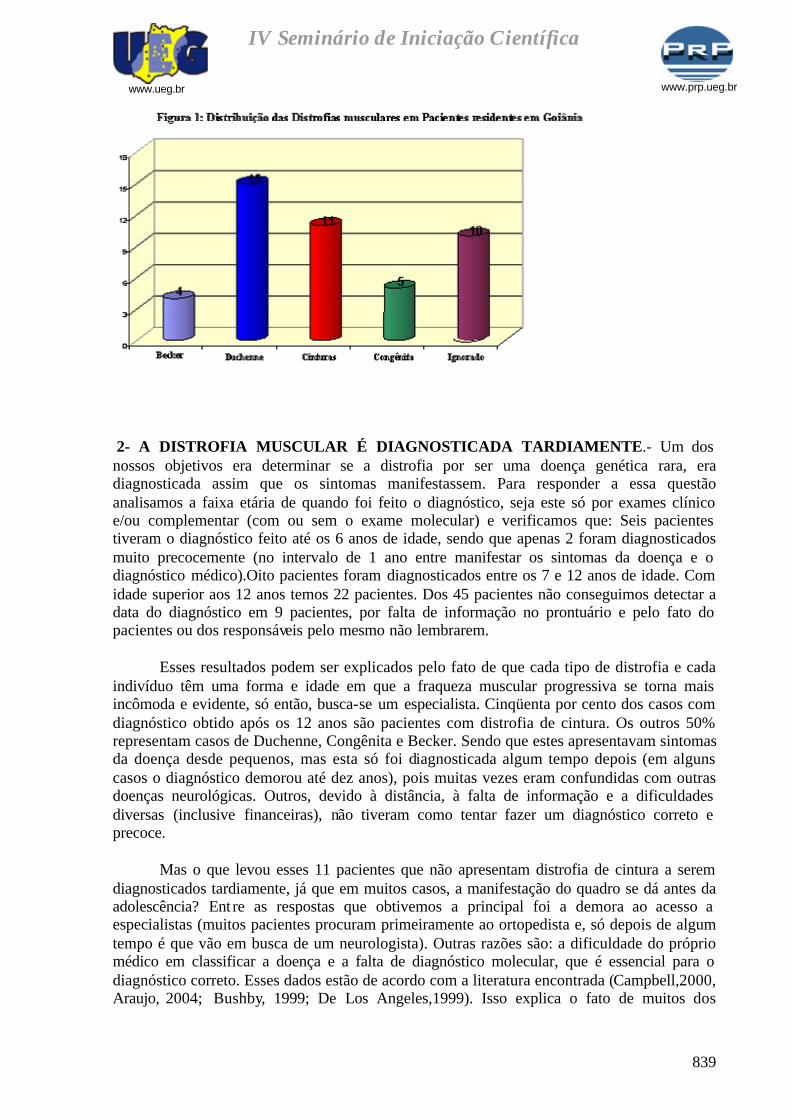
1. PERFIL DE DISTROFIAS DETECTADAS EM GOIÂNIA - Entre os 3283 prontuários analisados foram detectados 45 casos de distrofia. Sendo distribuídos da seguinte forma (Figura 1):4 apresentam distrofia do tipo Becker, 15 têm distrofia na forma de Duchenne, 11 pacientes são portadores da forma de Cinturas, 5 apresentam a forma Congênita, 10 foram classificados como casos ignorados por não terem um diagnóstico que especifique a forma da patologia.As freqüências de cada tipo da DMP são diferentes e independentes entre si. Cada uma tem uma forma diferente de se manifestar. As DMP podem ser autossômica dominante, autossômica recessiva ou podem ser ligadas ao sexo (formas mais comuns são a DMP de Duchenne e a DMP de Becker).
Dos 45 casos estudados, apenas 14 tinham familiares portadores da distrofia muscular. Isso indica que os demais casos, surgiram devido a novas mutações durante a formação do indivíduo.
IV Seminário de Iniciação Científica
839
www.ueg.br www.prp.ueg.br
2- A DISTROFIA MUSCULAR É DIAGNOSTICADA TARDIAMENTE.- Um dos nossos objetivos era determinar se a distrofia por ser uma doença genética rara, era diagnosticada assim que os sintomas manifestassem. Para responder a essa questão analisamos a faixa etária de quando foi feito o diagnóstico, seja este só por exames clínico e/ou complementar (com ou sem o exame molecular) e verificamos que: Seis pacientes tiveram o diagnóstico feito até os 6 anos de idade, sendo que apenas 2 foram diagnosticados muito precocemente (no intervalo de 1 ano entre manifestar os sintomas da doença e o diagnóstico médico).Oito pacientes foram diagnosticados entre os 7 e 12 anos de idade. Com idade superior aos 12 anos temos 22 pacientes. Dos 45 pacientes não conseguimos detectar a data do diagnóstico em 9 pacientes, por falta de informação no prontuário e pelo fato do pacientes ou dos responsáveis pelo mesmo não lembrarem.
Esses resultados podem ser explicados pelo fato de que cada tipo de distrofia e cada indivíduo têm uma forma e idade em que a fraqueza muscular progressiva se torna mais incômoda e evidente, só então, busca-se um especialista. Cinqüenta por cento dos casos com diagnóstico obtido após os 12 anos são pacientes com distrofia de cintura. Os outros 50% representam casos de Duchenne, Congênita e Becker. Sendo que estes apresentavam sintomas da doença desde pequenos, mas esta só foi diagnosticada algum tempo depois (em alguns casos o diagnóstico demorou até dez anos), pois muitas vezes eram confundidas com outras doenças neurológicas. Outros, devido à distância, à falta de informação e a dificuldades diversas (inclusive financeiras), não tiveram como tentar fazer um diagnóstico correto e precoce.
Mas o que levou esses 11 pacientes que não apresentam distrofia de cintura a serem diagnosticados tardiamente, já que em muitos casos, a manifestação do quadro se dá antes da adolescência? Ent re as respostas que obtivemos a principal foi a demora ao acesso a especialistas (muitos pacientes procuram primeiramente ao ortopedista e, só depois de algum tempo é que vão em busca de um neurologista). Outras razões são: a dificuldade do próprio médico em classificar a doença e a falta de diagnóstico molecular, que é essencial para o diagnóstico correto. Esses dados estão de acordo com a literatura encontrada (Campbell,2000, Araujo, 2004; Bushby, 1999; De Los Angeles,1999). Isso explica o fato de muitos dos

IV Seminário de Iniciação Científica
840
www.ueg.br www.prp.ueg.br
pacientes estudados (vinte seis) não terem um diagnóstico conclusivo sobre que tipo de distrofia muscular portam.
3. A maioria dos Pacientes com Distrofia Muscular não apresentam diagnóstico molecular - Os exames moleculares são imperativos para a detecção e classificação do tipo de distrofia muscular. Nestes casos, esse exame é o único capaz de “fechar” um diagnóstico (Campbell, 2000 Flanigan,2003; Van Essen,1997). Em nosso trabalho, detectamos que 26 dos 45 pacientes não foram submetidos a um diagnóstico molecular. Não conseguimos informação de três pacientes (classificados como ignorados) e 16 pacientes fizeram o exame, que foi feito em diversos centros de diagnóstico no país. Alguns pacientes haviam feito os exames na USP, outros no Centro de Estudos do Genoma Humano, outros os fizeram no Hospital SARAH de Brasília, na PUC de São Paulo e na Escola Paulista de Medicina (EPM).
O diagnóstico molecular é conclusivo, portanto verificamos aqui um alto índice de pacientes (57,8%) que, ou não foram indicados a fazer esse exame ou, foram indicados e não o fizeram. Isso reflete a falta de informação que muitos médicos têm sobre a necessidade do diagnóstico molecular e sobre como encaminhar seus pacientes para centros de diagnóstico, que fazem o exame gratuitamente, através do Programa de Tratamento Fora de Domicílio, que é custeado pela Secretaria Municipal de Saúde. Reflete também a falta de comunicação entre o médico e o paciente, pois o médico deveria tentar explicar ao paciente a importância dos exames.
4 - Pacientes Distróficos iniciam tratamento fisioterapêutico tardiamente - Em nossos resultados, detectamos que 39 pacientes fazem fisioterapia, dois não fazem e os demais pacientes não conseguimos determinar por falta de informações. Portanto, a maioria dos pacientes (86,7%) faz tratamento fisioterapêutico. Mas será que esses pacientes iniciaram esse tratamento assim que tiveram um laudo médico sobre sua doença?
Avaliamos, portanto neste item, o tempo que o paciente levou entre o diagnóstico e o início do seu tratamento fisioterapêutico e o porquê deste intervalo (Figura 2). Observamos os seguintes resultados entre os pacientes que recebem tratamento fisioterapêutico: 38,5 % iniciaram o tratamento imediatamente após o diagnóstico, 12,8 % iniciaram a terapia após um ano do diagnóstico, 38,5 % iniciaram após dois anos de diagnóstico, 10,2% têm o dado ignorado por não saberem a data de diagnóstico ou de início dos tratamentos.
Considerando a progressão da patologia, temos que, um intervalo de tempo superior a um ano já é considerado longo e extremamente prejudicial à estrutura e às funções do paciente. Constatamos que a maioria dos pacientes demora de um a dois anos para iniciar o tratamento (51,3 %), sendo que existem aqueles que chegaram a demorar mais de cinco anos. Os pacientes, que estão classificados como tendo um intervalo menor que um ano, são aqueles que receberam o diagnóstico já tardio e que estavam já em quadro avançado (pela própria demora em buscar o diagnóstico). Quando questionamos aos pacientes, ou familiares, o porquê da demora de início tivemos diversas respostas, mas a mais comum foi a de que os pacientes que residiam em cidades do interior não sabiam onde buscar o tratamento ou porque suas cidades não tinham o serviço de fisioterapia disponível de forma gratuita, ou seja, por motivos financeiros. Uns poucos pacientes (cerca de dois deles) relataram um descaso inicial

IV Seminário de Iniciação Científica
841
www.ueg.br www.prp.ueg.br
com relação à necessidade deste tipo de tratamento. Um outro paciente não quis aderir ao tratamento. A falta de fisioterapia é preocupante visto que a mesma é uma das poucas formas de se tentar prolongar a vida do paciente (mesmo que seja por alguns meses mais) e dar- lhe uma melhor qualidade de vida e condições de vivê-la.
CONCLUSÃO:
Neste trabalho constatamos que não houve um diagnóstico precoce da Distrofia Muscular na maioria dos casos levantados, bem como poucos pacientes tiveram um diagnóstico molecular. Isso ocorre principalmente devido a falta de informação e de encaminhamento dos pacientes a centros de diagnóstico molecular, pois não sabem que podem obter esse diagnóstico de forma gratuita. A falta de laboratórios em Goiânia, que realizem esse diagnóstico, também contribui para não se fazer o exame molecular. Todos os pacientes investigados apresentam comprometimento motor. A maioria realiza tratamento fisioterapêutico, porém demoram em iniciar essa terapia, não por falta de indicações médicas, mas por falta de informações sobre os centros de tratamento e sobre as “melhorias” que a terapêutica pode gerar. Vale ressaltar que a fisioterapia é importante para evitar que o quadro de distrofia já estabelecido se torne mais rapidamente grave, portanto não é um tratamento que cura, porém melhora a qualidade de vida dos pacientes, prolongando suas vidas. Quanto mais tardiamente o paciente inicia o tratamento pior será a qualidade de vida deste.
AGRADECIMENTOS:
Aos pacientes que se prontificaram em participar da pesquisa.
Aos funcionários da Clínica Escola da Eseffego, do CORAE, do CEAD e do CRER que nos auxiliaram colocando a nossa disposição os prontuários.

IV Seminário de Iniciação Científica
842
www.ueg.br www.prp.ueg.br
Ao Prof. Dr. João Alírio, que permitiu estabelecermos uma parceria entre o CRER e a ESEFFEGO , permitindo então a realização deste trabalho.
REFERÊNCIA BIBLIOGRÁFICA.
ALVAREZ LEAL M, MORALES AGUILERA A, PEREZ ZUNO JA, SEGURA ROMERO S, QUEIROZ GONGORA MC, PAREDES GARCIA A. Relations between delayed diagnosis and forms of onset in Duchenne Muscular Dystrophy. Gac Med Mex 1994; 130:459-64.

IV Seminário de Iniciação Científica
843
www.ueg.br www.prp.ueg.br
ARAUJO,A, DECO,M.; KLÔH,B; GÓIS, FERNANDA AND GUIMARÃES, A.C.M. Diagnosis delay of Duchenne Muscular Distrophy. Rev. Bras. Saúde Mat. Infantil. 2004 Jun (4) n2: 177-184.
BARTON, E. R. Impact of sarcoglycan complex on mechanical signal transduction in murine skeletal muscle. Am. J. Physiol 2006, 290: 411 – 419.
BOLAND BJ, SILBERT PL, GROOVER RV, WOLLAN PC, SILVERSTEIN MD. Skeletal, cardiac, and smooth muscle failure in Duchenne Muscular Dystrophy. Pediatr Neurol 1996; 14: 7-12
BUSHBY KM, HILL A, STEELE JG. Failure of early diagnosis in symptomatic Duchenne Muscular Dystrophy. Lancet 1999; 353: 557-8.
CAMPBELL, K.P. AND COHAN R.D. Molecular basis of muscular dystrophies. Muscle &Nerve, 2000, vol 23, , 1456-1471
DE LOS ANGELES AVARIA M, KLEINSTEUBER K, HERRERA L, CARVALLO P. Delayed diagnosis of Duchenne Muscular Dystrophy in Chile. Rev Med Chil 1999; 127: 65-70.
ESSEX, C; ROPER, H. Lesson of the week: late diagnosis of Duchenne's Muscular Dystrophy presenting as global developmental delay. BMJ 2001; 323: 37-8.
FLANIGAN KM, VON NIEDERHAUSERN A, DUNN DM, ALDER J, MENDELL JR, WEISS RB. Rapid Direct Sequence Analysis of the Dystrophin Gene. Am. J. Hum. Genet.2003 Mar; 72:931-939.
GRAIN L., CORTINA-BORJA M., FORFAR C. HILTON-JONES D., HOPKIN J., BURCH M. Cardiac abnormalities and skeletal muscle weakness in carriers of Duchenne and Becker muscular dystrophies and controls. Neuromuscul. Disord. 2001, vol 11 :186-191.
Mc NALLY, E.M.; KAKKAR, R. and LAPIDOS, K.A. The Dystrophin Glycoprotein Complex. Circulation Research 2004, 94: 1023.
SÉGALAT, L. AND MARIOL, M. Muscular degeneration in the absence of dystrophin is a calcium-dependent process. Current Biology, 2001., vol 11, 1691-1694.
SHIM JY, KIM TS. Relationship between utrophin and regenerating muscle fibers in duchenne muscular dystrophy. Yonsei Med J. 2003 Feb; 44 (1):15-23.
SMITH RA, SIBERT JR, WALLACE SJ, HARPER PS. Early diagnosis and secondary prevention of Duchenne Muscular Dystrophy. Arch Dis Child 1989; 64: 787-90
VAN ESSEN AJ, KNEPPERS AL, VAN DER HOUT AH, SCHEFFER H, GINJAAR IB, TEN KATE LP, VAN OMMEN GJ, BUYS CH, BAKKER E. The clinical and molecular genetic approach to Duchenne and Becker Muscular Dystrophy: an updated protocol. J Med Genet 1997; 34: 805-12.





























