Embed Size (px)
Citation preview

UNIVERSIDADE ESTADUAL DE CAMPINAS
FACULDADE DE CIÊNCIAS MÉDICAS
MARCELO BURLAMARQUE NUNES
DISTONIA NA DOENÇA DE MACHADO-JOSEPH: ASPECTOS CLÍNICOS,
TERAPÊUTICOS E BASES ANATÔMICAS
CAMPINAS
2016

MARCELO BURLAMARQUE NUNES
DISTONIA NA DOENÇA DE MACHADO-JOSEPH: ASPECTOS CLÍNICOS,
TERAPÊUTICOS E BASES ANATÔMICAS
Tese apresentada à Faculdade de Ciências Médicas da Universidade
Estadual de Campinas como parte dos requisitos exigidos para a
obtenção do título de Doutor em Ciências
ORIENTADOR: MARCONDES CAVALCANTE FRANCA JUNIOR
COORIENTADORA: ANELYSSA CYSNE FROTA D’ABREU
ESTE EXEMPLAR CORRESPONDE À VERSÃO
FINAL DA TESE DEFENDIDA PELO
ALUNO MARCELO BURLAMARQUE NUNES, E ORIENTADO PELO
PROF. DR. MARCONDES CAVALCANTE FRANCA JUNIOR
CAMPINAS
2016


BANCA EXAMINADORA DA DEFESA DE DOUTORADO
MARCELO BURLAMARQUE NUNES
ORIENTADOR: PROF. DR. MARCONDES CAVALCANTE FRANCA JUNIOR
COORIENTADORA: PROF. DRA. ANELYSSA CYSNE FROTA D’ABREU
MEMBROS:
1. PROF. DR. MARCONDES CAVALCANTE FRANCA JUNIOR
2. PROFA. DRA. ANAMARLI NUCCI
3. PROF. DR. MARCIO LUIZ FIGUEREDO BALTHAZAR
4. PROF. DR. JOSÉ LUIZ PEDROSO
5. PROF. DR. ORLANDO GRAZIANI POVOAS BARSOTTINI
Programa de Pós-Graduação em Fisiopatologia Médica da Faculdade de Ciências
Médicas da Universidade Estadual de Campinas.
A ata de defesa com as respectivas assinaturas dos membros da banca
examinadora encontra-se no processo de vida acadêmica do aluno.
Data: 12/02/2016

DEDICATÓRIA
Aos meus pais, minha esposa Bruna, meus filhos
João Marcelo e Benjamim e ao Senhor.

AGRADECIMENTOS
Aos meus pais, Eurialdo e Fátima, que desde a tenra infância me ensinaram a
busca do saber.
A minha esposa Bruna, companheira em todos os momentos e guerreira em
todas as nossas lutas.
A meus filhos, João e Benjamim, estímulo a cada chamado de “pai”.
Ao meu amigo Alberto Martinez, essencial para a realização desse trabalho e a
quem considero como irmão e exemplo.
Ao meu amigo Thiago Rezende, um grande exemplo de humildade,
competência e companheirismo.
Ao meu professor Marcondes, a quem mais admiro e referencio neste mundo.

RESUMO
Introdução: Distonia é frequente na Doença de Machado-Joseph (DMJ), mas
muitos aspectos importantes dessa associação ainda não foram definidos,
como características clinicas, resposta a terapêutica e substrato anatômico.
Objetivos: descrever a distonia em seus aspectos clínico, anatômico e
terapêutico em pacientes com DMJ/SCA3. Métodos: Nós avaliamos 75
pacientes consecutivos com DMJ e identificamos aqueles com distonia. A
Escala de avaliação da distonia de Burke-Marsden-Fahn foi aplicada para
quantificar a gravidade da distonia nesse subgrupo de pacientes. Pacientes
com distonia receberam 600 mg/dia de Levodopa por 2 meses e um vídeo foi
gravado antes e após o tratamento. Um avaliador cego analisou a distonia nos
vídeos. Pacientes que não apresentaram resposta a Levodopa receberam
toxina botulínica. Finalmente, imagens volumétricas ponderadas em T1 e por
tensor de Difusão (DTI) foram obtidas usando uma ressonância magnética de
3T para identificar as áreas de substância branca e cinzenta seletivamente
lesadas no grupo distônico. Resultados: foram identificados 21 pacientes com
distonia (28%): 9 classificados como tendo a forma generalizada e 12 com
apresentação segmentar/focal. Pacientes com distonia tinham início mais
precoce e maior expansão (CAG) quando comparados aos sem distonia (28,9
± 11.7 vs 40,6 ± 11.4 anos; p < 0,001 e 75 vs 70; p < 0,001, respectivamente).
Não observamos benefício significativo com uso da levodopa na análise de
grupos (p= 0,07), mas alguns pacientes tiveram melhora. Além disso, dez
pacientes receberam toxina botulínica, resultando em uma importante mudança
nos escores de avaliação após 4 semanas (p = 0,03). Na análise de imagem,
verificamos que os pacientes com distonia tinham atrofia no córtex pré e
paracentral, enquanto que os pacientes não-distônicos tinham atrofia occipital.
Ambos os grupos apresentaram redução volumétrica dos gânglios da base,
mas a atrofia talâmica, da substância branca cerebelar e no diencéfalo ventral
foi desproporcionalmente maior no grupo distônico. Conclusão: Distonia na
DMJ é frequente e muitas vezes severa, mas pode responder a Levodopa. É
associada predominantemente com anormalidades estruturais próximo ao
córtex motor e no tálamo.

Palavras-chave: Distonia, Doença de Machado-Joseph, ataxia, Levodopa,
Toxina botulínica, ressonância magnética

ABSTRACT
Introduction: Dystonia is frequent in Machado-Joseph disease, but several
important aspects are not yet defined, such as the detailed clinical profile,
response to treatment and anatomical substrate. Objectives: To describe
dystonia in their clinical, anatomical and therapeutic aspects in patients with
MJD/SCA3. Methods: We screened 75 consecutive patients and identified
those with dystonia. The Burke-Marsden-Fahn Dystonia Rating Scale was
employed to quantify dystonia severity. Patients with dystonia received
levodopa 600 mg/day for 2 months and were videotaped before and after
treatment. A blinded evaluator rated dystonia in the videos. Patients with
disabling dystonia who failed to respond to levodopa treatment received
botulinum toxin. Finally, volumetric T1 and diffusion tensor imaging sequences
were obtained in the dystonic group using a 3T-MRI scanner to identify areas of
gray and white matter that were selectively damaged. Results: There were 21
patients with dystonia (28%): 9 classified as generalized and 12 as focal/
segmental. Patients with dystonia had earlier onset and larger (CAG)
expansions (28,9 ± 11,7 vs 40,6 ± 11,4; p < 0,001 and 75 vs 70; p < 0,001,
respectively). Although group analyses failed to show benefit on levodopa (p =
0,07), some patients had objective improvement. In addition, ten patients
received botulinum toxin resulting in a significant change in dystonia scores
after 4 weeks (p = 0,03). Patients with dystonia had atrophy at pre- and
paracentral cortices; whereas, non-dystonic patients had occipital atrophy.
Basal ganglia volume was reduced in both groups, but atrophy at the thalami,
cerebellar white matter and ventral diencephali was disproportionately higher in
the dystonic group. Conclusion: Dystonia in Machado-Joseph disease is
frequent and often disabling, but may respond to levodopa. It is associated
predominantly with structural abnormalities around the motor cortices and in the
thalami.
Key words: Dystonia. Machado-Joseph Disease. Ataxia. Levodopa. Botulinum
toxin. MRI

LISTA DE TABELAS
Tabela 1 - Dados clínicos e genéticos de pacientes com DMJ/SCA3 com e sem
distonia -------------------------------------------------------------------------------------------33
Tabela 2 - Áreas que apresentaram alteração na espessura cortical: (A)
pacientes com distonia (dDMJ/SCA3) comparados com seus respectivos
controles saudáveis; (B) pacientes sem distonia (cDMJ/SCA3) comparados
com seus respectivos controles (nível de significância 0,001) ---------------------34
Tabela 3 - Estruturas subcorticais que apresentaram redução volumétrica
significativa em pacientes com dDMJ/SCA3 e cDMJ/SCA3 quando
comparados com seus respectivos controles (nível de significância 0,001) ---36
Tabela 4. Comparação entre estruturas atrofiadas em ambos os grupos, mas
mais atrofiadas no grupo distônico através do z-score (nível de significância
0,001) ---------------------------------------------------------------------------------------------37

LISTA DE FIGURAS
Figura 1 - Circuitos neuronais envolvidos na produção e modulação do
movimento ---------------------------------------------------------------------------------------21
Figura 2 - Desenho do estudo -------------------------------------------------------------25
Figura 3 - Resultados de TBSS que mostram áreas de anisotropia fracional
(FA) reduzida e aumento da difusividade média (MD), difusividade axial (AD) e
difusividade radial (RD) em pacientes com DMJ / SCA3 após a comparação
com a idade e sexo controle- pareados. -------------------------------------------------38

ABREVIATURAS E SIGLAS
DMJ – Doença de Machado Joseph
dMDJ/SCA3 – Doença de machado Joseph com Distonia
cDMJ/SCA3 - Doença de machado Joseph sem Distonia
SCA 3 - Ataxia Espinocerebelar tipo 3
SCAs - Ataxias Espinocerebelares
RM – Ressonância Magnética
3T – 3 Tesla
DTI – Imagem por Tensor de Difusão
SB – Substância Branca
SC – Substância Cinzenta
ATXN3 – Ataxina 3

TBSS – Tract-Based Spatial Statistics
VITe - Volume Intracraniano Total Estimado
AF - anisotropia fracionada
DM - difusividade média
DR- difusividade radial
DA- difusividade axial

SUMÁRIO
INTRODUÇÃO...................................................................................................16
OBJETIVOS......................................................................................................23
METODOLOGIA................................................................................................24
RESULTADOS..................................................................................................32
DISCUSSÃO GERAL........................................................................................40
CONCLUSÃO....................................................................................................46
REFERÊNCIAS.................................................................................................47
ANEXOS............................................................................................................57

16
INTRODUÇÃO
I. DOENÇA DE MACHADO JOSEPH
A Doença de Machado Joseph ou Ataxia Espinocerebelar tipo 3 (DMJ/SCA3)
foi inicialmente descrita na década de 1970, em famílias de ancestrais portugueses
residentes nos Estados Unidos, e atualmente é considerada a ataxia autossômica
dominante mais comum no mundo e corresponde a 60% dos casos de ataxia no
Brasil [Coutinho, 1970; Sequeiros, 1993; Lopes-Cendes, 1997; de Castilhos, 2014].
A primeira descrição da Doença de Machado-Joseph foi dada por Nakano em
1972, neurologista atuante do Hospital Peter Bent Brigham em Masssachusets, que
observou uma família luso-americana com o que pensava ser uma “neuropatia
periférica herdada de maneira autossômica dominante. Ele a chamou de “Doença de
Machado”, já que a doença havia sido diagnosticada na família “Guilherme
Machado”, um açoreano da região da então Bretanha que havia emigrado da ilha de
São Miguel. Naquele mesmo ano, 12 pessoas da família “José Tomás”, procedente
da região de Flores, também dos Açores, foram diagnosticas com a mesma doença,
para a qual Woods deu o nome de “Degenerescência nigroespinodentada com
oftalmoplegia nuclear”. Em 1975, na Califórnia foram reunidas 100 pessoas no
Children´s Hospital de Oakland para investigação de uma doença neurológica de
“degeneração estriatonígrica”, a partir de então denominada “Doença de Joseph”,
nome da família acometida [Sequeiros, 1993]. Em 1976, Paula Coutinho e Corino
Andrade vão aos Açores estudar a doença e em 1980 há a unificação dos nomes já
dados a doença que passa a ser chamada de Doença de Machado-Joseph. Os
primeiros casos no Brasil, foram descritos por Helio Teive em 1991, que descreveu o
acometimento de 5 pessoas da mesma família que moravam em Florianópolis,
Santa Catarina [Teive, 1991].
Trata-se de uma doença neurodegenerativa de início tardio, cujo substrato
genético é a expansão do tripleto CAG no 10º exon do gene ATXN3, localizado no
cromossomo 14q32.1 [Kawagushi, 1994]. O alelo mutante apresenta 56 a 84
repetições CAG, que resulta na expressão de uma cadeia anormalmente longa de
aminoácidos glutamina na proteína formada, a ataxina 3. Há uma correlação entre o
tamanho da expansão do (CAG) e algumas manifestações clínicas da doença, como

17
a idade de início, achado também encontrado em outras doenças por expansão de
tripletos [Jardim, 2001]. A proteína mutante tem ação neurotóxica por mecanismo
ainda não completamente estabelecido [Bettencourt, 2011]. A expressão patológica
da neurodegeneração consiste de perda neuronal/gliose no cerebelo, tratos
medulares, substância negra, núcleo rubro, núcleo subtalâmico, núcleos de nervos
cranianos e nervos periféricos [Durr, 1996]. Isso vem reverberar tão somente o fato
de a ataxina 3 ser expressa difusamente nos neurônios do sistema nervoso central e
periférico [Rub, 2008].
Com esse padrão de acometimento do Sistema Nervoso, a heterogeneidade
fenotípica tem sido reconhecida como uma das características marcantes da
doença. Atualmente, há 5 subtipos clínicos descritos: tipo 1 [ “tipo Joseph”] – início
precoce (média de 24 anos) com ataxia e Oftalmoplegia Externa Progressiva (OEP)
associados a sinais piramidais e distonia; tipo 2 [ tipo “Thomas” ] – início
intermediário (média de 40 anos) com ataxia “pura”. Eventualmente, há sinais
piramidais, mas com sinais extrapiramidais e periféricos tênues; tipo 3 [“tipo
Machado”]- início mais tardio (média de 46 anos) com ataxia, OEP e sinais
periféricos, mas com sinais piramidais e extrapiramidais leves [Coutinho, 1992]; tipo
4 - com achados parkinsonianos proeminentes e apenas leve ataxia cerebelar [Suite,
1986 ]; e tipo 5 – síndrome de paraparesia espástica praticamente isolada [Sakai,
1996].
Sinais extrapiramidais são mais frequentes em DMJ/SCA3 do que em
qualquer outra ataxia hereditária e incluem parkinsonismo, coréia, tremor, mioclonias
e distonia. Recentemente, acatisia foi descrita como um distúrbio do movimento
também associada a DMJ/SCA3 [Pedroso, 2011]. Especificamente, distonia é rara
na maioria dos demais tipos de SCAs, mas se presente sugere fortemente o
diagnóstico de DMJ/SCA3 [Schols, 2000; van Galen, 2011; Moro, 2014;
Jhunjhunwala, 2014]. Outra condição em que distonia também é comumente
encontrada é na Ataxia Espinocerebelar tipo 17 (SCA17), o terceiro genótipo mais
comum entre as SCAs em algumas regiões. Em um estudo, distonia esteve presente
em até 50% dos pacientes avaliados com SCA 17, juntamente com os típicos sinais
Huntigton-like, oculomotores e cognitivos [Mariot, 2007]. Em estudo recente,
achados extra-cerebelares foram encontrados em uma coorte de pacientes com
SCA2, dentre os quais 27,27% tinham distonia [Pedroso, 2016].

18
II. DISTONIA
Distonia é um distúrbio de movimento descrito inicialmente em 1901 por
Joseph Destarac para descrever uma jovem de 17 anos com o que ele chamou de
torcicollis spasmodique, fenômeno que, segundo observou, melhorava com repouso
e algumas manobras [Destarac, 1901]. Oppenheim, em 1911 usou o termo dystonia
musculorum deformans, usando assim pela primeira vez o termo distonia, para
descrever o que ele julgou ser uma doença do tônus muscular em quatro jovens
pacientes [Oppenheim, 1911]. Porém, distonia só foi definida em 1984, e teve sua
definição ampliada em 2013, pela International Consensus Committee of the
European Federation of Neurologic Societies. Segundo esse Comitê, ela pode ser
definida como um distúrbio de movimento caracterizado por uma contração muscular
sustentada ou intermitente causando movimentos ou posturas anormais,
frequentemente repetitivos. Movimentos distônicos são tipicamente padronizados,
torcionais e podem ser associados a tremor. É frequentemente iniciada ou piorada
pela ação voluntária e associada com ativação muscular sinérgica [Steeves, 2012].
Distonia tem sido classificada de várias maneiras ao longo dos últimos anos.
Marsden, Fahn e Calne classificaram-na de acordo com a etiologia, distribuição e
idade de início. Conforme a etiologia, é subdividida em: primária (com ou sem
padrão hereditário) ou secundária (devido a outra condição neurológica hereditária
ou a alguma causa ambiental). Conforme distribuição, isto é, de acordo com o
padrão de acometimento dos seguimentos corporais, como: focal (quando apenas
um seguimento do corpo é afetado, como por exemplo, no blefaroespasmo e nas
distonias tarefa-específica); segmentar (quando dois segmentos contíguos são
afetados, como por exemplo na distonia cervical, com acometimento de pescoço e
ombro); generalizada (quando são afetados os membros inferiores e algum outro
segmento corporal) e hemidistonia, quando a manifestação distônica ocorre de
forma dimidiada. Por fim, a distonia também pode ser classificada conforme a idade
de inicio dos sintomas em precoce (indivíduos com menos de 26 anos) ou tardia
(indivíduos com mais de 26 anos) [Fahn, 1987]. Pela utilidade clínica dessa
classificação, ela continua sendo usada, mesmo com o advento de novas

19
classificações baseadas em novos conhecimentos, principalmente genéticos sobre a
doença. Bressman et al classificou a distonia em primária (autossômica dominante
ou outras causas genéticas) e distonia secundária (incluindo formas adquiridas,
distonia-plus e causas degenerativas) [Bressman, 2004]. Atualmente, porém, o
guideline da Sociedade Européia distingue a distonia em Primária,
Heredodegenerativa ou Secundária, classificação mais utilizada na atualidade
[Albanese, 2013].
Importante também é o entendimento dos termos aplicados à síndrome. Por
distonia primária, entende-se um fenótipo puro (não associado a outros achados
neurológicos e sem evidências de outras anormalidades patológicas). Tremor,
entretanto, é considerado como parte integrante da distonia primária, juntamente
com a presença de sintomas psiquiátricos comumente associados. Distonia-plus foi
um termo cunhado para descrever distonia associada a outro achado neurológico,
mais comumente, mioclonia ou parkinsonismo. Por distonia secundária, entende-se
uma distonia com etiologia conhecida e/ou adquirida. Distonia heredodegenerativa
se refere àquelas de substrato hereditário e/ou degenerativo como o próprio nome
subscreve [Albanese, 2013].
Os movimentos distônicos são comumente debilitantes do ponto de vista
funcional e social [Tarsy, 2006]. Pacientes com distonia podem ter desde dificuldade
para enxergar (naqueles com blefaroespasmo) até limitação para realizar tarefas
manuais (como escrever ou fazer uso de objetos) ou mesmo deambular (naqueles
com distonia em membros). Além disso, outros sintomas, como dor, estão também
frequentemente associados nos pacientes distônicos. Como já amplamente descrito,
dor é um dos principais limitantes da qualidade de vida desses pacientes [Peall,
2015; Jinnah, 2015]. No caso da distonia cervical, por exemplo, dor é a principal
causa para se procurar assistência médica e está presente em 90% dos pacientes
com esse tipo de distonia em alguns estudos [Camfield, 2002; Brin, 2003; Charles,
2014].
O diagnóstico de distonia é clínico e esta é uma condição que pode advir de
diversas circunstâncias, incluindo enfermidades genéticas, doenças
neurodegenerativas (como doença de Wilson, doença de Huntington e as
neuroferritinopatias), e outras condições secundárias como traumas cranianos,
injúrias cerebrais perinatais, encefalites, tumores no sistema nervoso central,

20
induzidas por drogas, paraneoplasias e eventos vasculares (esses levando
geralmente a hemidistonias) [Phukan, 2011].
A fisiopatologia desta condição pode ser explicada por uma série de
alterações na produção e modulação do movimento (Figura 2). A perda de inibição
muscular levando a co-contração de músculos agonistas e antagonistas de
determinado movimento se dá a nível cortical, tronco cerebral e medular [Hallet,
2011]. Anormalidades na aferência sensorial tem sido apontadas como tendo papel
importante no desencadeamento do movimento distônico e explicam o alívio parcial
muitas vezes obtido com a realização dos truques sensoriais (tentativa de inibir a
distonia através de posturas ou gestos que normalmente envolvem estímulo tátil
próximo à região comprometida) [Tinazzi, 2000; Molloy, 2003]. Estudos também têm
mostrado que o córtex motor hiperexcitável tem papel na fisiopatologia dos
movimentos distônicos, algo que não se restringe a áreas motoras primárias, mas
também às áreas suplementares ligadas ao planejamento motor [Rona, 1998].
Porém, dentre todos os elementos do circuito de produção/modulação do
movimento, os núcleos da base e tálamo são apontados como sendo os maiores
responsáveis pela disfunção na distonia. Alterações nessa região levam a perda de
inibição cortical e resultam no quadro hipercinético [Vidailhet, 2005].
A associação fisiopatológica entre os movimentos distônicos e os núcleos da
base são, portanto, bem estabelecidas. Nos últimos anos, porém, atenção especial
tem sido dispensada ao papel do cerebelo na fisiopatologia de pelo menos alguns
tipos de distonias, principalmente no que diz respeito a adaptação comportamental
ao movimento distônico [Malone, 2014]. O cerebelo também parece participar de
maneira importante na integração sensório-motor, importante na fisiopatologia das
distonias focais [Avanzino, 2015]. Entretanto, há estudos que mostram que
alterações cerebelares puderam ser encontradas em até 14% de pacientes com
distonia cervical/segmentar [Batla,2015]

21
Figura 1. Circuitos neuronais envolvidos na produção e modulação do
movimento
No que diz respeito ao tratamento da distonia, muito se tem procurado por
estratégias terapêuticas. Medicações orais, uso de toxina botulínica, estimulação
cerebral profunda (DBS) e estimulação transcraniana tem sido as principais
estratégias propostas [Albanese, 2015]. Geralmente, pacientes com distonia
recebem um curso de levodopa para avaliação de dopa-responsividade. A dose
média utilizada é de 300 a 600 mg/dia e reavaliações são realizadas para alteração
de dose conforme a resposta. A opção terapêutica, na falha da levodopa é a toxina
botulínica, com dose definida conforme locais e gravidade da distonia [Albanese,
2006].
Córtex Motor Primário
Área Motora Suplementar Córtex Pré-Motor
Abreviações: Gpe: Globo Pálido Externo; Gpi: Globo Pálido Interno; NST: Núcleo Subtalâmico

22
III. DISTONIA ASSOCIADA A DOENÇA DE MACHADO JOSEPH
Distonia associada a DMJ/SCA3 é um achado frequente. Em um estudo
recente com uma coorte de 378 pacientes brasileiros com ataxia, distonia esteve
presente em 5,5% dos casos, principalmente nos pacientes com DMJ/SCA3 [Moro,
2014]. No estudo de Schols et al. aproximadamente 12,5% dos pacientes tinham
DMJ/SCA3 associada a distonia, mas essa estimativa varia de 5,5 a 33% em
diferentes séries [Moro, 2014; Jhunjhunwala, 2014]. Nesses estudos, os pacientes
com DMJ/SCA3 eram mais jovens (frequentemente tinham menos de 20 anos) e
tinham uma expansão de CAG maior [Schols 2000; Jardim, 2001; Moro, 2014]. Em
relação à terapia, há escassos relatos de pacientes com SCA3/DMJ e distonia que
tiveram resposta excelente a terapia com levodopa, usada geralmente na dose de
300mg/dia [Nadangopal, 2004].
Entretanto, muitos aspectos da distonia associada à DMJ/SCA3 ainda não
foram esclarecidos. Por exemplo, a frequência e impacto clínico dessa associação
ainda não foram estudados. A avaliação do substrato neuroanatômico desse
subgrupo de pacientes ainda não foi avaliado, aspecto particularmente importante
levando-se em consideração que a neurocirurgia funcional é uma opção terapêutica
efetiva desde que sejam identificados os alvos terapêuticos corretos para
neuromodulação. Além disso, existem poucos estudos que avaliam a resposta à
terapia com levodopa nesse grupo de pacientes e o padrão de resposta a terapia
convencional da distonia: toxina botulínica.

23
OBJETIVOS
OBJETIVO GERAL
Descrever a distonia em seus aspectos clínico, anatômico e terapêutico em
pacientes com DMJ/SCA3.
OBJETIVOS ESPECÍFICOS
I. Determinar a frequência e o perfil clínico da distonia em pacientes com
DMJ/SCA3.
II. Avaliar a resposta terapêutica à Levodopa em pacientes distônicos com
DMJ/SCA3.
III. Avaliar a resposta terapêutica à toxina botulínica em pacientes distônicos
com DMJ/SCA3.
IV. Caracterizar as estruturas anatômicas comprometidas de forma seletiva
nos pacientes com DMJ/SCA3 e distonia em comparação aos pacientes
com DMJ/SCA3 sem distonia.

24
METODOLOGIA
SELEÇÃO DE PACIENTES
Esse estudo foi aprovado pelo comitê de ética em pesquisa da UNICAMP
(protocolo: 157.822 – anexo I) e todos os pacientes que participaram da pesquisa
assinaram um Termo de Consentimento Livre e Esclarecido.
Foram selecionados um total de 75 pacientes consecutivos, sintomáticos, com
diagnóstico molecular confirmado de DMJ/SCA3, regularmente acompanhados no
ambulatório de Neurogenética da Universidade Estadual de Campinas (UNICAMP)
do período de 2008 a 2015, que aceitaram participar da pesquisa. As avaliações
clínicas e de imagem foram realizadas, porém, do período de 2012 a 2015.
Foram incluídos pacientes com mais de 18 anos de ambos os sexos.
Foram excluídos:
1) Pacientes que não tiverem diagnóstico confirmado de DMJ/SCA3 por teste
molecular;
2) Pacientes com contraindicação para realização de estudo por imagem ou uso
de Levodopa.
3) Pacientes que não assinaram o termo de consentimento livre e esclarecido.
Cada paciente, então, foi avaliado por um dos quatro médicos envolvidos
diretamente na pesquisa: Dr Marcelo Burlamarque Nunes, Dr Alberto Rolim Muro
Martinez, Dra Anelyssa D’Abreu ou Dr Marcondes Cavalcante França Jr.
Os pacientes com qualquer tipo de distonia foram submetidos a avaliações
subsequentes: exame clínico detalhado, ensaio clínico com levodopa e aquisição de
uma Ressonância Magnética (Figura 1). Todos esses procedimentos serão
detalhados abaixo.

25
Figura 2. Desenho do estudo
AVALIAÇÃO CLÍNICA
Na avaliação de cada paciente, foram coletados dados sobre a idade de início
da distonia e da ataxia; os pacientes foram questionados sobre a percepção dos
movimentos distônicos, se usavam truques sensoriais (embora esses sejam
característicos das distonias primárias) e se esses tinham algum impacto no alívio
dos sintomas. A presença de alterações funcionais percebidas pelos pacientes
também foi questionada.
Pacientes com DMJ/SCA3 avaliados
(n=75)
Pacientes com DMJ/SCA3 sem distonia
(n= 54)
Pacientes com DMJ/SCA3 com distonia
(n= 21) - Análise clinica (n=21)
-3recusaram a usar
Levodopa
-2 tiveram ef.colaterais
-2 perderam
seguimento
Análise por RM
(n=52)
Analise Clínica
(n=21)
Ensaio com Levodopa
(pacientes com distonia)
(n=14)
- 9 não permitiram
aquisição
- 1 estava grávida
- 5 imagens com
“artefatos”
- 8 perderam follow
up

26
Cada paciente foi submetido a exame neurológico detalhado e a escala de
Burke-Marsden-Fahn foi aplicada para quantificar a gravidade da distonia [Burke et
al, 1985], conforme Anexo II. A gravidade da ataxia, por sua vez, foi avaliada
aplicando a escala validada para essa finalidade: a Scale for assessment and rating
of ataxia (SARA) [Schmitz-Hübsch, 2006; Braga-Neto, 2010], conforme Anexo III. A
dor relacionada ao movimento distônico também foi avaliada, utilizando-se para isso
o Inventário Breve de Dor [Ferreira, 2011], conforme Anexo IV.
O tamanho da expansão CAG foi pesquisada para cada paciente e utilizada
para análises posteriores.
ENSAIO CLÍNICO COM LEVODOPA
Todos os pacientes com distonia associada a DMJ/SCA3 foram convidados a
fazer uso da Levodopa-benzerazida 600mg/dia durante 2 meses. Essa é a dose
padrão utilizada para tratamento das distonias dopa-responsivas de uma maneira
geral [Wilder-Smith, 2003; Fasano, 2014].
Um vídeo padronizado de 5,5 minutos proposto por Louis et al [Louis, 1999]
foi então realizado para cada paciente antes e após o tratamento com Levodopa.
O vídeo consistia dos seguintes componentes:
- 45s: paciente sentado e lê uma passagem padrão em voz alta
- 45s: paciente sentado olhando para a câmera
- 4 minutos: paciente anda no corredor enquanto é filmado
- 1 minuto focado nas mãos
-1 minuto focado nos pés
-1 minuto no corpo inteiro
-1 minuto no corpo inteiro com paciente tentando suprimir o
movimento mais grave

27
Os pacientes que não conseguiram deambular tiveram o vídeo gravado
sentados com câmera focada na região de interesse como seria se deambulando.
Essas imagens foram encaminhadas de modo aleatório e então avaliadas de
um modo cego por um especialista em Distúrbios de Movimento (Dr Joseph
Friedman da Brown University/USA), que não conhecia os pacientes e não tinha
conhecimento se cada vídeo analisado tinha sido filmado antes ou após o uso da
Levodopa. A gravidade foi avaliada usando escala proposta pelo mesmo autor que
propôs o vídeo, conforme anexo V.
Foram perdidas informações de alguns pacientes durante o curso de
tratamento com Levodopa: três se recusaram a usar a medicação, 2 tiveram efeitos
adversos que os motivaram a interromper o uso e 2 perderam o seguimento. No
total, 14 pacientes foram avaliados quanto à resposta a Levodopa.
AVALIAÇÃO DA TOXINA BOTULÍNICA
Pacientes que não responderam a Levodopa receberam tratamento com
Toxina Botulínica do tipo A (apresentação com 500Ui por frasco), com doses padrão
para cada músculo envolvido e conforme localização da distonia. No total, 10
pacientes receberam a aplicação de toxina.
Foram avaliadas resposta ao uso da medicação através da aplicação da
Escala de Burke-Marsden-Fahn antes e após uso da toxina, porém isso não foi feito
de modo cego, como para avaliar os efeitos da Levodopa. Além disso, foram
avaliados os efeitos adversos após a aplicação da toxina.
AVALIAÇÃO DAS IMAGENS
AQUISIÇÃO DAS IMAGENS
Foram adquiridas imagens de 33 pacientes adultos com DMJ/SCA3 sem
distonia (27 homens) e de 19 dos 21 pacientes com DMJ/SCA3 com distonia.

28
Imagens de 52 controles saudáveis foram adquiridas (27 homens). Nós não
conseguimos obter/avaliar imagens de RM de 21 pacientes com DMJ/SCA3 sem
distonia e de 2 pacientes com DMJ/SCA3 com distonia. As razões para isso foram:
recusa a realizar o exame (n=9); gravidez (n=1); artefatos de movimento (n=5) e
perda de seguimento (n=8).
Geralmente, pacientes não-distônicos com DMJ/SCA3 são mais velhos e tem
maior duração de doença do que os pacientes distônicos. Essas são duas variáveis
que poderiam interferir diretamente na comparação volumétrica direta entre os dois
grupos (distônicos e não distônicos). Por isso, em todas as análises, optamos por
realizar comparações pareadas: pacientes distônicos foram comparados ao seu
grupo controle e o mesmo procedimento foi feito com pacientes não-distônicos.
Foram feitos, então, 2 mapas estatísticos que mostravam regiões anormais em cada
grupo de pacientes. Por fim, esses mapas foram sobrepostos com o objetivo de
encontrar estruturas e tratos anormais apenas no grupo distônico.
Inicialmente, porém, todas as imagens foram revisadas, a fim de excluir as
imagens de pacientes com outras anormalidades não relacionadas a DMJ/SCA3
(p.ex.: doenças de substância branca, pequenos AVCs, etc).
Posteriormente, imagens volumétricas ponderadas em T1 foram usadas para
análise no software Freesurfer, enquanto imagens em spin-eco DTI foram usadas
para estudo de substância branca através da metodologia Tract Based Spatial
Statistics (TBSS).
Todos os pacientes foram submetidos a um protocolo de aquisição de RM em
equipamento de 3T Achieva-Intera PHILIPS, usando uma bobina padrão de 8
canais. As imagens foram adquiridas segundo o protocolo descrito abaixo:
1. Seqüência T1 volumétrica (3D) do crânio: orientação sagital, espessura entre
os cortes de 1 mm, Tempo de eco (TE)=3.2ms, Tempo de repetição (TR)=7.1ms,
ângulo de excitação (flip angle) 8o, voxels isotrópicos de 1.0 x 1.0 x 1.0 mm e FOV =
240x240.
2. Seqüência DTI spin-eco: tamanho do voxel adquirido de 2x2x2 mm³,
interpolada a 1x1x2 mm³; matriz reconstruída 256x256; 70 cortes; TE/TR 61/8500
ms; ângulo de excitação de 90°; 32 direções de gradiente; fator b max = 1000 s/mm².

29
ANÁLISE DA SUBSTÂNCIA CINZENTA
FreeSurfer
O software Freesurfer v.5.3 foi usado para medir a espessura cortical e o
volume subcortical nesse estudo. O método é capaz de realizar uma análise
automatizada, fazer a volumetria individual, permitir a análise de áreas específicas e
fornecer dados quantitativos [Fischl, 2012]. Além disso, esse foi um método utilizado
com êxito na análise de alterações estruturais em outras doenças
neurodegenerativas [Bauer, 2014; Zhang, 2014; Schuster, 2013].
Para conseguir isso, imagens de RM foram corrigidas para inomogeneidade
do campo magnético, e em seguida, alinhadas a um atlas específico; o osso foi
então removido da imagem e os voxels foram segmentados em Substância cinzenta
(SC), Substância branca (SB) e líquido cefaloraqueano (LCR). A espessura cortical
foi então calculada baseada na menor distância entre as duas superfícies criadas a
partir dos voxels processados: a superfície branca que é a interface entre a SC e a
SB, e a superfície pial. Um filtro Gaussiano de 10 mm foi usado para suavizar a
superfície em todas as análises. O volume Intracraniano total Estimado (VITe) foi
também aferido [Desikan, 2006]. Todas as imagens processadas foram checadas
quanto a possíveis erros na segmentação ou na aferição da fronteira com o LCR e,
se houvesse necessidade, uma intervenção manual seria feita. Entretanto, isso não
foi necessário para nenhuma das imagens. Anormalidades da espessura cortical e
do volume subcortical entre pacientes distônicos e não –distônicos com DMJ/SCA3
e seus respectivos controles foram avaliadas usando um modelo linear generalizado
que usou idade, gênero e VITe como co-variáveis. FreeSurfer possibilita também o
cálculo da espessura cortical de macrorregiões baseadas, por exemplo, em atlas
anatômico tal como proposto por Desikan et al [Desikan, 2006]. Todos os resultados
foram corrigidos para múltiplas comparações usando a correção de Dunn-Sidak
(nível de significância α= 0.001). Essa análise foi realizada usando o software
SYSTAT v13.0 (San José, CA).
A análise descrita acima permite a identificação de regiões com atrofia em
áreas específicas no grupo de pacientes distônicos. Entretanto, também havia
interesse em estudar áreas que apresentassem atrofia nos dois grupos (distônicos e
não-distônicos). Acreditamos que possa haver áreas comprometidas nos dois

30
grupos (especialmente na região subcortical), mas de forma proporcionalmente mais
intensa no grupo distônico. Para avaliar isso, foi realizada para cada região atrófica
uma normalização dos dados volumétricos “brutos” com a média dos respectivos
grupos controles usando o z-score. Então, após cada normalização com a média,
esses valores dos grupos foram comparados entre si usando o test T de Student
com Correção de Dunn-Sidak para múltiplas comparações (α =0.01).
Além disso, avaliamos também a correlação de estruturas atrofiadas
específicas com a gravidade da distonia, quantificada pela escala de Marsdenn-
Fahn (antes do uso da Levodopa). Para tanto, realizamos uma regressão com
múltiplas variáveis, usando idade, gênero e VITe como covariáveis.
ANÁLISE DA SUBSTÂNCIA BRANCA (SB)
Tract based spatial statistics (TBSS)
O TBSS é um método utilizado para avaliar de forma automatizada alterações
nos tratos de substância branca entre grupos de indivíduos utilizando imagens de
DTI. Para isso é utilizado o conceito de difusão de moléculas de água. Em um meio
homogêneo, as moléculas tendem a se difundir de forma idêntica em todas as
direções, portanto apresentam coeficiente de difusão chamado isotrópico. Em
tecidos biológicos, como os tractos neurais, o coeficiente de difusão é diferente nas
várias direções (apresenta sentido preferencial), sendo chamado anisotrópico
[Kingsley, 2006]. Usando o tensor de difusão, consegue-se extrair as direções de
difusão das moléculas e sua magnitude. Índices então são calculados e mapas são
criados, sendo os principais os assim chamados: anisotropia fracionada (AF),
difusividade média (DM), difusividade radial (DR) e difusividade axial (DA).
Quantifica-se assim o coeficiente de difusão das moléculas de cada voxel em várias
direções, permitindo inferências sobre organização, compactação e grau de
mielinização de fibras que compõem os tratos de substância branca cerebrais
[Ameis, 2011].
O toolbox FMRIB do software FSL v.4.1.4 foi usado com essa finalidade
nesse estudo e foram realizadas comparações entre pacientes e controles usando o
algoritmo do TBSS [Smith, 2006]. Os seguintes passos precederam a análise

31
estatística: imagens em AF foram alinhadas uma a outra usando um registro não-
linear e uma média da AF foi criada; isso possibilitou a criação de um esqueleto
médio que representa todos os tratos compartilhados entre os sujeitos no estudo.
Depois disso, imagens de AF pré-processadas de cada paciente foram “projetadas”
sobre o esqueleto médio. Para visualizar o mapa estatístico de DM, DA e DR esses
parâmetros foram aplicados sobre o esqueleto médio de AF [Hosseinbor, 2012].
Para análise estatística um Teste T de Student para 2 amostras foi usado
para avaliar diferenças entre pacientes e controles nos parâmetros de AF, DM, DA e
DR . Usamos uma correção baseada em “cluster” para múltiplas comparações
(p<0.05). Além disso, para identificar os tratos na substância branca afetados, o
Johns Hopkins white matter DTI based atlas, encontrado no software FSL software,
foi usado.
ANÁLISE ESTATÍSTICA
Os dados clínicos e genéticos dos pacientes e controles foram detalhados
com estatística descritiva.
Para avaliar a diferença nos escores clínicos antes e após tratamento com
levodopa, foi empregado o teste de Wilcoxon.
O nível de significância foi de α=0.05 para todas as comparações. A análise
estatística foi realizada com o software SYSTAT versão 9.0.

32
RESULTADOS
AVALIAÇÃO CLÍNICA
Nós encontramos distonia em 21 dos 75 pacientes avaliados com DMJ/SCA3
(28%). Em dois pacientes a distonia foi a primeira manifestação da doença.
Pacientes com distonia tinham início mais precoce da ataxia, maiores expansões de
(CAG) e um maior escore na avaliação da ataxia pela escala SARA em comparação
aos pacientes sem distonia (Tabela 1).
Os achados clínicos da distonia nos pacientes avaliados não eram
homogêneos. Nove pacientes tinham distonia generalizada e os demais tinham
distonia segmentar ou focal. Neste último grupo, nós encontramos: seis pacientes
com distonia isolada em membro inferior do tipo inversão assimétrica dos pés; dois
tinham blefaroespasmo e apresentavam “caretas” ao falar; dois tinham distonia
crânio-facial ( blefaroespasmo associado a retrocolo ) e dois tinham distonia facial
associada a distonia em pés. De modo geral, blefaroespasmo (n= 13) e inversão do
pé (n=15) foram as duas apresentações distônicas mais frequentes. Dois pacientes
que apresentavam distonia facial tinham ataxia muito leve e uma disfemia severa.
Sempre quando tentavam falar movimentos involuntários da face e disfemia
apareciam simultaneamente, sugerindo assim que isso poderia corresponder a um
outro fenômeno distônico.
Distonia era frequentemente grave e o escore médio pela escala de Burke-
Marsden-Fahn foi de 15,9 (variando de 4 – 36,5). Dezessete dos vinte e um
pacientes identificavam todos os movimentos distônicos e usavam truques
sensoriais para aliviá-los. Entretanto, apenas dois deles referiam melhora com essa
medida.
Dor relacionada a distonia foi frequente (14/21 = 70%) e muitas vezes intensa
(escore médio no Inventário Breve de Dor de 31,9 mas variando de 2 a 70).
Em nossa coorte, 3 pacientes apresentavam parkinonismo: 1 no grupo
distônico e 2 no grupo não distônico (tabela 1).
Nós realizamos eletroneuromiografia em 15 dos 21 pacientes com distonia e
DMJ/SCA3 e em 33 dos 54 pacientes com DMJ/SCA3 sem distonia. Encontramos

33
evidência de alteração no sistema nervoso periférico em 6 dos pacientes distônicos
(6/15 = 40%) e em 17 dos pacientes sem distonia (17/33 = 51%). A maioria dos
pacientes com neuropatia periférica tinham predominantemente anormalidades
sensoriais.
Tabela 1. Dados clínicos e genéticos de pacientes com DMJ/SCA3 com e sem
distonia.
Pacientes com
distonia
(n = 21)
Pacientes sem
distonia
(n = 54) p
Idade (média ± DP, anos) 39,7± 13,9 50,0 ± 11,1 0,005
Sexo (M/F) 13/8 25/29 0,305
Início (média ± DP, anos) 28,9 ± 11,7 40,6 ± 11,4 <0,001
Expansão CAG (média ± DP) 75,3 ± 3,6 70,9 ± 3,0 <0,001
Escore SARA (média ± DP) 18,8 ± 8,5 13,7 ± 6,5 0,021
Parkinsonismo (%) 1/21 2/54 1,000
p<0,001
TRATAMENTO COM LEVODOPA E TOXINA BOTULÍNICA
Quatorze pacientes completaram o tratamento com Levodopa. O escore
médio antes e após o tratamento com levodopa foi 21,0 ± 12,0 vs 16,2 ± 9,1
(Wilcoxon p = 0,075). O escore diminuiu em sete pacientes, permaneceu estável em
três e aumentou em quatro pacientes. Entretanto, três pacientes tiveram melhora
significativa com o uso da Levodopa (declínio maior que 50% em relação ao escore
pré-tratamento) – dois tinham distonia generalizada e um tinha distonia focal em
perna. O único paciente com parkinsonismo concomitante a distonia melhorou com
levodopa, mas não dramaticamente (escore pré =8 e escore pós =6).
Dez pacientes receberam toxina botulínica, incluindo os sete que tinham
permanecido com escore inalterado ou com aumento após o tratamento com
levodopa; dois que tiveram benefícios apenas leves com o uso da levodopa; e um
que não havia tolerado a medicação. Nós usamos dose padrão de Toxina Botulínica
A (apresentação de 500Ui por frasco) de acordo com cada músculo. Para evitar

34
efeitos sobrepostos ao uso da levodopa nós aplicamos a toxina ao menos 4
semanas após a descontinuação do uso da medicação. Considerando o grupo como
um todo houve uma melhora significativa nos escores antes e após 4 semanas do
tratamento com toxina (Wilcoxon p = 0,03). Sete pacientes referiram melhora
subjetiva.
Embora nós tenhamos seguido técnica estrita de aplicação houve alta
incidência de eventos adversos (3/10 = 30%). Dois pacientes tiveram diplopia e
ptose após o tratamento para blefaroespasmo (usando 20UI de Toxina Botulínica A
em cada olho), e um paciente apresentou piora dramática da disfagia requerendo
uso de sonda nasoenteral após tratamento do torcicolo espasmódico (usando 350UI
de Toxina Botulínica A no trapézio, esplênios e esternocleidomastóideo). Dois dos
três pacientes que tiveram efeitos adversos tinham sinais eletroneuromográficos de
desnervação crônica em membros.
ANÁLISE DAS IMAGENS
Análise da Substância Cinzenta (Freesurfer)
A análise do Freesurfer mostrou diferença significativa no padrão de atrofia
entre pacientes com DMJ/SCA3 com e sem distonia. Pacientes distônicos tinham
redução volumétrica no córtex motor primário e em ambos os hipocampos, com
algum envolvimento no córtex motor secundário (Tabela 2). Em contraste, o grupo
com DMJ/SCA3 sem distonia apresentava essencialmente atrofia cortical nos lobos
occipitais (Tabela 2). Ambos os grupos apresentavam redução volumétrica nos
gânglios da base (Tabela 3).
Tabela 2. Áreas que apresentaram alteração na espessura cortical: (A) pacientes
com distonia (dDMJ/SCA3) comparados com seus 34espectivos controles
saudáveis; (B) pacientes sem distonia (cDMJ/SCA3) comparados com seus
respectivos controles (nível de significância 0,001).

35
Estrutura Média dDMJ/SCA3 (mm) Média Controle (mm)
(A)
Hemisfério Esquerdo
Lobo e sulco paracentral 1,977 ± 0,187 2,148 ± 0,120
Giro Precentral 2,417 ± 0,177 2,613 ± 0,206
Sulco Temporal Superior 2,163 ± 0,180 2,331 ± 0,125
Córtex paracentral 2,049 ± 0,178 2,252 ± 0,141
Córtex Precentral 2,221 ± 0,138 2,380 ± 0,142
Cortex supramarginal 2,285 ± 0,160 2,352 ± 0,160
Hemisfério Direito
Lobo e sulco paracentral 1,957 ± 0,183 2,146 ± 0,139
Parte triangular do Giro
frontal inferior 2,316 ± 0,232 2,527 ± 0,175
Giro Precentral 2,374 ± 0,272 2,596 ± 0,288
Córtex paracentral 2,074 ± 0,176 2,263 ± 0,152
Córtex Precentral 2,188 ± 0,192 2,379 ± 0,189
Estrutura Média cDMJ/SCA3 (mm) Média Controle (mm)
(B)
Hemisfério Esquerdo
Sulco calcarino 1,690 ± 0,114 1,809 ± 192
Parte inferior do sulco
precentral 2,111 ± 0,149 2,242 ± 0,175
Córtex Occipital Lateral 1,923 ± 0,166 2,059 ± 0,149
Hemisfério Direito
Lobo e sulco paracentral 1,991 ± 0,175 2,175 ± 0,191
Giro Occipital Superior 1,881 ± 0,202 2,052 ± 0,198
Sulco calcarino 1,714 ± 0,120 1,862 ± 0,215
Sulco Central 1,609 ± 0,122 1,727 ± 0,130
Sulco occipito-tempora
medial e sulco lingual 2,200 ± 0,178 2,352 ± 0,189
Córtex Paracentral 2,137 ± 0,172 2,300 ± 0,178

36
Quando nós comparamos os grupos com DMJ/SCA3 com e sem distonia
entre si, os pacientes distônicos tinham volume menor em ambos os tálamos (tabela
3). A análise por z-score entre estruturas que estavam atrofiadas nos dois grupos,
mostrou que o tálamo, diencéfalo ventral e cerebelo estavam mais atrofiadas no
grupo distônico (tabela 4).
Nós não encontramos correlação entre o escore na escala de Burke-Marsden-
Fahn e os achados da RM (espessura cortical e volume subcortical).
Tabela 3. Estruturas subcorticais que apresentaram redução volumétrica
significativa em pacientes com dDMJ/SCA3 e cDMJ/SCA3 quando comparados
com seus respectivos grupos controles (nível de significância 0,001).
Estrutura
Média
dDMJ/SCA3
(mm3)
Média controle
dDMJ/SCA3
(mm3)
Média
cDMJ/SCA3
(mm3)
Média Controle
cDMJ/SCA3
(mm3)
Hemisfério esquerdo
SB Cerebelo 8388 ± 2194 15126 ± 1929 8445 ± 1904 15292 ± 2475
SC Cerebelo 38338 ± 6587 45441 ± 7058 37566 ± 4267 44682 ± 5395
Tálamo 5935 ± 734 7546 ± 880 6237 ± 821 7381 ± 1097
Caudado 2994 ± 497 3511 ± 455 2955 ± 331 3441 ± 427
Putamen - - 5031 ± 544 5771 ± 929
Pallidum 1174 ± 184 1410 ± 274 1145 ± 219 1383 ± 256
Hipocampo 3871 ± 463 4356 ± 472 - -
Diencéfalo
Ventral 2862 ± 382 3772 ± 337 2991 ± 382 3738 ± 388
Hemisfério direito
SB Cerebelo 8330 ± 2203 15230 ± 1666 8196 ± 1747 15572 ± 2324
SC Cerebelo 39053 ± 6491 46502 ± 6802 38024 ± 4424 45292 ± 5116
Tálamo 5692 ± 659 6780 ± 603 5771 ± 669 6583 ± 783
Caudado 3110 ± 461 3705 ± 728 3144 ± 394 3660 ± 423
Putamen 5067 ± 678 5795 ± 920 5072 ± 558 5700 ± 789
Pallidum 1150 ± 170 1513 ± 245 1190 ± 169 1586 ± 237
Hipocampo 4004 ± 418 4524 ± 504 - -

37
Diencéfalo
Ventral 2877 ± 415 3868 ± 357 3049 ± 350 3856 ± 499
Tronco
Cerebral 13670± 2497 20919 ± 2173 14486 ± 2345 21035 ± 2549
P<0,001
Tabela 4. Comparação entre estruturas atrofiadas em ambos os grupos, mas mais
atrofiadas no grupo distônico através do z-score (nível de significância 0,001).
Estrutura cDMJ/SCA3 media
(mm)
dDMJ/SCA3 media
(mm)
“p”
Hemisfério Esquerdo
Tálamo 1,83±0,83 1,04±0,75 <0,001
Cerebelo 3,49±1,14 2,77±0,77 <0,001
Diencéfalo Ventral 2,70±1,33 1,92±0,98 0,004
Hemisfério Direito
Tálamo 1,81±1,09 1,04±0,85 <0,001
Cerebelo 4,14±1,32 3,17±0,75 <0,001
Diencéfalo Ventral 2,77±1,16 1,62±0,70 0,005
P<0,001
Análise da Substância Branca (TBSS)
A análise por TBSS identificou anormalidades em SB tanto no grupo distônico
como no grupo sem distonia, mas com um padrão similar de acometimento (Figura
1).
Figura 3. Resultados de TBSS que mostram áreas de anisotropia fracional (AF)
reduzida e aumento da difusividade média (DM), difusividade axial (DA) e
difusividade radial (DR) em pacientes com DMJ / SCA3 após a comparação com
controles pareados por idade e sexo. Em cada linha, dois mapas estatísticos são

38
mostrados nos mesmos cortes: a superior mostra pacientes com distonia vs
respectivos controles, e os pacientes não-distônicos vs respectivos controles. Os
resultados são apresentados no template MNI152 e abaixo são representados os
valores de p.
Esses resultados foram publicados na revista “Parkinsonism and related
disorders” em outubro de 2015, conforme anexo VI.

39
0,001 0,05 P-valor

40
DISCUSSÃO GERAL
Análise dos achados clínicos
Esse estudo mostrou que distonia no contexto de DMJ/SCA3 é
frequente, geralmente limitante, não melhora com truques sensoriais e
frequentemente está associada a dor. Isso está de acordo com estudos
prévios que investigaram a frequência de distúrbios do movimento em
pacientes com ataxia espinocerebelar [Jhunjhunwala, 2014; Moro, 2014].
Distonia, portanto, deve ser pesquisada, avaliada e tratada nesse grupo de
pacientes.
Em alguns dos nossos pacientes, a distonia era mais limitante do que a
própria ataxia. Além disso, sendo uma condição potencialmente tratável, a
abordagem correta da distonia pode agregar uma melhor qualidade de vida a
esses pacientes.
A informação de que dor é um achado frequente é coerente com a
associação comumente relatada na literatura de dor e distonia [Comella, 2015].
Dor é um sintoma muito descrito, por exemplo, nos casos de distonia cervical
[Kutvonen, 1997; Barbanti, 2005; Brin, 2003; Charles, 2014]. Nossos pacientes
foram questionados sobre dor associada ao movimento distônico e 70% deles
a referiram, chegando a escores tão elevados como 70 no Inventário Breve de
Dor. Considerando-se que essa é uma condição tratável, também com um
importante impacto na qualidade de vida, ela deve ser pesquisada, quantificada
e tratada nos pacientes com DMJ/SCA3 e distonia. Esses achados mostram,
portanto, que não apenas distonia deve ser pesquisada em pacientes com
DMJ/SCA3, mas também a presença de dor associada a ela[Camargo, 2015].
Nesse estudo, não houve diferença de prevalência da distonia entre os
gêneros (8 mulheres e 13 homens; p=0,305). A literatura mostra que, de
maneira geral, distonia é um distúrbio de movimento mais prevalente em
mulheres [Jankovic, 1983].
Além disso, o fenótipo distônico na DMJ/SCA3 não se mostrou
homogêneo. Em nossa coorte de pacientes, blefaroespasmo e inversão

41
unilateral (ou assimétrica) do pé foram as duas apresentações distônicas mais
comuns. Essas duas localizações implicam especial atenção já que ambas
limitam a funcionalidade: o blefaroespasmo levando a alteração visual e
significativo constrangimento social e a inversão do pé implicando em piora da
marcha de modo independente à ataxia. No que diz respeito, portanto, a
localização da distonia nesse subgrupo, isso difere do que é encontrado na
epidemiologia das distonias em geral, onde distonia cervical é o tipo mais
prevalente [Jinah, 2015].
Outros distúrbios do movimento podem estar associados a distonia.
Pacientes com ataxia hereditária podem apresentar vários tipos de distúrbios
de movimento, como tremor postural, bradicinesia e outros sinais
parkinsonianos [Jhunjhunwala, 2014]. Na série de pacientes desse estudo,
houve apenas um que tinha parkinsonismo e distonia simultaneamente.
Um achado interessante é que dois pacientes avaliados apresentavam
ataxia muito leve, mas distonia em face (tipo “careta”) associada a gagueira
quando tentavam falar. Esse achado era associado a blefaroespasmo e isso
sugeria que a gagueira correspondesse a um outro componente distônico
nesse subgrupo de pacientes (com DMJ/SCA3), algo até então não relatado na
literatura. Vale ressaltar que esses pacientes não apresentavam sintomas
atáxicos preponderantes, sendo esses sinais distônicos descritos como a
primeira manifestação da doença.
Os distônicos eram mais jovens, tinham idade de inicio da ataxia mais
precoce e tinham um CAG maior. Esses dados são compatíveis com os
relatados na literatura [Schols 2000; van Gallen, 2011]. A escala SARA usada
para quantificar a gravidade da ataxia mostrou que os pacientes distônicos
tinham maior gravidade (p=0.021). Possivelmente, isto é um reflexo da
combinação ataxia e distonia, que acarreta maior incapacidade motora e,
portanto, maior pontuação em diversos itens da escala SARA.
No que diz respeito ao tratamento, existiam apenas alguns relatos
ocasionais de distonia associada a DMJ/SCA3 que eram dopa-responsivos na
literatura [Wilder-Smith, 2003; Nadangopal, 2004]. Porém, nenhuma avaliação
sistemática em uma coorte maior de pacientes havia sido realizada até então.

42
A frequência de dopa-responsividade na distonia relacionada a DMJ/SCA3 não
havia ainda sido definida em um subgrupo maior de casos.
Nesse estudo, os pacientes receberam tratamento com levodopa nas
doses habituais para o tratamento das distonias dopa-responsivas e então,
realizamos uma análise cega do impacto desse tratamento na gravidade da
distonia. Acreditamos que esse tipo de abordagem - controlada e com
casuística maior - pode permitir conclusões mais robustas acerca do real papel
da Levodopa na patogênese da distonia. Nessa análise, não ficou demonstrado
benefício do uso da Levodopa na comparação entre os grupos (p=0.07). É
possível que o trial tenha sido incapaz de detectar pequenas diferenças no
status clínico dos pacientes que fizeram uso da medicação e uma coorte ainda
maior de pacientes fosse necessária para provar esse fato. Porém, chama
atenção o fato de que ao menos 3 dos pacientes (21%) tiveram melhora
objetiva na nossa série. Esse achado sugere que disfunção dopaminérgica
pode desempenhar um papel ao menos em um subgrupo de pacientes
distônicos com DMJ/SCA3. Vários estudos em neuroimagem, principalmente
com o uso do TRODAT, já apontam para essa hipótese [Karimi, 2015; Stoessl,
2014; Alongi, 2014].
Em termos clínicos, embora a melhora da distonia em pacientes com
DMJ/SCA3 com o uso de levodopa não seja usual, entendemos que essa
droga deva ser oferecida aos pacientes nesse cenário e uma avaliação de
resposta deve ser avaliada.
A maioria dos nossos pacientes que não melhoraram com o uso da
levodopa tiveram alguma melhora com o uso da toxina botulínica. Isso segue o
que a literatura tem demonstrado para as distonias de uma maneira geral.
Devido a sua comprovada eficácia no tratamento da distonia, essa tem sido a
medicação de escolha na maioria dos casos que necessitam de tratamento
[Albanese, 2015].
Um achado importante que tivemos em nossa série de pacientes foi a
alta frequência de eventos adversos após as injeções da toxina botulínica
(30%), incluindo uma complicação séria (piora da disfagia com necessidade de
uso de sonda nasoentérica para alimentação). Achado similar foi descrito por

43
Tuite e Lang [Tuite&Lang, 1996]. De uma maneira geral, a taxa de
complicações associadas ao uso da Toxina botulínica A é de 5 a 12% [Kessler,
1999; Haussermann, 2004]. Esse achado pode ser justificado pelo fato de que
pacientes com DMJ/SCA3 frequentemente têm acometimento de nervos
periféricos e isso faz com que suas junções neuromusculares sejam mais
sensíveis aos efeitos da toxina botulínica [França Jr, 2009]. De fato, 2 dos 3
pacientes que tiveram efeitos adversos tinham sinais eletroneuromiográficos
leves, mas claros, de desnervação crônica em membros. Esses dados em
conjunto indicam que a toxina botulínica deve ser considerada no tratamento
da distonia nos pacientes com DMJ/SCA3, mas que doses mais baixas devem
ser tentadas inicialmente e atenção estrita a técnica de aplicação deve ser
dispensada.
Análise das Imagens
DMJ/SCA3 é caracterizada por um dano neuronal em várias regiões que
envolvem o controle motor, como o cerebelo, os gânglios da base, o tronco
cerebral e o córtex motor [D’Abreu, 2012; Guimarães, 2013; Rezende, 2015].
Semelhantemente, distonia primária está relacionada também com disfunção
em ampla rede neuronal que envolve os núcleos da base, córtex motor,
cerebelo, tronco cerebral e medula espinhal e a avaliação de lesão em circuitos
específicos tem sido cada vez mais estudada, com achados recentes, por
exemplo, de disfunção na rede córtico-palidal [Vidailhet, 2005]. O DBS
(estimulação cerebral profunda) é tratamento efetivo em muitos casos de
distonia primária, muito embora os mecanismos desse efeito não tenham sido
ainda totalmente compreendidos. [Neumann, 2015].
Entretanto, pouco é conhecido sobre as estruturas que são lesadas
seletivamente em pacientes distônicos com DMJ/SCA3. Esse estudo buscou
avançar na avaliação dessa questão já que essa é uma informação importante,
pois ela poderia trazer um entendimento a mais sobre a patogênese da distonia
e, consequentemente, alvos terapêuticos.

44
Com base nisso, nós empregamos uma avaliação multimodal para
análise da substância branca e substância cinzenta, a fim de serem
encontradas áreas anormais exclusivamente no grupo de pacientes distônicos.
No que diz respeito à avaliação de estruturas corticais, verificamos que
pacientes distônicos com DMJ/SCA3 tem atrofia no córtex pré e paracentral,
enquanto os pacientes com DMJ/SCA3 sem distonia tem atrofia occipital. Isso
demonstra uma alteração objetiva não apenas em áreas de modulação do
movimento, mas também em áreas envolvidas com o comando inicial/
planejamento do processo motor na fisiopatologia da distonia.
A análise da substância cinzenta subcortical mostrou volumes reduzidos
tanto no grupo distônico como no grupo não-distônico. Entretanto, redução
volumétrica no tálamo, diencéfalo ventral e substância branca cerebelar foram
significativamente mais pronunciadas nos pacientes com DMJ/SCA3
distônicos. Isso é particularmente relevante, pois os pacientes com DMJ/SCA3
não-distônicos eram mais velhos, tinham maior duração da ataxia e, portanto,
esperava-se atrofia mais intensa nessas estruturas discriminadas nesse grupo
em relação aos pacientes distônicos.
Em contraste, nós encontramos alterações difusas de substância
branca, mas com padrão similar tanto no grupo distônico como no não-
distônico, não havendo, portanto, diferença entre os grupos. Esse achado pode
estar relacionado com limitações inerentes a abordagem da imagens
empregada nesse estudo. O protocolo de aquisição do DTI utilizado permite
uma adequada avaliação dos grandes tractos de substância branca, entretanto
pode haver uma limitação para análise de tractos menores, como por exemplo
os tractos eferentes do cerebelo. Além disso, em regiões onde há cruzamento
de tractos a estimativa dos parâmetros do DTI também pode ser
comprometida. Uma abordagem que vem sendo proposta recentemente para
contornar essas restrições é o uso de sequências chamadas HARDI (High
Angular Resolution Diffusion Tensor Image) que tem mais de 100 direções de
tensor e se mostram apropriadas para análise de tractos menores [Presseau,
2015].

45
Nossos resultados indicam que distonia na DMJ/SCA3 é associada com
anormalidades estruturais nos gânglios da base e no córtex cerebral. Nós
encontramos atrofia talâmica desproporcional no grupo distônico, o que está de
acordo com um estudo prévio do nosso grupo que empregou o método de
volumetria manual [D’Abreu, 2011]. Esse achado é interessante já que o tálamo
está envolvido tanto com o controle motor quanto com a integração sensorio-
motora que são dois importantes componentes na patogênese da distonia. Já é
bem estabelecido que lesões talâmicas focais levam a distonia contralateral em
ao menos 30% dos casos [Lehericy, 2001]. Um achado novo nesse estudo foi a
presença de atrofia cortical nas proximidades do sulco central nos pacientes
distônicos. Esse achado está alinhado com estudos de neuroimagem que
mostram alterações funcionais e estruturais no córtex sensório-motor em
diferentes formas de distonia primária [Ceballos-Baumann, 1995; Butterworth,
2003; Piccinin, 2015]. Nesse contexto, a extensão desse trabalho com o uso de
protocolo com RM funcional poderia ajudar a definir qual a contribuição
específica das estruturas corticais e subcorticais na origem da distonia ligada a
DMJ/SCA3.

46
CONCLUSÕES
Nós mostramos que distonia é frequente, geralmente debilitante e
clinicamente heterogênea em pacientes com DMJ/SCA3.
Ela pode responder a levodopa e a injeções de toxina botulínica, sendo
necessário cuidado especial na aplicação desta pela maior possibilidade de
efeitos colaterais.
O padrão de acometimento cerebral é diferente entre pacientes
distônicos e não-distônicos com DMJ/SCA3. Os córtices precentral e
paracentral são acometidos somente no grupo distônico. Atrofia talâmica,
entretanto, está presente em ambos os grupos de pacientes, porém ela é muito
mais intensa em pacientes distônicos.

47
REFERÊNCIAS BIBLIOGRÁFICAS
Lima L, Coutinho P. Clinical criteria for diagnosis of Machado-Joseph
disease: report of a non-Azorena Portuguese family. Neurology
1980;30:319-22.
Lopes-Cendes I, Teive HG, Calcagnotto ME, et al. Frequency of the
different mutations causing spinocerebellar ataxia (SCA1, SCA2,
MJD/SCA3 and DRPLA) in a large group of Brazilian patients. Arq
Neuropsiquiatr 1997;55:519-29.
Sequeiros J, Coutinho P. Epidemiology and clinical aspects of Machado-
Joseph disease. Adv Neurol 1993;61:139-53.
de Castilhos RM, Furtado GV, Gheno TC, Schaeffer P, Russo A,
Barsottini O, Pedroso JL, Salarini DZ, Vargas FR, de Lima MA, Godeiro
C, Santana-da-Silva LC, Toralles MB, Santos S, van der Linden H Jr,
Wanderley HY, de Medeiros PF, Pereira ET, Ribeiro E, Saraiva-Pereira
ML, Jardim LB; Rede Neurogenetica. Spinocerebellar ataxias in Brazil--
frequencies and modulating effects of related genes. Cerebellum
2014;13:17-28.
Kawaguchi Y, Okamoto T, Taniwaki M, et al. CAG expansions in a novel
gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet.
1994;8:221-228.
Jardim LB, Pereira ML, Silveira I, Ferro A, Sequeiros J, Giugliani R.
Neurologic findings in Machado-Joseph disease: relation with disease
duration, subtypes, and (CAG)n. Arch Neurol 2001;58:899–904.
Bettencourt C, Lima M. Machado-Joseph Disease: from first descriptions
to new perspectives. Orphanet J Rare Dis. 2011 Jun 2;6:35.

48
Dürr A, Stevanin G, Cancel G, Duyckaerts C, Abbas N, Didierjean O et
al. Spinocerebellar ataxia 3 and Machado-Joseph disease: clinical,
molecular, and neuropathological features. Ann Neurol. 1996;39:490-9.
Rüb U, Brunt ER, Deller T. New insights into the pathoanatomy of
spinocerebellar ataxia type 3 (Machado-Joseph disease). Curr Opin
Neurol 2008;21:111-116.
Coutinho P. Doença de Machado-Joseph: Tentativa de definição. PhD
Dissertation, Instituto de Ciências Biomédicas Abel Salazar, Porto; 1992.
Suite ND, Sequeiros J, McKhann GM: Machado-Joseph disease in a
Sicilian-American family. J Neurogenet 1986;3:177-182.
Sakai T, Kawakami H: Machado-Joseph disease: A proposal of spastic
paraplegic subtype. Neurology 1996;46:846-847.
Pedroso JL, Braga-Neto P, Felício AC, Barsottini OG, Jardim
LB, Saraiva-Pereira ML. Akathisia: An unusual movement disorder
in Machado-Joseph disease. Parkinsonism Relat Disord. 2011
Nov;17(9):712-3.
Schols L, et al. Extrapyramidal Motor signs in degenerative ataxias. Arch
Neurol 2000;57:1495-1500.
van Gaalen J, Giunti P, van de Warrenburg BP. Movement disorders in
spinocerebellar ataxias. Mov Disord 2011;26:792-800.
Moro A, Munhoz RP, Moscovich M, Arruda WO, Raskin S, Teive HA.
Movement disorders in spinocerebellar ataxias in a cohort of Brazilian
patients. Eur Neurol 2014;72:360-362.
Jhunjhunwala K, Netravathi M, Purushottam M, Jain S, Pal PK. Profile of
extrapyramidal manifestations in 85 patients with spinocerebellar ataxia
type 1, 2 and 3. J Clin Neurosci 2014;21:1002-1006.

49
Mariotti C, Alpini D, Fancellu R, Soliveri P, Grisoli M, Ravaglia S, Lovati C,
Fetoni V, Giaccone G, Castucci A, Taroni F, Gellera C, Di Donato
Spinocerebellar ataxia type 17 (SCA17): oculomotor phenotype and clinical
characterization of 15 Italian patients. J Neurol 2007; 254:1538-1546.
Pedroso JL, Braga-Neto P, Escorcio-Bezerra ML, et al. Non-motor and
Extracerebellar Features in Spinocerebellar Ataxia Type 2.
Cerebellum. 2016;1-6.
Destarac L. Torticollis spasmodique et spasmes fonctionelles. Rev.
Neurol 1901;9:591–597.
Oppenheim H. Neurologisches Centralblatt. Volume 30. Veit; Leipzig,
Germany: 1911. Über eine eigenartige Krampfkrankheit des kindlichen
und jugendlichen Alters (Dysbasia lordotica progressiva, Dystonia
musculorum deformans) pp. 1090–1107.
Steeves TD, Day L, Dykeman J, Jette N, Pringsheim T. The prevalence
of primary dystonia: A systematic review and meta-analysis. Mov
Disord 2012;27:1789–1796.
Fahn, S.; Marsden, CD.; Calne, DB. Classification and investigation of
dystonia. In: Marsden, CD.; Fahn, S., editors. Movement disorders. Vol.
2. London: Butterworths; 1987. p. 332-58.
Bressman SB. Dystonia genotypes, phenotypes, and classification. Adv
Neurol 2004; 94:101–107.
Albanese A, Asmus F, Bhatia KP, Elia AE, Elibol B, Filippini G, et al.
EFNS guidelines on diagnosis and treatment of primary dystonias. Eur J
Neurol 2011; 18:5–18.
Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS,
Hallett M, Jankovic J, Jinnah HA, Klein C, et al. Phenomenology and

50
classification of dystonia: A consensus update. Mov Disord
2013;28:863–873.
Tarsy D, Simon DK. Dystonia. N Engl J Med 2006;355:818-829.
Peall KJ, Kuiper A, de Koning TJ, Tijssen MA. Non-motor symptoms in
genetically defined dystonia: Homogenous groups require systematic
assessment. Parkinsonism Relat Disord 2015;21:1031-1040.
Jinnah HA, Factor SA. Diagnosis and treatment of dystonia. Neurol Clin
2015;33:77-100.
Camfield L, Ben-Shlomo Y, Warner TT. Epidemiological Study of
Dystonia in Europe Collaborative Group. Impact of cervical dystonia on
quality of life. Mov Disord 2002;17:838–841.
Brin M, Brashear A, Mordaunt J. Effect of botulinum toxin type A (BoNT-
A) therapy on pain frequency and intensity in patients with cervical
dystonia. Cephalalgia 2003;23:743.
Charles PD, Adler CH, Stacy M, Comella C, Jankovic J, Manack Adams
A, Schwartz M, Brin MF. Cervical dystonia and pain: Characteristics and
treatment patterns from CD PROBE (Cervical Dystonia Patient Registry
for Observation of OnabotulinumtoxinA Efficacy). J
Neurol 2014;261:1309–1319.
Phukan J, Albanese A, Gasser T, Wagner T. Primary dystonia and
dystonia-plus syndromes: clinical characteristics, diagnosis, and
pathogenesis. Lancet Neurol 2011;12: 1074–1085.
Hallett M. Neurophysiology of dystonia: the role of inhibition. Neurobiol
Dis 2011; 42: 177–184.
Tinazzi M, Priori A, Bertolasi L et al. Abnormal central integration of a
dual somatosensory input in dystonia. Evidence for sensory overflow.
Brain 2000;123: 42–50.

51
Molloy F, Carr T, Zeuner K, Drambosia J, Hallett M. Abnormalities of
spatial discrimination in focal and generalized dystonia. Brain, 2003; 126:
2175–2182.
Rona S, Berardelli A, Vacca L, Inghilleri M, Manfredi M. Alterations of
motor cortical inhibition in patients with dystonia. Mov Disord
1998;13:118–124.
Vidailhet M, Vercueil L, Houeto JL, et al. Bilateral deep-brain stimulation
of the globus pallidus in primary generalized dystonia. N Engl J Med
2005; 352:459–467.
Malone A, Manto M, Hass C. Dissecting the links
between cerebellum and dystonia. Cerebellum. 2014 Dec;13(6):666-8.
Avanzino L, Tinazzi M, Ionta S, Fiorio M. Sensory-motor integration in
focal dystonia. Neuropsychologia. 2015 Dec;79(Pt B):288-300.
Batla A, Sánchez MC, Erro R, et al.. The role of cerebellum in patients
with late onset cervical/segmental dystonia?--evidence from the clinic.
Parkinsonism Relat Disord. 2015 Nov;21(11):1317-22.
Albanese A, Romito LM, Calandrella D. Therapeutic advances
in dystonia. Mov Disord 2015;30:1547-1556.
Albanese A, Barnes MP, Bhatia KP, Fernandez-Alvarez E, Filippini G,
Gasser T, Krauss JK, Newton A, Rektor I, Savoiardo M, Valls-Solé J. A
systematic review on the diagnosis and treatment of primary (idiopathic)
dystonia and dystonia plus syndromes: report of an EFNS/MDS-ES Task
Force. Eur J Neurol 2006;13:433–444.
Nandagopal R, Moorthy SGK. Dramatic levodopa responsiveness of
dystonia in a sporadic case of spinocerebellar ataxia type 3. Postgrad
Med J 2004;80:363–365.

52
Burke RE, Fahn S, Marsden CD, Bressman SB, Moskowitz C, Friedman
J. Validity and reliability of a rating scale for the primary torsion
dystonias. Neurology 1985;35:73-77.
Schmitz-Hübsch T, du Montcel ST, Baliko L, et al. Scale for the
assessment and rating of ataxia: development of a new clinical scale.
Neurology 2006;66:1717-1720.
Braga-Neto P, Godeiro-Junior C, Dutra LA, Pedroso JL, Barsottini OG.
Translation and validation into Brazilian version of the Scale of the
Assessment and Rating of Ataxia (SARA). Arq Neuropsiquiatr.
2010;68:228-230.
Ferreira KA, de Andrade DC, Teixeira MJ. Development and validation of
a Brazilian version of the short-form McGill pain questionnaire (SF-MPQ).
Pain Manag Nurs 2013;14:210-219.
Wilder-Smith E, Tan EK, Law HY, Zhao Y, Ng I, Wong MC.
Spinocerebellar ataxia type 3 presenting as an L-DOPA responsive
dystonia phenotype in a Chinese family. J Neurol Sci 2003;213: 25-28.
Fasano A, Bove F, Lang AE. The treatment of dystonic tremor: a
systematic review. J Neurol Neurosurg Psychiatry 2014;85:759-769.
Louis ED, Lee P, Quinn L, Marder K. Dystonia in Huntington's disease:
prevalence and clinical characteristics. Mov Disord 1999; 14:95-101.
B. Fischl. FreeSurfer. NeuroImage 2012;62:774-781.
Bauer CM, Cabral HJ, Killiany RJ. It is unclear if adjusting cortical
thickness for changes in gray/white matter intensity ratio improves
discrimination between normal aging, MCI, and AD. Alzheimer’s Disease
Neuroimaging Initiative. Brain Imaging Behav 2014;8:133-140.

53
Zhang Y, Zhang J, Xu J, Wu X, Zhang Y, Feng H, Wang J, Jiang T.
Cortical gyrification reductions and subcortical atrophy in Parkinson's
disease. Mov Disord 2014;29:122-126.
Schuster C, Kasper E, Machts J, Bittner D, Kaufmann J, Benecke R,
Teipel S, Vielhaber S, Prudlo J. Focal thinning of the motor cortex mirrors
clinical features of amyotrophic lateral sclerosis and their phenotypes: a
neuroimaging study. J Neurol 2013;260:2856-2864.
Desikan RS, Segonne F, Fischl B, et al. An automated labeling system
for subdividing the human cerebral cortex on mri scans into gyral based
regions of interest. NeuroImage 2006; 31:968-980.
Kingsley PB. Introduction to Diffusion Tensor Imaging Mathematics: Part
I. tensors, rotations and eigenvectors. Concepts Magn Reson
2006;28A:101-122.
Ameis SH, Fan J, Rockel C, Voineskos AN, Lobaugh NJ, Soorya L,
Wang AT, Hollander E, Anagnostou E. Impaired structural connectivity of
socio-emotional circuits in autism spectrum disorders: a diffusion tensor
imaging study. PLoS One 2011;6:e28044.
Beckmann CF, Jenkinson M, Woolrich MW, Behrens TE, Flitney DE,
Devlin JT, Smith SM. Applying FSL to the FIAC data: model-based and
model-free analysis of voice and sentence repetition priming. Hum Brain
Mapp 2006;27:380-391.
Hosseinbor AP, Chung MK, Wu YC, Fleming JO, Field
AS, Alexander AL. Extracting quantitative measures from EAP: a small
clinical study using BFOR. Med Image Comput Comput Assist
Interv 2012;15:280-287.
Comella C, Bhatia K. An international survey of patients with cervical
dystonia. J. Neurol 2015;262:837–848.

54
Kutvonen O, Dastidar P, Nurmikko T. Pain in spasmodic torticollis. Pain
1997;69:279–286.
Barbanti P., Fabbrini G., Pauletti C., Defazio G., Cruccu G., Berardelli A.
Headache in cranial and cervical dystonia. Neurology 2005;64:1308–
1309.
Camargo CH, Cattai L, Teive HA. Pain Relief in Cervical Dystonia with
Botulinum Toxin Treatment. Toxins 2015;7:2321-2335.
Jankovic J, Ford J. Blepharospasm and orofacial-cervical dystonia:
clinical and pharmacological findings in 100 patients. Ann Neurol
1983;13:402–411.
Jinnah HA, Factor SA. Diagnosis and treatment of dystonia. Neurol
Clin 2015;33:77-100.
Karimi M, Perlmutter JS.. The role of dopamine and dopaminergic
pathways in dystonia: insights from neuroimaging. Tremor Other
Hyperkinet Mov 2015;5:280.
Stoessl AJ, Lehericy S, Strafella AP. Imaging insights into basal ganglia
function, Parkinson's disease, and dystonia. Lancet 2014;384:532-544.
Alongi P, Iaccarino L, Perani D. PET Neuroimaging: Insights
on Dystonia and Tourette Syndrome and Potential Applications. Front
Neurol 2014;5:183.
Albanese A, Abbruzzese G, Dressler D, et al. Practical guidance for CD
management involving treatment of botulinum toxin: a consensus
statement. J Neurol 2015;262:2201-2213.
Tuite PJ, Lang AE. Severe and prolonged dysphagia complicating
botulinum toxin A injections for dystonia in Machado–Joseph disease.
Neurology 1996; 46: 846.

55
Kessler KR, Skutta M, Benecke R. Long-term treatment of cervical
dystonia with botulinum toxin A: Efficacy, safety, and antibody frequency.
German Dystonia Study Group. J Neurol 1999;246:265–274.
Haussermann P, Marczoch S, Klinger C, Landgrebe M, Conrad B,
Ceballos-Baumann A. Long-term follow-up of cervical dystonia patients
treated with botulinum toxin A. Mov Disord 2004;19:303–308.
França MC Jr, D'abreu A, Nucci A, Cendes F, Lopes-Cendes I.
Prospective study of peripheral neuropathy in Machado-Joseph disease.
Muscle Nerve 2009;40:1012-1018.
D'Abreu A, França MC Jr, Yasuda CL, Campos BA, Lopes-Cendes
I, Cendes F. Neocortical atrophy in Machado-Joseph disease: a
longitudinal neuroimaging study. J Neuroimaging 2012;22:285-291.
Guimarães RP, D'Abreu A, Yasuda CL, França MC Jr, Silva
BH, Cappabianco FA, Bergo FP, Lopes-Cendes IT, Cendes F. A
multimodal evaluation of microstructural white matter damage in
spinocerebellar ataxia type 3. Mov Disord 2013;28:1125-1132.
de Rezende TJ, D'Abreu A, Guimarães RP, Lopes TM, Lopes-Cendes
I, Cendes F, Castellano G, França MC Jr. Cerebral cortex involvement in
Machado-Joseph disease. Eur J Neurol 2015;22:277-83, e23-4.
Neumann WJ, Jha A, Bock A, et al. Cortico-pallidal oscillatory
connectivity in patients with dystonia. Brain 2015;138:1894-1906.
Presseau C, Jodoin PM, Houde JC, Descoteaux M. A new compression
format for fiber tracking datasets. Neuroimage. 2015;109:73-83.
D'Abreu A, França MC Jr, Yasuda CL, Souza MS, Lopes-Cendes
I, Cendes F. Thalamic volume and dystonia in Machado-Joseph disease.
J Neuroimaging 2011;21:e91-3.

56
Lehericy S, Grand S, Pollak P, et al. Clinical characteristics and
topography of lesions in movement disorders due to thalamic lesions.
Neurology 2001;57:1055-1066.
Ceballos-Baumann AO, Passingham RE, Warner T, Playford
ED, Marsden CD, Brooks DJ. Overactive prefrontal and underactive
motor cortical areas in idiopathic dystonia. Ann Neurol 1995;37:363-372.
Butterworth S, Francis S, Kelly E, McGlone F, Bowtell R, Sawle GV,
Abnormal cortical sensory activation in dystonia: an fMRI study. Mov
Disord 2003;18:673-682.
Piccinin CC, Piovesana LG, Santos MC, et al. Diffuse decreased gray
matter in patients with idiopathic craniocervical dystonia: a voxel-based
morphometry study. Front Neurol 2015;5:283.

57
ANEXOS
ANEXO I: Parecer Consubstanciado do Comitê de Ética
PARECER CONSUBSTANCIADO DO CEP
DADOS DO PROJETO DE PESQUISA
Título da Pesquisa:DISTONIA NA DOENÇA DE MACHADO JOSEPH:
ASPECTOS CLINICOS, DE NEUROIMAGEM, ELETROFISIOLÓGICOS E
TERAPÊUTICOS
Pesquisador: MARCELO BURLAMARQUE NUNES
Área Temática: Versão: 2
CAAE: 05052912.3.0000.5404
Instituição Proponente: Hospital de Clínicas - UNICAMP
DADOS DO PARECER
Número do Parecer: 157.822
Data da Relatoria: 05/12/2012
Apresentação do Projeto:
O pesquisador esclarece que, dentre as várias manifestações clínicas da ataxia
(falta de coordenação muscular), algumas tem origem genética. Dentre essas
manifestações, de origem hereditária, a SCA3 (ataxia espinocerebelar tipo 3,
ou doença de Machado-Joseph, DMJ) é a mais comum. Referente às
manifestações clínicas desses últimos, os portadores de SCA3/DMJ

58
apresentam com frequência a manifestação de sinais extrapiramidais, e
notadamente uma frequência elevada da manifestação de distonias (definidas
como um distúrbio de movimento caracterizado por movimentos anormais
sustentados do tipo torsional, comumente debilitantes e frequentemente
acompanhados de dor).Pacientes portadores de distonias ocasionadas por
outros motivos encontram como opção de tratamento a levedopa (fármaco
utilizado em síndromes parkinsonianas) e/ou injeções de toxina botulínica nos
grupos musculares acometidos). No entanto, dado a relativa raridade da
SCA3/DMJ, os relatos de pacientes com SCA3/DMJ que tiveram resposta
excelente à terapia com levodopa, quando no caso da manifestação de
distonias, são escassos. No ambulatório de neurogenética doHC-
UNICAMP alguns pacientes foram identificados como portadores da
SCA3/DMJ, e é intenção do projeto avaliar a reposta desses pacientes à
terapia com levodopa e com toxina botulínica. Outros pacientes do mesmo
ambulátorio, portadores de SCA3/DMJ mas que não apresentem diagnóstico
clínico de distonias, assim como pessoas sem manifestações de alterações
neurológicas, serão utilizadas como 'controle'. Pelo projeto, os voluntários
selecionados do Ambulatório de Neurogenética serão avaliados clinicamente
para determinar o padrão e intensidade da distonia, bem como a resposta
terapêutica a levodopa e a toxina botulínica. Em seguida, serão submetidos a
estudo de ressonância magnética encefálica e estudo neurofisiológico dos
reflexos de tronco ("blink reflex") com objetivo de determinar o substrato
estrutural e funcional da distonia. Os dados clínicos, de imagem e
neurofisiológicos dos pacientes distônicos com SCA3/DMJ serão comparados
com pacientes com SCA3/DMJ sem distonia e com controles saudáveis. Como

59
objetivo, pretende-se obter um melhor entendimento dos mecanismos da
doença, em particular da correlação entre locais de lesão, genótipo e diferentes
fenótipos presentes. Exames neurológicos propostos aos voluntários: aquisição
de imagens de ressonância magnética (sequências T1 volumétrica e T2 multi-
echodo crânio), e estudo neurofisiológico (eletroneuromiografia na face),
específico para os portadores de SCA3/DMJ, a ser executado no setor de
eletrofisiologia do HC-UNICAMP. Número de sujeitos da pesquisa: 60 (20
SCA3/DMJ com manifestação de distonia, SCA3/DMJ sem distonia e 20
controles saudáveis). Critérios de exclusão: Pacientes com contra-
indicação para realização de estudo por imagem ou eletrofisiológico.
Objetivo da Pesquisa:
Descrever a distonia em seus múltiplos aspectos - clínico, eletrofisiológico, de
imagem e terapêutico - nos pacientes com SCA3/DMJ.
Avaliação dos Riscos e Benefícios:
Os pesquisadores esclarecem que as avaliações clínicas não implicam em
nenhum risco aos sujeitos de pesquisa. Quanto da realização da ressonância
magnética os principais incômodos serão barulho intermitente durante a
aquisição das imagens e o pequeno espaço no qual o paciente é colocado, o
que pode ser desconfortável para indivíduos claustrofóbicos. A
eletroneuromiografia traz alguns desconfortos inerentes ao procedimento,
incluindo dor. Em relação aos benefícios diretos aos sujeitos de pesquisa, os
resultados eventualmente obtidos com esse estudo podem auxiliar no manejo
clínico da distonia relacionada a doença de Machado Joseph.

60
Comentários e Considerações sobre a Pesquisa:
Projeto apresentado pelo Dr. Marcelo Burlamarque Nunes para a obtenção do
título de Doutor em Fisiopatologia Médica (orientador: Dr. Marcondes
Cavalcante França Junior).
Considerações sobre os Termos de apresentação obrigatória:
Folha de rosto assinada pelo pesquisador responsável e pelo responsável da
instituição proponente (superintendente do HC/Unicamp). Projeto de pesquisa
gerado pela Plataforma Brasil com todos os itens adequadamente preenchidos,
incluindo cronograma e orçamento, sendo ainda complementado por protocolo
do estudo conforme orientação do CEP. TCLE detalhado e reformulado após
pendências na avaliação inicial pelo CEP.
Recomendações:
1.Lembramos que o TCLE deve ser elaborado em duas vias, sendo uma retida
pelo sujeito da pesquisa ou por seu representante legal e uma arquivada pelo
pesquisador (resolução 196/96 CNS/MS, artigo IV.2 ).
2.Se o TCLE tiver mais de uma página, o sujeito de pesquisa ou seu
representante, quando for o caso, e o pesquisador responsável deverão
rubricar todas as folhas desse documento, apondo suas assinaturas na última
página do referido termo (Carta Circular nº. 003/2011/CONEP/CNS).

61
Conclusões ou Pendências e Lista de Inadequações:
As readequações no TCLE, anteriormente sugeridas, foram implementadas.
Aprovado após resposta a pendências.
Situação do Parecer:
Aprovado
Necessita Apreciação da CONEP:
Não
Considerações Finais a critério do CEP:
CAMPINAS, 29 de Novembro de 2012
Assinador por: Carlos Eduardo Steiner
(Coordenador)

62
ANEXO II: Escala de Burke-Marsden-Fahn
Região Fatores
desencadeantes
Gravidade peso Escore
Olhos 0. Sem distonia
1. Distonia somente
com ação
específica
2. Distonia com
diversas ações
3. Distonia de ação de
parte remota do
corpo ou
intermitente no
repouso
4. Distonia presente
no repouso
0. Sem distonia
1. Leve. Piscamento
ocasional.
2. Leve. Piscamentos
freqüentes, sem
espasmos prolongados
de fechamento ocular.
3. Moderado.
Fechamento ocular
prolongado, mas olhos
abertos a maior parte
do tempo.
4. Severo. Olhos
fechados por pelo
menos 30% o tempo.
0.5

63
Boca 0. Sem distonia
1. Distonia somente
com ação
específica
2. Distonia com
diversas ações
3. Distonia de ação de
parte remota do
corpo ou
intermitente no
repouso
4. Distonia presente
no repouso
0. Sem distonia
1. Leve. Movimentos de
língua e mandíbulas
ocasionais.
2. Leve. Movimento
presente menos de
50% do tempo.
3. Movimentos ou
contrações distônicas
moderadas presentes a
maior parte do tempo.
4. Movimentos ou
contrações distônicas
severas presentes a
maior parte do tempo.
0.5
Fala e
deglutiç
ão
1. Ocasional, uma ou
ambas
2. Uma destas
freqüente
3. Uma desta
freqüente e outra
ocasional
4. Ambas freqüentes
0. Normal
1. Leve, fala fácil de
entender ou engasgos
ocasionais.
2. Alguma dificuldade
para entender fala ou
engasgos freqüentes
3. Dificuldade importante
para entender fala ou
inabilidade de engolir
comidas sólidas
4. Anartria, ou dificuldade
grave para engolir
comidas pastosas e
líquidas.
1
Pescoç
o
0. Sem distonia
1. Distonia somente
com ação
0. Sem distonia
1. Leve. Repuxo
ocasional.
0.5

64
específica
2. Distonia com
diversas ações
3. Distonia de ação de
parte remota do
corpo ou
intermitente no
repouso
4. Distonia presente
no repouso
2. Torcicolo óbvio, porém
leve.
3. Repuxo moderado.
4. Repuxo severo.
Braço D 0. Sem distonia
1. Distonia somente
com ação
específica
2. Distonia com
diversas ações
3. Distonia de ação de
parte remota do
corpo ou
intermitente no
repouso
4. Distonia presente
no repouso
0. Sem distonia
1. Distonia leve.
Clinicamente
insignificante.
2. Leve, distonia óbvia
porém não
incapacitante.
3. Moderada, consegue
segurar, alguma
função manual.
4. Grave. Sem função
manual.
1
Braço E 0. Sem distonia
1. Distonia somente
com ação
específica
2. Distonia com
diversas ações
3. Distonia de ação de
parte remota do
corpo ou
intermitente no
0. Sem distonia
1. Distonia leve.
Clinicamente
insignificante.
2. Leve, distonia óbvia
porém não
incapacitante.
3. Moderada, consegue
segurar, alguma
função manual.
1

65
repouso
4. Distonia presente
no repouso
4. Grave. Sem função
manual.
Perna D 0. Sem distonia
1. Distonia somente
com ação
específica
2. Distonia com
diversas ações
3. Distonia de ação de
parte remota do
corpo ou
intermitente no
repouso
4. Distonia presente
no repouso
0. Sem distonia
1. Distonia leve.
Clinicamente
insignificante.
2. Distonia leve. Anda
sem assistência em
boa velocidade.
3. Distonia moderada.
Requer assistência ou
marcha gravemente
afetada.
4. Grave. Não consegue
ficar em pé, ou andar
na perna afetada.
1
Perna E 0. Sem distonia
1. Distonia somente
com ação
específica
2. Distonia com
diversas ações
3. Distonia de ação de
parte remota do
corpo ou
intermitente no
repouso
4. Distonia presente
no repouso
0. Sem distonia
1. Distonia leve.
Clinicamente
insignificante.
2. Distonia leve. Anda
sem assistência em
boa velocidade.
3. Distonia moderada.
Requer assistência ou
marcha gravemente
afetada.
4. Grave. Não consegue
ficar em pé, ou andar
na perna afetada.
1

66
Tronco 0. Sem distonia
1. Distonia somente
com ação
específica
2. Distonia com
diversas ações
3. Distonia de ação de
parte remota do
corpo ou
intermitente no
repouso
4. Distonia presente
no repouso
0. Sem distonia presente.
1. Inclinação lateral leve.
Clinicamente
insignificante.
2. Inclinação clara, mas
não interfere com
marcha e posição
supina.
3. Inclinação moderada.
Interfere na marcha e
manutenção da posição
supina.
4. Inclinação grave.
Paciente não consegue
andar ou manter-se em
pé.
1

67
ANEXO III: Scale for Assessment and Rating of Ataxia (SARA)
1) Marcha
O paciente é solicitado (1) a andar em uma distância segura paralela a uma
parede e dar uma meia-volta (meia volta para direção oposta da marcha) e (2)
andar pé-ante-pé sem apoio.
0 Normal, sem dificuldade para andar, virar-se ou andar na posição pé-
ante-pé (até um erro aceito)
1 Discretas dificuldades, somente visíveis quando anda 10 passos
consecutivos na posição pé-ante-pé
2 Claramente anormal, marcha na posição pé-ante-pé impossível com 10
ou mais passos
3 Consideravelmente cambaleante, dificuldades na meia-volta, mas ainda
sem apoio
4 Marcadamente cambaleante, necessitando de apoio intermitente da
parede
5 Gravemente cambaleante, apoio permanente com uma bengala ou apoio
leve de um braço
6 Marcha > 10 m somente possível com apoio forte (2 bengalas especiais
ou um andador ou um acompanhante)
7 Marcha < 10 m somente possível com apoio forte (2 bengalas especiais
ou um andador ou um acompanhante)
8 Incapaz de andar mesmo com apoio
Pontuação:

68
2) Postura
O paciente é solicitado a permanecer (1) na posição natural, (2) com os pés
juntos e em paralelo (dedões juntos) e (3) em pé-ante-pé (ambos os pés em
uma linha, sem espaço entre os tornozelos e os dedos). Deve-se retirar os
sapatos e olhos permanecerem abertos. Para cada condição, três tentativas
são permitidas. A melhor resposta é considerada.
0 Normal, consegue permanecer em pé na posição pé-ante-pé por > 10 s
1 Capaz de permanecer em pé com os pés juntos sem desvios, mas não
na posição de pé-ante-pé por >10 s
2 Capaz de permanecer em pé com os pés juntos por >10 s, mas somente
com desvios
3 Capaz de permanecer em pé por > 10 s sem apoio na posição natural,
mas não com os pés juntos
4 Capaz de permanecer em pé por > 10 s na posição natural somente com
apoio intermitente
5 Capaz de permanecer em pé por >10 s na posição natural somente com
apoio constante de um braço
6 Incapaz de permanecer em pé por > 10 s mesmo com apoio constante
de um braço
Pontuação:

69
3) Sentar
O paciente é solicitado a sentar na cama de exame sem apoio dos pés, olhos
abertos e braços esticados na frente.
0 Normal, sem dificuldades em sentar > 10 s
1 Discretas dificuldades, desvios leves
2 Desvios constantes, mas capaz de sentar > 10 s sem apoio
3 Capaz de sentar > 10 s somente com apoio intermitente
4 Incapaz de sentar > 10 s sem um apoio constante
Pontuação:
4) Distúrbios da fala
A fala é avaliada durante uma conversação normal
0 Normal
1 Sugestivo de alteração na fala
2 Alteração na fala, mas fácil de entender
3 Ocasionalmente palavras difíceis de entender
4 Muitas palavras difíceis de entender
5 Somente palavras isoladas compreensíveis
6 Fala initelígivel / anartria

70
Pontuação:
5) Teste de perseguição do dedo
Cada lado avaliado isoladamente
O paciente permanece confortavelmente sentado. Se necessário, é permitido o
apoio dos pés e do tronco. O examinador senta em frente do paciente e realizar
5 movimentos consecutivos inesperados e rápidos de apontar em um plano
frontal, a mais ou menos 50% do alcance do paciente. Os movimentos deverão
ter uma amplitude de 30 cm e uma freqüência de 1 movimento a cada 2
segundos. O paciente é solicitado a seguir os movimentos com o índex, o mais
preciso e rápido possível. É considerada a execução dos 3 últimos
movimentos.
0 Ausência de dismetria
1 Dismetria, não atingir ou ultrapassar o alvo<5 cm
2 Dismetria, não atingir ou ultrapassar o alvo < 15 cm
3 Dismetria, não atingir ou ultrapassar o alvo > 15 cm
4 Incapaz de realizar os 5 movimentos
Pontuação direito: Pontuação esquerdo:
Média dos dois lados (D + E /2):
6) Teste index-nariz
Cada lado avaliado isoladamente

71
O paciente permanece confortavelmente sentado. Se necessário, é permitido o
apoio dos pés e do tronco. É solicitado que o paciente aponte repetidamente
seu índex em seu nariz para o dedo do examinador, que esta a cerca de 90%
do alcance do paciente. Os movimentos são realizados a uma velocidade
moderada. A execução do movimento é graduada de acordo com a amplitude
do tremor de ação.
0 Ausência de tremor
1 Tremor com uma amplitude < 2 cm
2 Tremor com uma amplitude < 5 cm
3 Tremor com uma amplitude > 5 cm
4 Incapaz de realizar os 5 movimentos
Pontuação direito: Pontuação esquerdo:
Média dos dois lados (D + E /2):
7) Movimentos alternados e rápidos das mãos
Cada lado avaliado isoladamente
O paciente deve permanecer confortavelmente sentado. Se necessário, é
permitido o apoio dos pés e do tronco. É solicitado que o paciente realize 10
ciclos com alternação pronação e supinação em suas coxas o mais rápido e
preciso possível. O movimento é demonstrado ao paciente há
aproximadamente 10 ciclos em 7 segundos. O tempo exato para execução do
movimento deverá ser obtido.
.
0 Normal, sem irregularidades (realiza <10s)

72
1 Discretamente irregular (realiza <10s)
2 Claramente irregular, difícil de distinguir movimentos individuais ou
interrupções relevantes, mas realiza <10 s
3 Muito irregular, difícil de distinguir movimentos individuais ou
interrupções relevantes, realiza > 10s
4 Incapaz de completer 10 ciclos
Pontuação direito: Pontuação esquerdo:
Média dos dois lados (D + E /2):
8) Manobra calcanhar-joelho
Cada lado avaliado isoladamente
O paciente deita na cama de exame, sem conseguir visualizar suas pernas. É
solicitado que levante uma perna, aponte com o calcanhar no outro joelho
,deslize pela tíbia até o tornozelo e retorne a perna em repouso na cama. A
tarefa é realizada 3 vezes. O movimento de deslizamento deverá ser feito em 1
s. Se o paciente deslizar sem o contato com a tíbia em todas as três tentativas,
gradue como 4.
0 Normal
1 Discretamente anormal, contato com a tíbia mantido
2 Claramente anormal, saída da tibia mais do que 3 vezes durante 3 ciclos
3 Gravemente anormal, saída da tibia 4 ou mais vezes durante 3 ciclos
4 Incapaz de realizar a tarefa
Pontuação direito: Pontuação esquerdo:
Média dos dois lados (D + E /2):
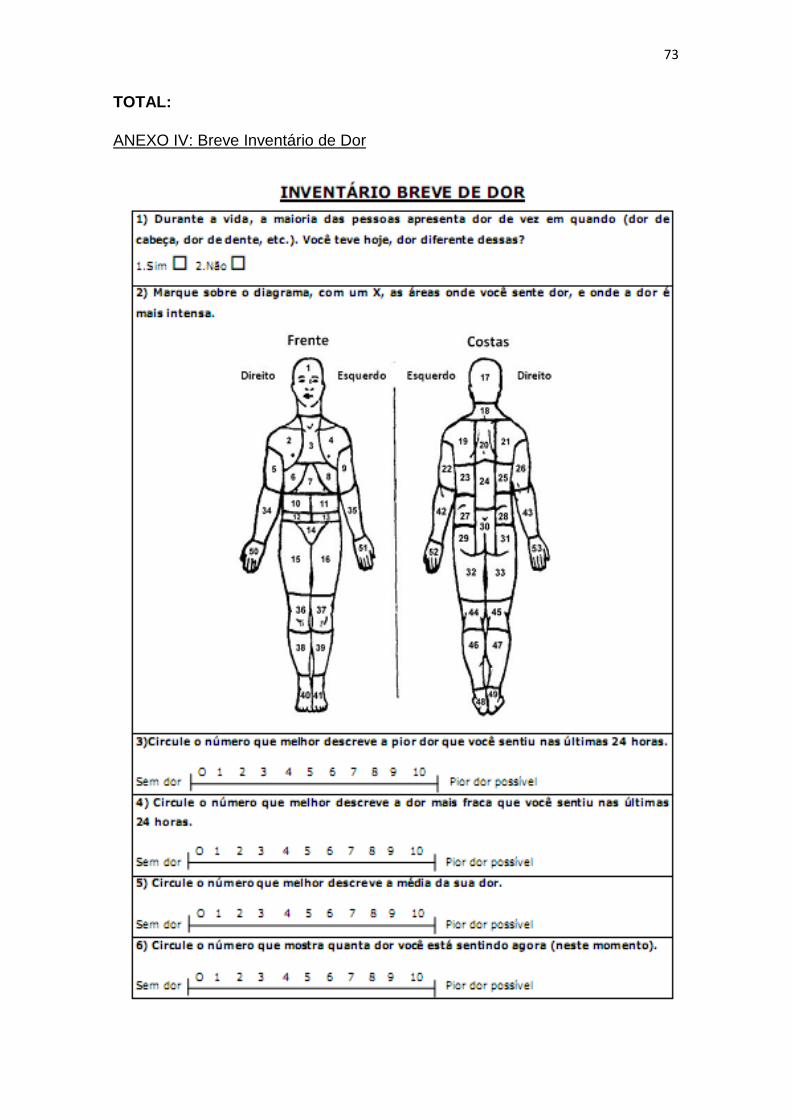
73
TOTAL:
ANEXO IV: Breve Inventário de Dor

74

75
ANEXO V: Avaliação da Distonia segundo proposta por Louis et al.
1. SEVERIDADE ( máxima amplitude do movimento ou extensão até a qual a parte do corpo envolvida desvia-se da posição normal ). (0): nenhum (1): Leve desvio da parte do corpo (2): moderado desvio (3): severo desvio com movimento máximo ou próximo ao máximo
2. CONSTÂNCIA ( porcentagem de tempo na análise em que o movimento esteve presente ).
(0): nunca presente (1): raramente presente (2): presente em menos da metade do tempo do vídeo (3): presente em mais da metade do tempo do vídeo (4): quase sempre ou sempre presente durante o vídeo
3. SUPRESSIBILIDADE ( paciente tenta suprimir em uma etapa da gravação do vídeo o movimento mas severo ):
(0): não suprimível (1): parcialmente suprimível por menos de 1 minuto (2): completamente suprimível por 1 minuto

76
ANEXO VI: Artigo publicado na “Parkinsonism and related disorders” em
Outubro/2015.

77

78

79

80

81

82









![[Achados da Semana] Looks Infantis - 04/11/2016](https://img.document.onl/doc/110x75/589ad5a91a28abc93a8b626f/achados-da-semana-looks-infantis-04112016.jpg)






![[Achados da Semana] Looks Infantis 2017](https://img.document.onl/doc/110x75/58ee942a1a28ab23428b45a9/achados-da-semana-looks-infantis-2017.jpg)







![[Achados da Semana] Looks Infantis Verão 2017](https://img.document.onl/doc/110x75/58750a941a28ab29208b6f03/achados-da-semana-looks-infantis-verao-2017.jpg)


![[Achados da Semana] Lookinhos para Meninos e Meninas](https://img.document.onl/doc/110x75/589d360f1a28abd17d8b575f/achados-da-semana-lookinhos-para-meninos-e-meninas.jpg)
![[Achados da Semana] Conjuntos para Meninos e Meninas](https://img.document.onl/doc/110x75/589a49bb1a28ab040e8b4d23/achados-da-semana-conjuntos-para-meninos-e-meninas.jpg)
