Embed Size (px)
Citation preview

Capítulo I
Introdução

2
1.1 Perspectiva ambiental
1.1.1 Água
A água representa cerca de dois terços do corpo humano e é essencial para o funcionamento
do mesmo. Por essa razão, devemos beber água todos os dias, de preferência, cerca de um
litro. Mas, nem sempre a água que sai das nossas torneiras está nas melhores condições, como
descrevem alguns artigos. O alumínio, nitratos, arsénio, hidrocarbonetos aromáticos
polinucleares (HAPs) e bactérias são apenas alguns dos problemas que têm sido encontrados
(1).
A melhoria da qualidade de vida das populações e uma maior preocupação ambiental tem
levado a que sejam desenvolvidos sistemas de recolha e tratamento de águas residuais
domésticas e industriais. Por princípio, e de acordo com o número de habitantes, estes caudais
de água poluída devem ser transportados para uma estação de tratamento centralizada. O
destino final do efluente tratado nestas estações depende da legislação em cada país, sendo
utilizados critérios por exemplo o número de habitantes, o local de descarga e o tipo de
concentração do poluente a remover (1)
.
1.2. Hidrocarbonetos aromáticos polinucleares – importância ambiental e
na saúde humana
Os poluentes orgânicos persistentes (POPs), como hidrocarbonteos aromáticos polinucleares
são de grande preocupação devido à sua persistência e efeito tóxico (2)
.
A poluição da água, por compostos orgânicos, tem causado uma preocupação crescente em
todo o mundo. Entre esses compostos, os HAPs tem recebido uma atenção considerável por
causa da sua carcinogenicidade. Estes compostos são potencialmente tóxicos e, portanto, a sua
presença deve ser monitorizada, tanto em águas ambientais como na água para consumo
humano (3)
.
Os HAPs podem ser encontrados em plantas, tanto terrestres como aquáticas, no solo, em
águas. Dezasseis destes compostos estão incluídos como poluentes prioritários pela Agência
Norte-Americana de Protecção Ambiental – EPA (4)
.
Os HAPs são formados a partir de processos de combustão incompleta a altas temperaturas.
Quando a matéria orgânica é queimada, em geral, é formada uma grande variedade de HAPs
em diferentes níveis de concentração, a complexidade das misturas de HAPs dependem das

3
fontes emissoras. Entre as inúmeras fontes, podemos citar as de origem natural, as que
ocorrem espontaneamente, como por exemplo, os incêndios florestais e as erupções
vulcânicas e as de origem antropológicas (resultantes da actividade humana), tais como:
pirólise de madeira para produção de carvão, durante a operação de transporte e refinação de
petróleo, incineração de resíduos domésticos industriais, queima de matéria orgânica de
campos e florestas, geração de energia via queima de combustíveis fósseis, emissão de gases
de veículos, particularmente a diesel, fumo de cigarro, incêndios e processos industriais, por
exemplo, a produção de alumínio e em alguns tipos de processamento durante as seguintes
etapas torrefacção, secagem, em alguns casos defumação e processo de embalamento (2) (3) (5)
(6) (7) (8).
No âmbito do Codex Alimentarius, houve a necessidade de estabelecer limites para HAPs em
alimentos, sendo este tema considerado prioritário dentro do Comité do Codex para Aditivos
Alimentares e Contaminantes (CCFAC). Em Abril de 2005, o mesmo comité recomendou que
fossem elaboradas estratégias de modo a minimizar a contaminação durante os processos de
secagem e defumação de alimentos (7) (8)
.
Desta maneira, perante estes potenciais riscos da exposição humana a HAPs é necessário
utilizar métodos de análise que sejam capazes de detectar concentrações bastante baixas
destas moléculas (3) (5) (6)
.
Os HAPs são sólidos à temperatura ambiente e altamente lipofílicos, apresentando elevada
solubilidade em solventes orgânicos e uma baixa solubilidade em água que tende a diminuir
com o aumento da massa molecular. No entanto, a solubilidade em água pode aumentar
sensivelmente na presença de outros compostos orgânicos e de detergentes aniónicos,
facilitando a passagem dos HAPs tanto para o meio ambiente como para a cadeia alimentar (9)
.
Quimicamente inertes, os HAPs no ambiente estão sujeitos a reacções de foto-decomposição e
reacções com óxidos de nitrogénio, acido nítrico, óxidos de enxofre, acido sulfúrico e radicais
hidroxilo (10)
.
1.3 Vias de exposição
1.3.1 Ar
A inalação de partículas de HAPs presentes no ar é uma importante via de exposição a estes
compostos(11) (12)
(13)
.

4
Figura nº 1.1 - HAPs, adaptado da Refª (73)
1.3.2 Alimentos
Os HAPs podem ser encontrados em diferentes grupos de alimentos e bebidas incluindo
águas, vegetais, frutas, carnes, óleos e gorduras, cereais e derivados, produtos lácteos, café,
chá, entre outros. Os HAPs já foram detectados em alimentos brutos e processados. O grau de
contaminação depende do modo como os alimentos são processados, preservados e
armazenados (7) (11) (12) (13)
.
1.3.3 Água
De acordo com a Agência Internacional para Pesquisas do Cancro (IARC) os HAPs com
poder carcinogénico que podem aparecer na água são os benzofluoranteno, benzo[a]pireno,
benzo[a]antraceno, dibenzo[a,h]antraceno e indeno[1,2,3-cd]pireno. Destes, o benzo[a]pireno
(BaP), é considerado o mais cancerígeno. Segundo a U.S. Environmental Protection Agency
(U.S. EPA), o BaP em quantidades superiores a 0.2 ppb na água potável causa problemas à
saúde. A exposição prolongada a concentrações superiores a 2 ppb causa um efeito potencial
ao desenvolvimento de cancro. Na maioria das águas e sedimentos, o BaP resiste ao ataque
por microrganismos ou substâncias químicas reactivas, podendo, contudo, evaporar ou ser
degradado por exposição à luz solar. O BaP bioconcentra-se em organismos aquáticos que
não o metaboliza, incluindo o plâncton, as ostras e alguns peixes. Devido à baixa solubilidade
e elevada afinidade para a matéria particular, os HAPs não são, normalmente, encontrados na
água em elevadas concentrações. A maior fonte de contaminação dos HAPs em água potável
é devido ao revestimento que é usado para proteger os canos da água potável da corrosão.
Esta situação faz com que os valores de HAPs na nossa água aumentem, e deste modo vemos
também os valores aumentados na nossa comida, devido à água que usamos para a
confeccionar, assim como o uso de utensílios contaminados com revestimento (11) (12)
(13)
.

5
1.3.4 Solo
Os HAPs são também encontrados à superfície dos solos. Estes compostos são absorvidos
para as folhas das plantas e depois são transferidos para o solo das florestas, mas nas áreas
metropolitanas estes valores são muito mais elevados, devido à queima de combustíveis
fósseis (11) (12)
(13)
.
1.4 Toxicocinética
1.4.1 Absorção
Na realidade, e devido ao seu carácter lipofílico, os HAPs podem ser absorvidos por todas as
vias de exposição, inalação, exposição oral e dérmica, sendo rapidamente distribuídos no
organismo (9) (11) (12)
(13)
.
1.4.2 Distribuição
Os HAPs distribuem-se em quase todos os tecidos. O armazenamento ocorre principalmente
nos rins, fígado e tecido adiposo, com pequenas quantidades no baço, glândulas adrenais e
ovários. Existe uma acumulação transiente de HAPs no tecido mamário e outros tecidos
gordos, estes são depósitos de armazenagem importantes mas devido ao rápido metabolismo
não há acumulação significativa.
Níveis detectáveis de HAPs podem ser observados, principalmente no fígado, desde minutos a
horas após a exposição (11) (12)
(13)
.
1.4.3 Metabolização
O metabolismo dos HAPs torna-os mais hidrosolúveis e assim mais rapidamente excretáveis,
mas no entanto, podem ser convertidos em metabolitos mais tóxicos e carcinogénicos. Os
HAPs são convertidos em todos os tecidos do corpo que contém gordura e envolvendo várias
vias possíveis (11) (12)
(13)
.
1.4.4 Excreção
Quando absorvidos directamente da fase gasosa, os HAPs são rapidamente metabolizados e
eliminados pelo organismo (o Benzo[a]pireno, por exemplo, é eliminado em cerca de 1 hora).

6
(mainlib) Anthracene
20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 1900
50
100
39 5063
76 89
98 126 139
152
178
Entretanto, quando estão associados a partículas respiráveis, esta eliminação é bem mais
demorada podendo levar semanas.
A eliminação destas substâncias após metabolismo hepático, são as fezes e em níveis muito
baixos, a urina (11) (12)
(13)
.
1.5 Efeitos dos HAPs na saúde
Quando os HAPs são absorvidos nas vias respiratórias podem provocar irritação, tosse
crónica e bronquite, gastrointestinal, dermatológico, imunológico, reprodutivo, nível do
sistema cardiovascular, carcinogenecidade e inflamatório (11) (12)
(13)
.
1.6 Características Físico-químicas
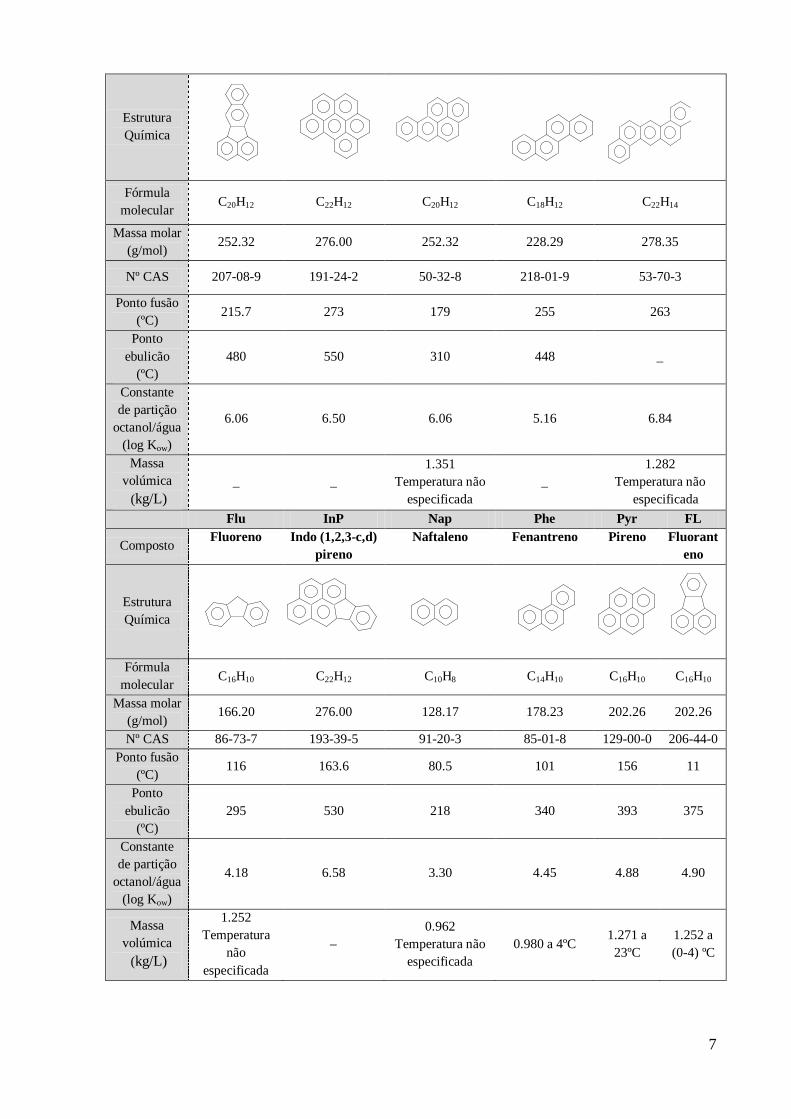
Tabela nº1.1 – Principais características físico-químicas dos HAPs estudados (14)
(15) (16)
.
AcP AcPy Ant BaA BbFL
Composto Acenafteno Acenaftileno Antraceno
1,2-
Benzoantrace
no
Benzo
(b)
fluoranteno
Estrutura
Química
Fórmula
molecular C
12H
10 C12H8 C14H10 C18H12 C20H12
Massa molar
(g/mol) 154.21 152.20 178.23 228.00 252.32
Nº CAS 83-32-9 208-96-8 120-12-7 56-55-3 205-99-2
Ponto fusão
(ºC) 95 92 216 159 168
Ponto
ebulição
(ºC)
96.2 265 340 435.0 _
Constante
de partição
octanol/água
(log Kow)
3.98 4.07 4.45 5.61 6.04
Massa
volúmica
(kg/L)
1.225 a 0ºC _ _ 1.274 a 20ºC _
BkFL BghiP BaP Chr DBA
Composto
Benzo
(k)
fluoranteno
1,12-
Benzoperileno
Benzo(a)
pireno Criseno
1,2:5,6-
Dibenzoantraceno
(mainlib) Acenaphthene
10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 1700
50
100
27 3951
63
76
87 98126
153
(mainlib) Acenaphthylene
20 30 40 50 60 70 80 90 100 110 120 130 140 150 1600
50
100
39 51
63
76
86 98 126 141
152
(mainlib) Benz[a]anthracene
20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 2400
50
100
28 63 75 88101
114
150 174200
228
(mainlib) Benz[e]acephenanthrylene
20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 2600
50
100
100
113
126
200224
252
7
(mainlib) Benzo[a]pyrene
40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 2600
50
100
51 63 74 8499
113
126
200224
252
(mainlib) Chrysene
30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 2400
50
100
39 50 63 74 88
101
114
200
228
(mainlib) Dibenz[a,h]anthracene
20 40 60 80 100 120 140 160 180 200 220 240 260 2800
50
100
32
4456 71 83 97
113125
139
149224 250
278
(mainlib) Fluorene
10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 1800
50
100
27 39 5063
6974
83
8798 115
126
139
166
(mainlib) Indeno[1,2,3-cd]pyrene
20 40 60 80 100 120 140 160 180 200 220 240 260 2800
50
100
28
111124
138
224 248
276
(mainlib) Naphthalene
20 30 40 50 60 70 80 90 100 110 120 130 1400
50
100
28 39
51 63 74 7787
102
128
(mainlib) Phenanthrene
20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 1900
50
100
39 5063
7689
98 126 139
152
178
(mainlib) Pyrene
30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 2100
50
100
88
101
150 174
202
(mainlib) Fluoranthene
20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 2100
50
100
28 50 63 75
88
101
110 150 174
202
Estrutura
Química
Fórmula
molecular C20H12 C22H12 C20H12 C18H12 C22H14
Massa molar
(g/mol) 252.32 276.00 252.32 228.29 278.35
Nº CAS 207-08-9 191-24-2 50-32-8 218-01-9 53-70-3
Ponto fusão
(ºC) 215.7 273 179 255 263
Ponto
ebulicão
(ºC)
480 550 310 448 _
Constante
de partição
octanol/água
(log Kow)
6.06 6.50 6.06 5.16 6.84
Massa
volúmica
(kg/L)
_ _
1.351
Temperatura não
especificada
_
1.282
Temperatura não
especificada
Flu InP Nap Phe Pyr FL
Composto Fluoreno Indo (1,2,3-c,d)
pireno
Naftaleno Fenantreno Pireno Fluorant
eno
Estrutura
Química
Fórmula
molecular C16H10 C22H12 C10H8 C14H10 C16H10 C16H10
Massa molar
(g/mol) 166.20 276.00 128.17 178.23 202.26 202.26
Nº CAS 86-73-7 193-39-5 91-20-3 85-01-8 129-00-0 206-44-0
Ponto fusão
(ºC) 116 163.6 80.5 101 156 11
Ponto
ebulicão
(ºC)
295 530 218 340 393 375
Constante
de partição
octanol/água
(log Kow)
4.18 6.58 3.30 4.45 4.88 4.90
Massa
volúmica
(kg/L)
1.252
Temperatura
não
especificada
0.962
Temperatura não
especificada
0.980 a 4ºC 1.271 a
23ºC
1.252 a
(0-4) ºC
(mainlib) Benzo[ghi]perylene
10 30 50 70 90 110 130 150 170 190 210 230 250 270 2900
50
100
18 111124
138
252
276
(mainlib) Benzo[k]fluoranthene
70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 2600
50
100
74 87 99
113
126
200224
252

8
Devido às suas propriedades físico-químicas, especialmente os HAPs de elevada massa molar,
dificilmente são degradados e tendem a acumular-se em diferentes compartimentos
ambientais. Comparando os HAPs com menos de 4 anéis com os que contém quatro ou mais
anéis, estes últimos são menos voláteis e menos solúveis em água (17)
.
A constante de partição octanol/água é um parâmetro que se correlaciona com a partição do
analito entre solventes orgânicos e soluções aquosa e com a sua mobilidade ambiental,
nomeadamente a sua tendência para adsorção a solos e sedimentos e a sua bioconcentração. O
log Kow está directamente correlacionado com o carácter hidrofóbico do analito e portanto
com a sua tendência para ser extraído por um solvente orgânico apolar. Dos analitos incluídos
neste trabalho, o menos hidrofóbico é, portanto o naftaleno seguido do acenafteno, enquanto o
mais hidrofóbico é o 1,2:5,6-dibenzoantraceno (136) (14) (16)
.
1.7 Método Analítico
As propriedades físicas e químicas das moléculas de HAPs permitem que estas sejam
analisadas tanto por cromatografia gasosa (GC) e espectrometria de massa (ver Figura nº1.2)
como por cromatografia líquida de alta eficiência (HPLC). É importante ressalvar que neste
tipo de análises é fundamental utilizar um método analítico com baixos limites de detecção. O
detector de espectrometria de massa tem sido muito utilizado na análise de HAPs em matrizes
ambientais, por ser um detector que para além de apresentar baixos limites de detecção,
oferece informação estrutural dos compostos (2) (3) (4)
.
A cromatografia líquida de alta eficiência é utilizada para a determinação de compostos
polares não voláteis, cuja análise em cromatografia gasosa não é possível.
A cromatografia gasosa foi inventada em 1952 por James e por Martin (18)
e o conceito chave,
que está por detrás desta técnica, é o conceito da volatilização dos compostos quando estes
são aquecidos.
O processo de volatilização ocorre a uma dada temperatura definida no injector sendo os
analitos introduzidos na coluna analítica, sob a forma gasosa, com o auxílio de um gás de
arraste (hidrogénio, hélio ou azoto). Ao entrar na coluna que está normalmente a uma
temperatura muito mais baixa que o injector, os analitos (e parte do solvente que foi
injectado) condensam, sendo retidos pela fase estacionária. O efeito conjunto do gás de arraste
(fase móvel) e do aumento da temperatura do forno vão promover a eluição dos analitos,
idealmente a diferentes velocidades, de forma a atingir a separação de todos os componentes
da mistura injectada.

9
A distinção entre os analitos depende da capacidade de distribuição que estes têm entre a fase
estacionária e a fase móvel, havendo portanto um equilíbrio de distribuição entre fases, que é
determinante para a separação cromatográfica dos solutos.
À saída da coluna os analitos são detectados num detector sensível à sua massa ou a outras
características estruturais e cuja resposta é proporcional à concentração do analito no gás de
arraste (19) (20) (21)
.
O gás de arraste é um componente essencial do sistema cromatográfico pois tem a função de
eluir os compostos na coluna através de um equilíbrio de partição (fase estacionária ↔ fase
móvel) que depende da densidade de cada gás, afectando por sua vez, a velocidade de difusão
das moléculas de soluto através da fase estacionária. Assim a escolha do gás deve depender
do tipo de coluna e do tipo de detector usado na análise cromatográfica (21)
.
As características principais de um gás de arraste devem ser a inércia e a fraca absorção pela
fase estacionária (21)
.
O gás mais usado para colunas com fases estacionárias mais finas é o hélio devido à sua baixa
densidade, conseguindo velocidades de difusão altas para a maioria dos analitos. Contudo o
azoto e o hidrogénio podem também ser utilizados, embora apresentem algumas
desvantagens: no caso do azoto, a baixa capacidade de difusão na fase estacionária quando se
utilizam colunas espessas; no caso do hidrogénio, a alta velocidade linear e baixa densidade
pode provocar pontos activos de absorção dos analitos na fase estacionária.
O injector é outro componente determinante do sistema cromatográfico, sobretudo quando se
pretendem analisar resíduos, como é o caso das determinações de HAPs (22) (23) (24) (25)
.
A escolha adequada do injector e das condições de injecção é essencial para conseguir
efectuar uma boa transferência dos analitos para a coluna.
Outros parâmetros de injecção fundamentais para optimizar a sensibilidade dos analitos
detectados e a resolução dos picos cromatográficos são a temperatura do alinhador e da
coluna, a pressão na cabeça da coluna, o solvente da amostra, o volume de injecção e a
velocidade de injecção e ainda o volume do alinhador (23)
.
Também a temperatura do forno onde está inserida a coluna cromatográfica, deve ser
programada em modo de gradiente ou isocrático, de modo a que os componentes de uma
mistura sejam devidamente separados e com picos bem resolvidos, considerando a natureza
das moléculas de soluto e da complexidade da amostra. Normalmente o modo de temperatura
mais escolhido é o da temperatura programada porque reduz o tempo de análise e melhora a
separação e consequente detecção das moléculas de soluto, que possuem tempos de retenção
muito próximos e que não são devidamente separados no modo de temperatura isocrática.

10
Outras das vantagens do uso deste modo, é o facto de haver uma limpeza gradual da coluna
com o aumento linear da temperatura (22) (23) (24) (25)
.
A coluna é o componente essencial de qualquer técnica separativa. Existem assim dois tipos
de colunas dependendo da natureza da fase estacionária:
Colunas de empacotamento
Colunas capilares
As mais usadas são as colunas capilares devido à sua capacidade de resolução e alta eficiência
separativa. Todavia, a eficiência das colunas capilares depende de vários parâmetros como
sendo o gás de arraste, o comprimento e diâmetros interno da coluna e da espessura de filme
da fase estacionária (23)
.
A escolha das características da coluna tem consequências decisivas no tipo de separação
cromatográfica que se poderá efectuar (23)
.
As fases estacionárias mais comuns usadas para análise de hidrocarbonetos aromáticos são
compostas por 5% fenil/ e 95% dimetilsiloxano (DB-5).
A detecção em cromatografia gasosa, é um parâmetro fundamental como em qualquer outra
técnica analítica, pois condiciona directamente a eficiência e a especificidade da análise (21),
(22), (24).
1.8 Legislação
A Directiva 98/83/CE, de 3 de Novembro de 1998, relativa à qualidade da água destinada ao
consumo humano, transposto para a legislação nacional, com o Decreto-Lei n.º 243/2001 de 5
de Setembro tem por objectivo proteger a saúde humana dos efeitos nocivos resultantes de
qualquer contaminação da água destinada ao consumo humano, assegurando a sua salubridade
e limpeza (26) (27)
.
Directiva 2006/11/CE do Parlamento Europeu e do Conselho de 15 de Fevereiro de 2006,
relativa à poluição causada por determinadas substâncias perigosas lançadas no meio aquático
da Comunidade, que inclui os hidrocarbonetos, cuja escolha é feita principalmente com base
na sua toxicidade, persistência e bioacumulação, com excepção dos que são biologicamente
inofensivos ou que se transformam rapidamente em substâncias biologicamente inofensivos
(28).
O Decreto-Lei n.º 306/2007, de 27 de Agosto, apresenta um programa de controlo da
qualidade da água para consumo humano, mostrando qual deve ser a frequência de
amostragem e quais os valores paramétricos que devem ser mantidos, evitando deste modo,

11
situações que comportem riscos para a saúde humana (tabela nº1.2). Refere que a realização
de análises deve ser feita, preferencialmente em laboratórios de ensaios credenciados (29)
.
É da responsabilidade da autoridade competente, isto é, a Entidade Reguladora dos Serviços
de Águas e Resíduos deve assegurar o controlo analítico da qualidade das águas destinadas a
consumo humano, permitindo por sua vez, que este decreto-lei seja cumprido (29)
.
Tabela nº 1.2- Apresenta o valor guia que deve ser considerado como valor paramétrico para
os HAPs (29)
.
Parâmetro Valor
paramétrico Unidade Notas
Hidrocarbonetos
aromáticos policíclicos
0,10 g/L
Soma das concentrações dos seguintes
compostos: Benzo[b] fluorateno;
Benzo[k] fluoranteno; Benzo[ghi] perileno;
Indeno [1,2,3-cd] pireno.
Benzo(a) pireno 0,010 g/L _
Decreto-Lei n.º 351/2007, visa estabelecer um valor alvo para as concentrações de
benzo(a)pireno no ambiente de modo a evitar, prevenir ou limitar os efeitos nocivos dos
HAPs na saúde humana e no ambiente na sua globalidade (30)
.
O Decreto-Lei 103/2010 de 24 de Setembro, transpôs a Directiva 2008/105/CE do Parlamento
Europeu, de 16 Dezembro 2008, estabelece um quadro de acção comunitária no domínio da
política da água, visando a protecção das águas de superfície interiores, das águas de
transição, das águas costeiras e das águas subterrâneas que previne a degradação dos
ecossistemas aquáticos europeus, promove um consumo de água sustentável, assegura a
redução das descargas de poluentes para os ecossistemas e assim contribui para reduzir a
poluição de águas superficiais e subterrâneas (31)
(tabela nº1.3).

12
Tabela nº 1.3 - Refere-se à concentração máxima admissível para os HAPs. (MA média
anual; CMA concentração máxima admissível e Unidade: [μg/L]) (31)
.
Nome da substância
NQA-MA (1)
Águas de
superfície
interiores
(2)
NQA-MA (1)
Outras
águas de
superfície
NQA-CMA (3)
Águas de
superfície
interiores
(2)
NQA-CMA (3)
Outras
águas de
superfície
Identificada
como
substância
perigosa
prioritária
Antraceno 0,1 0,1 0,4 0,4 X
Benzo(a)pireno 0,05 0,05 0,1 0,1 X
Benzo(b)fluoranteno Σ = 0,03 Σ = 0,03
N.A N.A X
Benzo(k)fluoranteno N.A N.A X
Benzo(g,h,i)-perileno Σ = 0,002 Σ = 0,002
N.A N.A X
Indeno(1,2,3-cd)-pireno N.A N.A X
(1) Este parâmetro constitui a NQA expressa em valor médio anual (NQA-MA). Salvo indicação em contrário, aplica-se à concentração total de todos os isómeros. (2) As águas de superfície interiores compreendem os rios, lagos e todas as massas de água artificiais ou fortemente modificadas com eles relacionadas.
(3) Este parâmetro constitui a NQA expressa em concentração máxima admissível (NQA-CMA). Quando se indica «não aplicável» nas colunas, significa que se considera que os valores NQA-MA protegem contra picos de poluição de curta duração em descargas contínuas, visto que são significativamente inferiores aos valores determinados com base na toxicidade aguda. (4) N.A.- Não aplicável.
A Agência de Protecção Ambiental (EPA) e a Organização Mundial de Saúde (OMS)
recomendam ensaios de rotina ao benzo[a]pireno. Segundo a EPA o nível de concentração
máximo não deve ser superior a 200 ng/L, enquanto a OMS estabeleceu que o valor máximo
admissível é de 700 ng/L (32)
(33)
.
Embora mais de 100 HAPs já foram caracterizados na natureza, somente dezasseis HAPs têm
sido seleccionados pela United States Environmental Protection Agency (USEPA) e dezassete
HAPs pela National Institute for Occupational Safety and Health (NIOSH), para serem
monitorizados rotineiramente para fins reguladores. São eles: naftaleno, acenaftileno,
acenafteno, fluoreno, fenantreno, antraceno, fluoranteno, pireno, benzo[a]antraceno, criseno,
benzo[e]pireno, benzo[b]fluoranteno, benzo[k]fluoranteno, benzo[a]pireno,
dibenzo[a,h]antraceno, benzo[g,h,i]perileno e indeno[1,2,3-cd]pireno. Este facto deve-se à sua
elevada toxicidade carcinogénica e mutagénica e sua persistência no ambiente (34) (35)
.
Na Europa apresenta-se uma lista de seis HAPs (fluoranteno, benzo[a]pireno,
benzo[b]fluoranteno, benzo[k]fluoranteno, benzo[g,h,i]perileno e indeno(1,2,3-cd pireno)
para controlo em águas superficiais usada para fins de potabilidade (17)
.

13
1.9 Determinação de HAPs em águas
1.9.1 Métodos de extracção
Os HAPs são compostos não polares e que têm por isso uma solubilidade em água muito
baixa. No entanto a sua elevada toxicidade faz com que mesmo em concentrações muito
reduzidas estes compostos constituam um factor de risco ambiental grave. Assim um passo
crítico da determinação de HAPs em águas é a sua extracção e concentração.
Esta operação era efectuada por extracção líquido-líquido (LLE), utilizando grandes volumes
de água de forma a atingir os limites de detecção requeridos na legislação. Para extrair estes
grandes volumes de água há que utilizar também grandes volumes de solvente orgânico
obtendo-se assim os HAPs sob a forma de soluções muito diluídas, que devem ser
concentradas para atingir os limiares de detecção do equipamento analítico. Desta forma este
processo de extracção líquido-líquido envolve custos elevados em solventes e tempo de
execução, além de poder apresentar recuperações baixas para os HAPs mais voláteis que
podem ser facilmente perdidos durante a evaporação do solvente. Assim nos últimos anos
desenvolveram-se diferentes técnicas alternativas que visam simplificar o procedimento de
extracção, torná-lo mais eficiente e reduzir o consumo de solventes orgânicos (36) (37)
:
Extracção em fase sólida (SPE)
Microextracção em fase sólida (SPME)
Extracção adsorptiva em barra de agitação (SBSE)
Microextracção líquido-líquido dispersiva (DLLME)
A microextracção em fase sólida, utilizando uma barra magnética revestida com a fase
estacionária polimérica (designada extracção adsorptiva em barra de agitação, SBSE), surgiu
como alternativa à SPME.
As técnicas de microextracção em fase líquida (LPME) permitem extrair compostos orgânicos
a partir de matrizes aquosas recorrendo a volumes de solventes orgânicos na gama dos
microlitros, com tempos de extracção curtos e com uma boa eficiência de extracção. Os
volumes de amostra utilizados são tipicamente baixos (5 a 10 mL) e não requerem a utilização
de consumíveis caros como as colunas de SPE, as fibras de SPME ou as barras de SBSE.
Apresentam-se de seguida as descrições sucintas de cada um destes métodos de extracção e
exemplos da sua aplicação à determinação de HAPs em águas.
1.9.1.1 Extracção líquido-líquido (LLE)
A extracção líquido-líquido é o processo clássico de extracção de compostos orgânicos a
partir de matrizes aquosas, sendo muitas vezes utilizado como termo de comparação com

14
técnicas mais recentes (38) (39)
. É uma técnica utilizada tradicionalmente em áreas como a
química, petroquímica, farmacêutica e agricultura (40)
.
O princípio subjacente à LLE é a partilha dos analitos entre a fase aquosa e uma fase orgânica
imiscível na qual os analitos se deverão dissolver preferencialmente. O parâmetro que
descreve a partilha de um analito A entre as duas fases em equilíbrio é a constante de
distribuição ou de partição (KD) que pode ser calculada pela seguinte fórmula (40) (41)
:
onde [A]org é a concentração do analito na fase orgânica e [A]aq é a concentração do analito na
fase aquosa. A extensão da extracção é avaliada pela percentagem de recuperação (R%) que
pode ser definida pela seguinte fórmula:
onde ne é o número de moles ou a massa de analito presentes na fase orgânica, após a
extracção e ni é o número de moles ou a massa de analito existentes na fase aquosa, antes da
extracção. Normalmente são necessárias mais de duas extracções descontínuas sucessivas
para se obterem percentagens de recuperação superiores a 99%. Quando os valores de KD são
muito baixos pode optar-se por extracção líquido-líquido contínua de forma a obter uma
percentagem de recuperação elevada sem ter que efectuar um grande número de extracções
líquido-líquido sucessivas.
Apesar de a LLE ser o método de referência utilizado em inúmeras normas internacionais, é
um método que como já foi referido, requer a utilização de elevadas quantidades de solventes
orgânicos (geralmente mais do que 100 mL), envolve vários passos e um período de tempo
considerável sobretudo nas etapas de concentração da amostra que têm que ser efectuadas por
procedimentos suaves (evaporadores Kuderna-Danish ou equivalentes, evaporação em
corrente de azoto), para evitar perdas dos solutos mais voláteis.
1.9.1.2 Extracção em fase sólida (SPE)
A extracção em fase sólida (com discos de extracção ou com colunas de extracção) consiste
na adsorção selectiva dos analitos a extrair numa fase estacionária sólida, denominada
adsorvente. A fase aquosa (ou outra amostra líquida) é filtrada sob vácuo, através do disco ou
coluna de extracção. Os analitos são retidos pelo adsorvente e posteriormente eluídos com
alguns mililitros de um solvente orgânico adequado. O extracto é finalmente concentrado até
um volume adequado à sua análise (41)
.

15
Genericamente, a SPE é um processo de retenção dos analitos de interesse de uma amostra
sobre um suporte sólido, designado por enchimento e posterior eluição através da passagem
de um líquido eluente, através do mesmo. A retenção dos diversos analitos depende da
afinidade que cada um apresenta para a fase estacionária (42)
.
A extração em fase sólida tem as seguintes vantagens: menor consumo de solventes orgânicos
em comparação com extracção LLE, elevada recuperação, elevada reprodutibilidade em
relação a outras técnicas de extracção e sem formação de espuma durante a formação de
emulsão. Apesar de todas as vantagens acima referido, a SPE é uma técnica demorada e
relativamente cara. Os cartuchos de SPE são fabricados para
uma única utilização (43) (44)
.
Esta técnica é mais rápida que a LLE, utiliza menores volumes de solventes orgânicos e pode
ser automatizada reduzindo o tempo dispendido pelo utilizador (41) (45)
.
Existem diversas fases estacionárias (sílica, alumina, florisil, carbono e resinas) cuja selecção
depende da natureza dos analitos a analisar e às quais correspondem diferentes tipos de
interacção analito-fase estacionária (41) (45) (36)
.
a) Fase Reversa envolve interacções de analitos presentes em matrizes polares com uma
fase estacionária apolar. Como exemplos tem-se as fases constituídas por cadeias C8 e C18
ligadas a grupos silanol de sílica.
b) Fase Normal envolve interacções entre uma fase estacionária polar e analitos presentes
em matrizes apolares. Os exemplos de fases polares são a sílica, a alumina ou a silica
funcionalizada com grupos polares tais como, CN, NH2 ou diol.
c) Permuta Iónica envolve interacções iónicas entre a fase estacionária (positiva ou
negativa) e os solutos carregados. Uma diversidade de resinas aniónicas ou catiónicas
podem ser usadas neste tipo de interacções.
d) Absorção envolve interacções entre analitos e materiais não modificados, ficando os
compostos de interesse retidos no adsorvente, geralmente um material poroso.

16
Figura 1.2 - Colunas de extracção em fase sólida (a) e sistema de filtração sob vácuo (b)
(Adaptado da Refª (45)).
O procedimento de SPE inicia-se pelo condicionamento da fase estacionária seguindo-se a
filtração da amostra; poderá incluir-se um passo de lavagem da coluna de extracção com um
solvente fraco, para remover impurezas, ou um passo de secagem da coluna em corrente de
azoto, para remover água; por fim efectua-se a eluição dos analitos com um solvente ou
solventes adequados (Figura 1.3).
Figura 1.3 – Etapas do processo de extracção em fase sólida (Adaptado da Refª (42)).
A utilização de novas fases estacionárias na extracção de diversos contaminantes orgânicos
tem sido uma área de inovação em SPE: os nanotubos de carbono ou um polímero de
(a)
(b)

17
metiltetradecilsiloxano (46)
são exemplos de novas fases estacionárias propostas para a
extracção de hidrocarbonetos.
A combinação da técnica de SPE-HLLME-GC-ECD foi introduzida pela primeira vez por
Assadi e os seus colaboradores para extrair e determinar os clorofenóis de matrizes altamente
salinas (47)
.
Em 2009, Liu e os seus colaboradores desenvolveram SPE-HLLME-GC-ECD para a
determinação de éteres difenis polibromados (PBDEs) em amostras de água (44)
.
Rodriguez e os seus colaboradores associaram a técnica de SPE-HLLME para a determinação
de sete fungicidas em amostras de vinho (48)
.
1.9.1.3 Microextracção em Fase Sólida (SPME)
A microextracção em fase sólida revolucionou o campo da análise de compostos orgânicos
em água ao permitir efectuar extracção e concentração dos analitos num só passo e na
ausência de solventes orgânicos (49)
. Algumas limitações iniciais da técnica, como a baixa
resistência mecânica das fibras ou alguma variabilidade entre lotes, foram sendo ultrapassadas
e hoje em dia as fibras de SPME apresentam, além da capacidade de efectuarem extracções
muito selectivas, uma boa estabilidade mecânica (fibras Stableflex) e portanto um tempo de
vida mais longo bem como boa reprodutibilidade para ensaios feitos com diferentes lotes.
A microextracção em fase sólida (SPME) permite efectuar a concentração de compostos
orgânicos voláteis ou não voláteis, expondo a fibra adsorvente à fase gasosa em equilíbrio
com a amostra líquida (HS-SPME) ou directamente à amostra (DI-SPME). Os compostos
retidos na fibra são posteriormente desadsorvidos termicamente ou por solventes adequados
no injector do GC ou do HPLC (Figura 1.4).
Figura 1.4 - Microextracção em fase sólida (Adaptado da Refª (158)).

18
A escolha da fase estacionária da fibra é uma etapa fundamental da optimização de um
processo de SPME, existindo actualmente uma gama variada de fases que incluem o
polidimetilsiloxano (PDMS), o divinilbenzeno (DVB), o carboxen (CAR) e o poliacrilato
(PA), bem como combinações destes polímeros de forma a obter as características de
adsorção adequadas a analitos com diferentes volatilidades e polaridades (50)
.
O equilíbrio de partição dos analitos entre a amostra e a fibra é descrito por uma constante de
partição K, que é definida como a razão entre a concentração de analitos na fibra e a
concentração de analitos na amostra.
A técnica de SPME pode no entanto, apresentar alguma variabilidade para resultados obtidos
ao longo do tempo da fibra pois cada vez que esta é exposta à temperatura do injector perde
algum do seu revestimento polimérico pelo que na extracção seguinte não apresentará
exactamente a mesma eficiência.
1.9.1.4 Extracção Adorptiva em Barra de Agitação (SBSE)
A extracção adsorptiva com barra de agitação magnética (SBSE) é uma técnica relativamente
recente, proposta em 1999 por P. Sandra e colaboradores (51)
.
A criação de barras magnéticas revestidas com polímeros, idênticos aos utilizados na
fabricação das fibras de SPME (Twister, Gerstel (52)
), abriu a possibilidade de efectuar
extracções selectivas, também na ausência de solvente, mas com maior capacidade de
retenção de analitos do que em SPME, devido à maior quantidade de polímero em contacto
com a amostra.
Figura 1.5 – Ilustração da barra magnética: 1- haste de aço inox; 2- revestimento de vidro;
3- fase extracção adsorptiva com fibras de SPME, onde ocorre a extracção dos analitos
(Adaptado da Refª (53)).
Após a extracção dos analitos estes podem ser desadsorvidos termicamente ou com um
solvente adequado (Figura 1.6).

19
Figura 1.6 – Desorpção dos analitos com solvente após SBSE com Twister
(Adaptado da Refª (54)).
Esta técnica tem sido aplicada à determinação de pesticidas e outros contaminantes presentes
em águas tanto em associação com GC (55)
, como com HPLC (56)
.
1.9.1.5 Microextracção em Fase Líquida (LPME)
Microextracção em fase líquida (LPME) é uma designação genérica que engloba diversas
técnicas de microextracção, cujo denominador comum é o facto de utilizarem volumes de
solvente de extracção na gama dos microlitros. Outro factor comum é o facto de não ser
necessário utilizar as fibras, colunas ou barras de extracção necessárias aos diversos métodos
de extracção em fase sólida, reduzindo assim consideravelmente os custos do processo.
É possível encontrar na literatura algumas revisões à aplicação da técnica de LPME na
determinação de HAPs (57) (58)
.
1.9.1.6 Microextracção líquido-líquido com gota flutuante (SFDME)
Numa variante das técnicas de microextracção em fase líquida, utilizam-se solventes
imiscíveis com água, menos densos que a água e com um ponto de congelação elevado (acima
de 0ºC). Alguns microlitros de solvente são adicionados à solução aquosa que contém os
analitos. A fase aquosa é submetida a uma agitação mecânica vigorosa para favorecer a
migração dos analitos através da interface. Após o período de extracção as fases em equilíbrio
são arrefecidas a 0ºC para congelar o solvente orgânico. Uma vez no estado sólido o solvente
de extracção pode ser simplesmente removido com uma pinça metálica ou outro instrumento
adequado (Figura 1.7).
Esta técnica recente descrita por Zanjani e pelos seus colaboradores (59)
tem sido utilizada para
a extracção de HAPs, metais e pesticidas organoclorados (60) (61)
.
3

20
Figura 1.7 - Microextracção líquido-líquido com gota flutuante.
Esta técnica permitiu obter resultados interessantes do ponto de vista da sensibilidade e
precisão mas o desenvolvimento de novas aplicações está condicionado pelo número limitado
de solventes com propriedades físico-químicas adequadas.
1.9.1.7 Microextracção Líquido-Líquido Dispersiva (DLLME)
A microextracção líquido-líquido dispersiva é de entre as várias técnicas de LPME aquela que
regista um maior número de aplicações. O conceito fundamental desta técnica é a mistura de
uma fase aquosa (5 a 10 mL) que contém os analitos, com alguns microlitros do solvente de
extracção (apolar, imiscível com água e tipicamente mais denso que a água), e um pequeno
volume (0.25 mL a 1 mL) do solvente de dispersão (um solvente orgânico miscível com
água). A proporção volumétrica dos três componentes desta mistura ternária deve ser
seleccionada de modo a que o sistema se situe na região de transição entre o estado
monofásico e o estado difásico. Assim existem duas fases em equilíbrio: uma fase constituída
pelo solvente de extracção apolar e que pode conter pequenas quantidades do solvente de
dispersão e uma outra fase aquosa na qual está dissolvido o solvente de dispersão. No entanto,
como as fases em equilíbrio estão próximo da transição para o estado monofásico, as suas
densidades são bastante próximas o que permite formar emulsões estáveis. A função do
solvente de dispersão é pois modificar as densidades da fase aquosa e da fase orgânica de
forma a permitir a dispersão do solvente de extracção na fase aquosa de modo eficaz e
persistente. Ao dividir o volume de solvente de extracção num grande número de pequenas
gotículas, maximiza-se a interface de contacto entre as fases o que facilita a partição dos
analitos entre as fases, acelerando o processo de extracção. Ao manter o volume de solvente
de extracção na ordem de alguns microlitros atinge-se um factor de enriquecimento muito
elevado. Assim, a técnica de DLLME permite atingir factores de enriquecimento comparáveis
ou superiores aos obtidos com as técnicas de microextracção líquido-líquido com gota

21
flutuante ou com membrana mas com um tempo de extracção muito mais reduzido e sem
necessidade de agitação pois a dispersão de solvente de extracção remove as barreiras
cinéticas à transferência de massa entre as fases. Após a extracção as fases são separadas por
centrifugação obtendo-se assim uma fase orgânica de alguns mililitros que, como é
habitualmente mais densa que a fase aquosa se designa por fase sedimentada.
Os solventes de extracção mais utilizados em microextracção líquido-líquido dispersiva são o
tetracloreto de carbono, o clorobenzeno, o tricloroetileno, o clorofórmio, o tetracloroetileno, o
tetracloroetano, o diclorometano, o 1,2-diclorobenzeno, o hexano e o 1,1,1-tricloroetano (62)
(63) (64) (65) (66) (67) (68) (69).
E os solventes de dispersão mais comuns são a acetona, o acetonitrilo, o metanol, o tert-
butilmetil éter e o tetrahidrofurano (62)- (69)
.
Na Figura 1.8 ilustram-se os passos fundamentais de um ensaio de DLLME. Os analitos são
adicionados à solução aquosa, numa determinada concentração (fortificação), em seguida
efectua-se a adição conjunta dos solventes de dispersão e de extracção à fase aquosa
(dispersão) e por fim procede-se à centrifugação para separar as fases. A fase sedimentada
pode então ser recolhida com uma microseringa.
Figura 1.8 - Microextracção líquido-líquido dispersiva (Adaptado da Refª (70)).

22
1.9.1.8 Microextracção Líquido-Líquido Dispersiva com Líquidos Iónicos (IL-
DLLME)
Os líquidos iónicos têm propriedades com interesse em vários domínios da Química: são
considerados menos poluentes para a atmosfera e menos perigosos para o utilizador, que os
solventes halogenados, pois não são tóxicos, não são inflamáveis, a sua volatilidade é quase
nula.
A técnica microextracção líquido-líquido dispersiva com líquidos iónicos para extracção de
HAPs em amostras de água foi desenvolvida pela primeira vez por M. Teresa Pena e os seus
colaboradores em 2009. A afinidade química entre o líquido iónico e os analitos permite a
extracção dos compostos da amostra de água, obtendo-se uma pré-concentração. Os limites de
quantificação obtidos em HPLC são de 0,1-0,7 ng/L. Os factores de enriquecimento obtidos
são na ordem 301-346 e a extracção dos diferentes HAPs variou entre 90,3% e 103% (71)
.
A extracção de diferentes compostos orgânicos (tais como aminas aromáticas ou pesticidas
organofosforados) (71)
a partir de amostras de água, recorrendo a IL-DLLME foi ainda
exemplificada.
O método consiste na adição dos analitos à solução aquosa, numa determinada concentração
(fortificação), depois efectua-se a adição do líquido iónico. De seguida, é feito um
aquecimento até à amostra estar homogénea, posteriormente um arrefecimento até ocorrer a
turvação, e por fim procede-se à centrifugação para separar as fases. A fase sedimentada pode
então ser recolhida com uma microseringa.
Os líquidos iónicos têm uma boa capacidade solvente em relação a uma grande variedade de
compostos orgânicos e inorgânicos, o que os torna interessantes como solventes de extracção
ou meios de reacção.
Como não são voláteis, as suas aplicações cromatográficas são sobretudo em cromatografia
líquida, apesar de recentemente ter surgido uma aplicação em cromatografia gasosa (72)
.

23
Figura 1.9 - Microextracção Líquido-Líquido Dispersiva com Líquidos Iónicos.
1.9.1.9 Microextracção líquido-líquido homogénea (HLLME)
Esta técnica surgiu para permitir a aplicação dos princípios da microextracção dispersiva a
amostras sólidas. Neste caso é efectuada uma pré-extracção dos analitos a partir da matriz
sólida onde estes se encontram, recorrendo a um solvente orgânico miscível com água. Um
pequeno volume (~1mL) do extracto obtido, com ou sem concentração, é utilizado como
solvente de dispersão num passo seguinte de DLLME. O solvente de extracção é adicionado
ao solvente de dispersão que contém os analitos, formando uma mistura homogénea que
favorece a solvatação dos analitos. A mistura é então injectada em alguns mililitros de água,
provocando a dispersão do solvente de extracção. Neste processo algumas impurezas polares
co-extraídas a partir da matriz sólida poderão dissolver-se predominantemente na fase aquosa
obtendo-se assim algum efeito de purificação do extracto (73) (74)
.
Este tipo de extracção pode ser usado em diversas matrizes (águas, solo) para a extracção de
vários grupos de compostos, entre estes encontram-se os pesticidas organofosforados, os
derivados benzénicos, os HAPs e os metais (73) (74) (75) (76)
.
Analise dos analitos
80ºC 0ºC
Centrifugação
Fortificação dos
analitos na fase
aquosa
Aquecimento Arrefecimento

24
Figura 1.10 - Microextracção Líquido-Líquido homogénea (Adaptado da Refª (74)).
1.9.1.10 Microextração líquido-líquido utilizando um agente dispersão não tóxico
(LT-DLLME)
A técnica microextracção líquido-líquido dispersiva com um agente dispersão não tóxico
utiliza solventes orgânicos menos tóxicos que a maior parte dos solventes halogenados.
Um exemplo desta aplicação é a extracção de HAPs a partir de água, por DLLME utilizando
como solvente de dispersão o 1-bromo-3-metilbutano. Este método foi desenvolvido pela
primeira vez por Mei-I Leong e os seus colaboradores em 2010, e os limites de quantificação
obtidos em GC-MS são de 0,0003 e 0,0078 μg/L. Os factores de enriquecimento obtidos são
da ordem 372-1308 e a extracção dos diferentes compostos variou entre 87 %-105% (77)
.
1.9.1.11 Técnica de Microextracção-Emulsificação coadjuvada por ultrasons
(USAEME)
Este método é uma variante de DLLME na qual se utilizam solventes orgânicos menos densos
que à água. Vários parâmetros foram optimizados: a natureza e o volume de solvente
orgânico, a temperatura, a força iónica e centrifugação.

25
Este procedimento proporciona inúmeras vantagens, tais como factores de enriquecimento
excelentes, simplicidade, estabilidade, facilidade de execução, baixo custo e consumo de
solventes orgânicos (78) (79)
.
Esta técnica visa ampliar a gama de solventes orgânicos que se podem utilizar em DLLME e
o recurso à ultrasonicação destina-se a melhorar a partição dos analitos, melhorando a mistura
das fases.
1.10 Aplicações recentes de DLLME à determinação de contaminantes
orgânicos em matrizes sólidas e líquidas
Esta técnica foi desenvolvida no ano de 2006 por Rezaee e seus colaboradores (80)
, testando
um conjunto de solventes de extracção e de dispersão para a extracção de hidrocarbonetos
poliaromáticos (HAPs) de amostras aquosas.
Desde então, têm-se multiplicado os trabalhos que exploram a aplicação desta técnica a
amostras de água ou outras matrizes aquosas como vinho, sumo de frutas, tendo-se obtido
factores de enriquecimento geralmente entre 500 a 1000 para a maior parte dos analitos
estudados.
Também foram efectuados ensaios em matrizes sólidas, mas em menor escala, pois é
necessário realizar um tratamento anterior. Exemplos de algumas matrizes sólidas são a
melancia, pepino, solo, maçã, banana, uva, ameixa e folhas de chá.
Na maioria dos ensaios recorreu se ao uso dos solventes halogenados, causando vários
problemas sendo o principal a toxicidade. No entanto a quantidade de solvente utilizado em
DLLME é muito reduzida (normalmente inferiores a 50 µL por extracção), as consequências
Figura 1.11 - Microextracção USAEME (Adaptado da Refª (78)).
Fortificação
dos analitos
na fase
aquosa
Adição do
solvente de
extracção à
fase aquosa
Adição do solvente
hidrofóbico de cor,
neste caso vermelha,
para distinguir o
solvente .Recolha
dos analitos
Centrifugação

26
ambientais da sua utilização é diferente da manipulação dos mesmos solventes em volumes
mais elevados, como é o caso por exemplo, na extracção líquido-líquido clássica.
Os produtos farmacêuticos são outro grupo importante, onde se fez ensaios recorrendo à
técnica de DLLME, como por exemplo, em antibióticos anti-inflamatórios, em anti-
depressivos e em agentes envolvidos na redução do colesterol (70)
.
A técnica DLLME também tem sido aplicada para a extracção de PCB e PBDEs. O Hu e os
seus colaboradores (81)
testaram um método para analisar os PCB em peixes e resultados
obtidos ao nível de LODs foram 0,12-0,35g/Kg. Mais tarde Lui e os seus colaboradores (82)
aplicaram o mesmo método para determinar os PBDEs em rãs, caracóis, tendo obtidos limites
de determinação ligeiramente maiores 2,4-4,9g/Kg.
A técnica de DLLME também foi usada para determinar antioxidantes por Farajzadeh e os
seus colaboradores (83)
.
Na tabela seguinte temos algumas aplicações recentes, recorrendo ao sistema de extracção
DLLME em pesticidas, produtos farmacêuticos, PCB e PBDEs e outras aplicações.
Tabela 1.4 – Aplicações utilizando o sistema de DLLME (Adaptado da Refª (70)).
Analitos Matriz Solvente de
extracção
Agente de
dispersão
Método
Analítico LOD
Recuperação
(%)
PESTICIDAS
Pesticidas
Organofosforados (84)
Melancia e
Pepino
Clorobenzeno
(27 µL)
Acetonitrilo
(1mL) GC-FPD
10 -
190
ng/kg
67 – 111
Metacrilato (85) Água Diclorometano
(116L)
Metanol
(0,565mL)
HPLC -
UV 1 µg/L 96.7 -101.8
Herbicidas Triazínicos
(86) Água _ _ GC-ITMS
21 –
120
ng/L
85.2- 119.4
Compostos
Organoestanhados
(88)
Água _ _ GC-FPD 0.2 – 1
ng/L 82.5 -104.7
Clorobenzenos (89) Água Clorobenzeno
(9,5 µL)
Acetona
(0,5mL) GC-ECD
0.0005- 0.05
g/L
71.1-81.3
Organoclorados (90) Água Tetracloroetileno
(5,2 L)
Metil-tert-
butil-éter
(MBTE) (7,8
L)
GC-MS 0.4-2.5
ng/L _
Fungicidas (65) Vinho
1,1,1-
tricloreoetano
(100 L)
Acetona
(1mL) GC-MS
40-250
ng/L 61-99

27
Herbicida de
sulfunilureia (91) Solo
Clorobenzeno
(60µL) Acetona (_)
HPLC-
DAD
0.5- 1.2 ng/g
30-55
Vários resíduos de pesticidas (92)
Sumo de
maçã
Tetracloreto de
carbono (25 µL)
Acetona
(0,4mL)
GC-GC-
MS
0.06- 2.20
g/L 60-105
FÁRMACOS
Cloranfenicol (93) Mel
1,1,2,2- Tetracloroetano
(30 L)
Acetonitrilo (1 mL)
HPLC-
UV
0.6 g/
kg 89.5-91.7
Drogas anti-
depressivas tricíclicas
(94)
Água e
plasma
humano
Tetracloreto de
carbono
(18 L)
Metanol
(1mL) GC-FID
0.005-0.01
g/
mL
54.76-74.02
Clenbuterol (87) Água Tetracloroetileno
(25 L)
Acetona (0.5 mL)
HPLC-
UV
4.9
µg/L 97
“Quinolonas” (95) Músculo do
porco
Diclorometano
(300 L) Acetonitrilo
(1.5 mL)
HPLC-
DAD
5.6-
23.8
g/kg 93.0-102.6
“Aminoflunitrazepam”
(96) Urina
Diclorometano
(250 L)
Alcool isopropilico
(0.5 mL)
HPLC-
MS/MS
0.025
ng/mL _
Drogas Psicotrópicas
(97) Urina
Tetracloreto de carbono
(20 L)
Acetonitrilo (0.5 mL)
HPLC-
UV
3-8
ng/mL _
Anti-inflamatórios
(98) Urina
[BMIm][PF6]
(280 L) Metanol
(0.720 mL)
HPLC-
UV
8.3-
32.0
ng/mL
99.6-107.0
Lovastatina e Simvastatina (99)
Água [HMIm][PF6]
(500 L) _
HPLC-
UV
0.17- 0.29
ng/mL
80.5-112.0%
PCBs e PBDEs
Éteres difenílicos
polibromados (100)
Água /
Lixiviados
Tetracloroetano
(20 L)
Acetonitrilo (1 mL)
HPLC-
DAD
12 – 56
ng/L 87 - 119.1
Éteres difenílicos
polibromados (101)
Tecido dos
animais
aquáticos
Clorobenzeno
(33 L)
Acetona (0,75 mL)
GC-MS 2.4-4.9
g/kg 75.3-127.8
OUTRAS APLICAÇÕES
Clorofenóis (82) Água Clorobenzeno
(10 µl)
Acetona (0.5 mL)
GC-ECD
10 -
2000
ng/L
80.8 -117.9
Ésteres ftálicos
(102) Água
Clorobenzeno
(9,5µl)
Acetona
(0,5mL) GC-MS
2-8
mg/L 68.1-88.9
Bisfenol (103)
Agua Clorobenzeno
(30 L)
Acetona
(0,5 mL) GC-MS
0.01
g/L -
Antioxidantes (Irganox 1010,
Irganox 1076 e Irgafos 168)
(104)
Agua
Tetracloreto de carbono
(40 L)
Acetonitrilo (2 mL)
HPLC-
DAD
3-7
ng/mL 78-110
Hidrocarbonetos
Aromáticos (83)
Resíduos
sólidos de
pirólise
Tetracloreto de carbono
(50L)
Acetona (0.5 mL)
GC-MS
1.02- 24.6
ng/L _
28
Aminas Biogénicas
(105) Vinho
1-octanol
(50 L) Acetonitrilo
(0.4 mL)
HPLC-
FD
0.02-5
ng/mL 95.42-104.56
Ácidos Gordos (106) Agua do
mar
Tetracloreto de carbono
(10 L)
Acetona (0.96 mL)
GC-FID
0.67- 14.5
g/L
90-110
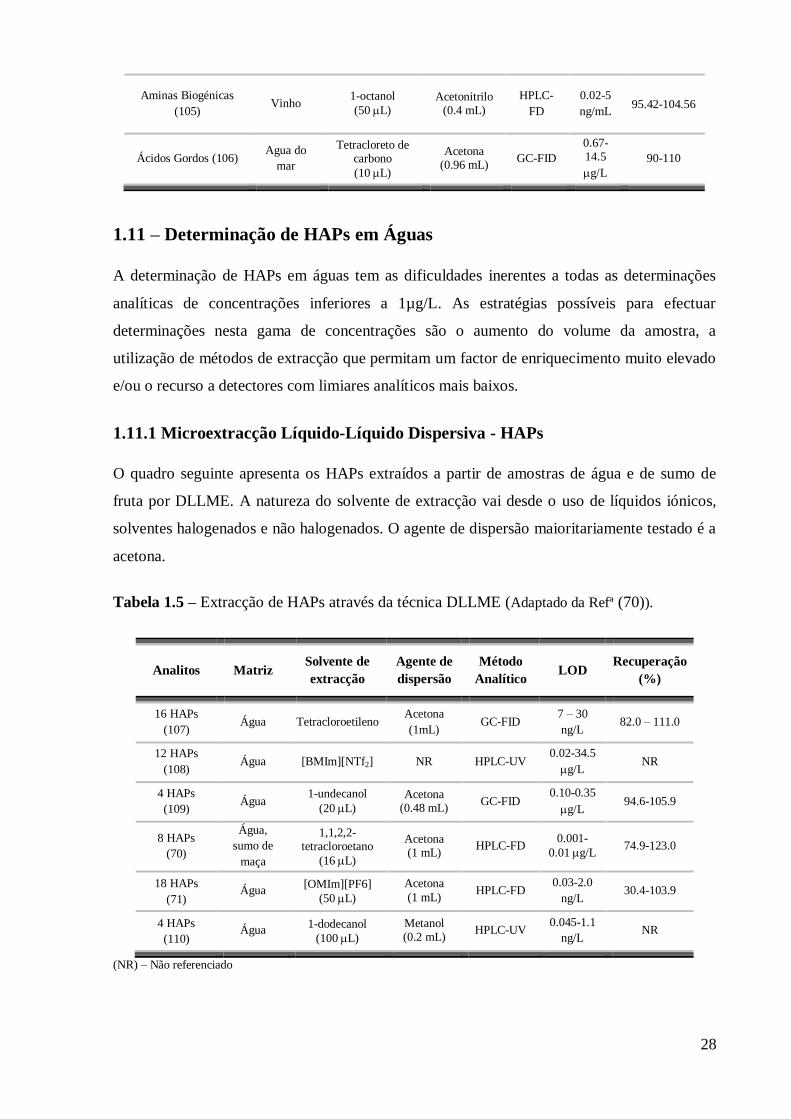
1.11 – Determinação de HAPs em Águas
A determinação de HAPs em águas tem as dificuldades inerentes a todas as determinações
analíticas de concentrações inferiores a 1µg/L. As estratégias possíveis para efectuar
determinações nesta gama de concentrações são o aumento do volume da amostra, a
utilização de métodos de extracção que permitam um factor de enriquecimento muito elevado
e/ou o recurso a detectores com limiares analíticos mais baixos.
1.11.1 Microextracção Líquido-Líquido Dispersiva - HAPs
O quadro seguinte apresenta os HAPs extraídos a partir de amostras de água e de sumo de
fruta por DLLME. A natureza do solvente de extracção vai desde o uso de líquidos iónicos,
solventes halogenados e não halogenados. O agente de dispersão maioritariamente testado é a
acetona.
Tabela 1.5 – Extracção de HAPs através da técnica DLLME (Adaptado da Refª (70)).
Analitos Matriz Solvente de
extracção
Agente de
dispersão
Método
Analítico LOD
Recuperação
(%)
16 HAPs
(107) Água Tetracloroetileno
Acetona
(1mL) GC-FID
7 – 30
ng/L 82.0 – 111.0
12 HAPs
(108) Água [BMIm][NTf2] NR HPLC-UV
0.02-34.5
g/L NR
4 HAPs
(109) Água
1-undecanol
(20 L)
Acetona (0.48 mL)
GC-FID 0.10-0.35
g/L 94.6-105.9
8 HAPs
(70)
Água,
sumo de
maça
1,1,2,2- tetracloroetano
(16 L)
Acetona (1 mL)
HPLC-FD 0.001-
0.01 g/L 74.9-123.0
18 HAPs
(71) Água
[OMIm][PF6]
(50 L)
Acetona
(1 mL) HPLC-FD
0.03-2.0
ng/L 30.4-103.9
4 HAPs
(110) Água
1-dodecanol
(100 L)
Metanol (0.2 mL)
HPLC-UV 0.045-1.1
ng/L NR
(NR) – Não referenciado
29
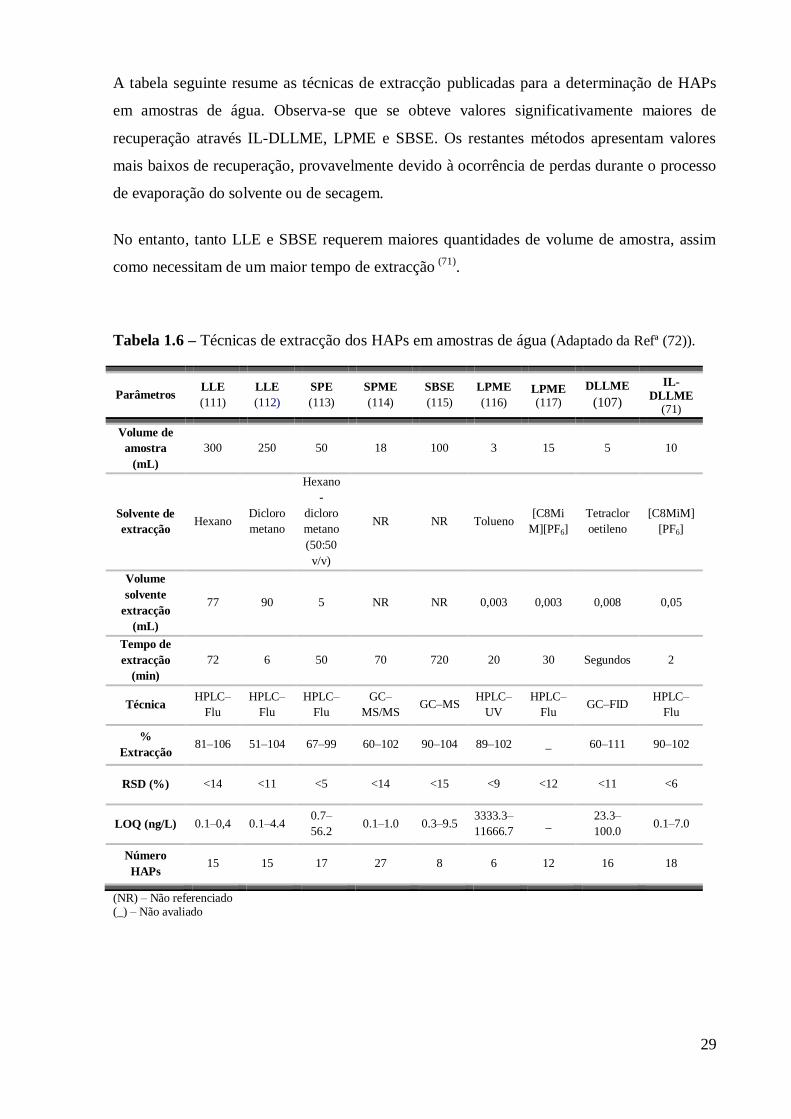
A tabela seguinte resume as técnicas de extracção publicadas para a determinação de HAPs
em amostras de água. Observa-se que se obteve valores significativamente maiores de
recuperação através IL-DLLME, LPME e SBSE. Os restantes métodos apresentam valores
mais baixos de recuperação, provavelmente devido à ocorrência de perdas durante o processo
de evaporação do solvente ou de secagem.
No entanto, tanto LLE e SBSE requerem maiores quantidades de volume de amostra, assim
como necessitam de um maior tempo de extracção (71)
.
Tabela 1.6 – Técnicas de extracção dos HAPs em amostras de água (Adaptado da Refª (72)).
Parâmetros LLE
(111)
LLE
(112)
SPE
(113)
SPME
(114)
SBSE
(115)
LPME
(116) LPME (117)
DLLME
(107)
IL-
DLLME (71)
Volume de
amostra
(mL)
300 250 50 18 100 3 15 5 10
Solvente de
extracção Hexano
Dicloro
metano
Hexano
-
dicloro
metano
(50:50
v/v)
NR NR Tolueno [C8Mi
M][PF6]
Tetraclor
oetileno
[C8MiM]
[PF6]
Volume
solvente
extracção
(mL)
77 90 5 NR NR 0,003 0,003 0,008 0,05
Tempo de
extracção
(min)
72 6 50 70 720 20 30 Segundos 2
Técnica HPLC–
Flu
HPLC–
Flu
HPLC–
Flu
GC–
MS/MS GC–MS
HPLC–
UV
HPLC–
Flu GC–FID
HPLC–
Flu
%
Extracção 81–106 51–104 67–99 60–102 90–104 89–102 _ 60–111 90–102
RSD (%) <14 <11 <5 <14 <15 <9 <12 <11 <6
LOQ (ng/L) 0.1–0,4 0.1–4.4 0.7–
56.2 0.1–1.0 0.3–9.5
3333.3–
11666.7 _
23.3–
100.0 0.1–7.0
Número
HAPs 15 15 17 27 8 6 12 16 18
(NR) – Não referenciado (_) – Não avaliado
30
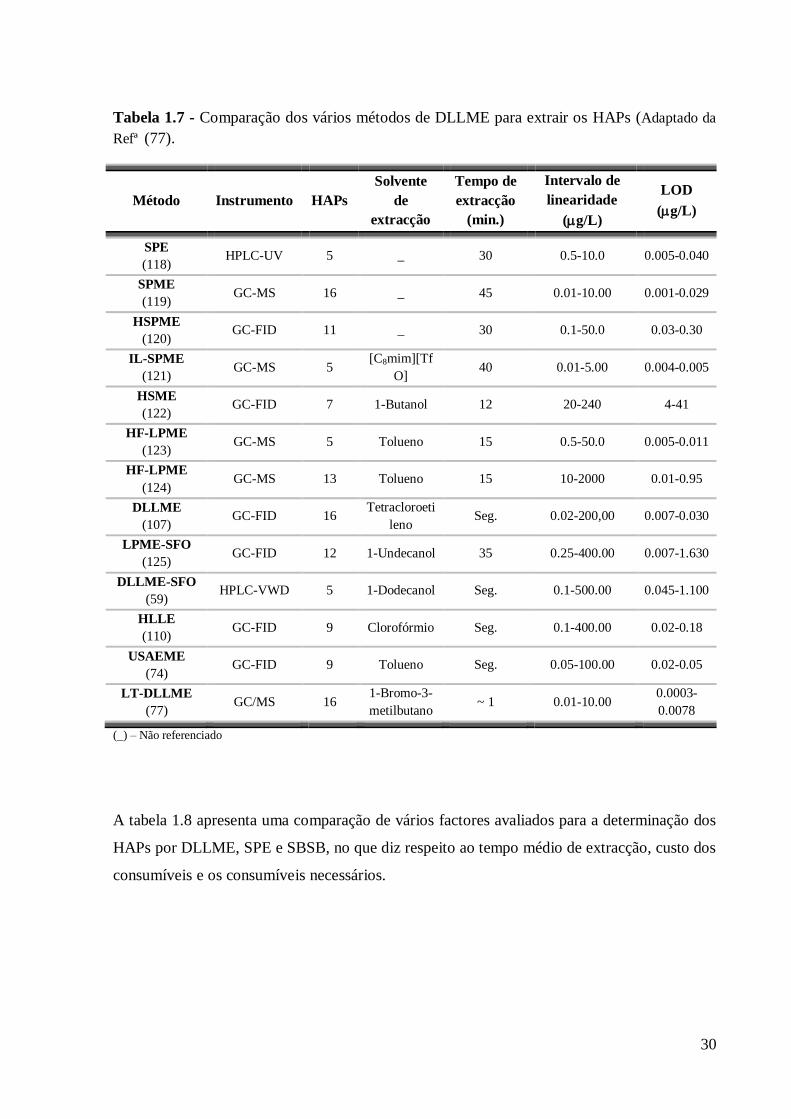
Tabela 1.7 - Comparação dos vários métodos de DLLME para extrair os HAPs (Adaptado da
Refª (77).
Método Instrumento HAPs
Solvente
de
extracção
Tempo de
extracção
(min.)
Intervalo de
linearidade
(g/L)
LOD
(g/L)
SPE
(118) HPLC-UV 5 _ 30 0.5-10.0 0.005-0.040
SPME
(119) GC-MS 16 _ 45 0.01-10.00 0.001-0.029
HSPME
(120) GC-FID 11 _ 30 0.1-50.0 0.03-0.30
IL-SPME
(121) GC-MS 5
[C8mim][Tf
O] 40 0.01-5.00 0.004-0.005
HSME
(122) GC-FID 7 1-Butanol 12 20-240 4-41
HF-LPME
(123) GC-MS 5 Tolueno 15 0.5-50.0 0.005-0.011
HF-LPME
(124) GC-MS 13 Tolueno 15 10-2000 0.01-0.95
DLLME
(107) GC-FID 16
Tetracloroeti
leno Seg. 0.02-200,00 0.007-0.030
LPME-SFO
(125) GC-FID 12 1-Undecanol 35 0.25-400.00 0.007-1.630
DLLME-SFO
(59) HPLC-VWD 5 1-Dodecanol Seg. 0.1-500.00 0.045-1.100
HLLE
(110) GC-FID 9 Clorofórmio Seg. 0.1-400.00 0.02-0.18
USAEME
(74) GC-FID 9 Tolueno Seg. 0.05-100.00 0.02-0.05
LT-DLLME
(77) GC/MS 16
1-Bromo-3-
metilbutano ~ 1 0.01-10.00
0.0003-
0.0078
(_) – Não referenciado
A tabela 1.8 apresenta uma comparação de vários factores avaliados para a determinação dos
HAPs por DLLME, SPE e SBSB, no que diz respeito ao tempo médio de extracção, custo dos
consumíveis e os consumíveis necessários.

31
Tabela 1.8 - Tempo médio gasto e custos associados a cada técnica extracção dos HAPs a
partir de amostras de água.
Factores
Avaliados
Técnicas de Extracção
DLLME
(Refª (66)) SPE
(Refª (126)) SBSB
(Refª (52))
Tempo médio (min.) 6-10 15-30 720
Custos de
consumíveis/unidade (€) 0.04 2.63 70.00
Consumíveis
Contabilizados
Ciclohexano
Acetona
Cartuchos com fase
RP-C18 a 500 mg em tubos com volume de
6 mL de amostra
Ciclohexano Acetona
Barra
magnética
Acetonitrilo Tetracloreto
de carbono
Como se pode constatar com este exemplo, a adopção de métodos de DLLME pode
representar uma vantagem significativa tanto em termos de tempo de análise como em termos
dos custos por análise, para além de poder apresentar uma sensibilidade superior à atingida
com as técnicas de SPE ou SBSB para os mesmos analitos.
1.12 Validação do método analítico
Na validação do método analítico pretende-se determinar as características analíticas do
método desenvolvido e a sua aplicabilidade a amostras reais, produzindo resultados fiáveis e
interpretáveis (127)
.
Os parâmetros cuja determinação é necessária para proceder à validação completa de um
método analítico são os seguintes (128)
:
1) Selectividade /Especificidade
2) Calibração analítica
3) Intervalo de Linearidade Analítica
4) Limites de Detecção e de Quantificação
5) Sensibilidade
6) Precisão
7) Robustez
8) Exactidão
9) Eficiência e Extracção

32
1.12.1 Selectividade/ Especificidade
Define a capacidade que o método tem em identificar e distinguir inequivocamente um analito
na presença de outros solutos ou interferentes que possam estar presentes na matriz,
respondendo aos analitos de interesse, sem se pretender uma análise quantitativa.
Um método que produz resposta para apenas um analito é chamado específico, por outro lado,
um método que produz respostas para vários analitos, mas que pode distinguir a resposta de
um analito da resposta de outros, independentemente das características dos interferentes, é
considerado selectivo. Estes dois parâmetros estão correlacionados e permitem ser avaliados
pela realização de testes de recuperação e pela análise de padrões com concentrações
conhecidas. Assim deste modo, o método é considerado específico e selectivo quando se
verificam taxas de recuperação próximas de 100% (127)
.
Existem parâmetros e testes que permitem avaliar cada um destes factores individualmente.
No caso da especificidade há que considerar os seguintes parâmetros: resolução, retenção
relativa (factor de separação), factor de capacidade (factor de retenção), factor de simetria e
número de pratos teóricos. Na análise da selectividade os testes mais comuns são o teste de
Snedecor e o teste de homegeneidade de variâncias.
1.12.2 Calibração analítica
A calibração analítica relaciona a resposta de um sistema de medida, como por exemplo a
área ou altura do pico cromatográfico, isto é, um factor quantitativo ou numérico com a
concentração do analito que originou esse sinal. Este tipo de calibração pode ser feito a partir
de (127) (129)
:
Curva de Calibração com padrão externo ou interno
Curva de Calibração pelo método de adição de padrão à amostra
Factor de resposta
Cada opção é adequada a diferentes casos, devendo o laboratório seleccionar a mais adequada
para a análise em causa.
No caso de se recorrer à opção de efectuar uma curva de calibração, é necessário preparar um
conjunto de soluções-padrão com várias concentrações conhecidas que serão analisadas nas
mesmas condições a aplicar à análise das amostras. Para este tipo de calibração recomenda-se
o uso da norma ISO 8466-1 (130)
como referência, designadamente para efectuar regressões
lineares pelo método dos mínimos quadrados.
O cálculo das curvas de calibração aplica o método dos mínimos quadrados, que permite
encontrar o melhor ajuste possível para o conjunto de dados tentando minimizar a soma dos

33
quadrados das diferenças entre a curva ajustada e os dados. Neste método, define-se que o
eixo vertical (eixo yy) representa a medida instrumental do equipamento (área ou altura do
pico cromatográfico) e que no eixo horizontal (eixo xx) está representada a concentração dos
padrões utilizados; considera-se que os erros associados ao eixo dos xx são desprezáveis
relativamente aos valores do eixo dos yy. Assume-se que os erros apresentam uma
distribuição normal e que existe homogeneidade de variâncias ao longo da curva de
calibração. Segundo a norma referida, são recomendados dez pontos de calibração não
podendo ser utilizados menos de cinco pontos de calibração; a sua distribuição deve ser
homogénea no intervalo de concentração considerado. De acordo como o método analítico e
considerando os critérios laboratoriais, os valores para o coeficiente de determinação (R2) das
curvas de calibração devem ser o mais próximo possível da unidade, e não devem ser
inferiores a 0,990 de forma a traduzir uma fraca dispersão dos resultados e um bom ajuste dos
pontos experimentais à recta de calibração.
Para uma calibração linear (y = a+ bx) o declive (b) é dado pela seguinte expressão (127) (131)
:
Σ
E o desvio padrão residual (Sy) que representa a dispersão das medidas efectuadas a partir da
função de calibração é dado pela seguinte fórmula (127) (131)
:
Σ
Sendo
O desvio padrão entre os pontos no eixo dos xx é designado por Sx0 ou S0 e é dado por:
O coeficiente de variação (CV) relativo ao desvio padrão Sxo é dado por:

34
1.12.3 Intervalo de Linearidade Analítica
A linearidade é entendida como a capacidade que o método tem em fornecer resultados
directamente proporcionais à concentração do soluto num determinado intervalo de
concentrações conhecidas, isto é, num intervalo de linearidade em que a variação nas
respostas do detector deve ser inferior a 5%.
Assim este intervalo pode ser avaliado pela aplicação de um modelo estatístico conforme o
que está descrito na norma ISO 8466-1, designado por Fisher/Snedecor ou Teste de Mandel.
Todavia a análise da linearidade também pode ser avaliada a partir da análise de resíduos, que
descreve a distância entre os valores dos dados observados e os valores estimados através da
regressão linear, no eixo dos yy (127) (130) (132)
.
A gama de trabalho pode ser definida como sendo o intervalo entre a concentração inferior e a
concentração superior do analito na solução padrão ou na amostra, para o qual deve haver um
nível adequado de precisão e exactidão na definição do procedimento analítico. A norma ISO
8466-1 avalia a linearidade para modelos lineares, enquanto a norma ISO 8466-2 avalia o
mesmo parâmetro mas em modelos polinomiais de 2º grau (130) (133)
.
1.12.4 Limites Analíticos
Os limites analíticos de um método são definidos pelo limite de detecção (LOD) e pelo limite
de quantificação (LOQ). O limite de detecção para um determinado analito deve ser entendido
como uma concentração que é traduzida por um sinal instrumental diferente do sinal do
branco num dado intervalo de confiança estatística (normalmente 95%). Este limite situa-se
acima do somatório do sinal médio do branco (x0) somando três vezes o desvio-padrão do
branco (Sx0) e isto é:
O limite de quantificação corresponde à menor concentração da gama de trabalho, em que é
possível quantificar o analito na amostra, em que o coeficiente de variação do sinal está
reduzido a valores inferiores a 10%. Assim o primeiro padrão da gama de trabalho deve de ser
superior ao limite de quantificação.
Este limite situa-se dez vezes superior ao desvio-padrão do branco, ou seja (129)
:
Existem outras formas de calcular os limiares analíticos. O limiar analítico é o teor mínimo
medido, a partir do qual é possível detectar a presença do analito com uma certeza estatística

35
razoável. Este limiar analítico corresponde a mais pequena quantidade de substancia a analisar
que pode ser detectada numa amostra, mas não necessariamente quantificada como valor
exacto.
Para uma correcta analise dos limiares analiticos, existem outros métodos de cálculo que
podem ser utilizados para medir o limite de detecção e o limite de quantificação.
Método de Cálculo – a partir da Curva de Calibração
Método de Cálculo - Razão Sinal Ruído (S/N)
Método de Cálculo – Desvio padrão de Brancos ou padrão vestigial
1.12.5 Sensibilidade
A sensibilidade traduz-se pela resposta que o detector fornece por unidade de concentração ou
por unidade de massa entre dois analitos.
Esta grandeza é expressa pela quantidade mínima detectável, definida por uma concentração
do analito para a qual a razão sinal/ruído seja superior a quatro. No caso de o método envolver
uma calibração linear, este parâmetro pode ser visto como o declive da curva de calibração (b)
em que no eixo dos yy está a resposta do detector (área ou altura do pico) e no eixo dos xx
estão as concentrações dos analitos, sendo portanto o seu valor constante ao longo da gama de
trabalho (134) (132) (127)
.

36
1.12.6 Precisão
A precisão do método analítico define-se como o grau de concordância entre os vários
resultados dos ensaios laboratoriais individuais, quando o método envolve leituras de
replicadas de padrões ou de amostras (norma ISO 3534) (135)
. É avaliado pelo desvio-padrão
(SD) ou pelo desvio-padrão relativo (RSD) ou coeficiente de variação (CV), que avaliam a
discrepância entre os dados que podem estar associados a erros aleatórios ou contínuos
(operador, equipamento, factores ambientais como a temperatura, humidade, tempo entre
medições). O teste de homogeneidade de variâncias permite avaliar se os padrões da gama
considerada linear (um da gama de concentrações mais baixa e outro da gama de
concentrações mais alta) têm valores de variâncias muito diferentes entre si. Também o teste
da análise de resíduos permite avaliar a precisão do método, discriminando os valores
considerados fora da gama de calibração (127) (131)
.
Para avaliar a precisão há que considerar três parâmetros: a repetibilidade, a precisão
intermédia e a reprodutibilidade. O primeiro parâmetro expressa a precisão de um método
realizado a partir de várias medições sucessivas da mesma amostra em condições de operação
idênticas (metodologia, operador, equipamento, reagentes, laboratório, factores ambientais,
tempo entre repetições) num curto intervalo de tempo. Este parâmetro pode ser avaliado
quantitativamente através da dispersão de resultados produzidos numa análise de soluções-
padrão, material de referência ou de brancos a várias concentrações.
A precisão intermédia é definida com base na avaliação da precisão numa dada amostra ou em
amostras idênticas, utilizando o mesmo procedimento experimental, no mesmo laboratório,
mas definindo previamente quais as condições que variam (período de tempo, operador ou
equipamento). Este tipo de precisão é usada para assegurar que há resultados concordantes
para amostras idênticas analisadas em períodos de tempo distintos por outros operadores e/ou
equipamentos.
Por fim, a reprodutibilidade avalia se a precisão de um método é constante ao longo do tempo,
usando um procedimento analítico que pode apresentar diferentes condições de operação
(diferentes laboratórios, operadores, equipamentos, condições de análise), e que é geralmente
avaliado através da participação de ensaios interlaboratoriais ou de ensaios de normalização.
Contudo, a reprodutilidade também pode ser avaliada entre métodos iguais, como é o caso dos
ensaios de aptidão onde se pretende avaliar qual o método que apresenta melhores resultados.

37
1.12.7 Robustez
A robustez é definida como a capacidade de um método analítico resistir a pequenas
alterações experimentais. O método é considerado robusto se não for afectado por ligeiras
alterações dos seus parâmetros (temperatura, caudal, entre outros), sendo que este parâmetro é
avaliado na fase de desenvolvimento do método e depende do tipo de processo em estudo. A
avaliação da robustez é importante quando se pretende avaliar que problemas podem afectar a
resposta dada pelo equipamento ao método estudado. Este parâmetro pode ser calculado
recorrendo ao teste de Youden. Este teste permite não só avaliar a robustez do método, como
permite também, avaliar a variação entre os dados e permite comparar resultados
interlaboratoriais para o mesmo tipo de amostras.
É importante salientar que quanto maior for a robustez de um método, maior será a sua
precisão (131)
.
1.12.8 Exactidão
A exactidão é definida como a concordância entre o resultado de um ensaio e o valor de
referência anteriormente aceite (norma ISO 3534). A avaliação da exactidão é portanto a
avaliação da combinação de erros sistemáticos e aleatórios.
A exactidão pode ser avaliada através de ensaios de recuperação analítica, permitindo
comparar valores esperados com valores observados pelo próprio método. No entanto, este
parâmetro também pode ser avaliado a partir de ensaios interlaboratoriais ou através de
comparação de resultados com os materiais de referência certificados (MRC). Os ensaios de
recuperação também permitem avaliar a exactidão de um método.
1.12.9 Eficiência de Extracção
A eficiência de extracção (E) pode ser descrita como uma função que avalia o nível de
modificação que a natureza da amostra introduz na calibração analítica (127)
.
A eficiência de extracção de um determinado analito pode ser estimada pela análise de uma
amostra, adicionando uma quantidade conhecida desse analito, isto é, fortificando a amostra
com uma concentração conhecida de um determinado padrão do analito.
Para avaliar a recuperação desse analito em amostras reais, devem-se estudar as recuperações
do mesmo analito em três situações distintas: numa concentração próxima do limite de
quantificação, numa concentração próxima da concentração máxima admissível e numa

38
concentração que esteja a meio da gama de trabalho. Assim teremos o cálculo da recuperação
como:
Onde,
C1 é a concentração determinada na amostra fortificada
C2 é a concentração determinada na amostra não fortificada
C3 é a concentração de fortificação

39
Capítulo II
Determinação de HAPs em águas de consumo ou
superficiais por extracção em fase sólida associada a
microextracção líquido-líquido dispersiva e análise por
cromatografia gasosa e espectrometria de massa

40
2.1 Considerações gerais
2.1.1 Estudo do processo de Microextracção Líquido-Líquido Dispersiva
(DLLME) de HAPs
A extracção líquido-líquido dispersiva tem como base o equilíbrio de fases em sistemas
ternários de água, um solvente de extracção e um co-solvente (solvente de dispersão). Para
que se consiga efectuar com sucesso uma extracção líquido-líquido dispersiva têm portanto
que reunir-se as seguintes condições:
Efectuar uma mistura dos três componentes do sistema, sendo a água o componente
maioritário, o solvente de extracção o componente minoritário e o solvente de
dispersão numa quantidade tal que permita a formação de uma emulsão estável. Por
outras palavras, as proporções dos três componentes do sistema têm que ser tais que o
equilíbrio de fases se situe na zona de transição entre o estado monofásico e o estado
difásico.
Após a separação de fases, o volume da fase orgânica deverá ser o menor possível
para maximizar o factor de enriquecimento.
O volume de fase orgânica sedimentada deve ser suficientemente elevado para ser
reprodutível e será desejável que possa ser injectado automaticamente.
O factor de enriquecimento dos analitos na fase orgânica deverá ser elevado.
A percentagem de extracção deverá estar situada entre 50% e 120% pois para valores
fora desta gama observa-se geralmente uma perda de repetibilidade.
O objectivo deste trabalho foi o desenvolvimento e optimização de um método de DLLME
adequado à determinação de um conjunto de HAPs (naftaleno, acenaftileno, acenafteno,
fluoreno, fenantreno, antraceno, fluoranteno, pireno, criseno, 1,2-benzoantraceno,
benzo(k)fluoranteno, benzo(b)fluoranteno, benzo(a)pireno, indeno(1,2,3-c,d)-pireno, 1,2:5,6-
dibenzoantraceno, 1,12-benzoperileno) numa gama de concentrações que incluísse o limite
máximo admissível para a soma de quatro hidrocarbonetos aromáticos policíclicos é de
0,10g/L e que o benzo(a) pireno é de 0,010g/L segundo a Directiva 98/83/CE, de 3 de
Novembro de 1998 (26)
.
Cada um dos HAPs apresenta dois ou mais anéis na sua estrutura. Os HAPs que contém
quatro ou mais anéis são menos voláteis e menos solúveis em água, logo permanecem no
ambiente por um longo período de tempo. Por essa razão, é importante monitorizar os HAPs
com mais anéis em amostras ambientais.

41
Tabela 2.1 - Solubilidade em água dos vários HAPs (136) (14) (16)
.
Composto
Solubilidade
a 25 ºC
(mg/L)
Naftaleno 3.000
Acenaftileno 3.980
Acenafteno 1.930
Fluoreno 1.680-1.980
Fenantreno 1.200
Antraceno 0.076
Fluoranteno 0.200-0.260
Pireno 0.077
Criseno 2.8x10-3
1,2-Benzoantraceno 0.010
Benzo(k)fluoranteno 7.6x10-4
Benzo(b)fluoranteno 0.0012
Benzo(a)pireno 2.3x10-3
Indeno(1,2,3-c,d)-pireno 0.062
1,2:5,6-Dibenzoantraceno 5.0x10-4
1,12-Benzoperileno 2.6x10-4
No que se refere à solubilidade em água, o acenaftileno tem um valor particularmente
elevado, enquanto o menos solúvel é o 1,12-benzoperileno.
A determinação de HAPs em águas por DLLME foi a 1ª aplicação desta técnica (107)
; o
solvente seleccionado foi o tetracloroetileno e nas condições óptimas obteve-se um volume
sedimentado de 5 µL. No entanto, o método proposto é de difícil reprodução e o baixo volume
sedimentado dificulta a separação de fases quantitativa, bem como a injecção automática.
Assim, procurou-se estudar a aplicação de DLLME ao grupo de HAPs incluídos neste
trabalho, utilizando tanto solventes halogenados como solventes não halogenados e
seleccionaram-se as condições que permitiam a recolha de volumes reprodutíveis de fase
sedimentada (tipicamente acima de 10 µL).
2.1.2 SPE-HLLME de HAPs
A associação do HLLME à técnica de extracção em fase sólida (SPE) permite conjugar a
grande capacidade extractiva da microextracção líquido-líquido à pré-concentração resultante
da extracção em fase sólida e atingir assim a ultraconcentração dos analitos na fase orgânica
recolhida após a separação de fases (137)
(138)
.
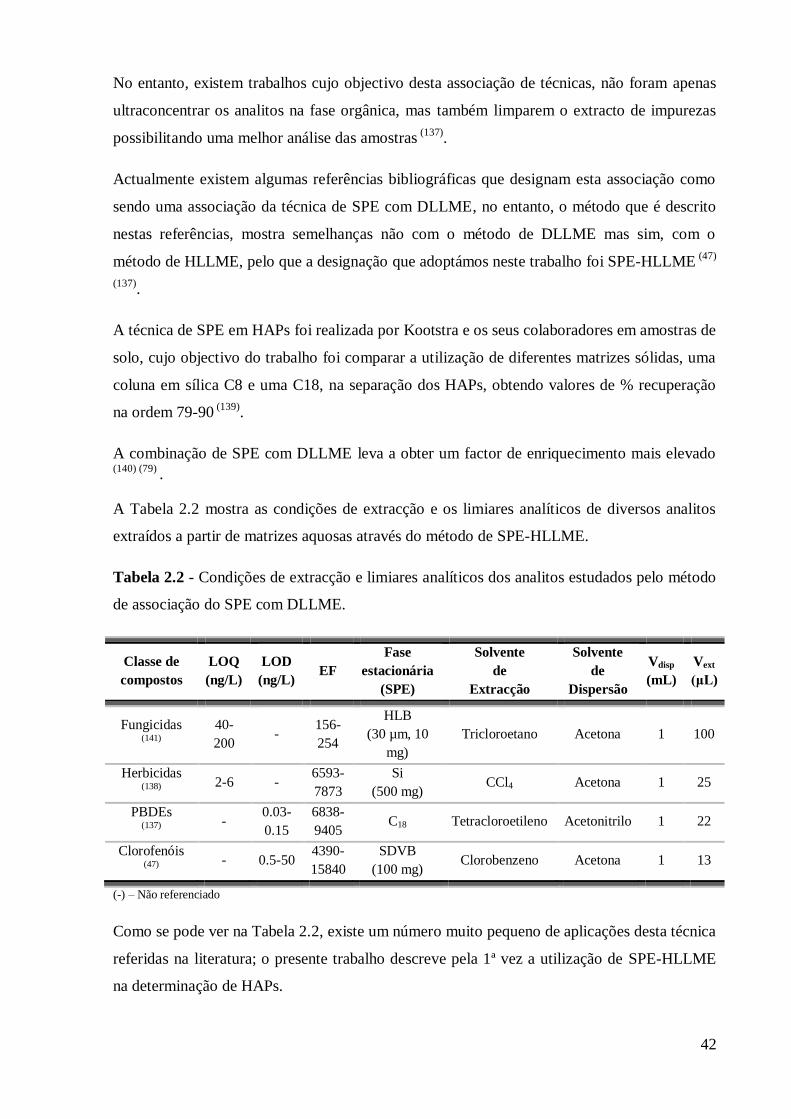
42
No entanto, existem trabalhos cujo objectivo desta associação de técnicas, não foram apenas
ultraconcentrar os analitos na fase orgânica, mas também limparem o extracto de impurezas
possibilitando uma melhor análise das amostras (137)
.
Actualmente existem algumas referências bibliográficas que designam esta associação como
sendo uma associação da técnica de SPE com DLLME, no entanto, o método que é descrito
nestas referências, mostra semelhanças não com o método de DLLME mas sim, com o
método de HLLME, pelo que a designação que adoptámos neste trabalho foi SPE-HLLME (47)
(137).
A técnica de SPE em HAPs foi realizada por Kootstra e os seus colaboradores em amostras de
solo, cujo objectivo do trabalho foi comparar a utilização de diferentes matrizes sólidas, uma
coluna em sílica C8 e uma C18, na separação dos HAPs, obtendo valores de % recuperação
na ordem 79-90 (139)
.
A combinação de SPE com DLLME leva a obter um factor de enriquecimento mais elevado (140) (79)
.
A Tabela 2.2 mostra as condições de extracção e os limiares analíticos de diversos analitos
extraídos a partir de matrizes aquosas através do método de SPE-HLLME.
Tabela 2.2 - Condições de extracção e limiares analíticos dos analitos estudados pelo método
de associação do SPE com DLLME.
Classe de
compostos
LOQ
(ng/L)
LOD
(ng/L) EF
Fase
estacionária
(SPE)
Solvente
de
Extracção
Solvente
de
Dispersão
Vdisp
(mL)
Vext
(μL)
Fungicidas
(141)
40-
200 -
156-
254
HLB
(30 µm, 10
mg)
Tricloroetano Acetona 1 100
Herbicidas
(138) 2-6 - 6593-
7873
Si
(500 mg) CCl4 Acetona 1 25
PBDEs (137) -
0.03-
0.15
6838-
9405 C18 Tetracloroetileno Acetonitrilo 1 22
Clorofenóis
(47) - 0.5-50 4390-
15840
SDVB
(100 mg) Clorobenzeno Acetona 1 13
(-) – Não referenciado
Como se pode ver na Tabela 2.2, existe um número muito pequeno de aplicações desta técnica
referidas na literatura; o presente trabalho descreve pela 1ª vez a utilização de SPE-HLLME
na determinação de HAPs.

43
A associação entre extracção em fase sólida (SPE) e microextracção líquido – líquido
homogénea (HLLME) na determinação de HAPs em águas foi estudada neste trabalhado,
tendo sido testadas as possíveis combinações de diferentes solventes de dispersão (acetona,
metanol, isopropanol) e diferentes solventes de extracção não halogenados (ciclohexano,
tolueno e octano).
O método optimizado foi posteriormente validado de acordo com os procedimentos
detalhados na Norma ISO 3534 (135)
.
2.2 Procedimento Experimental
2.2.1 Solventes e Reagentes
Utilizou-se em todos os ensaios água ultra pura (Milli-Q, modelo Milipore, Molsheim,
França).
O ciclohexano (99%), a acetona (99.5%), o metanol (99.9%), o hexano (99%), foram
fornecidos por Lab-scan (Dublin, Irlanda); o octano (99%) foi fornecido por Koch-Light
Laboratory (Cambridge, Reino Unido); o isopropanol (≥99.5%) foi adquirido à Fluka
(Steinheim, Suiça); o clorofórmio (99.8%), o tetracloreto de carbono (99,9%) e o
tetracloroetileno (99,9%) à Aldrich (Steinheim, Alemanha) e o Tolueno (99,5%) à Panreac
(Barcelona, Espanha).
2.2.2 Amostras Reais
Para efeitos de validação da técnica de SPE-HLLME foram realizados ensaios de recuperação
em água ultra pura e em diferentes matrizes, nomeadamente água subterrânea (subterrânea
Odivelas), da rede pública de abastecimento (água da torneira, Monte de Caparica) e água da
nascente (mineral).
2.2.3 Materiais
Nos ensaios de microextracção líquido-líquido dispersiva utilizaram-se vials cónicos de vidro
e com rosca e tampa com septo de PTFE, com capacidades de 15 mL (Refªs 27039 e 27479,
Supelco).
Na medida e transferência dos solventes de extracção e de dispersão, bem como da fase
sedimentada utilizaram-se microseringas de vidro, graduadas, com a capacidade de: 10µL

44
(SGE, Australia); 25 µL, 50 µL, 100 µL e 250 µL (Agilent, Australia); 1000 µL (Ruthe,
Portugal).
Os extractos foram colocados em redutores de volume de 100 µL (Refª 110501) e 300 µL
(Refª 110502) (142)
, para frascos de capacidade 1,5 mL (Refª CVP1-C02-100) (143)
, com tampa
de capsular, do injector automático.
Durante o trabalho efectuado utilizou-se material de vidro comum como erlenmeyers, funis,
balões volumétricos, pipetas volumétricas, gobelés, etc.
Todo o material de vidro utilizado é descontaminado com acetona após a sua utilização
seguindo-se uma primeira lavagem em banho de água com um detergente (Deconex 21) e
ácido (Deconex 26 Mineral acid) apropriados. De seguida o material é lavado numa máquina
de lavagem de material de laboratório, equipada com sistema de passagem final por água
destilada (Miele Professional, modelo G 7883). O material volumétrico é posteriormente seco
numa estufa a 40 ºC (Memmert, Schwabach, Alemanha) enquanto o material não volumétrico
é seco numa estufa a 100ºC (Memmert, Schwabach, Alemanha).
As microseringas são lavadas com acetona e hexano sob vácuo e secas à temperatura
ambiente.
Os frascos onde se efectua a microextracção são descontaminados com acetona, lavados com
detergente aniónico apropriado (Sodosil RM03, Riedel-de-Haën, Seelze, Alemanha), e
lavados sucessivamente com água corrente, água bidestilada e acetona; em seguida são secos
em estufa a 40ºC.
2.2.4 Preparação de soluções-padrão e construção e rectas de calibração
Os dezasseis HAPs foram adquiridos sob a forma de uma solução da mistura em metanol,
com uma concentração de 1000 mg/L (Chem Service Inc., West Chester, USA, Refª PPH-
10RPM, lot:372-93A).
A partir desta solução concentrada prepararam-se, por diluição em metanol, soluções de 100
mg/L e de 2 mg/L. Estas soluções foram transferidas para frascos de 2 mL, que foram
capsulados e armazenados em caixas plásticas opacas, a uma temperatura inferior a -10 ºC.
Durante a optimização do método de SPE-HLLME foram construídas rectas de calibração nas
gamas de 100 µg/L a 800 µg/L, por diluição da solução de 2 mg/L, com metanol.
As soluções de fortificação utilizadas nos ensaios de SPE-HLLME tinham a concentração de
2 µg/L.
A solução dos HAPs em metanol, com a concentração de 2 mg/L, foi diluída com metanol até
concentrações finais de 100 µg/L até 800 µg/L. Estas soluções metanólicas foram então

45
adicionadas a amostras de água, nas quantidades adequadas para preparar os padrões das
rectas de calibração na gama de 100 ng/L a 800 ng/L e nos ensaios de recuperação efectuados
a 100 ng/L.
2.2.5 Microextracção líquido-líquido dispersiva (DLLME)
A amostra de água (5 mL) é colocada num frasco de fundo cónico de (5 - 10 mL) utilizando
uma pipeta volumétrica.
Num copo de 5 mL coloca-se o volume adequado de solvente de dispersão (1mL), utilizando
microseringas de 1000 µL. Adiciona-se em seguida ao mesmo copo o solvente de extracção
(50 µL), utilizando microseringas de 50 µL. A mistura do solvente de dispersão e solvente de
extracção é então transferida do copo de mistura para o frasco com a amostra de água,
utilizando uma seringa de 1000 µL. A ponta da agulha é colocada a cerca de metade da altura
do frasco e a adição da mistura de solventes à amostra de água é efectuada de forma rápida,
obtendo-se assim uma boa dispersão dos solventes orgânicos na fase aquosa.
2.2.5.1 Solventes Halogenados
Após a dispersão as fases são separadas por centrifugação (Centrífuga Modelo Hettich-EBA
20) durante 6 min, a 3000 rpm.
A fase sedimentada é removida com uma microseringa de capacidade apropriada (10 µL a
100 µL) e colocada num redutor de volume de 100 µL ou de 300 µL; à fase sedimentada é
adicionado um pequeno volume (50 µL a 200 µL) da fase aquosa com a qual estava em
equilíbrio de forma a evitar a evaporação do solvente de extracção durante o processo de
injecção automática. O redutor de volume é colocado num frasco de 1.5 mL que é
imediatamente capsulado e colocado no tabuleiro do injector automático do cromatógrafo,
para se proceder à sua análise.
2.2.5.2 Solventes não Halogenados
Após a dispersão as fases são separadas por centrifugação durante 6 min, a 3000 rpm, retira-se
alguma fase aquosa com uma microseringa de vidro de capacidade apropriada (250 µL),
realiza-se uma nova centrifugação durante 8 min a 3000rpm, volta-se a retirar a restante fase
aquosa. O eluato obtido é removido com uma microseringa de capacidade (10 µL a 50 µL) e
colocada num redutor de volume de 100 µL ou de 300 µL. O redutor de volume é colocado

46
num frasco de 1.5 mL que é imediatamente capsulado e colocado no tabuleiro do injector
automático do cromatógrafo, para se proceder à sua análise.
2.2.6 Método de associação de extracção em fase sólida com microextracção
líquido-líquido homogénea (SPE-HLLME)
As amostras de água são colocadas em balões volumétricos de 100 mL, 200 mL, 500 mL e
1000 mL, fortificada com 2 μg/L de HAPs. De seguida, é feita a eluição da amostra através do
método de SPE, utilizando-se cartuchos com fase RP-C18 a 500 mg em tubos com volume de
3 mL de amostra (J.T.Baker). Foi feito um condicionamento prévio da fase estacionária com a
eluição dos solventes de acordo com a sua polaridade. Deste modo, eluiu-se em primeiro
metanol (99.9% pureza) num volume de 5 mL, seguido de água ultra pura com o mesmo
volume. De seguida e após a secagem da fase estacionária e com o auxílio de um recaudal de
água, (fez-se a eluição da amostra). A eluição dos analitos foi feita com o solvente de
dispersão adequado (acetona, metanol, isopropanol) e com volumes entre 1 mL - 2 mL
utilizando pipetas volumétricas, sendo o eluato recolhido nos tubos de centrífuga onde se vai
efectuar o passo de SPE. Após a etapa de SPE, efectuou-se a passagem na coluna RP-18, de
dois volumes (0,5mL) de forma sucessiva do solvente de dispersão optimizado.
Adicionou-se o solvente de extracção (ciclohexano, tolueno, octano) num volume de
extracção (50µL a 100 µL), com auxílio de microseringas de vidro 50 µL e de 100 µL. A
adição de água, utilizando uma pipeta volumétrica (5 mL a 10mL), é feita após a adição do
solvente de extracção, causando a turvação da mistura de solventes.
Após a dispersão as fases são separadas por centrifugação, o eluato obtido é removido com
uma microseringa e colocado num redutor. O redutor de volume é colocado num frasco de 1.5
mL que é imediatamente capsulado e colocado no tabuleiro do injector automático do
cromatógrafo, para se proceder à sua análise.
2.2.7 Análise cromatográfica
A análise cromatográfica dos extractos foi efectuada num cromatógrafo gasoso (modelo
Focus GC, Thermo Scientific) equipado com injector com e sem repartição (split-splitless),
uma coluna analítica TR-V1-cyanopropylphenyl polysiloxane (30 m x 1,4 µm x 0.25 mm
i.d.), (Thermo Scientific) e acoplado a um espectrómetro de massa (Polaris Q, Thermo
Scientific). A injecção foi efectuada em modo automático utilizando um injector automático
(AS2000). O extracto (1 µL) foi injectado a uma temperatura de 260ºC, em modo sem
repartição durante 1 minuto, velocidade de 30 mL/minuto e com um splitess time de 1 min..

47
O caudal do gás de arraste (Hélio com 99.9999% de pureza), foi mantido a 1 mL/minuto.
Utilizaram-se temperaturas da interface e da fonte de ionização de 290ºC e 260ºC,
respectivamente.
A temperatura do forno foi programada da seguinte forma: temperatura inicial de 40ºC
durante 3 min.; aquecimento a 20ºC/min. até 150ºC; aquecimento a 5ºC/min. até 200ºC,
aquecimento a 10ºC/min. até 230ºC, por fim um aquecimento a 3ºC/min. até 300ºC mantendo-
se a essa temperatura durante 20 min. O tempo total de análise foi de 65 minutos.
A detecção foi feita em modo de varrimento completo numa gama de massas de 45 a 400 m/z.
Contudo, para uma detecção mais selectiva, optou-se por escolher o modo de selecção do pico
base para cada analito (extracção de ião individual), correspondendo ao ião mais intenso do
espectro de massa de cada HAPs.
No entanto a coluna analítica utilizada, apresentou um sangramento quando se procedeu a um
aumento da temperatura de eluição acima dos 290ºC, por outro lado sempre que a temperatura
do injector subia acima dos 270ºC, observou-se uma diminuição das áreas dos analitos, apesar
deste comportamento não ser o mais esperado. Portanto, devido a esta situação os seguintes
analitos (Indeno (1,2,3-c,d)-pireno, 1,2:5,6-Dibenzoantraceno, 1,12-Benzoperileno), não
foram considerados na elaboração deste trabalho.
2.2.8 Cálculos
O factor de enriquecimento, parâmetro que avalia a partição dos analitos entre as fases, sob
diferentes condições de microextracção, foi calculado a partir da seguinte expressão:
0C
CEF sed
onde Csed é a concentração de analitos na fase sedimentada e C0 a concentração inicial de
analitos na fase aquosa.
A avaliação da extensão do processo de microextracção é efectuada através do cálculo da
percentagem de extracção, utilizando a seguinte expressão:
onde Csed é a concentração de analitos na fase sedimentada, C0 a concentração inicial de
analitos na fase aquosa, Vsed é o volume de fase sedimentada e Vaq o volume da amostra de
água.

48
2.3 Resultados e Discussão
2.3.1 Optimização do processo de separação cromatográfica
A selecção das melhores condições de separação cromatográfica dos analitos foi efectuada em
modo de varrimento total, utilizando uma solução de HAPs em metanol com a concentração
de 2 mg/L.
Em seguida procedeu-se à selecção dos iões mais adequados à detecção selectiva dos analitos
em modo de pico base (método de extracção de ião individual). Foram seleccionados os iões
com maior razão sinal/ruído que em alguns casos não foi o ião molecular (Tabela 2.3, Figura
2.1).
Tabela 2.3 – Iões seleccionados para detecção selectiva e tempos de retenção dos HAPs.
Analitos
Tempo de
retenção
(min.)
Ião
seleccionado
(m/z)
Naftaleno 13.91 128
Acenaftileno 20.08 152
Acenafteno 20.61 153
Fluoreno 22.59 166
Fenantreno 27.14 178
Antraceno 27.38 178
Fluoranteno 34.38 202
Pireno 35.92 202
Criseno 44.83 228
1,2-Benzoantraceno 45.12 228
Benzo(k)fluoranteno 55.92 252
Benzo(b)fluoranteno 56.15 252
Benzo(a)pireno 60.32 252
Indeno(1,2,3-c,d)-pireno 51.25 276
1,2:5,6-Dibenzoantraceno 51.74 278
1,12-Benzoperileno 57.65 276
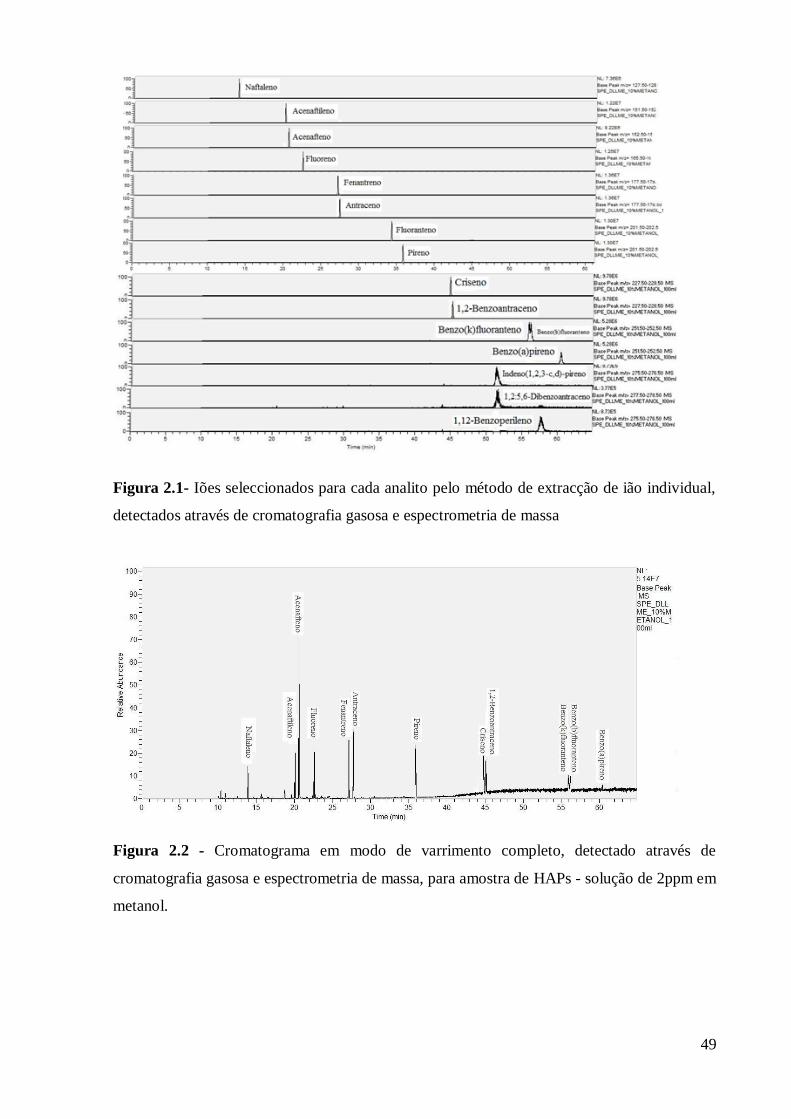
49
Figura 2.1- Iões seleccionados para cada analito pelo método de extracção de ião individual,
detectados através de cromatografia gasosa e espectrometria de massa
Figura 2.2 - Cromatograma em modo de varrimento completo, detectado através de
cromatografia gasosa e espectrometria de massa, para amostra de HAPs - solução de 2ppm em
metanol.

50
Assim neste trabalho adoptamos a estratégia de abordar, em primeiro lugar, as questões
relacionadas com o processo de microextracção líquido-líquido dispersiva recorrendo,
inicialmente, ao uso de solventes halogenados, posteriormente, aos solventes não
halogenados, assim como o equilíbrio e fases para as combinações dos três componentes do
sistema e estudar as questões relacionadas com a partição dos analitos.
2.3.2 Estudo do processo de DLLME (Microextracção Líquido-Líquido
Dispersiva)
2.3.2.1 Solventes halogenados
A utilização de solventes halogenados em DLLME é a opção mais comum encontrada na
literatura, e tem as vantagens óbvias de permitir que a fase a recolher seja sedimentada no
fundo cónico do frasco de extracção facilitando assim a separação de fases e a solubilidade da
água nestes solventes ser suficientemente baixa para permitir a obtenção de uma fase orgânica
praticamente isenta de água dispensando portanto, um passo adicional de secagem. Por outro
lado, apesar de serem solventes com maior toxicidade do que os solventes não halogenados,
sendo as quantidades utilizadas normalmente inferiores a 50 µL por extracção, as
consequências ambientais da sua utilização é diferente da manipulação dos mesmos solventes
em volumes mais elevados, como é o caso, por exemplo, na extracção líquido-líquido
clássica.
Seleccionaram-se três solventes de extracção halogenados (tetracloreto de carbono,
clorofórmio e tetracloroetileno), que se caracterizam por possuir diferentes densidades,
solubilidades em água momento dipolar e constante dieléctrica (Tabela 2.4). Estes quatro
parâmetros podem afectar directamente tanto o equilíbrio e separação de fases, como a
partição dos analitos entre a fase orgânica e a fase aquosa.
Tabela 2.4 - Densidades, solubilidades em água, momentos dipolares e constantes dieléctricas
dos solventes halogenados incluídos neste estudo (14)
.
Solvente
Densidade
a 20 ºC
(g/mL)
Solubilidade em
água a 25ºC (g/L)
Momento
Dipolar
(D)
Constante
Dieléctrica
Tetracloreto de Carbono 1.593 0.65 0 2.24
Clorofórmio 1.489 8.00 1.04 4.81
Tetracloroetileno 1.623 0.21 0 2.27

51
Como se pode observar na Tabela 2.4, os solventes seleccionados exibem uma gama de
densidades desde o clorofórmio com a densidade mais próxima da água (1.489 g/mL) até ao
tetracloroetileno com uma densidade de 1.623 g/mL. Esta propriedade deverá ter um reflexo
directo na qualidade da emulsão formada e na subsequente separação de fases. Para solventes
com uma densidade muito superior à da água, terá que ser utilizada uma maior de quantidade
de solvente de dispersão para provocar a emulsificação das fases, enquanto os solventes com
densidade mais próxima da água, poderão formar uma emulsão estável com uma menor
quantidade do solvente de dispersão, e podem mesmo apresentar interfaces instáveis após a
centrifugação.
A solubilidade do solvente de extracção em água determina qual o volume de solvente de
extracção sedimentado e pode afectar a partição dos analitos. Estes dois factores afectam de
forma inversa a eficiência do processo de extracção, nos seus parâmetros factor de
enriquecimento (EF) e percentagem de extracção (%E).
Se a solubilidade do solvente de extracção em água for relativamente elevada, a fase aquosa
pode conter uma quantidade de moléculas orgânicas suficiente para promover uma razoável
solvatação das moléculas dos solutos, e desta forma, impedir a sua migração eficiente para a
fase orgânica, mas por outro lado o volume sedimentado será menor o que resulta num melhor
factor de enriquecimento dos analitos na fase orgânica.
O inverso deverá ocorrer quando a solubilidade do solvente de extracção em água for baixa
tendo, como resultado, um maior volume sedimentado, um menor factor de enriquecimento
mas, geralmente uma maior percentagem de recuperação.
Os solventes seleccionados têm também uma diversa gama de solubilidades em água, desde o
tetracloroetileno com 0.21 g/L até ao clorofórmio com uma solubilidade de 8.0 g/L (Tabela
2.4).
O momento dipolar e a constante dielétrica de cada solvente são parâmetros que condicionam
a sua capacidade de solvatação de outras moléculas pelo que considerámos interessante
incluir neste estudo solventes com valores bastante diferentes destes parâmetros.
Tendo em conta a densidade e a solubilidade em água de cada solvente considerado, é
possível calcular a quantidade desse solvente que se dissolveria num determinado volume de
água a 25ºC. A massa e volume de saturação em 5 mL de água, a 25ºC, são apresentados na
Tabela 2.5, para os solventes halogenados incluídos neste estudo. Estes valores são apenas
indicativos do comportamento real do solvente de extracção, pois não é contabilizado o efeito
do solvente de dispersão que pode afectar significativamente a solubilidade do solvente de

52
extracção na fase aquosa. Este cálculo permite no entanto estimar qual a quantidade de
solvente de extracção que deverá utilizar para obter um determinado volume de fase
sedimentada.
Tabela 2.5 – Massa (msol) ou volume (Vsol) de solvente halogenado necessário para saturar 5
mL de água, a 25ºC.
Solvente msol / 5 mL de água
(mg) Vsol / 5 mL de água
(µL)
Tetracloreto de Carbono 3.25 2.04
Clorofórmio 40.00 26.86
Tetracloroetileno 1.05 0.65
Os solventes de dispersão seleccionados para este estudo foram: a acetona, o metanol e o
isopropanol. A acetona e o metanol são os solventes de dispersão mais frequentemente
utilizados. A selecção do isopropanol foi motivada pelo facto de serem solventes menos
polares que o metanol. Na Tabela 2.6 apresentam-se os valores de densidade, solubilidade em
água, momento dipolar e constante dieléctrica dos solventes de dispersão seleccionados.
Tabela 2.6 - Densidades, solubilidades, momentos dipolares e constantes dieléctricas dos
solventes de dispersão incluídos neste estudo (14)
.
Solvente de
Dispersão
Densidade
a 20 ºC
(g/mL)
Solubilidade em
água a 25ºC
(g/L)
Momento
Dipolar
(D)
Constante
Dieléctrica
Acetona 0.7845
Miscível
2.88 21.01
Metanol 0.7914 1.70 33.00
Isopropanol 0.7809 1.58 20.18
Os solventes de dispersão seleccionados apresentam densidades semelhantes, situadas na
gama de 0.7809 g/mL a 0.7914 g/mL, sendo o metanol o mais denso.
Os momentos dipolares dos solventes de dispersão seleccionados situam-se na gama de 1.58
D até 2.88 D, enquanto as suas constantes dieléctricas variam entre 20.18 e 33.00. A acetona é
o solvente com maior momento dipolar, enquanto o metanol apresenta uma maior constante
dieléctrica, por conseguinte o isopropanol é o solvente com menor valor para estes dois
parâmetros.

53
2.3.2.1.1 Avaliação do equilíbrio de fases
As diferentes combinações de solventes de extracção e solventes de dispersão foram testadas
quanto à qualidade da dispersão produzida e quanto ao volume de fase sedimentada,
utilizando as seguintes condições de DLLME: volume de água = 5 mL; volume do solvente
de dispersão =1000 L; volume de solvente de extracção 50 L.
Adicionaram-se então ao tubo de centrífuga, 5mL da amostra aquosa, fortificada com 2 μg/L
de HAPs. Em seguida o solvente de extracção misturado com o solvente de dispersão,
efectuou-se a centrifugação das amostras a uma velocidade de 3000 rpm durante 6 minutos.
A dispersão foi classificada como boa quando se formou uma emulsão fina, provocando a
turvação homogénea da fase aquosa e essa emulsão permaneceu estável e inalterada durante
períodos iguais ou superiores a cinco minutos. Quando a solução aquosa ficava límpida
alguns segundos após a adição dos solventes orgânicos, considerou-se que a dispersão foi
fraca (Tabela 2.7). As combinações de solventes que deram origem a dispersões boas foram
seleccionadas para os ensaios seguintes pois em qualquer destes casos ocorre dispersão que se
mantém durante alguns segundos e portanto é possível extrair os analitos. A estabilidade da
dispersão não é um factor decisivo na eficácia de extracção pois, como já foi referido, a
transferência de analitos entre as fases ocorre de forma praticamente instantânea devido à
grande superfície de contacto entre as fases. Já no caso da dispersão fraca, não chega a ocorrer
a formação de emulsão pelo que essas combinações de solventes não foram seleccionadas
para os ensaios subsequentes.
De acordo com estes critérios foram considerados adequados do ponto de vista de equilíbrio
de fases todos os solventes de extracção e de dispersão.
Tabela 2.7 - Avaliação da qualidade da dispersão para diferentes combinações de solvente de
extracção e solvente de dispersão através processo de extracção DLLME
Solventes de Dispersão
Solventes de Extracção
Tetracloreto de
carbono Clorofórmio Tetracloroetileno
Volume do solvente de
extracção (µL) 50 50 50
Acetona Boa NE NE
Metanol Boa Boa Boa
Isopropanol Boa Boa Boa
NE - ensaio não efectuado

54
Tabela 2.8 - Volume de fase orgânica sedimentada para as diferentes combinações de solvente de
extracção e solvente de dispersão, através do método de extracção DLLME
Solventes de
Dispersão
Volume Sedimentado (µL) Volume de
Extracção
(µL)
Tetracloreto
de carbono Clorofórmio Tetracloroetileno
Medido Medido Medido
Acetona 28 NE NE 50
Metanol 32 _ 31 50
Isopropanol 32 10 31 50
NE - ensaio não efectuado
(_)- não se obteve volume sedimentado
Tabela 2.9 - Factores de enriquecimento dos HAPS em DLLME efectuada com diversas combinações
de solventes.
Analitos
Solvente
de
Dispersão
Solvente de Extracção
Tetracloreto
de carbono Clorofórmio Tetracloroetileno
Naftaleno
Ace
ton
a
0 NE NE
Acenaftileno 20 NE NE
Acenafteno 55 NE NE
Fluoreno 0 NE NE
Fenantreno 133 NE NE
Antraceno 162 NE NE
Fluoranteno 69 NE NE
Pireno 34 NE NE
Criseno 0 NE NE
1,2-Benzoantraceno 0 NE NE
Benzo(k)fluoranteno 0 NE NE
Benzo(b)fluoranteno 0 NE NE
Benzo(a)pireno 0 NE NE
Naftaleno
Met
an
ol
7 Não extraiu 7
Acenaftileno 4 Não extraiu 5
Acenafteno 2 Não extraiu 6
Fluoreno 6 Não extraiu 7
Fenantreno 4 Não extraiu 5
Antraceno 3 Não extraiu 5
Fluoranteno 4 Não extraiu 6
Pireno 4 Não extraiu 7
Criseno 6 Não extraiu 7
1,2-Benzoantraceno 11 Não extraiu 7
Benzo(k)fluoranteno 0 Não extraiu 0
Benzo(b)fluoranteno 0 Não extraiu 0
Benzo(a)pireno 0 Não extraiu 0

55
Naftaleno
Iso
prop
an
ol
6 1 4
Acenaftileno 5 1 4
Acenafteno 6 2 4
Fluoreno 7 1 5
Fenantreno 5 1 4
Antraceno 5 1 3
Fluoranteno 7 1 5
Pireno 8 1 6
Criseno 9 1 5
1,2-Benzoantraceno 9 1 7
Benzo(k)fluoranteno 0 0 0
Benzo(b)fluoranteno 0 0 0
Benzo(a)pireno 0 0 0
NE - ensaio não efectuado
Relativamente aos factores de enriquecimento com a utilização dos solventes halogenados,
através da sua aplicação em DLLME, os valores mais elevados foram obtidos para o sistema
tetracloreto de carbono e acetona, mas é necessário mencionar que a maioria dos analitos não
foi extraída. Os analitos benzo(k)fluoranteno, benzo(b)fluoranteno e benzo(a)pireno não
foram extraídos com estas combinações de solventes de dispersão e extracção.
2.3.2.2 Solventes não halogenados
Os solventes não halogenados têm vantagens relativamente aos solventes halogenados devido
à sua menor toxicidade tornando o seu uso prioritário na área da chamada “Química Verde” e
suscitando portanto o interesse da sua aplicação em DLLME.
O primeiro problema a defrontar na aplicação de solventes não halogenados em DLLME é o
facto de, sendo a fase orgânica menos densa que a aquosa, se obter após a centrifugação das
fases, não uma fase sedimentada no fundo cónico do frasco, mas sim um filme fino de fase
orgânica sobrenadante; a remoção da fase orgânica, na posição de fase superior no sistema
difásico resultaria em erros grosseiros na determinação do volume de fase orgânica separado
após a extracção.
Assim propôs-se um sistema alternativo de recolha da fase orgânica sobrenadante que permite
a sua separação mais eficiente relativamente à fase aquosa e uma medida mais exacta do seu
volume: após a centrifugação a fase aquosa é removida quase na totalidade, recorrendo a uma
seringa de 5 mL equipada com uma agulha fina e longa; em seguida repete-se a centrifugação
(8 min, 3000 rpm) para promover a acumulação máxima da fase orgânica no fundo cónico do
frasco de extracção; remove-se então o que resta de fase aquosa ficando no frasco de
extracção apenas a fase orgânica, que é de seguida removida com uma microseringa adequada

56
à determinação do seu volume. Esta técnica permite a recolha de volumes reprodutíveis de
fase sedimentada com diversas combinações de solventes de extracção e de dispersão pelo
que foi adoptada nos ensaios de DLLME com solventes não halogenados.
Os solventes de extracção seleccionados foram o ciclohexano, o tolueno e o octano. Na
Tabela 2.10 apresentam-se algumas propriedades físicas e químicas dos solventes não
halogenados seleccionados para os ensaios de DLLME.
Tabela 2.10 – Densidade, solubilidade em água a 25 ºC, momento dipolar e constante
dieléctrica dos solventes não halogenados seleccionados para este trabalho (14)
.
Solventes
Densidade
a 20 ºC
(g/mL)
Solubilidade em
água a 25ºC
(g/L)
Momento
Dipolar
(D)
Constante
Dieléctrica
Ciclohexano 0.7790 0.05800 0.00 2.0243
Tolueno 0.8647 0.53000 0.31 2.3800
Octano 0.7030 0.00073 0.00 1.9480
A solubilidade em água a 25ºC e a constante dieléctrica são mais baixas do as mesmas
propriedades no caso dos solventes halogenados estudados anteriormente pelo que estes três
solventes não halogenados têm um bom potencial como solventes de extracção em DLLME.
A massa e volume de cada um destes solventes necessários para saturar 5 mL de água
(volume típico das amostras em DLLME) são apresentados na Tabela 2.11. Como se pode
verificar os volumes de solvente de extracção retidos na fase aquosa estarão na gama de 0.005
µL a 3.065 µL, ou seja, é expectável que a maior parte do solvente de extracção seja recolhido
na fase sedimentada.
Tabela 2.11 – Massa (msol) ou volume (Vsol) de alguns solventes não halogenados necessário
para saturar 5 mL de água, a 25ºC.
Solvente msol / 5 mL de água
(mg) Vsol / 5 mL de água
(µL)
Ciclohexano 0.290 0.372
Tolueno 2.650 3.065
Octano 0.004 0.005

57
2.3.2.2.1 Avaliação do equilíbrio de fases
O equilíbrio de fases obtido em DLLME com misturas de solventes não halogenados foi
avaliado tal como foi efectuado para os solventes halogenados por apreciação da qualidade de
dispersão e do volume de fase sedimentada obtida nas diferentes combinações de solventes de
extracção e de dispersão. Utilizou-se um volume de amostra de água 5mL, um volume de
solvente de dispersão de 1mL e um volume de solvente de extracção de 50 µL.
Os solventes de dispersão seleccionados para os ensaios com solventes de extracção não
halogenados foram os mesmos que se utilizaram com os solventes halogenados: acetona,
metanol e isoproponal.
As dispersões obtidas com os solventes não halogenados foram no geral, boas exceptuando o
caso da mistura octano/metanol.
Tabela 2.12 - Avaliação da qualidade da dispersão para diferentes combinações de solvente
de extracção e solvente de dispersão, através do método de extracção DLLME
Solventes de Dispersão Solventes de Extracção
Ciclohexano Tolueno Octano
Volume do solvente de
extracção (µL) 50 50 50
Acetona Boa Boa Boa
Metanol Boa Boa Má
Isopropanol Boa Boa Boa
Tabela 2.13 - Volume de fase orgânica sedimentada para as diferentes combinações de
solvente de extracção e solvente de dispersão, através do método de extracção DLLME
Solventes de
Dispersão
(1mL)
Volume Sedimentado (µL) Volume de
Extracção
(µL)
Ciclohexano Tolueno Octano
Medido (n=2) Medido
(n=1) Medido (n=2)
Acetona 20.0 30.0 24.0 50
Metanol 26.5 16.0 38.5 50
Isopropanol 24.0 22.0 41.5 50

58
Os factores de enriquecimento dos analitos na fase orgânica foram testados, para todas as
possíveis combinações dos solventes de extracção ciclohexano, tolueno e octano com os
solventes de dispersão acetona, metanol e isopropanol.
Tabela 2.14 - Factores de enriquecimento dos HAPs em DLLME efectuada com diversas
combinações de solventes.
Analitos
Solvente
de
Dispersão
Solvente de Extracção
Ciclohexano Tolueno Octano
Naftaleno
Ace
ton
a
115 199 121
Acenaftileno 55 98 28
Acenafteno 89 131 101
Fluoreno 86 135 102
Fenantreno 29 77 106
Antraceno 140 78 85
Fluoranteno 78 82 0
Pireno 185 0 0
Criseno 0 0 53
1,2-Benzoantraceno 0 0 97
Benzo(k)fluoranteno 8 472 59
Benzo(b)fluoranteno 16 0 10
Benzo(a)pireno 212 0 0
Naftaleno
Met
an
ol
176 1 395
Acenaftileno 70 1 112
Acenafteno 21 2 212
Fluoreno 94 1 192
Fenantreno 53 1 160
Antraceno 81 1 164
Fluoranteno 50 1 181
Pireno 92 1 0
Criseno 0 2 0
1,2-Benzoantraceno 0 2 0
Benzo(k)fluoranteno 0 3 54
Benzo(b)fluoranteno 0 2 0
Benzo(a)pireno 0 0 0
Naftaleno
Isop
rop
an
ol
Não extraíu 209 148
Acenaftileno Não extraíu 89 58
Acenafteno Não extraíu 116 106
Fluoreno Não extraíu 120 80
Fenantreno Não extraíu 81 80
Antraceno Não extraíu 78 85
Fluoranteno Não extraíu 90 115
Pireno Não extraíu 0 0
Criseno Não extraíu 8 19
1,2-Benzoantraceno Não extraíu 17 20
Benzo(k)fluoranteno Não extraíu 5 8
Benzo(b)fluoranteno Não extraíu 0 0
Benzo(a)pireno Não extraíu 0 0

59
As combinações de solventes para as quais se obteve um maior factor de enriquecimento
foram: ciclohexano-acetona; tolueno-acetona; octano-acetona; octano-metanol; tolueno-
isopropanol; isopropanol-octano. O analito benzo(a)pireno só foi extraído pela combinação de
solventes ciclohexano/acetona.
Em suma, é possível afirmar a aplicação deste método de DLLME à extracção dos HAPs de
matriz é adequado para a maioria dos analitos estudados, no entanto, os factores de
enriquecimento obtidos foram relativamente baixos, não permitindo atingir para todos os
analitos testados o limite de quantificação de 0,1 µg/L, requerido na legislação. No entanto,
tendo-se constatado através da injecção regular de padrões de HAPs que a sensibilidade
analítica do GC-MS sofreu flutuações significativas durante a realização deste trabalho não é
de excluir que a aplicação do método de DLLME estudado, em condições de sensibilidade
analítica elevada possa permitir atingir os limites de quantificação requeridos nas normas para
águas.
Ainda assim procurou-se testar um método cuja maior capacidade de concentração dos
analitos permitisse ultrapassar as limitações de sensibilidade do equipamento analítico.
Efectuou-se então o estudo da técnica hifenada de SPE-HLLME que ao associar dois
processos de concentração de analitos permite atingir condições de ultraconcentração de
compostos orgânicos a partir de matrizes aquosas.
2.4 Estudo do processo de SPE-HLLME
2.4.1 Avaliação do equilíbrio de fases
Apesar da selecção de condições em DLLME sejam adequadas à determinação de HAPs,
estas tiveram que ser previamente testadas. A associação entre SPE e HLLME introduz
algumas alterações no equilíbrio de fases uma vez que a água presente na coluna de SPE após
a extracção da amostra é eluída pelo solvente de dispersão e afecta portanto os volumes
relativos dos componentes do sistema. Assim procurou-se testar a eficácia das diferentes
combinações de solventes não halogenados e solventes de dispersão na determinação de
HAPs por SPE-HLLME.
Avaliou-se a qualidade da dispersão produzida e o volume de fase sedimentada, utilizando as
seguintes condições de HLLME: volume de água = 5mL; volume de solvente de dispersão =
1mL; volume de solvente de extracção 50 µL.

60
O processo de extracção em fase sólida foi efectuado utilizando colunas com fase estacionária
C18, que foram previamente condicionadas. Esse condicionamento foi feito passando pela
coluna e em vácuo, 5 mL do solvente metanol e água. A amostra aquosa (100mL) foi
fortificada com 2 μg/L de HAPs.
Após o condicionamento da coluna, foi efectuada a filtração das amostras, sob vácuo, a um
caudal médio de 10 mL/min. Em seguida efectuou-se a eluição dos analitos utilizando 1,0 mL
de solvente de eluição/dispersão, que foi recolhido no tubo de centrífuga onde foi efectuado o
passo de HLLME. Adicionaram-se, então, ao tubo de centrífuga de solvente de extracção,
seguidos de 5 mL de água, seguidamente efectuou-se a centrifugação das amostras a uma
velocidade de 3000 rpm durante 6 minutos. Após a centrifugação, com auxílio de uma
microseringa de vidro e com uma agulha comprida, retirámos alguma fase aquosa e voltámos
a centrifugar a 3000 rpm durante 8 minutos. Volta-se a retirar a restante fase aquosa. O eluato
obtido é removido com uma microseringa e colocado num redutor. O redutor é colocado num
frasco e imediatamente capsulado e colocado no tabuleiro do injector automático do
cromatógrafo, para se proceder à sua análise.
As dispersões obtidas com os solventes não halogenados foram no geral, boas exceptuando o
caso da mistura octano/metanol.
Tabela 2.15 - Avaliação da qualidade da dispersão para diferentes combinações de solvente
de extracção e solvente de dispersão, através do método de extracção SPE-HLLME
Solventes de
Dispersão
Solventes de Extracção
Ciclohexano Tolueno Octano
Volume do solvente
de extracção
(µL)
50 50 50
Acetona Boa Boa Boa
Metanol Boa Boa Má
Isopropanol Boa Boa Boa
Na Tabela 2.16, apresentam-se os volumes sedimentados obtidos para cada combinação de
solventes após a centrifugação e o volume sedimentado teórico que seria obtido considerando
apenas a solubilidade de cada solvente de extracção em água pura a 25ºC. O volume
sedimentado teórico é a diferença entre o volume adicionado e o volume solúvel em 5 mL de
água a 25ºC. O volume sedimentado medido é, geralmente, inferior ao valor teórico pois a

61
presença do solvente de dispersão pode aumentar ligeiramente a solubilidade do solvente de
extracção na fase aquosa.
Tanto o ciclohexano, como o tolueno e o octano permitem obter fase sedimentada em todos os
ensaios realizados, sendo o octano o solvente de extracção onde se obteve maior volume
sedimentado.
Tabela 2.16 - Volume de fase orgânica sedimentada para as diferentes combinações de
solvente de extracção e solvente de dispersão, através do método de extracção SPE-HLLME;
(volume de solvente de extracção = 50 µL, volume de solvente de dispersão = 1 mL, volume
de água = 5 mL, caudal médio de 10 mL/min, amostra aquosa (100mL) foi fortificada com 2
μg/L de HAPs)
Solventes de
Dispersão
Volume Sedimentado (µL)
Ciclohexano Tolueno Octano
Medido Teórico Medido Teórico Medido Teórico
Acetona 18 49.628 20 46.935 26 49.995
Metanol 16 49.628 17 46.935 23 49.995
Isopropanol 12 49.628 20 46.935 41 49.995
A presença do solvente de dispersão induz uma solubilização adicional do solvente de
extracção na fase aquosa pelo que os volumes sedimentados obtidos são 24% a 82% do que
seriam na ausência do solvente de dispersão.
Os factores de enriquecimento dos analitos na fase orgânica foram testados, para todas as
possíveis combinações dos solventes de extracção ciclohexano, tolueno e octano com os
solventes de dispersão acetona, metanol e isopropanol. As combinações de solventes para as
quais se obtiveram um maior factor de enriquecimento foram para as combinações
ciclohexano/acetona; ciclohexano/isopropanol e octano/isopropanol. O sistema
ciclohexano/acetona foi seleccionado para os ensaios seguintes como o melhor sistema para
extrair os HAPs, pois tem factores de enriquecimento superiores a todos os outros sistemas.

62
Tabela 2.17 - Factores de enriquecimento dos HAPs em SPE-HLLME efectuada com
diversas combinações de solventes (volume de solvente de extracção = 50 µL, volume de
solvente de dispersão = 1 mL, volume de água = 5 mL, caudal médio de 10 mL/min, amostra
aquosa (100mL) foi fortificada com 2 μg/L de HAPs).
Analitos
Solvente
de
Dispersão
Solvente de Extracção
Ciclohexano Tolueno Octano
Naftaleno
Ace
ton
a
3169 2176 2036
Acenaftileno 2703 1512 1369
Acenafteno 5547 2777 2875
Fluoreno 2756 1327 1307
Fenantreno 2522 1076 1034
Antraceno 2474 1043 1047
Fluoranteno 2325 863 862
Pireno 2324 799 822
Criseno 2200 752 843
1,2-Benzoantraceno 2134 785 840
Benzo(k)fluoranteno 1811 671 738
Benzo(b)fluoranteno 1528 660 548
Benzo(a)pireno 1536 623 325
Naftaleno
Met
an
ol
2256 2409 1183
Acenaftileno 1565 1584 690
Acenafteno 2364 1916 991
Fluoreno 975 757 368
Fenantreno 584 414 201
Antraceno 447 252 143
Fluoranteno 195 76 60
Pireno 115 34 36
Criseno 112 55 33
1,2-Benzoantraceno 88 51 25
Benzo(k)fluoranteno 0 0 0
Benzo(b)fluoranteno 0 0 0
Benzo(a)pireno 0 0 0
Naftaleno
Isop
rop
an
ol
3824 3554 3626
Acenaftileno 2891 2341 2357
Acenafteno 5509 4217 4551
Fluoreno 2606 1671 1938
Fenantreno 2080 1085 1424
Antraceno 1835 958 1115
Fluoranteno 1175 620 830
Pireno 985 523 679
Criseno 628 431 488
1,2-Benzoantraceno 504 330 385
Benzo(k)fluoranteno 451 285 400
Benzo(b)fluoranteno 500 387 366
Benzo(a)pireno 367 268 286

63
2.4.2. Avaliação dos factores de enriquecimento normalizados para um volume de
fase orgânica sedimentada de 20 μL
Apesar de os volumes sedimentados apresentados na Tabela 2.16, serem relativamente
homogéneos e próximos de 20 μL, para algumas combinações de solvente de extracção e
solvente de dispersão obtiveram-se volumes sedimentados distintos por excesso ou por
defeito. Como este parâmetro é determinante na concentração final obtida e portanto no factor
de enriquecimento, decidiu-se recalcular os factores de enriquecimento considerando que a
eficiência de extracção se mantinha constante (mesma massa de analitos extraída), mas num
volume de fase sedimentada normalizada para um valor de 20 μL. Esta normalização
pressupõe que nos situamos numa região de equilíbrio de fases na qual a partição dos analitos
não é afectada por variações no volume de fase sedimentada que produzem apenas um efeito
de concentração ou diluição dos analitos. Este pressuposto requer obviamente a confirmação
através de medidas experimentais mas permite estimar qual a máxima variação nos factores
de enriquecimento que se poderia obter com uma determinada variação do volume do
solvente de extracção.
Os factores de enriquecimento obtidos nesta série de cálculos encontram-se representados na
tabela seguinte.
Tabela 2.18 - Factores de enriquecimento dos HAPs em SPE-HLLME efectuada com
diversas combinações de solventes, com fixação do volume de fase orgânica sedimentada em
20 μL.
Analitos
Solvente
de
Dispersão
Solvente de Extracção
Ciclohexano Tolueno Octano
Naftaleno
Ace
ton
a
3521 2176 1566
Acenaftileno 3003 1512 1053
Acenafteno 6163 2777 2211
Fluoreno 3062 1327 1006
Fenantreno 2802 1076 795
Antraceno 2749 1043 805
Fluoranteno 2584 863 663
Pireno 2582 799 633
Criseno 2445 752 649
1,2-Benzoantraceno 2372 785 646
Benzo(k)fluoranteno 2013 671 568
Benzo(b)fluoranteno 1697 660 421
Benzo(a)pireno 1706 623 250

64
Naftaleno
Met
an
ol
2821 2835 1029
Acenaftileno 1956 1864 600
Acenafteno 2955 2255 861
Fluoreno 1218 890 320
Fenantreno 730 487 175
Antraceno 559 297 125
Fluoranteno 244 90 52
Pireno 144 40 32
Criseno 140 64 28
1,2-Benzoantraceno 109 60 22
Benzo(k)fluoranteno 0 0 0
Benzo(b)fluoranteno 0 0 0
Benzo(a)pireno 0 0 0
Naftaleno
Isop
rop
an
ol
6374 3554 1769
Acenaftileno 4819 2341 1150
Acenafteno 4470 2021 1075
Fluoreno 4343 1671 945
Fenantreno 3467 1085 695
Antraceno 3058 958 544
Fluoranteno 1958 620 405
Pireno 1641 523 331
Criseno 1047 431 238
1,2-Benzoantraceno 840 330 188
Benzo(k)fluoranteno 752 285 195
Benzo(b)fluoranteno 833 387 178
Benzo(a)pireno 612 268 140
Os factores de enriquecimento normalizados para um volume sedimentado de 20 μL foram
mais elevados para a maior parte dos analitos, no caso do sistema ciclohexano/acetona; este
sistema apresentou factores de enriquecimento substancialmente maiores para os analitos
menos voláteis em particular para o benzo(a)pireno.
A comparação dos factores de enriquecimento obtidos na extracção dos HAPs a partir de
amostras de água utilizando os sistemas ciclohexano/acetona e ciclohexano/isopropanol é
ilustrada na Figura 2.3.
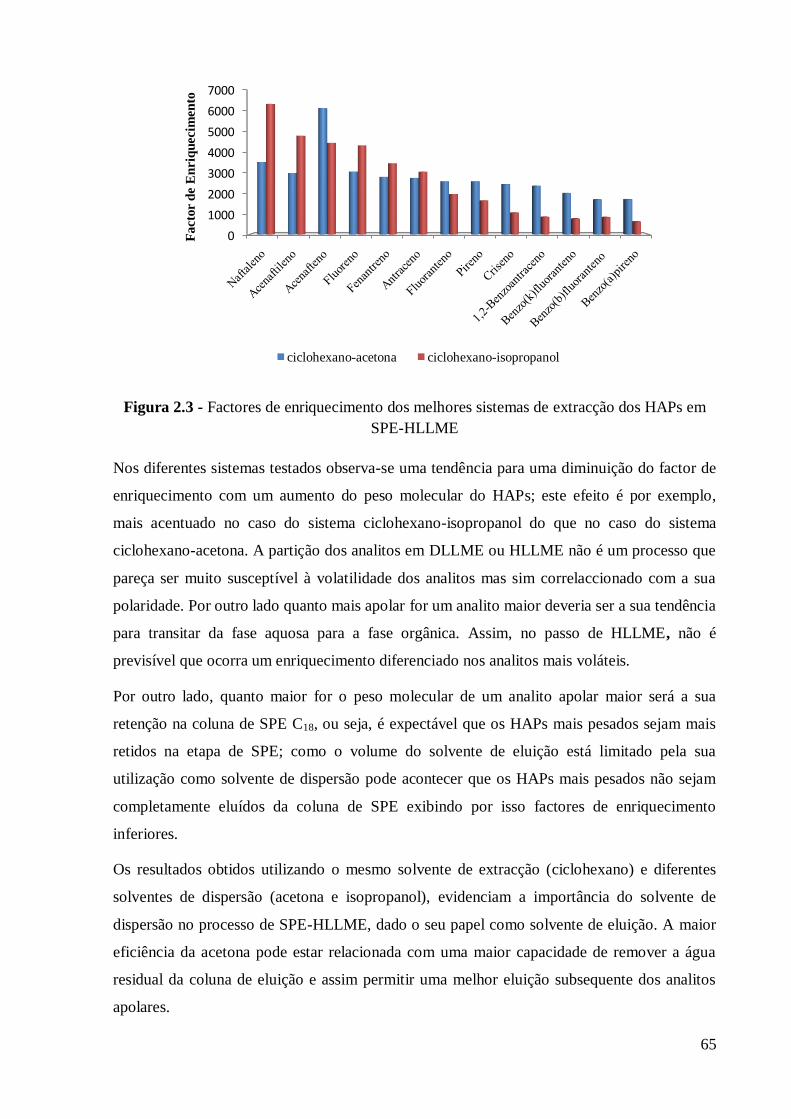
65
Figura 2.3 - Factores de enriquecimento dos melhores sistemas de extracção dos HAPs em
SPE-HLLME
Nos diferentes sistemas testados observa-se uma tendência para uma diminuição do factor de
enriquecimento com um aumento do peso molecular do HAPs; este efeito é por exemplo,
mais acentuado no caso do sistema ciclohexano-isopropanol do que no caso do sistema
ciclohexano-acetona. A partição dos analitos em DLLME ou HLLME não é um processo que
pareça ser muito susceptível à volatilidade dos analitos mas sim correlaccionado com a sua
polaridade. Por outro lado quanto mais apolar for um analito maior deveria ser a sua tendência
para transitar da fase aquosa para a fase orgânica. Assim, no passo de HLLME, não é
previsível que ocorra um enriquecimento diferenciado nos analitos mais voláteis.
Por outro lado, quanto maior for o peso molecular de um analito apolar maior será a sua
retenção na coluna de SPE C18, ou seja, é expectável que os HAPs mais pesados sejam mais
retidos na etapa de SPE; como o volume do solvente de eluição está limitado pela sua
utilização como solvente de dispersão pode acontecer que os HAPs mais pesados não sejam
completamente eluídos da coluna de SPE exibindo por isso factores de enriquecimento
inferiores.
Os resultados obtidos utilizando o mesmo solvente de extracção (ciclohexano) e diferentes
solventes de dispersão (acetona e isopropanol), evidenciam a importância do solvente de
dispersão no processo de SPE-HLLME, dado o seu papel como solvente de eluição. A maior
eficiência da acetona pode estar relacionada com uma maior capacidade de remover a água
residual da coluna de eluição e assim permitir uma melhor eluição subsequente dos analitos
apolares.
0
1000
2000
3000
4000
5000
6000
7000
Facto
r d
e E
nriq
uecim
en
to
ciclohexano-acetona ciclohexano-isopropanol
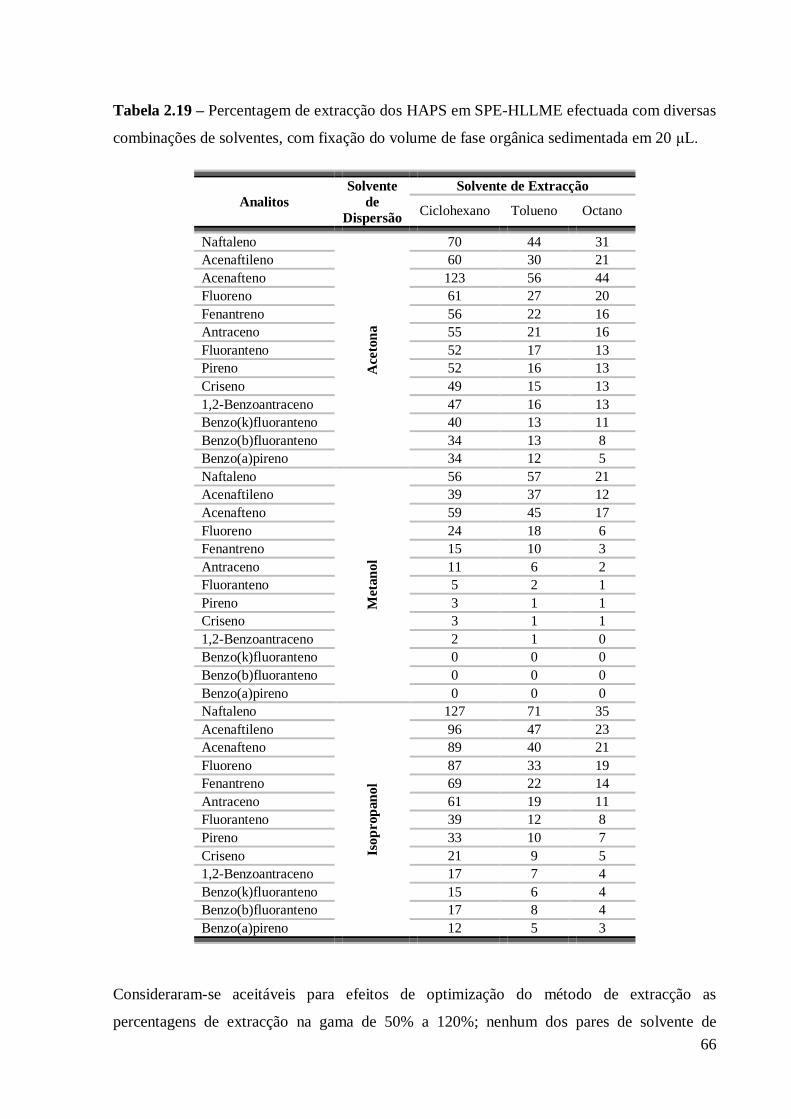
66
Tabela 2.19 – Percentagem de extracção dos HAPS em SPE-HLLME efectuada com diversas
combinações de solventes, com fixação do volume de fase orgânica sedimentada em 20 μL.
Analitos
Solvente
de
Dispersão
Solvente de Extracção
Ciclohexano Tolueno Octano
Naftaleno
Ace
ton
a
70 44 31
Acenaftileno 60 30 21
Acenafteno 123 56 44
Fluoreno 61 27 20
Fenantreno 56 22 16
Antraceno 55 21 16
Fluoranteno 52 17 13
Pireno 52 16 13
Criseno 49 15 13
1,2-Benzoantraceno 47 16 13
Benzo(k)fluoranteno 40 13 11
Benzo(b)fluoranteno 34 13 8
Benzo(a)pireno 34 12 5
Naftaleno
Met
an
ol
56 57 21
Acenaftileno 39 37 12
Acenafteno 59 45 17
Fluoreno 24 18 6
Fenantreno 15 10 3
Antraceno 11 6 2
Fluoranteno 5 2 1
Pireno 3 1 1
Criseno 3 1 1
1,2-Benzoantraceno 2 1 0
Benzo(k)fluoranteno 0 0 0
Benzo(b)fluoranteno 0 0 0
Benzo(a)pireno 0 0 0
Naftaleno
Isop
rop
an
ol
127 71 35
Acenaftileno 96 47 23
Acenafteno 89 40 21
Fluoreno 87 33 19
Fenantreno 69 22 14
Antraceno 61 19 11
Fluoranteno 39 12 8
Pireno 33 10 7
Criseno 21 9 5
1,2-Benzoantraceno 17 7 4
Benzo(k)fluoranteno 15 6 4
Benzo(b)fluoranteno 17 8 4
Benzo(a)pireno 12 5 3
Consideraram-se aceitáveis para efeitos de optimização do método de extracção as
percentagens de extracção na gama de 50% a 120%; nenhum dos pares de solvente de

67
extracção / solvente de dispersão atingiram esta gama para todos os analitos, no entanto a
combinação de solventes que mais se aproximou destes valores foi o sistema ciclohexano-
acetona com percentagens de extracção a variar na gama de 34% a 123%.
Embora nos trabalhos envolvendo DLLME ou HLLME se obtenham tipicamente
percentagens de extracção próximas de 100% porque a extracção dos analitos é potenciada
pela grande superfície de contacto entre as fases, em SPE-HLLME não é sempre possível
utilizar o volume de solvente de eluição que seria adequado à etapa de SPE, pois este volume
seria excessivo para o passo de HLLME seguinte. Assim, algum potencial de enriquecimento
adicional em SPE é sacrificado de forma a ter um volume de dispersão adequado em HLLME.
A extracção incompleta não é em si própria um problema analítico inaceitável pois esta é uma
característica de algumas técnicas como o SPME, e tal como neste caso, o que é essencial é
garantir a reproductibilidade da extracção. O enriquecimento adicional que se poderia obter
utilizando um maior volume de solvente de eluição pode ser atingido no caso de analitos não
voláteis, efectuando um passo adicional de concentração do eluente para um volume
adequado à etapa de HLLME.
Em face dos resultados obtidos, o sistema ciclohexano-acetona foi seleccionado para
optimização de alguns parâmetros do processo de SPE-HLLME.
2.4.3 Desenvolvimento e optimização do processo de SPE-HLLME
2.4.3.1 Alteração dos volumes dos solventes de extracção e de dispersão e do
volume de água na etapa de HLLME
O efeito do aumento dos volumes dos solventes de extracção e de dispersão e do volume de
água (etapa de HLLME) na eficiência do processo global foi estudado de forma conjugada,
para um volume de amostra de água de 100 mL extraída por SPE a um caudal de 10 mL/min.
A concentração de fortificação foi de 2 µg/L e o volume de água utilizado na etapa de
HLLME, variou entre 5 mL e 10 mL. O volume do solvente de extracção variou entre 50 µL e
100 µL enquanto o volume do solvente de dispersão variou entre 1 mL e 2 mL.

68
Tabela 2.20 – Avaliação da qualidade de dispersão, para diferentes combinações de volumes
de solvente de dispersão (acetona) e solvente de extracção (ciclohexano), através do método
de extracção SPE-HLLME
Solventes de
Dispersão (mL)
Solventes de Extracção
Ciclohexano Ciclohexano Ciclohexano
Volume do solvente
de extracção
(µL)
50 75 100
Acetona (mL) 1.0 1.5 2.0
Volume sedimentado
(µL) 18 24 34
Dispersão Boa Boa Boa
Tabela 2.21 - Variação dos factores de enriquecimento (EF) dos HAPs em função do volume
de acetona (Vdisp), volume de solvente de extracção (Vextr) e de volume de água Milli Q (V H20
MILLI Q)
Analitos
Factor de enriquecimento
Vdisp = 1,0 mL
V H20 MILLI Q=
5,0ml
Vextr= 50l
Vdisp = 1,5 mL
V H20 MILLI Q= 7,5ml
Vextr= 75l
Vdisp = 2,0 mL
V H20 MILLI Q= 10
ml
Vextr= 100l
Vsed=
18l
Vsed*=
20l
Vsed=
18l
Vsed*=
20l
Vsed=
18l
Vsed*=
20l
Naftaleno 3169 2852 1614 1937 850 1445
Acenaftileno 2703 2433 1435 1722 673 1144
Acenafteno 5547 4992 3173 3808 1514 2574
Fluoreno 2756 2480 1637 1964 781 1328
Fenantreno 2522 2270 1528 1834 776 1319
Antraceno 2474 2227 1602 1922 791 1345
Fluoranteno 2325 2093 1492 1790 770 1309
Pireno 2324 2092 1493 1792 796 1353
Criseno 2200 1980 1479 1775 787 1338
1,2-Benzoantraceno 2134 1921 1445 1734 775 1318
Benzo(k)fluoranteno 1811 1630 1397 1676 697 1185
Benzo(b)fluoranteno 1528 1375 1221 1465 637 1083
Benzo(a)pireno 1536 1382 1130 1356 649 1103
Vsed*- volume sedimentado normalizado para um volume de 20l
Os factores de enriquecimento dos HAPs foram mais elevados para um volume de acetona de
1 mL e 50 µL de ciclohexano para um volume de 5 mL de água mili-Q; mesmo efectuando a
normalização do volume sedimentado para um valor de 20 µL, o efeito do aumento nos
volumes de solvente de extracção e de dispersão traduziu-se numa diminuição destes factores
(Figura 2.4).

69
Figura 2.4 - Factores de enriquecimento para um aumento volume dos solventes de extracção
e de dispersão e do volume água para o sistema ciclohexano/acetona
O aumento do volume do solvente de eluição em SPE não se traduziu num aumento da
eficiência global do processo apesar das eficiências de extracção anteriormente determinadas
quando se utilizou um volume de ciclohexano de 50 µL e um volume de acetona de 1 mL,
terem sido para muitos analitos da ordem de 50% ou menores. Este resultado parece indicar
que apesar do potencial melhoramento da etapa de SPE, a utilização de maiores volumes de
solvente de extracção e de dispersão pode afectar a partição dos analitos em HLLME, pois ao
aumentar significativamente a quantidade de solventes orgânicos na água é favorecida a
solvatação dos analitos nesta fase e desfavorecida a sua migração para a fase orgânica
(solvente de extracção).
2.4.3.2 Variação do caudal no processo SPE-HLLME, para o sistema
acetona/ciclohexano
O efeito do caudal de filtração da amostra na eficiência do processo global foi estudado na
gama de 10 mL/min a 40 mL/min, para um volume de amostra de água de 100 mL (Tabela
2.22, Figura 2.5). A amostra de água foi fortificada com a mistura dos analitos num nível de 2
µg/L e foi eluída com 1 mL de acetona. No passo de HLLME utilizaram-se 50 µL de
ciclohexano e 5 mL de água.
0
1000
2000
3000
4000
5000
6000
Facto
r d
e E
nriq
uecim
en
to
1,0 mL 1,5 mL 2,0 mL

70
Tabela 2.22 - Variação dos factores de enriquecimento dos HAPs em função do caudal de
passagem da amostra
Analitos
Factor de enriquecimento
10mL/min 20mL/min 30mL/min 40mL/min
Vsed=18µL Vsed=12,5 µL Vsed=13 µL Vsed=12 µL
Naftaleno 3169 3918 2785 2507
Acenaftileno 2703 3504 2310 2018
Acenafteno 5547 3036 2160 1903
Fluoreno 2756 3172 2134 1815
Fenantreno 2522 3217 1939 1776
Antraceno 2474 3057 1882 1719
Fluoranteno 2325 3963 2089 2074
Pireno 2324 4125 2062 2112
Criseno 2200 4948 1821 2202
1,2-Benzoantraceno 2134 4916 1824 2155
Benzo(k)fluoranteno 1811 5104 1472 1564
Benzo(b)fluoranteno 1528 4731 1392 1419
Benzo(a)pireno 1536 4737 974 1394
Figura 2.5 - Factores de enriquecimento para um caudal de passagem
da amostra de 10-40 mL/min
Uma vez que os valores dos factores de enriquecimento obtidos apresentam algumas
diferenças entre os quatro caudais de passagem da amostra estudada, seleccionou-se o caudal
20mL/min, o qual se traduz num maior valor de factores de enriquecimento para todos os
analitos à excepção acenafteno.
0
1000
2000
3000
4000
5000
6000
Fa
cto
r d
e E
nriq
uecim
en
to
10 mL/min 20 mL/min 30 mL/min 40 mL/min

71
Para caudais superiores a 20 mL/min observou-se um decréscimo nos factores de
enriquecimento que pode estar relacionado com uma retenção incompleta dos analitos na
coluna de SPE. Quando a amostra foi eluida a um caudal de 10 mL/min também se obtiveram
factores de enriquecimento mais baixos do que a 20 mL/min o que não está de acordo com a
tendência esperada de ocorrer uma maior retenção quanto menor fosse a velocidade de
filtração da amostra. O volume sedimentado que se obteve no ensaio realizado com o caudal
de 10 mL/min foi maior do que nos restantes ensaios, o que provocou um efeito de diluição
sem o qual os factores de enriquecimento dos analitos mais voláteis seriam superiores aos
obtidos a 20 mL/min; no entanto este factor não explica os resultados obtidos para os analitos
mais pesados que continuam a ter factores de enriquecimento a 10 mL/min inferiores aos que
apresentam a 20 mL/min, mesmo quando é efectuada a normalização do volume sedimentado.
Uma possível explicação para este comportamento é a eventual migração dos analitos do seio
da solução aquosa para as paredes do recipiente de vidro onde a amostra é colocada durante o
processo de eluição, quando o tempo de residência da amostra nesse recipiente aumenta
muito.
Tabela 2.23 - Percentagens de extracção para o sistema de solventes optimizados
(ciclohexano/acetona), variando o caudal de passagem da amostra.
Analitos % Extracção
10mL/min 20mL/min 30mL/min 40mL/min
Naftaleno 57 49 36 30
Acenaftileno 49 44 30 24
Acenafteno 100 38 28 23
Fluoreno 50 40 28 22
Fenantreno 45 40 25 21
Antraceno 45 38 24 21
Fluoranteno 42 50 27 25
Pireno 42 52 27 25
Criseno 40 62 24 26
1,2-Benzoantraceno 38 61 24 26
Benzo(k)fluoranteno 33 64 19 19
Benzo(b)fluoranteno 27 59 18 17
Benzo(a)pireno 28 59 13 17
As percentagens de extracção obtidas parecem estar de acordo com as hipóteses enunciadas
pois de uma forma geral estas percentagens aumentam com a diminuição do caudal da
amostra excepto para os analitos menos voláteis no ensaio efectuado ao menor caudal (10
mL/min).

72
2.4.3.3 Aumento do volume de amostra no passo de SPE-HLLME
Nesta fase da optimização do método de SPE-HLLME, estudou-se o efeito da variação do
volume da amostra numa gama de 100 mL a 1000 mL e para uma concentração dos analitos
na água de 2 µg/L. O passo de HLLME foi efectuado utilizando o sistema de solventes
acetona/ciclohexano, nas condições previamente optimizadas. A filtração das amostras foi
efectuada sob vácuo, a um caudal médio de 20 mL/min.
A comparação dos factores de enriquecimento obtidos nesta série de ensaios (Tabela 2.24 e
Figura 2.6), mostra que, como seria expectável, este parâmetro aumenta com o aumento do
volume da amostra, atingindo factores de enriquecimento entre 3932-18562 para um volume
de água de 1000 mL.
O volume de fase sedimentada, variou entre 10 µL e 18 µL. Em particular, no ensaio
efectuado com um volume de água de 1000 mL no qual se obteve um volume sedimentado de
10 µL, pode-se ter afectado a determinação do factor de enriquecimento por um ventual erro
cometido na determinação do volume sedimentado.
Podemos observar que o aumento de volume, no passo de SPE, não foi muito favorável para
os últimos três analitos.
Tabela 2.24 - Variação dos factores de enriquecimento dos HAPs num aumento de volume de
amostra no passo SPE
Analitos
Factor de enriquecimento
100mL 250mL 500mL 1000mL
Vsed=18µL Vsed=18µL Vsed=16µL Vsed=10µL
Naftaleno 3918 4704 9141 15923
Acenaftileno 3504 3944 8777 18455
Acenafteno 3036 3705 7737 17887
Fluoreno 3172 3354 7454 18562
Fenantreno 3217 2951 6831 15992
Antraceno 3057 2735 6421 15255
Fluoranteno 3963 2899 7084 13827
Pireno 4125 2899 6909 12922
Criseno 4948 2750 6187 8666
1,2-Benzoantraceno 4916 2720 6005 8203
Benzo(k)fluoranteno 5104 2354 4120 4333
Benzo(b)fluoranteno 4731 2040 4846 4115
Benzo(a)pireno 4737 1907 4006 3932

73
Figura 2.6 - Efeito da variação do volume de amostra nos factores de enriquecimento
Tabela 2.25 - Percentagens de extracção dos HAPs, num aumento de volume de amostra no
SPE
Analitos % Extracção
100mL 250mL 500mL 1000mL
Naftaleno 71 34 29 16
Acenaftileno 63 28 28 18
Acenafteno 55 27 25 18
Fluoreno 57 24 24 19
Fenantreno 58 21 22 16
Antraceno 55 20 21 15
Fluoranteno 71 21 23 14
Pireno 74 21 22 13
Criseno 89 20 20 9
1,2-Benzoantraceno 88 20 19 8
Benzo(k)fluoranteno 92 17 13 4
Benzo(b)fluoranteno 85 15 16 4
Benzo(a)pireno 85 14 13 4
O efeito do aumento do factor de enriquecimento quando se aumenta o volume de amostra é
particularmente evidente no caso dos analitos mais voláteis. A concentração dos analitos mais
pesados pode estar a ser limitada pela sua adsorção irreversível à coluna de SPE ou pela sua
migração para o vidro do recipiente onde a amostra é colocada durante o processo de
filtração.
0
5000
10000
15000
20000
Facto
r d
e E
nriq
uecim
en
to
100 mL 250 mL 500 mL 1000 mL

74
Quando o volume de amostra aumenta a quantidade de analitos retidos na coluna de SPE deve
aumentar proporcionalmente mas não temos a garantia de que todos os analitos presentes na
amostra tenham sido retidos e sobretudo que sejam completamente eluídos.
2.4.3.4 Variação do volume de metanol no passo de SPE-HLLME
O efeito do metanol no passo de SPE, foi avaliado através da adição de metanol à amostra de
água de forma a obter concentrações finais de metanol de 0%, 5%, 10%, 15%, 20 %, 25% e
30%. Amostra aquosa (100mL) fortificada com 2 μg/L de HAPs, com as concentrações de
metanol referidas. Após o condicionamento da coluna, foi efectuada a filtração das amostras,
sob vácuo, a um caudal médio de 20 mL/min. De seguida efectuou-se a eluição dos analitos
utilizando 1 mL de acetona, seguindo-se a etapa de HLLME realizada com 50 μL de
ciclohexano e 5 mL de água.
Os factores de enriquecimento aumentam quando a concentração de metanol varia entre 0% e
10% diminuindo para concentrações superiores. O aumento na quantidade de metanol até uma
concentração de 10% inibe a migração dos analitos para a parede de vidro do recipiente
diminuindo um pouco a constante dieléctrica da fase aquosa.
Para concentrações de metanol superiores a 15% o volume de fase sedimentada obtida foi
substancialmente inferior ao obtido nos ensaios anteriores; esta observação pode resultar do
facto uma quantidade apreciável de metanol ficar retido na coluna de SPE quando se efectua a
eluição da amostra sendo depois eluído conjuntamente com os analitos. Assim o passo de
HLLME pode ser afectado pela inclusão de um outro componente no equilíbrio de fases. Por
outro lado a presença de quantidades crescentes de metanol na amostra a ser eluída pode
conduzir à menor retenção ou eluição precoce dos analitos durante o processo de adsorção da
amostra (arrastamento ou “carryover”).

75
Tabela 2.26 - Variação dos factores de enriquecimento (EF) dos HAPs na variação de
metanol no passo de SPE.
Analitos
Factor de enriquecimento (EF)
0% 5% 10% 15% 20% 25% 30%
Vsed=
18µL
Vsed=
16µL
Vsed=
17µL
Vsed=
10µL
Vsed=
5µL
Vsed=
9µL
Vsed=
5µL
Naftaleno 3918 7141 8527 1710 2634 3034 5411
Acenaftileno 3504 9034 10589 1787 3451 3781 7014
Acenafteno 3036 8104 9032 1851 3521 3853 7510
Fluoreno 3172 9753 11913 1882 3798 3882 7501
Fenantreno 3217 9000 10237 1810 3776 3698 7643
Antraceno 3057 8264 9955 1758 3623 3656 7323
Fluoranteno 3963 7976 9610 1622 3523 3419 6813
Pireno 4125 8189 9515 1653 3403 3333 6714
Criseno 4948 7257 8969 1247 2578 2216 5324
1,2-Benzoantraceno 4916 7254 8775 1283 2499 2052 4973
Benzo(k)fluoranteno 5104 7130 9088 1306 2572 2039 4792
Benzo(b)fluoranteno 4731 5507 8820 1190 2289 1920 3599
Benzo(a)pireno 4737 5948 8454 1204 2188 1705 3622
Tabela 2.27 - Percentagens de extracção dos HAPs na variação de metanol no passo de SPE.
Analitos % Extracção
0% 5% 10% 15% 20% 25% 30%
Naftaleno 71 114 145 17 13 27 27
Acenaftileno 63 145 180 18 17 34 35
Acenafteno 55 130 154 19 18 35 38
Fluoreno 57 156 203 19 19 35 38
Fenantreno 58 144 174 18 19 33 38
Antraceno 55 132 169 18 18 33 37
Fluoranteno 71 128 163 16 18 31 34
Pireno 74 131 162 17 17 30 34
Criseno 89 116 152 12 13 20 27
1,2-Benzoantraceno 88 116 149 13 12 18 25
Benzo(k)fluoranteno 92 114 155 13 13 18 24
Benzo(b)fluoranteno 85 88 150 12 11 17 18
Benzo(a)pireno 85 95 144 12 11 15 18
As percentagens de extracção obtidas com adição de metanol à amostra de água nas
concentrações de 5% e de 10% foram superiores às obtidas sem adição de metanol. Utilizando
uma concentração de metanol de 10% obtiveram-se factores de enriquecimento e
percentagens de extracção superiores às obtidas com qualquer das outras concentrações de
metanol e superiores às obtidas sem adição de metanol. As percentagens de extracção obtidas

76
com a concentração de 10 % de metanol apresentam valores substancialmente superiores a
100 % o que pode denotar a presença de vestígios de água na fase sedimentada.
Para concentrações de metanol igual ou superiores a 15 % os factores de enriquecimento e as
percentagens de extracção baixam muito significativamente ao mesmo tempo que o volume
sedimentado baixa para valores entre 5 µL a 10 µL. Este comportamento parece indicar uma
mudança significativa no equilíbrio de fases que ocorre quando a concentração de metanol
ultrapassa um determinado valor; a presença de uma elevada quantidade de metanol altera a
solubilidade do solvente de extracção na fase aquosa o que provoca simultaneamente o
decréscimo do volume sedimentado e a diminuição na transferência dos analitos para a fase
orgânica.
Figura 2.7 - Efeito do metanol da fase aquosa no factor de enriquecimento dos HAPs, no
sistema ciclohexano-acetona-água.
0
2000
4000
6000
8000
10000
12000
Fa
cto
r d
e E
nriq
uecim
en
to
sem metanol 5% metanol 10% metanol 15% metanol 20% metanol 25% metanol 30% metanol

77
2.5 Validação do método de SPE-HLLME-GC-MS para a determinação de
HAPs em águas
2.5.1 Estudo da linearidade e gama de trabalho
A gama de trabalho foi seleccionada tendo em conta os factores de enriquecimento obtidos
anteriormente para amostras de água com 100 mL; a sensibilidade do método permitiu incluir
nesta gama o valor de 100 ng/L (valor limite da legislação para detecção de HAPs em águas
de consumo). Assim estabeleceu-se uma gama de trabalho situada entre 800 ng/L e 100 ng/L
e traçaram-se rectas de calibração para cada analito, com seis pontos homogeneamente
distribuídos nesta gama. A Tabela 2.28 mostra os coeficientes de correlação que se situam
acima de 0.99, excepto para o antraceno, o criseno, 1,2-benzoantraceno, cujo valor se situa
um pouco mais abaixo. Apresentam-se, também, na Tabela 2.30 os limites de detecção e de
quantificação calculados através das rectas de calibração, através da razão sinal/ruído e do
desvio padrão.
Os LODs e os LOQs calculados pela razão sinal ruído, estão na gama dos ng/L. Tanto os
limites de detecção como os de quantificação calculados a partir das rectas, situam-se acima
da primeira concentração da gama de trabalho. Os coeficientes de variação do método foram
extremamente baixos, com valores entre 3.52% e 11.28%.
Tabela 2.28- Equações das rectas de calibração e respectivos coeficientes de determinação
para os HAPs estudados, na gama de 100 ng/L a 800 ng/L, com n=5
Analitos Equação da recta de calibração Coeficiente de
determinação (R2)
Naftaleno y = 1014.08 x + 77122.12 0.9970
Acenaftileno y = 1332.60 x + 78891.00 0.9921
Acenafteno y = 1276.10 x + 13567.00 0.9980
Fluoreno y = 1328.30 x + 33544.00 0.9928
Fenantreno y = 1990.30 x + 6219.20 0.9918
Antraceno y = 1802.90 x + 8605.60 0.9814
Fluoranteno y = 1297.20 x + 156970.00 0.9918
Pireno y = 1802.50 x + 46378.49 0.9920
Criseno y = 1257.10 x + 44066.00 0.9813
1,2-Benzoantraceno y = 721.76 x + 128933.39 0.9800
Benzo(k)fluoranteno y = 1976.50 x + 263251.00 0.9918
Benzo(b)fluoranteno

78
Benzo(a)pireno y = 1163.90 x – 61341.00 0.9917
Tabela 2.29 – Gamas de trabalho e coeficientes de variação do método validado.
Analitos Gama de trabalho
(ng/L)
CVm
(%)
Naftaleno 100-800 4.63
Acenaftileno 100-800 7.01
Acenafteno 100-800 3.52
Fluoreno 100-800 6.72
Fenantreno 100-800 7.15
Antraceno 100-800 10.84
Fluoranteno 100-800 7.18
Pireno 100-800 6.99
Criseno 100-800 10.86
1,2-Benzoantraceno 100-800 11.28
Benzo(k)fluoranteno 100-800 7.16
Benzo(b)fluoranteno
Benzo(a)pireno 100-800 7.22
CVm- Coeficiente de variação do método
2.5.2 Determinação dos limites de detecção e de quantificação
O limite de detecção e o limite de quantificação de cada HAPs foram determinados por três
métodos diferentes: a partir da recta de calibração, a partir da razão sinal/ruído e através do
desvio padrão (Tabela 2.30).
Tabela 2.30 – Limites de detecção e limites de quantificação dos HAPs através do método
SPE-HLLME.
Analitos LODcal
(ng/L) LODS/N
(ng/L) LOD
(ng/L)
LOQcal
(ng/L) LOQS/N
(ng/L) LOQ
(ng/L)
Naftaleno 58.30 9.39 36.70 194.34 31.30 122.34
Acenaftileno 88.31 11.19 28.94 294.35 37.30 96.48
Acenafteno 44.36 11.64 22.47 147.86 38.80 74.91
Fluoreno 84.72 8.04 27.29 282.42 26.80 90.97
Fenantreno 90.13 7.41 34.11 300.44 24.70 113.71
Antraceno 136.54 5.52 45.54 455.15 18.40 151.79
Fluoranteno 90.45 11.10 55.63 301.52 37.00 185.45
Pireno 88.13 11.73 44.87 293.76 39.10 149.56
Criseno 136.85 10.65 64.92 456.18 35.50 216.41
1,2-Benzoantraceno 142.13 10.47 129.36 473.75 34.90 431.18
Benzo(k)fluoranteno 90.24 8.40 127.85 300.78 28.00 426.17
Benzo(b)fluoranteno
Benzo(a)pireno 90.97 3.69 81.45 303.24 12.30 271.49
LODcal – Limite de detecção calculado a partir da curva de calibração; LOQcal – Limite de quantificação
calculado a partir da curva de calibração; LODS/N – Limite de detecção estimado a partir de 3x da razão S/N;
LOQS/N- Limite de quantificação estimado a partir de 10x da razão S/N; LOD – Limite de detecção calculado a

79
partir do desvio padrão = + 3xdesvio padrão; LOQ – Limite de quantificação calculado a partir do desvio
padrão= + 10 xdesvio padrão
Quando o LODS/N foi calculado pela razão sinal/ruído obtiveram-se valores situados entre
3.69 ng/L e 11.73 ng/L. No que diz respeito ao cálculo do limite de quantificação (LOQ), este
parâmetro há que considerar que os LOQS/N calculados pelas razões sinal/ruído são menores
que o padrão mais baixo das respectivas gamas de trabalho e os valores situam-se entre 12.30
e 39.10, o mesmo não ocorre com os LOQcal estimados a partir das rectas de calibração. Por
outras palavras, se seleccionássemos os LOQcal calculados a partir das rectas de calibração
teríamos que ajustar as gamas de trabalho ou considerar como valor mais baixo da gama de
trabalho para cada HAPs o respectivo LOQ.
Em relação aos LOD calculados obtiveram-se valores situados entre 22.47 ng/L e 129.36
ng/L e os LOQ variaram entre 74.91 ng/L e 426.17 ng/L, estes valores são calculados a partir
da recta de calibração, logo teríamos de ajustar as gamas de trabalho.
Podemos observar se os valores obtidos através LOD e LOQ se situam entre os limites de
detecção e quantificação calculados através da recta de calibração e da razão sinal/ruído.
Assim tendo em conta os resultados obtidos com padrões vestigiais, concluímos que os
limites de detecção e de quantificação calculados a partir da curva de calibração obtiveram
valores superiores aos reais e que os limites de quantificação e de detecção calculados a partir
da razão sinal ruído, apresentaram valores baixos, o que será pouco provável que os analitos
se consigam detectar em limites tão baixos.
2.5.3 Ensaios de Repetibilidade
2.5.3.1 Precisão do método analítico: análise da Repetibilidade
Para analisar a precisão do método de SPE-HLLME-GC-MS estudou-se o parâmetro da
repetibilidade na concentração de 100 ng/L e de 800 ng/L. Efectuaram-se cinco a seis
replicadas no processo de SPE-HLLME em ambas as concentrações, nas mesmas condições
anteriormente optimizadas e determinou-se a concentração dos analitos nos extractos obtidos.
A precisão foi avaliada como o desvio padrão relativo da concentração calculada pelas rectas
de calibração (Tabela 2.28). O método demonstrou ter uma boa precisão (< 10%) para os
alguns analitos.

80
Os ensaios de repetibilidade de cada HAPs foram realizados em duas gamas 100 ng/L (n=5) e
800 ng/L limite (n=6) (Tabela 2.31).
Tabela 2.31- Precisão do método de SPE-HLLME-GS-MS
Analitos % R.S.D.
(100 ng/L)
% R.S.D.
(800 ng/L)
Naftaleno 9.21 10.75
Acenaftileno 8.87 11.69
Acenafteno 8.90 8.36
Fluoreno 9.22 10.40
Fenantreno 11.09 6.69
Antraceno 15.41 6.28
Fluoranteno 15.06 7.13
Pireno 16.38 8.66
Criseno 17.60 7.93
1,2-Benzoantraceno 17.79 9.48
Benzo(k)fluoranteno 19.91 19.31
Benzo(b)fluoranteno
Benzo(a)pireno 23.38 19,91
R.S.D.– Desvio padrão relativo
2.5.4 Ensaios de recuperação em amostras reais
Para verificar a precisão do método de SPE-HLLME em amostras reais, efectuaram-se
ensaios de recuperação dos analitos em três amostras de águas destinadas a consumo humano
(água da rede de distribuição pública, água subterrânea) e uma amostra de água de mineral.
As amostras foram fortificadas com a mistura dos HAPs a um nível de concentração de 100
ng/L e extraídas por SPE-HLLME sendo os correspondentes extractos analisados por GC-MS.
Verifica-se através dos resultados obtidos na Tabela 2.32, que as recuperações são
maioritariamente superiores a 50%. Os HAPs cuja a determinação parece ser mais
influenciada pelo efeito matriz são benzo(k)fluoranteno e benzo(b)fluoranteno. Das três
amostras reais, foi na água mineral onde se verificou um menor valor de recuperação para os
HAPs.

81
Tabela 2.32 - Recuperações das três amostras reais estudadas e fortificadas com 100 ng/L de
cada analito.
Analitos
R.(%) ± S.D. (n=2)
Água subterrânea
(Odivelas) Água da Torneira
Água mineral
(Fastio)
Naftaleno 65.5 ± 4.5 563.6 ± 582.8 59.7 ± 10.9
Acenaftileno 68.2 ± 2.4 148.8 ± 24.0 48.6 ± 4.5
Acenafteno 97.7 ± 6.3 159.0 ± 19.4 55.0 ± 0.2
Fluoreno 90.6 ± 1.3 164.2 ± 21.2 52.8 ± 10.3
Fenantreno 122.7 ± 6.4 207.2 ± 1.8 50.5 ± 17.2
Antraceno 108.7 ± 4.2 198.3 ± 17.9 43.6 ± 15.4
Fluoranteno 49.9 ± 6.9 168.7 ± 35.2 40.6 ± 25.3
Pireno 88.1 ± 9.7 170.2 ± 11.5 38.3 ± 27.1
Criseno 104.7 ± 13.9 151.0 ± 26.4 29.7 ± 38.5
1,2-Benzoantraceno 65.2 ± 6.7 107.3 ± 52.4 37.5 ± 28.3
Benzo(k)fluoranteno 33.7 ± 19.5 88.1 ± 26.1 6.8 ± nd
Benzo(b)fluoranteno
Benzo(a)pireno 65.5 ± 10.1 226.5 ± 28.7 59.7 ± nd
S.D- Desvio padrão; %.R- % Recuperação.
A seguir apresentam-se os cromatogramas das amostras reais fortificadas com 100 ng/L de
cada HAPs nas condições de microextracção anteriormente optimizadas.
Figura 2.8 – Cromatograma em modo de varrimento completo, detectado através de
cromatografia gasosa e espectrometria de massa, para extracção dos HAPs a partir de uma
amostra de água subterrânea, utilizando o método SPE-HLLME-GS-MS.
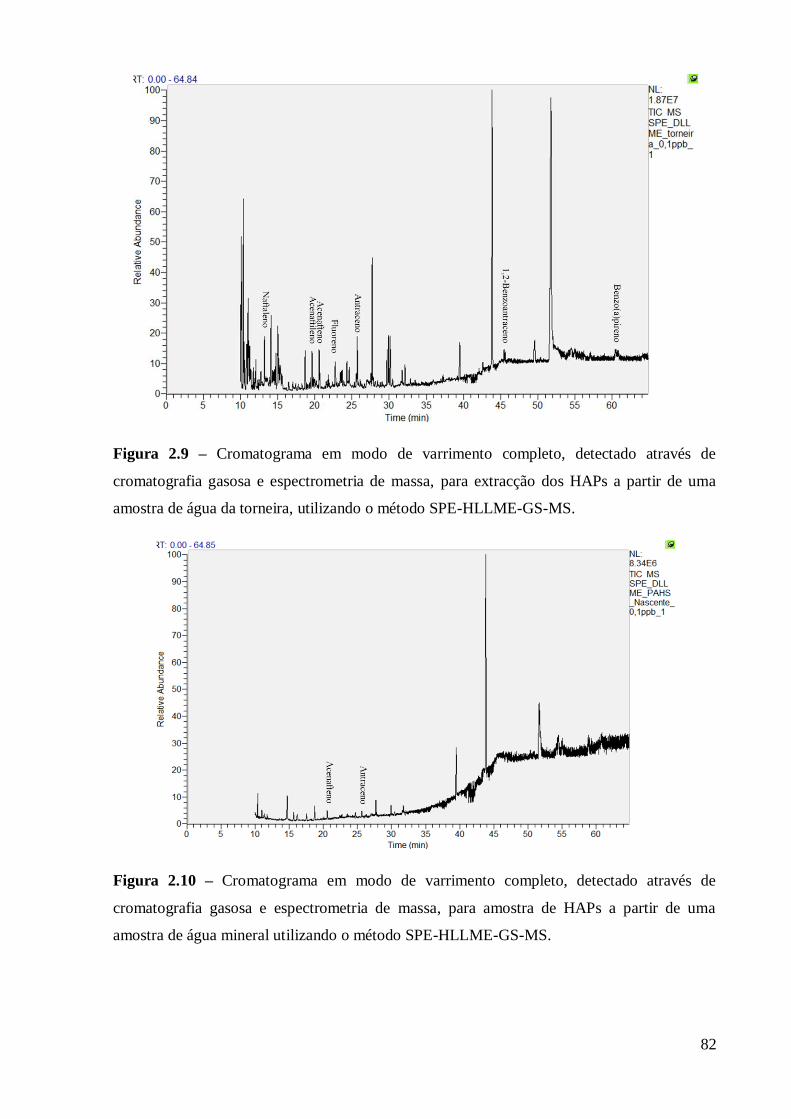
82
Figura 2.9 – Cromatograma em modo de varrimento completo, detectado através de
cromatografia gasosa e espectrometria de massa, para extracção dos HAPs a partir de uma
amostra de água da torneira, utilizando o método SPE-HLLME-GS-MS.
Figura 2.10 – Cromatograma em modo de varrimento completo, detectado através de
cromatografia gasosa e espectrometria de massa, para amostra de HAPs a partir de uma
amostra de água mineral utilizando o método SPE-HLLME-GS-MS.

83
Em suma, é possível afirmar que a aplicação deste método de SPE-HLLME à extracção dos
HAPs de matrizes aquosas é adequada para a maioria dos analitos estudado.
Os ensaios de recuperação foram afectados pelo estado da coluna cromatográfica que
começou a exibir algum sangramento o que afectou a detecção tanto pela presença de iões
interferentes como pela perda de sensibilidade. Assim, é provável que em condições
cromatográficas mais favoráveis estes resultados possam ainda ser melhorados.

84
Capítulo III
Microextracção líquido-líquido dispersiva (DLLME)
como passo de concentração após extracção adsorptiva
com barra de agitação magnética (SBSE) – aplicação à
determinação de analitos orgânicos em amostras de
água

85
3.1 Considerações gerais
A técnica SBSE, aparece como uma das técnicas de extracção mais promissoras, devido à sua
simplicidade e às suas potencialidades analíticas, para a análise de compostos orgânicos
voláteis e semi-voláteis. Na verdade, esta técnica permite um aumento de cerca de 500 vezes
no factor de enriquecimento, em relação à técnica de extracção SPME, na medida em que
utiliza uma fase estacionária análoga mas em muito maior quantidade (144)
.
Alguns exemplos da aplicação desta técnica são o caso de análises com fungicidas, os
pesticidas Organofosforados, carbamatos, compostos fenólicos, compostos voláteis do vinho,
triazinicos e HAPs (144)
.
3.1.1. Extracção dos analitos a partir da barra de SBSE – desorpção térmica
versus desorpção com solventes.
O processo de SBSE envolve duas etapas: a extracção dos analitos a partir da amostra de água
e a sua desorpção e análise cromatográfica. A desorpção dos analitos pode ser efectuada
térmicamente ou recorrendo ao uso de solventes (144)
.
No caso da desorpção térmica, algumas das variáveis operacionais estudadas são o tempo e a
temperatura de desorpção, bem como o caudal do gás de arraste (145)
. A temperatura de
desorpção é normalmente avaliada no intervalo de 150-300ºC (144) (146) (147) (148)
.
Em geral, observa-se uma associação positiva entre a temperatura de desorpção e a
sensibilidade do método (149)
(150)
, embora alguns autores optem por não trabalhar com valores
de temperatura elevados para aumentar a vida útil do twister (147)
.
O tempo de desorpção, mostra geralmente um efeito positivo na sensibilidade do método (147)
no entanto em alguns trabalhos esta correlacção não foi verificada(148)
. Este parâmetro é
frequentemente fixado em valores elevados, a fim de minimizar a desorpção incompleta (151)
;
alguns trabalhos mostram que o tempo de desorpção óptimo situa-se na gama de 1 a 15 min.
Além disso, alguns autores preferem associar longos tempos de desorpção a temperaturas de
desorpção mais baixas com o objectivo de preservar a vida do twister (151)
.
A temperatura de criofocagem é também uma das variáveis da etapa de desorpção térmica,
uma vez que garante a concentração dos analitos no topo da coluna cromatográfica. Na
análise de MeHg e compostos organoestanhados em matrizes ambientais (146)
, foi necessário,
para observar o pico de MeHg recorrer a temperaturas tão baixas como -140°C.

86
Quando se efectua a desorpção dos analitos com solventes orgânicos as variáveis mais
frequentemente estudadas são o volume de solvente e o tempo de desorpção. Os solventes
mais utilizados são ACN e metanol (144)
.
Após a extracção, o twister é removido, lavado com água destilada, para remover os sais,
açúcares, proteínas ou outros componentes da amostra. A etapa de lavagem é extremamente
importante quando os analitos são termicamente desadsorvidos a fim de evitar a formação de
material não-volátil que pode obstruir a unidade de desorpção. Além disso, a lavagem não
deve causar perda significativa de solutos uma vez que, estes estão adsorvidos na fase de
polímero (144)
.
O número de publicações indexadas na WEB of Science com o termo SBSE tem aumentado
regularmente desde 1999 (figura 3.1)
Anos
Figura 3.1- Publicações envolvendo SBSE, indexadas na Web of Science, no período de
1999-2010 (Adaptado Ref. (152))
A Tabela 3.1 mostra as condições de extracção e os limiares analíticos de diversos analitos
extraídos a partir de matrizes aquosas através do método de SBSE.
N.º
de
publi
caçõ
es
87
Tabela 3.1- Condições de extracção e limiares analíticos dos analitos estudados pelo método
de associação do SBSE (144)
.
Classe de
compostos
Quantidade
de amostra Tempo
Método
Analítico
Fase
estacionária
Tipo de
desorpção LODs
%
Recuperação
Organofosforados (153) 10mL 50min
CGC-
AED
PDMS
10mm×0.5mm
Desorpção
térmica
0.8–
15.4
ng/L
2.5–15
HAPs (154) 10mL 60min HPLC
PDMS
10mm×0.5mm
Desorpção
com
solventes
0.3–2
ng/L _
HAPs (155) 10mL 210min GC–MS
PDMS
10mm×0.5mm
Desorpção
térmica
0.2–2.0
ng/L 3–15
Fenóis (156) 10mL 45min GC–MS
PDMS
10mm×0.5mm
Desorpção
térmica
0.1–
0.4g/L 6–27
Triazinas (157) 20mL 60min GC–MS
PDMS 10mm×0.5mm
Desorpção térmica
0.2–3.4 ng/L
1.8–7
Como se pode ver na Tabela anterior, existe um número muito pequeno de aplicações desta
técnica referidas na literatura; o presente trabalho descreve pela 1ª vez a utilização de SBSE
na determinação de HAPs utilizando a desorpção com solventes através do método analítico
GC-MS.
Neste trabalho efectuou-se um conjunto de ensaios de avaliação do potencial da técnica de
HLLME como método de desorpção dos analitos na sequência de SBSE.
Os analitos utilizados neste estudo foram os HAPs utilizados anteriormente nesta dissertação
e a estratégia seguida foi a adsorção dos analitos presentes numa amostra de água fortificada
utilizando o Twister seguindo-se a sua desorpção num volume reduzido de um solvente
orgânico miscível com água (metanol, acetonitrilo, acetona, isopropanol e uma mistura de
50% acetona e 50% metanol). De seguida esta solução contendo os analitos foi submetida a
HLLME através da adição de um solvente de extracção apropriado bem como de um volume
de água susceptível de provocar a separação de fases. Os analitos são assim concentrados num
pequeno volume de solvente orgânico adequado à análise cromatográfica.

88
3.2 Parte Experimental
3.2.1 Materiais e Solventes
Utilizou-se em todos os ensaios água ultra pura (Milli-Q, modelo Milipore, Molsheim,
França).
O ciclohexano (99%), a acetona (99.5%), o metanol (99.9%) e o acetonitrilo (99,9%), foram
fornecidos por Lab-scan (Dublin, Irlanda); o clorobenzeno (99%) foi adquirido à Panreac
(Barcelona, Espanha), o octano (99%) foi fornecido por Koch-Light Laboratory (Cambridge,
Reino Unido); o isopropanol (≥99.5%) foi adquirido a Fluka (Steinheim, Suiça); o heptano
(99%) foi adquirido à Merck (Darmstadt, Alemanha); o clorofórmio (99.8%), o tetracloreto de
carbono (99,9%) e o tetracloroetileno (99,9%) foram adquiridos pela Aldrich (Steinheim,
Alemanha) Tolueno (99,5%) à Panreac (Barcelona, Espanha).
3.2.2 Amostras Reais
Para o estudo dos factores de enriquecimento obtidos por fortificação em amostras reais da
desorpção dos analitos a partir do Twister, foram realizados ensaios de recuperação em água
ultra pura e água da torneira.
3.2.3 Materiais
Nos ensaios de SBSE utilizaram-se tubos de centrífuga de vidro com fundo cónico, com
tampa roscada e septo de PTFE, com capacidades de 15 mL (Corning Life Sciences). Na
medida e transferência dos solventes de extracção, bem como da fase sedimentada utilizaram-
se microseringas de vidro, graduadas, com a capacidade de: 10µL (SGE, Australia); 25 µL, 50
µL, 100 µL (Agilent, Australia).
Os extractos foram colocados em redutores de volume de 100 µL (Refª 110501) e 300 µL
(Refª 110502), para frascos de capacidade 1,5 mL (Refª CVP1-C02-100), com tampa de
capsular, do injector automático.
Durante o trabalho efectuado, utilizou-se material de vidro comum como copos de vidro de
capacidade 5mL, provetas, funis, frascos de vidro de fundo plano (Supelco), pipetas
volumétricas, gobelés, etc. A descontaminação de todo este material, dos frascos de 1,5 mL e
dos redutores de volume utilizados, foi feita de acordo com o procedimento anteriormente
descrito na parte experimental do Capítulo II (secção 2.2.3).

89
Foi necessário recorrer ao uso de algum equipamento durante a realização deste trabalho
como: vortex (Heidolph); banho de ultra-sons (Sonorex); placa de agitação magnética (Velp
Scientific); banho a 50 ºC (Fischer).
3.2.4 Extracção adsorptiva com barra de agitação magnética (SBSE) associada à
Microextracção líquido-líquido dispersiva (DLLME)
A amostra de água (50 mL) é colocada em frascos de vidro com fundo plano de capacidade de
50 mL (Supelco), fortificada com uma mistura dos 16 HAPs, numa concentração final de 2
g/L. Coloca-se a barra de agitação magnética (SBSE) e por fim um septo de PTFE e a tampa
de alumínio. O frasco é então colocado sob uma placa de agitação, durante o período de
tempo de 1 hora de agitação. Após a agitação, o Twister é removido com auxílio de uma pinça
e colocado num copo de 5mL onde se adicionou 0,5mL de solvente de dispersão e deixou-se a
agitar numa placa de agitação (vortex, banho de ultra-sons, banho a 50ºC) durante um período
de tempo 3min. Este solvente de dispersão é removido com auxílio de uma microseringa de
1000 µL e colocado num tubo de centrífuga. Ao twister adicionou-se novamente 0,5mL de
solvente de dispersão e voltou-se agitar (placa de agitação, vortex, banho de ultra-sons, banho
a 50ºC).
O solvente de dispersão, obtido na segunda extracção, é colocado no tubo de centrífuga
adicionando 50 µL do solvente de extracção. À mistura anterior acrescenta-se 5mL de água
utilizando uma seringa de 5 mL. A ponta da agulha é colocada dentro da mistura do solvente
de extracção e de dispersão, a adição da água é efectuada de forma rápida, obtendo-se assim
uma boa dispersão dos solventes orgânicos na fase aquosa.
Após a dispersão, as fases são separadas por centrifugação, e a fase orgânica obtida é
removida com uma microseringa e colocada num frasco de 1,5 mL equipado com um redutor
de 100 µL. O frasco é imediatamente capsulado e colocado no tabuleiro do injector
automático do cromatógrafo, para se proceder à sua análise.
A figura 3.2 mostra o esquema de desorpção dos analitos após extracção adsorptiva com barra
de agitação magnética associado a uma microextracção líquido-líquido dispersiva.

90
Figura 3.2 – Extracção adsorptiva com barra de agitação magnética (SBSE) associada à
microextracção líquido-líquido dispersiva (DLLME)
3.2.5 Análise cromatográfica
As condições da análise cromatográfica durante a realização de alguns ensaios preliminares
entre HLLME e a extracção adsorptiva com barra de agitação magnética (SBSE), foram as
mesmas utilizadas na optimização e validação do método de SPE-HLLME no Capítulo II
(secção 2.2.7).
Para o estudo dos factores de enriquecimento em amostras reais da desorpção dos analitos a
partir do Twister, utilizou-se uma coluna analítica TJ 17MS -cyanopropylphenyl polysiloxane
(30 m x 0.25 µm x 0.25 mm i.d.), (Thermo Scientific).

91
3.2.6 Cálculos
Os cálculos efectuados durante a optimização do método SBSE foram feitos de acordo com as
equações referidas na parte experimental do Capítulo II (secção 2.2.8).
3.3 Resultados e Discussão dos Resultados
3.3.1 - Extracção de hidrocarbonetos aromáticos polinucleares a partir de
amostras de água
3.3.1.1 – Estudo da influência da natureza do solvente de extracção
Efectuou-se SBSE utilizando 50 mL de água fortificada com a mistura dos 16 HAPs numa
concentração final de 2 g/L. Após a extracção os analitos foram desadsorvidos do Twister
por duas extracções sucessivas com 500 µL de acetonitrilo. O acetonitrilo foi seleccionado
como solvente de desorpção por ser frequentemente utilizado para esta função em SBSE. Aos
extractos combinados adicionaram-se 50 L de diferentes solventes de extracção e à mistura
de extracto de acetonitrilo com o solvente de extracção foram adicionados 5 mL de água de
forma a promover a transferência dos analitos do solvente de desadsorção/dispersão para o
solvente de extracção em HLLME. Foram testados solventes halogenados (clorobenzeno,
clorofórmio, tetracloroetileno, 1,2-dicloroetano e tetracloreto de carbono) e solventes não
halogenados (ciclohexano, tolueno, octano e heptano). Nas Tabelas 3.2 e 3.3 apresentam-se os
resultados obtidos com os solventes de extracção halogenados e não halogenados
respectivamente.

92
Tabela 3.2 – Factores de enriquecimento (normalizados para um volume sedimentado de 20
L) obtidos nos ensaios de SBSE-HLLME com diferentes solventes de extracção
halogenados.
Hidrocarbonetos Aromáticos
Polinucleares
Factores de Enriquecimento
Clorobenzeno Clorofórmio Tetracloroetileno Tetracloreto
de carbono
Naftaleno 410 172 283 615
Acenaftileno 466 178 286 618
Acenafteno 619 207 340 817
Fluoreno 581 221 365 854
Fenantreno 650 254 422 1007
Antraceno 681 260 446 741
Fluoranteno 914 344 560 1276
Pireno 994 364 601 1293
Criseno 588 213 361 888
1,2-Benzoantraceno 290 108 157 557
Benzo(k)fluoranteno 587 160 253 918
Benzo(b)fluoranteno 187 0 22 290
Benzo(a)pireno 393 92 211 288
Em particular, o tetracloreto de carbono apresenta uma capacidade de concentração dos
analitos muito superior à de qualquer dos outros solventes testados. Assim o tetracloreto de
carbono foi o solvente de extracção halogenado seleccionado para os ensaios de SBSE-
HLLME posteriores.
Tabela 3.3 – Factores de enriquecimento (normalizados para um volume sedimentado de 20
L) obtidos nos ensaios de SBSE-HLLME com diferentes solventes de extracção não
halogenados.
Hidrocarbonetos Aromáticos
Polinucleares
Factores de Enriquecimento
Octano Ciclohexano Heptano Tolueno
Naftaleno 188 129 203 195
Acenaftileno 203 145 233 227
Acenafteno 275 178 329 278
Fluoreno 293 203 361 309
Fenantreno 345 240 432 363
Antraceno 377 257 477 384
Fluoranteno 497 294 589 482
Pireno 552 296 646 516
Criseno 324 114 449 390
1,2-Benzoantraceno 162 57 249 263
Benzo(k)fluoranteno 294 50 445 311
Benzo(b)fluoranteno 57 16 87 114
Benzo(a)pireno 203 34 188 212

93
Os dois solventes que apresentaram maior capacidade de concentração dos HAPs foram o
heptano e o tolueno. O heptano apresenta melhores resultados para os HAPs mais voláteis
desde o naftaleno até ao criseno e também para o benzo(k)fluoranteno. O tolueno permite
atingir maiores concentrações para o 1,2-benzoantraceno, o benzo(b)fluoranteno e o
benzo(a)pireno. Seleccionou-se, como melhor solvente de extracção não halogenado, o
heptano tendo em conta que este solvente produz melhores factores de enriquecimento para a
maior parte dos analitos testados e que nos casos em que apresenta menores factores de
enriquecimento a diferença é inferior a 24%.
Os factores de enriquecimento obtidos com os solventes halogenados foram, de uma forma
geral, superiores aos obtidos com os solventes não halogenados.
3.3.1.2 – Natureza do solvente de dispersão (DLLME) e de desadsorção (SBSE)
Utilizando o solvente de extracção para o qual se obtiveram os melhores factores de
enriquecimento na mistura com acetonitrilo (tetracloreto de carbono), procedeu-se então ao
estudo do efeito da natureza do solvente de dispersão (DLLME) e de desadsorção (SBSE).
Este parâmetro influencia decisivamente a extracção dos analitos a partir do Twister e por
outro lado também condiciona o equilíbrio de fases e a partição dos analitos em DLLME.
Os solventes de dispersão testados foram o acetonitrilo, a acetona, o metanol, o propanol, o
isopropanol, a mistura metanol:acetona (50% v/v).
Tabela 3.4 – Factores de enriquecimento obtidos nos ensaios de SBSE-HLLME com
diferentes solventes de dispersão.
Hidrocarbonetos
Aromáticos
Polinucleares
Factores de Enriquecimento
Acetonitrilo Acetona Metanol Isopropranol
Mistura
Metanol:Acetona
(50% v/v)
Naftaleno 615 324 569 369 374
Acenaftileno 618 291 551 385 391
Acenafteno 817 395 711 500 976
Fluoreno 854 413 743 524 493
Fenantreno 1007 439 858 612 506
Antraceno 741 386 658 506 464
Fluoranteno 1276 474 1110 848 523
Pireno 1293 467 1119 882 505
Criseno 888 208 623 645 281
1,2-Benzoantraceno 557 98 285 397 202
Benzo(k)fluoranteno 918 149 457 823 242
Benzo(b)fluoranteno 290 2 6 0 130
Benzo(a)pireno 288 27 158 242 99

94
3.3.1.3 – Estudo de diferentes condições de desorpção dos analitos a partir do
Twister utilizando como solvente acetonitrilo
As condições de agitação e temperatura, durante a desorpção dos analitos com acetonitrilo
foram estudadas, tendo sido comparado o efeito das seguintes operações: a) agitação em
vortex durante 1 min, b) agitação em banho de ultrasons durante 5 min, c) agitação magnética
à temperatura ambiente durante 3 min e d) agitação magnética a 50 ºC durante 3 min.
Tabela 3.5 – Factores de enriquecimento obtidos nos ensaios de SBSE-HLLME na mistura de
acetonitrilo/ tetracloreto em diferentes condições de desorpção dos analitos a partir do twister.
Hidrocarbonetos Aromáticos
Polinucleares
Factores de Enriquecimento
Agitação
em vortex
durante
1min.
Agitação
em banho
de
ultrasons
durante
5min.
Agitação
magnética á
temperatura
ambiente
durante
3min.
Agitação
magnética a
50ºC
durante
3min.
Naftaleno 133 220 615 317
Acenaftileno 130 231 618 351
Acenafteno 162 255 817 412
Fluoreno 172 266 854 423
Fenantreno 205 240 1007 438
Antraceno 210 204 741 377
Fluoranteno 267 206 1276 395
Pireno 285 205 1293 396
Criseno 205 75 888 199
1,2-Benzoantraceno 147 36 557 118
Benzo(k)fluoranteno 160 106 918 151
Benzo(b)fluoranteno 0 0 290 33
Benzo(a)pireno 77 0 288 93
A desorpção efectuada por agitação em vórtex ou em banho de ultrasons produziu resultados
inferiores à agitação magnética a qualquer das temperaturas testadas.
3.3.1.4 – Estudo dos factores de enriquecimento obtidos por fortificação em
amostras reais da desorpção dos analitos a partir do Twister
Efectuou-se SBSE utilizando 50 mL de água fortificada com a mistura dos 16 HAPs. Após a
extracção os analitos foram desadsorvidos do Twister por duas extracções sucessivas com
1000 µL de metanol e 500 µL de acetona. Ao extracto adicionou-se 50 L do solvente de
extracção, o tetracloreto de carbono, foi adicionado 5 mL de água de forma a promover a
transferência dos analitos do solvente de desadsorção/dispersão para o solvente de extracção

95
em DLLME. As condições foram as seguintes agitação magnética à temperatura ambiente
durante 3min.
Para verificar a precisão do método de SBSE-HLLME em amostras reais, efectuaram-se
ensaios de recuperação dos analitos em uma amostra de água destinada ao consumo humano
(água da rede de distribuição pública) e uma amostra de água Milli Q. As amostras foram
fortificadas com a mistura dos HAPs a um nível de concentração de 100 ng/L e extraídas por
SBSE-HLLME sendo os correspondentes extractos analisados por GC-MS.
Tabela 3.6 – Factores de enriquecimento obtidos nos ensaios de SBSE-HLLME em amostras
reais.
Hidrocarbonetos Aromáticos
Polinucleares
Factores de Enriquecimento
Água Milli Q
(n=2)
Água da
Torneira
(n=2)
Naftaleno 818 783
Acenaftileno 474 420
Acenafteno 686 599
Fluoreno 820 564
Fenantreno e Antraceno 824 742
Fluoranteno 1102 514
Pireno 963 517
Criseno 1248 652
1,2-Benzoantraceno 1324 813
Benzo(k)fluoranteno 1906 742
Benzo(b)fluoranteno 1882 683
Benzo(a)pireno 2098 834
Observando a tabela 3.6, verifica-se que os factores de enriquecimento obtidos na água da
Milli Q são mais elevados do que os obtidos com água da torneira pelo que se pode concluir
que os HAPs são melhor extraídos a partir da água Milli Q, pois a partir da matriz água da
torneira existem contaminantes que podem influenciar um decréscimo nos factores de
enriquecimento.

96
Tabela 3.7 – Percentagens de extracção dos HAPs obtidas nos ensaios de SBSE-HLLME em
amostras reais.
Hidrocarbonetos Aromáticos
Polinucleares
% Extracção
Água Milli Q
(n=2)
Água da
Torneira
(n=2)
Naftaleno 56 55
Acenaftileno 32 29
Acenafteno 46 42
Fluoreno 56 40
Fenantreno e Antraceno 56 53
Fluoranteno 76 36
Pireno 66 36
Criseno 86 46
1,2-Benzoantraceno 91 57
Benzo(k)fluoranteno 131 52
Benzo(b)fluoranteno 130 47
Benzo(a)pireno 142 59
3.4 - Extracção de organofosforados a partir de amostras de água
Efectuou-se SBSE utilizando 50 mL de água fortificada com a mistura dos organofosforados
numa concentração final de 2 g/L. Após a extracção os analitos foram desadsorvidos do
Twister por duas extracções sucessivas com 500 µL de acetonitrilo. Aos extractos
combinados adicionaram-se 50 L de tetracloreto de carbono, como solvente de extracção e 5
mL de água, de forma a promover a transferência dos analitos do solvente de
desadsorção/dispersão para o solvente de extracção em DLLME.
Tabela 3.8 – Factores de enriquecimento obtidos nos ensaios de SBSE-HLLME na mistura de
acetonitrilo/ tetracloreto em diferentes condições de desorpção dos analitos a partir do twister
com uma mistura de organofosforados.
Organofosforados Factores de Enriquecimento
Tetracloreto de carbono
Profos 183
Diazinao 345
Clorpirifos metil 388
Metil paratiao 217
Fenclorfos 400
Clorpirifos metil 314
TBPT 24

97
Todos os ensaios de avaliação da associação entre SBSE e HLLME foram efectuados numa
fase de utilização intensiva do GC-MS por diversos operadores, com amostras de
concentração e volatilidade muito diversa e portanto com consequências na variação da
sensibilidade do equipamento. Assim os resultados deste capítulo foram avaliados
criticamente e são incluídos nesta dissertação apenas a título de exemplificação do potencial
desta técnica em sistemas não optimizados e numa perspectiva de comparação
semiquantitativa.
Mesmo nestas condições desfavoráveis de sensibilidade, foi possível registar alguns factores
de enriquecimento superiores a 1000 para uma fortificação de 2 µg/L o que nos leva a
concluir que será possível noutras condições atingir limites de detecção inferiores a 2 ng/L.
Esta associação parece-nos um método elegante e inovador de completar a extracção de SBSE
e se for testado em condições cromatográficas adequadas poderá permitir atingir resultados
competitivos com as metodologias existentes para desorpção dos analitos do Twister.

98
Capítulo IV
Conclusões e perspectivas de futuro

99
Conclusões e Perspectivas para o Futuro
Após a optimização de diversos parâmetros do método de microextracção líquido-líquido
dispersiva, concluiu-se que utilizando os mesmos solventes de dispersão (acetona, metanol e
isopropanol) mas solventes de extracção diferentes, não halogenados e halogenados,
obtiveram-se factores de enriquecimento mais elevados, para a extracção de HAPs, aquando
da utilização dos solventes de extracção não halogenados.
No estudo na determinação de HAPs por DLLME utilizando solventes não-halogenados, os
factores de enriquecimento obtidos não permitiram atingir limites de quantificação inferiores
a 100 ng/L pelo que se considerou útil testar métodos mais sensíveis na determinação de
HAPs.
O sistema de extracção em fase sólida associada a microextracção líquido-líquido dispersiva e
análise por cromatografia gasosa e espectrometria de massa, foi testado e após a optimização
de diversos parâmetros deste método, concluiu-se que as condições óptimas de extracção dos
HAPs de matrizes aquosas se atingiram utilizando um volume de amostra de 100 mL, usando
o ciclohexano como solvente de extracção e a acetona como solvente de dispersão, para os
volumes de 50 µL e 1 mL, respectivamente.
Nas condições seleccionadas, este foi o sistema que ao longo do trabalho, apresentou boa
reprodutiilidade e um volume de fase sedimentada que possibilitou a automatização do
processo, minimizando os erros de injecção de volume e de repetibilidade que pudessem daí
advir.
Este método permite determinar treze HAPs ao nível de 100 ng/L (limite máximo admissível
para águas de consumo), num tempo de preparação de amostra aproximadamente 30 minutos
e com um consumo mínimo de solventes orgânicos.
Por outro lado obtiveram-se eficiências de extracção mais homogéneas para os vários HAPs
testados, em particular conseguiu-se com estes solventes efectuar a extracção do
benzo(a)pireno de forma reprodutível e com bons factores de enriquecimento.
A utilização destes solventes em SPE-HLLME-GC-MS não está descrita na literatura pelo
que os resultados obtidos neste trabalho abrem novas perspectivas de substituição dos
solventes halogenados na extracção desta matriz de HAPs.
O método de SPE-HLLME-GC-MS optimizado para a extracção dos HAPs, foi proposto para
validação, demostrando um bom desempenho analítico na gama de concentrações de 100 ng/L

100
a 800 ng/L. Nomeadamente, este método apresentou limites de detecção calculado a partir da
curva de calibração; 90.97 ng/L situando-se abaixo da legislação definida para águas
superficiais destinadas a consumo humano, cujo limite se situa na concentração de 100 ng/L,
para o benzo(a)pireno. No entanto, para os analitos, o antraceno, criseno, 1,2-benzoantraceno
este limite não foi abaixo de 143 ng/L, situando-se acima do limite legislado.
Para os restantes analitos, os limites de detecção ficaram abaixo de 100 ng/L que é a
concentração limite referida na legislação para a detecção de HAPs em águas superficiais e
subterrâneas destinadas à produção de água para consumo humano.
Os limites de quantificação estimados a partir da curva de calibração situaram-se entre 147.86
ng/L e 473.75 ng/L, estando estas concentrações acima da primeira concentração da gama de
trabalho.
Os limiares analíticos calculados a partir da razão sinal/ruido apresentaram valores bastante
inferiores (limites de detecção entre 3.69 ng/L e 11.73 ng/L e limites de quantificação entre
12.30 ng/L e 39.10 ng/L) que estão claramente abaixo da primeira concentração da gama de
trabalho para todos os analitos.
Em relação aos LOD calculados obtiveram valores situados entre 22.47 ng/L e 129.36 ng/L e
os LOQ variaram entre 74.91 ng/L e 426.17 ng/L, estes valores são calculados a partir da
recta de calibração, logo teríamos de ajustar as gamas de trabalho.
A repetibilidade foi testada em duas concentrações da gama de trabalho, ou seja, numa
concentração referida na legislação (100 ng/L) e numa concentração da gama alta (800 ng/L),
mostrando valores de R.S.D entre 8.87 % e 23.38 %, entre 6.28% a 19.91% respectivamente.
Estes dados confirmam que existiu precisão do método de extracção dos HAPs de matrizes
aquosas.
As recuperações obtidas para as amostras reais, apresentaram valores entre 33.70% a 122.70%
para a água do subterrânea, onde se demonstra que existe um efeito de matriz, em alguns dos
analitos, pois apresentam valores acima de 100%.
As recuperações obtidas, utilizando a água da torneira como matriz, apresentaram valores
entre 88.10% a 563.60% . Enquanto que para a água mineral os valores variaram entre 6.80%
e 59.70%. Portanto, das três amostras reais, foi na água mineral onde se verificou um menor
valor de recuperação para os HAPs.
Relativamente ao futuro no que diz respeito a técnica de SPE-HLLME optimizada para a
extracção dos HAPs, poderiamos efectuar a sua validação completa no mesmo sistema

101
cromatográfico e em condições melhores de sensibilidade o que permitirá certamente
melhorar ainda os resultados obtidos no que diz respeito à repetibilidade e à recuperação.
No estudo de optimização do método de microextracção líquido-líquido dispersiva (DLLME)
como passo de concentração após extracção adsorptiva com barra de agitação magnética
(SBSE) a aplicação na determinação de analitos orgânicos em amostras de água é importante
de salientar, que nesta parte do trabalho pretendeu-se desenvolver um método estudando a
influência de alguns parâmetros como a natureza do solvente de extracção recorrendo ao uso
de solventes halogenados e de solventes não halogenados, a natureza do solvente de
dispersão, estudo de diferentes condições de desorpção e a recuperação em amostras reais.
Os ensaios realizados apesar de fornecerem uma informação escassa revelam o potencial
desta associação de técnicas para atingir uma elevada concentração dos analitos após SBSE
sem recorrer à desorpção térmica, certamente eficiente mas que requer equipamento muito
caro e que pode ser prejudicial no caso de analitos termolábeis.
Relativamente ao futuro poderia validar-se o método de SBSE-HLLME, como sendo uma
técnica em expansão e uma das mais promissoras, devido à sua simplicidade e às suas
potencialidades analíticas, para a análise de compostos orgânicos voláteis e semi-voláteis.

102
Referências Bibliográficas

103
Bibliografia
1. Lemos P., "Engenharia do processo de remoção biológica de fosfato"; Tese de
Doctouramento, Universidade Nova de Lisboa, (2001), 3.
2. Cao Z., Y. Wang, Y. Ma, Z. Xu, G. Shi, Y. Zhuang, T. Zhu,"Occurrence and distribution of
polycyclic aromatic hydrocarbons in reclamimed water and surface water in of Tianjin Chin",
Journal of Harzardous Materials A, 122, (2005), 51-59.
3. Manoli E., C. Samara,"Polycyclic aromatic hydrocarbons in natural waters: sources,
occurrence and analysis", Trends Anal. Chem, 18, (1999), 417.
4. Cristale J., F. S. Silva, M. R. R Marchi,"Desenvolvimento e aplicação de método GC-
MS/MS para análise simultânea de 17 HAPs em material particulado atmosférico", Quím, 33,
(2008), 69-78.
5. Doong R., S. Chang, Y. Sun,"Solid-phase microextraction for determining the distribution
of sixteen US Environmental Protection Agency polycyclic aromatic hydrocarbons in water
samples", Journal of Chromatography A, 879, (2000), 177-188.
6. American Water Works Association:Water Quality & Treatment - A Handbook of
Community Water Supplies, 5 Ed. New York, McGraw-Hill Handbooks, (1999),Vol. 2.
7. Camargo M., C. R. Toledo, "Determinação de hidrocarbonetos policíclicos aromáticos
(HAPs) em guaraná em pó", Ciênc. Tecnol. Aliment, 26, (2006), 1, 230-234.
8. Camargo M., C. R. Toledo, M. C. Figueiredo, "Chá-mate e café como fontes de
hidrocarbonetos policíclicos aromáticos (HAPs) na dieta da população de Campinas", Ciênc.
Tecnol. Aliment, 22, (2002), 1, 49-53.
9. Lo M., E. Sandi, "Polycyclic aromatic hydrocarbons (polynuclears) in food", Residue
Review, 69, (1978), 35-56.
10. Andelman J.B., M. J. Suess, "Polynuclear aromatic hydrocarbons in the water
environment", Bulletin World Health Organization, 43, (1970), 479-508.
11. Luch A., "The Carcinogenic Effects of Polycyclic Aromatic Hidrocarbons", London,
Imperial College Press, (2005).
12. Fetzer J.C., "The Chemistry and Analysis of the Large Polycyclic Hydrocarbons", Wiley,
(2000).
13. Boström C. E., P. Gerde, A. Hanberg, B. Jernström, C. Johansson, T. Kyrklund, A.
Rannug, M. Törnqvist, K. Victorin, R. Westerholm, "Cancer Risk Assessment, Indicators, and
Guidelines for Polycyclic Aromatic Hydrcarbons in the Ambient Air", Environmental Health
Perspectives, 110, (2002), 3, 451-488.
14. Lide D. R.,"Handbook of Chemistry and Physics", 72th Ed, CRC Press, (1992).

104
15. Toxicological profile for polycyclic aromatic hydrocarbons - U.S. Department of health
and human services- Public Health Service - Agency for Toxic Substances and Disease
Registry, (1995).
16. Portal do inchem : http://www.inchem.org/documents/icsc/icsc/eics0667.htm.
17. Cotta J., A. O. Rezende, M. O. Oliveira, M. D. Landgraf, "Avaliação de solventes de
extração por ultrassom usando-se cromatografia líquida de alta eficiência para a determinação
de hidrocarbonetos policíclicos aromáticos em solos contaminados" Quími. Nova, 32, (2009),
8, 2026-2033.
18. James A. T., A. J. P. Martin, "Gas-liquid partition chromatography: the separation and
micro-estimation of volatile fatty acids from formic acid to dodecanoic acid", Biochemical
Journal, 50, (1952), 679-690.
19. W.H.O (World Health Organization),”Guidelines for Drinking Water Quality”, Chemical
Aspects, 2ª Ed., Genéve, (1993), 145-196.
20. W.H.O (World Health Organization),”Guidelines for Drinking-Water Quality”,3ª Ed.
Genéve, 1, (2006).
21. Kitson F.G., B.S. Larsen, C.N. McEwen,"Gas Cromatography and Mass Spectrometry",
Academic Press , (1996).
22. Günzler H., A. Williams," Handbook of Analytical Techniques", Wiley-VCH, Vol.I.
(2001).
23. Grob R.L.,"Modern practice of gas chromatography" 3ªEd., John Wiley & Sons Inc,
(1995).
24. Fowlis I.A.,"Gas Chromatography", 2ª Ed., John Wiley and Sons, (1995).
25. Agilent Technology Inc.,"Fundamentals of Gas Chromatography", Manual Part Number
G1176-90000.(2002).
26. Directiva 98/83/CE do Parlamento Europeu e do Conselho de 3 de Novembro de 1998,
relativa à qualidade da água destinada ao consumo humano, Jornal Oficial das Comunidades
Europeias, L 330/32, 5.12.98.
27. Decreto-Lei n.º 243/2001, Diário da República Série I-A, N.º 206, 2001-09-05.
28. Directiva 2006/118/CE do Parlamento Europeu e do Conselho de 12 de Dezembro de
2006 relativa à protecção das águas subterrâneas contra a poluição e a deterioração, Jornal
Oficial da União Europeia, L 372/19, 27.12.2006.
29. Decreto-Lei N.º 306/2007, Diário da República, Série I, N.º 164, 5747, 27-08-2007.
30. Decreto-Lei n.º 351/2007, Diário da República, 1.ª série, N.º 204, 23.10. 2007.

105
31. Directiva 2008/105/CE do Parlamento Europeu e do Conselho de 16 de Dezembro de
2008, relativa a normas de qualidade ambiental no domínio da política da água, Jornal Oficial
das Comunidades Europeias, L 348/84, 24.12.2008.
32. Portal da EPA, http://www.epa.gov/safewater.
33. WHO (World Health Organization), "Guidelines for Drinking Water Quality", First
Addenndum to Third Edition, Geneva, (2006).
34. Chen B. H., C. Y. Wang, C. P. Chiu, "Evaluation of analysis of polycyclic aromatic
hydrocarbons in meat products by liquid chromatography", Journal of Agriculture and Food
Chemistry, 44, (1996), 8, 2244-2251.
35. Dabestani R., I.N Ivanov, "A compilation of physical, spectroscopic and photophysical
properties of polycyclic aromatic hydrocarbons", Photochemistry and Photobiology, 70,
(1999), 1, 10-34.
36. Nolett L.M.L.,"Food Analysis by HPLC", Marcel Dekker Inc., 2ª Ed., (2000).
37. Alves A.C.H.,"Determinação de pesticidas em águas por microextracção em fase líquida
associada á cromatografia gasosa e espectrometria de massa" Tese de Mestrado; Faculdade de
Ciências e Tecnologia Universidade Nova de Lisboa, (2010) 20-40.
38. Guillot S., M.T. Kelly, H. Fenet, M. Larroque “Evaluation of solid-phase microextraction
as an alternative to the official method for the analysis of organic micro-pollutants in drinking
water”, Journal of Chromatography A, 1101 (2006) 46–52.
39. Barrionuevo W. R., F.M. Lanças “Comparison of Liquid–Liquid Extraction, Solid-Phase
Extraction, and Solid-Phase Microextraction for Pyrethroid Pesticides Analysis from Enriched
River Water”, Bull. Environ. Contam. Toxicol., 69 (2002) 123-128.
40. Frank T. C., L. Dahuron, B. S. Holden,W. D. Price, A. F. Seibert, L. C. Wilson Eds,
Perry’s chemical engineer’s handbook, 8th Edition, McGraw-Hill, (2008).
41. Dean J. R., Extraction methods for environmental analysis, Wiley, (1998).
42. Henriques M.L.G.S., “Hormonas Naturais e de Síntese, Bisfenol A, Octifenol e
Nonilfenol em Águas para Consumo Humano: Optimização do Método de Análise por SPE-
LC-ESI-MS/MS”, Tese de Mestrado, Faculdade de Farmácia (2008) 1-158.
43. Novakova L.; Vickova, H., "A review of current trends and advances in modern bio-
analytical methods: Chromatography and sample preparation" Analytica Chimica Acta, 656
(2009) 8–35.
44. Liua X., J. Li, Z. Zhao,W. Zhang, K. Lin, C. Huang, X. Wang, "Solid-phase extraction
combined with dispersive liquid–liquid microextraction for the determination for
polybrominated diphenyl ethers in different environmental matrices", Journal of
Chromatography A, 1216 (2009) 2220–2226.
45. Portal da Sigma Aldrich: http://www.sigmaaldrich.com/.

106
46. Faria AM, L. Maldaner, CC Santana, IC Jardim, CH Collins.
“Poly(methyltetradecylsiloxane) immobilized onto silica for extraction of multiclass
pesticides from surface waters”, Analytica Chimica Acta, 582, (2007) 34-40.
47. Fattahia N., S. Samadia, Y. Assadia, M. R. M. Hosseinia, "Solid-phase extraction
combined with dispersive liquid–liquid microextraction-ultra preconcentration of
chlorophenols in aqueous samples", Journal of Chromatography A, 1169, (2007), 1-2, 63-69.
48. Montes R., I. Rodríguez, M. Ramil, E. Rubí, R. Cela. "Solid-phase extraction followed by
dispersive liquid–liquid microextraction for the sensitive determination of selected fungicides
in wine ", Journal of Chromatography A, 1216, (2009), 29, 5459-5466.
49. Pawliszyn J., Solid Phase Microextraction-Theory and Practice, Wiley-VCH, (1997).
50. SPME fibers and Holders, Portal da Sigma-Aldrich,
http://www.sigmaaldrich.com/analytical-chromatography/analytical-
products.html?TablePage=9645336.
51. Baltussen E., P. Sandra, F. David, C. Cramers,"Stir bar sorptive extraction (SBSE), a
novel extraction technique for aqueous samples: Theory and principles", Journal of
Microcolumn Separations, 11, (1999), 10, 737-747.
52. Twister – Stir bar sorptive extraction, Portal da Gerstel,
http://www.gerstel.com/en/twister-stir-bar-sorptive-extraction.htm.
53. Chaves A. R., M. E. C. Queiroz,"Extração sortiva em barra deagitação para análise de
fármacos em fluidos biológicos", Quim. Nova, 31, (2008), 7, 1814-1819.
54. Prietoa A., O. Basauria, R. Rodilb, A. Usobiagaa, L.A. Fernándeza, N. Etxebarriaa, O.
Zuloagaa,"Stir-bar sorptive extraction: A view on method optimisation, novel applications,
limitations and potential solutions", Journal of Chromatography A, 1217, (2010).
55. Sánchez-Avila J., J. Quintana, F. Ventura, R.Tauler, C. M. Duarte, S. Lacorte, “Stir bar
sorptive extraction-thermal desorption-gas chromatography–mass spectrometry: An effective
tool for determining persistent organic pollutants and nonylphenol in coastal waters in
compliance with existing Directives", Marine Pollution Bulletin., 60, (2010) 103-112.
56. Huanga X., J. Lina, D. Yuana, R. Hu, “Determination of steroid sex hormones in
wastewater by stir bar sorptive extraction based on poly(vinylpyridine-ethylene
dimethacrylate) monolithic material and liquid chromatographic analysis”, Journal of
Chromatography A, 1216, (2009), 3508-3511.
57. Hou L., H.K. Lee,"Application of static and dynamic liquid-phase microextraction in the
determination of polycyclic aromatic hydrocarbons", Journal of Chromatography A, 976,
(2002), 377.
58. Liu J., G. Jiang, Y. Chi, Y. Cai, Q. Zhou, J. Hu, "Use of Ionic Liquids for Liquid-Phase
Microextraction of Polycyclic Aromatic Hydrocarbons", Analytical Chemistry, 75, (2003),
5870.

107
59. Zanjani M. R. K., Y. Yamini, S. Shariati, J.A. Jönsson, “A new liquid-phase
microextraction method based on solidification of floating organic drop”, Analytica Chimica
Acta, 585, (2007), 286–293.
60. Dadfarnia S., A. M. Salmanzadeh, A. M. H. Shabani, “A novel
separation/preconcentration system based on solidification of floating organic drop
microextraction for determination of lead by graphite furnace atomic absorption
spectrometry”, Analytica Chimica Acta, 623, (2008) 163-167.
61. Yamini Y., M. Rezaee, A. Khanchi, M. Faraji, A. Saleh, “Dispersive liquid-liquid
microextraction based on solidification of floating organic drop followed by inductively
coupled plasma–optical emission spectrometry as a fast technique for the simultaneous
determination of heavy metals", Journal of Chromatographaphy A, 1217, (2009), 16, 2358-
64.
62. Cortada C., L. Vidal, R. Pastor, N. Santiago, A. Canals, “Determination of organochlorine
pesticides in water samples by dispersive liquid-liquid microextraction coupled to gas
chromatography-mass spectrometry”, Analytica Chimica Acta, 649, (2009), 218-221.
63. Moinfar S., M. R. M. Hosseini, “Development of dipersive liquid-liquid method for the
analysis of organophosphorous pesticides in tea”, Journal of Hazardous Materials, 169,
(2009), 907-911.
64. Jahromi E. Z., A. Bidari , Y. Assadi , M. R. M. Hosseini, M.R. Jamali, “Dispersive liquid–
liquid microextraction combined with graphite furnace atomic absorption spectrometry Ultra
trace determination of cadmium in water samples”, Analytica Chimica Acta, 585, (2007), 305-
311.
65. Tsai W.C, S. Huang, “Dispersive liquid-liquid microextraction with little solvent
consumption combined with gás chromatography-mass spectrometry for the pretreatment of
organochlorine pesticides in aqueous samples”, Journal of Chromatography A, 1216, (2009)
5171-5175.
66. Berijani S., Y. Assadi, M. Anbia, M. R. M. Hosseini, E. Aghaee, “Dispersive liquid-liquid
microextraction combined with gas chromatography-flame photometric detection – Very
simple, rapid and sensitive method for the determination or organophosphorous pesticides in
water", Journal of Chomatography A, 1123, (2006), 1-9.
67. Zhao E., W. Zhao, L. Han, S. Jiang , Z. Zhou, “Application of dispersive liquid–liquid
microextraction for the analysis of organophosphorus pesticides in watermelon and
cucumber”, Journal of Chromatography A, 1175, (2007), 137–140.
68. López M. G., I. Rodríguez, R. Cela, “Development of a dispersive liquid–liquid
microextraction method for organophosphorus flame retardants and plastizicers determination
in water samples”, Journal of Chromatography. A, 1166, (2007), 9–15.
69. Fu L., X. Liu, J. Hu, X. Zhao, H. Wang, X. Wang, “Application of dispersive liquid–
liquid microextraction for the analysis of triazophos and carbaryl pesticides in water and fruit
juice samples”, Analytica Chimica Acta, 632, (2009), 289–295.

108
70. Herrara-Herrera A, M. A. Ramos, J. H. Borges, M. R. Delgado, "Dispersive liquid-liquid
microextraction for determination of organic analytes", Trends in Analytical Chemistry,
29,(2010), 7 .
71. Pena M. T., M. C. Casais, M. C. Mejuto, R. Cela, "Development of an ionic dispersive
liquid-liquid microextraction method for the analysis of polycyclic aromatic hydrocarbons in
water samples", Journal of Chromatography A, 1216, (2009), 6356–6364.
72. Wald G., D. Albers, T. McCabe, “Analysis by desorption of volatile impurities from an
ionic liquid solution in an unmodified gas chromatograph inlet”, Journal of Chromatography
A, 1216, (2009), 5927-5930.
73. Portal da wilkipédia, http://en.wikipedia.org/wiki/Polycyclic_aromatic_hydrocarbon.
74. Tavakoli L., Y.Yamini, H. Ebrahimzadeh, S.Shariati, “Homogeneous liquid-liquid
extraction for preconcentration of polycyclic aromatic hydrocarbons using a
water/methanol/chloroform ternary component system”, Journal of Chromatography A, 1196-
1197, (2008), 133-138.
75. Ebrahimzadeh H., Y. Yamini, F. Kamarei, S. Shariati, “Homogeneous liquid-liquid
extraction of trace amounts of mononitrotoluenes from waste water samples”, Analytica
Chimica Acta, 594, (2007), 93-100.
76. Ghiasvand A. R., S. Shadabi, E. Mohagheghzadeh, P. Hashemi, “Homogeneous liquid–
liquid extraction method for the selective separation and preconcentration of ultra trace
molybdenum”, Talanta, 66, (2005), 912-916.
77. Leong M.-I., C.-C. Chang, M.-R. Fuh, S.-D. Huang, "Low toxic dispersive liquid-liquid
microextraction by using halosolvents for extraction of polycyclic aromatic hydrocarbons in
water samples", Journal of Chromatography A, 1217, (2010), 34, 5455-5461.
78. Saleh A, Y Yamini, M Faraji, M. Rezaee, M. Ghambarian, "Ultrasound-assisted
emulsification microextraction method based on applying low density organic solvents
followed by gas chromatography analysis for the determination of polycyclic aromatic
hydrocarbons in water samples", Journal of Chromatography A, 1216, (2009), 6673-6679.
79. Rezaee M, Y. Yamini, M. Faraji, "Evolution of dispersive liquid-liquid microextraction
method", Journal Chromatography A, 1217, (2010), 2342-2357.
80. Rezaee M., Y. Assadi, M. M. Hosseini, E. Aghaee, F. Ahmadi, S. Berijani,
“Determination of organic compounds in water using dispersive liquid–liquid
microextraction”, Journal of Chromatography A, 1116, (2006), 1–9.
81. Hu J., Y. Li, W. Zhang, H. Wang, C. Huang, M. Zhang, X. Wang, "Dispersive liquid-
liquid microextraction followed by gas chromatography–electron capture detection for
determination of polychlorinated biphenyls in fish", Journal of Separation Science, 32 (2009),
2103-2108.
82. Liu X., J. Hu, C. Huang, H. Wang, X. Wang, "Determination of polybrominated diphenyl
ethers in aquatic animal tissue using cleanup by freezing-dispersive liquid–liquid

109
microextraction combined with GC-MS", Journal of Separtion Science, 32, (2009), 4213–
4219.
83. Bernardo M. S., M. Gonçalves, N. Lapa, R. Barbosa, B. Mendes, F. Pinto, I.Gulyurtlu,
"Determination of aromatic compounds in eluates of pyrolysis solid residues using HS-GC–
MS and DLLME–GC–MS", Talanta, 80, (2009), 1, 104-108.
84. Zhao E., W. Zhao, L. Han, S. Jiang, Z. Zhou, "Application of dispersive liquid–liquid
microextraction for the analysis of organophosphorus pesticides in watermelon and
cucumber", Journal of Chromatography A, 1175, (2007), 1, 137-140.
85. Xia J., B. Xiang, W. Zhang, "Determination of metacrate in water samples using
dispersive liquid–liquid microextraction and HPLC with the aid of response surface
methodology and experimental design", Analytica Chimica Acta, 625, (2008), 1, 28-34.
86. Nagaraju D., S.-Da Huang, "Determination of triazine herbicides in aqueous samples by
dispersive liquid–liquid microextraction with gas chromatography–ion trap mass
spectrometry", Journal of Chromatography A, 1161, (2007), 1-2, 89-97.
87. Melwanki M.B., M. Fuh, "Dispersive liquid–liquid microextraction combined with semi-
automated in-syringe back extraction as a new approach for the sample preparation of
ionizable organic compounds prior to liquid chromatography" Journal of Chromatography A,
1198-1199, (2008), 1-6.
88. Birjandi A. P., A. Bidari, F. Rezaei, M. R. M. Hosseini, Y. Assadi, "Speciation of butyl
and phenyltin compounds using dispersive liquid–liquid microextraction and gas
chromatography-flame photometric detection", Journal of Chromatography A, 1193, (2008),
1-2, 19-25.
89. Kozani R. R., Y. Assadi, F. Shemirani, M. R. M. Hosseini, M.R. Jamali, "Part-per-trillion
determination of chlorobenzenes in water using dispersive liquid–liquid microextraction
combined gas chromatography–electron capture detection", Talanta, 72, (2007), 2, 387-393.
90. Tsai W.-C., S.-D. Huang, "Dispersive liquid–liquid microextraction with little solvent
consumption combined with gas chromatography–mass spectrometry for the pretreatment of
organochlorine pesticides in aqueous samples", Journal of Chromatography A, 1216, (2009),
27,5171-5175.
91. Wu Q., C. Wang, Z. Liu, C. Wu, X. Zeng, J. Wen, Z. Wang, "Dispersive solid-phase
extraction followed by dispersive liquid–liquid microextraction for the determination of some
sulfonylurea herbicides in soil by high-performance liquid chromatography", Journal of
Chromatography A,1216, (2009), 29,5504-5510.
92. Cunha S. C., J. O. Fernandes, M. B. P. P. Oliveira, "Fast analysis of multiple pesticide
residues in apple juice using dispersive liquid–liquid microextraction and multidimensional
gas chromatography–mass spectrometry", Journal of Chromatography A,1216, (2009), 51,
8835-8844.

110
93. Chen H., J. Ying, H. Chen, J. Huang, L. Liao, "LC Determination of Chloramphenicol in
Honey Using Dispersive Liquid–Liquid Microextraction", Chromatographia, 68, (2008), 7,
629-634.
94. Yazdi A. S., N. Razavi, S. R Yazdinejad, "Separation and determination of amitriptyline
and nortriptyline by dispersive liquid–liquid microextraction combined with gas
chromatography flame ionization detection", Talanta, 75, (2008), 5,1293-1299.
95. Tsai W.-H., H.-Y. Chuang, H.-H. Chen, J.-J. Huang, H.-C. Chen, S.-H. Cheng., T.-C
Huang, "Application of dispersive liquid–liquid microextraction and dispersive micro-solid-
phase extraction for the determination of quinolones in swine muscle by high-performance
liquid chromatography with diode-array detection", Analytica Chimica Acta, 656, (2009), 1-
2,56-62.
96. Melwanki M. B., W.-S. Chen, H.-Y. Bai, T.-Y. Lin, M.-R. Fuh, "Determination of 7-
aminoflunitrazepam in urine by dispersive liquid–liquid microextraction with liquid
chromatography–electrospray-tandem mass spectrometry", Talanta, 78, (2009), 2, 618-622.
97. Xiong C., J. Ruan, Y. Cai, Y. Tang, "Extraction and determination of some psychotropic
drugs in urine samples using dispersive liquid–liquid microextraction followed by high-
performance liquid chromatography", Journal of Pharmaceutical and Biomedical Analysis,
49, (2009), 2, 572-578.
98. Cruz M. V., R. Lucena, S. Cárdenas, M. Valcárcel, "One-step in-syringe ionic liquid-
based dispersive liquid–liquid microextraction", Journal of Chromatography A, 1216, (2009),
37, 6459-6465.
99. Mao T., B. Hao, J. He, W. Li, S. Li, Z. Yu, "Ultrasound assisted ionic liquid dispersive
liquid phase extraction of lovastatin and simvastatin: A new pretreatment procedure", Journal
of Separation Science, 32, (2009), 17, 3029–3033.
100. Li Y., G. Wei, J. Hu, X. Liu, X. Zhao, X. Wang, "Dispersive liquid–liquid
microextraction followed by reversed phase-high performance liquid chromatography for the
determination of polybrominated diphenyl ethers at trace levels in landfill leachate and
environmental water samples", Analytica Chimica Acta, 615, (2008), 1, 96-103.
101. Liu X., J. Hu., C. Huang, H. Wang, X. Wang, "Determination of polybrominated
diphenyl ethers in aquatic animal tissue using cleanup by freezing-dispersive liquid–liquid
microextraction combined with GC-MS", Journal of Separation Science, 32, (2009), 23-24,
4213–4219.
102. Farahani H., P. Norouzi, R. Dinarvand, M. R. Ganjali, "Development of dispersive
liquid–liquid microextraction combined with gas chromatography–mass spectrometry as a
simple, rapid and highly sensitive method for the determination of phthalate esters in water
samples ", Journal of Chromatography A, 1172, (2007), 2, 105-112.
103. Wang X., C.-P. Diao, R.-S. Zhao, "Rapid determination of bisphenol A in drinking water
using dispersive liquid-phase microextraction with in situ derivatization prior to GC-MS",
Journal of Separation Science, 32, (2009), 1, 154–159.

111
104. Farajzadeh M. A., M. Bahram, J.A. Jönsson, "Dispersive liquid–liquid microextraction
followed by high-performance liquid chromatography-diode array detection as an efficient
and sensitive technique for determination of antioxidants", Analytica Chimica Acta, 591,
(2007), 1, 69-79.
105. Huang K.-J., C.-Y. Wei, W.-L. Liu, W.-Z. Xie, J.-F. Zhang, W. Wang, "Ultrasound-
assisted dispersive liquid–liquid microextraction combined with high-performance liquid
chromatography-fluorescence detection for sensitive determination of biogenic amines in rice
wine samples", Journal of Chromatography A,1216, (2009), 38, 6636-6641.
106. Pusvaskiene E., B. Januskevic, A. Prichodko, V. Vickackaite, "Simultaneous
Derivatization and Dispersive Liquid–Liquid Microextraction for Fatty Acid GC
Determination in Water", Chromatographia, 69, (2009), 271-276.
107. Rezaee M., Y. Assadi, M.-R. M. Hosseini, E. Aghaee, F. Ahmadi, S. Berijani,
"Determination of organic compounds in water using dispersive liquid–liquid
microextraction", Journal of Chromatography A, 1116, (2006), 1-2, 1-9.
108. Yao C., J. L. Anderson,"Dispersive liquid–liquid microextraction using an in situ
metathesis reaction to form an ionic liquid extraction phase for the preconcentration of
aromatic compounds from water", Analytical and Bioanalytical Chemistry, 395, (2009), 5,
1491-1502.
109. Vickackaite V., E. Pusvaskiene, "Dispersion-solidification liquid–liquid microextraction
for volatile aromatic hydrocarbons determination: Comparison with liquid phase
microextraction based on the solidification of a floating drop", Journal of Separation Science,
32, (2009), 20, 3512–3520.
110. Xu H., Z. Ding, L. Lv, D. Song, Y.-Q. Feng, "A novel dispersive liquid–liquid
microextraction based on solidification of floating organic droplet method for determination
of polycyclic aromatic hydrocarbons in aqueous samples", Analytica Chimica Acta, 636,
(2009), 1, 28-33.
111. Brumm D. M., R. J. Cassella, A. D. P. Netto, "Multivariate optimization of a liquid–
liquid extraction of the EPA-HAPs from natural contaminated waters prior to determination
by liquid chromatography with fluorescence detection", Talanta, 74, (2008), 5,1392-1399.
112. Pensado L., E. Blanco, M. C. Casais, M. C. Mejuto, E. Martinez, A. M. Carro , R. Cela,
"Strategic sample composition in the screening of polycyclic aromatic hydrocarbons in
drinking water samples using liquid chromatography with fluorimetric detection", Journal of
Chromatography A, 1056, (2004), 1-2, 121-130.
113. Delhomme O., E. Rieb, M. Millet, "Solid-Phase Extraction and LC with Fluorescence
Detection for Analysis of HAPs in Rainwater", Chromatographia, 65, (2007), 3-4, 163-171.
114. Fernández-González V., E. Concha-Graña, S. Muniategui-Lorenzo, P. López-Mahía, D.
Prada-Rodríguez, "Solid-phase microextraction–gas chromatographic–tandem mass
spectrometric analysis of polycyclic aromatic hydrocarbons: Towards the European Union
water directive 2006/0129 EC", Journal of Chromatography A, 1176, (2007), 1-2, 48-56.

112
115. Huertas C., J. Morillo, J. Usero, I. Gracia-Manarillo, "Validation of stir bar sorptive
extraction for the determination of 24 priority substances from the European Water
Framework Directive in estuarine and sea water", Talanta, 72, (2007), 3, 1149-1156.
116. Hou L., H. K. Lee, "Application of static and dynamic liquid-phase microextraction in
the determination of polycyclic aromatic hydrocarbons", Journal of Chromatography A, 976,
(2002), 1-2, 377-385.
117. Liu J., G. Jiang, Y. Chi, Y. Cai, Q. Zhou, J. Hu, "Use of Ionic Liquids for Liquid-Phase
Microextraction of Polycyclic Aromatic Hydrocarbons", Anal. Chem., 75, (2003), 21, 5870–
5876.
118. Oliferova L., M. Statkus, G. Tsysin, O. Shpigun, Y. Zolotov, "On-line solid-phase
extraction and HPLC determination of polycyclic aromatic hydrocarbons in water using
fluorocarbon polymer sorbents", Analytica Chimica Acta, 538, (2005), 1-2, 35-40.
119. King A. J., J. W. Readman , J. L. Zhou, "Determination of polycyclic aromatic
hydrocarbons in water by solid-phase microextraction–gas chromatography–mass
spectrometry", Analytica Chimica Acta, 523, ( 2004), 2, 259-267.
120. Djozan D. J., Y. Assadi, "Monitoring of Polycyclic Aromatic Hydrocarbons in Water
Using Headspace Solid-Phase Microextraction and Capillary Gas Chromatography",
Microchemical Journal, 63, (1999), 2, 276-284.
121. Psillakis E., N. Kalogerakis, "Developments in liquid-phase microextraction", Trends in
Analytical Chemistry, 22, (2003), 9, 565-574.
122. Shariati-Feizabadi S., Y. Yamini, N. Bahramifar, "Headspace solvent microextraction
and gas chromatographic determination of some polycyclic aromatic hydrocarbons in water
samples". Analytica Chimica Acta, 489, Issue, (2003), 1, 21-31.
123. Charalabaki M., E. Psillakis, D. Mantzavinos, N. Kalogerakis, "Analysis of polycyclic
aromatic hydrocarbons in wastewater treatment plant effluents using hollow fibre liquid-phase
microextraction", Chemosphere, 60, (2005), 5, 690-698.
124. Ratola N, , A. Alves, N. Kalogerakis, E. Psillakis, "Hollow-fibre liquid-phase
microextraction: A simple and fast cleanup step used for HAPs determination in pine
needles", Analytica Chimica Acta, 618, (2008), 1, 70-78.
125. Zanjani M. R. K., Y. Yamini, S. Shariati, J. A Jönsson, "A new liquid-phase
microextraction method based on solidification of floating organic drop", Analytica Chimica
Acta, 585, (2007), 2, 286-293.
126. Huang Z., Y. Li, B. Chen, S. Yao, “Simultaneous determination of 102 pesticides
residues in Chinese teas by gas chromatography-mass spectrometry”, Journal of
Chromatography B, 853 (2007) 154-162.
127. Funk W., V. Dammann, G. Donnevert, Quality Assurance in Analytical Chemistry, VCH
(1995).

113
128. Fowlis I.A., Gas Chromatography, 2ª Ed., John Wiley and Sons (1995).
129. Instituto Português de Acreditação, Guia para acreditação de laboratórios químicos,
14.09.2005.
130. ISO 8466-1:1990, “Water Quality- Calibration and Evaluation of Analytical Methods
and Estimation of Performance Characteristics. Part 1: Statistical Evaluation of the Linear
Calibration Function”, (1990).
131. Miller J. C, J. N. Miller, Statistics for Analytical Chemistry, 3ª Ed., Ellis Hordwood PTr
Prentice Hall (1993).
132. Grob R. L., Modern practice of gas chromatography, 3ª Ed., John Wiley & Sons Inc.
(1995).
133. ISO 8466-2:1990, “Water Quality- Calibration and Evaluation of Analytical Methods
and Estimation of Performance Characteristics. Part 2: Calibration Strategy for Non-Linear
Calibration Function”, (1990).
134. Günzler H., A. Williams, Handbook of Analytical Techniques, Volume I, Wiley-VCH
(2001).
135. ISO 3534-1:2006, “Statistics- Vocabulary and Symbols. Part 1: General statistical terms
and terms used in probability”, (2006).
136. Toxicological Profile For Polycyclic Aromatic Hydrocarbons- U.S. Department Of
Health And Human Services- Public Health Service - Agency for Toxic Substances and
Disease Registry,(1999).
137. Liu X., J. Li, Z. Zhao, W. Zhang, K. Lin, C. Huang, X. Wang, "Solid-phase extraction
combined with dispersive liquid–liquid microextraction for the determination for
polybrominated diphenyl ethers in different environmental matrices", Journal of
Chromatography A, 1216, (2009), 12, 2220-2226.
138. Zhao R., C. Diao, Q. Chen, X. Wang, “Sensitive determination of amide herbicides in
envionmental water samples by combination of solid-phase extraction and dispersive liquid-
liquid microextraction prior to GC-MS”, Journal of Separation Science, 32 ,(2009), 1069-
1074.
139. Kootstra P.R., M. H. C. Straub, G. H. Stil, E. G. Van der Velde, W. Hesselink,C. C. J.
Land, "Solid-phase extraction of polycyclic aromatic hydrocarbons from soil samples",
Journal of Chromatography A, 697, (1995), 123-129
140. Fattahi N., S. Samadi , Y. Assadi , M. Reza, M. Hosseini, "Solid-phase extraction
combined with dispersive liquid–liquid microextraction-ultra preconcentration of
chlorophenols in aqueous samples", Journal of Chromatography A, 1169, (2007), 63–69
141. Montes R., I. Rodríguez, M. Ramil, E. Rubí, R. Cela, "Solid-phase extraction followed
by dispersive liquid–liquid microextraction for the sensitive determination of selected
fungicides in wine", Journal of Chromatography A, 1216, (2009), 29,5459-5466.

114
142. Specanalitica- Equipamentos Científicos, Lda:
http://www.specanalitica.com/scid/webspec/default.asp.
143. Labbox: http://www.labbox.com/es/.
144. Prieto A., O. Basauri, R. Rodil, A. Usobiaga, L. A. Fernández, N. Etxebarria, O.
Zuloaga, "Stir-bar sorptive extraction: A view on method optimisation, novel
applications,limitations and potential solutions", Journal of Chromatography A, 1217, (2010),
2642-2666.
145. Prieto A., O. Telleria, N. Etxebarria, L. A. Fernández, A. Usobiaga, O. Zuloaga,
"Simultaneous preconcentration of a wide variety of organic pollutants in water samples :
Comparison of stir bar sorptive extraction and membrane-assisted solvent extraction", Journal
of Chromatography A, 1214, (2008), 1-2, 1-10.
146. A. Prieto, O. Zuloaga, A. Usobiaga, N. Etxebarria, L. A. Fernández, C. Marcic, A.
Diego, "Simultaneous speciation of methylmercury and butyltin species in environmental
samples by headspace-stir bar sorptive extraction–thermal desorption–gas chromatography–
mass spectrometry", Journal of Chromatography A, 1185, (2008), 1, 130-138.
147. A. Prieto, O. Zuloaga, A. Usobiaga, N. Etxebarria, L. A. Fernández, "Development of a
stir bar sorptive extraction and thermal desorption–gas chromatography–mass spectrometry
method for the simultaneous determination of several persistent organic pollutants in water
sample", Journal of Chromatography A,1174,1-2 ,2007,40-49 .
148. MacNamara K., R. Leardi, F. McGuigan, "Comprehensive investigation and
optimisation of the main experimental variables in stir-bar sorptive extraction (SBSE)-thermal
desorption-capillary gas chromatography (TD-CGC)", Analytica Chimica Acta, 636, (2009),
2, 190-197 .
149. Snow J., K. Lokhnauth, H. Nicholas, "Stir-bar sorptive extraction and thermal
desorption-ion mobility spectrometry for the determination of trinitrotoluene and l,3,5-
trinitro-l,3,5-triazine in water samples" ,Journal of Chromatography A, 1105, (2006), 1-2, 33-
38.
150. Guerrero E. D., R. N. Marín, R. C. Mejías, C. G. Barroso, "Optimisation of stir bar
sorptive extraction applied to the determination of volatile compounds in vinegars", Journal
of Chromatography A,1104, (2006), 1-2,47-53 .
151. Moeder R., R. Monika, "Development of a method for the determination of UV filters in
water samples using stir bar sorptive extraction and thermal desorption–gas chromatography–
mass spectrometry", Journal of Chromatography A, 1179, (2008), 2, 81-88.
152. Portal da Web of Science:
http://isiwebofknowledge.com/products_tools/multidisciplinary/webofscience/.
153. Guan W., Y. Wang, F. Xu, Y. Guan, "Poly(phthalazine ether sulfone ketone) as novel
stationary phase for stir bar sorptive extraction of organochlorine compounds and
organophosphorus pesticides", Journal of Chromatography A, 1177, (2008), 1, 28-35 .

115
154. Popp P., C. Bauer, B. Hauser, P. Keil, L. Wennrich, "Extraction of polycyclic aromatic
hydrocarbons and organochlorine compounds from water: A comparison between solid-phase
microextraction and stir bar sorptive extraction", Journal of Separation Science,26,
(2003),961-967.
155. Kolahgar B., A. Hoffmann, A. C. Heiden, "Application of stir bar sorptive extraction to
the determination of polycyclic aromatic hydrocarbons in aqueous samples", Journal of
Chromatography A, 963, (2002), 1-2, 225-230 .
156. Montero L., S. Conradi, H. Weiss, P. Popp, "Determination of phenols in lake and
ground water samples by stir bar sorptive extraction–thermal desorption–gas
chromatography–mass spectrometry", Journal of Chromatography A, 1071, 1-2, (2005), 163-
169 .
157. Coelho E., R. Perestrelo, N. R. Neng, J. S. Câmara, M. A. Coimbra, J.M.F. Nogueira, S.
M. Rocha, "Optimisation of stir bar sorptive extraction and liquid desorption combined with
large volume injection-gas chromatography–quadrupole mass spectrometry for the
determination of volatile compounds in wines" Analytica Chimica Acta, 624, (2008), 1, 79-
89.
158. SPME mode, Portal da Brechbühler AG, http://www.brechbuehler.ch/SPME-
Mode.204.0.html.





























