Embed Size (px)
Citation preview

MAE0540-Genética de Populações
MAE5757-Métodos Estatísticos em
Genética e Genômica
Júlia Maria Pavan Soler
IME/USP - 1°Sem/2016

MotivaçãoMAPEAMENTO DE “GENES” NAS POPULAÇÕES MUNDIAIS
Já vimos: “Genoma é um espaço estruturado”
Inferências sobre “Efeitos Genéticos” em dados gerados de
delineamentos:
Estudos com indivíduos não relacionados e fenótipos quantitativos
Estudos Observacionais Caso-Controle
Estudos Observacionais com Famílias
Estudos de Expressão Gênica (Microarrays)
2
3
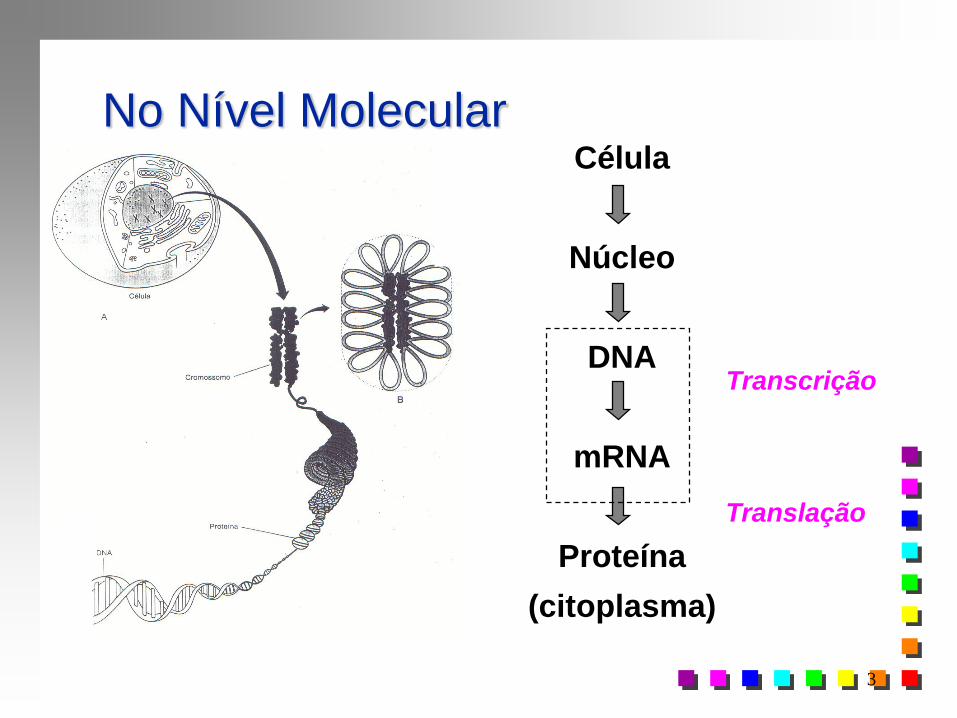
No Nível Molecular
Transcrição
Célula
Núcleo
DNA
mRNA
Proteína
(citoplasma)
Translação

4
Transcriptoma
Análise de Microarrays
Fonte: Doerge, R.W. Nature
Reviews 3, 2002
Do Gene ao Fenótipo
Variação Quantitativa em cada estágio “janelas” para a coleta de dados
Proteoma
Genoma
Análise de QTL’s
Dogma Central da Biologia

5
Coleta de Dados
...
...
Amostra da
População
Amostra do
Genoma
FAM INDIV PAI MÃE FENO1 FENO2 … FENOf SNP1 SNP2 … SNP2.882
REPL1 REPL2 REPL1 REPL2 … REPL1 REPL2
1 1
1 2
… …
1 14
2 15
… …
2 28
… …
14
14
14 ~200
FENOExp2 FENOExp3.554FENOExp1U.A. Fenótipos Genótipos Expressão de Genes

6
Motivação - Projetos InCor
Análise de QTL’s
e QTN´s
Estudos de
Associação (GWA´s)
Delineamentos com
Expressão Gênica
Delineamentos F2
Delineamentos com Famílias
Projeto Corações do Brasil
GH HJ
HH
Delineamentos com Trios
Estudos Caso-Controle
Grupo A AC
Total
Caso 60 40 100
Controle 40 60 100
Alelo Marcador
G U A A U C C U C
transcriptase
reversa
Experimentos com
Microarrays
Experimentos RT-PRC
(modelos animais)

7
Experimentos com Microarrays
Quantificar a Expressão de (Muitos) Genes (Simultaneamente)
DNA
(núcleo)
RNA (citoplasma)
Transcrição: “janela” para se observar
a Função Celular
Transcriptoma
Motivação: Medir a quantidade de
mRNA quais “genes” estão
sendo expressos ( quais “genes”
estão participando da função
celular)
GTAATCCTC
CATTAGGAG
mRNA

8
Experimentos com Microarrays
Transcrever DNA, Sintetizar DNA, Hibridizar,
DNA
(núcleo)
RNA (citoplasma)
GTAATCCTC
CATTAGGAG
mRNA
Transcrição
GUAAUCCUC
mRNA
Transcrição Reversa

9
Construção das Lâminas de Microarrays
Fragmentos de DNA de interesse são fixados nos spots em uma
lâmina de vidro
Tecnologia que permite a avaliação simultânea da expressão de
milhares de fragmentos
cDNA
cDNA: cada sequência é avaliada para dois canais de fluorescência
(Cy3=verde, Cy5=vermelho)
Oligonucleotídeos: sequências são avaliadas em um único canal (Cy3)
lâminas com 20, 30 mil fragmentos
sequências são geradas e spotadas
mecanicamente por um robô
Affymetrics: sequências curtas (20-25 bases), match/dismatch
Codelink: sequências longas

10
Clones cDNA
(probes)
printing
microarray Hibridização
Amostras mRNA
(Folha/raiz, raiz1/raiz2, …)
Experimentos com Microarrays de cDNA

11
Experimentos com Microarrays de cDNA
Construção das lâminasRobô fixa quantidades do fragmento de
cDNA nos spots nas lâminas
Volume de material na agulha: 100-250nl
Volume do spot: 0.2-1.0nl
Spot“Background”
• A anotação do cDNA
em cada spot deve ser
muito criteriosa
• Há diferentes
propostas para definição
da área do background

12
RNA 1(Cy 3)
RNA 2(Cy 5)
lavagem
hibridização
Scan 1 Scan 2
Imagem combinada
Imagem 1 Imagem 2
550 nm 650 nm
expressão do gene
na amostra 1
expressão do gene
na amostra 2
Experimentos com Microarrays de cDNA

13
Experimentos com Microarrays de cDNA
Hibridização
RNA 1(Cy 3)
RNA 2(Cy 5)

14
Experimentos com Microarrays de cDNA
Hibridização
Spot verde/vermelho: maior pico de leitura
Amarelo: mesma intensidade de sinal
Preto: não hibridização
Branco: “estouro” na leitura (saturação)
Escala da intensidade de fluorescência
igual em cy5 em cy3
Scaner
Leitura das intensidades de expressão
1=20 65.536=216

15
Quantificação da Expressão GênicaMedidas normalizadas
de expressão diferencial

16
Unidades amostrais: indivíduos ou pool
Spots: Replicação dentro das lâminas (“não concordantes”)
Intensidades variam com a distribuição dos spots
Muitos spots com nenhum sinal
Variação nos canais: Cy5: Intensidade fixação
Quantidade de cDNA (spot): variação
Propriedades de hibridação variam entre as sequências de cDNA
RNA 1(Cy 3)
RNA 2(Cy 5)
Análise dos
sinais R/G
Teoria de
Hibridização
Variação Biológica e Variação da Técnica
Medidas de expressão empíricas são imprecisas

17
Etapas da Análise de Dados de
Microarrays
Controle de fontes de
variação à priori
Controle de fontes de
variação à posteriori
Problema Biológico
Planejamento
Experimental
Experimento de Microarray
Pré-processamento dos
Dados
Análise Exploratória de DadosAnálise da Expressão
Diferencial entre Grupos
Predição de Redes
Regulatórias
Análise da Imagem Quantificação da Expressão
Correção pelo Background Normalização

18
Etapas da Análise de Dados de
Microarrays de cDNA
Planejamento de Experimentos (a Priori)
Experimento de Microarrays
Análise de Imagens (discordância entre Scaners)
Correção pelo Background
Controle de Fontes de Variação Externas (a Posteriori)
Normalizações (F.V. da Técnica)
Análise Estatística
Modelos ANOVA, Correção para Múltiplos Testes
Análises Exploratórias Multivariadas

19
Experimentos com Microarrays
Avaliação Simultânea da Expressão de “Muitos” Genes em
Diferentes Condições
Estudos Transversais
Grupo(s) Caso(s) x Grupo Controle(referência)
Experimentos Comparativos, Análise de Agrupamento e Discriminante
Estudos “Longitudinais”
Seguimento de etapas intermediárias de um sistema funcional
Inferência Funcional (células tronco, desenvolvimento)
Característica dos Experimentos “poucas” unidades amostrais
“muitos” genes (FV de interesse)
“muitas” fontes de variação sistemáticas

20
Experimentos com MicroarraysObjetivo: Comparar níveis/quantidades específicas de mRNA (expressão
de genes) em diferentes condições experimentais
Diferentes tecidos do mesmo organismo
raiz x folha, rim doente x rim não doente
Mesmo tecido, mesmo organismo, submetido a diferentes
intervenções raiz em stress hídrico x raiz em condição normal
Mesmo tecido, diferentes organismos
folha sp1 x folha sp mutante
...

21
Amostras de RNAm
Grupo SHR x Congenico Lâmina - Pool (n=3)
Controle SHR-Cy5 SHR/BN-Cy3 Rat 85
SHR-Cy3 SHR/BN-Cy5 Rat 89
NaCl SHR-Cy5 SHR/BN-Cy3 Rat 88
SHR-Cy3 SHR/BN-Cy5 Rat 86
Experimentos com Microarrays
Projeto InCor
Lâminas de cDNA : 26.912 Spots (2 réplicas dentro lâmina) Genes >> u.a.
inversão dos
corantes
SHRCSHR/BNC
SHRNaCl SHR/BNNaCl
comparações diretas

22
X
N1
SHRBN
SHR
X
SHR
X
50/50
25/75SHR
X
N2
N3
Em 20 gerações todo o material genético do BN é eliminado
12,5/87,5( . . . )
Geração de animais congênicos
SHR modificados: com as
regiões candidatas do animal BN

23
Lâmina RAT85: SHR-Cy5 SHR/BN-Cy3
26.912 Spots
32 quadrantes
(2 réplicas)

24
Amostras de RNAm
Grupo SHR x Congenico # lâminas (Pool:n=3)
Controle SHR-Cy5 SHR/BN-Cy3 1
SHR-Cy3 SHR/BN-Cy5 1
NaCl SHR-Cy5 SHR/BN-Cy3 1
SHR-Cy3 SHR/BN-Cy5 1
Experimento com Microarrays
Lâmina Spot Réplica Cy3 Cy5 T=Cy3/Cy5 log( T )
1 1 1
1 1 2
1 2 1
1 2 2
... ... ... ... ... ...
1 13000 1
1 13000 2
4676 8064 0,010 0,003
-285 890 0,045 0,020
Diferenças entre réplicas DENTRO de lâmina (13000 spots)
Médias entre
réplicas da
variável T:
menor
variabilidade

25
Planejamento de Experimentos com
Microarrays
Construção das matrizes (lâminas) de microarray
seleção de uma amostra de “genes” (fragmentos cromossômicos) que
seja representativa do genoma sob estudo (representativo dos possíveis
genes envolvidos na regulação do sistema biológico sob estudo)
replicações dentro da lâmina, spots controle negativo (brancos), spots
controle positivo
Amostras de mRNA
unidades amostrais: indivíduos ou pool de indivíduos
replicações biológicas
Atribuição dos corantes aos Grupos (Sistema de Duas Cores)
Lâmina : fator bloco (recebe ambos os tratamentos)
Genes (dentro da lâmina): é um fator adicional

26
Planejamento de Experimentos com
Microarrays
Atribuição dos corantes aos Grupos (Sistema de Duas Cores)
Lâmina : fator bloco
Estrutura de Tratamento: 1 fator em dois níveis (A e B)
A B
Cy3 Cy5
A B
Tipos de Delineamentos:
rOs tratamentos A e B são aplicados em r
lâminas de microarrays
A: recebeu Cy3 B: recebeu Cy5
Única situação de
blocos completos
Tamanho dos
blocos é 2

27
Planejamento de Experimentos com
Microarrays
A B
Cy3 Cy5
A B
Cy3 Cy5
Cy5 Cy3
Dye- swapp • Controlar a variabilidade devido ao corante
Lâmina Cy3 Cy5
1 A B
2 B A
r
Lâmina Cy3 Cy5
1 A B
2 A B
… … …
r A B
O efeito do
tratamento pode
estar confundido
com corante!
Aleatorizar a atribuição dos corantes
aos tratamentos tratamentos nas r
lâminas!
Atribuição dos corantes aos Grupos (Sistema de Duas Cores)

28
Experimentos com Microarrays
Comparações Diretas entre as Intensidades de Expressão
A B
Cy3 Cy5
A B
Dye- swapp
Representação Gráfica dos Experimentos
A B A B
1 lâmina
2 lâminas

29
Experimentos com Microarrays
Atribuição dos corantes aos Grupos (Sistema de Duas Cores)
C
BA2 lâminas: delineamento desbalanceado (A e B:1 réplica, C:2 réplicas)
Delineamento indireto: “log(A/B)” é estimado indiretamente por
“log(A/C) – log(B/C), mas com 1 réplica”
A
BC
(Yang and Speed, 2002)
Estrutura de Tratamento: 1 fator em três níves (A, B e C)
Delineamento em blocos incompletos
Possíveis Configurações:
Lâmina Cy3 Cy5
1 A B
2 B C
3 C A
Delineamentos em Loop:
em blocos incompletos
balanceados (2 réplicas)

30
Experimentos com Microarrays
Delineamento Looping
3 lâminas
Blocos Incompletos Balanceados
A
BC
A B B C C A
A
BC
Looping Dye Swapp

31
Experimentos com MicroarraysA
BC
2
2
2
R
BA C
Lâmina Cy3 Cy5
1 R A
2 R B
3 R C
4 R A
5 R B
6 R C
2 2 2
Lâmina Cy3 Cy5
1 A B
2 A B
3 C A
4 C A
5 B C
6 B C
Por simulação o delineamento Loop tem produzido erros padrão (em média)
menores que o de referência (Yang e Speed, 2001)
O Del. de Referência tem sido mais adotado. A amostra de mRNA de
referência tem que ser escolhida de forma criteriosa (ter alta expressão e não
ter especificidade)
É aconselhável aleatorizar a atribuição dos corantes

32
Planejamento de Experimentos
Replicação Biológica Replicação deve ser feita no nível das unidades amostrais que
compõem a população alvo (sobre a qual desejamos realizar inferências)
Deseja-se identificar genes diferencialmente expressos em qual
“população”? o pesquisador deve saber responder a qual população ele
deseja inferir seus resultados (para uma sp, para uma população F2, ...)
Em geral a fonte de variação ENTRE indivíduos é maior que outras fontes
de variação (corante, hibridização,...) ter arrays para diferentes u.a.
(tantas quanto possível para se ganhar precisão. Lembre-se que o efeito de
tratamento é a fonte de variação que se deseja mensurar.)
Pool como unidade amostral (recomendado para populações
homogêneas, F2) reduz variabilidade biológica (o que é a medida de
expressão de uma amostra de mRNA combinada de muitos indivíduos?)
Replicação (do spot) dentro do array útil para normalização

33
Experimentos com Microarrays

34
Experimentos com Microarrays
Nos Experimentos de Microarrays não existe um delineamento
ótimo universal. Os princípios de Planejamento de
Experimentos devem nortear a decisão do pesquisador e
assegurar que estaremos acumulando e atualizando nosso
conhecimento e não simplesmente gerando uma enorme
quantidade de dados.
(Churchill e Oliver, 2001)

35
Etapas da Análise de Dados de
Microarrays
Problema Biológico
Planejamento
Experimental
Experimento de Microarray
Pré-processamento dos
Dados
Análise Exploratória de DadosAnálise da Expressão
Diferencial entre Grupos
Predição de Redes
Regulatórias
Análise da Imagem Quantificação da Expressão
Correção pelo Background Normalização

36
Pré-Processamento dos Dados
Dados amostrais sujeitos a muitas fontes de variações
(conhecidas e desconhecidas):
Leituras dos scaners
Spots com nenhum sinal ou saturação
Variação espacial
Variação do canal de fluorescência
Outras F.V. sistemáticas
Eliminar efeitos indesejáveis calibrar, normalizar,
transformar, ponderar os dados encontrar uma “escala”
apropriada que permita comparar os níveis de expressão
entre e dentro das lâminas

37
Pré-Processamento dos Dados
Visualização cuidadosa dos dados de intensidade de expressão
Correções nos dados de expressão observados são necessários
por diferentes razões. Possíveis correções:
b
aYY
*
MeYYYa Aparadak ,,,
1
2
n
aYb
Distribuição de Y
Y: intensidade de expressão
YYY ˆ*
*YY

38
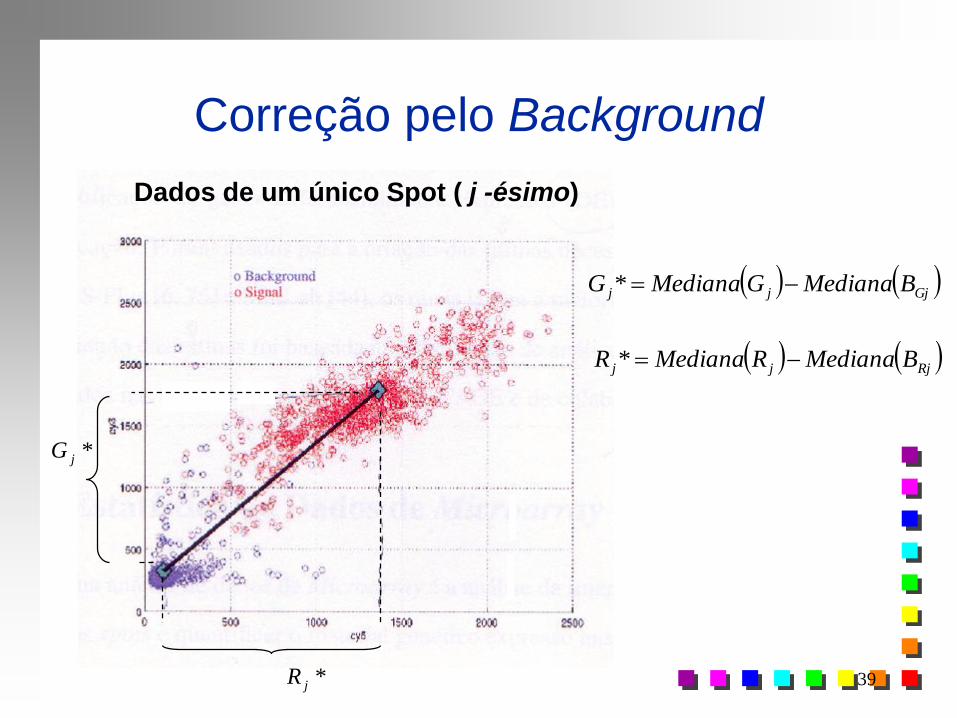
Correção pelo Background
Rj Gj (Red, Green)
Spot j (“ Gene j ” )
Spot verde/vermelho: maior pico de leitura
Amarelo: mesma intensidade de sinal
Preto: não hibridização
Branco: “estouro” na leitura (saturação)
Leitura da intensidade
de expressão de um
spot abre uma grade de
pontos 50x50 (pixels)
Background
Foreground
39
Correção pelo Background
Dados de um único Spot ( j -ésimo)
*jR
Gjjj BMedianaGMedianaG *
*jG
Rjjj BMedianaRMedianaR *

40
Visualização da Expressão Diferencial
Os spots (pontos no gráfico) fora (abaixo ou acima) das retas indicadas
têm expressão diferencial entre as amostras de mRNA (segundo um ponto
de corte arbitrário adotado)
A anotação dos spots é muito importante identificação do fragmento
cromossômico e de sua função em algum sistema biológico
Diagrama de Dispersão das Intensidades de Expressão

41
Visualização da Expressão
Diferencial

42
Normalizações
Ainda há fontes de variação conduzindo a “erros/vícios” nas
medidas de intensidade de expressão:
Eliminar Vícios Sistemáticos
Ocorrência de “outliers”
Tendências: Variância crescendo com a média das
intensidades, não Normalidade (assimetria)
Dados de cada Spot já normalizados pelo
Background:
Cy3 Cy5 *jR*jG

43
log Gj
log Rj

44
Análise Baseada na Razão(Chen et al., 1997)
Assume para o g-ésimo gene:
kR e ~ i.i.d. Normais homocedásticaskG
kkk GRT /
22
2
22
2
12
1exp
21
11
tc
t
tc
tttfk
c: coeficiente de variação estimado dos dados
Outras alternativas são usadas para distribuições
assimétricas.

45
Transformação Log
Modelo de Efeitos Multiplicativos Linearizar
Distribuição Assimétrica Positiva Normalizar
Log2 escala conveniente
jC
j ey

46
Análise Baseada no Log
Cy5 R
Cy3 G
Log2 R
Log2 G
T= R/G
Log2 T = Log2 R - Log2 G
dados de cada Gene
Corrigir as medidas de expressão gênica para muitas
fontes de variação possíveis Normalização

47
Normalização
Cy5 Rj
Cy3 Gj
Normalização Não Paramétrica x Normalização Paramétrica
Normalização: Eliminar Vícios Sistemáticos
Controle de Qualidade das Medidas
dados de cada Spotj
j
jG
RT
G
RLogGLogRLogTLog 2222
RLog2
GLog2
cTLogTLogTLog 222 *
A expressão
diferencial é avaliada
na escala Logaritmica
(base 2)

48
Transformação Razão: R/G
Há algum padrão
(espacial) na distribuição
das razões ?
Visualizar a
distribuição de
G
RLogTLog 22

49
Normalização Não Paramétrica
Normalização pela Energia Total (Global)
cG
RT
j
j
j
2
*
2 loglog
j
j
G
RMédiac 2log
Mediana, Média Aparada
j-ésimo Spot
tais correções também
podem ser feitas por sub-
array.

50
Normalização Não Paramétrica
Normalização dependente da Intensidade de Expressão
j
j
j
jjj
AcG
R
AcTT
2
2
*
2
log
loglog
A x M Gráfico do através estimado:jAc
j-ésimo Spot j=1, 2, ..., n
jj TT 2log de )(suavizado ajustadovalor :ˆ
Neste gráfico
podemos visualizar
a distribuição de
log2T.

51
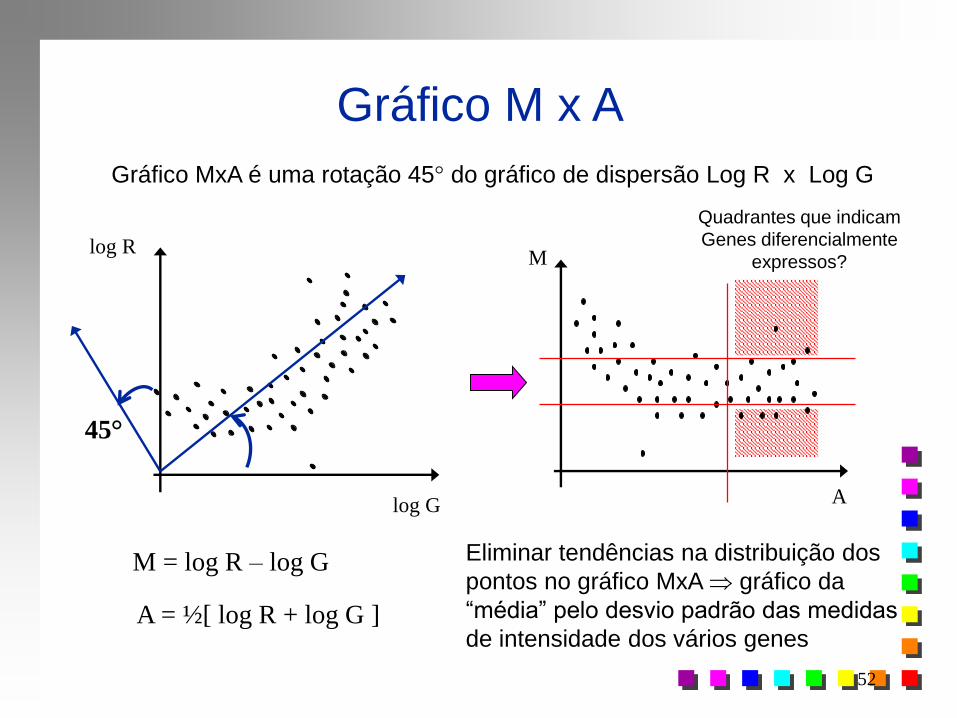
Gráfico M x A
A
M
log G
log R
45°
Gráfico MxA é uma rotação 45° do gráfico de dispersão Log R x Log G
M = log R – log G
M = Minus
A = ½[ log R + log G ]
A = Add
Dependência funcional entre
média e variância?
52
Gráfico M x A
A
M
log G
log R
45°
Gráfico MxA é uma rotação 45° do gráfico de dispersão Log R x Log G
M = log R – log G
A = ½[ log R + log G ]
Quadrantes que indicam
Genes diferencialmente
expressos?
Eliminar tendências na distribuição dos
pontos no gráfico MxA gráfico da
“média” pelo desvio padrão das medidas
de intensidade dos vários genes

53
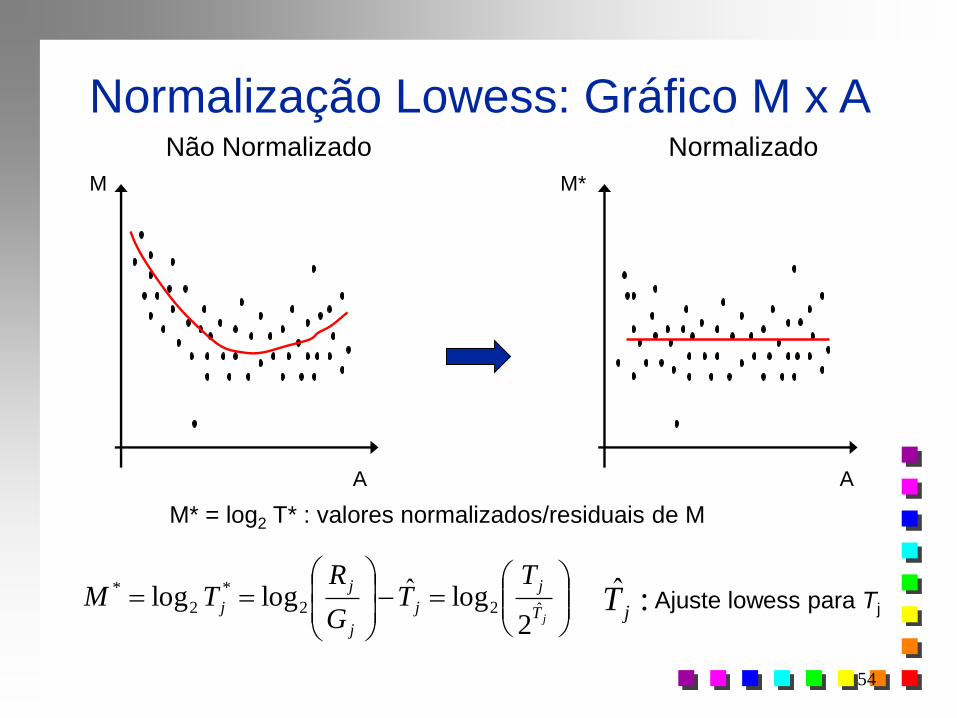
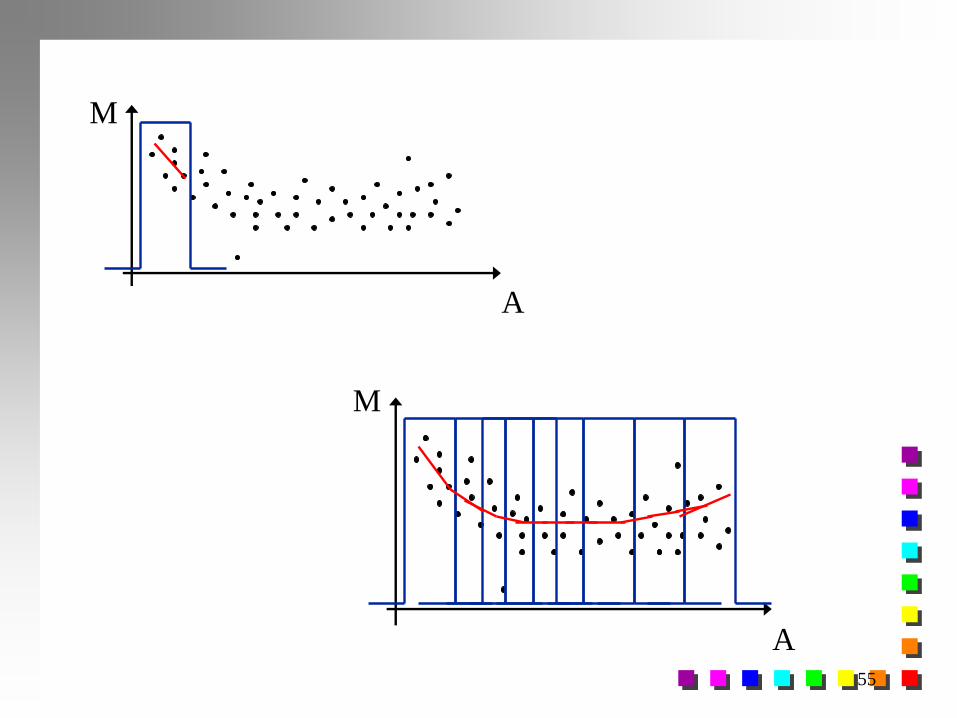
Normalização: Gráfico M x A
Não Normalizado Normalizado
M
A
M*
A
Eliminar tendência
Obter uma regressão robusta para MxA obter M* : valores residuais
jjj MMM ˆ* j-ésimo Spot j=1, 2, ..., n
Normalização por Lowess (locally weighted scatterplot smoothing)
Normalização por Splines
54
Normalização Lowess: Gráfico M x A Não Normalizado Normalizado
M* = log2 T* : valores normalizados/residuais de M
M M*
A A
jT
j
j
j
j
j
TT
G
RTM
ˆ22
*
2
*
2logˆloglog :ˆ
jT Ajuste lowess para Tj
55
A
M
A
M

56
A
M
Tamanho da janela (f= 0,20 - 0,40 )
Consultar Mestrado de Adèle Ribeiro, IME/USP/2014: Estimativas
melhoradas da expressão diferencial (valor M)

57
Normalização Splines Smoothing
Normalização dependente da Intensidade de Expressão mais
adaptativo, escolha ótima dos parâmetros de suavização
j
j
j
j AsG
RT
2
*
2 loglog
jj TT de )(suavizado ajustadovalor :ˆ
j-ésimo Spot j=1, 2, ..., 26912
jA para splines ajuste do estimado:jAs
resíduo
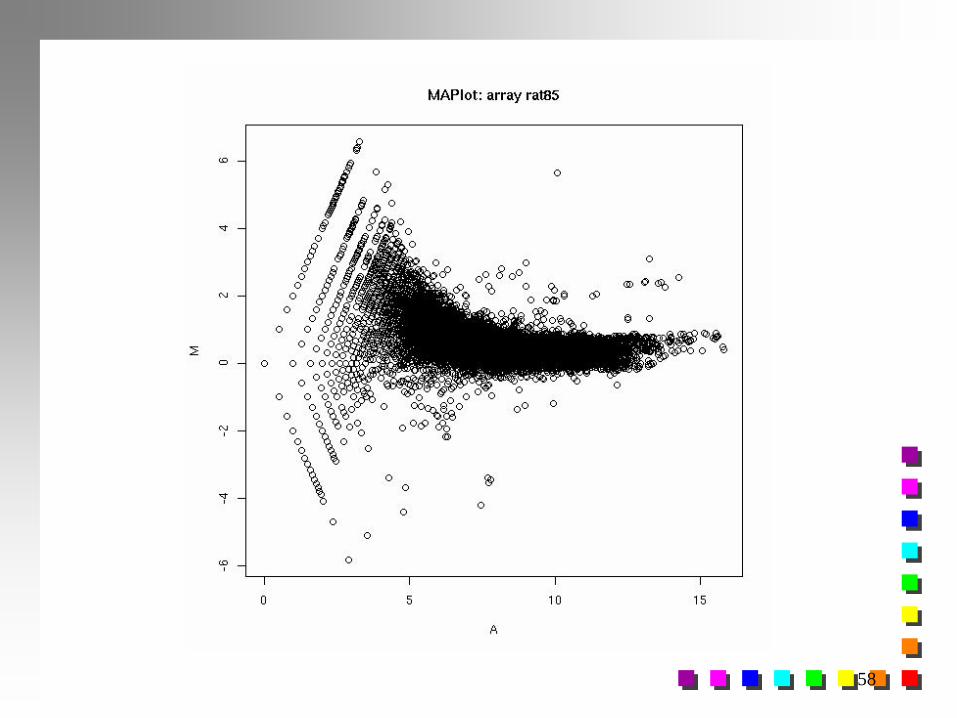
58

59
Normalização: Gráfico M x A
Não Normalizado Normalizado (Lowess por Subarray)
Note como a variabilidade é maior para spots com baixos níveis de expressão
A significância estatística de uma diferença de expressão depende da escala
Lâmina
Rat85

60
Dados Normalizados
Global por Subarray Lowess por Subarray Splines por Subarray
Global Lowess Splines Lâmina
Rat85

61
Dados Normalizados
Lâminas:
Rat85, Rat86,
Rat88, Rat89 2 réplicas (dye
swap):
Média: Rat85
e Rat89
Média: Rat86
e Rat88
Controle:
Rat85, Rat89
Sal:
Rat88, Rat89

62
Seleção de Genes Candidatos
Normalizações para Inversão de Corantes (“dye swap”)
como combinar as lâminas?
22
2121 AAA
MMM
Gráfico M x A:
ControleNaCl
|M|>1 A>10

63
Microarrays de Oligonucleotídeos
Sequência mRNA 3’ ____________________________ 5’
Probes DNA ___
______
______
______ ___
___
Construção das Lâminas:
Oligonucleotídeos com 25 pares de bases (pequenos)
Há um conjunto de probes para representar cada sequência de interesse
cerca de 16 – 20 pares (PM,MM)
PM: seq. de interesse MM: seq. Controle negativo
PM (perfect match): A-C-T-G-T-T-T-A-C-G-C-T -C- A-G-T-G-C-C-T-C-T-A-A-T
MM (mismatch): A-C-T-G-T-T-T-A-C-G-C-T -A- A-G-T-G-C-C-T-C-T-A-A-T
troca de base no meio da
sequência
Plataformas de UM ÚNICO canal de Fluorescência Affymetrix

64
Microarrays de Oligonucleotídeos
Sequência mRNA 3’ ____________________________ 5’
Oligos são gerados por fotolitografia diretamente na lâmina
Cada lâmina é hibridizada com UM ÚNICO canal de fluorescência
Oligonucleotídeos com 25 pares de bases (pequenos)
Conjunto de probes : cerca de 16 – 20 pares (PM,MM)
hibridização cruzada: muitos genes podem hibridizar o mesmo spot
a expressão de cada gene é medida usando a informação do conjunto
de probes que o representam
Microarray com 5K - 20K conjuntos de probes
PM (perfect match): A-C-T-G-T-T-T-A-C-G-C-T -C- A-G-T-G-C-C-T-C-T-A-A-T
MM (mismatch): A-C-T-G-T-T-T-A-C-G-C-T -A- A-G-T-G-C-C-T-C-T-A-A-T

65
Microarrays de Oligonucleotídeos
Outras plataformas de UM ÚNICO canal Codelink
Os spots contêm “grandes” fragmentos cromossômicos
Há réplicas dentro da lâmina (em geral, 2)
Estrutura da lâmina semelhante àquela de cDNA, exceto o # de canais
PM
MM
Plataforma Affymetrics
K
MMPM
Y
K
k
jkjk
j
1
K entre 16 e 20: número de representantes de
cada fragmento sob estudo

66
Experimentos com Microarrays
Objetivo: Identificar “genes” diferencialmente expressos sob
diferentes condições experimentais
SHRCSHR/BNC
SHRNaCl SHR/BNNaCl
Experimento com Animais Congênicos (cDNA)
Experimento Veia/Artéria (Plataforma Codelink)
Paciente Venoso (Controle) Arterial (Experimental)
1 A11 A12
2 A21 A22
3 A31 A32
4 A41 A42
Um único canal
8 lâminas
Amostras pareadas
Normalização ENTRE
Dois canais: “blocos”
4 lâminas
Amostras independentes
Normalização DENTRO da lâmina

67
Normalização Quantílica - Entre Lâminas
Generalização do Q-Q Plot de ² para n (de 2 para n lâminas)
Considere os dados de expressão de p fragmentos e n arrays
dispostos na matriz Xpxn
Ordene os dados de cada coluna: XSORT
Calcule as médias de cada linha e substitua pelos valores das
linhas: XSORT-MEAN
Retorne as linhas para as posições originais: X*pxn matriz de dados
normalizados
Array1 Array2
-
-
-
-
-
-
-
-
Array2
Array1Min Max
Min
Max
2
1
*
212
1;,,
j
kjkkkk
kqualtil
kkk qqqqqqqq
Pareamento entre lâminas
pelos quantis: Q-Q Plot
Gene
-
-
-
-
Xpx2

68
Normalização Quantílica (Entre)
Tratamento Venoso
Tratamento Arterial
Dados Normalizados
Dados Normalizados
Tentativa 1: Normalização quantílica das 4 lâminas de cada tratamento (NÃO leva em
conta a estrutura de pareamento dos pacientes): há muita variabilidade entre lâminas
?

69
Dados Veia/Arteria
Identificação de Genes Diferencialmente expressos
(Análise Exploratória)
Gráfico MxA: média das 4 lâminas
Normalização quantílica
entre lâminas (sem
considerar o pareamento
entre os indivíduos).
Não há indicação de
nenhum gene com
expressão diferencial entre
as amostras de mRNA

70
Normalização (Entre)
Não Normalizado Normalizado (Spline Global)
Opção 2: Gráficos MxA dos Pareamentos entre Tratamentos (Veia/Arteria)

71
Dados Veia/Artéria
A normalização das
respostas de
intensidade ENTRE
lâminas do mesmo
indivíduo
(pareamento) resultou
em um ganho em
precisão, tanto que foi
possível identificar
genes
diferencialmente
expressos.
Lista dos 20 genes mais diferencialmente expressos

72
Outras Normalizações
Normalização Paramétrica
Seguida de procedimentos de inferência sobre os genes
diferencialmente expressos

73
Normalização Paramétrica
(Wolfinger et al., 2000; Kerr e Churchill, 2001)
Cy5 Rj
Cy3 Gj j
j
jG
RT cTLogTLogTLog jjj 222 *
jAc
Já vimos diferentes normalizações não paramétricas dos dados:
:;ˆˆijjijiji YYYe Log2 intensidade de expressão gênica observada
no spot j sujeito ao canal de coloração i
É um valor residual
Uma alternativa à normalização não paramétrica é obter o valor
residual de Y, resultante do ajuste de modelos paramétricos que
incluem fontes de variação indesejáveis mas conhecidas.

74
Modelos Paramétricos de Normalização e
Seleção de Genes
Fontes de Variação nos Experimentos de Microarrays:
1. D: canal de fluorescência (Dye)
2. A: lâmina (Array)
3. S: spot (considerado como unidade amostral)
4. G: gene
5. T: tratamento
6. etc.
(Wolfinger et al., 2000; Kerr e Churchill, 2001)
ijklmlkjiijklm eGTDAy ...

75
Modelos ANOVA: Normalização e
Seleção de Genes Diferencialmente
Expressos
yijklm log2 da medida de intensidade corrigida pelo background
Ai : efeito do array Dj : efeito do Dye ADij : efeito de interação
Gil : efeito do Gene TGkl : efeito de interação (o de maior interesse)
AGil : efeito do Spot DGjl : efeito de interação do Dye e Gene
e ijklm ~ N (0, ²e ) efeito residual (variância constante ?)
Normalização e seleção de genes em um único ajuste ?
ijklmjlilkllijjiijklm eDGAGTGGADDAy
(Kerr e Churchill, 2001)

76
Modelos ANOVA: Normalização e Seleção de
Genes Diferencialmente Expressos
ijklmjlilmkllijjiijklm eDGAGTGGADDAy
Quais são os fatores (fontes de variação) indesejáveis (que serão usados
para normalização) e os desejáveis (que identificam genes diferencialmente
expressos)?
Efeito de Interesse (interação TG), os demais termos fazem o papel
de normalização dos dados
A expressão do gene l é a mesma para cada nível de tratamento?
LlH Klll ,...,2,1...: 21 Modelo de efeitos fixos
Uso de todos os dados (spots e lâminas) em um único ajuste
Desvantagem: Modelo com muitos parâmetros (baixa precisão)
(Kerr e Churchill, 2001)

77
Modelos Paramétricos de Normalização e
Seleção de Genes (Wolfinger et al., 2000)
Proposta de um Modelo (Misto) em Dois Estágios:
1° Estágio: Modelo de Normalização
Correção das medidas de intensidade, eliminando-se o efeito de fatores
indesejáveis análise supervisionada na escolha do que deve ser
eliminado (diferentemente do procedimento não paramétrico)
Uso de todas as medidas de intensidade de expressão (de todos os
genes e de todas as lâminas) para o ajuste do modelo
2° Estágio: Modelo Genético:
Identificação dos genes com expressão diferencial significante: uso do
resíduo co modelo do 1° estágio como var resposta
O modelo paramétrico é ajustado para cada gene independentemente

78
Modelos Paramétricos de Normalização e
Seleção de Genes
1º Estágio: Ajuste do Modelo de Normalização
yijg log2 da medida de intensidade corrigida pelo background
Ti efeito do tratamento i Aj ~ N (0, ²A ) efeito do array j
(T*A)ij ~ N (0, ²TA ) efeito do tratamento i no array j
e ijg ~ N (0, ²e ) efeito residual; i=1,...,I; j=1,...,J; g=1,...,G
Expressão Gênica Normalizada
(Wolfinger et al., 2000)
ijgijjiijg eATATy )(
Efeitos Sistemáticos a serem eliminados
ijgijgijgijg yyer ˆˆ
Gg ,...,2,1
Modelo considera
todos os genes

79
Ajustes separados para cada gene (spot) família de valores p
Correção para múltiplos testes
rijg : expressão gênica normalizada
Sig efeito do spot g no array j (bloco) ijg ~ N (0, ²g ) efeito residual
Efeito de interesse
2º Estágio: Ajuste do Modelo Paramétrico Genético
Para cada gene (spot) de um experimento cDNA:
ijgigjggijg TSr
(Wolfinger et al., 2000)
g = fixo; g =1, 2, ..., G
Modelo ajustado para os
dados de cada gene

80
Modelo Completo (ajuste em duas fases)
Yi j g log2 da medida de intensidade de expressão corrigida pelo background
Ti efeito do i-ésimo tratamento
Aj efeito do j-ésimo array
g média da intensidade de expressão do g-ésimo gene
(T*A)ij efeito do i-ésimo tratamento no j-ésimo array
Tig efeito do g-ésimo gene no i-ésimo tratamento
Sjg efeito do g-ésimo gene no j-ésimo array
yijg = + Ti + Aj + (T*A)ij + g + Sjg + Tig + ijg
yijg - ( + Ti + Aj + (T*A)ij ) = g + Sjg + Tig + ijg
ijg

81
Problema: O número de réplicas é pequeno e a variância da resposta é
grande, logo mesmo valores “altos” da razão de expressão não serão
significantes. Além disso, são realizados milhares destes testes e a correção
no nível descritivo dificultará ainda mais a chance de rejeitar H muitos
falsos negativos
rijg : expressão gênica normalizada ijg ~ N (0, ²g ) efeito residual
Efeito de interesse
2º Estágio: Ajuste do Modelo Paramétrico Genético
ijgigjggijg TSr
(Wolfinger et al., 2000)
g = fixo; g =1, 2, ..., G
iH gig :0
dados de cada gene
Rejeita H0 : gene diferencialmente expresso

82
Genes Diferencialmente Expressos(Wolfinger et al., 2000; Churchill, 2002)
Ggs
MT
g
g
g ,...,1
Estatística “t” regularizada: informação adicional de subconjuntos de
genes
Problema de Múltiplos Testes: correções no nível de significância
empírico (permutações, Bonferroni, FDR)
Estatísticas “t” : Teste Global (combinando “genes” / muitas réplicas) e
Teste Específico (para cada Gene / poucas réplicas)
0
00
s
MT
2
1
0
00
r
srs
MT
g
g
g
Nível descritivo: calculado
via permutações

83
Genes Diferencialmente Expressos
Múltiplos Testes
Suponha que 10 testes de Hipóteses independentes são realizados:
A probabilidade de um particular teste ser declarado significante
(quando na verdade não é) é igual a (1-0,95)=0,05 (valor α fixado)
Mas a probabilidade de pelo menos um teste dentre “10” ser declarado
significante (quando na verdade não é) é 1 – 0.9510 = 0.401
Se 20.000 destes testes são realizados esta probabilidade aumenta ...
Rej H0 N Rej H0
H0 V Erro Tipo I Nenhum Erro
H0 F Nenhum Erro Erro Tipo II
S
Decisão

84
Genes Diferencialmente Expressos
Múltiplos Testes
= 0.05 em média 5% dos genes que verdadeiramente não se
expressam diferencialmente serão considerados significantes
ocorrência de falsos positivos por chance se temos cerca de
20.000 testes é esperado 20.000 x 0,05 = 1000 falsos positivos ...
Correção de Bonferroni: A = /2g
FDR: False Discovery Rate
Permutações ou Bootstrap (como re-amostrar ?)
Gráfico de dispersão: -log(p) x log2 (R/G)
gráfico vulcano

85
Genes Diferencialmente Expressos
Múltiplos Testes Estatísticos Controle de Falsos Positivos e
Falsos Negativos
Análise Descritiva (Gráfico Vulcão)
Gráfico de Dispersão: -log(p) x log2 (R/G)
IIIII : genes não
significantes
estatisticamente mas
significativos factualmente
I: genes estatisticamente
significantes mas não
significativos factualmente
I
II III
Genes
estatísticamente
significantes
(p<10-5)

86
Experimentos com MicroarraysIdentificar “genes” diferencialmente expressos sob diferentes
condições experimentais
Delineamento Fatorial 5x2 com Animais Congênicos
Sal
Replic \ Cong. A B C D E A B C D E
R1
R2
R3
R4
R5
+ -
9
10
8
5
1
g
g
g
g
g
Y
Y
Y
Y
Y
11
9
10
1
2
g
g
g
g
g
Y
Y
Y
Y
Y
8
7
11
4
3
g
g
g
g
g
Y
Y
Y
Y
Y
7
11
9
2
4
g
g
g
g
g
Y
Y
Y
Y
Y
10
8
7
3
5
g
g
g
g
g
Y
Y
Y
Y
Y
3
4
2
11
7
g
g
g
g
g
Y
Y
Y
Y
Y
5
3
4
7
8
g
g
g
g
g
Y
Y
Y
Y
Y
2
1
5
10
9
g
g
g
g
g
Y
Y
Y
Y
Y
1
5
3
8
10
g
g
g
g
g
Y
Y
Y
Y
Y
4
2
1
9
11
g
g
g
g
g
Y
Y
Y
Y
Y
Tabela de dados de expressão para o probe g (g=1, 2,..., 35.129)
Fator Linhagem: A=SHR, B=C2a, C=C2c, D=C4, E=C16
Fator Exposição ao Sal: Sal (+), Controle (-) Estrutura de Blocos

87
Experimentos com Microarrays
A+ B+ C+
D+ E+
A- B- C-
D- E-
B+ D+ E+
C+ A+
B- D- E-
C- A-
E- A- D-
B- C-
E+ A+ D+
B+ C+
C- E- B-
A- D-
C+ E+ B+
A+ D+
D- C- A-
E- B-
D+ C+ A+
E+ B+
R1 R2 R3
R4 R5
Estrutura de Blocos: Hibridizações simultâneas dos 10 tratamentos

88
Experimentos com MicroarraysDelineamento Fatorial 5x2 com Animais Congênicos
Intensidades de Expressão do “gene” xxxx
Corrigidas pelo Background
Sal
Replic \ Cong. A B C D E A B C D E
R1
R2
R3
R4
R5
+ -
Normalização ENTRE lâminas, Respeitar a estrutura de Blocos
Normalização Não Paramétrica ?? (normalização quantílica)

89
Experimentos com MicroarraysDelineamento Fatorial 5x2 com Animais Congênicos
Tabela ANOVA para cada “Gene” (g=1, 2, …, 35.129)
F.V. g.l. S.Q. Q.M. F p
Linhagem 4
Sal 1
L*S 4
Bloco 4
Resíduo 36
Total 49
Obter CLASSES de genes de acordo com o efeito dos fatores
na variação das respostas de expressão gênica:
Genes com expressão diferencial no efeito de interação: FL*S “significante”
Genes com expressão diferencial no efeito principal de Sal: FL*S “signif.”
Genes com expressão diferencial no efeito principal de Linhagem: FL*S “signif.”

90
Genes Diferencialmente Expressos(Wolfinger et al., 2000; Churchill, 2002)
GgQMR
QMMF
g
g
g ,...,1
Estatística “F” regularizada: informação adicional de subconjuntos
de genes
Problema de Múltiplos Testes: correções no nível de significância
empírico (permutações, Bonferroni, FDR)
Estatísticas “F” : Teste Global e Teste Específico (para cada Gene)
0
0
0QMR
QMMF
2
0QMRQMR
QMMF
g
g
Rg Nível descritivo: análise de
permutações

91
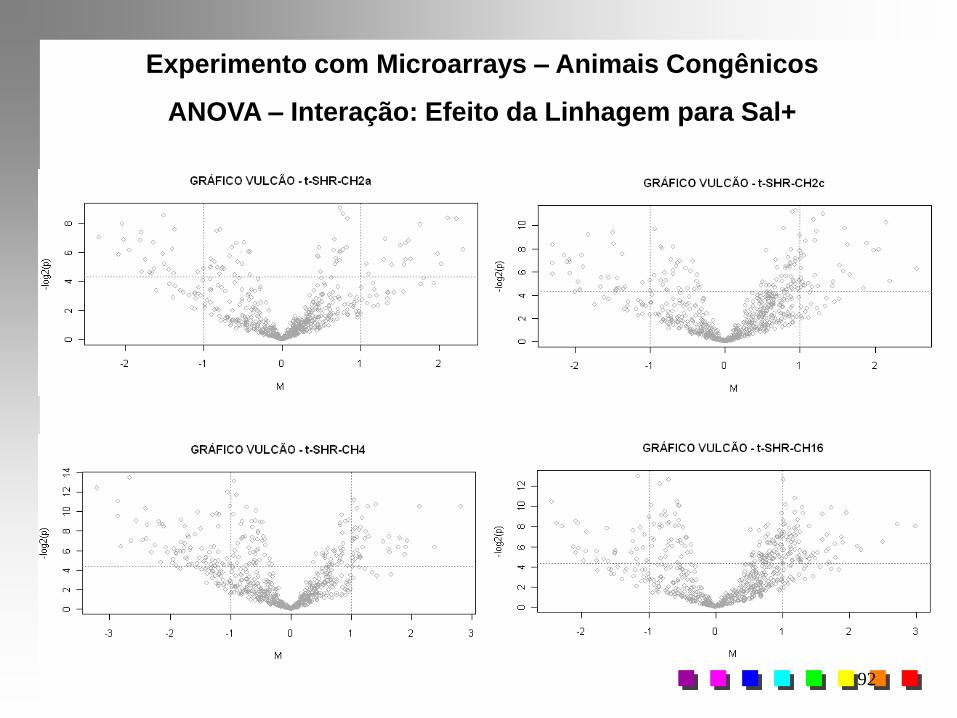
Experimentos com MicroarraysDelineamento Fatorial 5x2 com Animais Congênicos
Resultados da ANOVA – Efeito de Interação (L*S)
Contraste Estimativa E.P. "t" p
Controle
C2a-SHR
C2c-SHR
C4-SHR
C16-SHR
Sal
C2a-SHR
C2c-SHR
C4-SHR
C16-SHR
Linhagem
SHR
C2a
C2c
C4
C16
13 possíveis
contrastes de
interesse
92
Experimento com Microarrays – Animais Congênicos
ANOVA – Interação: Efeito da Linhagem para Sal+

93
Experimento com Microarrays – Animais Congênicos
ANOVA – Interação: Efeito da Linhagem para Sal-

94
Experimento com Microarrays – Animais Congênicos
ANOVA – Interação: Efeito do Sal em cada Grupo

95
Comparação entre Métodos de
Normalização
Normalização proposta por Wolfinger:
- PARAMÉTRICA
- Atribui pesos iguais às obs na obtenção dos resíduos (NORMAL)
- LINEAR (na escala log2)
- Os resíduos são correlacionados por construção e são modelados
como independentes resultados similares na prática (Wolfinger et
al., 2001)
Normalizações NÃO PARAMÉTRICAS:
- Parecem produzir resultados mais robustos e adaptativos
- Não há controle de quais efeitos estão sendo eliminados: análise
não supervisionada

Pré-Genômica/Genômica/Pós-Genômica
Mapeamento Genético Clássico
Análises de Ligação, QTL’s, L.D, QTN´s
Polimorfismo funcional (Diferenças no
DNA entre membros de uma População)
Genômica
Experimentos com Microarrays
Identificar genes diferencialmente
expressos (sob diferentes condições)
Genômica Genética
... a análise da expressão de genes encontra a Genética.
Análise de eQTL

97
qq Qq QQ
20
30
40
50
60
Modo Dominante de Herança
Média Y
qq Qq QQ
Média y
Intensidades de expressão são
altamente correlacionadas com fenótipos
clássicos (ex: estudos de sobrevivência)
A variação das respostas de expressão
(para um spot) entre indivíduos pode ser
explicada pelo efeito de Marcadores
Moleculares específicos
Validação dos Dados de MicroarraysShadt et al. (2003): Microarrays de Oligonucleotídeos
Ye
Y
Análises Combinadas: Genética Genômica (Análise de eQTL)
Análise da Expressão Relativa de Genes: Quantitativo RT-PCR

98
Genética Genômica... onde a análise da expressão de genes encontra a Genética
(Jansen and Nap, 2001; Darvasi, 2003)
(Shadt et al., 2003)
Tratar as medidas de intensidade de expressão gênica
como traços quantitativos em uma análise de QTL
30
eQTL
jj EY 2log novo traço
eQTL (ou eQTN) estão na mesma
localização cromossômica dos
genes que estão sendo expressos
a variação na expressão gênica
não é ruído aleatório

99
FAM INDIV PAI MÃE FENO1 FENO2 … FENOf SNP1 SNP2 … SNP2.882
REPL1 REPL2 REPL1 REPL2 … REPL1 REPL2
1 1
1 2
… …
1 14
2 15
… …
2 28
… …
14
14
14 ~200
FENOExp2 FENOExp3.554FENOExp1
Genética Genômica
Banco de Dados
Estrutura
das u.a.
Fenótipos Genótipos
Mapa de SNP’s
Fenótipos de
Expressão
Quantos Locos? Quantos
Transcripts?
Cruzamentos controlados
Famílias
Indivíduos Não relacionados

100
Genética Genômica – Interação Genética
eQTN1
chr5
eQTN2
chr8
bp
LOD
10
20
30 __ Probe 1
__ Probe 2
**
* **
**
**
**
*
*
** *
** * *
** * *
**
**
**
*
*
* *
*
**
EProbe1
EProbe2
BB
eQTL BS
SS
entender o padrão
de correlação*
*
*
Shadt et al. (2003)

101
Genética Genômica – Interação Genética
eQTL1
chr5
eQTL2
chr8
cM
LOD
10
20
30 __ Probe 1
__ Probe 2
**
* **
**
**
**
*
*
** *
** * *
** * *
**
**
**
*
*
* *
*
**
EProbe1
EProbe2 eQTL2
eQTL1 BB BS SS
BB * * *
BS * * *
SS * * *
*
*
*

102
Perspectivas
Para medir a Expressão Gênica pode-se ajustar um modeloestatístico linear para as respostas de intensidades(i) corrigidas pelo background, (ii) normalizadas e (iii) log-transformadas ...
O sistema de regulação celular, gerador das medidas de intensidade de
expressão gênica, parece ser tão complexo para ser completamente
descrito por modelos estatísticos clássicos!
A resposta de expressão gênica tem uma base herdável de variação
(mais uma fonte de variação??)
Por que log-transformar os dados sem qualquer análise exploratória
prévia?
Modelos Estocásticos Robustos
Modelos de Efeitos Aleatórios (generalização sobre o conjunto de
probes amostrados)

103
Perspectivas
Métodos de normalização/calibração:
- Caso não paramétrico utilizar métodos cada vez mais adaptativos
- Caso paramétrico adotar distribuições mais gerais
Identificação de Genes diferencialmente expressos (modelos ANOVA):
- Modelo Genético Misto
- Incorporar heterocedasticidade (entre genes)
- Múltiplos testes: controle da taxa de falso positivo
Validação da análise de Microarrays
- Intensidades de expressão são altamente correlacionadas com
fenótipos clássicos
- Análises Combinadas: Genética Genômica (Análise de eQTN)

104
Recursos Computacionais
MAANOVA (R)
MANMADA (SAS)
http://statgen.ncsu.edu/ggibson/Manual.htm
BIOCONDUCTOR (R)
www.bioconductor.org

105
Consolidando Conhecimento
Como planejar cada experimento ?
Como a Teoria de Ligação entre Locos é utilizada no mapeamento genético ?
Como estão definidos os fenômenos de Ligação e Desequilíbrio de Ligação ?
Como a informação do QTL é predita da informação de Marcadores ?
Como decidir sobre modelos de efeitos fixos ou aleatórios do gene ? Cite exemplos.
Qual a diferença entre modelos poligênicos e oligogênicos ?
O que são efeitos de pleiotropia e epistasia ?
Como está definida a matriz de covariância entre unidades amostrais nos
experimentos com cruzamentos controlados e nos familiares ?
Que diferenças podem ser estabelecidas considerando mapeamento genético
clássico e experimentos de expressão gênica ?
Por que realizar normalizações dentro e entre lâminas nos experimentos de
expressão gênica ?
Como construir as lâminas de cDNA ? Como o número limitado de réplicas biológicas
pode ser compensado considerando o grande número de genes avaliados ?
…

106
Bibliografia
• Churchill, G.A. (2002). Fundamentals of experimental design for cDNA
mocroarrays. Nature Genetics Suppl 32: 490-495.
• Durbin, B and Rocke, DM. (2002). Exact and approximate variance-stabilizing
transformations for two-color microarrays. Bioinformatics, 18: S105-S110.
• Kerr, M.K.; Churchill, G.A. (2001). Statistical Design and the Analysis of Gene
Expression Micro Array Data. Genet. Res. 77: 123-128.
• Speed, T (Ed) (2003). Statistical of gene expression microarray data. Chapman
& Hall.
• Storey, JD and Tibshirani, R. (2003). Statistical significance for genomewide
studies. PNAS 100(16): 9440-9445.
• Yang, HY; Dudoit, S; Luu, P and Speed, TP. (2001) Normalization for cDNA
Microarray Data. Em www.citeseer.nj.nec.com/406329.
• Wolfinger, R.D.; Gibson, G.; Wolfinger, E.D.; Bennett, L.; Hamadeh, H.; Bushel,
P.; Afshari, C.; Paules, R.S. (2001). Assessing Gene Significance from cDNA
Microarray Expression Data via Mixed Models. J. Compu. Biol. 8(6): 625-637.

107
Seleção de Genes Diferencialmente
Expressos
n
i
ggiBg MMmn
s1
22
1
1
Caso de 2 Tratamentos com réplicas dentro da Lâmina (Gordon et al.,2005):
Para o g-ésimo gene (g=1,…,G):
Lâmina Spot A B
1 1
1 2
2 1
2 2
3 1
3 2
4 1
4 2
ts
Mt
gM
g
g ~
n
i
m
j
gigijW g MMmn
s1
22
)1(
1
Variância ENTRE lâminas
Variância DENTRO da lâminan=4 m=2
gggg m
nmNM 11
1;~ 2
ˆ1
)1(
ˆ)1(1
)1(
1
122
2 W gBg
M
smn
m
sn
mns
g
?2
gMs





![SERVIÇO DE PSICOLOGIA HOSPITALAR AVALIAÇÃO PSICOLÓGICA ( SEBASTIANI, R.W. & FONGARO, M.L - in ANGERAMI, V. A. [org] -"E a Psicologia Entrou no Hospital",](https://img.document.onl/doc/110x75/570638561a28abb8238fb568/servico-de-psicologia-hospitalar-avaliacao-psicologica-sebastiani-rw.jpg)























