Embed Size (px)
Citation preview

I
UNIVERSIDADE FEDERAL DE GOIÁS
INSTITUTO DE CIÊNCIAS BIOLÓGICAS
PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS BIOLÓGICAS
O PAPEL DO POLIMORFISMO METABÓLICO DE GSTM1 E GSTT1
NA SUSCEPTIBILIDADE A NEFROPATIA DIABÉTICA
RAYANE MENDES DE LIMA
GOIÂNIA-GO
2016

II
RAYANE MENDES DE LIMA
O PAPEL DO POLIMORFISMO METABÓLICO DE GSTM1 E GSTT1
NA SUSCEPTIBILIDADE A NEFROPATIA DIABÉTICA
Dissertação apresentada ao Programa de Pós-
Graduação em Ciências Biológicas do Instituto de
Ciências Biológicas da Universidade Federal de
Goiás, como requisito parcial para obtenção do
título de Mestre em Ciências Biológicas.
Área de Concentração: Biologia Celular e Molecular Orientador: Prof. Dra. Angela Adamski da Silva
Reis
Co-orientadora: Prof. Dra. Aline Helena da Silva
Cruz
GOIÂNIA-GO
2016

III
RAYANE MENDES DE LIMA
O PAPEL DO POLIMORFISMO METABÓLICO DE GSTM1 E GSTT1
NA SUSCEPTIBILIDADE A NEFROPATIA DIABÉTICA
BANCA EXAMINADORA
_____________________________________________
Prof. Dra. Angela Adamski da Silva Reis
Universidade Federal de Goiás
_____________________________________________
Prof. Dr. Gustavo Rodrigues Pedrino
Universidade Federal de Goiás
Prof. Dra. Aline Helena da Silva Cruz
Faculdade Alfredo Nasser
Prof. Dr. Rodrigo da Silva Santos
Faculdade Alfredo Nasser
Aprovada em: 02/03/2016

IV
Dedico esse trabalho a Deus e a todos aqueles que me apoiaram durante essa longa e árdua jornada.

V
Agradecimentos
Gostaria de começar agradecendo a Deus, por me permitir chegar aonde
cheguei e por durante essa jornada ter colocado pessoas tão especiais em meu
caminho.
Meus pais também tiveram um papel de suma importância nesse caminho,
sempre me apoiaram mesmo naqueles momentos difíceis.
A minha orientadora, a professora Drª. Angela Adamski da Silva Reis, por
toda paciência, apoio, toda dedicação e por ter me mostrado o lado bom da
pesquisa e da docência, mesmo em meio a tantos obstáculos e dificuldades que
encontramos ao longo do caminho, ela nunca desistiu. Foi um honra ser sua
primeira aluna oficial de mestrado.
A minha co-orientadora Dra. Aline Helena da Silva Cruz e ao Dr. Rodrigo da
Silva Santos por compartilharem tantas historias e alegrias durante os experimentos
e por sua contribuição à minha formação como pesquisadora.
Aos meus colegas e amigos de laboratório, em especial a colombiana Ayda e
ao Thales, muito obrigada por tudo. Também aqueles que tiveram uma rápida
passagem, mas que de alguma forma contribuíram durante esse período, seja com
uma ideia ou apenas uma conversa no final de um experimento. Obrigada por me
apoiarem.
Aos funcionários e pacientes da Nefroclínica que me acolheram e permitiram
que eu participasse dessa família, da rotina e conhecer um pouquinho das suas
histórias.
Não poderia deixar de agradecer também as agências de fomento, CAPES e
FAPEG, permitindo que essa pesquisa fosse feita, financiando os materias do
laboratório e também permitiram que eu pudesse me dedicar integralmente a esse
projeto.
Aos meus amigos e namorado por aguentar minhas crises de ansiedade, que
eu sei que não foram poucas.
Aqui não caberiam todos os agradecimentos. De um modo geral, muito
obrigada a todos.

VI
“O importante é nunca parar de questionar. A curiosidade tem sua própria
razão para existir. Uma pessoa não pode deixar de se sentir reverente ao
contemplar os mistérios da eternidade, da vida, da maravilhosa estrutura da
realidade. Basta que a pessoa tente apenas compreender um pouco mais
desses mistérios a cada dia. Nunca perca uma sagrada curiosidade”.
Albert Einsten.

VII
SUMÁRIO
LISTA DE ABREVIATURAS E SIGLAS
LISTA DE ANEXOS
LISTA DE FIGURAS
LISTA DE TABELAS
RESUMO
ABSTRACT
1. INTRODUÇÃO
1.1. Diabetes Mellitus
1.2. Nefropatia diabética
1.3. Estresse oxidativo e a nefropatia diabética
1.4. Sistema Glutationa S-transferase
1.5. Polimorfismo de Deleção de GSTT1 e GSTM1 na nefropatia diabética
2. JUSTIFICATIVA
3. OBJETIVOS
3.1. Objetivo Geral
3.2. Objetivos Específicos
4. METODOLOGIA
5.1 Pacientes e Controles
5.2 Obtenção das Amostras
5.3 Extração e Quantificação de DNA
5.4 Análise do polimorfismo de deleção de GSTT1 e GSTM1
5.5 Análise Estatística
5. RESULTADOS
5.1. Distribuição genotípica e associação do polimorfismo de GSTT1 e
GSTM1 com DM2
5.2. Influência do polimorfismo de deleção de GSTT1 e GSTM1 sobre
variáveis clínicas e laboratoriais.
6. DISCUSSÃO
7. CONCLUSÃO
8. REFERÊNCIAS
ANEXOS
16
37
41
22
48
45
43
42
41
41
40
41
40
40
39
34
28
22
50
56
57
78
XIV
XIII
XII
XI
X
15
VIII
44

VIII
LISTA DE ABREVIATURAS E SIGLAS
ADA Associação Americana de Diabetes (American Diabetes Association)
AGEs Produtos Finais de Glicação Avançada
BRA Bloqueador de Receptor de Angiotensina
CAT Catalase
CDKAL1 Gene que codifica quinase dependente de ciclina 5 subunidade reguladora
associada a proteína 1
DCVs Doenças Cardiovasculares
DM Diabetes mellitus
DM 1 Diabetes mellitus tipo 1
DM 2 Diabetes mellitus tipo 2
DMG Diabetes mellitus gestacional
dNTP Desoxinucleotídio Trifosfato
DRC Doença Renal Crônica
EFDR Estágio Final de Doença Renal
EUA Excreção de Albumina Urinária
EROs Espécies Reativas do Oxigênio
FABP2 Proteína de Ligação de Ácidos Graxos 2
GLUT4 Transportador de Glicose Tipo 4
GSH Glutationa
GST Glutationa S-transferase
GSTM1 Isoforma M1 da família Glutationa S- transferase
GSTT1 Isoforma T1 da família Glutationa S-transferase
HAS Hipertensão Arterial Sistêmica
HbA1c Hemoglobina Glicada
HLA Antígeno Humano Leucocitário
IC95% Intervalo de Confiança 95%
IDF Federação Internacional de Diabetes (International Diabetes Federation)
IMC Índice de Massa Corpórea
JAK-STAT Janus Quinase e ativador de transcrição
IkB Inibidor de NFkB
MAO Monoamina Oxidase
MAPK Miogênico Ativador de Proteína quinase

IX
MHC Complexo de Histocompatibilidade principal
NDP Nicotinamida adenina difosfato
NADH Nicotinamida adenina difosfato reduzida
ND Nefropatia Diabética
NFkB Fator nuclear kB
OMS Organização Mundial da Saúde
OR Odds ratio
P Probabilidade
Pb Pares de base
PKC Proteína Quinase C
qPCR PCR em Tempo Real
RER Retículo Endoplasmático Rugoso
R.P.M. Rotações Por Minuto
SBD Sociedade Brasileira de Diabetes
SBEM Sociedade Brasileira de Endocrinologia e Metabologia
SLC30A8 Gene codificador da proteína transportadora de zinco (gene encodes zinc
transporter protein member 8)
SOD Superóxido Desmutase
SNPs Polimorfismo de Nucleotídeo Único (Single-Nucleotide Polymorphisms)
TCF7L2 Fator de Transcrição 7-semelhante a 2 (Transcription factor 7-like 2)
TGF β Fator transformador de crescimento β
TFG Taxa de Filtração Glomerular
χ2 Qui- quadrado
α-KGDH Alfa cetoglutarato desidrogenase

X
LISTA DE ANEXOS
ANEXO I – Termo de Consentimento Livre e Esclarecido
ANEXO II – Parecer do Comitê de Ética Institucional
ANEXO III – Formulário de Preenchimento de Dados de Pacientes
ANEXO IV – Formulário de Preenchimento de Dados de Controles
ANEXO V – Carta de submissão do artigo científico em revista qualis A1
87
83
81
78
91

XI
LISTA DE FIGURAS
Figura 1. Esquema demonstrando o mecanismo da ação da insulina nos
diferentes tipos de diabetes.
Figura 2. Descrições de moléculas inflamatórias e vias de sinalização na
nefropatia diabética.
Figura 3. Representação esquemática do mecanismo que unifica as vias de
dano celular induzida pela hiperglicemia.
Figura 4. Esquema representativo da via dos polióis.
Figura 5. Desenho esquemático ilustrando a deleção de GSTM1.
Figura 6. Desenho esquemático ilustrando a deleção de GSTT1.
Figura 7. Curva de melting da qPCR multiplex (SYBR Green).
43
36
36
32
31
28
18

XII
LISTA DE TABELAS
Tabela 1. Estágios clínicos da nefropatia diabética
Tabela 2. GSTs citosólicas humanas
Tabela 3. Sequências de primers utilizados nas reações de amplificação por
qPCR multiplex
Tabela 4. Condições de termociclagem das reações de amplificação por
qPCR multiplex
Tabela 5. Características da população estudada e comparação entre os
grupos caso e controle
Tabela 6. Distribuição de frequências genotípicas para GSTM1 e GSTT1 na
população estudada e analise de risco para nefropatia diabética
Tabela 7. Distribuição das frequências genotipicas combinadas entre GSTM1
e GSTT1 nos grupos caso e controle e o risco para o desenvolvimento de
nefropatia diabética em pacientes em EFDR.
Tabela 8. Distribuição das frequências genotípicas de GSTT1 e GSTM1 e das
combinações genotípicas entre GSTT1 e GSTM1 nos grupos caso
e controle
Tabela 9. Associação entre genótipos nulos e presentes de GSTT1 com
variáveis clínicas em pacientes com DM2-EFRD.
Tabela 10. Associação entre genótipos nulos e presentes de GSTM1 com
variáveis clínicas em pacientes com DM2-EFRD.
26
35
42
42
44
46
47
47
48
49

XIII
RESUMO
A nefropatia diabética é a principal causa de Estágio Final de Doença Renal (ESRD)
em países desenvolvidos e evidências tem apontado o estresse oxidativo como
unificador de várias vias de dano celulares em condições de hiperglicemia, que
resultaria no desenvolvimento e complicações da doença. As glutationa-S-
transferases (GSTs) são uma família de enzimas multifuncionais que desempenham
um papel importante na desintoxicação celular e eliminação de numerosas
substâncias e também pode funcionar como um dos antioxidantes. O polimorfismo
genético de deleção nos genes GSTT1 e GSTM1, quando em homozigose,
apresentam ausência de atividade dessas isoformas, conhecido como genótipo nulo.
Procurando estabelecer uma possível relação entre nefropatia diabética e os
polimorfismos acima mencionados, foi feito um estudo por caso-controle e a
genotipagem por meio de PCR em tempo real (qPCR) e curva de melting. Dados
clínico-laboratoriais de 65 pacientes (diagnosticados com nefropatia diabética e que
estavam em hemodiálise) e 90 controles foram coletados, por meio de entrevistas e
consulta a prontuários (pacientes) e a resultados de exames recentes (controles).
Foi verificado que no grupo caso existe um risco associado ao polimorfismo de
deleção, onde o genótipo GSTT1-nulo (p=0,0230) provoca um risco aumentado de
aproximadamente 2,9 vezes em desenvolver a doença (nefropatia diabética) em
relação aos portadores do genótipo GSTT1-presente. Não houve associação de
GSTM1 (p=0,3860) com a susceptibilidade a doença na população estudada. A
análise da influência da deleção de GSTT1 e GSTM1 sobre as alterações
bioquímicas e clínicas no grupo caso não resultou em uma associação significativa
de nenhuma das variáveis clínicas analisadas. Estes resultados sugerem que o
polimorfismo de deleção de GSTT1 pode estar associado ao risco de
desenvolvimento da doença, mas não com as alterações bioquímicas que foram
analisadas. Novos estudos poderão esclarecer a relação desse polimorfismo com a
nefropatia diabética e auxiliar no tratamento desta doença.
Palavras-Chave: Diabete mellitus; Nefropatia diabética; Polimorfismos; GSTT1;
GSTM1.

XIV
ABSTRACT
Diabetic nephropathy is the leading cause of end-stage renal disease (ESRD) in
developed countries and in the literature shows as oxidative stress possibly
contributes to the development of the diseases. Glutathione S-transferases (GSTs)
are a family of multifunctional enzymes that play an important role in the cellular
detoxification and excretion of numerous substances and can also work as one of the
antioxidants. The genetic polymorphism of deletion in GSTT1 and GSTM1 gene,
when homozygous, show lack of activity of these isoforms, known as null genotype.
Looking for a possible relationship between diabetic nephropathy and polymorphisms
mentioned above, this study was made for the case-control and genotyping using
real-time PCR (qPCR) and melting curve. Clinical and laboratorial data of 65 patients
(diagnosed with diabetic nephropathy and were on hemodialysis) and 90 controls
were collected through interviews and consultation with medical records (patients)
and the results of recent surveys (controls). It was found that in the group if there is a
risk associated with deletion polymorphism, where the GSTT1-null genotype (p =
0,0230) causes an increased risk of about 2,9 times in developing the disease
(diabetic nephropathy) compared to carriers of the genotype GSTT1-present. There
was no association of GSTM1 (p = 0.3860) with susceptibility to disease in this
population. Analysis of the influence of the deletion of GSTT1 and GSTM1 about the
biochemical and clinical changes in the group case did not result in a significant
association in any of the clinical variables analyzed. These results suggest that the
GSTM1 deletion polymorphism may be associated with risk of developing the
disease, but not with the biochemical changes that were analyzed. Further studies
may clarify the relationship of this polymorphism with diabetic nephropathy and help
in the treatment of this disease.
Keywords: Diabetes mellitus; Diabetic nephropathy; Polymorphism; GSTT1;
GSTM1.

15
1. INTRODUÇÃO
O diabetes mellitus (DM) é considerado uma das principais síndromes de
evolução crônica que acometem o homem moderno em qualquer idade, condição
social e localização geográfica (revisado por Oliveira, 2004). E é definido como um
conjunto de doenças metabólicas caracterizadas por hiperglicemia decorrente de
problemas no mecanismo de produção e/ou ação da insulina em tecidos periféricos
(ADA, 2012).
Essas alterações metabólicas, com destaque para dislipidemia e hipertensão
arterial sistêmica podem levar ao desenvolvimento de complicações micro e
macrovasculares, com acometimento de olhos, rins, nervos, coração e artérias
(ADA, 2013), destacando a complicação denominada de nefropatia diabética (ND),
uma das principais complicações microvasculares decorrentes da DM, sendo uma
importante causa de Insuficiência Renal Crônica (IRD).
A patogênese e a evolução da ND estão relacionadas a vários fatores, como
por exemplo, as alterações hemodinâmicas. A presença de fatores genéticos
predisponentes associados com essas alterações, bem como a glicemia não
controlada, corpos cetônicos, hormônio de crescimento, ingestão protéica, sistema
renina-angiotensina entre outros, acarretariam no desenvolvimento de lesões
glomerulares (Moreira et al., 2008).
Estudos têm tentado fazer uma triagem de genes candidatos para melhor
identificar a susceptibilidade genética (Huo et al., 2015; Ma et al., 2015; Dabhi et al.,
2015; Zhou et al., 2015; Rhee, 2015; Houldsworth et al., 2015). Por ser uma doença
multifatorial, vários genes estão envolvidos na gênese da doença, entre eles,
destacam-se o polimorfismo de inserção / deleção do gene da Enzima conversora
de angiotensina (ECA) e o gene das enzimas que auxiliam na eliminação do
estresse oxidativo, como por exemplo, as Glutationas S-tranferase (GSTs), que tem
demonstrado resultados controversos em diferentes populações.
O estresse oxidativo tem sido estudado na susceptibilidade no
desenvolvimento e progressão da ND por ser um unificador de várias vias de dano
celulares em condições de hiperglicemia, entre elas o aumento da ativação das vias
de polióis, aumento da formação dos Produtos Avançados de Glicação (AGEs),
aumento da ativação da proteína quinase C (PKC) entre outros (Brownlee, 2001).

16
Existem vários mecanismos que tentam promover a defesa celular contra o
estresse oxidativo, entre eles podemos destacar as enzimas superóxido desmutase
(SOD), catalase (CAT) e o sistema glutationa, que tem como componente as GSTs
(Ukkola et al., 2001; Meister, 1994).
Estudos realizados por Doney et al. (2005) demonstraram um aumento do
risco de morbidade e mortalidade cardiovascular, sendo essa caracterizada como
uma progressão da ND em pacientes com DM, com os polimorfismos de deleção no
gene que codifica a isoforma Glutationa S-transferase-Theta (GSTT1) na população
escocesa, enquanto Kim et al em 2005 encontrou associação entre o genótipo
GSTM1-nulo na população coreana.
A necessidade desse estudo consiste em que há poucos dados da população
brasileira, mais especificamente na população goianiense, relacionando esses
polimorfismos com o desenvolvimento da ND. Na literatura observamos dados
conflitantes em diferentes populações da participação desses polimorfismos e o
desenvolvimento dessa patologia. A fisiopatologia e os fatores genéticos que
envolvem essa doença, ainda não são completamente esclarecidos, por se tratar de
uma progressão que depende de vários fatores, destacando assim a necessidade de
estudos nessa área para uma melhor compreensão, permitindo assim um
prognóstico definido a esses pacientes.
1.1. Diabetes Mellitus
O DM é conceituado pelo Ministério da Saúde como um grupo de doenças
metabólicas que resultam da dificuldade de excreção ou atividade da insulina,
envolvendo mecanismos nocivos específicos, como a destruição das células beta
pancreáticas, resistência à ação da insulina e distúrbios na sua secreção (Brasil,
2006).
O DM configura-se atualmente como uma epidemia mundial, despertando o
interesse de muitos profissionais de saúde e da população em geral. É considerada
uma patologia crônica, traduzindo-se em um grande desafio para os sistemas de
saúde de todo o mundo. Esse número crescente de indivíduos diabéticos deve-se ao
crescimento e envelhecimento populacional, maior urbanização, crescente
prevalência de obesidade e sedentarismo, e também da maior sobrevida de
pacientes com DM (Diretrizes da Sociedade Brasileira de Diabetes, 2015). Além

17
disso, têm sido observado uma crescente proporção de pessoas afetadas em grupos
etários mais jovens (International Diabetes Federation, 2013).
Cerca de 8,3% da população mundial têm DM, sendo que 46% das pessoas
no mundo não sabem que a possui (International Diabetes Federation, 2013). De
acordo com a 6ª edição do Atlas de Diabetes da International Diabetes Federation
(IDF) de 2014, no Brasil os casos de diabetes em indivíduos na faixa etária de 20 a
79 anos ultrapassam 11 milhões de pessoas e ainda que aproximadamente 3,2
milhões de pessoas nessa mesma faixa etária apresentam a doença, mas não foram
diagnosticadas. Os mesmos autores descrevem que quase 120 mil pessoas morrem
por complicações decorrentes da doença, sendo que 41,7% dessas mortes
relacionadas à doença são em pacientes abaixo de 60 anos.
Existe uma dificuldade para realizar essa estimativa, porque na declaração de
óbito não é mencionado a DM como a causa da morte, e sim as suas complicações,
particularmente as cardiovasculares e cerebrovasculares. De acordo com o
Ministério da Saúde a cidade brasileira onde foi identificado o menor percentual da
população com diabetes é Palmas com 2,7% da população, seguida por Goiânia (4,1%)
e Manaus (4,2%) que estão na segunda e terceira posição, respectivamente, com os
menores índices (Brasil, 2012).
Atualmente, essa patologia é classificada conforme a etiologia. A classificação
da Organização Mundial da Saúde (OMS) e da Associação Americana de Diabetes
(ADA) inclui, basicamente, quatro classes clínicas: diabetes mellitus tipo 1 (DM 1),
diabetes mellitus tipo 2 (DM 2), diabetes mellitus gestacional (DMG) e outros tipos
específicos de diabetes. Ainda existem duas categorias referidas como pré-diabetes,
que são a glicemia de jejum alterada e a tolerância diminuída à glicose, que não são
entidades clínicas, mas consideradas como fatores de risco para o desenvolvimento
de DM e doenças cardiovasculares (DCVs) (SBD, 2014).
A figura 1 demostra o mecanismo de ação do hormônio insulina nos
diferentes tipos de diabetes.

18
Figura 1. Esquema demonstrando o mecanismo da ação da insulina nos diferentes tipos de
diabetes. Na situação normal, o hormônio insulina é produzido normalmente pelas células beta pancreáticas e reconhecido pelos seus receptores nas células. A diabetes do tipo 1 ocorre quando o corpo não consegue produzir insulina suficiente ou não consegue utilizar a insulina de forma eficaz. Em contra partida, na diabetes do tipo 2 e na diabetes gestacional, a secreção da insulina é normal, mas existe uma deficiência nos receptores celulares, que levará ao aumento dos níveis plasmáticos de glicose. Sigla: GLUT4 – Transportador de Glicose Tipo 4.
A insulina humana é um hormônio peptídico produzido pelas células β
pancreáticas, e é constituída por duas cadeias de resíduos de aminoácidos. A sua
síntese tem início no retículo endoplasmático rugoso (RER), na forma de pré-insulina
(Greco & Stabenfeldt, 2007; Martin & Crump, 2003), que após perder o peptídeo
sinal durante a passagem pela membrana do retículo origina a proinsulina (Martin &
Crump, 2003), molécula esta constituída pelas cadeias A e B da insulina ligadas pelo
peptídeo C (Andrade & Marco, 2006; Azevedo, 2006). A conversão em insulina
ocorre por remoção proteolítica do peptídeo C, ficando ambos armazenados no
interior de grânulos de secreção das células β pancreáticas (Andrade & Marco,
2006).

19
O processo de secreção tem como estímulo mais importante a concentração
da glicose no interstício, sendo que o aumento da glicemia (níveis de glicose no
sangue) causa a elevação da secreção de insulina, que irá agir em diferentes
tecidos do organismo permitindo a entrada de glicose, resultando em uma
diminuição da glicemia (Machado et al., 2012).
A pessoa sem DM tem uma produção normal desse hormônio pelas células
beta pancreáticas. Em pacientes com DM 1, não ocorre uma produção normal ou o
corpo não consegue utilizar a insulina, levando o aumento de glicose na corrente
sanguínea ocasionando assim, danos em outros tecidos. Já na DM 2, existe uma
produção normal ou em alguns casos um defeito na secreção de insulina, mas as
células não conseguem fazer o transporte da glicose para o seu interior, por um
defeito na ação desse hormônio o que, assim como na DM 1, vai levar a um
aumento dos níveis plasmáticos de glicose.
A DM 1 está presente em 5% a 10% dos casos (ADA, 2012). Na maioria das
vezes, a destruição das células beta-pancreáticas ocorre por um mecanismo de
autoimunidade, porém existem situações em que não há evidências deste processo
sendo, portanto, caracterizado como forma idiopática do DM 1 (ADA, 2012). As
razões para o crescente número de pessoas que desenvolvem a doença continuam
desconhecidas, mas acredita-se que envolvam fatores de risco ambientais, causas
genéticas, alimentação ou infecções virais.
A DM 1 tem seu desenvolvimento repentino, tendo como alguns sintomas:
sede anormal e boca seca, micção frequente, falta de energia e cansaço extremo,
aumento do apetite, perda repentina de peso, visão borrada, entre outros. Pessoas
diagnosticadas com essa patologia podem levar uma vida normal e saudável através
da combinação de uma terapia diária de insulina, dieta saudável, prática de exercício
físico regular e disciplina do paciente.
A DM 2 apresenta prevalência de 90% a 95% dos casos de DM, geralmente
atingindo adultos, mas tem-se observado um crescente número de crianças e
adolescentes diagnosticados (ADA, 2012). As razões para o desenvolvimento da
doença continuam desconhecidas, tendo vários fatores de risco, que incluem:
obesidade, dieta, sedentarismo, idade avançada, histórico na família entre outros.
O indivíduo portador desse tipo de diabetes desenvolve resistência à ação da
insulina, isso ocorre quando as células não usam a insulina para transportar a
glicose para o meio intracelular. O pancrêas em um primeiro momento aumenta a

20
produção desse hôrmonio, com o tempo, a quantidade produzida não é suficiente,
aumentando os níveis plasmáticos de glicose (NIH, 2013). Muitas pessoas com DM
2 permanecem desconhecedoras da doença, pois os sintomas podem demorar a
aparecer ou serem identificados, sendo somente diagnosticados quando
complicações decorrentes da doença já foram desencadeadas.
A American Diabetes Association (ADA) também menciona que grande parte
dos pacientes com essa forma de DM apresenta sobrepeso (Índice de Massa
Corpórea em kg/m2, IMC: 25 a 29,9) ou obesidade (IMC: 30 a 34,9), sendo estes
fatores contribuintes para a diminuição da sensibilidade à insulina nos tecidos
periféricos.
Na DMG a mulher desenvolve resistência à insulina em consequência dos
altos níveis de glicose durante a gravidez. Tende a ocorrer por volta da 24ª semana
de gravidez, tendo como consequência o bloqueio da ação da insulina, sendo
induzido provavelmente pelos hormônios produzidos pela placenta (ADA, 2013).
O risco para o bebê não é tão grave como para aqueles cuja mãe tenha DM 1
ou DM 2 antes da gravidez. No entanto, o diabetes gestacional não controlado
podem desenvolver consequências graves para a mãe e o seu neonato como:
macrossomia (excesso de peso de recém-nascidos), trauma ao nascimento,
icterícia, cesariana prematura e pré-eclâmpsia (aumento repentino da pressão
sanguínea) (National Institute of Health Consensus Development Conference
Statement, 2013; Crowther et al., 2005). Após o parto, 15-50% das mulheres, podem
desenvolver DM2 (Practice Bulletin, 2013), além do que as crianças e adolescentes
com macrossomia tem um risco aumentado em desenvolver diabetes e obesidade
(Wei et al., 2007; Hillier et al., 2007).
Atualmente são três os critérios definidos pela American Diabetes Association
(ADA, 2013) para o diagnostico de DM com utilização da glicemia: sintomas de
poliúria (aumento na produção de urina), polidipsia (sede excessiva) e perda de
peso acrescidos de glicemia casual > 200 mg/dl a; glicemia de jejum ≥ 126 mg/dl (7
mmol/l)b e o teste de glicemia após duas horas pós-sobrecarga de 75 g de glicose
anidra > 200 mg/dl ou quando a hemoglobina glicada (HbA1c) é ≥ 6,5% para
a: Compreende-se por glicemia casual aquela realizada a qualquer hora do dia, independentemente do horário das refeições; b: Em caso de pequenas elevações da glicemia, o diagnostico deve ser confirmado pela repetição do teste em outro dia;

21
indivíduos com alto risco para o desenvolvimento de diabetes adota-se HbA1c entre
5,7% e 6,4%.
Muitas das complicações do diabetes podem ser de origem microvascular
(devido a danos aos pequenos vasos sanguíneos) e macrovasculares (devido a
danos em vasos de grande calibre) (Melendez-Ramirez et al., 2010). O principal
determinante dessas complicações é a hiperglicemia sustentada, que acarreta
anormalidades bioquímicas e estruturais de olhos, rins, coração, vasos sanguíneos e
nervos periféricos (Aguiar et al., 2007).
As complicações microvasculares em longo prazo incluem retinopatia com perda
potencial de visão, sendo esta a principal causa de cegueira adquirida na população;
nefropatia diabética (principal causa de insuficiência renal crônica e hemodiálise em
nosso país); neuropatia periférica com risco de úlceras nos pés e amputações; e
neuropatia autonômica causando sintomas gastrointestinais, geniturinários,
cardiovasculares, e disfunções sexuais. Esses pacientes também apresentam uma
incidência de aterosclerose aumentada e anormalidades do metabolismo de
lipoproteínas (ADA, 2014).
As principais complicações macrovasculares incluem: doença arterial
periférica (representa a principal causa de amputação não traumática na população
adulta, decorrente de um mau controle glicêmico e o hábito do tabagismo) e doença
arterial coronariana (pode manifestar-se por episódios de angina e por infarto agudo
do miocárdio constituindo-se a principal causa de morte nos pacientes diabéticos)
(SBEM, 2015).
Embora ainda as causas de desenvolvimento de DM 2 não são conhecidos,
há vários fatores de risco importantes, que podem ser divididos em dois grupos:
fatores de risco modificáveis, que são aqueles possíveis de controlar, e os fatores de
risco não modificáveis, ou seja, aqueles que não se pode controlar (SBD, 2001).
Nos fatores de risco modificáveis podemos destacar a hipertensão arterial,
obesidade, privação de sono e o sedentarismo (Cercato et al., 2004; Brasil, 2006).
Entre os não modificáveis estão as doenças do pâncreas ou doenças endócrinas,
que levará a problemas na produção ou na eficácia da insulina, e a predisposição
genética (Marks & Raskin, 2000; James et al., 2007; Schwarz et al., 2008; Sanz et
al., 2010; Sinclair et al., 2011;).
Já se sabe que a DM 1 tem herança mendeliana a partir de genes do
Complexo Humano de Histocompatibilidade (MHC), incluindo HLA e outros genes

22
(Maahs et al., 2010). No entanto, o DM 2 ainda precisa de identificação dos
componentes genéticos. A identificação de genes susceptíveis para DM 2 tem sido
investigada, em geral por meio de estudos populacionais caso-controle,
comparando-se a frequência de determinado alelo entre as populações doente e
sadia.
Neste sentido, recentes avanços no entendimento da base genética e na
suceptibilidade da DM 2 revelaram a participação de novos genes e de
Polimorfismos de Nucleotídeo Único (SNPs) associados a esse risco (Farbstein et
al., 2010; Sladek et al., 2007; Taylor et al., 2007) como o gene TCF7L2, que é o
responsável por codificar o fator de transcrição 4, que está envolvido no
desenvolvimento e função das células beta pancreáticas, afetando o metabolismo de
glicose (Prunier et al., 2004; Lyssenko et al., 2007; Liu et al., 2015 ); e o gene
CDKAL1 onde a perda funcional desse gene resultaria em defeitos na tradução das
proteínas, causando uma síntese anormal de insulina que provocaria um estresse no
retículo endoplasmático das células beta pancreáticas (Tuerxunyiming et al., 2015) e
o grupo de Peng et al (2013) em sua meta analise concluíram que o polimorfismo
rs7754840 relacionado a esse gene aumenta o risco de DM2.
Também podemos citar o gene SLC30A8 que possiu a função de codificar um
fator transportador de zinco, apresentando um importante papel na síntese e
secreção de insulina (Chimienti et al., 2005), podendo estar associado ao
desenvolvimento de DM por afetar a atividade das células beta pancreáticas
(Chimienti et al., 2006), sendo o polimorfismo rs13266634 associado ao risco de
DM2 nas populações asiáticas, européias e africanas (Fan et al., 2016).
Vários polimorfismos de genes responsáveis pela alteração hemodinâmica,
endotelial, pressão sanguínea intraglomerular e exposição ao estresse oxidativo
estão associados às complicações microvasculares da DM 2 que levam a disfunção,
dano ou falência de vários órgãos, incluindo nosso objeto de estudo, a ND.
1.2. Nefropatia diabética
A DM foi listada como a principal causa de insuficiência renal mundial, em
44% de todos os novos casos em 2011, sendo que aproximadamente 230 mil
pessoas de todas as idades com falência renal decorrentes da diabetes,
necessitaram de diálise crônica ou de transplante renal, nos Estados Unidos, e 7 de

23
cada 10 novos casos de EFDR tiveram como principal causa a DM (National
Diabetes Statistics Report, 2014). De acordo com a Sociedade Brasileira de
Diabetes (SBD), essa complicação é considerada progressiva e irreversível,
atingindo até 80% dos diabéticos (Diretrizes da Sociedade Brasileira de Diabetes,
2009). Em relação ao diagnóstico da doença renal primária, os mais frequentes, em
2013, foram hipertensão arterial (35%) e diabetes (30%) (Sesso et al., 2014).
Em seus estudos, Murussi et al. (2008), destaca a necessidade de se
identificar as lesões renais de maneira precoce, o que permite um prognóstico mais
favorável, do que naqueles casos em que o diagnóstico acontece quando o paciente
já se encontra em fases mais avançadas. Ele ainda propõe a realização do exame
de excreção urinária de albumina (EUA) anualmente, o que iria ajudar na detecção e
em um melhor prognóstico do estágio no qual aquele paciente se encontra.
A ADA em 2012 lançou suas recomendações para a triagem da ND, entre
elas podemos destacar o aperfeiçoamento do controle glicêmico e da pressão
arterial, o que permitiria reduzir o risco ou retardar a progressão da doença; a
realização do teste anual para avaliar a EUA, e a medida da creatinina séricab pelo
menos anualmente em todos os adultos com DM, independente do grau de EUA.
Em relação ao tratamento eles propõem uma redução do consumo de
proteínas em pacientes com DM, principalmente aqueles que apresentam DRC; e a
continuação do monitoramento da EUA para avaliar a resposta terapêutica e
progressão da doença, além da utilização de inibidores de ECAc (IECA) ou
Bloqueadores dos Receptores da Angiotensinac (BRAs) em pacientes não graves
com micro ou macroalbuminúria (ADA, 2012).
Esses medicamentos são usados por evidências demonstrarem um
retardamento na progressão da ND, por ajudar no controle da pressão sanguínea e
diminuir a quantidade de proteína excretada pela urina, através da inibição da ação
da angiotensina II nos receptores AT1, resultando na diminuição da pressão de
filtração intraglomerular (Kunz et al., 2008; Montero et al., 2015). Apesar de
possuírem mecanismo de ação diferentes, os IECAS diminuindo a produção de
angiotensina II e os BRAs impedindo a ligação desse peptídeo aos receptores AT1,
b Deve ser utilizado para estimar a taxa de filtração glomerular (TFG) e fase o nível de doença renal crônica (DRC), se estiver presente; c Importante que se monitore níveis séricos de creatinina e potássio para que não desenvolva um aumento da creatinina e hipercalemia;

24
o que em condições normais resulta em vasoconstrição e consequente aumento da
pressão sanguínea (Hilgers & Mann, 2002; Brenner et al., 2001).
Em estudos in vitro e em animais observou-se uma redução da expansão
mesangial, glomeruloesclerose, inflamação e fibrose tubulointersticial, por meio do
bloqueia do sistema Renina-angiotensina-aldosterona (Yamagishi & Matsui, 2010;
Doi et al., 2008; Liao et al., 2003;).
Um estudo mostrou que o aumento da albumina excretada na urina em
pacientes diabéticos, aumenta em 50 vezes os riscos de óbito comparado com
aqueles pacientes que não desenvolveram nefropatia, afirmando ainda a
participação de fatores genéticos e não genéticos, destacando a glicemia alterada,
hipertensão arterial sistêmica (HAS) e as dislipidemias (Alves et al., 2011).
De acordo com Mogensen et al., (1983) a ND pode ser caracterizada por
cinco estágios clínicos. Estes estágios são classificados com base nos valores da
taxa de filtração glomerular (TFG), EUA, e a pressão sanguínea sistêmica. As lesões
estruturais discretas no parênquima renal e na vasculatura geralmente tornam-se
mais grave com o avanço dos estágios clínicos, mas o diagnóstico geralmente é
feito por motivos clínicos, sem a necessidade de biopsia renal.
A fase I em pacientes com DM 1 é caracterizado por nefromegalia e
hipertrofia glomerular, que são seguidos por vasodilatação da arteríola aferente,
hiperperfusão renal e hiperfiltração glomerular. Microscopicamente, há
espessamento das membranas basais glomerulares e tubulares. A taxa de excreção
de albumina nesta fase é normal (30 mg / dia ou, 20 µg / min), mas, ocasionalmente,
pode existir um aumento transitório ("microalbuminúria transitória"). Tipicamente não
existe aumento da pressão arterial, porém ele pode ocorrer (140/90 mm Hg) e a TFG
é aumentada em 20 a 40%, podendo ser ainda mais elevada em casos em que não
existe o controle glicêmico (Rigalleau, et al., 2010). Em pacientes com DM 2 pode se
observar nefromegalia e hiperfiltração (Nelson et al., 1997).
Na fase II ou nefropatia incipiente ou latente é definida pelo aparecimento de
microalbuminúria (UAE de 30 a 300 mg / dia ou 20 a 200 g / min), sendo quase
sempre acompanhada por um aumento da pressão arterial (140 / 90 mm Hg), tanto
em pacientes com DM 1 como com DM 2 (Mogensen et al., 1995). Uma das
possíveis causas para o aumento da EUA é a hipertensão renovascular. Mesmo
durante esta fase clinicamente "silenciosa" da doença, pode existir uma significativa
expansão da matriz mesangial ou glomeruloesclerose difusa, espessamento das

25
membranas basais glomerulares e tubulares e algum grau de perda de podócitos,
sendo que a TFG permanece elevada ou pode diminuir para valores "normais" (100
120 ml / min) (revisado em Sheldon et al., 2013).
A fase III é caracterizada pelo desenvolvimento de proteinúria evidente
(excreção total de proteína de 0,500 mg / dia) ou microalbuminúria (EUA 300 mg / d)
(Mogensen et al., 1995). No tipo 1 isso ocorre após uma média de 15 anos de
diabetes. A hipertensão é quase sempre presente, e um controle pobre pode levar a
um declínio de TFG. Observa-se na biópsia renal glomeruloesclerose difusa e/ou
nodular, perda de podócitos com áreas focais, hialinose arteriolar, nas aferentes e
eferentes, e graus variados de fibrose túbulo-intersticial. A ampliação progressiva do
mesângio glomerular provoca uma redução na área de superfície de filtração
glomerular (Mauer et al., 1984). Existe uma perda nefrótica devido à fibrose túbulo-
intersticial sendo esta uma das principais causas da redução da TFG. No doente não
tratado, a TFG cai a uma taxa de cerca de 1 ml / min / mês, mas esta taxa de queda
pode variar significativamente de doente para doente (Sheldon et al., 2013). À
medida que a TFG cai, a creatinina no soro pode permanecer normal ou ser
ligeiramente elevada (Sheldon et al., 2013).
Na fase IV, os pacientes não tratados evoluem para um quadro de nefropatia
avançada, caracterizado por proteinúria nefrótica (3,5 g / d), uma piora da
hipertensão que se torna difícil de controlar, e um declínio progressivo da TFG. Na
verdade, a nefropatia diabética é a causa mais comum da síndrome nefrótica na
população adulta. Lesões vasculares e do parênquima se tornam mais graves. A
taxa de declínio da TFG é constante ao longo de um período de meses, mas é
variável de paciente para paciente e depende do grau de elevação da pressão
sanguínea, bem como a quantidade de EUA (Sheldon et al., 2013).
A fase V é a fase final, onde ocorre a evolução para um quadro de
insuficiência renal progressiva, com diminuição da TFG para 15 ml / min ou inferior.
O tempo médio de progressão para o estágio final de doença renal (EFDR) a partir
do momento do diagnóstico de diabetes é de cerca de 20 a 30 anos, com um curso
mais rápido em pacientes com hipertensão não controlada e / ou proteinúria grave.
Muitos pacientes, especialmente aqueles com DM2, ao atingirem o quadro de IRC
têm o risco aumentado de mortalidade por problemas cardiovascular (Sheldon et al.,
2013). A Tabela 1, sumariza os 5 estágios da ND.

26
Tabela 1. Estágios clínicos da nefropatia diabética
TFG, Taxa de filtração glomerular; EUA, Excreção urinaria de albumina; EFDR, estágio final de doença renal.
Os custos para a saúde associados a essa patologia são enormes, por isso
na maioria dos casos o prognóstico pode ser desfavorável, apesar dos avanços
médicos no tratamento de doenças renais e cardiovasculares. Por exemplo, o
diabético com proteinúria tem de duas a quatro vezes maiores risco de morbidade e
mortalidade por doenças cardiovasculares (Hemmelgarn et al.,2010). Mesmo com
diálise crônica, a taxa de morte cardíaca de pacientes diabéticos é
aproximadamente 50% maior do que os pacientes não diabéticos. Já foi observado
que o risco de desenvolver essa complicação varia com a etnia (Remuzzi et al.,
2002).
Indivíduos afro-americanos, nativos americanos, asiáticos e hispânicos são
mais propensos ao desenvolvimento de DM 2 e ND do que os brancos não
hispânicos, indicando uma possível associação de fatores genéticos que podem
influenciar diretamente o desenvolvimento e a progressão da doença. Um suporte
para a transmissão genética é um estudo experimental mostrando que a medula
vermelha derivada de células mesangiais de doadores foram capaz de transmitir ND
dos doadores com DM 2 (camundongos db/db) para camundongos naive
normoglicêmicos (Zheng et al., 2004).
Entre os mecanismos de lesão renal relacionado à hiperglicemia crônica,
estão a glicação não enzimática e as alterações na via dos polióis. Os produtos de
glicação não enzimática podem causar alterações quantitativas e qualitativas nos
componentes da matriz extracelular, contribuindo para a ocorrência final de oclusão
glomerular (Murussi et al., 2003). A hiperglicemia promove também um aumento da
atividade dos polióis. Nessa via, a glicose é reduzida a sorbitol sob ação da aldose

27
redutase. O acúmulo do sorbitol ocasionaria estresse hiperosmótico para as células,
diminuição do mioinositol intracelular e da atividade da ATPase Na+/K+ dependente,
ocasionando dano celular (Murussi et al., 2003).
Novas vias para o desenvolvimento da ND envolvem processos inflamatórios
devido à hiperglicemia, sistema renina-angiotensina e stress oxidativo, envolvendo
uma infiltração renal com monócitos e linfócitos que aumentam a produção de
citocina pró-inflamatória, espécies reativas de oxigênio e danos nos tecidos (Duran-
Salgado & Rubio-Guerra, 2014; Kitada et al., 2014), resultando em uma maior
resposta inflamatória levando a lesão celular e fibrose do tecido do órgão (Figura 2).

28
Figura 2. Descrições de moléculas inflamatórias e vias de sinalização na nefropatia
diabética. Alterações promovidas pela diabetes agem nas células renais e ativam diversas cascatas de sinalizações intracelulares. A via da cinase lkB leva a ativação da NFkB. A via da MAPK controla a expressão do fator de crescimento endotelial vascular em células renais, sendo essencial na regulação de diversos processos celulares, como a inflamação, transcrição, ativação ou localização de proteínas individuais, e diferenciação celular. A sinalização promovida pela JAK-STAT medeia os efeitos dos fatores de crescimento, citocinas e angiotensina II, que resulta na expressão e ativação de TGF-β, produção de colágeno e fibronectina, e estimulação do crescimento de células mesangiais glomerulares e em células tubulointersticiais. A ativação destas vias resulta em infiltração de células inflamatórias, que aumenta e mantem o processo inflamatório nos rins, resultando no desenvolvimento e progressão de nefropatia diabética. Além disso, tem-se o aumento da proteinúria. Abreviaturas: IkB, inibidor de NFkB; JAK-STAT, Janus quinase e ativador de transcrição; MAPK, mitógeno ativadora de proteína quinase; NFkB, fator nuclear kB; TGF β, fator transformador de crescimento β.
1.3. Estresse oxidativo e a nefropatia diabética
Em células eucarióticas as espécies reativas de oxigênio (EROs) são
produzidos como uma consequência do metabolismo normal (Cossarizza et al.,
2009). Em condições fisiológicas normais, estes prooxidantes são contrabalançados
por mecanismos celulares antioxidantes. O estresse oxidativo é definido em geral
como o excesso de formação e / ou remoção insuficiente de moléculas altamente
reativas, tais como os EROs (Maritim et al., 2003), ou seja, irá existir um
desequilíbrio entre os pro-oxidantes e antioxidantes podendo resultar em lesões.

29
As EROs contribuem para a patogênese de várias doenças crônicas, incluindo DM
(Baynes, 1991).
A defesa antioxidante pode atuar basicamente em três níveis: 1) prevenção,
que consiste em mecanismos de proteção contra a formação de espécies reativas,
como a produção de pigmentos, a exemplo da melanina, e a quelação de metais por
proteínas, como a ferritina; 2) interceptação, envolvendo mecanismos enzimáticos
(peroxidases, catalases) e não enzimáticos (vitaminas) de desativação por interação
direta com os agentes potencialmente danosos; e 3) reparo, através da restituição
ou substituição do componente lesado, o que envolve a ação de sistemas
multienzimáticos especializados (Sies, 1993). Portanto, antioxidante pode ser
definido como qualquer substância que, quando presente em baixa concentração
comparada à do substrato oxidável, regenera o substrato ou previne
significativamente a oxidação do mesmo (Halliwell, 2000).
É bem documentado que o estresse oxidativo está aumentado em condições
diabéticas (Odetti et al., 1999; Davi et al., 1999; Telci et al., 2000; Ceriello et al.,
2002; Evans et al., 2002; Rahman, 2007) e provavelmente está envolvido na
disfunção das células β pancreáticas em particular, uma vez que estas células são
mais sensíveis ao esforço citotóxico em função de expressarem baixos níveis de
enzimas antioxidantes, como catalase e superóxido dismutase (Eizirik et al., 1996;
Tiedge et al., 1997; Robertson et al., 2003).
Desta forma, as células β apresentam maior risco de dano oxidativo do que
outros tecidos, tendo sido observado que o estresse oxidativo contribui na
diminuição da produção de insulina e destruição das células β (Eizirik et al., 1996;
Tiedge et al., 1997; Robertson et al., 2003). Isso sugere que o estresse oxidativo
aumentado possa desempenhar um papel relevante na patogênese do DM2 e
também no desenvolvimento das complicações clínicas relacionadas à doença
(Evans et al., 2002; Wang et al., 2006; Bid et al., 2010, Reis et al., 2011).
A geração de EROs tem recebido muita atenção no que diz respeito ao início
das complicações diabéticas microvasculares, incluindo a doença renal (Koya et al.,
2003). Como defendido por Brownlee (2001), o aumento da geração de EROs
envolve o aumento oxidativo mitocondrial, bem como diminuição da sua eliminação.
Existe certo número de fontes celulares que geram quantidades consideráveis de
EROs, sendo a mitocôndria a responsável pela geração intracelular dessas espécies

30
reativas, possuindo pelo menos 10 locais diferentes capazes de gerar EROs
(Cossarizza et al., 2009; Turrens, 2003).
Além da importante contribuição da cadeia transportadora de elétrons, uma
enzima do espaço intermembranar (p66Shc), a enzima de membrana monoamina
oxidase (MAO) (Migliaccio et al., 2006), a α-cetoglutarato desidrogenase (α-KGDH) e
o complexo enzimático do ciclo de Krebs, são capazes de alterar o pH da matriz
mitocondrial e o potencial de membrana (Sinha et al, 2013), podendo também
conferir efeitos consideráveis sobre a produção total de EROs celular (Pal et al.,
2013; Bhattacharyya et al., 2012).
A glicólise que ocorre intracelularmente resulta na produção de NADH e
piruvato. O NADH e o FADH2 mitocondrial fornecem energia para a produção de
ATP através da fosforilação oxidativa por meio da cadeia transportadora de elétrons.
Em resposta a elevação dos níveis extracelulares de glicose, as células endoteliais
geram EROs intracelular principalmente via NADH e FADH2 mitocondrial que são
gerados a partir do piruvato citosólico (Du et al., 2000). Os estudos em células
mesangiais humanas têm mostrado uma série de mudanças similares relacionadas
à geração de EROs mitocondrial (Kiritoshi et al., 2003).
O estresse oxidativo é considerado o elemento unificador das vias de dano
celulares, causados pela hiperglicemia, sendo as principais vias de danos: aumento
da ativação da via do poliol, aumento da formação dos produtos finais de glicação
avançada (AGEs), aumento da ativação da proteína quinase C e da via da
hexoamina (Figura 3).

31
Figura 3. Representação esquemática do mecanismo que unifica as vias de dano celular
induzida pela hiperglicemia. O excesso de radical superóxido (O2-) inibe parcialmente o
GAPDH, que resulta no aumento dos metabólitos formados antes da ação do GAPDH. Esses metabólitos são responsáveis pelo dano celular que existe em condições de hiperglicemia, aumentando a geração de O2
-. Siglas: NADPH- nicotinamida adenina difosfato reduzida, NADP - nicotinamida adenina difosfato, GAPDH - gliceraldeído-3-fosfato-desidrogenase, PKC – proteína quinase C, AGE – produtos finais de glicação avançada, Udp-GlcNAC – UDP-N-acetilglucosamina.
O aumento da ativação da via do polióis envolvendo o metabolismo da glicose
em experiências in vitro com células mesangiais, foi possível verificar que a atividade
da aldose-redutase (AR) aumenta (Henry et al., 1999). Enquanto que nas células
tubulares proximais esta atividade permanece inalterada, embora na presença de
NADPH, a aldose redutase catalise a redução de glicose para formar sorbitol na
fração citosólica (Lee & Chung, 1999).
Como podemos ver na Figura 4, o sorbitol sofre então um metabolismo
subsequente para formar frutose pela ação da enzima sorbitol desidrogenase,
usando NAD+ como cofator. Um elevado nível de sorbitol afeta a atividade de Na+ /
K+ - ATPase, que por sua vez resulta em disfunção do túbulo renal (disfunção
tubular proximal) (Dunlop, 2000; Manna et al., 2009;. Das & Sil, 2012). Acumulação
de sorbitol também resulta em uma redução de Na + / K + - ATPase nos glomérulos e
afeta a taxa de filtração glomerular, por causar um estresse osmótico, devido a baixa
difusibilidade do sorbitol (Brownlee, 2001). A Aldose redutase altera a atividade da

32
proteína kinase C (PKC) e da MAPK e aumenta a produção de fator de necrose
tumoral α (TNF-α).
Figura 4. Esquema representativo da via dos polióis, A hiperglicemia, com consequente
aumento de EROS leva a um aumento na ativação da aldose redutase. O aumento do fluxo pela via dos polióis, induzido pelo aumento de EROs, determina maior conversão de glicose a sorbitol, reduzindo NADPH e glutationa (antioxidante intracelular), aumentando assim o estresse oxidativo induzido pela hiperglicemia (Adaptado de Brownlee, 2001). Sigla: SDH- Sorbitol desidrogenase.
A ativação da PKC, induzido pelo aumento de síntese de novo diacilglicerol,
causa a diminuição da produção de óxido nítrico (NO), alteração na expressão de
fatores de crescimento, como o TGF-β1, que no quadro de hiperglicemia induz
aumento da matriz extracelular em células mesangiais e glomérulos (Bertoluci et al.,
1996; Murphy et al., 1999); ativação do fator de transcrição NFΚβ (aumenta a
transcrição de várias proteínas, incluindo o fator de crescimento do endotélio
vascular e moléculas de adesão como a ICAM-1) (Goldin et al., 2006; Yan et al.,
1994), e da enzima NADPH oxidase, sendo essa última a responsável por catalisar
a formação do radical superóxido (Brownlee, 2005; El-Benna et al., 2005).
Na via envolvendo o aumento da formação dos AGEs, sabe-se que a
membrana basal glomerular, as células mesangiais, os podócitos e as células
tubulares renais acumulam altos níveis de AGEs, sendo descritos três mecanismo
que podem danificar as células: modificações de proteínas intracelulares,
principalmente as envolvidas na regulação da transcrição gênica (Brownle, 2005); a
possibilidade de difusão para o meio extracelular dos precursores de AGE e que

33
levam a uma modificação das moléculas da matriz extracelular próxima (McLellan et
al., 1994), resultando assim, em uma alteração da sinalização entre a matriz e a
célula levando a uma disfunção celular; e o mecanismo que implica que os
precursores de AGE podem difundir para fora da célula e modificar proteínas
circulantes no sangue, tais como a albumina (Brownle, 2005).
Já na superativação da via da hexoamina, a glicose é convertida em frutose-
6-fosfato que será transformada em N-acetilglicosamina-6-fosfato. A sua conversão
em UDP-N-acetilglicosamina irá resultar em alterações patológicas na expressão
gênica, aumentando produção de citocinas inflamatórias e fatores de transcrição (Du
et al., 2000).
O estresse oxidativo também tem sido claramente observado em vários
modelos animais de doença renal diabética. Observou-se que animais tratados com
estreptozotocina, uma nitroamida utilizada como indutora de diabetes em animais,
que nos rins desses animais diabéticos, a produção de EROs ocorre através de um
número de diferentes processos, tais como a auto oxidação de glicose, AGEs,
atividade da xantina oxidase, deficiência na cadeia respiratória mitocondrial,
NADPH-oxidase e óxido nítrico sintase (Forbes et al., 2008).
A inibição de EROs com ácido α lipoíco (Melhem et al., 2002) ou através de
super expressão da superóxido desmutase citosólica (DeRubertis et al., 2004)
previne a doença renal decorrentes da diabetes em modelos de roedores. Tem sido
observado que a geração de EROs mitocondrial ocorre nas células endoteliais e
esse mecanismo provavelmente desempenha um importante papel na disfunção
endotelial generalizada na diabetes, incluindo o desenvolvimento da albuminúria.
Estudos identificaram uma nova forma de NADPH-oxidase no rim, a NOX-4, (Geiszt
et al., 2000), que sofre um aumento na regulação em modelos animais de doença
renal diabética (Etoh et al., 2003).
A administração de inibidores de NOX-4 tem mostrado uma diminuição na
hipertrofia glomerular e também resultou em uma diminuição da expressão de
fibronectina no córtex renal, mais não nos glomérulos. Além disso, NADPH oxidases
foram localizadas em podócitos e as isoformas NOX-1 e NOX-4 sofreram um
aumento em meio hiperglicêmico, tanto in vitro como in vivo (Eid et al., 2009). Sendo
assim, o estresse oxidativo pode estar contribuindo para as alterações patológicas
na ND, incluindo a perda de podócitos e a albuminúria. A fonte subcelular de EROs
na doença renal diabética ainda precisa ser esclarecida, e o papel da inibição das

34
vias de geração de EROs abrem caminho para uma linha de pesquisa que pode
ajudar a ditar terapias futuras para pacientes que desenvolverem o quadro de ND.
1.4. Sistema Glutationa S-transferase
As glutationa-S-transferase (GST) é uma família de enzimas multigênicas que
desintoxicam reativos eletrofílicos, produtos do estresse oxidativo e compostos
conhecidos ou suspeitos de serem cancerígenos através da conjugação com GSH
(Hayes et al., 2005). As GSTs apresentam uma ampla distribuição intracelular,
sendo localizada na mitocôndria (GSTA1), citosol (alfa, mu, pi, e zeta) e ligadas as
membranas celulares (MGST1) (Aniya & Imaizumi, 2011; Li et al., 2011). Estudos
recentes mostraram que a expressão de GSTA4 sofre uma regulação negativa
seletiva nos tecidos adiposos em camundongos obesos insulino resistentes e na
resistência à insulina associada à obesidade humana (Curtis et al., 2010). A função
mitocondrial em adipócitos de camundongos magros ou obesos com genótipo
GSTA4 nulo foi significantemente comprometida quando comparado com o grupo
controle que apresentava genótipo presente e foi acompanhado por um aumento na
produção do ânion superóxido.
A família de enzimas da GST é expressa como várias isoformas codificadas
por uma variedade de genes distribuídos amplamente em diferentes cromossomos
(Strange et al., 2001; Xu et al., 1998). Além disso, a maioria destes loci genéticos
são conhecidos por terem formas polimórficas dos genes (Seidegard et al., 1998;
Pemble et al., 1994). Alguns estudos têm mostrado que polimorfismos nos genes da
GST podem conduzir a alterações na expressão da enzima, quer qualitativamente
ou quantitativamente (Zhong et al., 2006) e, portanto, podem tornar os indivíduos
susceptíveis a várias doenças, incluindo nefropatias.
São classificadas com base em seu ponto isoelétrico, na similaridade da
sequência de aminoácidos e especificidade por substrato. Atualmente são
conhecidas oito classes de GSTs citosólicas humanas: Alpha, Kappa, Mµ, Omega,
Pi, Sigma, Theta e Zeta (Strange et al., 2001; Nebert & Vasiliou, 2004; Mcilwaim et
al., 2006) (Tabela 2).

35
Tabela 2 - GSTs citosólicas humanas
Classe Gene Localização Cromossômica
Alpha (α) GSTA1 6p12
GSTA2 6p12.2
GSTA3 6p12
GSTA4 6p12
GSTA5 6p12.1
Kappa (κ) GSTK1 7q35
Mu (μ) GSTM1 1p13.3
GSTM2 1p13
GSTM3 1p13.3
GSTM4 1p13.3
GSTM5 1p13.3
Omega (ω) GSTO1 10q25.1
GSTO2 10q25.1
Pi (π) GSTP1 11q13
Sigma (σ) GSTS1 4q21-22
Theta (θ) GSTT1 22q11.2
GSTT2 22q11.2
Zeta (z) GSTZ1 14q24.3
Adaptado de Mcilwaim et al, 2006.
Tanto GSTT1, quanto GSTM1 apresentam polimorfismo genético
caracterizado por deleção total do gene, resultando no genótipo homozigoto nulo
(GSTT1-nulo e GSTM1-nulo), caracterizado pela ausência de atividade enzimática
destas isoformas (Arruda et al., 1998; Koch et al., 2010; Reis, 2010). A deleção
aparentemente resulta de trocas homólogas desiguais envolvendo regiões
flanqueadoras contendo sequências com alta identidade (Figuras 5 e 6), que foram
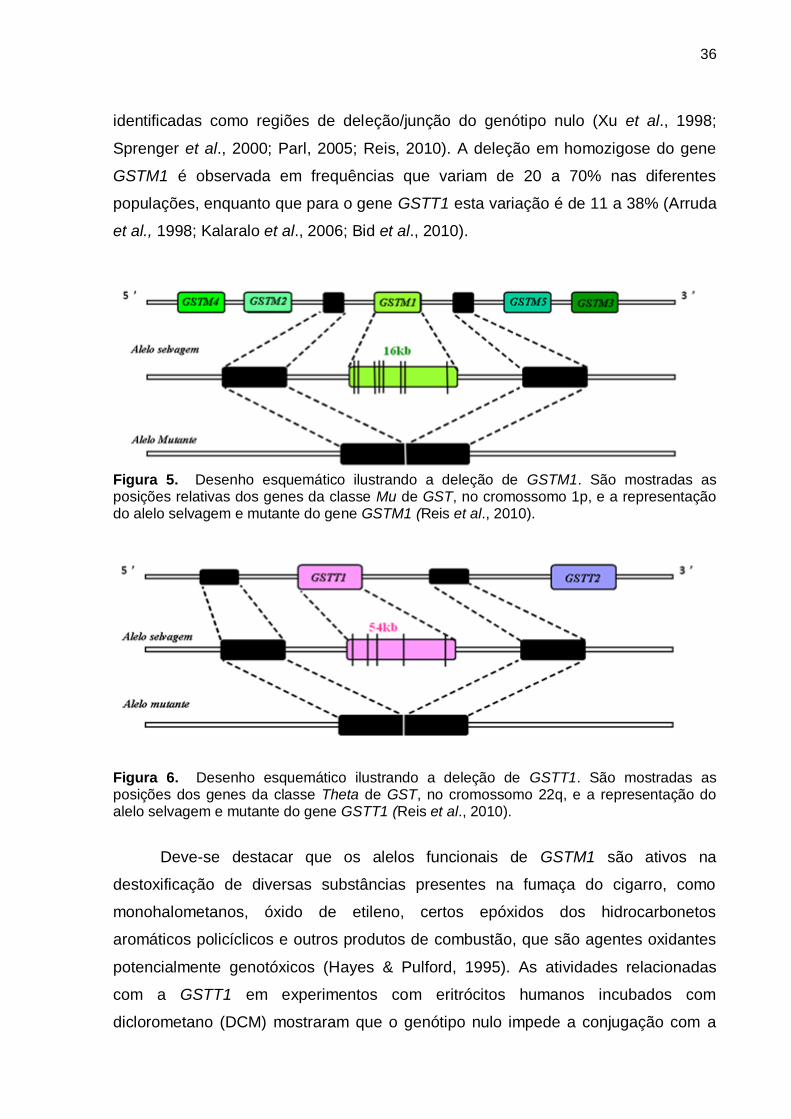
36
identificadas como regiões de deleção/junção do genótipo nulo (Xu et al., 1998;
Sprenger et al., 2000; Parl, 2005; Reis, 2010). A deleção em homozigose do gene
GSTM1 é observada em frequências que variam de 20 a 70% nas diferentes
populações, enquanto que para o gene GSTT1 esta variação é de 11 a 38% (Arruda
et al., 1998; Kalaralo et al., 2006; Bid et al., 2010).
Figura 5. Desenho esquemático ilustrando a deleção de GSTM1. São mostradas as posições relativas dos genes da classe Mu de GST, no cromossomo 1p, e a representação do alelo selvagem e mutante do gene GSTM1 (Reis et al., 2010).
Figura 6. Desenho esquemático ilustrando a deleção de GSTT1. São mostradas as posições dos genes da classe Theta de GST, no cromossomo 22q, e a representação do alelo selvagem e mutante do gene GSTT1 (Reis et al., 2010).
Deve-se destacar que os alelos funcionais de GSTM1 são ativos na
destoxificação de diversas substâncias presentes na fumaça do cigarro, como
monohalometanos, óxido de etileno, certos epóxidos dos hidrocarbonetos
aromáticos policíclicos e outros produtos de combustão, que são agentes oxidantes
potencialmente genotóxicos (Hayes & Pulford, 1995). As atividades relacionadas
com a GSTT1 em experimentos com eritrócitos humanos incubados com
diclorometano (DCM) mostraram que o genótipo nulo impede a conjugação com a

37
GSH, sendo, portanto não conjugadores (Kempkes et al., 1996). Também foi
observada uma atividade de peroxidade contra hidro peróxidos de fosfolipídios
(Hurst et al., 1998), hidro peróxido de cumeno (Meyer et al., 1991), mas essa
atividade não se estenderia a produtos secundários da peroxidação de lipídios
(Whittington et al., 1999).
De forma que o polimorfismo de deleção destas isoformas tem sido associado
a vários tipos de doenças em correlação com o hábito de fumar, com ênfase em
estudos de susceptibilidade ao câncer (Bell et al., 1993; London, et al., 2000;
Habdous et al., 2004; Lima-Jr et al., 2008).
Diante do exposto, pode-se demonstrar que a ausência de uma ou mais
formas de GST pode tornar a célula mais susceptível ao estresse químico e
oxidativo, os quais podem levar à disfunção celular, tornando de grande relevância a
caracterização da influência do polimorfismo de deleção de GSTT1 e GSTM1 no
risco e progressão de doenças (Wang et al., 2006; Zhang et al., 2014).
1.5. Polimorfismo de Deleção de GSTT1 e GSTM1 na nefropatia diabética
Dados da literatura demostram a associação do genótipo GSTM1 e GSTT1
nulo com diferentes doenças. Embora a maioria dos estudos iniciais concentrou-se
em seu papel no câncer, alguns autores têm mostrado a associação desses
polimorfismos genéticos em doenças multifatoriais e metabólicas, recebendo
destaque para a diabetes mellitus e suas complicações, como a nefropatia diabética
(Fujita et al., 2000; Yang et al., 2004; Kim et al., 2005; Manfredi et al., 2009; Koch et
al., 2010).
A identificação de genes marcadores de susceptibilidade a doenças tem sido
realizada, em geral, por meio de estudos caso-controle, comparando-se as
frequências de um determinado gene polimórfico entre as populações doente e
sadia (Reis, 2010). Esses genótipos, GSTM1 e GSTT1, são os que têm recebido
uma maior atenção, pois se sabe que esses genes quando apresentam o genótipo
de deleção (nulo) vai acarretar a uma não produção dessas isoformas da enzima.
Além disso, a prevalência de GSTM1 e GSTT1 nulos tem mostrado uma grande
variabilidade em diferentes grupos populacionais. Resultados de diferentes
populações asiáticas variam em relação à presença desses genótipos e a relação
com os casos de ND (Fujita et al., 2000; Yang et al., 2004; Kim et al., 2005). Na

38
população indiana ao avaliar a relação entre o polimorfismo no gene da GST e a
susceptibilidade ao EFDR, encontraram associação significativa para os genótipos
GSTM1-nulo como GSTT1-nulo (Agrawal et al., 2007).
Nos estudos realizados por Fujita et al (2000) na população japonesa em
pacientes com DM2, não foi encontrado associação entre o polimorfismo de deleção
no gene GSTM1 com a nefropatia diabética, enquanto Yang et al (2004) estabeleceu
que o genótipo GSTT1 nulo consiste em um fator de risco para o desenvolvimento
de ND na população chinesa estudada.
Em 2009, Datta et al., (2009) estudaram os polimorfismos no gene GST e
sugeriram que os genótipos GSTM1 e GSTT1 presente parecem ter um papel
protetor no desenvolvimento e progressão da ND em pacientes com DM2, enquanto
que os genótipos GSTM1 e GSTT1 nulos apareceram como fatores de risco para o
desenvolvimento dessa doença nesses mesmos pacientes.
Estudos realizados por Suvakov et al., em 2012 encontraram um risco
aumentado de desenvolvimento de EFDR e uma maior susceptibilidade ao estresse
oxidativo em pacientes que estavam em hemodiálise e possuíam o do genótipo
GSTM1 nulo. A presença do genótipo GSTT1 nulo nesses pacientes também
apresentou uma influência significativa nos danos resultantes da oxidação, mas não
tanto quanto o genótipo GSTM1.
É necessário que haja uma maior quantidade de estudos na população
brasileira para a susceptibilidade causada pelo polimorfismo de deleção dessas
isoformas descritas e a associação com a ND, já que podemos observar uma
quantidade significativa de dados que variam entre as etnias e as características
interindividuais.

39
2. JUSTIFICATIVA
Há evidências suficientes apoiando o conceito de susceptibilidade genética
para a nefropatia em pacientes com diabetes. Com o crescente aumento de
indivíduos portadores dessa doença existe uma maior necessidade de pesquisa que
permitam a descoberta de variantes genéticas que sustentem essa susceptibilidade,
o que poderia render importantes insights sobre essa condição.
O fato de pacientes diabéticos continuarem a desenvolver essa complicação,
mesmo na ausência dos fatores já conhecidos como o controle glicêmico, pressão
arterial, concentração de colesterol, e uma dieta proteica restritiva, levanta a
importância de mais estudos genéticos em diferentes grupos populacionais.
A participação do estresse oxidativo na fisiopatologia dessa complicação tem
sido bastante discutida na literatura, existindo ainda a necessidade de distinguir os
fatores que participariam desse desequilíbrio. As isoformas GSTM1 e GSTT1 são
candidatas por participarem do processo de detoxificação de radicais livres e o
polimorfismo genético que elas sofrem acarretarem a um genótipo nulo, ou seja, a
não produção dessas enzimas.
Essas descobertas permitiriam a identificação de pacientes com risco de
nefropatia logo após o diagnóstico de diabetes, em vez de mais tarde, quando o
prognóstico se torna ainda mais desfavorável a esses pacientes. Em segundo lugar,
a necessidade da descoberta de novos genes associados a uma susceptibilidade
aumentada ao desenvolvimento e progressão dessa complicação, ajudando assim
um melhor entendimento das vias relacionadas e ao desenvolvimento de novas
terapias, acarretando assim uma melhora nas condições de vida desses pacientes e
diminuindo os gastos em tratamentos caros como a diálise.

40
3. OBJETIVOS
3.1. Objetivo geral
Avaliar a associação dos polimorfismos de deleção dos genes GSTM1 e
GSTT1 com a susceptibilidade no desenvolvimento de nefropatia diabética na
população goianiense.
3.2. Objetivos específicos
Estabelecer as frequências alélicas e genotípicas dos genes GSTT1 e GSTM1 na
população estudada;
Correlacionar o polimorfismo dos genes GSTM1 e GSTT1 com a susceptibilidade
no desenvolvimento de nefropatia diabética;
Avaliar a influência do polimorfismo dos genes GSTM1 e GSTT1 com os
parâmetros clínicos;

41
4. METODOLOGIA
4.1. Pacientes e Controles
Para estabelecer os perfis alélicos dos genes GSTT1 e GSTM1 o grupo
amostral foi constituído por 155 amostras no total, sendo os grupos classificados em
caso e controle. O grupo caso foi constituído de 65 pacientes diagnosticados e
acompanhados pelo Serviço de Endocrinologia do Hospital das Clínicas da
Universidade Federal de Goiás (UFG) e de pacientes da Nefroclínica Goiânia que
foram diagnosticados com nefropatia diabética e encontravam-se em tratamento de
hemodiálise. O grupo controle foi constituído de 90 indivíduos saudáveis
selecionados da população geral de nossa região.
A participação individual no presente estudo teve caráter voluntário para
ambos, casos e controles, na qual a participação foi voluntária e todos os
participantes assinaram o termo de consentimento livre e esclarecido [Anexo I]. Para
a realização do presente estudo obteve-se o parecer favorável do Comitê de Ética
institucional (Nº195/11 datado de 27 de Junho de 2011) [Anexo II]. Dados
demográficos e clínico-laboratoriais (relativos a condições de saúde gerais,
acometimento por doenças, hábitos de consumo de bebidas alcoólicas e tabagismo,
entre outros) foram obtidos por meio de entrevistas e consulta a prontuários
(pacientes) [Anexo III] e a resultados de exames laboratoriais (controles) [Anexo IV].
4.2. Obtenção das Amostras
A coleta de amostra dos pacientes participantes foi realizada no Hospital das
Clínicas da UFG durante as visitas para acompanhamento e na Nefroclinica, clínica
colaboradora do estudo situada em Goiânia-GO, durante o procedimento de diálise.
As amostras dos controles foram obtidas durante a realização de exames de rotina
no Laboratório da Área de Saúde da Pontifícia Universidade Católica de Goiás
(PUC-GO). Destas, foram coletados 4 mL de sangue periférico em tubos a vácuo
heparinizados, sendo posteriormente preservados a -20°C, para posterior extração
de DNA.
4.3. Extração e Quantificação de DNA
A extração e purificação de DNA das amostras obtidas foram realizadas com
uso do kit comercial de extração PureLink™ Genomic DNA Mini Kit, seguindo o

42
protocolo sugerido pelo fabricante (Invitrogen - Carlsbad, CA, USA). As amostras de
DNA foram quantificadas em espectrofotômetro NanoDrop (Thermo Scientific®) e em
seguida foram diluídas com Água Milli-Q Estéril resultando em uma concentração
final de 10 ng/μL para a análise dos polimorfismos de GSTT1 e GSTM1 utilizando a
técnica de amplificação por PCR em tempo real (qPCR).
4.4. Análise do polimorfismo de deleção de GSTT1 e GSTM1
Na genotipagem dos polimorfismos de GSTT1 e GSTM1 por ensaios de
qPCR multiplex foi utilizado o fluoróforo SYBR ®
Green I, com discriminação dos
genótipos nulo e presente por análise das curvas de melting geradas após as
reações de amplificação. Os primers utilizados foram previamente sugeridos por
Voso et al., 2002, sendo o primer reverse de GSTT1 sugerido por Marín et al. (2010)
(tabela 3). Foi realizada a coamplificação da região RH92600, uma região de
microssatélite de 135pb localizado em 6q13, usada como controle endógeno da
reação, para validar os genótipos nulos. As condições de ciclagem podem ser
observadas na tabela 4.
Tabela 3 - Sequências de primers utilizados nas reações de amplificação por qPCR
multiplex
Primer Sequência 5’→3’ Amplicon (pb)
GSTM1 F-GAA CTC CCT GAA AAG CTA AAG C R-GTT GGG CTC AAA TAT ACG GTG G
215
GSTT1 F-TTC CTT ACT GGT CCT CAC ATC TC R-GGA AAA GGG TAC AGA CTG GGG A
257
RH92600 F-GCA ATT CCG CAT TTA ATT CAT GG R-AAA CAG GCC ACG TAA AGC AAC
135
Tabela 4 - Condições de termociclagem das reações de amplificação por qPCR multiplex
Ciclagem Etapas Temperatura (°C) Tempo
Início Desnaturação 95 10 min. Desnaturação 95 10 s 33 Ciclos Anelamento 60 20 s Extensão 72 25 s
Foi utilizado um volume final de 25 μL nas reações, contendo
aproximadamente 10 ng de DNA das amostras previamente quantificadas e diluídas,

43
0,3 μM de cada primer GSTT1, 0,4 μM de cada primer GSTM1 e 0,5 μM de cada
primer RH92600 (Tabela 3), 1X de SYBR ® Green qPCR Master Mix (Fermentas®),
com adição de 1,5 mM de MgCl2. A programação para realização da curva de
melting foi: 95°C por 10 segundos, 65°C por 1 minuto e aumento para 95°C, com 5
aquisições por °C. A figura 7 demonstra a curva de melting para a caracterização
dos genótipos de GSTT1 e GSTM1.
Figura 7. Curva de melting da qPCR multiplex (SYBR Green). Os picos correspondem aos genótipos presentes de GSTM1 (77,5°C), GSTT1 (82,5°C) e RH92600 (74°C). A ausência dos picos correspondentes a GSTM1 e/ou GSTT1, na presença do controle endógeno (RH92600), permite a identificação dos respectivos genótipos nulos.
4.5. Análise Estatística
Foi empregado o software BioEstat® versão 5.3 (disponível para download no
link: http://www.mamiraua.org.br/pt-br/downloads/programas/bioestat-versao-53/). O
teste do qui-quadrado foi utilizado para comparar as frequências genotípicas, tendo
sido aplicado o teste exato de Fisher quando necessário.
O cálculo de Odds Ratio, com intervalo de confiança 95%, foi utilizado para
avaliar o genótipo de risco e a susceptibilidade a nefropatia diabética, por regressão
logística múltipla com controle de fatores de confundimento relacionados à
distribuição por sexo, hábito de fumar e etilismo.
O teste t foi empregado para comparar as variáveis clínicas entre os grupos
analisados. O valor de p ≤ 0,05 foi considerado estatisticamente significante.

44
5. RESULTADOS
Foram avaliadas características clínico-laboratoriais na população estudada,
os resultados obtidos estão demonstrados na tabela 5. O grupo caso foi constituído
por 65 pacientes (33 masculinas e 32 femininos) que se encontrava em EFDR e em
tratamento de hemodiálise, sendo que a média de idade dos indivíduos foi de
aproximadamente 62,3 anos, foram encontrados valores médios significativos
alterados para as variáveis clinico laboratoriais analisadas no grupo caso quando
comparado com o grupo controle. O grupo controle foi composto de 90 indivíduos
(51 masculinos e 39 femininos), apresentando 61,5 anos como média de idade entre
os participantes desse grupo. Foi feita uma avaliação dos níveis de glicose,
creatinina e da taxa de filtração glomerular.
Tabela 5. Características da população estudada e comparação entre os grupos
caso e controle.
Dados são reportados como média ± desvio padrão. Análise estatística por teste t ou teste do Qui-quadrado. *Diferença significativa entre os grupos (p ≤ 0,05). #TFG- Taxa de Filtração Glomerular estimado pela formula de Cockcroft-Gault.
A análise estatística da distribuição por sexo e idade não apresentou
diferenças significativas, indicando que há homogeneidade entre os grupos
estudados. A proporção de indivíduos fumantes e etilistas também não apresentou
Variáveis Caso (N=65) Controle (N=90) P
Sexo (M/F) 33/32 51/39 0.5729
Idade (anos) 62.43 ± 10.66 61.52 ± 9.43 0.5763
IMC (kg/m²) 26.04 ± 4.73 27.35 ± 5.19 0.1087
Glicemia de jejum 197.45 ± 85.47 88.23 ± 8.76 < 0.0001*
Creatinina (mg/dl) 5.79 ± 3.97 0.75 ± 0.20 < 0.0001*
TFG# (ml/min) 39.94 ± 49.87 108.05 ± 18.02 < 0.0001*
Etilismo (S/N) 28/37 34/56 0.6182
Tabagismo (S/N) 13/52 26/64 0.2842

45
diferenças significativas entre os dois grupos pela comparação realizada pelo teste
do qui-quadrado.
5.1. Distribuição genotípica e associação do polimorfismo de GSTT1 e
GSTM1
Um total de 155 indivíduos (sendo 65 pacientes e 90 controles) foram
genotipados para o polimorfismo de deleção das duas isoformas de GST. Nos
pacientes nefropatas, a frequência de GSTM1-nulo foi de 32,3% e de GSTT1-nulo
foi de 24,6%. No grupo controle as frequências foram de 42,2% e 11,1%
respectivamente (Tabela 6).
No grupo caso, a frequência de GSTT1-nulo foi significativamente mais
elevada do que nos controles (p = 0,0230), já para GSTM1 não houve diferença
significativa (0,3860) na distribuição de genótipos nulos e presente entre os grupos
caso e controle (Tabela 6).
A distribuição por sexo, tabagismo e etilismo foram as variáveis de
confundimento utilizadas na análise por regressão logística múltipla, havendo
homogeneidade na distribuição por sexo, tabagismo e etilismo entre os grupos em
todas as análises realizadas.
Foi verificado na análise de risco associado ao polimorfismo de deleção, que
o genótipo GSTT1-nulo (p=0,0230) provoca um risco aumentado de
aproximadamente 2.9 vezes em desenvolver a doença (nefropatia diabética) em
relação aos portadores do genótipo GSTT1-presente. Não houve associação de
GSTM1 (p=0.3860) com a susceptibilidade a doença na população estudada.

46
Tabela 6. Distribuição de frequências genotípicas para GSTM1 e GSTT1 na
população estudada e analise de risco para nefropatia diabética.
Caso Controle
Genótipo n (%) n (%) OR (IC 95%) P
GSTM1
Presente (+) 44 (67.7) 52 (57.8) 0.73 (0.37-1.47) 0.3860
Nulo (-) 21 (32.3) 38 (42.2)
GSTT1
Presente (+) 49 (75.4) 80 (88.9) 2.88 (1.16 - 7.17) 0.0230*
Nulo (-) 16 (24.6) 10 (11.1)
Total 65 (100) 90 (100)
Analise por regressão logística múltipla para obter valores ajustados de odds ratio (OD) e intervalos de confiança (95%IC). *Diferença significativa entre os grupos (p ≤ 0,05).
Foi verificada uma baixa frequência de indivíduos com genótipo duplo nulo (- /
-), tanto no grupo caso (4,6%), quanto no grupo controle (3,3%), com predominância
de indivíduos com genótipo duplo presente (+ / +) nos dois grupos (47,7% e 50%,
respectivamente).
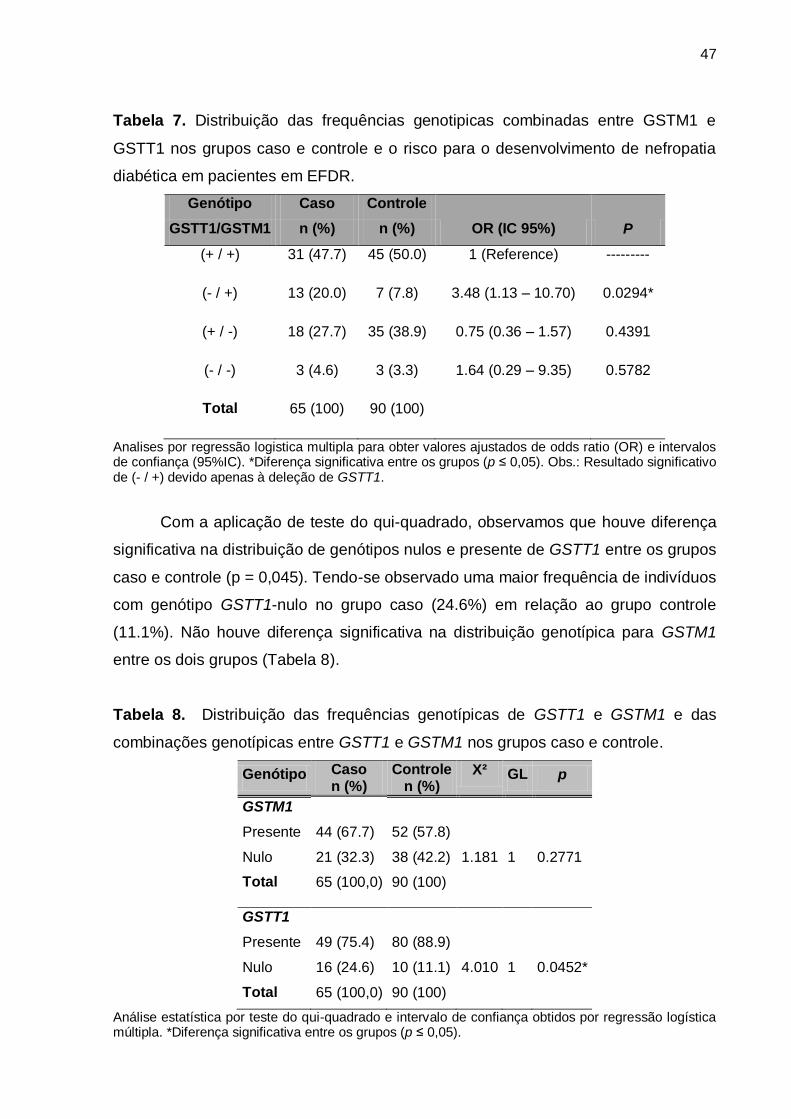
Na análise dos genótipos combinados de GSTT1 e GSTM1 por regressão
logística múltipla foi verificado que a combinação de GSTT1-nulo com GSTM1-
presente (- / +) conferiu um risco aumentado de 3.5 vezes ao desenvolvimento da
doença. As demais combinações genotípicas não trouxeram associações
significativas (Tabela 7).
47
Tabela 7. Distribuição das frequências genotipicas combinadas entre GSTM1 e
GSTT1 nos grupos caso e controle e o risco para o desenvolvimento de nefropatia
diabética em pacientes em EFDR.
Genótipo Caso Controle
GSTT1/GSTM1 n (%) n (%) OR (IC 95%) P
(+ / +) 31 (47.7) 45 (50.0) 1 (Reference) ---------
(- / +) 13 (20.0) 7 (7.8) 3.48 (1.13 – 10.70) 0.0294*
(+ / -) 18 (27.7) 35 (38.9) 0.75 (0.36 – 1.57) 0.4391
(- / -) 3 (4.6) 3 (3.3) 1.64 (0.29 – 9.35) 0.5782
Total 65 (100) 90 (100)
Analises por regressão logistica multipla para obter valores ajustados de odds ratio (OR) e intervalos de confiança (95%IC). *Diferença significativa entre os grupos (p ≤ 0,05). Obs.: Resultado significativo de (- / +) devido apenas à deleção de GSTT1.
Com a aplicação de teste do qui-quadrado, observamos que houve diferença
significativa na distribuição de genótipos nulos e presente de GSTT1 entre os grupos
caso e controle (p = 0,045). Tendo-se observado uma maior frequência de indivíduos
com genótipo GSTT1-nulo no grupo caso (24.6%) em relação ao grupo controle
(11.1%). Não houve diferença significativa na distribuição genotípica para GSTM1
entre os dois grupos (Tabela 8).
Tabela 8. Distribuição das frequências genotípicas de GSTT1 e GSTM1 e das
combinações genotípicas entre GSTT1 e GSTM1 nos grupos caso e controle.
Genótipo Caso n (%)
Controle n (%)
X² GL p
GSTM1
Presente
44 (67.7) 52 (57.8)
1.181 1 0.2771 Nulo 21 (32.3) 38 (42.2)
Total 65 (100,0) 90 (100)
GSTT1
Presente
49 (75.4) 80 (88.9)
4.010 1 0.0452* Nulo 16 (24.6) 10 (11.1)
Total 65 (100,0) 90 (100)
Análise estatística por teste do qui-quadrado e intervalo de confiança obtidos por regressão logística múltipla. *Diferença significativa entre os grupos (p ≤ 0,05).

48
5.2. Influência do polimorfismo de deleção de GSTT1 e GSTM1 sobre
variáveis clínicas e laboratoriais.
Também foi feita uma analise da influência da deleção de GSTT1 e GSTM1
sobre as alterações clínicas e bioquímicas no grupo de pacientes estudados pela
comparação entre os indivíduos com genótipo nulo e genótipo presente (Tabela 9 e
Tabela 10). Observou-se que não ocorreram diferenças significativas quanto à
distribuição por sexo, tabagismo e etilismo entre os grupos, inferindo assim que há
homogeneidade entre os pacientes estudados para a comparação das variáveis
analisadas.
Com relação ao polimorfismo de GSTT1, não houve associação significativa
de nenhuma das variáveis clínicas analisadas com os genótipos nulos e presente
(Tabela 9), apesar de esse polimorfismo ter apresentado diferença significativa
quando comparado com o grupo controle.
Em relação ao genótipo GSTM1 não foram observadas diferenças
significativas tanto nas análises comparando grupo controle com grupo de pacientes,
como nas analises estatísticas das variáveis clínicas (Tabela 10).
Tabela 9. Associação entre genótipos nulos e presentes de GSTT1 com variáveis
clínicas em pacientes com nefropatia diabética.
GSTT1 Presente (N = 49) Nulo (N = 16) P
Sexo (M/F) 23/26 10/6 0.4278
Idade (anos) 63.47 ± 10.42 59.25 ± 11.08 0.1709
IMC (kg/m²) 26.38 ± 4.59 24.98 ± 5.14 0.3071
Glicemia de jejum (mg/dl) 189.44 ± 74.21 220.93 ± 112.28 0.2374
Creatinina (mg/dl) 5.34 ± 3.82 7.19 ± 4.24 0.1053
TFG# (ml/min) 44.05 ± 51.38 27.34 ± 44.03 0.2475
Etilismo (+/-) 10/39 3/13 1.0000
Tabagismo (+/-) 20/29 8/8 0.7238
Os dados são apresentados como média ± desvio padrão. A análise estatística pelo teste t, qui-quadrado ou teste exato de Fisher. Nível de significância (p ≤0,05). # TFG-Taxa de filtração glomerular estimada pela formula de Cockcroft-Gault.

49
Tabela 10. Associação entre genótipos nulos e presentes de GSTM1 com variáveis
clínicas em pacientes com Nefropatia diabética.
GSTM1 Presente (N = 44) Nulo (N = 21) P
Sexo (M/F) 24/20 9/12 0.5377
Idade (anos) 62.32 ± 10.64 62.67 ± 10.97 0.9030
IMC (kg/m²) 26.45 ± 4.91 25.17 ± 4.29 0.3133
Glicemia de jejum (mg/dl) 199.58 ± 86.38 193.42 ± 85.93 0.8020
Creatinina (mg/dl) 5.99 ± 3.99 5.37 ± 4.01 0.5596
TFG# (ml/min) 40.87 ± 52.77 37.98 ± 44.34 0.8290
Etilismo (+/-) 22/22 6/19 0.0630
Tabagismo (+/-) 8/36 5/16 0.7416
Os dados são apresentados como média ± desvio padrão. A análise estatística pelo teste t, qui-quadrado ou teste exato de Fisher. Nível de significância (p ≤0,05). # TFG-Taxa de filtração glomerular estimada pela formula de Cockcroft-Gault.

50
6. DISCUSSÃO
A ND caracteriza-se por ser uma das principais complicações microvasculares
da DM, sendo uma das principais causas do estágio final de doença renal (EFRD)
(Tuttle et al., 2014). Em um levantamento feito pela União Européia estimou que os
custos de tratamentos destinados a pacientes em EFDR seria de aproximadamente
51 mil euros no primeiro ano por indivíduo (Haller et al., 2011). Na literatura
observamos dados controversos em diferentes populações a respeito dos fatores
genéticos que estariam associados à susceptibilidade e evolução para estágios
clínicos mais avançados dessa patologia.
A patogênese da ND tem o envolvimento de vários fatores, incluindo
alterações hemodinâmicas e metabólicas, e o estresse oxidativo. Mecanismos
bioquímicos, como a alteração na via dos polióis, a formação de espécies reativas
de oxigênio e a formação dos AGEs, têm sido propostos para explicar as alterações
estruturais e funcionais relacionados com o quadro de hiperglicemia sustentada nos
tecidos vasculares, com indícios de que a capacidade endógena antioxidante nesses
indivíduos esteja prejudicada, o que dificultaria a remoção desses radicais livres
formados (Santini et al., 1997).
As GSTs constituem uma família de enzimas que têm grande importância na
defesa aos danos oxidativos, assim como na biotransformação de xenobióticos
(Koch et al., 2010; Reis, 2010), destacando as isoformas GSTT1 e GSTM1 que
apresentam polimorfismo de deleção, o que resulta na ausência de atividade
enzimática (London et al., 2000). Os membros da família das GSTs possuem assim,
um papel importante no mecanismo de defesa que neutralizam alterações
bioquímicas que ocorrem durante o estado urêmico, agindo tanto na conjugação
com a glutationa como na atividade antioxidante enzimática (Simic et al., 2009;
Hayes & Strange, 1995).
No presente estudo, foi feita uma análise do polimorfismo envolvendo esses
genes e a susceptibilidade em desenvolver ND em pacientes que se encontravam
em tratamento de hemodiálise, sendo encontrado um risco aumentado de 2,9 vezes
em desenvolver a doença para os portadores do genótipo GSTT1-nulo, resultado
esse confirmado com a análise dos genótipos combinados, onde GSTM1-presente
não reduziu o efeito de GSTT1-nulo sobre o risco a desenvolver nefropatia.

51
Na comparação entre os grupos caso e controle, o achado de diferenças
significativas nas variáveis clínicas analisadas (RFG, creatinina e glicemia), pode ser
explicado pelo elevado grau de descompensação metabólica dos pacientes
envolvidos no estudo. Os níveis tão alterados de creatinina podem ser explicados
por serem pacientes que se encontram em hemodiálise, ou seja, já apresentam um
grau avançado de lesão renal, e sabe-se que uma pessoa que apresenta nível alto
desse elemento no sangue, significa que a função dos rins já está cerca de 50%
comprometida. Em relação ao TFG, este foi estimado pela fórmula matemática que
leva em consideração os níveis de creatinina, peso e idade, e novamente por serem
pacientes que se encontram em um estágio avançado de doença renal, existe uma
diminuição dessa taxa.
O TFG foi estimado pela fórmula de Cockcroft-Gault, que não representa o
TFG real com a mesma fidelidade de outros métodos, como o clearance de inulina
(Stevens et al., 2006). Um dos problemas que recebe destaque na utilização de
fórmulas para o cáculo do TFG em pacientes diabéticos é a não detecção da fase
preococe de ND, por essa razão TFG menores que 90 mL/min já podem representar
perda da função renal (KDOQI Clinical Practice Guidelines, 2007).
Entre os polimorfismos estudados e analisados neste trabalho, apenas o
genótipo GSTT1 nulo apresentou uma associação significativa em desenvolver o
quadro de ND em pacientes que estavam em hemodiálise, em relação aos
portadores do genótipo GSTT1-presente. Estes dados vão de acordo com os
encontrados na literatura, onde o genótipo GSTT1 duplo nulo apresentou um fator
de risco para desenvolvimento de EFDR em pacientes diabéticos em Taiwan, não
sendo encontrada associação entre a deleção homozigótica de GSTM1 nesses
pacientes (Yang et al., 2004). Vojtková et al. (2013), encontrou o mesmo risco ao se
comparar GSTT1 nulo com M1 presente em crianças e adolescentes eslovacas
portadores de DM1.
Fujita et al. (2000), sugeriu que o genótipo nulo de GSTM1 não contribui para
o desenvolvimento da nefropatia diabética em pacientes com DM2 na população
japonesa. Datta et al. (2010), também estabeleceram a existência de influências do
genótipo GSTT1 nulo com um maior risco para o desenvolvimento de doença renal
terminal. Agrawal et al, relatou que os genótipos GSTP1-313 e os alelos de GSTM1
e GSTT1 são considerados fatores de risco predisponentes, mas não esclareceram
se os pacientes em EFDR eram diabéticos.

52
Estudo realizado por Orhan et al., (2014) para avaliar a participação desses
polimorfismos com o risco aumentado no desenvolvimento de DM em gestantes não
encontraram resultados estatisticamente significativos, concluindo que os genes
GSTM1 e GSTT1 não influenciariam na DMG, além disso a presença de uma das
isoformas não apresentou efeito protetor contra a ocorrência da doença.
Suvakov et al. (2012) encontrou em seus estudos que o nível de proteção
contra o estresse oxidativo na síndrome urêmica é influência pelos polimorfismos
nos genes de GSTs (A1, M1, P1 e T1), que determinam a quantidade e/ou atividade
dessas enzimas, que agem nos hidro peróxidos orgânicos e em compostos
prejudiciais que se acumulam no organismo. Lin et al. (2009) mostraram que
pacientes em hemodiálise com o genótipo GSTM1 nulo são mais vulneráveis aos
danos oxidativos e possuem maior risco de morte em comparação com aqueles que
possuem GSTM1 presente, resultado esse que não foi encontrado em nossos
estudos, possivelmente pelo fato de o n utilizado em nosso estudo ser mais limitado
do que o usado por Lin et al., (2009) (488 pacientes com insuficiência renal
recebendo tratamento de hemodiálise, sendo que destes 164 tinham como causa a
nefropatia diabética).
Ainda relacionado com o polimorfismo de GSTM1, estudos com resveratrol,
um polifenol que tem mostrado possuir efeitos antioxidantes, anti-inflamatórios e
efeitos cardioprotetores, (Miller & Rice-Evans, 1995; Hung et al., 2000; Das et al.,
2005) realizados por Jiang et al., (2013) em ratos com diabetes induzidos por
estreptozina e que apresentavam excreção urinária de proteínas aumentada e
alterações renais compatíveis com os estágios iniciais de ND, tratados com essa
substância mostraram redução na excreção de proteínas comparados com ratos
diabéticos não tratados.
Além disso, os seus resultados mostraram que a expressão de GSTM em
ratos diabéticos foi significantemente maior do que no grupo controle, sendo que,
com o tratamento com resveratrol a expressão de GSTM voltou aos níveis normais,
resultado semelhante ao encontrado por Fujita et al., (2001). Também foi observada
a ação dessa substância na redução da glicose plasmática, creatinina e uma
atenuação a hipertrofia renal em ratos, sugerindo que o mecanismo de regulação
negativa na expressão de GSTM pode ser uma das vias envolvidas no papel
renoprotetor do resveratrol. Apesar de não termos encontrado associação
significativa com o polimorfismo dessa isoforma no nosso estudo, não podemos

53
descartar a sua participação. Talvez essa associação não foi observada por o grupo
amostral ser pequeno, ou por serem pacientes que já se encontravam em estágios
clínicos mais avançadas da ND e não nos estágios inicias, o que poderia prejudicar
esse mecanismo de regulação.
Na análise de risco foi observada uma frequência mais elevada do genótipo
GSTT1-nulo (24,6%) no grupo caso do que em indivíduos não diabéticos do grupo
controle (11,1%). Aqueles que apresentam o genótipo de risco possuem uma
predisposição aumentada à doença de aproximadamente 2,9 vezes (p = 0.0230).
Portanto, os indivíduos podem apresentar defesas antioxidantes diminuídas quando
esta isoforma está deletada. Estes resultados sugerem que o polimorfismo de
deleção do gene GSTT1 pode desempenhar um papel importante na patogênese do
ND corroborando com os resultados achados nos estudos já citados.
Em relação ao polimorfismo de GSTM1, não foram encontradas diferenças
estatisticamente significativas nas proporções dos genótipos nulos entre os grupos
caso (32,3%) e controle (42,2%). Também foi verificado que não houve associação
da deleção de GSTM1 com a susceptibilidade a ND. Estes resultados estão de
acordo com estudo realizado por Fujita et al., (2000) na população japonesa (foi
utilizado um n de 105 pacientes com ND), que em seus estudos encontraram que a
frequência do genotipo nulo não foi significantemente maior no grupo de pacientes
com ND do que no grupo de pacientes sem, sugerindo assim que esse genótipo não
contribui para o desenvolvimento da doença.
As análises feitas por regressão logística múltipla indicaram que a
combinação GSTT1-nulo com GSTM1-presente (- / +) conferiu um risco aumentado
de 3.5 vezes ao desenvolvimento da doença. Porém, a análise log-linear não
mostrou haver contribuição do polimorfismo GSTM1 na susceptibilidade ao
desenvolvimento da ND (p = 0.386), portanto GSTM1-presente não é capaz de
suprir a falta causada pela deleção de GSTT1, já que podemos observar um risco
elevado de se desenvolver a doença.
Datta et al., (2010), em seus estudos, encontrou que entre a população
indiana saudável o aumento do estresse oxidativo leva a um aumento na expressão
de GST com o aumento dos genótipos GSTM1+/GSTT1+ comparado com outros
genótipos que também apresentam polimorfismo de deleção, implicando assim a
existência de mecanismos compensatórios. Ainda observaram que em pacientes
com doença crônica renal, esses genótipos presentes não são capazes de promover

54
esse mecanismo compensatório com o aumento do estresse oxidativo, indicando
assim um importante papel dos rins na atividade normal de GST.
E meta-análise publicada por Orlewski & Orlewska (2015) mostrou que os
indivíduos com os genótipos GSTM1-nulo ou GSTM1-nulo / GSTT1-nulo
apresentaram um risco significativamente aumentado de desenvolverem ND quando
comparados com indivíduos que não possuem estes genótipos, porém GSTT1 nulo
versus presente parece não estar relacionado associado à susceptibilidade em
desenvolver a doença. Além disso, foi encontrado um risco aumentado em
desenvolver a doença em indivíduos com o polimorfismo nulo para os dois genes em
comparação com indivíduos portadores do genótipo presente, sugerindo que pode a
existência de certos genótipos combinados pode aumentar o risco, do que apenas
um deles (Orlewski & Orlewska, 2015).
Nas análises de associação de variáveis clínico-laboratoriais com os
polimorfismos estudados não foram encontradas associações estatisticamente
significativas em nenhuma das variáveis analisadas com os genótipos nulos e
presente de GSTT1 e GSTM1. Pinheiro et al. (2013) em seus estudos estabeleceu
uma relação do genótipo GSTM1 nulo em pacientes com DM2, com aumento
significativo de Hb1c, glicemia de jejum e pressão arterial mais não com o risco de
desenvolver a DM2. Esses mesmos autores encontraram uma relação do genótipo
GSTT1 nulo em componentes do perfil lipídico. O fato de não termos encontrado
diferença significativa pode estar relacionado com o fato de serem pacientes que se
encontram em um estágio avançado de ND e não podemos descartar a
possibilidade de que os genótipos nulos podem estar influenciando em outras
variáveis que não foram analisadas.
Apesar do número reduzido de indivíduos no grupo caso, estes resultados
podem ser úteis, considerando poucos estudos que tratam do mesmo tema na
população brasileira, especificamente na região e Goiânia. Além disso, os resultados
que apresentaram níveis de significância são compatíveis com outros dados
apresentados na literatura. Destacamos ainda a necessidade de maiores estudos
visando esclarecer a relação entre esse polimorfismo e a susceptibilidade no
desenvolvimento da doença e o estresse oxidativo.
Os resultados obtidos em nosso trabalho sugerem que GSTT1 pode ser
considerado como um potencial marcador genético para auxiliar na identificação de
indivíduos com susceptibilidade aumentada em desenvolver ND e os estágios

55
avançados da doença. Os mecanismos que atuam nessa susceptibilidade e a
relação existente com o estresse oxidativo ainda necessitam de novos estudos para
uma melhor compreensão destes mecanismos. Portanto, os resultados obtidos no
presente trabalho proporcionam um direcionamento para novas pesquisas
relacionadas ao estudo da associação do polimorfismo de deleção de GSTT1 e a
ND. E consequentemente, melhorias no tratamento e prevenção da ND.

56
7. CONCLUSÃO
Este estudo mostrou evidências de que o polimorfismo de deleção da
isoforma T1 do sistema GST pode desempenhar um papel importante na
susceptibilidade no desenvolvimento de ND. As seguintes associações foram feitas:
1) GSTT1 teve influência sobre a susceptibilidade a ND, sendo que o genótipo
GSTT1-nulo provocou um risco aumentado de 2,9 vezes à doença;
2) GSTM1 não apresentou associação com a susceptibilidade a ND;
3) A análise dos genótipos combinados de GSTT1 e GSTM1 mostrou que o
genótipo GSTM1-presente não reduz o efeito da deleção de GSTT1 sobre o
risco à doença;
4) GSTT1-nulo com GSTM1-presente (- / +) conferiu um risco aumentado de 3.5
vezes ao desenvolvimento da doença
5) GSTT1-nulo e GSTM1-nulo não mostrou associação significativa com as
variáveis clinicas analisada;
Podemos concluir diante disso que o polimorfismo de deleção envolvendo o
genótipo GSTT1 pode ser considerado como um potencial marcador na identificação
de indivíduos com risco aumentado para o desenvolvimento de ND, mas não possui
influência nos parâmetros analisados nos portadores dessa complicação decorrente
da DM.

57
8. REFERÊNCIAS
1. American Diabetes Association [ADA]. Standards of Medical Care in
Diabetes. Diabetes Care, v. 35, n. 1, p. 11- 63, 2012.
2. American Diabetes Association [ADA]. Diagnosis and Classification of Diabetes
Mellitus. Diabetes Care, v. 36, s. 1, p. S67-74, 2013.
3. Aguiar LGK, Villela NR, Bouskela E. A microcirculação no diabetes: implicações
nas complicações crônicas e tratamento da doença. Arq Bras Endocrinol Metab
2007; 51: 204-211.
4. Agrawal S, Tripathi G, Khan F, Sharma R, Baburaj VP. Relationship between
GSTs gene polymorphism and susceptibility to end stage renal disease among North
Indians. Ren Fail 2007; 29:947-953.
5. Alves CMP, Lima CS, Oliveira FJL. Nefropatia diabética: avaliação dos fatores de
risco para seu desenvolvimento. Ver Bras Clin Med 2011; 9 (2):97-100.
6. Andrade MMJ; & Marco V. Insulina e hipoglicemiantes orais. In H. S. Spinosa, S.
L. Górniak & M. M. Bernardi (Eds.). Farmacologia aplicada à medicina veterinária.
Rio de Janeiro: Guanabara Koogan, 2006, 4ª ed., p. 395-405.
7. Aniya Y, and Imaizumi N. Mitochondrial glutathione transferases involving a new
function for membrane permeability transition pore regulation. Drug Metab Rev. 2011
May; 43(2):292-9 292–299.
8. Arruda VR, Grignolli CE, Gonçalves MS, Soares MC, Menezes R, Saad ST, Costa
FF. Prevalence of homozygosity for the deleted alleles of glutathione S-transferase
mu (GSTM1) and theta (GSTT1) among distinct ethnic groups from Brazil: relevance
to environmental carcinogenesis?. Clinical Genetics 1998; 54: 210-214.

58
9. Azevedo I. Insulina e outros antidiabéticos. In S. Guimarães, D. Moura & P. S.
Silva (Eds.), Terapêutica medicamentosa e suas bases farmacológicas. Porto,
Portugal: Porto Editora, 2006, 5ª ed., p. 574- 582.
10. Baynes JW. Role of oxidative stress in development of complications in diabetes.
Diabetes 1991; 40: 405-412.
11. Bell DA, Taylor JA, Paulson DF, Robertson CN, Mohler JL, Lucier GW. Genetic
risk and carcinogen exposure: a common inherited defect of the carcinogen
metabolism gene glutathione S-transferase M1 (GSTM1) that increases susceptibility
to bladder cancer. Journal of the National Cancer Institute 1993; 85:1159-1164.
12. Bertoluci MC, Schmid H, Lachat JJ, Coimbra TM. Transforming growth factor-
beta in the development of rat diabetic nephropathy. Nephron 1996; 74:189-96.
13. Bhattacharyya S, Ghosh J, Sil PC. Iron induces hepatocytes death via MAPK
activation and mitochondria-dependent apoptotic pathway: beneficial role of glycine.
Free Radical Res 2012; 46:1296–1307.
14. Bid HK, Konwar R, Saxena M, Chaudhari P, Agrawal CG, Banerjee M.
Association of glutathione S-transferase (GSTM1, T1 and P1) gene polymorphisms
with type 2 diabetes mellitus in north Indian population. J. Post. Grad. Med 2010;
56(3):176-181.
15. Brasil. Ministério da Saúde (MS). Secretaria de Atenção Básica à Saúde.
Departamento de Atenção Básica. Diabetes mellitus. Cadernos de Atenção Básica,
n. 16, 2006.
16. Brasil. Ministério da Saúde. Secretaria de Vigilância em Saúde. Secretaria de
Gestão Estratégica e Participativa. Vigitel-Brasil 2012: vigilância de fatores de risco e
proteção para doenças crônicas por inquérito telefônico. Brasília, 2012.

59
17. Brenner B, Cooper M, de Zeeuw D, et al. Effects of losartan on renal and
cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J
Med 2001;345:861-9.
18. Brownlee M. Biochemistry and molecular cell biology of diabetic complications.
Nature 2001; 414:813–820.
19. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism.
Diabetes 2005; 54(6):1615-1625.
20. Cercato C, Mancini MC, Arguello AMC, Passos VQ, Villares SMF, Halpern A.
Systemic Hypertension, Diabetes Mellitus, And Dyslipidemia in relation to Body Mass
Index: evaluation of a brazilian population, Rev. Hosp. Clín. Fac. Med. S. Paulo
2004; 59(3): 113-118.
21. Ceriello A, Quagliaro L, Catone B, Pascon R, Piazzola M, Bais B, Marra G,
Tonutti L, Taboga C, Motz E. Role of hyperglycemia in nitrotyrosine postprandial
generation. Diabetes Care 2002; 25:1439–43.
22. Chimienti F, Favier A, Seve M. ZnT-8, a pancreatic beta-cell-specific zinc
transporter. Biometals. 2005 Aug;18(4):313-7.
23. Chimienti F, Devergnas S, Pattou F, Schuit F, Garcia-Cuenca R, Vandewalle B,
Kerr-Conte J, Van Lommel L, Grunwald D, Favier A, Seve M. In vivo expression and
functional characterization of the zinc transporter ZnT8 in glucose-induced
insulinsecretion. J Cell Sci. 2006 Oct 15;119 (Pt 20):4199-206.
24. Cossarizza A, Ferraresi R, Troiano L, Roat E, Gibellini L, Bertoncelli L, Nasi M,
Pinti M. Simultaneous analysis of reactive oxygen species and reduced glutathione
content in living cells by polychromatic flow cytometry. Nat. Protoc. 2009; 4:1790–
1797.

60
25. Crowther CA, Hiller JE, Moss JR, et al. Effect of treatment of gestational diabetes
mellitus on pregnancy outcomes. N Engl J Med 2005; 352: 2477–2486.
26. Curtis JM, Grimsrud PA, Wright WS, Xu X, Foncea RE, Graham DW, et al..
Downregulation of adipose glutathione S-transferase A4 leads to increased protein
carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes. 2010 May;
59(5):1132-42.doi: 10.2337/db09-1105.
27. Dabhi B, Mistry KN, Patel H, Lal S. Vascular endotelial growth fator
insertion/deletion gene polymorphism in West Indian patients of type 2 diabetes and
diabtetic nephropathy. Indian J Biochem Biophys 2015; Apr, 52(2):209-12.
28. Das J, Sil PC. Taurine ameliorates alloxan-induced diabetic renal injury, oxidative
stress-related signaling pathways and apoptosis in rats. Amino Acids 2012;
43:1509–1523.
29. Das S, Alagappan VK, Bagchi D, Sharma HS, Maulik N, Das DK. Coordinated
induction of iNOS-VEGF-KDR-eNOS after resveratrol consumption: a potential
mechanism for resveratrol preconditioning of the heart. Vasc Pharmacol
2005;42:281–289.
30. Datta SK, Kumar V, Ahmed RS, Tripathi AK, Kalra OP, Banerjee BD. Role of
GSTM1 and GSTT1 double deletion in the development of diabetic nephropathy.
Proceedings of International Conference on Advances in Free Radical Research and
8th Annual meeting of the Society for Free Radical Research-India 2009; 87-88.
31. Davi G, Ciabattoni G, Consoli A, Mezzetti A, Falco A, Santarone S, Pennese
E, Vitacolonna E, Bucciarelli T, Costantini F, Capani F, Patrono C. In vivo formation
of 8-iso-prostaglandin-2-alpha and platelet activation in diabetes mellitus: effects of
improved metabolic control and vitamin E supplementation. Circulation 1999;99:224–
9.

61
32. DeRubertis FR, Craven PA, Melhem MF, Salah EM. Attenuation of renal injury in
db/db mice over expressing super oxide dismutase: evidence for reduced
superoxide-nitric oxide interaction.Diabetes 2004;53(3):762-8.
33. Diretrizes da Sociedade Brasileira de Diabetes. Sociedade brasileira de diabetes.
Ed. 3ª. Itapevi: A. Araújo Silva Farmacêutica, 2009.
34. Diretrizes da Sociedade Brasileira de Diabetes: 2014-2015/Sociedade Brasileira
de Diabetes; [organização José Egidio Paulo de Oliveira, Sérgio Vencio]. – São
Paulo: AC Farmacêutica, 2015.
35. Doi T, Mima A, Matsubara T, Tominaga T, Arai H, Abe H. The current clinical
problems for early phase of diabetic nephropathy and approach for pathogenesis of
diabetic nephropathy. Diabetes Res Clin Pract. 2008; 82(Suppl 1):S21–S24.
36. Doney AS, Lee S, Leese GP, Morris AD, Palmer CN. Increased cardiovascular
morbidity and mortality in type 2 diabetes is associated with the glutathione S
transferase theta-null genotype: A Go-DARTS study. Circulation 2005; 111(22):2927-
2934.
37. Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, Wu J,
Brownlee M. Hyperglycemia-induced mitochondrial superoxide overproduction
activates the hexosamine pathway and induces plasminogen activator inhibitor-1
expression by increasing Sp1 glycosylation. Proc Natl Acad Sci 2000;97(22):122-6.
38. Dunlop, M. Aldose reductase and the role of the polyol pathway in diabetic
nephropathy. Kidney Int 2000; 77:3–12.
39. Duran-Salgado MB, Rubio-Guerra AF. Diabetic nephropathy and inflammation.
World J Diabetes 2014; 5: 393-398.
40. Eid AA, Gorin Y, Fagg BM, Maalouf R, Barnes JL, Block K, Abboud HE.
Mechanisms of podocyte injury in diabetes: role of cytochrome P450 and NADPH
oxidases. Diabetes 2009;58(5):1201-11.

62
41. Eizirik DL, Flodstöm M, Karlsen AE, Welsh N. The harmony of the spheres:
inducible nitric oxide synthase and related genes in pancreatic beta cells.
Diabetologia 1996; 39:875–890.
42. El-Benna J. Dang PM, Gougerot-Pocidalo MA, Elbim C. Phagocyte NADPH
oxidase: a multicomponent enzyme essential for host defenses. Arch Immunol Ther
Exp (Warsz), Warszawa, 2005; 53:199-206.
43. Etoh T, Inoguchi T, Kakimoto M, Sonoda N, Kobayashi K, Kuroda J, Sumimoto H,
Nawata H. Increased expression of NAD(P)H oxidase subunits, NOX4 and
p22phox,in the kidney of streptozotocin-induced diabetic rats and its reversibity by
interventive insulin treatment. Diabetologia 2003;46(10): 1428-37.
44. Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative Stress and Stress-
Activated Signaling Pathways: A Unifying Hypothesis of Type 2 Diabetes. Endocrine
Reviews 2002; 23:599-622.
45. Fan M, Li W, Wang L, Gu S, Dong S, Chen M, Yin H, Zhen J, Wu X, Jin J, Jiang
X, Cai J, Liu P, Zheng C. Association of SLC30A8 gene polymorphism with type 2
diabetes, evidence from 46 studies: a meta-analysis. Endocrine. 2016 Feb 1.
46. Farbstein D, Levy A. The genetics of vascular complications in Diabetes mellitus.
Cardiol Clin 2010; 28:477-496.
47. Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in
kidney disease in diabetes. Diabetes 2008; 57:1446–1454.
48. Fujita H, Haseyama T, Kayo T, Nozaki J, Wada Y, Ito S, Koizumi A. Increased
expression of glutathione S-transferase in renal proximal tubules in the early stages
of diabetes: a study of type-2 diabetes in the Akita mouse model. Exp Nephrol
2001;9:380–386.

63
49. Fujita H, Narita T, Meguro H, Shimotomai T, Kitazato H, Kagaya E, Sugasawa H,
Hanyu O, Suzuki K, Ito S. No association of glutathione Stransferase M1 gene
polymorphism with diabetic nephropathy in Japanese type 2 diabetic patients. Ren
Fail 2000; 22: 479-486.
50. Geiszt M, Kopp JB, Varnai P, Leto TL. Identification of renox, an AD(P)H oxidase
in kidney. Proc Natl Acad Sci USA 2000;97(14):8010- 4.
51. Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end
products: sparking the development of diabetic vascular injury. Circulation
2006;114(6):597-605.
52. Greco DS, Stabenfeldt GH. Endocrinology. In J. G Cunningham & B. G. Klein
(Eds.), Textbook of veterinary physiology. St. Louis, Missouri: Saunders Elsevier,
2007, 4ª ed., p. 428-464.
53. Habdous M, Siest G, Herbeth B, Vincent-Viry M, Visvikis S. Glutathione S-
transferases genetic polymorphisms and human diseases: overview of
epidemiological studies. Ann. Biol. Clin. 2004; 62(1):15-24.
54. Haller M, Gutjahr G, Kramar R, Harnoncourt F, Oberbauer R. Cost-effectiveness
analysis of renal replacement therapy in Austria. Nephrol Dial Transplant 2011; 26:
2988–2995.
55. Halliwell B. The antioxidant paradox. Lancet 2000; 355:1179-80.
56. Hung LM, Chen JK, Huang SS, Lee RS, Su MJ. Cardioprotective effect of
resveratrol, a natural antioxidant derived from grapes. Cardiovasc Res 2000;47:549–
555.
57. Hayes JD, Pulford DJ. The glutathione S- transferase supergene family:
regulation of GST and the contribuition of the isoenzymes to cancer chemoprotetcion
and drug resistence. Crit. Rev. Biochem. Mol. Biol.1995; 30:445-600.

64
58. Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev
Pharmacol Toxicol. 2005;45:51-88.
59. Hemmelgarn B, Manns B, Lloyd A, et al. Relation between kidney function,
proteinuria, and adverse outcomes. JAMA 2010; 303:423-29.
60. Henry DN, Busik JV, Brosius FC 3rd, Heilig CW. Glucose transporters control
gene expression of aldose reductase, PKC alpha, and GLUT1 in mesangial cells in
vitro. Am J Physiol. 1999 Jul;277(1 Pt 2):F97-104.
61. Hilgers KF, Mann JF. ACE inhibitors versus AT (1) receptor antagonists in
patients with chronic renal disease. J Am Soc Nephrol. 2002;13:1100-08.
62. Hillier TA, Pedula KL, Schmidt MM, et al. Childhood obesity and metabolic
imprinting: the ongoing effects of maternal hyperglycemia. Diabetes Care 2007; 30:
2287–2292.
63. Houldsworth A, Hodgkinson A, Shaw S, Millward A, Demaine AG. Polymorphic
differences in the SOD-2 gene may affect the pathogenesis of nephropathy in
patients with diabetes and diabetic complications. Gene. 2015; 569(1):41-5.
64. Huo P, Zhang D, Guan X, Mei Y, Zheng H, Feng X. Association between genetic
polymorphism of ACE & eNOS and diabetic nephropathy. Mol Biol Rep 2015; 42:27-
33.
65. Hurst R, Bao Y, Jemth P, Mannervik B, Williamson G. Phospholipid
hydroperoxide glutathione peroxidase activity of human glutathione transferases.
Biochem J. 1998 May 15;332 ( Pt 1):97-100.
66. International Diabetes Federation. IDF Diabetes Atlas Group. Update of mortality
attributable to diabetes for the IDF Diabetes Atlas: estimates for the year 2011.
Diabetes Res Clin Pract. 2013 May; 100(2): 277-279.

65
67. International Diabetes Federation. IDF Diabetes Atlas update poster, 6th
edn. Brussels, Belgium: International Diabetes Federation, 2014.
68. James EG, Steven BH, Bernadette B, Ruud MB, Felix K, Thomas GP, Andrew
GR, Gary KZ, Dolores M. Sleep Duration as a Risk Factor for Diabetes Incidence in a
Large US Sample. . SLEEP. 2007, Vol. 30.
69. Jiang B, Guo L, Li BY, Zhen JH, Song J, Peng T, Yang XD, Hu Z, Gao HQ.
70. Resveratrol attenuates early diabetic nephropathy by down-regulating glutathione
s-transferases Mu in diabetic rats. J Med Food. 2013 Jun;16(6):481-6.
71. Kalaralo OP, Datta SK, Kumar S. Genetic Basis of Diabetic Nephropathy.
Medicine Update 2010; 20:695-701.
72. KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for
Diabetes and Chronic Kidney Disease. Am J Kidney Dis 2007 Feb;49(2 Suppl
2):S12-154.
73. Kempkes M, Wiebel FA, Golka K, Heitmann P, Bolt HM. Comparative genotyping
and phenotyping of glutathione S-transferase GSTT1. Arch Toxicol. 1996;70(5):306-
9.
74. Kim JH, Moon MK, Kim SW, et al. Glutathione S-transferase M1 gene
polymorphism is associated with type 2 diabetic nephropathy. J Korean Diabetes
Assoc 2005; 29: 315-32.
75. Kiritoshi S, Nishikawa T, Sonoda K, et al. Reactive oxygen species from
mitochondria induce cyclo oxygenase-2 gene expression in human mesangial cells:
potential role in diabetic nephropathy. Diabetes 2003; 52(10):2570-7.
76. Kitada M, Kanasaki K, Koya D. Clinical therapeutic strategies for early stage of
diabetic kidney disease. World J Diabetes 2014; 5:342-356.

66
77. Koch FP, Kämmerer PW, Kämmerer P, Al-Nawas B, Brieger J. Influence of
class M1 gluthationa s-transferase (GST M1) polymorphism on GSTM1 gene
expression level and tumor size in oral squamous cell carcinoma. Oral Oncology
2010; 46:128-33.
78. Koya D, Hayashi K, Kitada M, Kashiwagi A, Kikkawa R, Haneda M. Effects of
antioxidants in diabetes induced oxidative stress in the glomeruli of diabetic rats. J
Am Soc Nephrol 2003;14(8Suppl3):S250-3.
79. Kunz R, Friedrich C, Wolbers M, et al. Meta-analysis: Effects of Monotherapy and
Combination Therapy with Inhibitors of the Renin-Angiotensin System on Proteinuria
in Renal Disease. Ann Intern Med. 2008;148:30-48.
80. Lee AY, Chung SS. Contributions of polyol pathway to oxidative stress in diabetic
cataract. FASEB J. 1999; 13:23–30.
81. Liao J, Kobayashi M, Kanamuru Y, Nakamura S, Makita Y, Funabiki K, Horikoshi
S, Tomino Y. Effects of candesartan, an angiotensin II type 1 receptor blocker, on
diabetic nephropathy in KK/Ta mice. J Nephrol. 2003 Nov-Dec;16(6):841-9.
82. Lima-Jr MM, Oliveira MNL, Granja F, Trindade ACG, De Castro Santos LEM,
Ward LS. Lack of association of GSTT1, GSTM1, GSTO1, GSTP1 and CYP1A1
polymorphisms for susceptibility and outcome in brazilian prostate cancer patients.
Folia Biologica (Praha) 2008; 54:102-108.
83. Lin YS, Hung SC, Wei YH et al. GST M1 polymorphism associates with DNA
oxidative damage and mortality among hemodialysis patients. J Am Soc Nephrol
2009; 20: 405–415.
84. Li W, James MO, McKenzie SC, Calcutt NA, Liu C, Stacpoole PW. Mitochondrion
as a novel site of dichloroacetate biotransformation by glutathione transferase zeta 1.
J Pharmacol Exp Ther. 2011 Jan;336(1):87-94.

67
85. Liu XH, Xie CG, An Y, Zhang XX, Wu WB. Meta-analysis of the association
between the rs7903146 polymorphism at the TCF7L2 locus and type 2 diabetes
mellitus susceptibility. Genet Mol Res. 2015 Dec 14;14(4):16856-62.
86. London SJ, Yuan JM, Chung FL, Gao YT, Coetzee GA, Ross RK, Yu MC.
Isothiocyanates, glutathione S-transferase M1 and T1 polymorphisms, and lung-
cancer risk: a prospective study of men in Shanghai, China. Lancet 2000; 356:724-
729.
87. Lyssenko V, Lupi R, Marchetti P, Del Guerra S, Orho-Melander M, Almgren P, et
al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type
2 diabetes. J Clin Invest 2007; 117: 2155–2163. PMID: 17671651.
88. Ma H, Yu C, Wang R. Association of ACE polymorphism and diabetic nphropathy
susceptibility. Int J Clin Exp Med 2015 Feb 15; 8(2):2962-5.
89. Maahs DM, West NA, Lawerence JM, Mayer-Davis EJ. Epidemiology of type 1
diabetes. Endocrinol Met Clin N Am. 2010; 39:481-497.
90. Machado, UF; Carpinelli AR; Zechin HG; Saad MJA. Pâncreas Endócrino. In:
Margarida de Mello Aires. (Org.). Fisiologia. Rio de Janeiro: Guanabara Koogan,
2012, 4ª ed , p. 1098 – 1114.
91. Manfredi S, Calvi D, Del Fiandra M, Botto N, Biagini A, Andreassi MG.
Glutathione s-transferase T1- and M1-null genotypes and coronary artery disease
risk in patients with type 2 diabetes mellitus. Pharmacogenomics 2009; 10:29-34.
92. Manna P, Sinha M, Sil PC. Prophylactic role of arjunolic acid in response to
streptozotocin mediated diabetic renal injury: activation of polyol pathway and
oxidative stress responsive signaling cascades. Chem. Biol. Interact. 2009; 181:297–
308.

68
93. Marín F, García N, Muñoz X, Capellà G, González CA, Agudo A, Sala N.
Simultaneous genotyping of GSTT1 and GSTM1 null polymorphisms by melting
curve analysis in presence of SYBR Green I. J Mol Diagn. 2010 May;12(3):300-4.
94. Maritim AC, Sanders RA and Watkins JB, III. Diabetes, oxidative stress, and
antioxidants: a review. J. Biochem. Mol. Toxicol. 2003;17: 24-38.
95. Marks J.B., Raskin P. Cardiovascular risk in diabetes A brief
review. Elsevier. 2000 Mar-Apr;14(2):108-15.
96. Martin PA, & Crump MH. The endocrine pancreas. In M. H. Pineda & M. P.
Dooley (Eds.), McDonald`s veterinary endocrinology and reproduction. Iowa, EUA:
Iowa State Press, 2003, 5th ed., p. 141-160.
97. Mauer SM, Steffes MW, Ellis EN, Sutherland DE, Brown DM, Goetz FC.
Structural-functional relationships in diabetic nephropathy. J Clin Invest. 1984
Oct;74(4):1143-55.
98. Mcilwaim CC, Townsend DM, Tew KD. Glutathione S-transferase
polymorphisms: cancer incidence and therapy. Oncogene 2006; 25:1639–1648.
99. McLellan AC, Thornalley PJ, Benn J, Sonksen PH: Glyoxalase system in clinical
diabetes mellitus and correlation with diabetic complications. Clin Sci (Lond) 1994;
87:21–29.
100. Meister A. Glutathione, ascorbate and cellular protection. Cancer Res 1994 Apr
1; 54:(7 Suppl):1969-75.
101. Melendez-Ramirez LY, Richards RJ, Cefalu WT. Complications of type 1
diabetes. Endocrinology and Metabolism Clinics of North America, v. 39, p. 625-40,
2010.

69
102. Meyer DJ, Coles B, Pemble SE, Gilmore KS, Fraser GM, Ketterer B. Theta, a
new class of glutathione transferases purified from rat and man. Biochem J. 1991
Mar 1;274 ( Pt 2):409-14.
103. Melhem MF, Craven PA, Liachenko J, DeRubertis FR. Alpha-lipoic acid
attenuates hyperglycemia and prevents glomerular mesangial matrix expansion in
diabetes. J Am Soc Nephrol 2002;13(1):108-16.
104. Migliaccio E, Giorgio M, Pelicci PG. Apoptosis and aging: role of p66Shc redox
protein. Antioxid. Redox Signal 2006; 8:600–608.
105. Miller NJ, Rice-Evans CA: Antioxidant activity of resveratrol in red wine. Clin
Chem 1995;41:1789.
106. Mogensen CE,Christensen CK,Vittinghus E.The stages in diabetic renal
disease. With emphasis on the stage of incipient diabetic nephropathy. Diabetes
1983;32(Suppl2):64-78.
107. Mogensen CE, Vestbo E,Poulsen PL,et al. Microalbuminuria and potential
confounders. Are view and some observations on variability of urinary albumin
excretion. Diabetes Care 1995;18(4):572 -81.
108. Montero RM, Covic A, Gnudi L, Goldsmith D. Diabetic nephropathy: What does
the future hold? Int Urol Nephrol. 2015; 48:99–113.
109. Moreira, HG. et al. Diabetes mellitus, hipertensão arterial e doença renal:
estratégias terapêuticas e suas limitações. Rev. Brasileira de Hipertensão 2008; 15
(2):111-116.
110. Murphy M, Godson C, Cannon S, Kato S, Mackenzie HS, Martin F, et al.
Suppression subtractive hybridization identifies high glucose levels as a stimulus for
expression of connective tissue growth factor and other genes in human mesangial
cells. J Biol Chem 1999;274:5830-4.

70
111. Murussi M, Coester A, Gross JL, Silveiro SP. A nefropatia diabética em diabetes
mellitus tipo 2: fatores de risco e prevenção. Arq. Bras. Endocrinol. Metab 2003 Jun;
47(3).
112. Murussi M, Murussi N, Campagnolo N, Silveiro SP. Detecção Precoce da
Nefropatia Diabética. Arq Bras Endocrinol Metab 2008; 52(3).
113. National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the
United States, 2014. Centers for Disease Control and Prevention. Atlanta, GA: U.S.
Department of Health and Human Services; 2014.
114. National Institutes of Health Consensus Development Conference Statement.
Diagnosing Gestational Diabetes Mellitus. Obstet Gynecol 2013; 122: 358–369.
115. Nelson RG, Meyer TW, Myers BD, Bennett PH. Clinical and pathological course
of renal disease in non-insulin dependent diabetes mellitus: the Pi ma Indian
experience. Semin Nephrol 1997;17(2):124-31.
116. Nebert DW, Vasiliou V. Analysis of the glutathione S-transferase (GST) gene
family. Human Genomics 2004; 1(6):460–464.
117. NIH-National Institute of Diabetes and Digestive and Kidney Disease. Your
Guide to Diabetes: Type 1 and Type 2. December 2013. Minneapolis, Minnesota.
Disponível em: http://www.niddk.nih.gov/health-information/health-
topics/Diabetes/your-guide-diabetes/Documents/YourGuide2Diabetes_508.pdf
118. Odetti P, Garibaldi S, Noberasco G, Aragno I, Valentini S, Traverso N, Marinari
UM. Levels of carbonyl groups in plasma proteins of type 2 diabetes mellitus
subjects. Acta Diabetol 1999; 36:179–83.
119. ADAO Oliveira, J E P. Conceito, Classificação e Diagnóstico do Diabetes. In:
MILECH A. (Org.). Diabetes Mellitus Clínica, Diagnóstico, Tratamento
Multidisciplinar. 1. ed. São Paulo: Atheneu, 2004. p. 7-14.

71
120. Orhan O, Atalay MA, Orhan F, Karkucak M, Centinkaya Demir B, Yakut
T, Cengiz C. Glutathione s-transferase 1 and t1 gene polymorphisms are not
associated with increased risk of gestational diabetes mellitus development. West
Indian Med J. 2014 Aug;63(4):300-6.
121. Orlewski J & Orlewska E. Effects of genetic polymorphisms of glutathione S-
transferase genes (GSTM1, GSTT1, GSTP1) on the risk of diabetic nephropathy: a
meta-analysis. Pol Arch Med Wewn. 2015;125(9):649-58. Epub 2015 Aug 7.
122. Pal, PB, Sinha K, Sil PC. Mangiferin, a natural xanthone, protects murine liver in
Pb(II) induced hepatic damage and cell death via MAP kinase, NF- B and
mitochondria dependent pathways. PLoS ONE 2013; 8, e56894.
123. Parl FF. Glutathione S-transferase genotypes and cancer risk. Cancer Letters
2005; 221:123–129.
124. Pemble S, Schroeder KR, Spencer SR, Meyer DJ, Hallier E, Bolt HM, Ketterer
B, Taylor JB. Human glutathione S-transferase theta (GSTT1): cDNA cloning and
characterization of a genetic polymorphism. Biochem J 1994; 300: 271-276.
125. Peng F, Hu D, Gu C, Li X, Li Y, Jia N, Chu S, Lin J, Niu W. The relationship
between five widelyevaluated variants in CDKN2A/B and CDKAL1 genes and the risk
of type 2 diabetes: a meta-analysis. Gene 2013; 531: 435-443.
126. Pinheiro DS, Rocha Filho CR, Mundim CA. ; Marco Junior P, Ulhoa CJ, Reis
AAS, Ghedini PC. Evaluation of Glutathione S-Transferase GSTM1 and GSTT1
Deletion Polymorphisms on Type-2 Diabetes Mellitus Risk. Plos One 2013;
8:e76262.
127. Practice Bulletin No. 137: gestational diabetes mellitus. Obstet Gynecol 2013;
122: 406–416.

72
128. Prunier C, Hocevar BA, Howe PH. Wnt signaling: physiology and pathology.
Growth Factors. 2004 Sep;22(3):141-50.
129. Rahman K. Studies on free radicals, antioxidants, and co-factors. Clin. Interv. in
Aging 2007; 2 (2):219–236.
130. Reis AAS. Associação do polimorfismo genético em carcinomas da tiróide.
[Tese Doutorado] Universidade Federal de Goiás (UFG), Goiânia, GO, 2010.
131. Reis AAS, Pinheiro DS, Mundim CA, Jesuíno RSA, Ulhoa C. J. As implicações
do polimorfismo genético do gene GST na patogênese do diabetes mellitus tipo 2.
Rev. Univ. Vale do Rio Verde 2011; 9 (2):92-100.
132. Remuzzi G, Schieppati A, Ruggenenti P. Clinical practice. Nephropathy in
patients with type 2 diabetes. N Engl J Med 2002;346(15):1145 -51.
133. Rhee EP. NADPH Oxidase 4 at the Nexus of Diabetes, Reactive Oxygen
Species, and Renal Metabolism. J Am Soc Nephrol 2015 Jul:ASN 2015060698.
134. Rigalleau V, Garcia M, Lasseur C, et al. Large kidneys predict poor renal
outcome in subjects with diabetes and chronic kid- ney disease. BMC Nephrol
2010;11:3.
135. Robertson RP, Harmon J, Tran PO, Tanaka Y, Takahashi H. Glucose toxicity in
[beta]-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection.
Diabetes 2003; 52:581–587.
136. Santini SA, Marra G, Giardina B, Cotroneo P, Mordente A, Martorana GE,
Manto A, Ghirlanda G. Defective plasma antioxidant defenses and enhanced
susceptibility to lipid peroxidation in uncomplicated IDDM.
Diabetes.1997;46(11):1853-8.

73
137. Sanz C., Gautier J. F., Hanaire H. Physical exercise for the prevention and
treatment of type 2 diabetes. Diabetes Metab. 2010 Nov;36(5):346-51.
138. Sesso, Ricardo Cintra et al. Inquérito Brasileiro de Diálise Crônica 2013 -
Análise das tendências entre 2011 e 2013. J. Bras. Nefrol. 2014 Dec; 36(4): 476-
481.
139. Sociedade Brasileira de Diabetes (SBD). Consenso Brasileiro sobre Diabetes.
São Paulo (SP); 2001; p.50.
140. Sociedade Brasileira de Diabetes (SBD). Diretrizes da Sociedade Brasileira de
Diabetes: 2013-2014. São Paulo: AC Farmacêutica, 2014.
141. Sociedade Brasileira de Endocrinologia e Metabologia (SBEM). Oliveira R.
Complicações Crônicas do Diabetes Mellitus; 2015.
142. Schwarz P. E., Muylle F., Valensi P., Hall M. The European Perspective of
Diabetes Prevention. Hormone and Metabolic Research 2008 Aug;40(8):511-4.
143. Seidegard J, Vorachek WR, Pero RW, Pearson WR. Hereditary differences in
the expression of human glutathione transferase activity on trans-stilbene oxide are
due to a gene deletion. Proc Natl Acad Sci USA 1998; 85: 7293-7297.
144. Sheldon C, Khury C, Ziyadeh FN. Pathophysiology and Patogenesis of Diabetic
Nephropathy. In: Seldin and Giebich. The Kidney. Edição 5th. Academic Press 2013.
p. 2605-2632.
145. Sies H. Strategies of antioxidant defense. Europ. J. Biochem 1993; 215:213-19.
146. Simic T, Savic-Radojevic A, Pljesa-Ercegovac M, Matic M, Mimic-Oka J.
Glutathione S-transferases in kidney and urinary bladder tumors. Nat Rev Urol 2009;
6: 281–289.

74
147. Sinclair AJ, Paolisso G, Castro M, Bourdel-Marchasson I, Gadsby R, Rodriguez
Mañas L. European Diabetes Working Party for Older People 2011 clinical guidelines
for type 2 diabetes mellitus. Executive summary. Diabetes Metab 2011 Nov;37 Suppl
3:S27-38.
148. Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria-
dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol 2013
Jul;87(7):1157-80.
149. Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D et al. A genome-wide
association study identifies novel risk loci for type 2 diabetes. Nature 2007;445:881–
885.
150. Sprenger R, Schlagenhaufer R, Kerb R, Bruhn C, Brockmöller J, Roots I,
Brinkmann U. Characterization of the glutathione S-transferase GSTT1 deletion:
discrimination of all genotypes by polymerase chain reaction indicates a trimodular
genotype-phenotype correlation. Pharmacogenetics 2000; 10 (6):557-65.
151. Stevens LA, Coreseh J, Greene T, Levey AS. Assessing Kidney function
measured and estimated glomerular filtration rate. N Engl J Med 2006 Jun
8;354(23):2473-83.
152. Strange RC, Spiteri MA, Ramachandran S, Fryer AA. Glutathione S-transferase
family of enzymes. Mut Res / Fundamental and molecular mechanisms of
mutagenesis 2001; 482: 21-26.
153. Suvakov S, Damjanovic T, Stefanovic A, Pekmezovic T, Savic-Radojevic A,
Marija Pljesa-Ercegovac A, et al. Glutathione S-transferase A1, M1, P1 and T1 null or
low-activity genotypes are associated with enhanced oxidative damage among
haemodialysis patients. Nephrol Dial Transplant 2012; 0: 1–10.
154. Taylor KD, Norris JM, Rotter JI. Genome-wide association: which do you want
first: the good news, the bad news, or the good news?. Diabetes 2007; 56:2844-
2848.

75
155. Telci A, Cakatay U, Kayali R, Erdogän C, Orhan Y, Sivas A, Akçay T. Oxidative
protein damage in plasma of type 2 diabetic patients. Horm. Metab. Res 2000; 32:
40-3.
156. Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme
gene expression and antioxidative defense status of insulin producing cells. Diabetes
1997; 46:1733-42.
157. Tuerxunyiming M, Mohemaiti P, Wufuer H, Tuheti A. Association of rs7754840
G/C polymorphisms in CDKAL1 with type 2 diabetes: a meta-analysis of 70141
subjects. Int J Clin Exp Med. 2015 Oct 15;8(10):17392-405.
158. Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol
2003; 552:335–344.
159. Tuttle KR, Bakris GL, Bilous RW, Chiang JL, de Boer IH, Goldstein-Fuchs
J, Hirsch IB, Kalantar-Zadeh K, Narva AS, Navaneethan SD, Neumiller JJ, Patel
UD, Ratner RE, Whaley-Connell AT, Molitch ME. Diabetic kidney disease:
a report from an ADA Consensus Conference. Am J Kidney Dis. 2014 Oct;64(4):510-
33.
160. Ukkola o, Erkkila PH, Savolainen MJ, Kesaniemi YA. Lack of association
between polymorphisms of catalase, copper-zinc superoxide dismutase (SOD),
extracelular SOD and endotelial nitric oxide synthase genes and macroangiopathy in
patients with type 2 diabetes mellitus. J Intern Med 2001 May;249(5):451-9.
161. Vojtková J, Durdík P, Ciljaková M, Michnová Z, Turcan T, Babusíková E. The
association between gene polymorphisms of glutathione S- transferase T1/M1 and
type 1 diabetes in Slovakchildren and adolescents. Cent Eur J Public Health. 2013
Jun;21(2):88-91.

76
162. Voso MT, D'Alo' F, Putzulu R, Mele L, Scardocci A, Chiusolo P, Latagliata R,
Lo-Coco F, Rutella S, Pagano L, Hohaus S, Leone G. Negative prognostic value of
glutathione S-transferase (GSTM1 and GSTT1) deletions in adult acute
myeloidleukemia. Blood. 2002 Oct 15;100(8):2703-7.
163. Xu S, Wang Y, Roe B, Pearson WR. Characterization of the Human Class Mu
Glutathione S-Transferase gene cluster and the GSTM1 Deletion. J Biol Chem 1998;
273:3517–3527.
164. Wang G, Zhang L, Li, Q. Genetic polymorphisms of GSTT1, GSTM1, and NQO1
genes and diabetes mellitus risk in Chinese population. Biochem. Biophys. Res.
Commun 2006; 341:310-313.
165. Wei JN, Li HY, Sung FC, et al. Birth weight correlates differently with
cardiovascular risk factors in youth. Obesity (Silver Spring) 2007; 15: 1609–1616.
166. Whittington A, Vichai V, Webb G, Baker R, Pearson W, Board P. Gene
structure, expression and chromosomal localization of murine theta class glutathione
transferase mGSTT1-1. Biochem J. 1999 Jan 1;337 ( Pt 1):141-51.
167. Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS, Pinsky D, Stern
D. Enhanced cellular oxidant stress by the interaction of advanced glycation end
products with their receptors/binding proteins. J Biol Chem. 1994;269(13):9889-97.
168. Yang Y, Kao M, Chang C, et al. Glutathione S-transferase T1 deletion is a risk
factor for developing end-stage renal disease in diabetic patients. Intern J Mol Med
2004; 14: 855-859.
169. Yamagishi S, Matsui T. Advanced glycation end products, oxidative stress and
diabetic nephropathy. Oxid Med Cell Longev 2010; 3(2):101–108 35.
170. Zhang YF, Cheng Q, Tang NL, Chu TT, Tomlinson B, Liu F, Kwok TC. Gender
difference of serum angiotensin-converting enzyme (ACE) activity in DD genotype of

77
ACE insertion/deletion polymorphism in elderly Chinese. J Renin Angiotensin
Aldosterone Syst 2014 Dec;15(4):547-52.
171. Zheng F, Cornacchia F, SchulmanI, et al. Development of albuminuria and
glomerular lesions in normoglycemic B6 recipients of db/db mice boné marrow: the
role of mesangial cell progenitors. Diabetes 2004;53(9):2420-7.
172. Zhong SL, Zhou SF, Chen X, et al. Relationship between genotype and enzyme
activity of glutathione S-transferases M1 and P1 in Chinese. European J Pharm Sci
2006; 28: 77-85.
173. Zhou TB, Drummen GP, Jiang ZP, Li HY. Methylenetetrahydrofolate reductase
(MTHFR) C677T gene polymorphism and diabetic nephropathy susceptibility in
patients with type 2 diabetes mellitus. Ren Fail. 2015 Jul 10:1-13.

78
ANEXO I – Termo de Consentimento Livre e Esclarecido
TERMO DE CONSENTIMENTO LIVRE E ESCLARECIDO
Você está sendo convidado (a) para participar, como voluntário, em pesquisa.
Após ser esclarecido (a) sobre as informações a seguir e no caso de aceitar fazer parte do
estudo, assine ao final deste documento. Você deverá assinar duas vias, uma delas é sua e
a outra é do pesquisador responsável. Em caso de dúvidas sobre a pesquisa, você poderá
entrar em contato com o pesquisador responsável, Profª Drª Angela Adamski da Silva Reis
no telefone (62) 3521 1078.
INFORMAÇOES IMPORTANTES QUE VOCÊ DEVE SABER SOBRE A PESQUISA
A pesquisa é intitulada “Avaliação clínica e molecular do polimorfismo genético
na susceptibilidade às complicações do Diabetes mellitus" e tem como objetivo o
estudo da susceptibilidade genética às complicações do diabetes: nefropatia (insuficiência
renal), retinopatia (perda da visão) e cardiopatia (doenças cardiovasculares) comparado ao
grupo controle (pessoas saudáveis). O presente documento lhe será aplicado por um
pesquisador. Caso concorde em participar somente uma amostra de sangue será coletada
por integrantes da pesquisa. Desta forma a pesquisa não lhe causará nenhum prejuízo à
sua saúde. Todo material perfuro-cortante (Seringa + agulha) é descartável sem nenhum
risco de transmissibilidade de doenças infecto-contagiosa.
Não haverá nenhum tipo de despesa para participar desta pesquisa, bem como
nada será pago por sua participação. Ao participar desta pesquisa você não terá nenhum
benefício direto. Entretanto, esperamos que este estudo traga informações importantes
sobre às complicações do Diabetes, de forma que o conhecimento que será construído a
partir desta pesquisa possa auxiliar no diagnóstico precoce de outros pacientes. Sua
participação será limitada ao uso da amostra obtida durante a coleta de sangue, sendo que
os resultados lhe serão informados caso o voluntário solicite ao laboratório por meio do
telefone de contato já citado.
Todas as informações coletadas neste estudo são estritamente confidenciais.
Somente os pesquisadores terão conhecimento dos dados. Você tem liberdade de se
recusar a continuar participando em qualquer fase da pesquisa, sem qualquer prejuízo. Sua
amostra e dados clínicos serão utilizados apenas para este estudo e não serão
armazenados para futuros estudos.
Profª Drª Angela Adamski da Silva Reis Coordenadora da Pesquisa

79
TERMO DE CONSENTIMENTO LIVRE E ESCLARECIDO INFORMADO PARA INVESTIGAÇÃO
Dados de identificação ( ) Paciente ( ) Controle
Título do Projeto: Avaliação clínica e molecular do polimorfismo genético na susceptibilidade às complicações do diabetes mellitus Pesquisadora Responsável: Profa. Dra. Angela Adamski da Silva Reis Nome do voluntário: _______________________________________________
Telefone: ____________________________ Celular: ____________________
Idade: _______ anos R.G. _______________ Org. Exp. __________ Responsável legal (quando for o caso): _______________________________ Grau de parentesco: ____________________________________ R.G. Responsável legal: _________________________ Org. Exp. __________ 1. Este projeto tem o objetivo de avaliar clínica e molecularmente a susceptibilidade genética
nas complicações do Diabetes mellitus, nefropatia diabética.
2. Para tanto será necessário realizar os seguintes procedimentos:
- responder um questionário sobre seus hábitos de vida (com entrevistador);
- doação de 10 mL de amostra de sangue periférico (coletada por especialista)
3. Os procedimentos da pesquisa oferecem risco mínimo de ocorrência de algum dano
imediato ou tardio para o paciente (poderá ocorrer hematomas locais após a coleta de
sangue).
4. Sua amostra de sangue será testada com testes de DNA para avaliação a
susceptibilidade ao diabetes mellitus e/ou suas complicações clínicas.
5. A participação no estudo não acarretará custos para você e não será disponível nenhuma
compensação financeira.
6. Você será esclarecido (a) sobre a pesquisa em qualquer aspecto que desejar. Você é
livre para recusar-se a participar, retirar seu consentimento ou interromper a participação a
qualquer momento. A sua participação é voluntária e a recusa em participar não irá acarretar
qualquer penalidade ou perda em seu atendimento.
O(s) pesquisador(es) irá(ão) tratar a sua identidade com padrões profissionais de
sigilo e as informações colhidas serão utilizadas somente para fins científicos. Os resultados
da pesquisa serão entregues para você e permanecerão confidenciais. Seu nome ou o
material que indique a sua participação não será liberado sem a sua permissão. Você não
será identificado(a) em nenhuma publicação que possa resultar deste estudo.

80
Eu,_________________________________________________________________
_, RG nº _____________________ Org. Exp. __________ declaro ter sido
suficientemente esclarecido sobre os propósitos do estudo, os procedimentos a
serem realizados, seus desconfortos e riscos, as garantias de confidencialidade e de
esclarecimentos permanentes. Estou ciente de que todas as informações prestadas
durante a entrevista serão de caráter confidencial e os dados colhidos serão
utilizados somente para fins científicos. Concordo voluntariamente em participar
desta pesquisa, sendo que poderei retirar o meu consentimento a qualquer
momento, antes ou durante o mesmo, sem prejuízo ou perda de qualquer benefício
que eu possa ter adquirido durante o atendimento neste Serviço.
Ou,
Eu,_________________________________________________________________
_, RG nº__________________ Org. Exp. __________, responsável legal
por_____________________________________________________________
_____________________________________, RG nº ___________________, Org.
Exp. __________ declaro ter sido informado e concordo com a participação, como
voluntário, no projeto de pesquisa acima descrito.
Goiânia, _____ de ____________ de _______
___________________________________________________ Nome e assinatura do paciente ou seu responsável legal
______________________________________________________ Nome e assinatura do responsável por obter o consentimento
Assinatura Dactiloscópica :

81
ANEXO II – Parecer do Comitê de Ética Institucional

82

83
ANEXO III - Formulário de Preenchimento de Dados de Pacientes
Avaliação clínica e molecular, estilo de vida e hábitos alimentares de pacientes
com nefropatia diabética.
ENTREVISTA
INICIAIS: ________________ Data ________________ Nº ___________ DADOS PESSOAIS:
Nome: _______________________________________________________________ Data de nascimento:____/____/____ Idade: _____ Sexo:______________ Documento de Identidade:_____________________ Org. Exp. ___________ CPF: ________________ Profissão: _______________________________ Estado Civil: ______________________ Endereço:
_______________________________________________________________
Bairro: _____________________ Cidade: _____________ Estado: _______
CEP: _______________ Telefone: _______________ Celular: ____________ Naturalidade: _______________________ Nacionalidade: _______________ DADOS CLÍNICOS: Pressão Arterial: __________________ Classificação: _________________
Valores de Referência
Sistólica mmHg Diastólica mmHg
Ideal <120 <80
Alterada <130 <85
Hipertensão acima de 140 acima de 90 Anotações relevantes:
___________________________________________________________________
___________________________________________________________________
_______________________________________________________

84
Exames Clínicos: (obtidos pelo médico colaborador)
Exames clínicos Resultados Valores Referência
Glicose 70 - 99 mg/dL Triglicérides até 150 mg/dL
Colesterol desejável até 200 mg/dL
limítrofe 200 - 239
elevado acima 230 mg / dL HDL Acima de 50mg/dL
LDL inferior a 130 mg /dL
limítrofe 130-160 mg / dL
elevado acima 160 mg/dL
VLDL 2 a 35 mg/dL Hemoglobina Glicada 4.0 a 6.0 %
TGO Masculino: até 37 U/L
Feminino: até 31 U/L
TGP Masculino: até 41 U/L
Feminino: até 31 U/L Creatinina Masculino: 0,7 a 1,3 mg/dL
Feminino: 0,6 a 1,1 mg/dL Peso Corporal : ______________ Classificação IMC: __________________ Cálculo de Índice de Massa Corporal: IMC = __peso (Kg) altura 2
Questões a serem questionadas ao paciente (feitas pelo entrevistador)
1. Há quanto tempo é diabético? ______________, ano do diagnóstico_______
2. Possui familiares com diabetes?
( ) Não ( ) Sim, Qual grau de parentesco? ________________________
Observação: ______________________________________________________________________________________________________________________________
2. Uso de medicamentos
Para pressão arterial: ( ) Não ( ) Sim, qual? ____________________
Quem receitou o medicamento? (especialidade médico)___________________
Há quanto tempo utiliza? ___________________________________________
IMC Classificação Obesidade (grau)
Menor que 18,5
Magreza
0
Entre 18,5 e 24,9
Normal
0
Entre 25,0 e 29,9
Sobrepeso
I
Entre 30,0 e 39,9
Obesidade
II
Maior que 40,0
Obesidade Grave
III

85
Quais outros medicamentos utiliza?
( ) Analgésicos ( ) antibióticos ( ) anti-inflamatórios ( ) outro -
Qual?______________________
Para qual indicação clínica? Por quanto tempo?
___________________________________________________________________
___________________________________________________________
3. É fumante? ( ) Sim ( ) Não Se sim, por quanto tempo? _________
Já fumou?
( ) Sim ( ) Não
Se sim, por quanto tempo fumou?______________
Há quanto tempo parou de fumar?____________
4. Faz uso de bebida alcoólica? ( ) Sim ( ) Não Quanto tempo? ____________
Se sim, qual a frequência? ( )Diariamente ( )Socialmente ( ) Ocasionalmente
5. Possuía alguma doença antes do diagnóstico de diabetes? ( ) Sim ( ) Não Qual (is)? ______________________________ 6. Consumo de água. Quantos copos bebe de água diariamente? ___________
Possui alguma disfunção renal? ( ) sim ( ) não
Qual? ____________________ Há quanto tempo? ______________________
7.Utiliza algum sapato especial?
( ) sim ( ) não
Se sim, qual motivo? ______________________________________________
8. Hábitos alimentares.
Foi indicado a fazer dieta alimentar pelo médico? ( ) sim ( ) não Achou dificuldade em fazer a dieta alimentar ? ( ) sim ( ) não
por quê?________________________________________________________
9. Atividade física
Fazia atividade física antes do diagnóstico de diabetes? ( ) sim ( ) não
Qual? _____________________ Há quanto tempo? _____________________
Faz atividade física atualmente? ( ) sim ( ) não

86
Qual? __________________ Periodicidade? ___________________________
Só começou a fazer atividade física depois do diagnóstico? ( ) sim ( ) não
Foi recomendado pelo médico? ( ) sim ( ) não
Anotações Relevantes:
___________________________________________________________________
___________________________________________________________________
_______________________________________________________
_______________________________________________________________
___________________________________________________________________
___________________________________________________________________
_______________________________________________________
_______________________________________________________________
Data, ___________________ Local de Coleta:__________________________
Nome do Responsável pelo preenchimento do questionário e assinatura:
_______________________________________________________________
DADOS MOLECULARES (Análise no laboratório)
Polimorfismo Genótipo PCR em tempo real
GSTM1
GSTT1

87
ANEXO IV - Formulário de Preenchimento de Dados de Controles
Avaliação clínica e molecular, estilo de vida e hábitos alimentares de
indivíduos do grupo controle.
ENTREVISTA
INICIAIS: ________________ Data ________________ Nº ___________ DADOS PESSOAIS:
Nome: _______________________________________________________________ Data de nascimento:____/____/____ Idade: _____ Sexo:______________ Documento de Identidade:_____________________ Org. Exp. ___________ CPF: ________________ Profissão: _______________________________ Estado Civil: ______________________ Endereço:
_______________________________________________________________
Bairro: _____________________ Cidade: _____________ Estado: _______
CEP: _______________ Telefone: _______________ Celular: ____________ Naturalidade: _______________________ Nacionalidade: _______________ DADOS CLÍNICOS: Pressão Arterial: __________________ Classificação: _________________
Valores de Referência
Sistólica mmHg Diastólica mmHg
Ideal <120 <80
Alterada <130 <85
Hipertensão acima de 140 acima de 90

88
Exames Clínicos: (obtidos pelo médico colaborador)
Exames clínicos Resultados Valores Referência
Glicose 70 - 99 mg/dL Triglicérides até 150 mg/dL
Colesterol desejável até 200 mg/dL
limítrofe 200 - 239
elevado acima 230 mg / dL HDL Acima de 50mg/dL
LDL inferior a 130 mg /dL
limítrofe 130-160 mg / dL
elevado acima 160 mg/dL
VLDL 2 a 35 mg/dL Hemoglobina Glicada 4.0 a 6.0 %
TGO Masculino: até 37 U/L
Feminino: até 31 U/L
TGP Masculino: até 41 U/L
Feminino: até 31 U/L Creatinina Masculino: 0,7 a 1,3 mg/dL
Feminino: 0,6 a 1,1 mg/dL Peso Corporal : ______________ Classificação IMC: __________________ Cálculo de Índice de Massa Corporal: IMC = __peso (Kg) altura 2
Questões a serem questionadas ao paciente (feitas pelo entrevistador)
1. Há quanto tempo é diabético? ______________, ano do diagnóstico_______
2. Possui familiares com diabetes?
( ) Não ( ) Sim, Qual grau de parentesco? ________________________
Observação: ______________________________________________________________________________________________________________________________
2. Uso de medicamentos
Para pressão arterial: ( ) Não ( ) Sim, qual? ____________________
Quem receitou o medicamento? (especialidade médico)___________________
Há quanto tempo utiliza? ___________________________________________
IMC Classificação Obesidade (grau)
Menor que 18,5
Magreza
0
Entre 18,5 e 24,9
Normal
0
Entre 25,0 e 29,9
Sobrepeso
I
Entre 30,0 e 39,9
Obesidade
II
Maior que 40,0
Obesidade Grave
III

89
Quais outros medicamentos utiliza?
( ) Analgésicos ( ) antibióticos ( ) anti-inflamatórios ( ) outro -
Qual?______________________
Para qual indicação clínica? Por quanto tempo?
___________________________________________________________________
___________________________________________________________
3. É fumante? ( ) Sim ( ) Não Se sim, por quanto tempo? _________
Já fumou?
( ) Sim ( ) Não
Se sim, por quanto tempo fumou?______________
Há quanto tempo parou de fumar?____________
4. Faz uso de bebida alcoólica? ( ) Sim ( ) Não Quanto tempo? ____________
Se sim, qual a frequência? ( )Diariamente ( )Socialmente ( ) Ocasionalmente
5. Possuía alguma doença antes do diagnóstico de diabetes? ( ) Sim ( ) Não Qual (is)? ______________________________ 6. Consumo de água. Quantos copos bebe de água diariamente? ___________
Possui alguma disfunção renal? ( ) sim ( ) não
Qual? ____________________ Há quanto tempo? ______________________
7.Utiliza algum sapato especial?
( ) sim ( ) não
Se sim, qual motivo? ______________________________________________
8. Hábitos alimentares.
Consumo Quantidade Tipo Período
Doces
Carnes
Legumes e verduras
Frutas
Carboidratos e derivados
Outros (quais?)
Foi indicado a fazer dieta alimentar pelo médico? ( ) sim ( ) não Achou dificuldade em fazer a dieta alimentar ? ( ) sim ( ) não
por quê?________________________________________________________
Período: todos os dias =1 ; 1 a 3 vezes semana =2 4 a 6 vezes semana = 3 não consome = 4

90
9. Atividade física
Fazia atividade física antes do diagnóstico de diabetes? ( ) sim ( ) não
Qual? _____________________ Há quanto tempo? _____________________
Faz atividade física atualmente? ( ) sim ( ) não
Qual? __________________ Periodicidade? ___________________________
Só começou a fazer atividade física depois do diagnóstico? ( ) sim ( ) não
Foi recomendado pelo médico? ( ) sim ( ) não
Anotações Relevantes:
___________________________________________________________________
___________________________________________________________________
_______________________________________________________
_______________________________________________________________
___________________________________________________________________
___________________________________________________________________
_______________________________________________________
_______________________________________________________________
Data, ___________________ Local de Coleta:__________________________
Nome do Responsável pelo preenchimento do questionário e assinatura:
_______________________________________________________________
DADOS MOLECULARES (Análise no laboratório)
Polimorfismo Genótipo PCR em tempo real
GSTM1
GSTT1

91
ANEXO V – Carta de submissão do artigo em revista qualis A1





























