Embed Size (px)
Citation preview

UNIVERSIDADE FEDERAL DE SERGIPE
PRÓ-REITORIA DE PÓS-GRADUAÇÃO E PESQUISA
NÚCLEO DE PÓS-GRADUAÇÃO EM ENGENHARIA
QUÍMICA (PEQ-UFS)
ALEXANDRE CALDEIRA SILVA
CRAQUEAMENTO CATALÍTICO DE n-HEPTANO SOBRE
ZEÓLITAS ZSM-12 COM DIFERENTES RAZÕES SiO2/Al2O3
VISANDO OBTER ADITIVOS DE BAIXO CUSTO PARA
CATALISADORES DE FCC
São Cristóvão (SE)
2009

ALEXANDRE CALDEIRA SILVA
CRAQUEAMENTO CATALÍTICO DE n-HEPTANO SOBRE
ZEÓLITAS ZSM-12 COM DIFERENTES RAZÕES SiO2/Al2O3
VISANDO OBTER ADITIVOS DE BAIXO CUSTO PARA
CATALISADORES DE FCC
Dissertação de Mestrado
apresentada ao Programa de
Pós-Graduação em Engenharia
Química da Universidade
Federal de Sergipe, como
requisito parcial à obtenção do
título de Mestre em Engenharia
Química.
ORIENTADOR: Prof. Dr. Marcelo José Barros de souza
São Cristóvão (SE)
2009

“Dedico esta dissertação à minha querida, doce e
compreensível esposa Rosângela, aos meus pais,
Maria Caldeira e Zito, fontes de inspiração e
exemplos de honestidade, carinho e disposição, e à
minha filha Sarah, sol da minha vida”.

AGRADECIMENTOS
A Deus, que me deu forças, dia após dia, e proporcionou todas as condições que me
permitiram chegar até aqui.
Ao Prof. Dr. Marcelo José Barros de Souza, professor e orientador, pela sua
dedicação, simplicidade, entusiasmo e competência nas suas orientações, sempre me
indicando os caminhos e possibilidades na realização deste trabalho.
Ao laboratório de Catálise da Universidade Federal de Sergipe (LABCAT) e ao
Laboratório de Catálise e Petroquímica (LCP) da Universidade Federal do Rio Grande do
Norte pela realização dos experimentos deste trabalho.
A todos os professores do Núcleo de Pós-Graduação em Engenharia Química
(PEQ/UFS), pelos momentos felizes no compartilhamento do conhecimento.
Aos alunos da primeira turma do curso de mestrado, pela amizade e apoio em todas
as horas, foi uma grande satisfação compartilhar da companhia de todos vocês.
A todos os colegas da Petrobras/UN-SEAL, especialmente, Peixoto, Rômulo, Joel,
Felipe, Jorge, Paulo, Cleriston e Erenildo, sempre presentes, que contribuíram com idéias
e sugestões valiosas durante esta campanha.
A todos, indistintamente, que contribuíram para a realização desta dissertação.

RESUMO
O Petróleo é de vital importância para a sociedade devido aos seus derivados com
largo emprego nas atividades econômicas e principalmente sua utilização como fonte
energética. A expansão econômica e social do país, nas últimas décadas, vem exigindo
importante desenvolvimento da nossa infra-estrutura, notadamente no setor de energia. O
petróleo que se posiciona como a fonte de energia líder mundial, apresentou acentuada
oscilação do preço médio internacional ao longo dos últimos anos, atingindo no segundo
semestre do ano de 2008 o maior preço já praticado desde os primórdios da indústria
petrolífera. Neste cenário adverso, melhorias no processo de refino do petróleo
representam ganhos em larga escala para sociedade. O presente trabalho de pesquisa teve
como finalidade avaliar as características de eficiência de catálise de zeólitas ZSM-12
(Zeolite Socony Mobil) para serem utilizadas como aditivos de baixo custo para produção
de catalisadores heterogêneos microporosos para a etapa de craqueamento catalítico
(FCC), possibilitando futuras reduções no custo de refino do petróleo. As zeólitas são
aluminossilicatos cristalinos, que, na sua forma ácida, desempenham o papel de principal
componente ativo dos catalisadores de FCC. A zeólita ZSM-12 apresenta uma estrutura
formada por poros unidimensionais de abertura elíptica contendo 12 tetraedros SiO4 (ou
AlO4), com diâmetro de 5,5 x 5,9 Å. Nos experimentos foram utilizadas amostras de ZSM-
12 de razões SiO2/Al2O3 de 50, 80, 100 e 150, e foi possível observar que quanto maior a
cristalinidade da amostra (verificada por difração de raios-X) maior a conversão durante o
craqueamento. A região de máximo de conversão ocorre aproximadamente na razão
SiO2/Al2O3 de 80 a 100 e intervalos de tempo entre 0 e 20 minutos. Os resultados
mostraram-se bastante promissores para as razões SiO2/Al2O3 avaliadas, apresentando alta
conversão e seletividade (principalmente ao C1 e C3), apontando a ZSM-12 como
potencial aditivo zeolítico de baixo custo para catalisadores de FCC.
Palavras-chaves: Catalisadores Zeolíticos, FCC, Microporosos, Aditivos de Baixo Custo.

ABSTRACT
The oil is of vital importance to society due to its derivatives with large employment in
economic activities and especially its use as an energy source. The economic and social
expansion of the country in recent decades, it requires significant development of our
infrastructure, especially in the energy sector. The oil that is positioned as the energy
source worldwide leader showed marked increase in the average international price over
the past years. In this adverse scenario, improvements in the process of refining the oil
representing the large-scale gains for society. The present research study is to evaluate the
characteristics of efficiency of catalysis of zeolites ZSM-12 for use as additives for low-
cost production of heterogeneous microporous catalysts for catalytic cracking of step
(FCC), allowing further reductions in the cost of refining oil. The zeolites are crystalline
aluminosilicates, which, in its acid form, playing the role of the main active component of
FCC catalysts. In our study the zeolite ZSM-12 (zeolite Socony Mobil) a zeolite
synthesized with high SiO2/Al2O3 ratio that presents a structure formed by pores
dimensional elliptical opening 12 containing tetrahedral SiO4 (or AlO4) with diameter of
5.5 x 5.9 Å. The results were very promising for the SiO2/Al2O3 ratio assessed, with high
conversion and selectivity (mainly the C1 and C3), pointing to ZSM-12 that potential low
cost of zeolitic additive for FCC catalysts.
Keywords: Zeolitic Catalysts, FCC, Microporous, Low Cost additives.

ÍNDICE DE TABELAS
Tabela 3.1 – Classificação das ocorrências de petróleo segundo o grau API.................... 20
Tabela 3.2 – Refinarias brasileiras e dados de capacidade e produção.............................. 30
Tabela 3.3 – Dimensões de poros de algumas zeólitas e peneiras moleculares................. 35
Tabela 4.1 – Composição da mistura reacional para a síntese das zeólitas ZSM-12......... 43
Tabela 5.1 – Grau de cristalinidade das amostras de ZSM-12........................................... 53
Tabela 5.2 – Dados de conversão e seletividade dos produtos obtidos na reação de
craqueamento catalítico de n-heptano sobre a zeólita ZSM-12 com SiO2/Al2O3 = 50........
55
Tabela 5.3 – Dados de conversão e seletividade dos produtos obtidos na reação de
craqueamento catalítico de n-heptano sobre a zeólita ZSM-12 com SiO2/Al2O3 = 80........
55
Tabela 5.4 – Dados de conversão e seletividade dos produtos obtidos na reação de
craqueamento catalítico de n-heptano sobre a zeólita ZSM-12 com SiO2/Al2O3 = 100......
56
Tabela 5.5 – Dados relativos a conversão e seletividade dos produtos obtidos na reação
de craqueamento de n-heptano sobre a zeólita ZSM-12 com SiO2/Al2O3 = 150.................
56

ÍNDICE DE FIGURAS
Figura 3.1 – Distribuição das unidades de refino no país.................................................. 17
Figura 3.2 – Aumento da capacidade instalada de refino ao longo dos anos..................... 18
Figura 3.3 – Classificação de acordo com grau API das principais reservas brasileiras... 21
Figura 3.4 – Evolução da produção nacional de óleo e LGN............................................ 23
Figura 3.5 – Esquema simplificado de uma unidade de craqueamento térmico................ 28
Figura 3.6 – Esquema simplificado de uma unidade craqueamento catalítico de FCC..... 30
Figura 3.7 – Evolução do balanço entre a oferta e a demanda percentual de petróleo no
mundo nos últimos 40 anos..................................................................................................
31
Figura 3.8 – Representação da arquitetura de um catalisador de FCC.............................. 38
Figura 3.9 – Estrutura zeólita ZSM-12.............................................................................. 39
Figura 3.10 – Esquema representativo para formulação da lei de Bragg............................ 41
Figura 4.1 – Perfil de aquecimento do reator TCAT-10.................................................... 47
Figura 4.2 – Esquema representativo da unidade de avaliação catalítica TCAT-10.......... 49
Figura 4.3 – Foto da unidade de avaliação catalítica TCAT-10........................................ 50
Figura 5.1 – Difratograma de Raios-X da amostra de ZSM-12 com SiO2/Al2O3 = 50...... 51
Figura 5.2 – Difratograma de Raios-X da amostra de ZSM-12 com SiO2/Al2O3 = 80...... 52
Figura 5.3 – Difratograma de Raios-X da amostra de ZSM-12 com SiO2/Al2O3 = 100.... 53
Figura 5.4 – Difratograma de Raios-X da amostra de ZSM-12 com SiO2/Al2O3 = 150.... 54
Figura 5.5 – Comparação das conversões obtidas na reação de craqueamento catalítico
do n-heptano sobre ZSM-12 em função do tempo em diferentes razões SiO2/Al2O3........
58
Figura 5.6 – Relação entre o grau de cristalinidade das amostras de ZSM-12 e a razão
SiO2/Al2O3............................................................................................................................
59
Figura 5.7 – Conversão dos produtos obtidos na reação de craqueamento catalítico do
n-heptano em função da razão SiO2/Al2O3 e tempo de reação sobre a zeólita ZSM-12......
59
Figura 5.8 – Curva de nível mostrando a conversão em função do tempo e da razão
SiO2/Al2O3 na reação de craqueamento catalítico do n-heptano sobre a zeólita ZSM-12...
60

Figura 5.9 – Seletividade dos produtos obtidos na reação de craqueamento catalítico do
n-heptano sobre a zeólita ZSM-12 com SiO2/Al2O3 = 50....................................................
61
Figura 5.10 – Seletividade dos produtos obtidos na reação de craqueamento catalítico
do n-heptano sobre a zeólita ZSM-12 com SiO2/Al2O3 = 80...............................................
61
Figura 5.11 – Seletividade dos produtos obtidos na reação de craqueamento catalítico
do n-heptano sobre a zeólita ZSM-12 com SiO2/Al2O3 = 100.............................................
62
Figura 5.12 – Seletividade dos produtos obtidos na reação de craqueamento catalítico
do n-heptano sobre a zeólita ZSM-12 com SiO2/Al2O3 = 150.............................................
62
Figura 5.13 – Seletividade a compostos C1 obtidos na reação de craqueamento do
n-heptano em função da razão SiO2/Al2O3 e tempo de reação sobre a zeólita ZSM-12......
64
Figura 5.14 – Curva de nível mostrando a seletividade ao C1 em função do tempo e da
razão SiO2/Al2O3 na reação de craqueamento catalítico do n-heptano sobre ZSM-12.......
64
Figura 5.15 – Seletividade a compostos C2 obtidos na reação de craqueamento do
n-heptano em função da razão SiO2/Al2O3 e tempo de reação sobre a zeólita ZSM-12......
65
Figura 5.16 – Curva de nível mostrando a seletividade ao C2 em função do tempo e da
razão SiO2/Al2O3 na reação de craqueamento catalítico do n-heptano sobre ZSM-12........
65
Figura 5.17 – Seletividade a compostos C3 obtidos na reação de craqueamento do
n-heptano em função da razão SiO2/Al2O3e tempo de reação sobre a zeólita ZSM-12.......
66
Figura 5.18 – Curva de nível mostrando a seletividade ao C3 em função do tempo e da
razão SiO2/Al2O3 na reação de craqueamento catalítico do n-heptano sobre ZSM-12........
66
Figura 5.19 – Seletividade a compostos C4 obtidos na reação de craqueamento do
n-heptano em função da razão SiO2/Al2O3 e tempo de reação sobre a zeólita ZSM-12......
67
Figura 5.20 – Curva de nível mostrando a seletividade ao C4 em função do tempo e da
razão SiO2/Al2O3 na reação de craqueamento catalítico do n-heptano sobre ZSM-12........
67
Figura 5.21 – Seletividade a compostos C5 obtidos na reação de craqueamento do
n-heptano em função da razão SiO2/Al2O3 e tempo de reação sobre a zeólita ZSM-12......
68
Figura 5.22 – Curva de nível mostrando a seletividade ao C5 em função do tempo e da
razão SiO2/Al2O3 na reação de craqueamento catalítico do n-heptano sobre ZSM-12........
68

Figura 5.23 – Seletividade a compostos C6 obtidos na reação de craqueamento do
n-heptano em função da razão SiO2/Al2O3 e tempo de reação sobre a zeólita ZSM-12......
69
Figura 5.24 – Curva de nível mostrando a seletividade ao C6 em função do tempo e da
razão SiO2/Al2O3 na reação de craqueamento catalítico do n-heptano sobre ZSM-12........
69
Figura A – Exemplo de uma interpolação de dados unidimensional utilizando o método
de kriging...........................................................................................................................
81

SUMÁRIO
1. – Introdução................................................................................................................. 12
2. – Objetivos................................................................................................................... 14
2.1. – Objetivo geral.......................................................................................................... 14
2.2. – Objetivos específicos............................................................................................... 14
3. – Revisão da literatura................................................................................................ 15
3.1. – A indústria do petróleo no contexto nacional.......................................................... 15
3.2. – Refino de petróleo................................................................................................... 19
3.3. – Craqueamento de frações de petróleo..................................................................... 24
3.3.1. – Craqueamento térmico.................................................................................... 27
3.3.2. – Craqueamento catalítico.................................................................................. 29
3.4. – Zeólitas e peneiras moleculares............................................................................... 33
3.4.1. – Zeólitas como aditivos para catalisadores de FCC.......................................... 36
3.4.2. – Zeólita ZSM-12............................................................................................... 39
3.5. – Técnica para caracterização dos catalisadores zeolíticos: Difração de Raios-X..... 40
4. – Metodologia experimental....................................................................................... 43
4.1 – Obtenção das zeólitas ZSM-12 com diferentes razões SiO2/Al2O3......................... 43
4.2 – Caracterização via difração de Raios-X das zeólitas............................................... 44
4.3 – Testes catalíticos de craqueamento de n-heptano sobre ZSM-12 com diferentes
razões SiO2/Al2O3...............................................................................................................
45
5. – Resultados e Discussões............................................................................................ 51
5.1 – Características estruturais das zeólitas ZSM-12 c/ diferentes razões SiO2/Al2O3... 51
5.2 – Testes catalíticos de craqueamento de n-heptano sobre ZSM-12 com diferentes
razões SiO2/Al2O3..............................................................................................................
55
5.3 – Superfícies resposta para a relação entre conversão e seletividade em função do
tempo de reação, razão SiO2/Al2O3 e grau de cristalinidade das zeólitas ZSM-12...........
58
5.4 – Efeito do tempo de reação, razão SiO2/Al2O3 e grau de cristalinidade na
conversão catalítica do n-heptano......................................................................................
58
5.5 – Efeito do tempo de reação, razão SiO2/Al2O3 e grau de cristalinidade na
seletividade dos produtos de craqueamento do n-heptano.................................................
61

6. – Conclusões e sugestões para trabalhos futuros...................................................... 71
6.1 – Conclusões............................................................................................................... 71
6.2 – Sugestões para trabalhos futuros.............................................................................. 73
Referências bibliográficas................................................................................................... 73
Anexo.................................................................................................................................... 79
Anexo A - Interpolação pelo método de Kriging............................................................... 79
A.1 – Considerações acerca do método........................................................................... 79
A.2 – Equações gerais do método de Kriging................................................................. 81
A.3 – Equações básicas para o método de Kriging Simples........................................... 83

12
1. INTRODUÇÃO
O Petróleo é de crucial importância para a sociedade devido à diversidade de
derivados que dele podem ser obtidos, com largo emprego nas atividades econômicas,
principalmente na forma de combustíveis. No Brasil, o petróleo representa a fonte
energética mais importante, retratando a mesma situação da matriz enérgica mundial, na
qual a petróleo representa 34,2% do total da energia gerada, e o gás natural responde por
21,4% do total, ou seja, a combinação petróleo e gás natural, representa mais da metade de
toda a energia gerada no planeta. A expansão econômica e social do país, nas últimas
décadas, vem exigindo importante desenvolvimento da nossa infra-estrutura, notadamente
no setor de energia. Portanto, melhorias no processo de refino do petróleo representam
ganhos em larga escala para sociedade (consumidor final) devido ao efeito multiplicador
da cadeia produtiva. Descreve-se como o maior desafio deste estudo aliar dois fatores
aparentemente contraditórios: significativa eficiência de catálise e baixo custo de produção
de catalisadores heterogêneos microporosos para a etapa de craqueamento catalítico (FCC)
no refino do Petróleo. Uma vez que temos como premissa o estudo das características
atuais do processo de refino de petróleo, com foco na utilização de catalisadores
heterogêneos microporosos de baixo custo, os resultados deste estudo poderão ser
aplicados facilmente nas refinarias existentes, pois não há a necessidade de alteração dos
equipamentos e estruturas atualmente utilizadas no setor.
As zeólitas são aluminossilicatos cristalinos, que, na sua forma ácida,
desempenham o papel dos principais componentes ativos dos catalisadores de FCC. O
fenômeno da catálise é nanométrico por definição. O termo zeólita foi utilizado
inicialmente por Crönsted em 1756. A denominação do termo “zeólita” (grego: zein =
ferver e lithos = pedra) se deu ao verificar que quando calcinados a altas temperaturas
pareciam fundir e ferver ao mesmo tempo. Por definição, as zeólitas são compostos por
tetraedros SiO4 e AlO4 conectados pelos átomos de oxigênio dos vértices. A substituição
de Si+4 por Al+3 gera uma densidade de carga negativa estrutural que é balanceada por
cátions trocáveis (Na+, H+, etc.), assegurando a estabilidade do sólido. Existem
aproximadamente 40 tipos de zeólitas naturais, com composições químicas e estruturas
cristalográficas variáveis. Desde 1955, tentativas vêm sendo realizadas no sentido de

13
preparar zeólitas sinteticamente. A zeólita ZSM-5 (Zeolite Socony Mobil, sendo “5” a
abertura dos poros em Å), se caracteriza por apresentar um alto teor de silício e por
apresentar canais retilíneos e canais sinosuidais que se entrecruzam. Este tipo de zeólita
apresenta várias aplicações por sua alta seletividade e alta estabilidade térmica e ácida,
consistindo em um dos principais aditivos para catalisadores de FCC utilizados atualmente.
Em nosso trabalho apresentamos como objetivo principal o estudo da zeólita ZSM-
12 (da série ZSM - Zeolite Socony Mobil) que pertence à mesma família pentasil da ZSM-
5, porém apresenta apenas canais retos, portanto, de estrutura interna menos complexa. A
zeólita ZSM-12, trata-se de uma zeólita sintetizada com altas razões SiO2/Al2O3 que
apresenta estrutura formada por poros unidimensionais de abertura elíptica contendo 12
tetraedros SiO4 (ou AlO4), com diâmetro de 5,5 x 5,9 Å.
O uso da energia está profundamente ligado com o desenvolvimento econômico e
social. A energia é fundamental para alcançar objetivos de desenvolvimento sustentável. O
petróleo que se posiciona como a fonte de energia líder a nível mundial: Entre 2003 e 2007
o preço internacional do barril de petróleo aumentou em média 87%. Neste cenário
adverso, o resultado deste estudo poderá proporcionar uma expressiva redução de custo do
processo de refino de petróleo, especificamente na etapa de craqueamento catalítico,
possibilitando redução do preço final de seus derivados, gerando um efeito multiplicador
que beneficiará a sociedade como um todo.
Este presente trabalho visa estudar o potencial de zeólitas ZSM-12 de baixo custo
com diferentes razões SiO2/Al2O3 para sua utilização como aditivos para catalisadores de
FCC através de reações de craqueamento catalítico. Apresenta ainda como etapas
específicas a pesquisa bibliográfica acerca do tema proposto, a realização de reações de
craqueamento catalítico sobre zeólitas ZSM-12 de baixo custo com diferentes razões
SiO2/Al2O3 e o estudo da influência de alguns parâmetros operacionais, tais como tempo
de reação e razão SiO2/Al2O3 na conversão e distribuição de produtos obtidos
experimentalmente.

14
2. OBJETIVOS
2.1. Objetivo Geral
Estudar o potencial de zeólitas ZSM-12 (Zeolite Socony Mobil) com diferentes
razões SiO2/Al2O3 como aditivos para catalisadores de FCC através de reações de
craqueamento catalítico de hidrocarbonetos médios.
2.2. Objetivos Específicos
� Pesquisa bibliográfica acerca do tema proposto;
� Obtenção das amostras de zeólitas ZSM-12;
� Caracterização das zeólitas via Difração de Raios-X (DRX);
� Realizar reações de craqueamento catalítico de n-heptano sobre zeólitas ZSM-12
com diferentes razões SiO2/Al2O3 em micro-reator catalítico de leito fixo;
� Estudar a influência de alguns parâmetros operacionais, tais como tempo de reação
e razão SiO2/Al2O3 na conversão e distribuição de produtos;
� Estabelecer correlações entre a atividade catalítica nas reações de craqueamento de
n-heptano e características estruturais das zeólitas caracterizadas via DRX;

15
3. REVISÃO DA LITERATURA
3.1. A indústria do Petróleo no contexto nacional
A indústria do petróleo atualmente consiste num agregado de operações que de
acordo com a sua natureza se classificam em exploração, produção, refino, transporte,
armazenamento e distribuição. Para que essa indústria funcione de maneira satisfatória, é
necessário que essas operações estejam interligadas e em harmonia visando obter a
minimização dos custos de produção e maximização dos resultados (Szklo, 2005).
a) Exploração e Produção
O Brasil desfruta do reconhecimento internacional na área de exploração e
produção de petróleo, com tecnologia própria para águas ultra-profundas, possibilitando a
produção de petróleo a preços competitivos em campos offshore (marítimos) a
profundidades cada vez maiores, atraindo o interesse de companhias petrolíferas de todo
mundo, com algumas das quais já firmou acordos de parceria para a exploração e produção
de suas reservas na enorme plataforma submarina brasileira. A abertura da área de
exploração e produção para parceiros internacionais gerou, por sua vez, oportunidades de
participação brasileira em exploração e produção de petróleo em outras partes do mundo,
principalmente onde o “know how” desenvolvido para exploração de águas ultra-profundas
possa ser aplicado ( Szklo, 2005).
Aliado aos reconhecidos avanços da tecnologia nacional em exploração marítima,
também ocorrem avanços tecnológicos em exploração terrestre, principalmente no tocante
a campos maduros, alguns deles com mais de trinta anos em produção, e que devido às
novas técnicas, poderão atingir novamente valores de produção próximos aos registrados
no início de sua exploração ( Szklo, 2005).

16
b) Refino
O parque nacional de refino de petróleo vem acompanhando de perto as
transformações que a sociedade vivencia nos últimos anos, adequando-se ao novo modelo
de mercado do setor no Brasil. O desafio de processar a crescente produção de óleo pesado
brasileiro, permitindo a conversão para derivados de alto valor agregado, vem sendo
vencido com investimentos e grandes avanços tecnológicos. As refinarias brasileiras tem
batido sucessivos recordes de processamento, desenvolvendo tecnologias próprias e
possibilitando que o petróleo nacional, de característica mais pesada, possa render uma
percentagem maior de produtos nobres e aumentar a rentabilidade do negócio. Somente no
setor de refino foram investidos US$ 4,9 bilhões no período de 2002-2005.
Encontram-se em fase de implantação investimentos para da ampliação da
capacidade de processamento do parque nacional de refino, principalmente nas refinarias
dos estados da Bahia e São Paulo, além do início das obras de construção do novo pólo
petroquímico no estado do Rio de Janeiro - COMPERJ. Dentre os investimentos previstos
para o refino em 2009, cita-se como de destacada importância o início das obras de
construção da Refinaria do Nordeste, localizada no estado de Pernambuco.
Aproximadamente 20 anos após a construção de sua última refinaria (REVAP - Refinaria
Henrique Lage, no ano de 1980, no município de São José dos Campos - SP) o país volta a
investir na construção de uma nova refinaria que, quando estiver operando em capacidade
plena, contribuirá decisivamente para a obtenção da auto-suficiência brasileira em óleo
diesel, importante insumo para o desenvolvimento do país. A Figura 3.1 mostra a
distribuição das unidades de refino no país (Petrobras 2007).
Atualmente o Brasil possui 13 refinarias, sendo que destas, duas são privadas
(refinaria de Manguinhos e refinaria Ipiranga) e outra em parceria com a iniciativa privada
(REFAP). Existe uma forte concentração das refinarias brasileiras na região Sudeste (63%
da capacidade nominal total de refino) e na região Sul (20%) ( Szklo, 2005).
Verificamos que os próximos projetos de investimentos, elegeram a região
Nordeste como foco para a construção de novas refinarias.
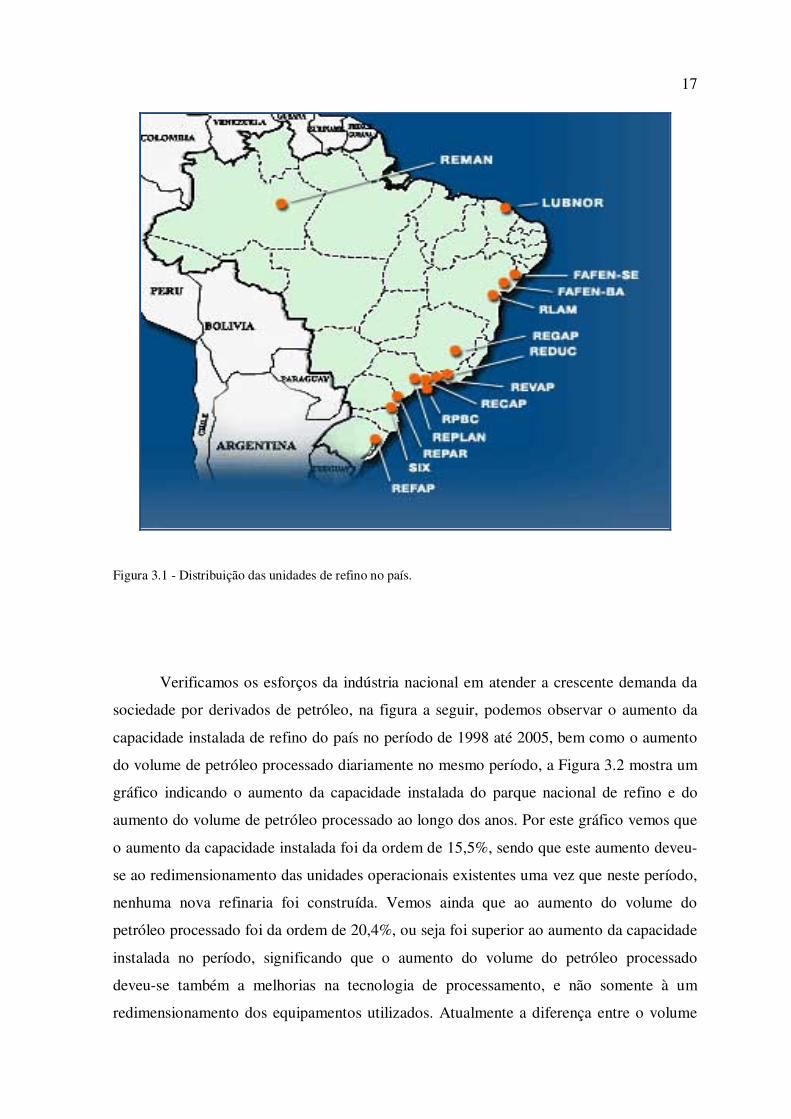
17
Figura 3.1 - Distribuição das unidades de refino no país.
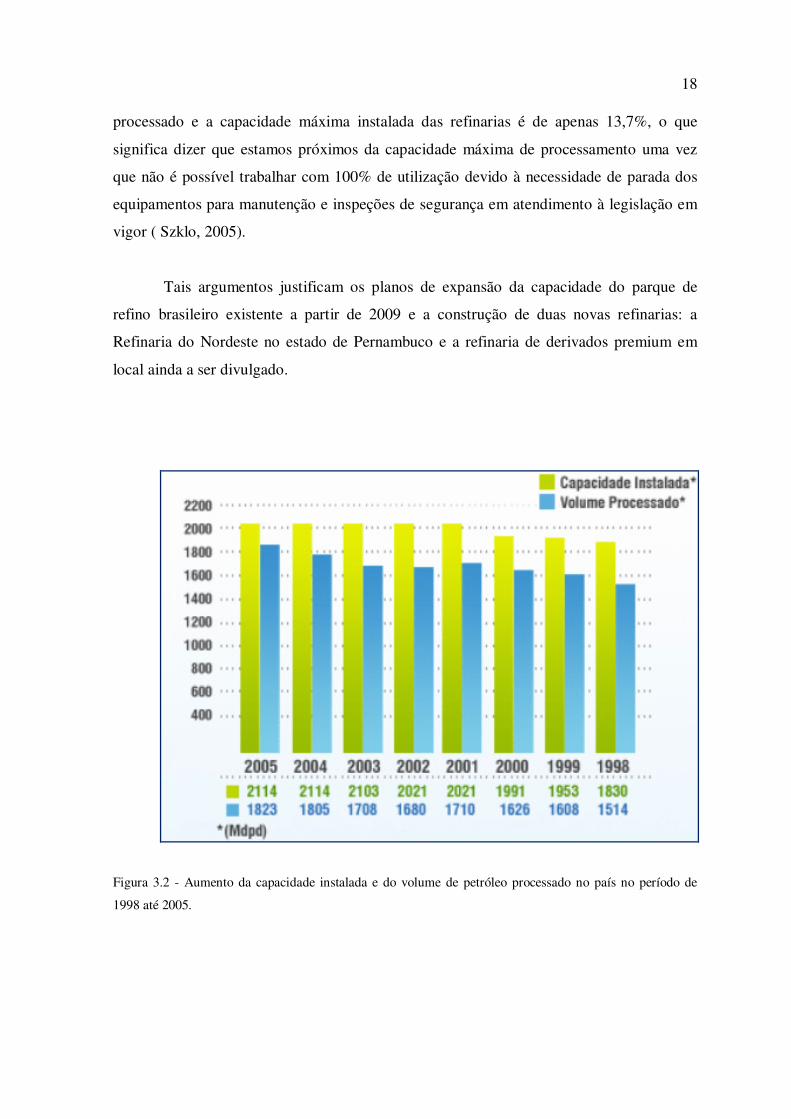
Verificamos os esforços da indústria nacional em atender a crescente demanda da
sociedade por derivados de petróleo, na figura a seguir, podemos observar o aumento da
capacidade instalada de refino do país no período de 1998 até 2005, bem como o aumento
do volume de petróleo processado diariamente no mesmo período, a Figura 3.2 mostra um
gráfico indicando o aumento da capacidade instalada do parque nacional de refino e do
aumento do volume de petróleo processado ao longo dos anos. Por este gráfico vemos que
o aumento da capacidade instalada foi da ordem de 15,5%, sendo que este aumento deveu-
se ao redimensionamento das unidades operacionais existentes uma vez que neste período,
nenhuma nova refinaria foi construída. Vemos ainda que ao aumento do volume do
petróleo processado foi da ordem de 20,4%, ou seja foi superior ao aumento da capacidade
instalada no período, significando que o aumento do volume do petróleo processado
deveu-se também a melhorias na tecnologia de processamento, e não somente à um
redimensionamento dos equipamentos utilizados. Atualmente a diferença entre o volume
18
processado e a capacidade máxima instalada das refinarias é de apenas 13,7%, o que
significa dizer que estamos próximos da capacidade máxima de processamento uma vez
que não é possível trabalhar com 100% de utilização devido à necessidade de parada dos
equipamentos para manutenção e inspeções de segurança em atendimento à legislação em
vigor ( Szklo, 2005).
Tais argumentos justificam os planos de expansão da capacidade do parque de
refino brasileiro existente a partir de 2009 e a construção de duas novas refinarias: a
Refinaria do Nordeste no estado de Pernambuco e a refinaria de derivados premium em
local ainda a ser divulgado.
Figura 3.2 - Aumento da capacidade instalada e do volume de petróleo processado no país no período de
1998 até 2005.

19
3.2. Refino de petróleo
Nas refinarias, o petróleo é submetido a diversos processos pelos quais se obtém
grande diversidade de derivados tais como: gás liquefeito de petróleo (GLP), gasolina,
naftas, óleo diesel, gasóleos, querosenes de aviação e de iluminação, óleo combustível,
asfalto, lubrificantes, solventes, parafinas, coque de petróleo e resíduos. As parcelas dos
derivados produzidos em determinada refinaria variam de acordo com o tipo de petróleo
processado. Assim, petróleos mais leves produzem maior quantidade de gasolina, GLP e
naftas, que são produtos leves. Já os petróleos pesados resultam em maiores volumes de
óleos combustíveis e asfaltos. No meio da cadeia estão os derivados médios, como o óleo
diesel e o querosene (Szklo, 2005).
Algumas propriedades físicas gerais são utilizadas para identificação dos petróleos,
como densidade relativa e viscosidade. Na comercialização, o ponto predominante e
altamente explorado é aquele que se refere ao teor de elementos leves, ou seja, que
produzem derivados mais rentáveis comercialmente.
O American Petroleum Institute - API classifica diversos tipos de petróleos quanto
ao teor de elementos leves, e para tal propósito adotou índice chamado Grau API. Quanto
maior o grau API do óleo, menor é a sua densidade relativa, o que equivale ao óleo ser
mais leve, portanto mais rico em voláteis (partes leves), ou seja, tem maior valor
comercial. Desta forma é desejável operar com petróleos de alto Grau API, pois além de
maior facilidade no processamento, pois são geralmente mais fluídos, possibilitam obter
produtos de alto valor agregado. A equação 3.1 demonstra o cálculo deste parâmetro de
classificação dos diversos tipos de petróleos (Szklo, 2005).
A maior parte das reservas brasileiras constitui-se de óleos pesados, de menor grau
API, necessitando de adequações do nosso parque de refino para o processamento do óleo
nacional, sendo necessário, por vezes, realizar blend da carga a ser processada com parte
do óleo importado árabe de alto grau API (Szklo, 2005).

20
Equação para obtenção do Grau API da amostra de petróleo:
(3.1)
Onde:
OAPI: Grau API
d: densidade relativa de petróleo à 60° F em relação à água também à 60° F.
Tabela 3.1 - Classificação das ocorrências de petróleo segundo o grau API.
DENSIDADE (GRAU API)
CLASSIFICAÇÃO
OAPI > 40 EXTRA-LEVE
40 > OAPI > 33 LEVE
33 > OAPI > 27 MÉDIO
27 > OAPI > 19 PESADO
19 > OAPI > 15 EXTRA-PESADO
OAPI < 15
ASFÁLTICO
De acordo com as características geológicas do local de onde é extraído, o petróleo
bruto pode variar quanto à sua composição química e ao seu aspecto. Quanto ao aspecto,
há petróleos pesados e viscosos, e outros leves e voláteis, segundo o número de átomos de
carbono existentes em sua composição, desta forma, o petróleo pode ter uma ampla gama
de cores, desde o amarelo claro, semelhante à gasolina, chegando ao verde, ao marrom e ao
preto. Com tão grande variedade de tipos de matéria-prima, a tarefa inicial no processo de
refino é determinar as características do petróleo a ser processado, por meio de análises de
laboratório. Existem, refinarias já projetadas para refinar determinado tipo de petróleo.
Atualmente, encontra larga aplicação uma etapa anterior ao processamento que é consiste
21
na obtenção de blend de petróleos de diferentes propriedades, visando obter determinadas
características, tais como: viscosidade, densidade, teores máximo de contaminantes, etc.
Esta etapa de blendagem possibilita a utilização de petróleos de piores propriedades, que,
anteriormente seriam preteridos no processo de refino (Szklo, 2005).
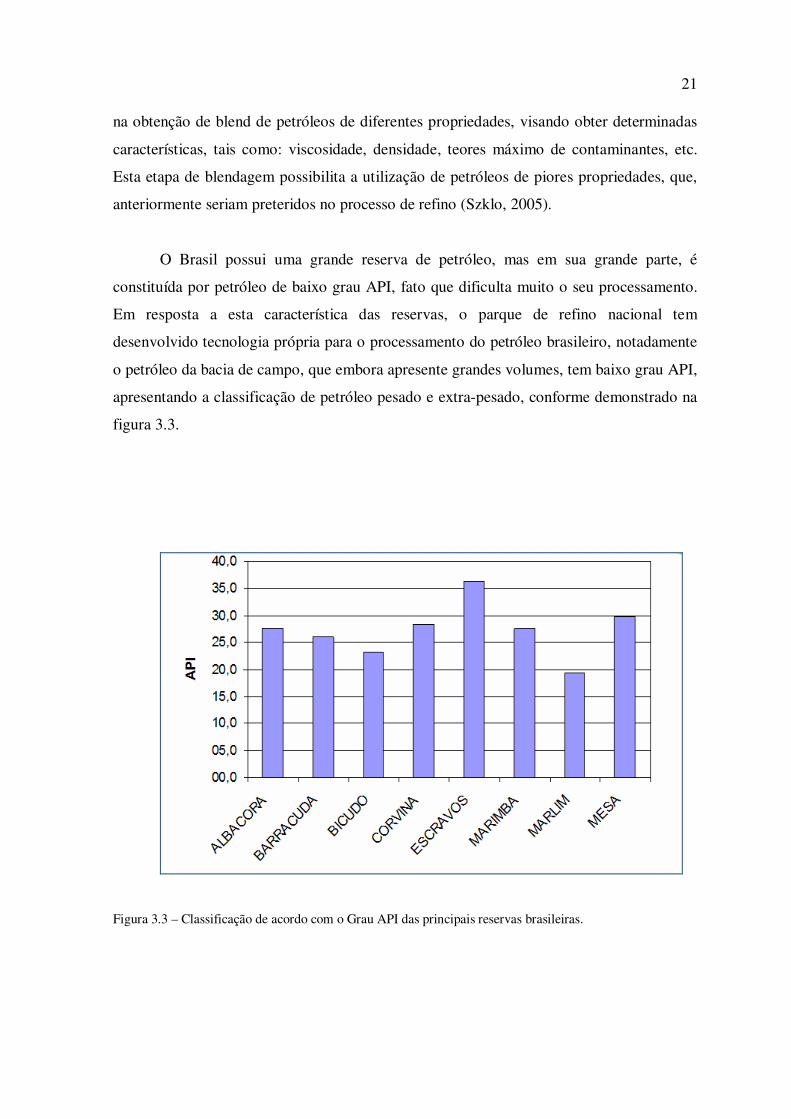
O Brasil possui uma grande reserva de petróleo, mas em sua grande parte, é
constituída por petróleo de baixo grau API, fato que dificulta muito o seu processamento.
Em resposta a esta característica das reservas, o parque de refino nacional tem
desenvolvido tecnologia própria para o processamento do petróleo brasileiro, notadamente
o petróleo da bacia de campo, que embora apresente grandes volumes, tem baixo grau API,
apresentando a classificação de petróleo pesado e extra-pesado, conforme demonstrado na
figura 3.3.
Figura 3.3 – Classificação de acordo com o Grau API das principais reservas brasileiras.

22
Fica evidenciando na figura 3.3 que, a maior parte das reservas do país recebem a
classificação de petróleo pelo critério do grau API, destacando-se ainda que o maior campo
atual de produção do Brasil, o campo de Marlim da bacia de Campos, apresenta grau API
de 18, ou seja é classificado como petróleo extra-pesado.
O desenvolvimento de rota de processamento para petróleos pesados e extra-
pesados, demonstra a vocação tecnológica do refino nacional, sendo esta tecnologia
responsável pelo aproveitamento de petróleos que não seriam processados em grande parte
das demais refinarias do mundo, as quais são adaptadas, em sua maioria, para petróleos de
grau API mais elevado de características médias e leves (Szklo, 2005).
A primeira etapa do processo de refino é a destilação atmosférica, pela qual passa
todo o óleo cru a ser beneficiado. Ela se realiza em torres de dimensões variadas, que
possuem, ao longo da coluna principal, uma série de pratos perfurados em várias alturas,
um para cada fração desejada. O petróleo é pré-aquecido e introduzido na metade da torre
de destilação. Como a parte de baixo da torre é mais quente, os hidrocarbonetos gasosos
tendem a subir e se condensar ao passarem pelos pratos. Nessa etapa, são recolhidos como
derivados da primeira destilação, principalmente, gás, gasolina, nafta e querosene. Essas
frações, retiradas nas várias alturas da coluna, ainda necessitam de novos processamentos e
tratamentos, para se transformarem em produtos ou servirem de carga para outros
derivados mais nobres (Szklo, 2005).
As frações mais pesadas do petróleo, que não foram separadas na primeira
destilação, descem para o fundo da torre e vão constituir o resíduo ou a carga para uma
segunda destilação, onde recebem mais calor, agora sob vácuo. O sistema é mais
complexo, mas segue o mesmo processo dos pratos que recolhem as frações menos
pesadas, praticamente o óleo diesel e o óleo combustível. Na parte de baixo, é recolhido
novo resíduo, que será usado para produção de asfalto ou como óleo combustível pesado
(Szklo, 2005).
A terceira etapa do refino consiste no craqueamento, que pode ser térmico ou
catalítico. O princípio desses processos é o mesmo, e se baseia na quebra de moléculas
longas e pesadas dos hidrocarbonetos, transformando-as em moléculas menores e mais
23
leves. O craqueamento térmico exige pressões e temperaturas altíssimas para a quebra das
moléculas, enquanto no catalítico o processo é realizado com o auxílio do catalisador,
agente que favorece a reação química, sem entrar como componente do produto. Uma série
de outras unidades de processo, transformam frações pesadas do petróleo em produtos
mais leves e colocam as frações destiladas nas especificações adequadas para consumo
(Bionda et al., 2000; Henshaw, 1998; Szklo, 2005).
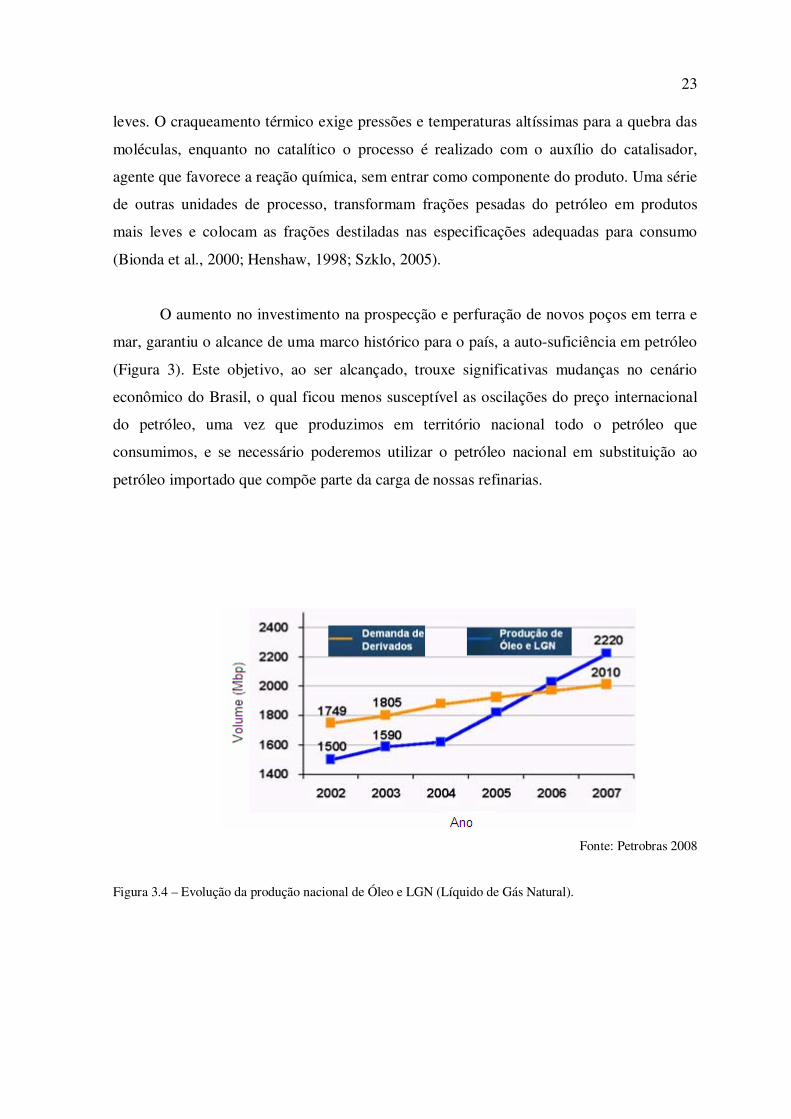
O aumento no investimento na prospecção e perfuração de novos poços em terra e
mar, garantiu o alcance de uma marco histórico para o país, a auto-suficiência em petróleo
(Figura 3). Este objetivo, ao ser alcançado, trouxe significativas mudanças no cenário
econômico do Brasil, o qual ficou menos susceptível as oscilações do preço internacional
do petróleo, uma vez que produzimos em território nacional todo o petróleo que
consumimos, e se necessário poderemos utilizar o petróleo nacional em substituição ao
petróleo importado que compõe parte da carga de nossas refinarias.
Fonte: Petrobras 2008
Figura 3.4 – Evolução da produção nacional de Óleo e LGN (Líquido de Gás Natural).

24
A figura 3.4 ilustra o acentuado aumento da demanda do país por derivados de
petróleo, porém devido aos investimentos em prospecção de novos campos produtores e
avanços tecnológicos aplicados na extração de petróleo, foi possível alcançar a auto-
suficiência em petróleo no ano de 2006. Para os anos seguintes, espera-se um incremento
na geração de divisas ao país por meio do aumento das exportações do excedente de
produção.
3.3. Craqueamento de frações de petróleo
Atualmente o craqueamento é um dos processos mais utilizados na indústria do
refino do petróleo. O objetivo do craqueamento é transformar hidrocarbonetos de alto peso
molecular em hidrocarbonetos leves de baixo peso molecular. Os fragmentos pequenos
apresentam alta volatilidade, sendo usados em grande escala como combustíveis. Neste
processo, as reações são tipicamente catalisadas por sólidos ácidos (Szklo, 2005).
As reações são basicamente constituídas por uma série de passos elementares,
bastante complexos, dado que os produtos de reação primários podem sofrer reações
secundárias (Haag et al., 1984; Shertukte et al., 1992).
Atualmente, é aceito que o craqueamento catalítico de hidrocarbonetos ocorre via a
formação de carbocátions como intermediários reacionais, que reagem ao estar em contato
com catalisadores possuindo acidez de Brönsted (centros doadores de prótons) ou de Lewis
(centros receptores de elétrons). Dois mecanismos reacionais foram estabelecidos como
possíveis para esta reação:
a) Mecanismo via a formação de íons carbênio (contêm um átomo de
carbono tri-coordenado e carregado positivamente) (Shertukte et al.,
1992).
b) Mecanismo via a formação de íons carbônio (contêm um átomo de
carbono penta-coordenado, carregado positivamente) ( Haag et al., 1984).

25
Os íons carbênio são formados por protonação de ligações insaturadas (olefínas ou
aromáticos) em sítios ácidos de Brönsted ou por saída de um hidreto em sítios ácidos de
Lewis. Os íons carbônio podem reagir, em seguida, através de uma série de etapas
reacionais que podem ocorrer simultaneamente (Haag et al., 1984; Shertukte et al., 1992):
• Perda de um próton gerando uma nova olefina.
• Transferência de hidreto intermolecular de um íon carbênio para uma parafina
(reação de propagação);
• Quebra de uma ligação C-C, do íon carbênio, em posição β.
• Iniciação
(Protonação)
(Oleofina) (Sítio de Brönsted) (Íon Carbênio)
(Deprotonação)
(Parafina) (Sítio Lewis) (Íon Carbênio)

26
• Propagação (Transferência de Hidrogênio)
(Transf. de Hidrogênio)
(Íon Carbênio) (Parafina) (Parafina) (Íon Carbênio)
• Cracking (Cisão β)
(Íon Carbênio) (Íon Carbênio) (Oleoafina)
Outro mecanismo alternativo foi proposto, para temperaturas acima dos 500ºC,
envolvendo a formação de íons carbônio penta-coordenados. Os íons carbônio são
formados nos sítios de Brönsted e sofrem uma cisãoβ, originando parafinas de menor
número de átomos de carbono e íons carbênio ou são convertidos diretamente em íons
carbênio através da perda de uma molécula de hidrogênio (Haag et al., 1984; Shertukte et
al., 1992):
(Parafina) (Sitio Brönsted) (Íon Carbênio)
a) Cisão β => (3.7)
b) Perda de H2 => (3.8)

27
Dado que estas reações são monomoleculares, são favorecidas em estruturas com
poros de pequenas dimensões (Por exemplo: zeólitas Y, ZSM-5, ZSM-12). Podem ocorrer
diversas reações secundárias, em que estão envolvidos quer centros ácidos como por
exemplo reações de transferência de hidrogênio: ciclização, aromatização, condensação,
isomerização da dupla ligação (isômeros cis- e trans-) e isomerização estrutural. As
reações de transferência de hidrogênio intermoleculares de naftenos para olefinas
(originando aromáticos e parafinas) são passos bimoleculares que podem igualmente
ocorrer, reduzindo o teor em olefinas no produto final. O aumento de espécies aromáticas
pode traduzir-se num aumento da formação de espécies poliaromáticas insaturadas
("coke") (Haag et al., 1984; Shertukte et al., 1992).
Atualmente na indústria do petróleo existem várias configurações para este
processo, dentre elas podemos citar como mais empregadas:
3.3.1 - Craqueamento Térmico
O craqueamento térmico é um dos processos mais antigos que existe. Este processo
tem como objetivo a redução do peso molecular da mistura de hidrocarbonetos pela
simples aplicação de calor sem sofisticações adicionais como por exemplo, a adição de
hidrogênio (Kanyanov et al., 1996; Yang et al., 1998). O craqueamento térmico pode
assumir 3 configurações básicas: quebra de viscosidade, craqueamento em fase vapor e
coqueamento (Henshaw, 1998).
A quebra de viscosidade é o tipo mais brando de craqueamento térmico (entre
450o C à 500o C). Este processo tem como objetivo abaixar a viscosidade de resíduos de
óleo cru pesado, provenientes de destilação atmosférica, bem como reduzir o ponto de
derramamento de ceras. Destilados médios também podem ser produzidos, dependendo da
demanda. Um esquema simplificado de uma unidade de craqueamento térmico pode ser
visualizado na Figura 3.5 abaixo.

28
Figura 3.5 – Esquema simplificado de uma unidade de craqueamento térmico (adaptado de Henshaw, 1998).
Outro tipo de craqueamento térmico consiste no craqueamento de correntes de
vapor. Esse processo é usado em refinarias para produzir hidrocarbonetos olefínicos de
várias cargas de alimentação para a manufatura de petroquímicos. A faixa de
hidrocarbonetos na alimentação varia desde etano até gás óleos. A alimentação mais
comum consiste de etano, butano e nafta. O craqueamento térmico de correntes de vapor
ocorre em temperaturas altas em torno de 800o C à 870o C e pressões levemente acima da
atmosférica (Szklo, 2005).
Já o coqueamento é a forma mais severa de craqueamento térmico, usada para
aumentar o nível de produtos leves ou destilados a partir de resíduos pesados. O
coqueamento produz uma estreita faixa de gasolina (nafta coqueada) e várias frações de
destilados médios usados na carga de craqueamento catalítico (Szklo, 2005).
Comumente os processos de craqueamento térmico apresentam rendimento da
ordem entres 10 a 15% de conversão de resíduos de vácuo em frações mais leves. A única

29
vantagem deste processo em relação ao craqueamento catalítico refere-se à inexistência de
problemas de contaminação do catalisador, sendo que, no processo de craqueamento
térmico, a carga a ser craqueada pode conter maior teor de contaminantes, metais e
compostos sulfurados. Estes contaminantes atacam e desativam o catalisador, impedindo o
craqueamento catalítico, como o processo de craqueamento térmico não utiliza
catalisadores, mas o efeito térmico, este processo não é sensível à presença destes
contaminantes (Szklo, 2005).
3.3.2 - Craqueamento Catalítico
No craqueamento catalítico, a quebra das cadeias longas de hidrocarbonetos se dá
tanto pelo efeito térmico, quanto pela presença de um catalisador, que vai atuar diminuindo
a energia de ativação, melhorando assim a seletividade a determinados produtos (Szklo,
2005).
Durante o processo de craqueamento catalítico ocorre uma desativação rápida do
catalisador, motivada pela deposição de coque, aumentando assim o custo do processo.
Isso geralmente era observado quando a reação se processava em grandes leitos catalíticos
fixos. Devido a isto estudos culminaram com o desenvolvimento do processo de
craqueamento realizado em leito fluidizado (FCC – Fluid Catalytic Cracking) (Hollander et
al., 1999; Bionda et al., 2000; Sugungun et al., 1998).
No processo FCC o catalisador é usado na forma de um fino pó (granulometria em
torno de 70 micra), sendo aquecido, para então entrar em contato contra corrente
diretamente com a carga, vaporizando-a e craqueando-a instantaneamente. Os gases
obtidos são separados do catalisador por intermédio de um ciclone e o catalisador
desativado é enviado a uma câmara de regeneração (atmosfera de ar e aquecimento), onde
sai novamente ativo e na temperatura necessária para craquear uma nova carga. Esse
processo é realizado de maneira contínua em tanques de grande dimensão, constituindo
quase sempre os maiores equipamentos existentes em uma refinaria (Nogueira et al.,
1984).

30
No Brasil, atualmente, existem unidades de FCC que operam com uma quantidade
de catalisador da ordem de 500 toneladas, com uma recirculação de catalisador da ordem
de 30 a 40 toneladas por minuto. Neste processo há um contato de 1 a 4 segundos da carga
de catalisador no “riser”, sendo este tempo suficiente para converter praticamente toda
fração pesada em frações mais leves (Henshaw, 1998). O esquema simplificado de uma
unidade de FCC é mostrado na Figura 6.
Figura 3.6 – Esquema simplificado de uma unidade de FCC (adaptado de Henshaw, 1998).
31
Os novos investimentos no setor de refino previstos para os próximos anos
contemplam também adaptação das refinarias existentes ao processamento do petróleo
pesado produzido no país. Novas unidades de craqueamento catalítico e coqueamento
retardado entraram em atividade na Refinaria Alberto Pasqualini (Refap), originalmente
projetada para operar com petróleo leve argentino; e uma unidade de coqueamento
começará a operar na Refinaria Duque de Caxias (Reduc) neste ano (Szklo, 2005).
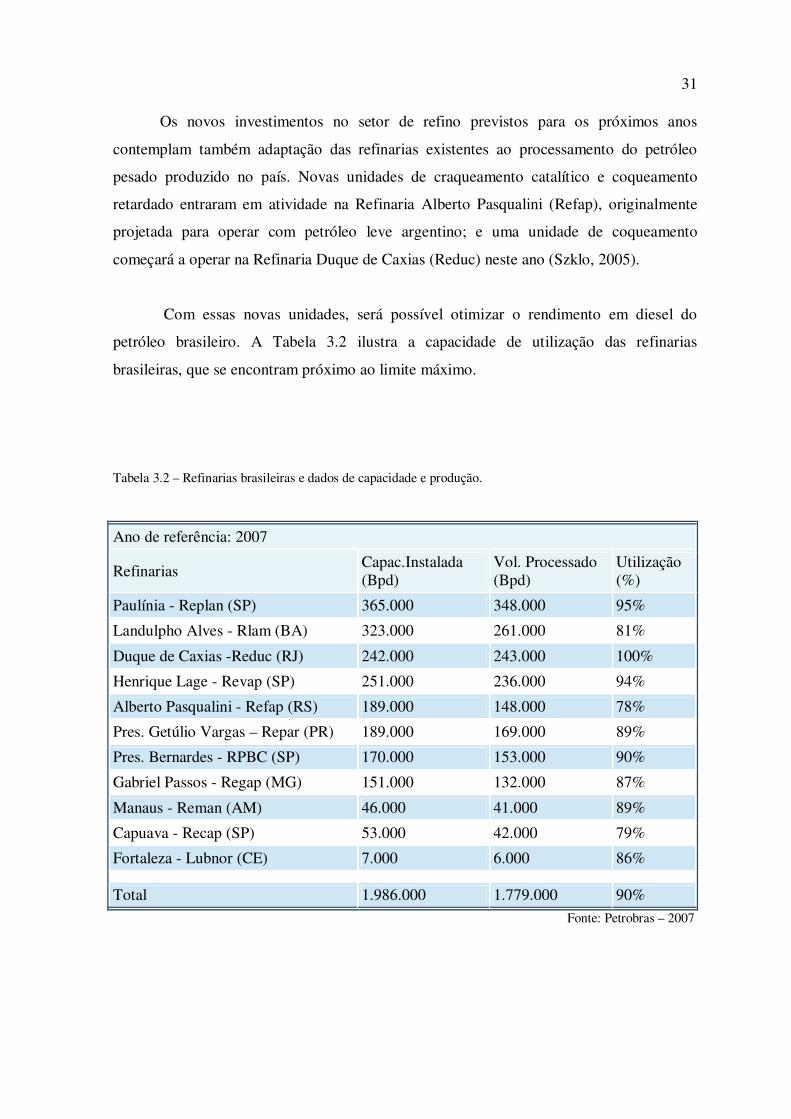
Com essas novas unidades, será possível otimizar o rendimento em diesel do
petróleo brasileiro. A Tabela 3.2 ilustra a capacidade de utilização das refinarias
brasileiras, que se encontram próximo ao limite máximo.
Tabela 3.2 – Refinarias brasileiras e dados de capacidade e produção.
Ano de referência: 2007
Refinarias Capac.Instalada (Bpd)
Vol. Processado (Bpd)
Utilização (%)
Paulínia - Replan (SP) 365.000 348.000 95%
Landulpho Alves - Rlam (BA) 323.000 261.000 81%
Duque de Caxias -Reduc (RJ) 242.000 243.000 100%
Henrique Lage - Revap (SP) 251.000 236.000 94%
Alberto Pasqualini - Refap (RS) 189.000 148.000 78%
Pres. Getúlio Vargas – Repar (PR) 189.000 169.000 89%
Pres. Bernardes - RPBC (SP) 170.000 153.000 90%
Gabriel Passos - Regap (MG) 151.000 132.000 87%
Manaus - Reman (AM) 46.000 41.000 89%
Capuava - Recap (SP) 53.000 42.000 79%
Fortaleza - Lubnor (CE) 7.000 6.000 86%
Total 1.986.000 1.779.000 90% Fonte: Petrobras – 2007

32
Em linha com o crescimento da produção nacional de petróleo, a refino tem em
curso dois grandes projetos: a Refinaria do Nordeste, no estado de Pernambuco, para 200
mil bpd, empreendimento de US$ 4,0 bilhões em parceria com a empresa estatal
venezuelana de petróleo; e a Refinaria Premium, em local a ser definido, para 500 mil bpd,
que será a maior refinaria do país.
Com entrada em operação prevista para 2012 e 2014 respectivamente, as novas
refinarias farão frente ao crescimento da demanda interna, reduzirão a importação de diesel
e assegurarão a exportação de derivados de alto valor agregado, valorizando os excedentes
de petróleo brasileiro.
Em todo o mundo o uso da energia está profundamente ligado com o
desenvolvimento econômico e social, desta forma a disponibilidade de energia é
fundamental para alcançar objetivos de desenvolvimento sustentável.
O petróleo que se posiciona como a fonte de energia líder a nível mundial, portanto
influencia profundamente o desenvolvimento do planeta.
Entre 2003 e 2007 o preço internacional do barril de petróleo aumentou em média
87%, reflexo da alta demanda por este importante recurso energético de origem fóssil,
portanto de reservas limitadas.
A figura 3.7 abaixo mostra a evolução do balanço entre a oferta e a demanda
percentual de petróleo no mundo nos últimos 40 anos, sendo visível o aumento da
demanda por este recurso energético na Ásia, em função da recente explosão de
industrialização nesta região principalmente dos chamados tigres asiáticos e também da
China e Japão, evidenciando a estreita relação entre desenvolvimento industrial e demanda
por petróleo.

33
Fonte: BP Amoco 2007
Figura 3.7 – Evolução do balanço entre a oferta e a demanda percentual de petróleo no mundo nos últimos 40
anos.
3.4. Zeólitas e peneiras moleculares
Zeólita é um aluminossilicato cristalino, com armação estrutural incluindo
cavidades ocupadas por cátions grandes e moléculas de água, ambos tendo considerável
liberdade de movimento, permitindo troca iônica e desidratação reversível (Smith, 1984;
Breck, 1974). A remoção das moléculas de água e a substituição dos cátions
intercambiáveis não alteram a estrutura básica das zeólitas (Dyer e Enamy, 1981).
As zeólitas foram descobertas em 1756 pelo cientista Sueco Cronstedt que verificou
a existência de um mineral (aluminossilicato, denominado de estilbita) que tinha a
capacidade de se intumescer quando submetido à chama, sendo assim batizado de
“zeolitho” (do grego, zeo = aquecer e litho = pedra).

34
Muitos anos mais tarde McBain (1932) introduziu o termo “peneira molecular”
para designar um grupo de zeólitas naturais que tinham a capacidade de separar grupos de
moléculas em função do seu diâmetro cinético ser inferior ou superior aos diâmetros dos
poros. Esta propriedade também definida como seletividade de forma, foi então tida como
a base da arquitetura de novos tipos de materias zeolíticos e muitos outros estudos foram
conduzidos no sentido de se obter e aplicar novas zeólitas sintéticas e naturais cada vez
mais específicas para determinados tipos de aplicação nas áreas de catálise e adsorção
(Weitkamp et al., 2000; Smith, 1984; Breck, 1974; Mascarenhas et al., 2001; Luna et al.,
2001; Braga, 2007).
Na década de 60, Donald W. Breck (1964) que desenvolvia pesquisas para a Mobil
Oil Corporation, conseguiu sintetizar uma zeólita do tipo Y. A descoberta desse material
revolucionou de forma marcante até os dias de hoje a indústria do refino do petróleo, visto
ser a zeólita Y um dos principais componentes do mais famoso catalisador típico de
craqueamento catalítico de frações de petróleo (catalisador de FCC). (Ciola, 1981; Breck,
1974; Dias, 1998; Figueiredo, 1987; Guisnet, 2004).
Outros avanços importantes foram realizados na década seguinte, quando Argauer e
Landolt (1972) sintetizaram a zeólita ZSM-5. Este material encontrou uso em vários
processos da petroquímica como isomerização de xilenos, alquilação,
desproporcionamento e conversão de álcoois em olefinas e parafinas nos processos MTG
(Metanol To Gasoline) e MTO (Metanol To Olefins) (Araujo et. al., 1996).
Na década de 80 vários outros materiais foram descobertos como, por exemplo, os
aluminofosfatos (ALPO´s) (Wilson et al., 1982) e silicoaluminofosfatos (SAPO´s) (Lok et
al., 1984). Até então pelo ponto de vista tradicional, as zeólitas e peneiras moleculares não
deveriam ter acima de 12 membros no anel principal. Essas características eram
importantes, principalmente tendo em vista suas potenciais aplicações no campo de refino
de petróleo e na petroquímica que não exigiam poros muito largos.
Somente na década de 80 e início dos anos 90 é que várias companhias
multinacionais e grupos de pesquisa começaram a direcionar seus esforços no sentido de
sintetizar materiais com poros maiores, principalmente em função do surgimento de muitos

35
processos de purificação de petróleo de compostos contendo heteroátomos como: enxofre,
nitrogênio e oxigênio, cujos compostos apresentavam diâmetros cinéticos elevados e
também em função do crescente aumento do peso das frações de petróleo. Os processos
catalíticos então ganharam agora novos rumos com a catálise ambiental que vinha para
caminhar lado a lado com os processos convencionais de refino de petróleo (Guisnet,
2004).
Em 1988, Davis sintetizou a VPI-5 com 18 membros de abertura no anel principal e
poros com diâmetro de 12,1 Å. Seguindo a mesma tendência vários outros cientistas
também realizaram suas pesquisas como: Dessau (1990) sintetizando o ALPO-8 com 14
membros no anel principal e poros de 7,8 por 8,7 Å e Jones (1993) sintetizando o JDF-20
com anéis de 20 membros e poros de 6,2 por 14,5 Å.
Atualmente existem mais de 200 tipos de estruturas diferentes de zeólitas e peneiras
moleculares. A maioria desses materiais e suas respectivas características estruturais
podem ser visualizadas no site da International Zeolite Association (IZA), sendo este site
uma importante referência de consulta no estudo de materiais zeolíticos. Em 1992
pesquisadores da Mobil Oil Corporation descobriram a família dos silicatos e
aluminossilicatos mesoposoros M41S (Beck et al., 1994; Kresge et al., 1992). Esses
materiais vieram então para revolucionar todo o cenário do mundo catalítico, visto que
apresentavam poros excepcionalmente largos da ordem de 2-10 nm.
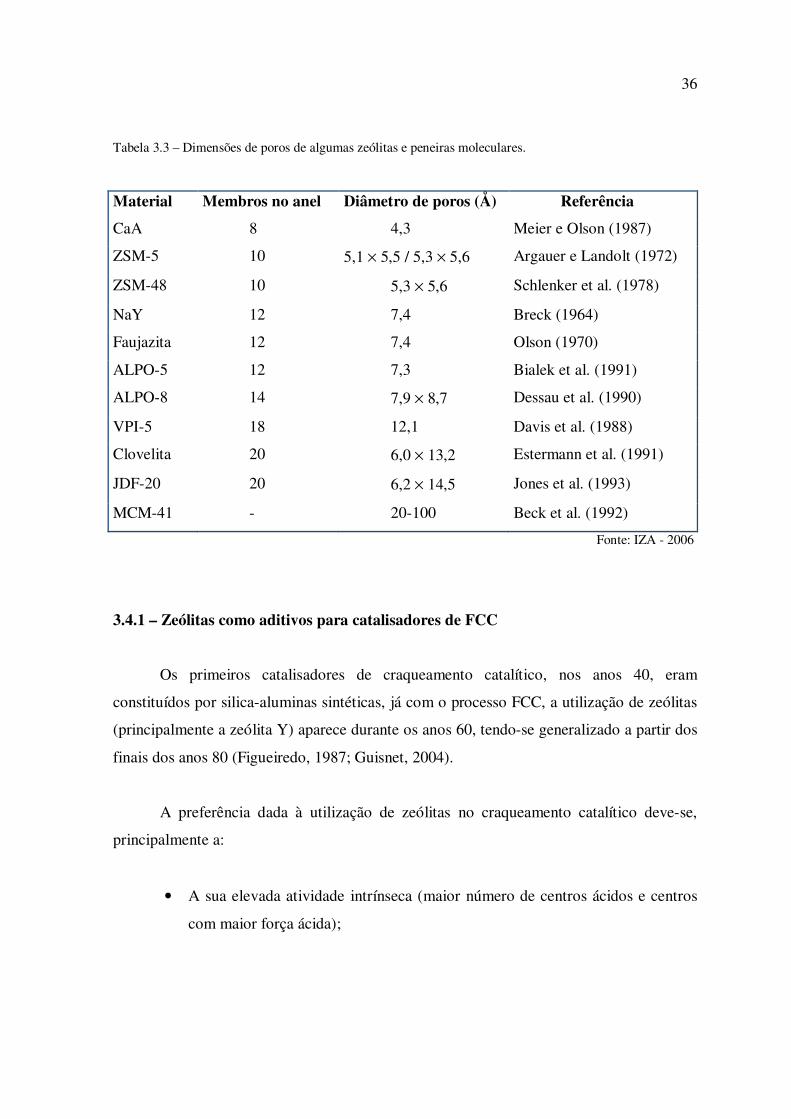
A Tabela 2 apresenta alguns exemplos de zeólitas e peneiras moleculares e suas
respectivas características estruturais. Segundo a IUPAC (Rouquerol et al., 1994) os
materiais porosos seguem uma classificação em relação aos diâmetros de seus poros:
Microporosos: dp < 2 nm (dp < 20 Å);
Mesoporosos: 2 < dp < 50 nm (20 < Dp < 500 Å);
Macroporosos: dp > 50 nm (dp > 500 Å).
36
Tabela 3.3 – Dimensões de poros de algumas zeólitas e peneiras moleculares.
Material Membros no anel Diâmetro de poros (Å) Referência
CaA 8 4,3 Meier e Olson (1987)
ZSM-5 10 5,1 × 5,5 / 5,3 × 5,6 Argauer e Landolt (1972)
ZSM-48 10 5,3 × 5,6 Schlenker et al. (1978)
NaY 12 7,4 Breck (1964)
Faujazita 12 7,4 Olson (1970)
ALPO-5 12 7,3 Bialek et al. (1991)
ALPO-8 14 7,9 × 8,7 Dessau et al. (1990)
VPI-5 18 12,1 Davis et al. (1988)
Clovelita 20 6,0 × 13,2 Estermann et al. (1991)
JDF-20 20 6,2 × 14,5 Jones et al. (1993)
MCM-41 - 20-100 Beck et al. (1992)
Fonte: IZA - 2006
3.4.1 – Zeólitas como aditivos para catalisadores de FCC
Os primeiros catalisadores de craqueamento catalítico, nos anos 40, eram
constituídos por silica-aluminas sintéticas, já com o processo FCC, a utilização de zeólitas
(principalmente a zeólita Y) aparece durante os anos 60, tendo-se generalizado a partir dos
finais dos anos 80 (Figueiredo, 1987; Guisnet, 2004).
A preferência dada à utilização de zeólitas no craqueamento catalítico deve-se,
principalmente a:
• A sua elevada atividade intrínseca (maior número de centros ácidos e centros
com maior força ácida);

37
• A sua boa seletividade para a produção de gasolinas, combinada com uma
menor produção de "coke" e de compostos leves C1-C3 (dada a estrutura
microporosa do zeólita).
Os catalisadores industriais de craqueamento contêm cerca de 5-40 % de zeólita Y,
dispersa numa matriz ativa, ligada por um ligante ("binder"). O tipo de matriz ativa
utilizada (alumina, sílica ou silica-alumina amorfa) condiciona a forma e a dimensão das
partículas de catalisador, atua como uma resistência à abrasão dos cristais da zeólita (1-5
µm) e fornece uma estrutura porosa adequada à difusão dos hidrocarbonetos (Figueiredo,
1987; Guisnet, 2004).
Ainda por (dependendo da sua composição) apresentar atividade catalítica
significativa para as reações de craqueamento, o que é de particular importância no
craqueamento de moléculas de hidrocarbonetos pesados, que se difundem mais facilmente
na estrutura aberta da matriz (poros de 50-150 Å) do que na microporosidade da zeólita
(7,4 Å). Na realidade as moléculas de cargas mais pesadas sofrem um "pré-craqueamento"
na estrutura da matriz, transformando-se em moléculas menores que, por fim, reagem nos
poros da zeólita (Figueiredo, 1987).
As matrizes ativas são ainda importantes dada a sua capacidade de reter metais que
entram na composição da alimentação (vanádio e níquel) e que são importantes venenos
das zeólitas; têm ainda a capacidade de reter moléculas de nitrogênio que vêm misturadas
na alimentação e que se adsorveriam fortemente nos centros da zeólita (Figueiredo, 1987).
São, no entanto, menos seletivas relativamente à produção de coque (2,5 a 6,5 %
em peso, se forem utilizadas cargas parafinicas ou aromáticas e/ou se tratem de matrizes de
baixa ou de alta área específica (Figueiredo, 1987).
Numa unidade de FCC a zeólita fresca é adicionada ao reator e sofre um
envelhecimento acelerado devido, principalmente, ao fato de passar mais de 80% da sua
vida útil no regenerador, onde se observam elevadas temperaturas de funcionamento (680-
750ºC), associadas a uma elevada pressão parcial de vapor de água (tipicamente 0,2 bar)
(Szklo, 2005).

38
Nestas circunstâncias a zeólita sofre uma desaluminação (perda de átomos de
alumínio da estrutura), que provoca uma diminuição do parâmetro de célula unitária e
conseqüentemente perda de atividade catalítica. Uma desaluminação em larga escala pode
levar mesmo ao colapso da estrutura zeolítica.
Um esquema mais detalhado da estrutura de catalisadores de FCC pode ser visto na
Figura 3, onde e observa que além dos componentes básicos, tais como a matriz de sílica-
alumina, o agente de granulometria, a parte zeolítica e o ligante, também que estes
possuem uma cobertura porosa e homogênea da superfície externa do catalisador, a base de
capturadores químicos de metais contaminantes,V, Ni, Fe (linha pontilhada vermelha -
nanofiltro de La2O3, BaTiO3). As principais zeólitas empregadas como aditivos são
atualmente as zeólitas Y.
Figura 3.8 – Representação da arquitetura de um catalisador de FCC (fonte: petrobras).

39
3.4.2 – Zeólita ZSM-12
A zeólita ZSM-12 é um membro da família pentasil, desenvolvida pela Mobil Co.
sendo as primeiras zeólitas a apresentar estruturas com anéis de cinco tetraedros como
unidade básica de formação de suas redes cristalinas (de onde se origina o nome de família
pentasil). Os membros mais conhecidos dessa família são a ZSM-5, ZSM-11, e a ZSM-12
(Szostak, 1989).
A zeólita ZSM-12 possui uma estrutura com poros unidimensionais de abertura
elíptica formadas por 12 tetraedros TO4, com diâmetro de 5,7 x 6,1 Angstrons (Figura 3.9)
sendo considerada uma zeólita de poros grandes (Meier,1996).
Na figura 3.9 abaixo podemos observam os poros de forma elíptica da zeólita ZSM-12, que, ao se sobreporem, formam canais retilíneos.
Figura 3.9 – Estrutura da Zeólita ZSM-12.

40
3.5 – Técnica para caracterização dos catalisadores zeolíticos: Difração de Raios-X
Em 1895 William Röntgen descobriu os raios-X, os quais foram definidos como
radiações eletromagnéticas cujo comprimento de onda varia de 0,1 a 100 Å. A técnica de
Difração de raios-X baseia-se no uso dessas radiações de forma controlada em um
equipamento para se obter informações sobre as propriedades de um determinado material
(Formoso et al., 1985).
Essa técnica tem muitas aplicações, dentre elas podemos citar:
a) Determinação da estrutura cristalina e grau de cristalinidade;
b) Identificação e análise quantitativa de fases;
c) Determinação de parâmetros da cela unitária;
d) Determinação da textura e tamanho dos cristalitos.
O material pode ser analisado na forma de sólidos em pó, monocristais, matrizes,
folhas e fibras. As amostras consistem em monocristais de 0,1 a 0,5mm de lado e pós (da
ordem gramas). Apesar de ser bastante empregada em catálise, principalmente na
determinação da estrutura cristalina de zeólitas e peneiras moleculares, a técnica apresenta
também suas limitações, dentre elas:
a) Usada apenas em materiais cristalinos. Materiais amorfos geralmente não
reproduzem difração proveitosa.
b) Picos sobrepostos podem atrasar a identificação na análise quantitativa.
c) Efeitos de matriz: materiais fortemente difratados podem encobrir os
fracamente difratados.
d) Amostras fluorescentes podem elevar a linha de difração ou pode causar
saturação em certos tipos de detectores.

41
A equação da lei de Bragg para a difração (Santos, 1989) e apresentada como:
nλ = 2dsin(θ) (3.9)
Onde n é a ordem de reflexão (n = {1,2,3,...}), λ é o comprimento de onda, d é a
distância interplanar e θ é o ângulo de incidência entre os planos reticulados.
A Equação 3.9 pode ser obtida pela análise matemática da Figura 3.10 que
representa o esboço um plano cristalino.
Figura 3.10 – Esquema representativo para formulação da lei de Bragg.
O princípio de obtenção dos raios-X consiste em se excitar átomos ou íons no
interior de uma fonte selada, mantida sobe alto vácuo (Formoso et al., 1985). Este tubo
consiste basicamente de um filamento aquecido (cátodo), geralmente de tungstênio,
funcionando como fonte de elétrons, e um alvo (ânodo) que pode ser formado por diversos
metais (cobre, molibdênio, cobalto, etc).
A aplicação de uma diferença de potencial entre o cátodo e o ânodo faz com que os
elétrons emitidos pelo filamento incandescente sejam acelerados em direção do ânodo,
quando estes colidem com metal do ânodo ocorre a transformação da energia cinética

42
adquirida pelos elétrons em calor e, em menor extensão em raios-X. Através de uma
pequena abertura, essa radiação primária deixa o tubo e segue em direção ao material a ser
analisado (Formoso et al., 1985).
Um método bastante empregado para a análise de raios-X é o método do pó (Settle,
1997), o qual é aplicado para materiais difíceis de preparar, tais como os monocristais. O
método consiste basicamente em uniformizar a amostra de modo a torná-la um pó fino e
homogêneo. Quando esse pó é colocado no porta-amostra do equipamento um grande
número de pequenos cristalitos é orientado em todas as direções possíveis. Dessa forma,
quando um feixe de raios-X atravessa o material, um número significante de partículas
estão orientadas de tal forma que a condição de Bragg para a reflexão de cada possível
distância interplanar seja obedecida (Equação 3.9).

43
4. MATERIAIS E MÉTODOS
4.1. Obtenção das zeólitas ZSM-12 com diferentes razões SiO2/Al2O3
As zeólitas ZSM-12 com diferentes razões SiO2/Al2O3 foram sintetizadas
hidrotermicamente (Araujo e colaboradores, 2004) usando-se sílica gel como fonte de
silício (Merck), pseudobohemita Catapal B (70% Al2O3 da Vista Chemical) como fonte de
alumínio, hidróxido de sódio como fonte de sódio (98%, Merck) e cloreto de
metiltrietilamônio como direcionador estrutural (97% da Sigma). As zeólita foram
preparadas com razões de SiO2/Al2O3 = 50, 80, 100 e 150. A composição da mistura
reacional está indicada na Tabela 3. A razão SiO2/Al2O3 é o dobro da razão Si/Al, visto que
nessa razão para cada mol de sílica se tem 1 mol de silício e para cada mol da alumina se
tem 2 mols de alumínio.
Tabela 4.1 - Composição da mistura reacional utilizada para síntese das zeólitas ZSM-12.
SiO2/Al2O3 Composição do Gel
50 20 MTEA : 10 Na2O : 1,0 Al2O3 : 50 SiO2 : 2000 H2O
80 20 MTEA : 10 Na2O : 1,0 Al2O3 : 80 SiO2 : 2000 H2O
100 20 MTEA : 10 Na2O : 1,0 Al2O3 : 100 SiO2 : 2000 H2O
150 20 MTEA : 10 Na2O : 1,0 Al2O3 : 150 SiO2 : 2000 H2O
A cristalização das amostras foi realizada em vasos de teflon revestidos por
autoclave de aço inox, em temperatura de 140 ºC por um período de 144 horas. O pH final
do sistema foi de 12-13. O sólido resultante do processo de cristalização hidrotérmica foi
separado do líquido sobrenadante por filtração a vácuo, lavado com água destilada e seco
em estufa a 100 oC por 12 horas. Estas condições de cristalização e recuperação do sólido

44
obtido foram idênticas em todas as sínteses realizada neste estudo. Esse material foi
submetido à calcinação em temperatura de 550 oC por 1 hora em atmosfera de N2. Em
seguida, o fluxo de N2 é substituído por atmosfera de ar mantendo-se a mesma temperatura
de 550 oC por 9 horas (Silva e colaboradores, 2005). Este procedimento de calcinação visa
a remoção do direcionador estrutural orgânico (MTEA) de maneira mais branda, evitando
que este seja queimado rapidamente pelo oxigênio. O que acarretaria na liberação de
grandes quantidades de calor dentro dos poros do catalisador podendo dessa forma, causar
danos à estrutura cristalina da zeólita.
Para obtenção das zeólitas na forma ácida as amostras ZSM-12 foram submetidas a
uma troca iônica com uma solução 1M de cloreto de amônio a 70 ºC por 2 horas. Em
seguida as zeólitas obtidas passam por uma nova etapa de calcinação para a obtenção da
forma ácida final.
4.2. Caracterização via Difração de Raios-X das zeólitas
A identificação das fases cristalinas presentes nas zeólitas foi realizada através das
análises de Difração de Raios-X (DRX). Os difratogramas de raios-X foram obtidos em um
difratômetro XRD-6000 da Shimadzu, utilizando CuKα como fonte de radiação, filtro de
níquel e 2θ na faixa de 1 a 50º.
Uma outra propriedade que pode ser obtida da difração de raios X é o grau de
cristalinidade dos materiais a partir da comparação com o difratograma de uma amostra
padrão obtido nas mesmas condições de análise. O grau de cristalinidade dos materiais em
estudo foi determinado por dois métodos: o primeiro baseado na comparação da soma das
áreas dos picos mais intensos do difratograma (2θ = 7,36º; 8,80º; 20,88º; 22,88º e 23,20º)
(Treacy et al., 1996) com a soma das áreas de uma amostra padrão; o segundo método
utiliza os mesmos picos só que ao invés da soma áreas, ele tem como base à soma das
intensidades. A partir destes dados foi possível verificar a influência da razão SiO2/Al2O3.

45
4.3. Testes Catalíticos de craqueamento de n-heptano sobre ZSM-12 com
diferentes razões SiO2/Al2O3
Os ensaios catalíticos de craqueamento de n-heptano sobre as zeólitas ZSM-12 com
diferentes razões SiO2/Al2O3 foram realizados em uma unidade de avaliação catalítica
modelo TCAT-10 à pressão atmosférica, instalada no Laboratório de Catálise da
Universidade Federal do Rio Grande do Norte - UFRN. Foram utilizadas as razões de
SiO2/Al2O3 de 50, 80, 100 e 150.
Como molécula modelo (molécula sonda) foi escolhida o n-heptano, que se
caracteriza juntamente com seus isômeros por ser uma substância com dimensões
moleculares adequadas de acessibilidade nos poros da zeólita, além de ser uma molécula
representativa de destilados médios de petróleo.
Todos os testes foram realizados à pressão atmosférica.
Os procedimentos empregados nos testes foram constituídos de:
a) Pesar cerca de 200 mg da amostra de zeólita e introduzir no reator em “U”
de vidro pyrex;
b) Aquecer o reator da temperatura ambiente até 450oC numa taxa de
aquecimento de 5oC min-1 em atmosfera dinâmica de N2 com fluxo de 30
mL min-1 (ativação);
c) Após atingir 450oC, a amostra permaneceu por mais 1 hora nessas
condições e em seguida o n-heptano contido num saturador foi utilizado
para saturar o N2 e a mistura foi direcionada ao leito catalítico.
d) Durante a reação o leito catalítico foi mantido a temperatura constante de
450o C através de um controlador de temperatura COEL HW1500.

46
e) Os produtos efluentes do reator foram sucessivamente injetados “on-line”
por uma válvula de dez vias em um cromatógrafo a gás Varian CP3800
com detector de condutividade térmica em intervalos de 15 minutos até
alcançar o estado estacionário. Utilizamos os tempos de: 0, 15, 30, 45, 60 e
75 minutos.
f) Os produtos foram separados e analisados numa coluna de sílica fundida de
60 m. A identificação dos produtos foi realizada através da comparação dos
tempos de retenção dos analíticos de cada cromatograma com os tempos de
retenção de padrões de n-heptano, gás natural e outros hidrocarbonetos tais
como, metano, etano, eteno, propano, propeno, butanos, pentanos e hexano,
levando em consideração as ordens de ebulição das substâncias através da
fase estacionária utilizada na coluna (separação baseada em pontos de
ebulição) conforme previsto pelo fabricante.
g) A quantificação dos cromatogramas foi realizada pelo método de padrões
externos analisados na faixa de linearidade do detector (106 unidades de
área) conforme recomendado para detectores de condutividade térmica
(Lanças, 1993).
h) Após a identificação dos cromatogramas foi possível determinar dados de
conversão e seletividade de produtos, sendo estes dados importantes na
estimativa de parâmetros cinéticos.
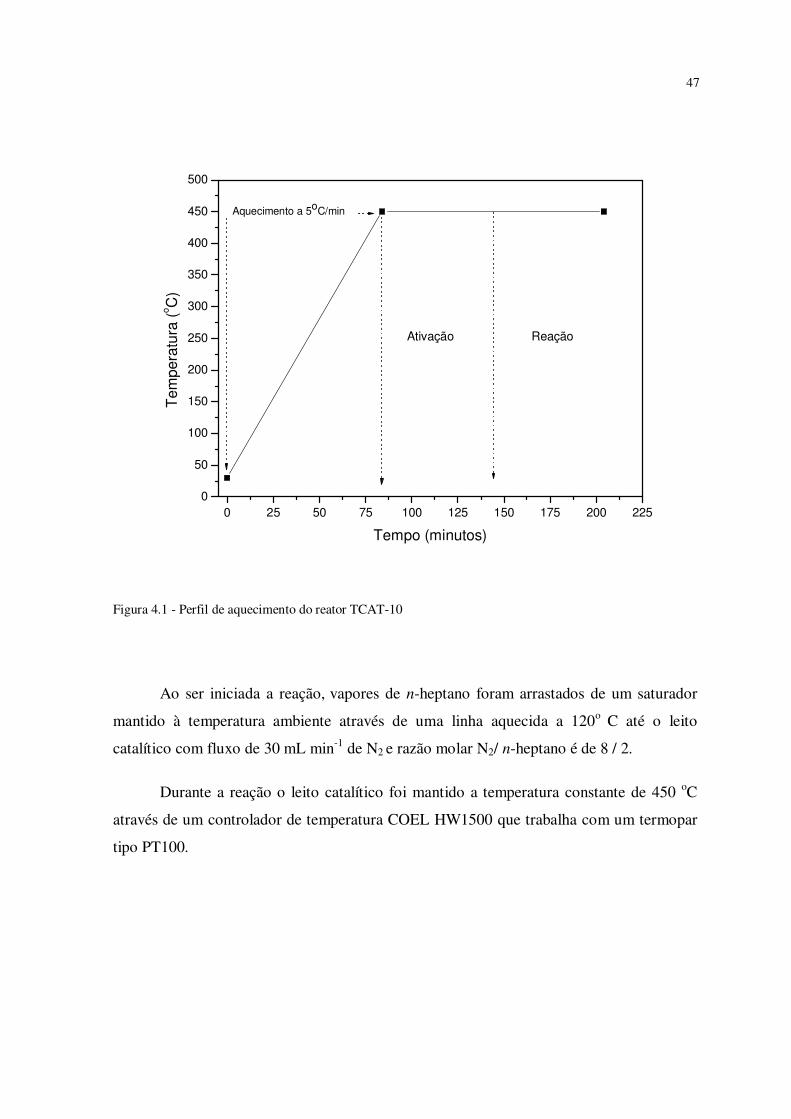
A etapa que consiste em manter amostra na temperatura de 450oC pelo tempo
mínimo de 1 hora, tem por objetivo permitir a ativação do catalisador zeolítico que será
utilizado, pois permite a ativação dos sítios ácidos do catalisador, com a remoção da água
fisissorvida na superfície das zeólitas. A Figura 4.1 mostra o perfil de aquecimento do
reator antes e durante a reação.
47
0 25 50 75 100 125 150 175 200 225
0
50
100
150
200
250
300
350
400
450
500
Reação
Tem
pera
tura
(oC
)
Tempo (minutos)
Aquecimento a 5oC/min
Ativação
Figura 4.1 - Perfil de aquecimento do reator TCAT-10
Ao ser iniciada a reação, vapores de n-heptano foram arrastados de um saturador
mantido à temperatura ambiente através de uma linha aquecida a 120o C até o leito
catalítico com fluxo de 30 mL min-1 de N2 e razão molar N2/ n-heptano é de 8 / 2.
Durante a reação o leito catalítico foi mantido a temperatura constante de 450 oC
através de um controlador de temperatura COEL HW1500 que trabalha com um termopar
tipo PT100.

48
As vazões volumétricas foram ajustadas à saída do reator por um medidor digital de
fluxo modelo ADM 1000 (Micronal). Com a operação à baixa pressão e relativamente alta
temperatura, considerou-se que os vapores afluentes do reator se comportam no estado de
gás ideal.
Os testes foram conduzidos com todos os catalisadores zeolíticos na forma de pós
(ca. 15 µm) de modo a minimizar os efeitos provenientes do transporte interno de massa,
uma vez que, para partículas micrométricas, o efeito de gradiente de massa pode ser
desconsiderado para fins de simplificação.
Foram também levados em consideração, os seguintes aspectos: reação isotérmica
em leito fixo, fase vapor em estado de gás ideal, escoamento ideal em fluxo pistonado,
porosidade uniforme e queda de pressão no leito desprezível sem a presença de efeitos de
dispersão axial.
A Figura 12 a seguir mostra um esquema ilustrativo do funcionamento da unidade
de avaliação catalítica TCAT-10 e a Figura 13 mostra uma foto da unidade utilizada para o
craqueamento catalítico do n-heptano neste experimento.
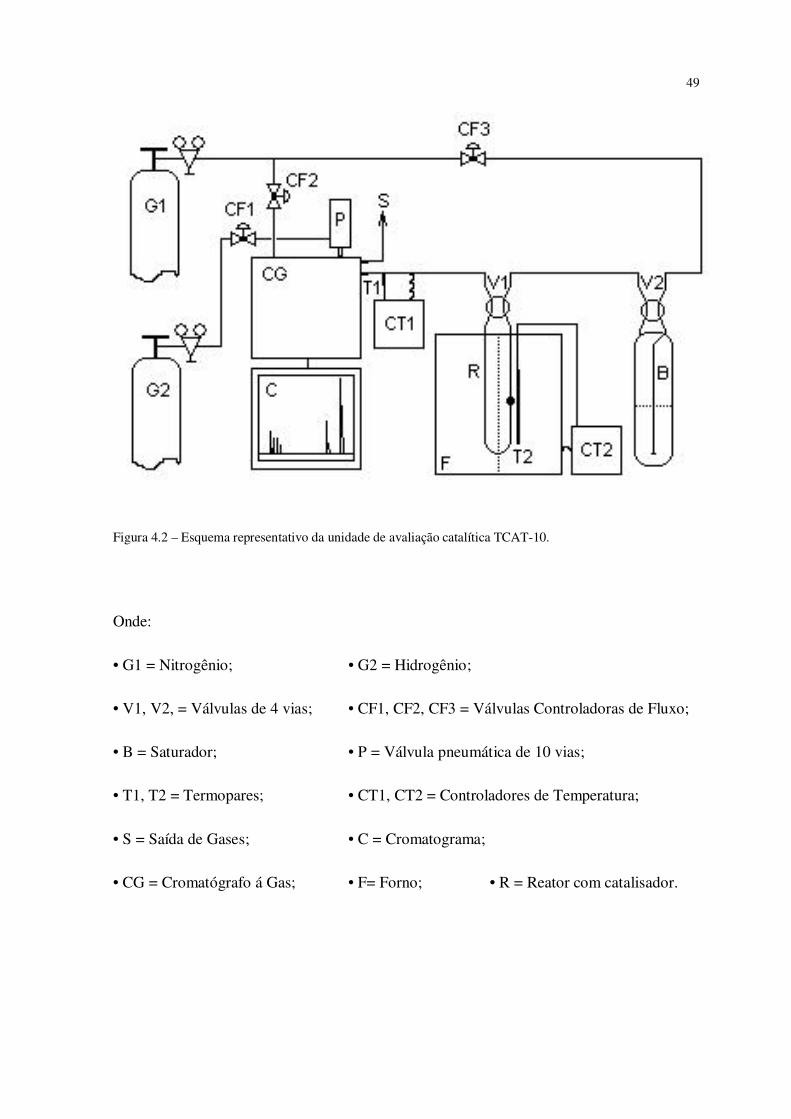
49
Figura 4.2 – Esquema representativo da unidade de avaliação catalítica TCAT-10.
Onde:
• G1 = Nitrogênio; • G2 = Hidrogênio;
• V1, V2, = Válvulas de 4 vias; • CF1, CF2, CF3 = Válvulas Controladoras de Fluxo;
• B = Saturador; • P = Válvula pneumática de 10 vias;
• T1, T2 = Termopares; • CT1, CT2 = Controladores de Temperatura;
• S = Saída de Gases; • C = Cromatograma;
• CG = Cromatógrafo á Gas; • F= Forno; • R = Reator com catalisador.

50
Figura 4.3 - Foto da unidade de avaliação catalítica TCAT-10.

51
5. RESULTADOS E DISCUSSÃO
5.1. Características estruturais das zeólitas ZSM-12 com diferentes razões
SiO2/Al2O3.
As análises de difração de raios X estão mostradas nas Figuras 11 a 14. Nos
difratogramas em que aparecem picos marcados com uma seta apontando para baixo (↓)
significa que estes picos são de fases contaminantes pertencentes a zeólita ZSM-5. Nas
Figuras de 11 a 14, com exceção da amostra com razão SiO2/Al2O3 = 50, que apresenta
uma pequena contaminação por ZSM-5, todos as demais amostras apresentam
difratogramas típicos de uma ZSM-12 pura. A presença de fases contaminantes em zeolitas
pode estar associado as condições de síntese, bem como a erros experimentais
potencialmente ocorridos, necessitando assim futuramente uma investigação maia ampla
no sentido de elucidar as causas prováveis.
Comparando-se todas as reflexões desses difratogramas a ficha padrão 47-0708 do
banco de dados JCPDS (Kuehl, G., 1984) e a outros dados da literatura (Kiniski et al.,
2002), pode-se observar que as demais amostras são formadas por um material monofásico
com estrutura do tipo ZSM-12.

52
Figura 5.1 – Difratograma de Raios-X da amostra de ZSM-12 com SiO2/Al2O3 = 50.

53
Figura 5.2 – Difratograma de Raios-X da amostra de ZSM-12 com SiO2/Al2O3 = 80.
Figura 5.3 - Difratograma de Raios-X da amostra de ZSM-12 com SiO2/Al2O3 = 100.

54
Figura 5.4 - Difratograma deRraios-X da amostra de ZSM-12 com SiO2/Al2O3 = 150.
O grau de cristalinidade das amostras foi calculado com base nas áreas e nas
intensidades dos cinco picos principais do difratograma. Os dados da Tabela 5 indicam que
o grau de cristalinidade aumenta à medida que a razão SiO2/Al2O3 no gel diminui, (exceto
na razão SiO2/Al2O3 de 80). Observou-se uma tendência de que uma maior quantidade de
alumínio na mistura reacional dificulta a cristalização da ZSM-12. Resultados similares
foram relatados por Ernst et al. (1987) e Gopal et al. (2002) para a síntese de ZSM-12.
Tabela 5.1 - Grau de cristalinidade das amostras de ZSM-12.
Grau de Cristalinidade (%) SiO2/Al2O3
Através da Área dos Picos Através da Intensidade dos Picos
50 82 77
80 100 100
100 87 83
150 99 96

55
5.2 - Testes Catalíticos de craqueamento de n-heptano sobre ZSM-12 com
diferentes razões SiO2/Al2O3
Os ensaios catalíticos de craqueamento de n-heptano sobre as zeólitas ZSM-12 com
razões SiO2/Al2O3 de 50, 80, 100 e 150, foram realizados em uma unidade de avaliação
catalítica modelo TCAT-10 à pressão atmosférica, à mesma temperatura de 450ºC. Foram
obtidas seis cromatogramas de cada uma das amostras de ZSM-12 em intervalos regulares
de 15 minutos, sendo este número de cromatogramas suficiente para ser verificar uma
estabilidade nos valores de conversão em função do tempo. As Tabelas 6 a 9 mostram os
dados experimentais de conversão (X) e seletividade (S) com função do tempo de reação e
razão SiO2/Al2O3. Os dados relativos a conversão foram obtidos através da Equação 5.1 e
os dados de seletividade através da Equação 5.2. Os cálculos dos números de mols iniciais
(nio) e em um tempo qualquer (ni) de cada espécie química envolvida na reação,
necessários aos cálculos de conversão e seletividade, foram realizados com base nas curvas
analíticas constantes no método do cromatográfico de análise, onde se relacionavam os
valores de resposta em área como função do volume injetado para cada substância para
cada cromatograma, seja em base volumar ou molar.
100(%) x
n
nn
X
io
iio−
= (5.1)
100(%)1
x
n
n
Si
i
i
∑= (5.2)
No caso da conversão, este conceito refere-se fração percentual do reagente
limitante de partida (n-heptano), que foi convertido em produtos. A utilização de diferentes
catalisadores e condições operacionais, pode originar várias reações simultaneamente,
originando diferentes produtos. Deste modo, o conceito de seletividade refere-se fração
percentual de cada produto específico que foi obtido a partir do craqueamento do reagente
limitante de partida (n-heptano) ou oriundos de outras reações paralelas.
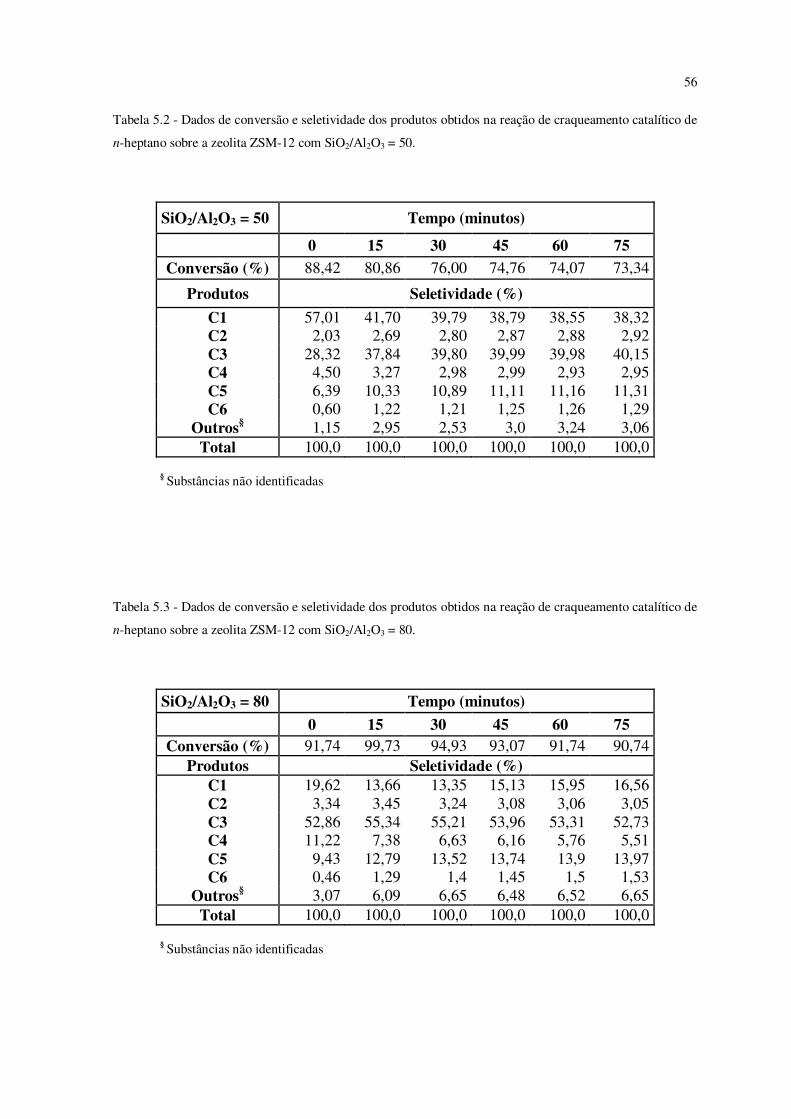
56
Tabela 5.2 - Dados de conversão e seletividade dos produtos obtidos na reação de craqueamento catalítico de
n-heptano sobre a zeolita ZSM-12 com SiO2/Al2O3 = 50.
SiO2/Al2O3 = 50 Tempo (minutos)
0 15 30 45 60 75 Conversão (%) 88,42 80,86 76,00 74,76 74,07 73,34
Produtos Seletividade (%)
C1 57,01 41,70 39,79 38,79 38,55 38,32 C2 2,03 2,69 2,80 2,87 2,88 2,92 C3 28,32 37,84 39,80 39,99 39,98 40,15 C4 4,50 3,27 2,98 2,99 2,93 2,95 C5 6,39 10,33 10,89 11,11 11,16 11,31 C6 0,60 1,22 1,21 1,25 1,26 1,29
Outros§ 1,15 2,95 2,53 3,0 3,24 3,06 Total 100,0 100,0 100,0 100,0 100,0 100,0
§ Substâncias não identificadas
Tabela 5.3 - Dados de conversão e seletividade dos produtos obtidos na reação de craqueamento catalítico de
n-heptano sobre a zeolita ZSM-12 com SiO2/Al2O3 = 80.
SiO2/Al2O3 = 80 Tempo (minutos)
0 15 30 45 60 75 Conversão (%) 91,74 99,73 94,93 93,07 91,74 90,74
Produtos Seletividade (%) C1 19,62 13,66 13,35 15,13 15,95 16,56 C2 3,34 3,45 3,24 3,08 3,06 3,05 C3 52,86 55,34 55,21 53,96 53,31 52,73 C4 11,22 7,38 6,63 6,16 5,76 5,51 C5 9,43 12,79 13,52 13,74 13,9 13,97 C6 0,46 1,29 1,4 1,45 1,5 1,53
Outros§ 3,07 6,09 6,65 6,48 6,52 6,65 Total 100,0 100,0 100,0 100,0 100,0 100,0
§ Substâncias não identificadas
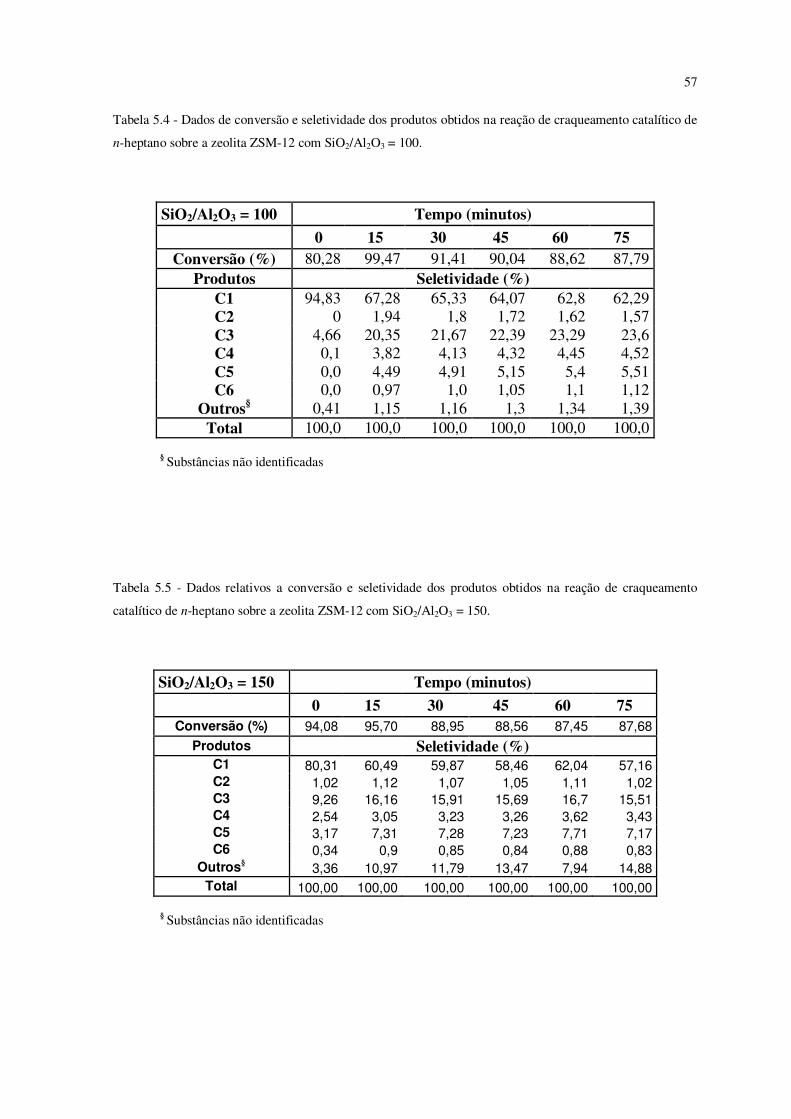
57
Tabela 5.4 - Dados de conversão e seletividade dos produtos obtidos na reação de craqueamento catalítico de
n-heptano sobre a zeolita ZSM-12 com SiO2/Al2O3 = 100.
SiO2/Al2O3 = 100 Tempo (minutos)
0 15 30 45 60 75 Conversão (%) 80,28 99,47 91,41 90,04 88,62 87,79
Produtos Seletividade (%) C1 94,83 67,28 65,33 64,07 62,8 62,29 C2 0 1,94 1,8 1,72 1,62 1,57 C3 4,66 20,35 21,67 22,39 23,29 23,6 C4 0,1 3,82 4,13 4,32 4,45 4,52 C5 0,0 4,49 4,91 5,15 5,4 5,51 C6 0,0 0,97 1,0 1,05 1,1 1,12
Outros§ 0,41 1,15 1,16 1,3 1,34 1,39 Total 100,0 100,0 100,0 100,0 100,0 100,0
§ Substâncias não identificadas
Tabela 5.5 - Dados relativos a conversão e seletividade dos produtos obtidos na reação de craqueamento
catalítico de n-heptano sobre a zeolita ZSM-12 com SiO2/Al2O3 = 150.
SiO2/Al2O3 = 150 Tempo (minutos)
0 15 30 45 60 75 Conversão (%) 94,08 95,70 88,95 88,56 87,45 87,68
Produtos Seletividade (%) C1 80,31 60,49 59,87 58,46 62,04 57,16 C2 1,02 1,12 1,07 1,05 1,11 1,02 C3 9,26 16,16 15,91 15,69 16,7 15,51 C4 2,54 3,05 3,23 3,26 3,62 3,43 C5 3,17 7,31 7,28 7,23 7,71 7,17 C6 0,34 0,9 0,85 0,84 0,88 0,83
Outros§ 3,36 10,97 11,79 13,47 7,94 14,88
Total 100,00 100,00 100,00 100,00 100,00 100,00
§ Substâncias não identificadas

58
5.3 – Superfícies resposta para a relação entre conversão e seletividade em
função do tempo de reação, razão SiO2/Al2O3 e grau de cristalinidade das zeólitas
ZSM-12.
As reações de craqueamento catalítico de n-heptano sobre a zeólita ZSM-12 com
diferentes razões SiO2/Al2O3 foram realizadas tendo como variáveis a razão SiO2/Al2O3 e o
tempo de reação. Foram utilizadas as razões SiO2/Al2O3 de 50, 80, 100 e 150 e os tempos
de reação de 0, 15, 30, 45, 60 e 75 minutos. Os dados de conversão e seletividade obtidos
em função da razão SiO2/Al2O3 e do tempo de reação foram interpolados numericamente
gerando uma matriz 100 por 100 pontos, através do método de Kriging (Anexo A) (Davis,
1986), gerando assim as superfícies de resposta que relacionam a conversão e seletividade
em função da razão SiO2/Al2O3 e do tempo de reação.
A geração de todos os gráficos deste trabalho, bem como o tratamento numérico
para a obtenção das superfícies de resposta que relacionam a conversão e seletividade com
a razão SiO2/Al2O3, tempo de reação e grau de cristalinidade das zeólitas, foram feitos
utilizando o software Microcal Origin® versão 6.0. Foi observado então que este pacote
computacional foi satisfatório para análise dos dados e possibilitou a determinação de
pontos e/ou regiões de otimização de variáveis de saída experimentais (conversão e
seletividade) para as reações de craqueamento de n-heptano.
5.4 – Efeito do tempo de reação, razão SiO2/Al2O3 e grau de cristalinidade na
conversão catalítica do n-heptano.
O gráfico que relaciona a conversão do n-heptano em função do tempo de reação,
em diferentes razões SiO2/Al2O3, e visualizado na Figura 15. Pode-se observar em todos os
casos que em tempos superiores à 60 minutos a conversão começa cair a estabilizar,
alcançando provavelmente o estado pseudo-estacionário. Nesse estado as variáveis do
sistema variam muito pouco significativamente com o tempo. A queda na conversão
também ocorre devido a fenômenos de desativação por formação de coque, forma de
desativação tipicamente encontrada em catalisadores de craqueamento. Observa-se que
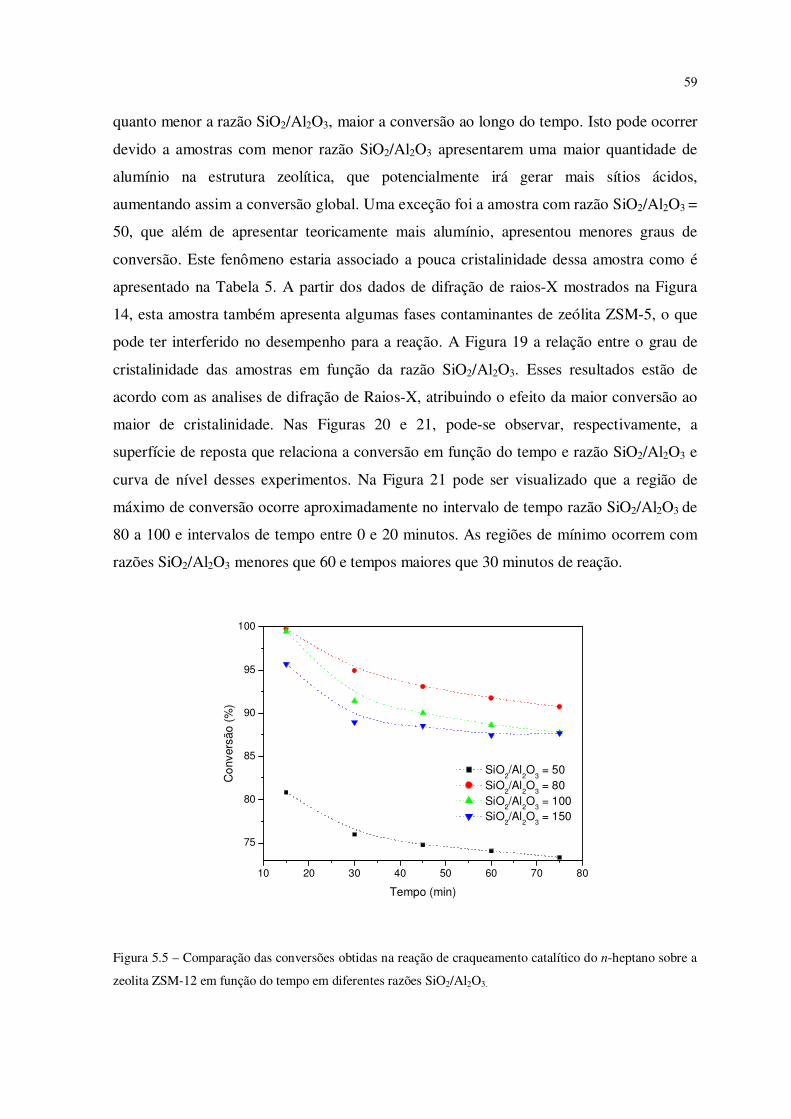
59
quanto menor a razão SiO2/Al2O3, maior a conversão ao longo do tempo. Isto pode ocorrer
devido a amostras com menor razão SiO2/Al2O3 apresentarem uma maior quantidade de
alumínio na estrutura zeolítica, que potencialmente irá gerar mais sítios ácidos,
aumentando assim a conversão global. Uma exceção foi a amostra com razão SiO2/Al2O3 =
50, que além de apresentar teoricamente mais alumínio, apresentou menores graus de
conversão. Este fenômeno estaria associado a pouca cristalinidade dessa amostra como é
apresentado na Tabela 5. A partir dos dados de difração de raios-X mostrados na Figura
14, esta amostra também apresenta algumas fases contaminantes de zeólita ZSM-5, o que
pode ter interferido no desempenho para a reação. A Figura 19 a relação entre o grau de
cristalinidade das amostras em função da razão SiO2/Al2O3. Esses resultados estão de
acordo com as analises de difração de Raios-X, atribuindo o efeito da maior conversão ao
maior de cristalinidade. Nas Figuras 20 e 21, pode-se observar, respectivamente, a
superfície de reposta que relaciona a conversão em função do tempo e razão SiO2/Al2O3 e
curva de nível desses experimentos. Na Figura 21 pode ser visualizado que a região de
máximo de conversão ocorre aproximadamente no intervalo de tempo razão SiO2/Al2O3 de
80 a 100 e intervalos de tempo entre 0 e 20 minutos. As regiões de mínimo ocorrem com
razões SiO2/Al2O3 menores que 60 e tempos maiores que 30 minutos de reação.
10 20 30 40 50 60 70 80
75
80
85
90
95
100
Co
nvers
ão (
%)
Tempo (min)
SiO2/Al
2O
3 = 50
SiO2/Al
2O
3 = 80
SiO2/Al
2O
3 = 100
SiO2/Al
2O
3 = 150
Figura 5.5 – Comparação das conversões obtidas na reação de craqueamento catalítico do n-heptano sobre a
zeolita ZSM-12 em função do tempo em diferentes razões SiO2/Al2O3.
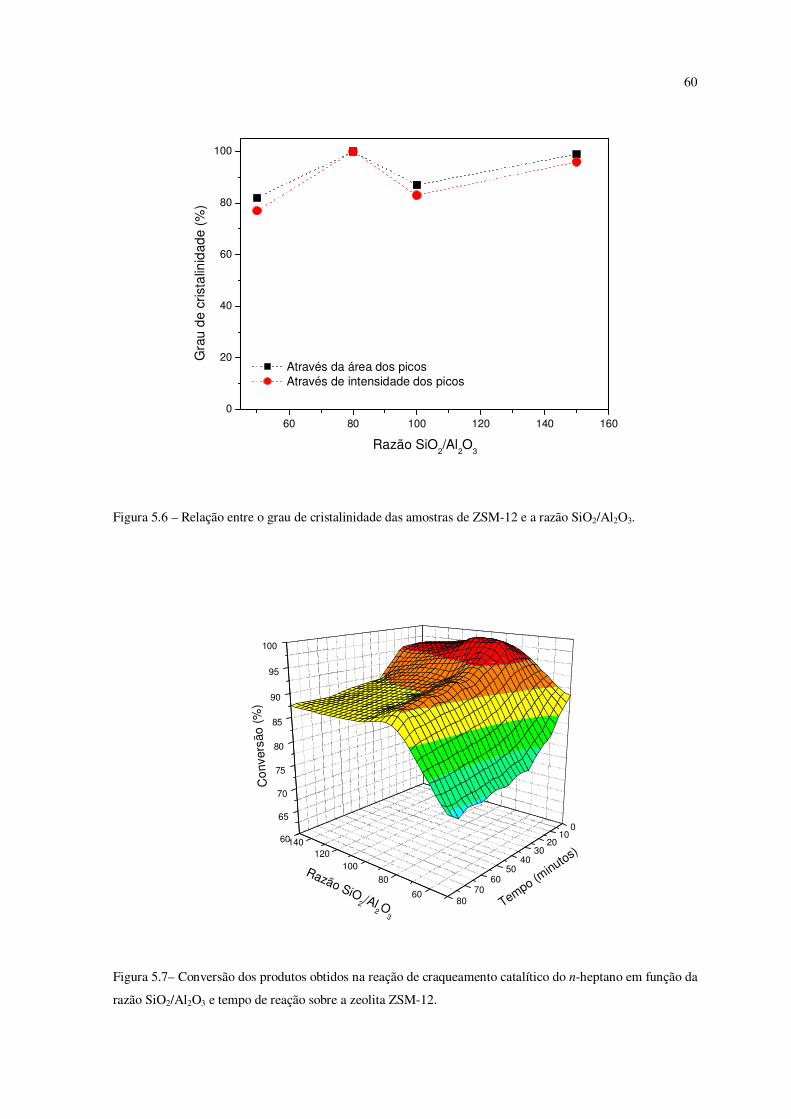
60
60 80 100 120 140 160
0
20
40
60
80
100
Gra
u d
e c
rista
linid
ad
e (
%)
Razão SiO2/Al
2O
3
Através da área dos picos Através de intensidade dos picos
Figura 5.6 – Relação entre o grau de cristalinidade das amostras de ZSM-12 e a razão SiO2/Al2O3.
010
2030
4050
6070
8060
80
100
12014060
65
70
75
80
85
90
95
100
Tempo (minutos)
Razão SiO2 /Al
2 O3
Co
nvers
ão
(%
)
Figura 5.7– Conversão dos produtos obtidos na reação de craqueamento catalítico do n-heptano em função da
razão SiO2/Al2O3 e tempo de reação sobre a zeolita ZSM-12.
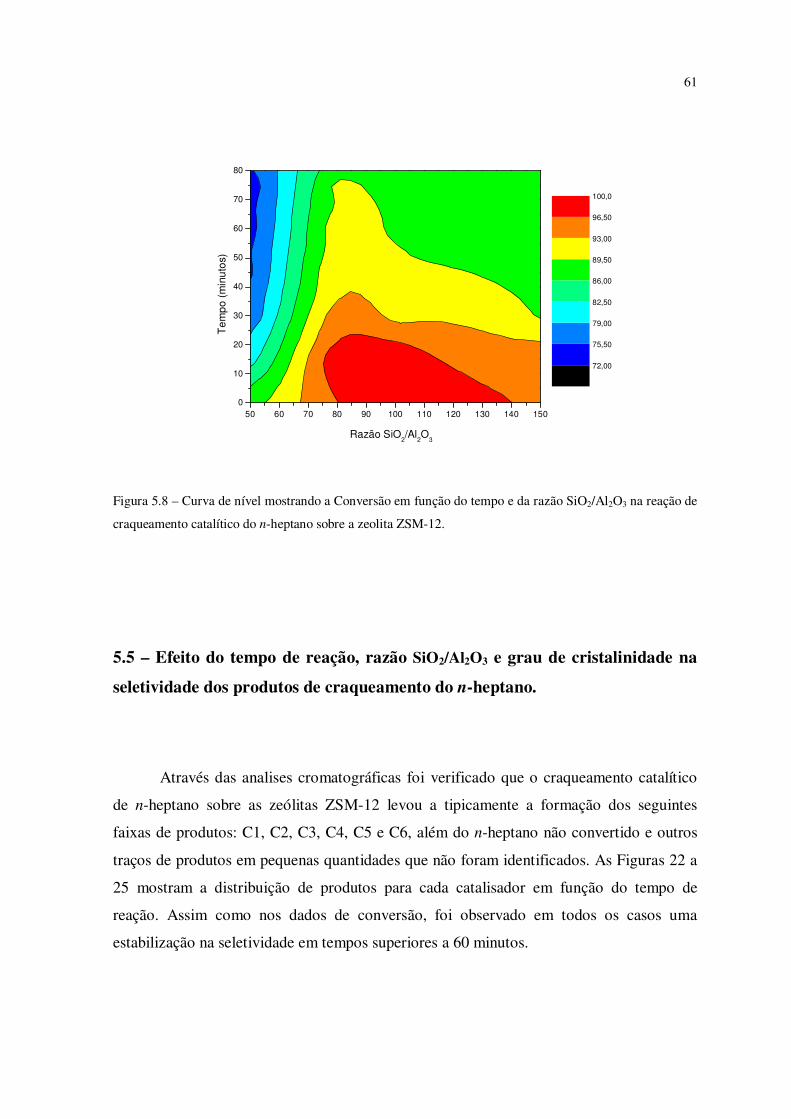
61
50 60 70 80 90 100 110 120 130 140 150
0
10
20
30
40
50
60
70
80
72,00
75,50
79,00
82,50
86,00
89,50
93,00
96,50
100,0
Razão SiO2/Al
2O
3
Te
mp
o (
min
uto
s)
Figura 5.8 – Curva de nível mostrando a Conversão em função do tempo e da razão SiO2/Al2O3 na reação de
craqueamento catalítico do n-heptano sobre a zeolita ZSM-12.
5.5 – Efeito do tempo de reação, razão SiO2/Al2O3 e grau de cristalinidade na
seletividade dos produtos de craqueamento do n-heptano.
Através das analises cromatográficas foi verificado que o craqueamento catalítico
de n-heptano sobre as zeólitas ZSM-12 levou a tipicamente a formação dos seguintes
faixas de produtos: C1, C2, C3, C4, C5 e C6, além do n-heptano não convertido e outros
traços de produtos em pequenas quantidades que não foram identificados. As Figuras 22 a
25 mostram a distribuição de produtos para cada catalisador em função do tempo de
reação. Assim como nos dados de conversão, foi observado em todos os casos uma
estabilização na seletividade em tempos superiores a 60 minutos.
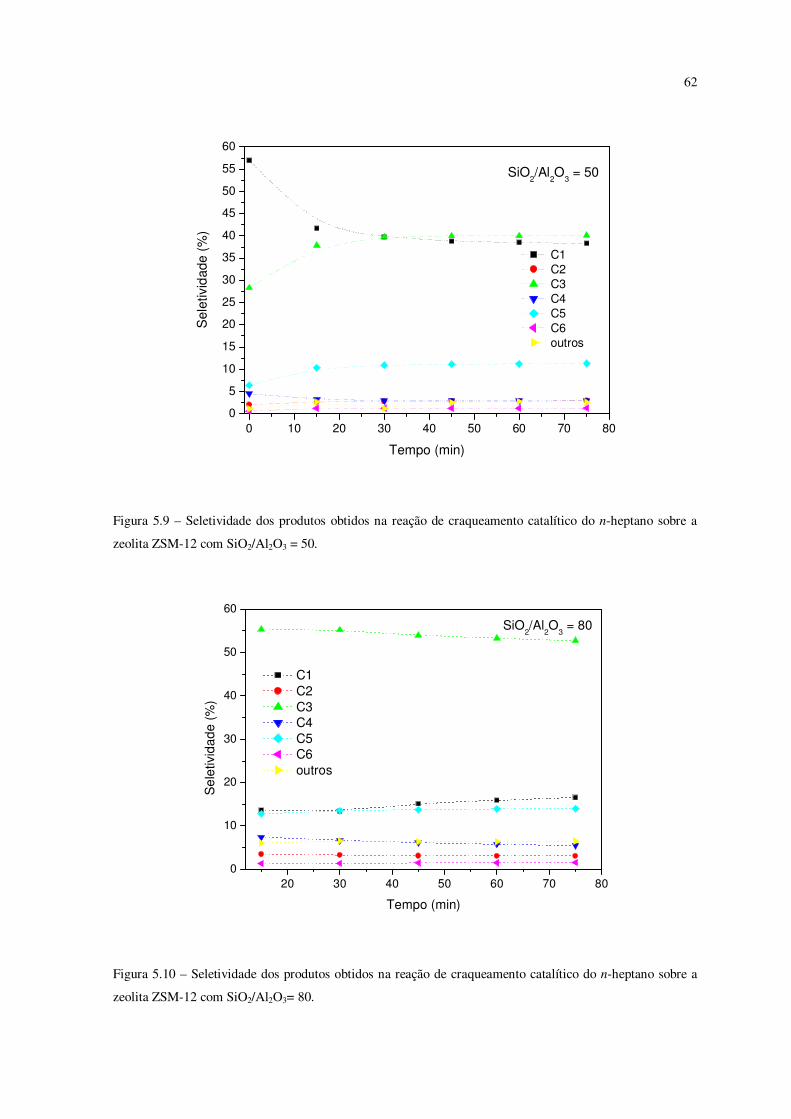
62
0 10 20 30 40 50 60 70 80
0
5
10
15
20
25
30
35
40
45
50
55
60
SiO2/Al
2O
3 = 50
Sele
tivid
ade
(%
)
Tempo (min)
C1 C2 C3 C4 C5 C6 outros
Figura 5.9 – Seletividade dos produtos obtidos na reação de craqueamento catalítico do n-heptano sobre a
zeolita ZSM-12 com SiO2/Al2O3 = 50.
20 30 40 50 60 70 80
0
10
20
30
40
50
60
SiO2/Al
2O
3 = 80
Se
letiv
ida
de (
%)
Tempo (min)
C1 C2 C3 C4 C5 C6 outros
Figura 5.10 – Seletividade dos produtos obtidos na reação de craqueamento catalítico do n-heptano sobre a
zeolita ZSM-12 com SiO2/Al2O3= 80.
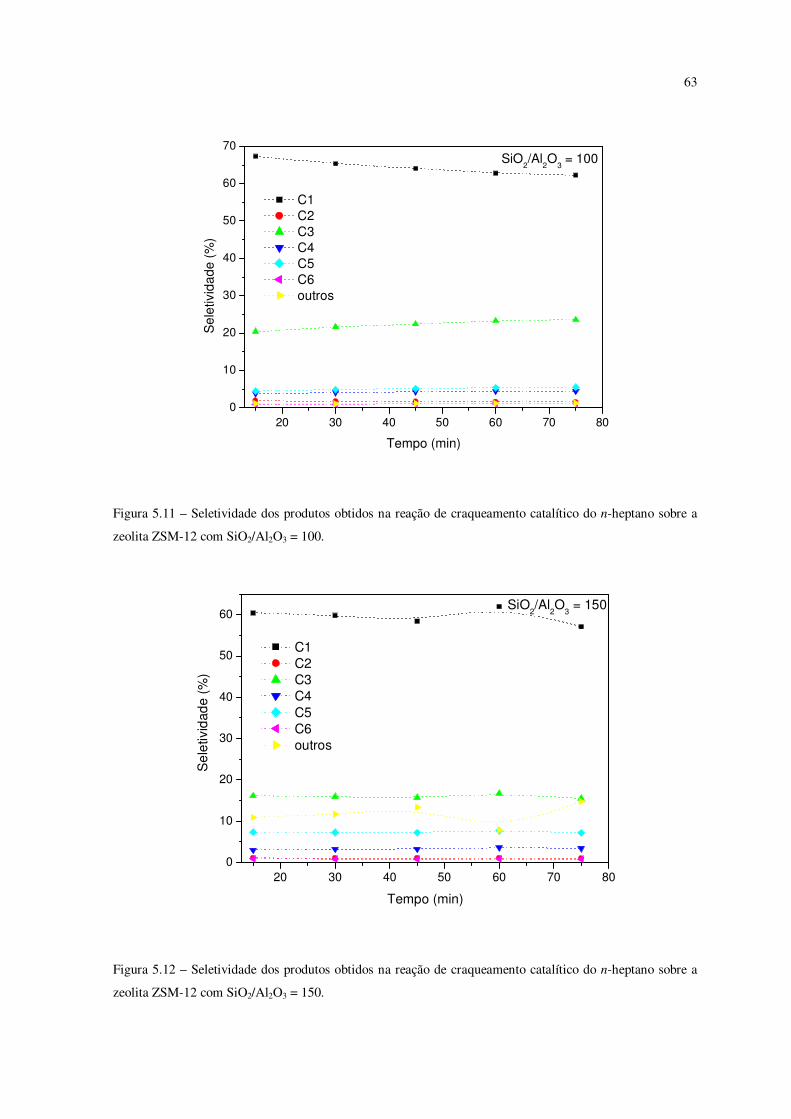
63
20 30 40 50 60 70 80
0
10
20
30
40
50
60
70SiO
2/Al
2O
3 = 100
Se
letiv
ida
de
(%
)
Tempo (min)
C1 C2 C3 C4 C5 C6 outros
Figura 5.11 – Seletividade dos produtos obtidos na reação de craqueamento catalítico do n-heptano sobre a
zeolita ZSM-12 com SiO2/Al2O3 = 100.
20 30 40 50 60 70 80
0
10
20
30
40
50
60SiO
2/Al
2O
3 = 150
Se
letivid
ad
e (
%)
Tempo (min)
C1 C2 C3 C4 C5 C6 outros
Figura 5.12 – Seletividade dos produtos obtidos na reação de craqueamento catalítico do n-heptano sobre a
zeolita ZSM-12 com SiO2/Al2O3 = 150.

64
Nas condições experimentais estudadas, foi observado que o craqueamento
catalítico de n-heptano sobre as zeólitas ZSM-12 levou a uma alta seletividade para C1 e
para C3, seguido de C5, C4, C2 e C6. Verificou-se que a seletividade para a fração C1, foi
máxima para a razão SiO2/Al2O3 de 100.
Como verificado por Corma et al., (1996), a força ácida nos aluminosilicatos
microporosos com estrutura zeolítica e sua atividade catalítica para o craqueamento de
moléculas pequenas, tais como o n-heptano, é menor quando comparada com a zeólita Y.
Entretanto os microporos do sistema unidimensional da ZSM-12 podem oferecer grandes
vantagens no craqueamento dessas moléculas devido a restringir um pouco a acessibilidade
nos poros, gerando um tempo de contato do reagente com a superfície zeolitica adequado
para o aumento da conversão e seletividade em hidrocarbonetos menores. (Reschetilowski
et al., 1997; Reschetilowski e Koch, 1998).
Com o decréscimo da razão SiO2/Al2O3 foi possível verificar um aumento
significativo da seletividade para a fração C3, atingindo o valor máximo para a razão
SiO2/Al2O3 = 80 em torno de 55 %. Considerarmos conjuntamente o somatório das frações
C1 e C3, para as quais as amostras de ZSM-12 apresentaram maior seletividade, obtemos
alta seletividade para essas frações. Como por exemplo, em torno de 70% para a razão de
SiO2/Al2O3 = 80 e em torno de 85% para a razão de SiO2/Al2O3 = 100. Foi observado que
frações mais pesadas (C6 e C5) são obtidas também, porém em quantidades menos
significativas. As Figuras 26 a 37 mostram alternadamente as superfícies de resposta e suas
respectivas curvas de nível, para a seletividade a cada produto. A análise das curvas
permite visualizar com mais clareza os limites experimentais de máximo e mínimo para a
seletividade a reação em estudo.
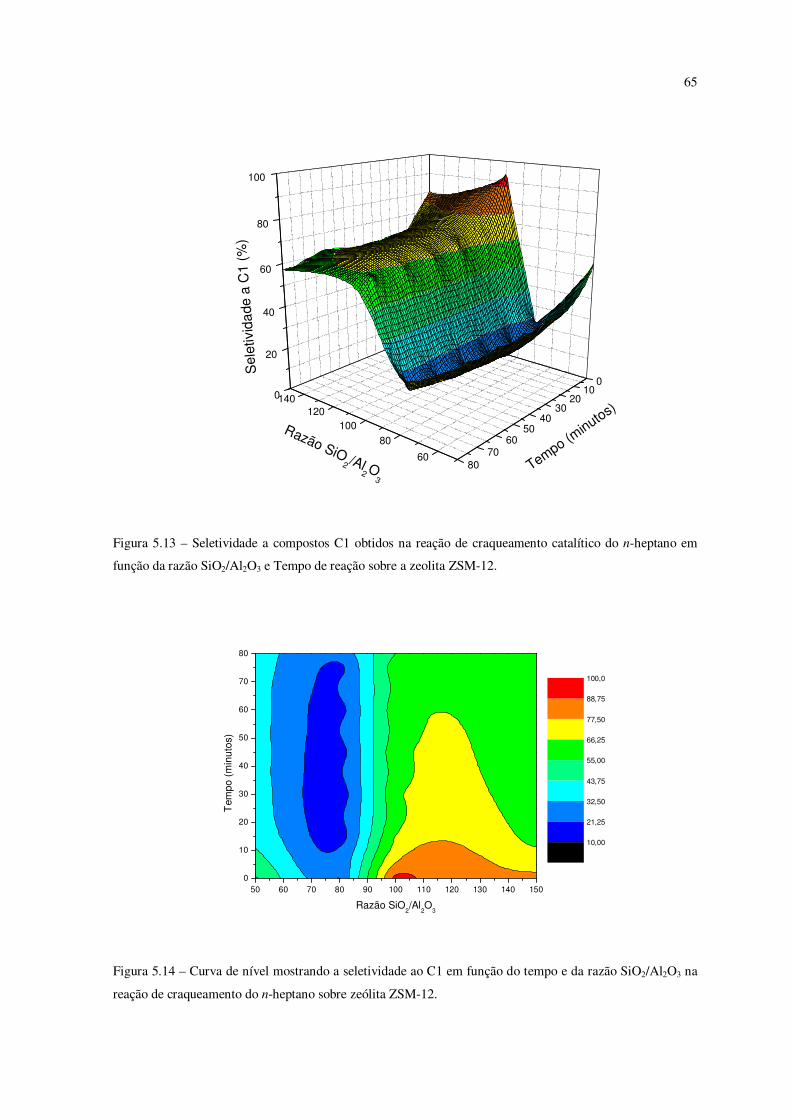
65
010
2030
4050
6070
8060
80
100
1201400
20
40
60
80
100
Tempo (m
inutos)
Razão SiO2 /Al
2 O3
Sele
tivid
ade a
C1 (
%)
Figura 5.13 – Seletividade a compostos C1 obtidos na reação de craqueamento catalítico do n-heptano em
função da razão SiO2/Al2O3 e Tempo de reação sobre a zeolita ZSM-12.
50 60 70 80 90 100 110 120 130 140 150
0
10
20
30
40
50
60
70
80
10,00
21,25
32,50
43,75
55,00
66,25
77,50
88,75
100,0
Razão SiO2/Al
2O
3
Tem
po
(m
inu
tos)
Figura 5.14 – Curva de nível mostrando a seletividade ao C1 em função do tempo e da razão SiO2/Al2O3 na
reação de craqueamento do n-heptano sobre zeólita ZSM-12.
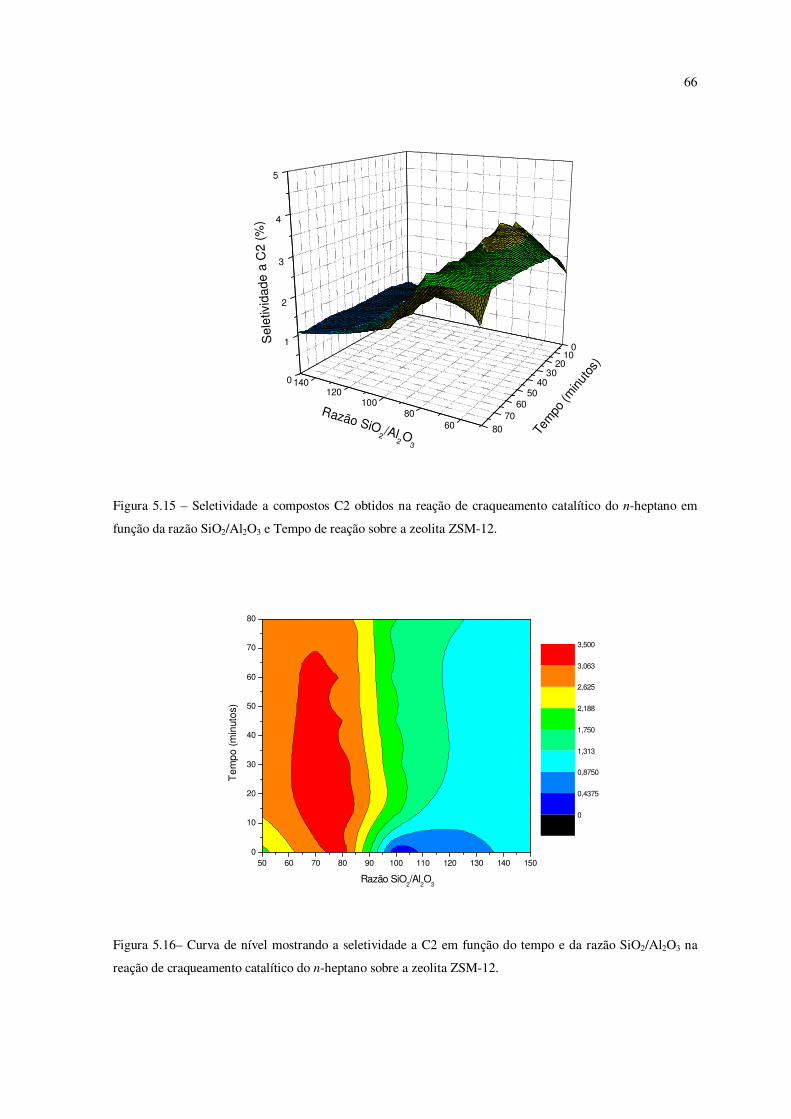
66
010
2030
4050
60
70
8060
80100
1201400
1
2
3
4
5
Tem
po (m
inut
os)
Razão SiO2 /Al
2 O3
Se
letiv
ida
de
a C
2 (
%)
Figura 5.15 – Seletividade a compostos C2 obtidos na reação de craqueamento catalítico do n-heptano em
função da razão SiO2/Al2O3 e Tempo de reação sobre a zeolita ZSM-12.
50 60 70 80 90 100 110 120 130 140 150
0
10
20
30
40
50
60
70
80
0
0,4375
0,8750
1,313
1,750
2,188
2,625
3,063
3,500
Razão SiO2/Al
2O
3
Te
mp
o (
min
uto
s)
Figura 5.16– Curva de nível mostrando a seletividade a C2 em função do tempo e da razão SiO2/Al2O3 na
reação de craqueamento catalítico do n-heptano sobre a zeolita ZSM-12.
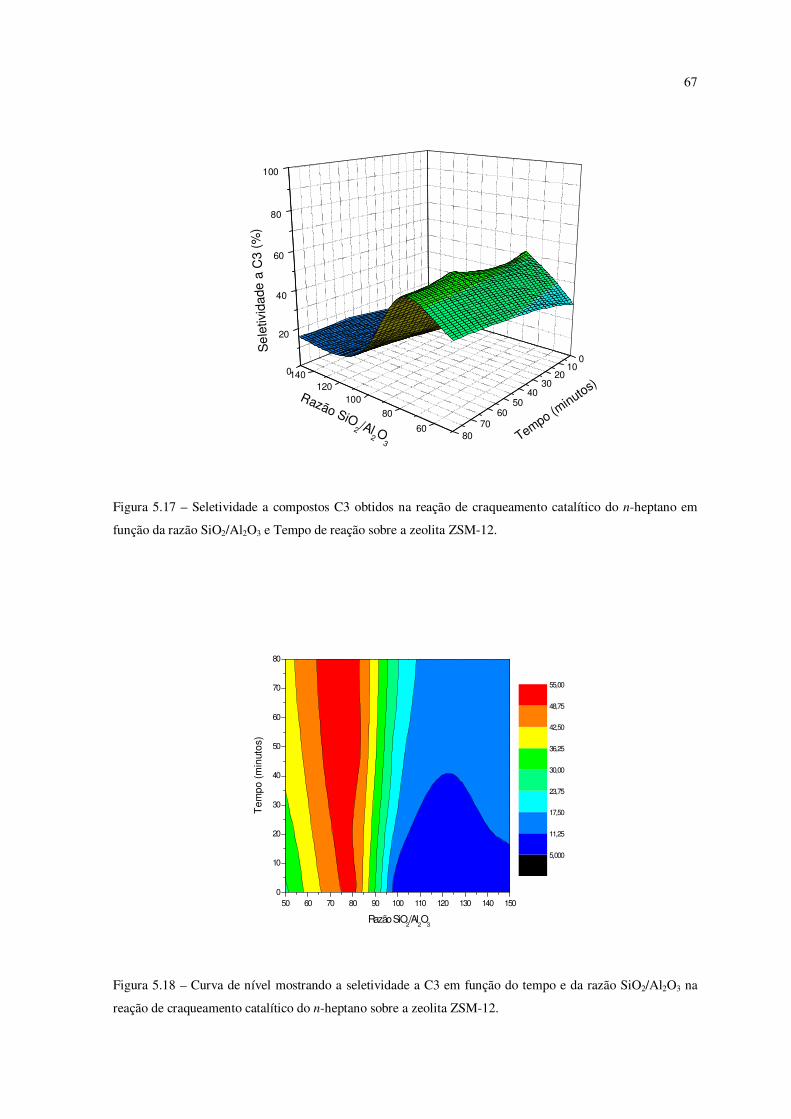
67
010
2030
4050
6070
8060
80
100
1201400
20
40
60
80
100
Tempo (m
inutos)
Razão SiO2 /Al
2 O3
Se
letivid
ade
a C
3 (
%)
Figura 5.17 – Seletividade a compostos C3 obtidos na reação de craqueamento catalítico do n-heptano em
função da razão SiO2/Al2O3 e Tempo de reação sobre a zeolita ZSM-12.
50 60 70 80 90 100 110 120 130 140 150
0
10
20
30
40
50
60
70
80
5,000
11,25
17,50
23,75
30,00
36,25
42,50
48,75
55,00
Razão SiO2/Al
2O
3
Te
mp
o (
min
uto
s)
Figura 5.18 – Curva de nível mostrando a seletividade a C3 em função do tempo e da razão SiO2/Al2O3 na
reação de craqueamento catalítico do n-heptano sobre a zeolita ZSM-12.
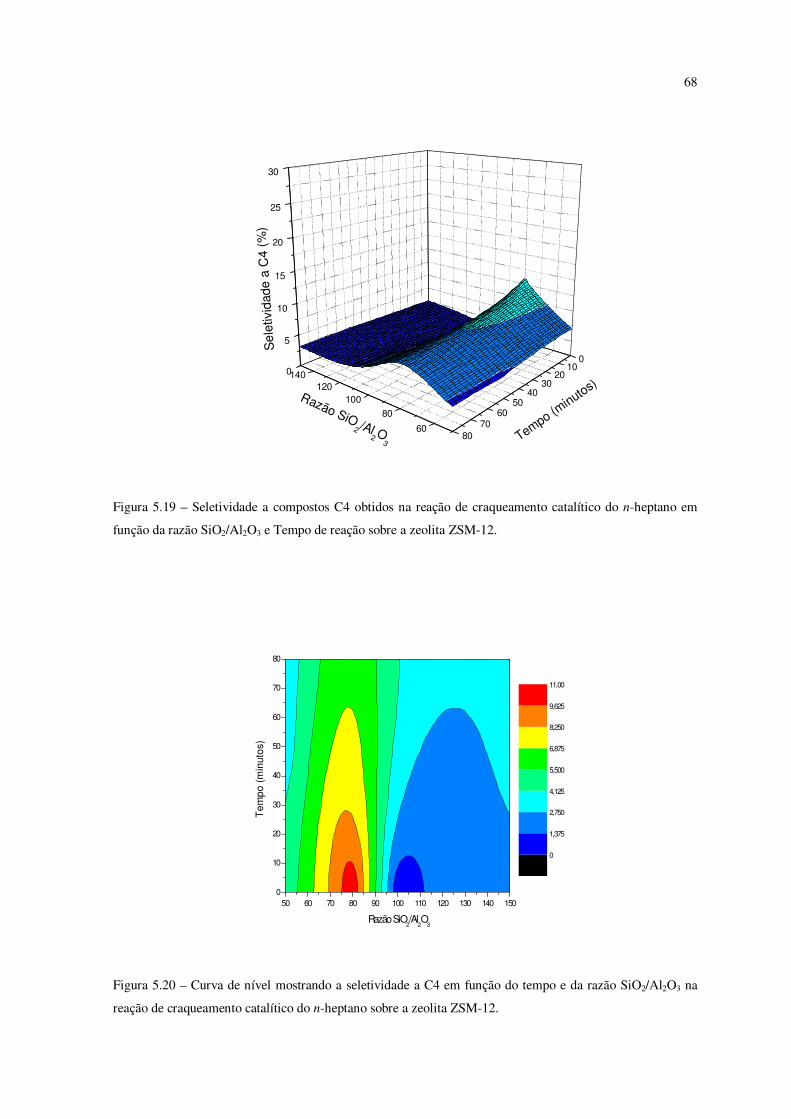
68
010
2030
4050
6070
8060
80
100
1201400
5
10
15
20
25
30
Tempo (m
inutos)
Razão SiO2 /Al
2 O3
Se
letivid
ade
a C
4 (
%)
Figura 5.19 – Seletividade a compostos C4 obtidos na reação de craqueamento catalítico do n-heptano em
função da razão SiO2/Al2O3 e Tempo de reação sobre a zeolita ZSM-12.
50 60 70 80 90 100 110 120 130 140 150
0
10
20
30
40
50
60
70
80
0
1,375
2,750
4,125
5,500
6,875
8,250
9,625
11,00
Razão SiO2/Al
2O
3
Te
mp
o (
min
uto
s)
Figura 5.20 – Curva de nível mostrando a seletividade a C4 em função do tempo e da razão SiO2/Al2O3 na
reação de craqueamento catalítico do n-heptano sobre a zeolita ZSM-12.
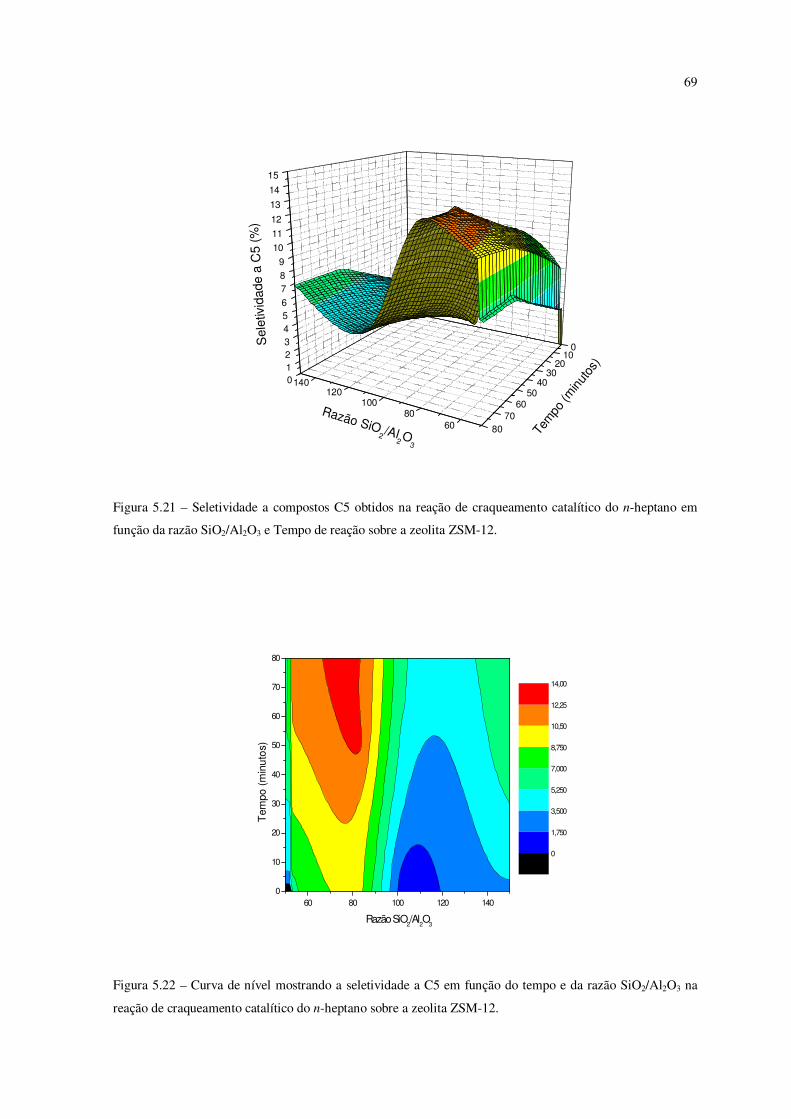
69
010
2030
4050
6070
806080
100120
1400
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
Tem
po (m
inut
os)
Razão SiO2 /Al
2 O3
Sele
tivid
ade a
C5 (
%)
Figura 5.21 – Seletividade a compostos C5 obtidos na reação de craqueamento catalítico do n-heptano em
função da razão SiO2/Al2O3 e Tempo de reação sobre a zeolita ZSM-12.
60 80 100 120 140
0
10
20
30
40
50
60
70
80
0
1,750
3,500
5,250
7,000
8,750
10,50
12,25
14,00
Razão SiO2/Al
2O
3
Tem
po
(m
inu
tos)
Figura 5.22 – Curva de nível mostrando a seletividade a C5 em função do tempo e da razão SiO2/Al2O3 na
reação de craqueamento catalítico do n-heptano sobre a zeolita ZSM-12.
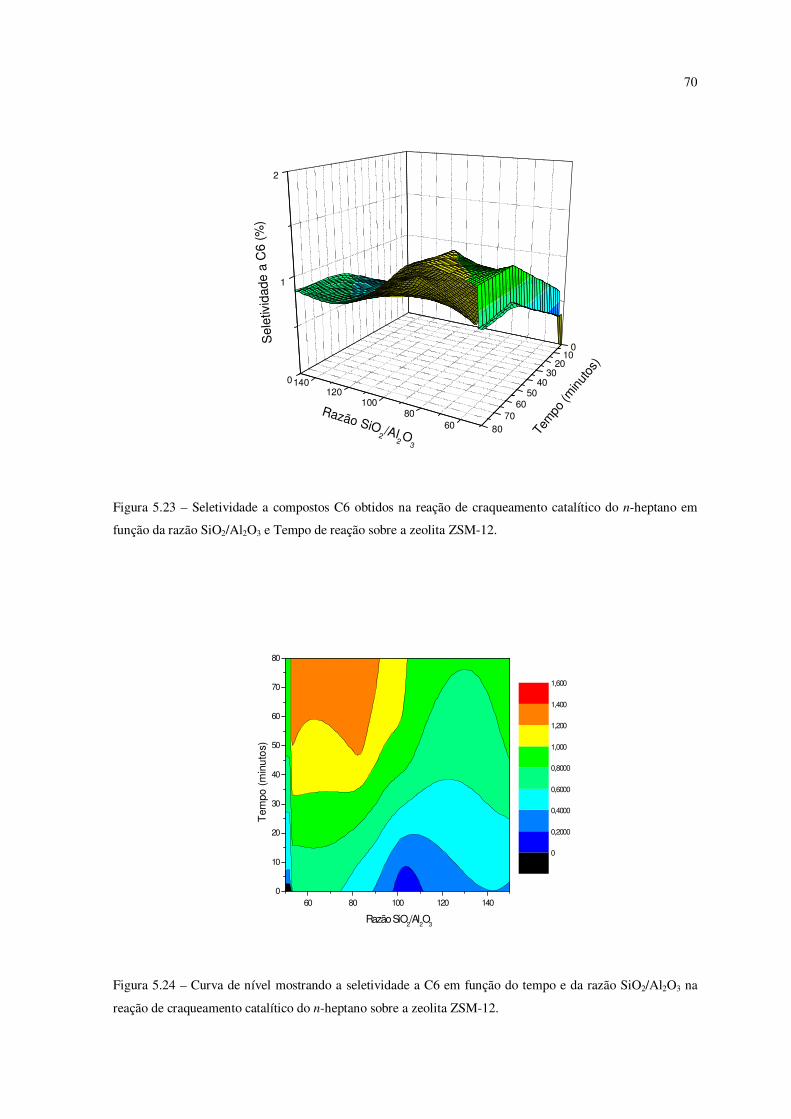
70
010
2030
4050
6070
806080
100120
1400
1
2
Tem
po (m
inut
os)
Razão SiO2 /Al
2 O3
Sele
tivid
ade a
C6 (
%)
Figura 5.23 – Seletividade a compostos C6 obtidos na reação de craqueamento catalítico do n-heptano em
função da razão SiO2/Al2O3 e Tempo de reação sobre a zeolita ZSM-12.
60 80 100 120 140
0
10
20
30
40
50
60
70
80
0
0,2000
0,4000
0,6000
0,8000
1,000
1,200
1,400
1,600
Razão SiO2/Al
2O
3
Tem
po
(m
inu
tos)
Figura 5.24 – Curva de nível mostrando a seletividade a C6 em função do tempo e da razão SiO2/Al2O3 na
reação de craqueamento catalítico do n-heptano sobre a zeolita ZSM-12.

71
6. CONCLUSÕES E SUGESTÕES PARA TRABALHOS FUTUROS
6.1. Conclusões
Neste trabalho, foram apresentados resultados experimentais sobre a reação de
craqueamento catalítico de n-heptano sobre catalisadores com estrutura zeolítica tipo ZSM-
12, com diferentes razões SiO2/Al2O3. Assim, podemos destacar como principais
conclusões:
Através das análises de difração de raios-X, foi observado as amostras de zeólitas
utilizadas neste trabalho, são formadas por um material monofásico com estrutura do tipo
ZSM-12 e alto grau de cristalinidade. Para a zeólitas ZSM-12 com SiO2/Al2O3 = 50,
foram observados alguns picos de pouca intensidade, relativos presença de uma fase
contaminante de ZSM-5.
Os ensaios catalíticos de craqueamento de n-heptano sobre as zeólitas ZSM-12 com
razões SiO2/Al2O3 de 50, 80, 100 e 150, foram promissores, pois todas as zeólitas
apresentaram reatividade para a reação de craqueamento;
Observa-se que quanto menor a razão SiO2/Al2O3, maior a conversão ao longo do
tempo e através da correlação com as analises de difração de raios-X, observou-se que
quanto maior a cristalinidade maior conversão. A região de máximo de conversão ocorre
aproximadamente no intervalo de tempo razão SiO2/Al2O3 de 80 a 100 e intervalos de
tempo entre 0 e 20 minutos. As regiões de mínimo ocorrem com razões SiO2/Al2O3
menores que 60 e tempos maiores que 30 minutos de reação
As analises por cromatografia gasosa revelaram que o craqueamento catalítico de n-
heptano sobre as zeólitas ZSM-12, levou a tipicamente a formação dos seguintes faixas de
produtos: C1, C2, C3, C4, C5 e C6, além do n-heptano não convertido e outros traços de
produtos em pequenas quantidades que não foram identificados;

72
Nas condições experimentais estudadas, foi observado que o craqueamento
catalítico de n-heptano sobre as zeólitas ZSM-12 levou a uma alta seletividade para C1 e
para C3, seguido de C5, C4, C2 e C6.
Ao observarmos as unidades de craqueamento catalítico industriais, verificamos
que aproximadamente 80% da vida útil do catalisador de FCC é passado na unidade de
regeneração de catalisador. A regeneração consiste na desobstruição dos microporos do
catalisador por meio de aquecimento e queima com ar aquecido do carbono aderido ao
catalisador durante sua utilização no craqueamento. Esta queima resulta em CO2 gasoso
que é extraído do regenerador restando apenas o catalisador regenerado e aquecido para ser
utilizado em mais um ciclo de craqueamento.
Um possível potencial ganho da zeólita em comparação com as zeólitas atualmente
utilizadas nos catalisadores de FCC é no tempo de permanência necessário para a
regeneração do catalisador após o ciclo de craqueamento, pois, por apresentar poros com
geometria mais simples, a ZSM-12, teoricamente, demandará menor tempo e energia para
o processo de regeneração, resultando em otimização do tempo de utilização efetiva do
catalisador (tempo em catálise), aumento no número de ciclos de utilização e
conseqüentemente menor custo do catalisador em comparação com os atualmente em
utilização. A avaliação detalhada da regeneração da zeólita ZSM-12 após utilização em
craqueamento consiste em estudo a ser realizado posteriormente.
Levando em consideração que as zeólitas são apenas parte do catalisador utilizado
em FCC. Portanto, faz-se necessária a continuidade dos estudos visando avaliar não apenas
as zeólitas, mas o catalisador como um todo, para permitir sua utilização em unidades
industriais de craqueamento catalítico de FCC, conforme descrevemos a seguir.
Concluímos que, a partir do craqueamento catalítico do n-heptano sobre ZSM-12
estudado podem ser produzidos hidrocarbonetos leves na faixa de C1-C6, que podem ser
usados para diversos fins industriais.

73
6.2 – Sugestões para trabalhos futuros
Com base nas técnicas empregadas e resultados obtidos nesse trabalho, podemos
sugerir para trabalhos futuros:
� Estudar a influência de outros tipos de zeólitas, como por exemplo, as zeólitas beta
e ferrierita, no craqueamento de n-heptano e outros hidrocarbonetos;
� Preparação amostras de catalisadores típicos de FCC com as zeólitas ZSM-12 mais
promissoras obtidas neste trabalho;
� Avaliar o desempenho de catalisadores de FCC contendo a zeólita ZSM-12 com
catalisadores de FCC convencionais, através de reações de craqueamento catalítico
em unidade de avaliação catalítica de leito fixo;
� Estudar a regeneração dos catalisadores e seu reuso em outros ciclos de reação;
� Caracterizar os catalisadores zeolíticos por outras técnicas, como por exemplo,
adsorção de nitrogênio e análise térmica, visando obter correlações para a estrutura
e a reatividade desses catalisadores.
� Determinar modelo cinético que represente os dados experimentais para as reações
de craqueamento, em função da conversão, seletividade e razão SiO2/Al2O3.

73
REFERÊNCIAS BIBLIOGRÁFICAS
ANDERSON, J. R.; PRATT, K. C: Introduction to characterization and testing of catalysts,
Sidney: Academic Press, p. 85, 1965.
ARAUJO, A. S.; JARONIEC, M. Thermogravimetric monitoring of the MCM-41
synthesis, Thermochim. Acta, v. 175, p. 363, 2000.
ARAUJO, A. S.; SOUZA, M. J. B. Conversão catalítica de metanol em hidrocarbonetos
usando zeólita HZSM-5 modificada por nióbio. Anais da Associaçâo Brasileira. Química,
São Paulo: ABQ, 45(2), 40, 1996.
BAUGIS, L.G. e TAN, M.H. Efeitos e moderação de contaminação metálica em
catalisadores de UFCC, 5°Encontro Sul-Americano de craqueamento catalítico. p. 188-
193, 2003.
BIALEK, R.; MEIER, W. M.; DAVIS, M. E. The synthesis and structure of SSZ-24, the
silica analogy of AlPO-5, Zeolites, v. 11, p. 438, 1991.
BOND, G. C: Heterogeneous catalysis: principles and applications, Oxford Science
Publications, New York, 2a. ed., 1987.
BRAGA, A. A. C.; MORGON, N. H., Descrições estruturais cristalinas de zeólitos,
Química Nova, v.30, p.11, 2007.
CIOLA; R.: Fundamentos da Catálise, 1ª ed. p.7-15, 68-74, 1981
CHILES, J.P.; DELFINER, P.: Geostatistics, Modeling Spatial uncertainty, Wiley Series in
Probability and statistics, p. 159, 1999.

74
CORMA, A.; MIGUEL, P. J.; ORCHILLES, A. V.: Product selectivity effects during
cracking of alkanes at very short and longer times on stream, Applied Catalysis A:
General, Amsterdam: Elsevier, v. 138(1), p. 57, 1996.
DAVIS, JOHN C.: Statistics and Data Analysys in Geology; John Wiley & Sons, Inc.
Segunda edição, pp. 383, 1987.
DESSAU, R. M. The First 14-ring Molecular Sieve, Zeolites, v. 10, p. 522, 1990.
DIAS, A.R., Catálise e catalisadores, Revista “Ciência, Tecnologia e Sociedade”, Portugal,
1998.
EVERETT, D. H: Characterization of Porous Solids, 1ª ed., Elsevier, Amsterdam, 1988.
FIGUEIREDO, J. L.; RIBEIRO, F. R.: Catálise Heterogênea; 1ª ed., Fundação Calouste
Gulbenkian, Lisboa, 1987,
FORMOSO, M. L. L.; TRESCASES, J. J.; DUTRA, C. V.; GOMES, C. B.: Técnicas
Analíticas Instrumentais Aplicadas à Geologia, São Paulo: Edgard Blücher, 1985
GATES, D. C.; KAÍZER, J. R.; SCHUIT, G. C. A., Chemistry of Catalytic Processes, New
York: McGraw-Hi1I, 1979.
GUISNET, M.; GNEP, N. S., Appl. Catal. A: General, 1996, 146, 33.
GIANETTO, G.: Zeolitas: Caracteristicas, Propriedades y Aplicaciones Industriales, Ed.
Innovatión Tecnológica, Caracas, 1990.
GORDON, S., J. Chem. Edu., v. 140, p. 87, 1963.
GUISNET, M.; RIBEIRO, F. R.: Zeólitos: Um Nanomundo ao Serviço da Catálise; 1º Ed.,
Fundação Calouste Gulbenkian, Lisboa, 2004.

75
HAAG, W. O.; DESSAU, R. M. Proceedings of 8th International Congress of Catalysis.
Berlin: Dechama, 1984.
JARONIEC, M.; KRUK, M.; SHIN, H. J.; RYOO, R.; SAKAMOTO, Y.; TERASAKI, O.,
Comprehensive characterization of highly ordered MCM-41 silicas using nitrogen
adsorption, thermogravimetry, X-ray diffraction and transmission electron microscopy,
Microporous Mesoporous Mater., v. 48, p. 127, 2001.
KINSKI, I.; DANIELS, P.; DEROCHE, C.; MARLER, B.; GIES, H. Structure and
properties of the composite zeolite silica-ZSM-12/para-nitroaniline. Microporous and
Mesoporous Materials, v. 56, p. 11-25, 2002.
KUEHL, G. US Patent 4,482,531, 1984.
LANÇAS, F. M.: Cromatografia em fase gasosa. Editora, Acta, São Carlos, 1993.
LEOFANTI, G. A.; TOZZOLA, G. A.; PADOVAN, M. A.; PETRINI, G. A.; BORDIGA,
S. B.; ZECCHINA, A. B. Catalyst characterization: characterization techniques; Catalysis
Today, v. 34, p.307-327, 1997.
LE PAGE, J. F.: Catalise de Contact, Paris, Technip, p. 441, 1978.
LIN, H. P.; MOU C. Y., Studies on mesoporous self-organizing aluminosilica, J. Cluster Science, v. 10, p. 271, 1999.
LUNA, F. J.; SCHUCHARDT, U., Modificação em Zeólitas para uso em Catálise,
Quimica Nova, v. 24, p. 885-892, 2001.
MASCARENHAS, A.; SANTOS, J.; OLIVEIRA, E. C.; PASTORE, H. O. Peneiras
Moleculares, Cadernos Temáticos de Química Nova na Escola; Edição especial, 2001.

76
MEIER, W. M.; OLSON, D. H.; BAERLOCHER, C. H., Atlas of zeolite structure types; 4th Ed., New York: Elsevier, 1996.
MIKHAIL, R.S.; BRUNAUER, S.; BODOR, E.E., Investigations of a complete pore structure analysis .i. analysis of micropores, J. Colloid Interface Sci., v. 26, p. 45, 1968.
MOKAYA, R., Al content dependent hydrothermal stability of directly synthesized aluminosilicate MCM-41, J. Phys. Chem., v. 104, p. 8279, 2000.
MOREIRA, Elizabeth Marques; PEREIRA Carlos Guerra; BORGES, Marcos Barboza;
Craqueamento Catalítico Fluído de GOP Caiúnas Desnitrificado: O impacto na Reduação
de Nitrogenados na Carga no Desempenho do FCC; Boletim Técnico Petrobras, Rio de
Janeiro, pag: 87-98, jul./dez., 2003.
MOTA, C. J. A., Química Nova, V. 18, P. 202, 1995.
PEIXOTO, D.; MENEZES, A.C.; ALVES, T.L.; MONTEIRO, J.L.F.; MOTA, C. J.A.
Craqueamento de n-pentano e isopentano sobre zeólita H-ZSM-5, Anais do 12º Congresso
Brasileiro de Catálise, P.307, 2001.
OLIVEIRA, H. M. T.; PEREIRA, M. M.; CERQUEIRA, H. S. Efeito do vanádio na
desativação de uma zeólita Y, Anais do 12° Congresso Brasileiro de catálise, p.1073-1077,
2003.
OLSON, D. H. A reinvestigation of the crystal structure of the zeolite NaX, J. Phys.
Chem., v. 74, p. 2758, 1970.
PEREGO, C.; VILLA, P. Catalyst preparation methods, Catalysis Today, v.34, p. 281-
305, 1997.
PEREIRA, C.G.; MOREIRA, E.M.; RULF, B.M. Processo de Craqueamento Catalítico
Utilizando Catalisadores de ZSM-5, Boletim Técnico. Petrobras, Rio de Janeiro, p. 120 -
132, 2004.

77
PEREIRA, C. G., MOREIRA, E. M., RULF, B. M., Processo Para Craqueamento
Catalítico Fluido De Carga De Hidrocarbonetos Pré-Tratada, Boletim Técnico. Petrobras,
Rio de Janeiro, v.45, p.274 -283, 2002.
PINNA, F. Supported metal catalysts preparation, Catal. Today, v. 41, p. 129,1998.
SCHUCHARDT, U.; GARCIA, C. M.; TEIXEIRA, S.; MARCINIUK, L. L.. Depto.
Química Inorgânica, Instituto de Química – Universidade Estadual de Campinas,
Campinas - SP, 2006.
SHERTUKTE, P. V.; MARCELIN, G.; SILL, G. A.; HALL, W. K. Study of the
mechanism of the cracking of small alkane molecules on HY zeolites. Journal of Catalysis,
New York: Academic Press, 136(2), 446, 1992.
SANTOS, P. S: Ciência e Tecnologia de Argilas, Edgard Blücher LTDA, São Paulo, 1988.
SETTLE, F.: Handbook of Instrumental Techniques for Analytical Chemistry: USA,
Prentice Hall, 1997.
RIVES, V. Characterization by thermal techniques, Catal. Today, v. 56, p. 357, 2000.
SMITH, J. V.: Definition of a zeolite, Zeolites, v. 4, p. 309-310, 1984.
SOUZA, M.J.B.; FERNANDES, F.A.N.; PEDROSA, A.M.G; ARAUJO, A.S. Selective
cracking of natural gasoline over HZSM-5 zeolite, Fuel Processing Technology, v. 15, p.
302, 2008.
SOUZA, M.J.B.; SILVA, A. O. S.; ARAUJO, A. S.; FERNANDES Jr., V. J., Revista
Técnica de Energia, Petróleo e Gás, v.1, p.74, 2002.
STOCKER, M.; OYE, G.; SJOBLOM, J., Synthesis, characterization and potential
applications of new materials in the mesoporous range, Adv. Colloid. Interfaces science, v.
89, p. 439, 2001.

78
SZKLO, A.: Fundamentos do Refino do Petróleo, 2ª ed., p.52-84, 112-154, 2005.
WEITKAMP, J. Zeolites and catalysis, Solid State Ionics, v.131, p. 175 –188, 2000.
Crystalline structures of zeolites, London, 2007.
Disponível em: <http://www.iucr.org/iucr-top>, Acessada em Março 2008.
ZHAO, X. S.; LU, M. G. Q.; MILLAR, G. J., Advances in mesoporous molecular sieve
MCM-41, Ind. Eng. Chem. Res., v. 35, p. 2075, 1996.
Evolução Histórica do Preço do Barril do Petróleo; RJ 2008.
Disponível <http://www.aepet.org.br> Acessado em Jul 2008.
Gerência de Tecnologia de FCC. Workshop sobre Nanotecnologia na industria do petróleo,
gás e energia, RJ, 2007.
Disponível em: <http://www.ufmg.br/prpq/nanociencia.pdf> Acessado em 02/02/2008.
MENDONÇA, B. Agencia Brasil, 2006.
Disponível em:
<www.agenciabrasil.gov.br/noticias/2006/12/23/materia.2006-12-23.6115763026/view -
26k > Acessado em 31/03/2008.

79
ANEXO A: Interpolação pelo método de Kriging
A.1. - Considerações acerca do Método
A teoria de Kriging foi desenvolvida pelo matemático francês Gerges Matheron
baseado na dissertação de mestrado de Daniel Gerhardus Krige. O verbo em inglês, em
homenagem ao cientista precursor, pode ser escrito como “to krige” e o adjetivo mais
comum é “kriging”. Assim, o método foi chamado originalmente de “krigeage”. O método
de krigeage pode ser classificado como uma predição linear. Parte do princípio que pontos
próximos no espaço tendem a ter valores mais parecidos do que pontos mais afastados.
(Davis, John C., 1987).
O termo Kriging abrange um conjunto de métodos, sendo os mais usuais os
seguintes:
a) Kriging Simples - Assume que as médias locais são relativamente constantes e
de valor muito semelhante à média da população que é conhecida. A média da população é
utilizada para cada estimação local, em conjunto com os pontos vizinhos estabelecidos
como necessários para a estimação. É o método mais usado em problemas de
geoestatística, e, é o método utilizado pelo Software Microcal Origin para o levantamento
das superfícies de resposta apresentadas neste trabalho (Davis, John C., 1987).
b) Kriging Ordinário - As médias locais não são necessariamente próximas da
média da população usando-se apenas os pontos vizinhos para a estimação.
c) Cokriging - É uma extensão do anterior à situações em que duas ou mais
variáveis são espacialmente dependentes e a variável que se quer estimar não está
amostrada com a intensidade com que estão as outras variáveis dependentes, utilizando-se
os valores destas e as suas dependências para estimar a variável requerida.

80
A técnica de Kriging assume que os dados recolhidos de uma determinada
população se encontram correlacionados no espaço. Como exemplo, podemos citar: se em
uma reserva mineral, a concentração de Zinco num ponto P é X%, é muito provável que se
encontrem resultados muito próximos de X%, quanto mais próximos se estiver do ponto P
(princípio da geoestatística). Porém, a partir de determinada distância de P, certamente não
se encontrarão valores aproximados de X% porque a correlação espacial pode deixar de
existir. (Davis, John C., 1987)
O método de Kriging é considerado pertencente à família dos algorítimos de
estimação linear dos mínimos quadrados. Como mostrado na Figura 8.1, o objetivo do
Kriging é calcular um valor real desconhecido de uma função f em um ponto x*, dados os
valores da função em outros pontos X1,...,Xn. Os quadrados indicam a posição dos pontos
originais e em vermelho é verde são mostrados, respectivamente, a linha de interpolação e
os intervalos de confiança (Davis, John C., 1987).
A estimação de kriging é dita é linear porque o valor predito f^(x*) é uma
combinação linear ponderada dos dados existentes e pode ser escrita como:
(8.1)
Os valores de λi são soluções de um sistema equações lineares, os quais são obtidos pela
consideração que f é uma função amostra de um processo randômico F(x), e que o erro de
predição é dado por:
(8.2)
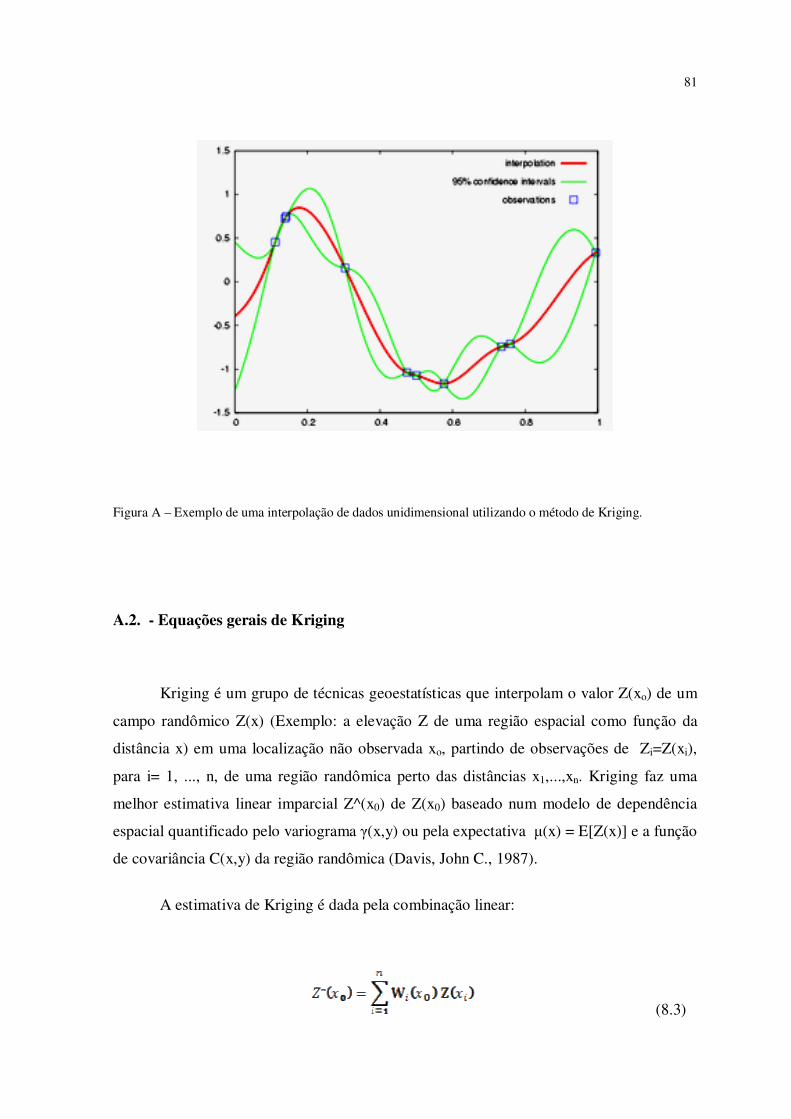
81
Figura A – Exemplo de uma interpolação de dados unidimensional utilizando o método de Kriging.
A.2. - Equações gerais de Kriging
Kriging é um grupo de técnicas geoestatísticas que interpolam o valor Z(xo) de um
campo randômico Z(x) (Exemplo: a elevação Z de uma região espacial como função da
distância x) em uma localização não observada xo, partindo de observações de Zi=Z(xi),
para i= 1, ..., n, de uma região randômica perto das distâncias x1,...,xn. Kriging faz uma
melhor estimativa linear imparcial Z^(x0) de Z(x0) baseado num modelo de dependência
espacial quantificado pelo variograma γ(x,y) ou pela expectativa µ(x) = E[Z(x)] e a função
de covariância C(x,y) da região randômica (Davis, John C., 1987).
A estimativa de Kriging é dada pela combinação linear:
(8.3)

82
Para os valores observados Zi = Z(xi) com pesos Wi(x0), para i= 1,....., n
escolhidos tal que a variância (também chamada de variância de Kriging ou erro de
Kriging):
(8.4)
A equação acima é minimizada sujeito à condição:
(8.5)
A variância de kriging não deve ser confundida com a variância de predição Z^(x0).
(8.6)

83
A.3. - Equações básicas para o Kriging simples
A equação básica para o sistema de equações Kriging simples é dada por:
(8.7)
A interpolação é dada por:
(8.8)
O erro é dado por:
(8.9)
O que conduz a versão generalizada dos mínimos quadrados do teorema de Gauss-
Markov (Chiles e Delfiner 1999):
(8.10)





























