Embed Size (px)
Citation preview

1
UNIVERSIDADE IGUAÇU
FACULDADE DE CIÊNCIAS BIOLÓGICAS E DA SAÚDE
CURSO DE GRADUAÇÃO EM MEDICINA
DIAGNÓSTICO E TRATAMENTO DAS SÍNDROMES POLIPÓIDES INTESTINAIS
NOVA IGUAÇU
2015

2
ADRIANO BRUNO DA FONSECA
ARTHUR LEONARDO CHAVIER GUIMARÃES
BIANCA GILI ARRIGONI MOREIRA
CLEVER EMANUEL F. CRUZ
FLÁVIO ANDRÉ SANTIAGO PRATA FILHO
KATYA ALVES SOUZA
LEONARDO HAYATO ARAÚJO OBA
MAURÍCIO BARBOSA FONSECA
RODRIGO ARAÚJO Q. DE MORAES
DIAGNÓSTICO E TRATAMENTO DAS SÍNDROMES POLIPÓIDES INTESTINAIS
Como requisito parcial de
obtenção de nota na disciplina
de, Clínica Cirúrgica II no
curso de medicina, na
Universidade Iguaçu.
Orientador :Prof°Dr°Thelles
NOVA IGUAÇU
2015

3
Fonseca, Adriano; Guimarães, Arthur; Moreira, Bianca; Cruz, Clever; Filho, Flávio; Souza, Kátya; Oba, Leonardo; Fonseca, Maurício; Moraes, Rodrigo.
Título: DIAGNÓSTICO E TRATAMENTO DAS SÍNDROMES POLIPÓIDES
INTESTINAIS
Nova Iguaçu, 2015
42 p.
Universidade Iguaçu - Faculdade de Ciências Biológicas e da Saúde - Curso de Medicina - Departamento de Cirurgia - Disciplina de Clínica Cirúrgica II.
Título em inglês: DIAGNOSIS AND TREATMENT OF INTESTINAL SYNDROMES POLYPOID
1. Fonseca, Adriano; 2.Guimarães, Arthur; 3.Moreira, Bianca; 4.Cruz, Clever; 5.Filho, Flávio; 6.Souza, Kátya; 7. Oba, Leonardo; 8.Fonseca; Maurício; 9.Moraes; Rodrigo.

4
“Renda-se, como eu me rendi.
Mergulhe no que você não conhece como
eu mergulhei. Não se preocupe em
entender, viver ultrapassa qualquer
entendimento."
Clarice Lispector

5
Dedicatória
“Dedicamos esse trabalho ao paciente,
que diante de nossa inexperiência, mostra-se
compreensivo. Que com seu sofrimento e pedido
de ajuda implícito neste, motiva a nós, mesmo na
condição de acadêmico, a encontrar soluções para
amenizar seus problemas.”

6
“Aos familiares pelo apoio sempre presente e aos
professores por nos proporcionar o conhecimento
não apenas racional, mas a manifestação do caráter e
afetividade da educação no processo de formação
profissional.”
Agradecimentos

7
RESUMO
Pólipo é toda estrutura tecidual que se projeta acima da superfície da camada mucosa do trato
digestivo, de forma regular e circunscrita, fazendo proeminência no seu lúmen. Os pólipos
podem apresentar-se macroscopicamente plano, séssil, subpediculado e pediculado. As
síndromes polipóides distinguem-se entre si em relação ao número de pólipos, à distribuição
dos pólipos pelo trato digestivo e histologia dos mesmos, que podem ser adenomatosos ou
hamartomatosos.
As síndromes polipóides intestinais são classificadas em familiares e não familiares. As
familiares são subdivididas em adenomatosas, como a polipose adenomatosa familiar (FAP) e
sua variante atenuada, a síndrome de Gardner e a síndrome de Turcot, e em hamartomatosas,
como a síndrome da polipose juvenil e de Peutz – Jeghers. As não familiares incluem a
síndrome de Cronkhite – Canadá, descrita em 1955. A principal síndrome é a Polipose
Adenomatosa Familiar no qual os pólipos predominam no cólon esquerdo e reto e que
manifestam-se geralmente na puberdade.
Palavras chaves: Síndromes polipóides. Pólipo. Familiar.

8
ABSTRACT
Polyps are all tissue structure protruding above the surface of the digestive tract mucous layer,
and circumscribed regular shape, making prominence in its lumen. Polyps can appear
macroscopically flat, sessile, subpediculado and pedicle. The polypoid syndromes are
distinguished from each other in the number of polyps, polyps the distribution of the digestive
tract and histology thereof, may be hamartomatous or adenomatous.
Intestinal polypoid syndromes are classified into familiar and unfamiliar. Familial
adenomatous are subdivided into as familial adenomatous polyposis (FAP) and its attenuated
embodiment, the Gardner syndrome and the Turcot syndrome, and hamartomatose like
syndrome and juvenile polyposis Peutz - Jeghers. Non family include the syndrome Cronkhite
- Canada described in 1955. The main syndrome is familial adenomatous polyposis, polyps in
which predominate in the left colon and rectum which usually manifest in puberty.
Key words: polypoid syndromes. Polyp. Family.

9
LISTA DE FIGURAS
Figura 1 – PÓLIPOS HAMARTOSOS.............................................................................................................18
Figura 2 – PIGMENTAÇÃO CARACTERÍSTICA DA SÍNDROME PJ................................ .......................20
Figura 3 – COLONOSCOPIA ..........................................................................................................................25
Figura 4 – PÁPULAS NA REGIÃO ORAL........ ...........................................................................................28
Figura 5 – HIPERPIGMENTACÃO DA PELE PENIANA.............................................................................29
Figura 6 – ASPECTO DA MUCOSA COLÔNICA NA PAF.........................................................................34
Figura 7 – HIPERTROFIA CONGÊNITA DO EP. PIGMENTAR DA RETINA...........................................36
Figura 8 – OSTEOMAS....................................................................................................................................37

10
ÍNDICE
1.Introdução.............................................................................................................................14
2.Objetivo.................................................................................................................................16
3.Metodologia..........................................................................................................................17
4.Revisão literária...................................................................................................................18
4.1.Polipose intestinal..............................................................................................................18
4.2.Síndromes polipóides........................................................................................................19
4.2.1.Poliposes hamartosas.....................................................................................................19
4.2.1.1.Síndrome de Preutz-Jeghers......................................................................................20
4.2.1.1.1.Aspectos histológicos e genéticos............................................................................20
4.2.1.1.2.Quadro clínico..........................................................................................................21
4.2.1.1.3.Potencial de malignidade.........................................................................................22
4.2.1.1.4.Tratamento...............................................................................................................23
4.2.1.2.Polipose juvenil............................................................................................................24
4.2.1.2.1.Potencial de malignidade.........................................................................................26
4.2.1.2.2.Tratamento...............................................................................................................27
4.2.1.3.Síndrome de Cowden..................................................................................................27
4.2.1.4.Síndrome de Bannayan-Riley....................................................................................28
4.2.1.5.Síndrome de Cronkhite..............................................................................................29
4.2.1.6.SPMH...........................................................................................................................31
4.2.2.Polipose adenomatosa...................................................................................................31
4.2.2.1.PAF...............................................................................................................................31
4.2.2.1.1.Aspectos morfológicos, clínicos e diagnósticos......................................................33
4.2.2.1.2.Manifestações extracolônicas..................................................................................35
4.2.2.1.3.Tratamento cirúrgico...............................................................................................39

13
5.Considerações finais.............................................................................................................43
6.Referências............................................................................................................................44

14
1.Introdução
Os pólipos intestinais são muito comuns e surgem em mais de 30% da população
adulta. Os pólipos são lesões benignas, porém, uma pequena parte tem potencial para se
transformar em câncer. Felizmente, através da colonoscopia é possível não só diagnosticar
como também remover os pólipos intestinais de forma completa e segura, impedindo – os de
se transformarem em um câncer do cólon.
O pólipo é uma pequena protuberância que cresce em cavidades revestidas por
mucosas. Podem surgir pólipos em varias regiões do nosso organismo, tais como estômago,
vesícula biliar, útero, cavidade nasal, intestinos e outros. No caso dos pólipos intestinais, o
local onde eles estão são mais comuns no intestino grosso ( cólon). O pólipo intestinal é um
tumor benigno que surge por um crescimento anormal das próprias células da mucosa do
intestino. Mal comparando, podemos dizer que são uma espécie de verruga do cólon. A maior
parte dos pólipos são lesões benignas e continuarão a sê-las para o resto da vida.
Não sabemos exatamente porque os pólipos surgem, mas alguns fatores de risco já são
bem conhecidos como, idade acima de 40 anos, doença inflamatória intestinal, historia
familiar de pólipos intestinais, sedentarismo, obesidade, dieta rica em gorduras saturadas,
dieta pobre em fibras, consumo excessivo de álcool, entre outros. Existem vários tipos de
pólipos, todavia, dois deles correspondem a imensa maioria:
- Pólipos hiperplásicos: são pólipos de tamanho pequeno, normalmente localizados na porção
terminal do cólon. Eles apresentam baixíssimo risco de transformação maligna e não
requerem tratamento na maioria dos casos.
- Adenomas: os pólipos adenomatosos são aqueles que apresentam risco de se transformar em
câncer. Felizmente, menos de 50% dos adenomas acabam por se transformar em um tumor
maligno. E, mesmo assim, um adenoma costuma demorar pelo menos 7 a 10 anos ate se
transformar em câncer.
Nem sempre é possível distinguir um pólipo hiperplásico de um adenomatoso com
base na aparência durante a colonoscopia, o qu e significa que muitos pólipos hiperplásicos
precisam ser removidos para que possam ser devidamente identificados através da
histopatologia. Na dúvida é melhor retirar e enviar o pólipo para o patologista. Em geral
qualquer pólipo com mais de 0,5 cm acaba sendo retirado para avaliação. Os adenomas são

15
divididos em três grupos de acordo com as características das sua células em, adenoma
tubular, viloso e túbulo – viloso. Todos os pólipos adenomatosos são displásicos, ou seja, são
lesões pré-malignas, porém uma minoria, como já foi dito, evoluem para câncer.
Existem algumas doenças raras, de origem genética, que se manifestam com dezenas
de pólipos no trato digestivo ainda na juventude, associados a outros sintomas em diversas
partes do corpo. Entre essas síndromes podemos citar: Lynch, Gardner, Turcot, Cronkhite,
Peutz-Jeghers e Cowden. As síndromes polipóides intestinais são classificadas em familiares
e não familiares. As familiares são subdivididas em adenomatosas, como a polipose
adenomatosa familiar (FAP) e sua variante atenuada, a síndrome de Gardner e a síndrome de
Turcot, e em hamartomatosas, como a síndrome da polipose juvenil e de Peutz-Jeghers. As
não familiares incluem a síndrome de Cronkhite.

16
2.Objetivo
O presente trabalho tem por objetivo facilitar o reconhecimento das síndromes
polipóides intestinais, por meio das condutas a ser tomadas em relação ao diagnóstico e
tratamento, devido à necessidade de intervenção médica adequada.

17
3.Metodologia
Baseada na revisão bibliográfica de trabalhos científicos de acordo com o tema
proposto, com ênfase em distinguir as variações sindrômicas polipóides e empregar os
procedimentos diagnósticos e/ou terapêutico no momento oportuno a fim de proporcionar
medida intervencionista adequada.

18
4. Revisão literária
4.1. Polipose intestinal
Os pólipos colorretais são massas de tecidos que se projetam para dentro da luz. Eles compreendem um
grupo heterogêneo de lesões sésseis, ou pediculadas, benignas ou malignas, mucosas, submucosas ou
musculares. “Pólipo” é um termo morfológico, sem que implique um diagnóstico histológico. Muitos adenomas
são tubulares, túbulo-vilosos, ou vilosos. Pólipos hiperplásicos são pequenas lesões mais frequentemente
encontradas no cólon esquerdo, já os hamartomas são incomuns.
As estimativas da incidência dos pólipos colônicos e retais na população geral variam de 9% a 60%. Os
adenomas polipóides são encontramos aproximadamente em 25% dos adultos assintomáticos que se submetem
à colonoscopia de triagem. A prevalência de adenomas é de 30% aos 50 anos de idade, 40% aos 60 anos, 50%
aos 70 anos e 55% aos 80 anos. A idade média é de 55 anos, aproximadamente 5 a 10 anos mais jovem que a
idade média dos pacientes com câncer colorretal. Aproximadamente 50% dos pólipos ocorrem no sigmoide ou
reto. Cerca de 50% dos pacientes com adenoma apresentam mais de uma lesão, e 15% apresentam mais de
duas lesões.
Os pólipos inflamatórios não apresentam potencial maligno, sendo raro o câncer que se desenvolve em
associação com hamartomas. Os pólipos hiperplásicos não são neoplásicos e, portanto, não se tornam malignos.
Foi sugerido que os pólipos hiperplásicos no cólon esquerdo são marcadores de pólipos neoplásicos em
qualquer local no cólon, porém o peso das evidencias vai contra esse conceito. Os adenomas são uma lesão pré
maligna. Acredita-se que a grande maioria dos adenocarcinomas do intestino grosso na América do Norte e na
Europa evolui a partir de adenomas, embora, no Japão, pareça que os pequenos canceres podem surgir de novo,
sem evidência de uma fase neoplásica benigna. Nas populações ocidentais, adenomas e o câncer aumentam em
incidência com a idade, e a distribuição dos adenomas e do câncer no intestino é similar.
Cerca de 25% dos pacientes que possuem cinco ou mais pólipos adenomatosos apresentam um câncer
de cólon sincrônico na colonoscopia inicial. Aproximadamente um terço das amostras colônicas e retais
ressecadas para o câncer também aloja adenomas; se uma amostra cirúrgica contém dois ou mais carcinomas
sincrônicos, a incidência de adenomas associados é de 75%. Todas as gradações de malignidade podem ser
observadas nas neoplasias colônicas; por outro lado, os cânceres menores que 0,5 cm de diâmetro e que não
contém adenoma benigno são extremamente raro. O suporte adicional para o potencial maligno dos adenomas é
o seguinte: (1) Paciente com polipose adenomatosa familial morrem de câncer em uma idade jovem, a menos
que o cólon seja removido. (2) Os carcinógenos químicos produzem adenomas e cânceres indiscriminadamente

19
nos cólons de cobaia. (3) A remoção rotineira dos adenomas, a partir do cólon, reduz a incidência de câncer retal
subsequente.
O potencial maligno de um adenoma depende do tamanho, padrão de crescimento e o grau de atipia
epitelial. O câncer é encontrado em 1% dos adenomas com menos de 1 cm de diâmetro, em 10% dos adenomas
com a 1 a 2cm de comprimento e em até 45% dos adenomas com mais de 2 cm. Os chamados adenomas
planos, pequenos adenomas planos ou deprimidos que tendem a ocorrer no cólon direito, podem ser uma
exceção a estas orientações; eles podem tornar-se malignos quando apresentam apenas alguns milímetros de
diâmetro. Os três padrões histológicos do adenoma são variações de um processo neoplásico; aproximadamente
5% dos adenomas tubulares, 22% dos adenomas túbulo-vilosos e 40% dos adenomas vilosos se tornam
malignos. O potencial para a transformação cancerosa aumenta com os graus crescentes de displasia epitelial. As
sésseis estão mais propensas a ser malignas que pediculadas. Provavelmente, transcorrem pelo menos cinco
anos, e com maior frequência de 10 a 15 anos, para que um adenoma se torne maligno.
4.2. Síndromes polipóides
4.2.1. Poliposes hamartomatosas
As poliposes hamartomatosas reúnem um grupo de doenças em que a lesão cólon retal básica
corresponde a um hamartoma, com as síndromes de Peutz-Jeghers (SPJ), da polipose juvenil (PJ), de Cowden
(CS), de Bannayan- Riley-Ruvalcaba (BRR) e de Cronkhite-Canada (CC). Com exceção da extremamente rara
Cronkhite-Canada, as polipóses hamartomatosas também são herdadas por mecanismo autossômico
dominante, constituindo um grupo diversificado de doenças com variados graus de sobreposição fenotípica e
genética. O acúmulo de conhecimento sobre suas base genéticas propiciou informações adicionais para a
classificação dessas síndromes. Os hamartomas se formam a partir de um crescimento desregulado de tecido
mesenquimal ou estromal e a carcinogênese pode ser resultado direto de uma deleção em gene supressor de
tumor ( síndromes de Peutz-Jeghers e Cowden) ou de mutações estromais por gene implicado na transdução de
sinais (polipose juvenil).
A síndrome de Peutz-Jeghers é associada a mutações do gene LKB1 que codifica uma cinase
serinatreonina multifuncional. Essas mutações ocorrem no componente epitelial, sugerindo um efeito supressor
de tumor direto. Há aumento de risco para tumores intestinais em outras localizações ( pâncreas, mama, ovários,
testículos e cervical). Multações germinativas do gene PTEN são responsáveis pela síndrome de Cowden na
qual há maior risco de tumores de mama e tireóide. A síndrome de Bannayan-Riley é menos comum, existindo
uma controvérsia ser ela uma variação da CS. A polipose juvenil resulta de mutação germinativa nos genes

20
SMAD4 e BMPR1A, usados para confirmar o diagnóstico. O gene SMAD4 codifica uma enzima implicada na
transdução de sinais para fator beta de transformação do crescimento (TGF-beta-tranforming growth fator beta).
O aumento do risco neoplásico advém de mutações SMAD4 no componente estromal, que estimula displasia
epitelial e progressão para invasão maligna.
O conhecimento dos mecanismos moleculares ainda está evoluindo nos aspectos heterogeneidade, vias
de sinalizações e maneira pela qual esses genes funcionam no desenvolvimento das respectivas síndromes.
Essas síndromes expõem os indivíduos afetados a maior risco de desenvolver neoplasias em vários órgãos e
sistemas (gastrointestinal, urogenital, mamas e tireóide). Assim, o conhecimento das bases genéticas e das
diferentes formas de expressão fenotípica permitirá formular táticas uniformes de rastreamento, vigilância e
tratamento, que devem ser distintas daquelas da população geral.
4.2.1.1. Síndrome de Peutz-Jeghers
4.2.1.1.1. Aspectos históricos e genéticos
A associação de pigmentação mucocutânea e polipose gastrointestinal foi primeiramente descrita pelo
médico inglês Sir Jonathan Huchinson, em 1896. Ao longo do tempo, a doença recebeu diferentes
denominações como síndrome de Hutchinson-Weber-Peutz, polipose intestinal II, síndrome da polipose
intestinal-pigmentação cutânea, síndrome de Jeghers, síndrome lentigio-polipose digestiva e síndrome de Peutz-
Touraine, dentre outras. Entretanto somente após os trabalhos do holandês Peutz (1886-1897), em 1921 e do
americano Jeghers (1944), que firmaram os caracteres da doença, essa associação passou a ser denominada de
síndrome de Peutz-Jeghers.
Inicialmente, os pólipos foram descritos como lesões adenomatosa pré-malignas, impressão logo
contestada em revisão de Bartholomew. Os primeiros a observar que tais pólipos apresentavam figuras
FIGURA 1: PÓLIPOS HAMARTOSOS

21
histológicas compatíveis com hamartomas. Os pólipos gastrointestinais da SPJ têm características distintas
daquelas dos encontrados em outras síndromes hamartomatosas, como a presença de componente muscular liso
infiltrando o tecido conectivo em padrão de ramificações. Constitui a afecção familiar transmitida por gene
anômalo autossômico dominante responsável tanto pela polipose como pela pigmentação cutaneomucosa. No
entanto, alguns casos isolados já foram descritos. Esses distúrbios apresenta incidência de 1:120 mil, afetando
igualmente os sexos.
A mutação genética ocorre no gene supressor que codifica a proteína serina/treonina cinase, localizado
no cromossomo 19p13.3. Mutações germinativas do gene levam à formação de hamartomas e as mutações
somáticas deste e de outros genes transformam os hamartomas em adenomas e, depois, em carcinomas. As
múltiplas mutações identificadas no gene LKB1 são responsáveis pela variabilidade fenotípica da SPJ, incluindo
desenvolvimento de casos mais agressivos e outros que nunca desenvolvem câncer. O gene STK11 é o primeiro
exemplo de gene de suscetibilidade para uma síndrome tumoral cuja mutação provoca inativação da atividade
de sua enzima, ao contrario do que acontece com os oncogenes que ativam as funções.
4.2.1.1.2. Quadro clínico
A SPJ é uma entidade clínica caracterizada pela tríade pigmentação melânica mucocutânea, polipose
intestinal e histórico familiar. As alterações pigmentares benignas de pele e mucosa fizeram com que a SPJ fosse
classificada em conjunto com outras síndromes lentiginosas, como a BRR.
A pigmentação manifesta-se por manchas pretas ou azuis ao redor de lábios, olhos e extremidades
(palma das mãos e planta dos pés), sendo encontrada também no pescoço, tórax e períneo. Formadas por
deposito de melanina, assumem formas arredondadas ou ovais, raramente confluentes, lisas e de no máximo 1
cm. Podem aparecer desde o período neonatal ou após o inicio dos sintomas gastrointestinais, não apresentando
potencial maligno.
As manifestações clínicas mais importantes da SPJ são secundárias aos pólipos. Estes tem tamanho
variável, geralmente são múltiplos e afetam o intestino delgado (95%), cólon (27%), estômago (24%) e reto
(24%); o jejuno costuma está mais implicado do que o duodeno e íleo. Pólipos escassos ou solitários tem
ocorrência excepcional, porque pequenos pólipos gástricos e colônicos raramente determinam sintomas,
também não sendo diagnosticados pelos exames radiológicos convencionais.
O quadro clínico varia dependendo de localização, tamanho e numero de pólipo. Os sintomas surgem
em qualquer idade, predominando na segunda e terceira décadas. Os pacientes se apresentam com dor em cólica
recorrente, resultado de hiperperistaltismo ou invaginação de pólipos do intestino delgado. Lesões grandes
podem causar sintomas obstrutivos ou “prolapsa” pelo reto. Outros apresentam hemorragia discreta ou oculta,
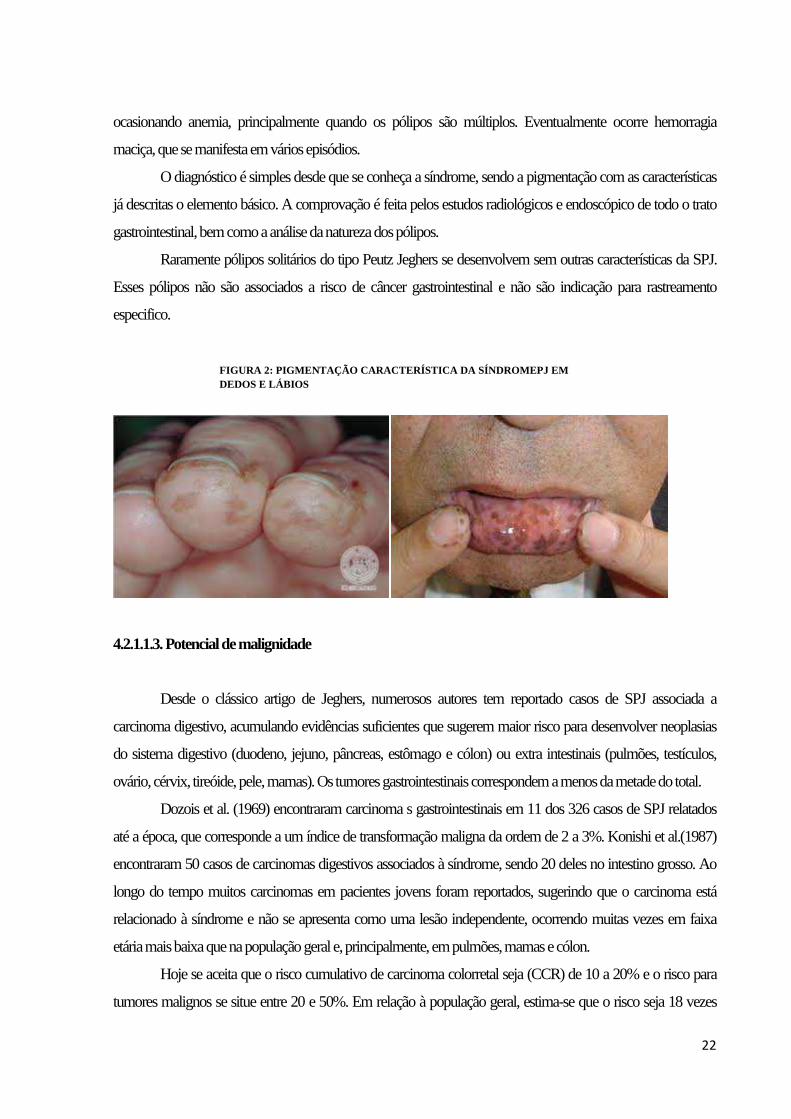
22
ocasionando anemia, principalmente quando os pólipos são múltiplos. Eventualmente ocorre hemorragia
maciça, que se manifesta em vários episódios.
O diagnóstico é simples desde que se conheça a síndrome, sendo a pigmentação com as características
já descritas o elemento básico. A comprovação é feita pelos estudos radiológicos e endoscópico de todo o trato
gastrointestinal, bem como a análise da natureza dos pólipos.
Raramente pólipos solitários do tipo Peutz Jeghers se desenvolvem sem outras características da SPJ.
Esses pólipos não são associados a risco de câncer gastrointestinal e não são indicação para rastreamento
especifico.
4.2.1.1.3. Potencial de malignidade
Desde o clássico artigo de Jeghers, numerosos autores tem reportado casos de SPJ associada a
carcinoma digestivo, acumulando evidências suficientes que sugerem maior risco para desenvolver neoplasias
do sistema digestivo (duodeno, jejuno, pâncreas, estômago e cólon) ou extra intestinais (pulmões, testículos,
ovário, cérvix, tireóide, pele, mamas). Os tumores gastrointestinais correspondem a menos da metade do total.
Dozois et al. (1969) encontraram carcinoma s gastrointestinais em 11 dos 326 casos de SPJ relatados
até a época, que corresponde a um índice de transformação maligna da ordem de 2 a 3%. Konishi et al.(1987)
encontraram 50 casos de carcinomas digestivos associados à síndrome, sendo 20 deles no intestino grosso. Ao
longo do tempo muitos carcinomas em pacientes jovens foram reportados, sugerindo que o carcinoma está
relacionado à síndrome e não se apresenta como uma lesão independente, ocorrendo muitas vezes em faixa
etária mais baixa que na população geral e, principalmente, em pulmões, mamas e cólon.
Hoje se aceita que o risco cumulativo de carcinoma colorretal seja (CCR) de 10 a 20% e o risco para
tumores malignos se situe entre 20 e 50%. Em relação à população geral, estima-se que o risco seja 18 vezes
FIGURA 2: PIGMENTAÇÃO CARACTERÍSTICA DA SÍNDROMEPJ EM DEDOS E LÁBIOS

23
maior e o risco de neoplasias pancreáticas seja 100 vezes superior. Em estudo de metanálise para avaliar o risco
de vários tumores na SPJ, Giardiello et al. reuniram os dados de 107 homens e 106 mulheres de 79 famílias,
estimando risco cumulativos de 57% em mama, 39% em cólon, 36% em pâncreas, 29% em estômago e 21%
em ovário.
Outra questão importante é a origem dos carcinomas nesses pacientes: se a partir dos pólipos
hamartomatosos ou de adenomas pré-existentes. Foi proposta uma sequencia progressiva de hamartoma para
adenoma e para carcinoma, com base no modelo de carciogênese cclorretal. Admite-se ainda, que os
carcinomas digestivos possam ter origem em zonas de replicação celular ou de displasias em pólipos
hamartomatosos de várias localizações.
4.2.1.1.4. Tratamento
A conduta na SPJ baseia-se no tratamento de condições benignas sintomáticas, pólipos de grandes
dimensões e na vigilância quanto a tumores malignos. Tratamento expectante deve ser reservado aos pacientes
sintomáticos ou poucos sintomáticos. Havendo intussuscepção, obstrução ou hemorragia, indica-se tratamento
cirúrgico a fim de o evitar em situação emergencial.
A tática cirúrgica deve ser a mais econômica possível, variando desde a polipectomia até a enterectomia
segmentar. Os pólipos do reto e os pediculados do cólon podem ser ressecados endoscopicamente. Pólipos
sesseis, de maior tamanho e acima da reflexão peritoneal, são mais bem abordados por laparotomia, podendo ser
retirados por colostomia ou ressecções segmentares. Excepcionalmente, há necessidade de ressecar todo o
cólon.
A realização de enteroscopia durante laparotomia no manuseio da SPJ foi relatada por Van Coevorden
et al. (1986), com objetivo de obter maior “limpeza” dos pólipos e, assim, reduziu o número de laparotomias
subsequentes. Edwards et al. (2003) reportaram a experiência do St. Mark’s Hospital’s, de Londres, em 35
pacientes nos quais a enteroscopia identificou 350 pólipos não detectados pela palpação ou transiluminação,
reduzindo de maneira significativa a necessidade de enterotomias adicionais e a frequência de laparotomias
nesse grupo.
Embora a maioria das operações seja indicada por condições benignas, o aspecto mais importante na
abordagem da SPJ refere-se ao diagnóstico e tratamento de neoplasias malignas. Todos os pólipos maiores de
1,5 cm devem ser removidos, mesmo em pacientes assintomáticos, este risco não justifica porém, que sejam
necessárias ressecções profiláticas de segmentos intestinais afetados pela polipose. No entanto, pacientes tratados
conservadoramente ou operados devem se submeter periodicamente a exames clínicos, radiológicos e
endoscópicos a partir dos 30 anos. A investigação deve incluir exame endoscópico bienal dos sistemas

24
digestivos superior e inferior, ultrassonografia anual das regiões pélvica, testicular e pancreática r mamografia
anual, após os 25 anos. Todos os membros da família devem ser investigados.
4.2.1.2. Polipose Juvenil
As características patológicas de lesões polipoides em crianças foram descritas na metade do século
passado, simultaneamente à adoção do termo “pólipo juvenil” por Horrilleno em 1957. Poucos anos depois,
Morson (1962) classificou os pólipos juvenis como hamartomas, cabendo a McColl (1964) a distinção entre
poliopose juvenil (PJ) e outras síndromes.
Os pólipos hamartosos são redondos ou ovais, apresentando superfície lisa que contrasta com a dos
adenomas, geralmente granular. São constituídos os tecidos com estroma vascular abundante, infiltrado com
células inflamatórias no qual predominam os eosinófilos. O tecido ainda apresenta espaço cístico característico
de diferentes tamanhos, alinhados por células epiteliais secretoras de muco.
Os pólipos juvenis costumam ser encontrados no sigmoide e no reto, provocando sangramento retal ou
presença de sangue nas fezes. Predominam no sexo masculino. A maioria dos pólipos juvenis é pedunculada e
com frequência de auto amputa. O sangramento retal recorrente se deve a torção do pedúnculo, inflamação ou
ulceração. Intussuscepção e prolapso através do ânus são menos frequentes.
Pólipos juvenis podem ocorrer de forma isolada ou múltipla. Pólipos esporádicos aparecem
isoladamente, são diagnosticados predominantemente na infância, estimando-se que possam ser assintomáticos
em ate 1% de crianças, sendo o risco de transformação maligna extremamente baixa em tal situação. Por outro
lado, a síndrome da polipose juvenil (PJ) é uma condição ainda mais rara, caracterizada por múltiplos pólipos
juvenis no trato gastrointestinal.
Sachatello et al. (1974) sugeriram que o diagnóstico da PJ poderia ser feito pelo achado de uma das
seguintes situações: (1) mais de 10 pólipos juvenis no cólon; (2) pólipos juvenis ao longo do trato
gastrointestinal; (3) qualquer numero de pólipos juvenis em individuo com histórico familiar de PJ. Outros
propõem menor numero de pólipos para essa caracterização (Giardiello-1991).
Nessa síndrome distinguem-se três formas de apresentação clínica: a PJ da infância, a PJ generalizada e
a PJ do cólon. A primeira se manifestar ao redor dos quatro anos, associada a diarreia, hemorragia,
intussuscepção, prolapso retal e enteropatia perdedora de proteínas. Acomete todo trato gastrointestinal, sendo o
prognostico dependente desse envolvimento. Eventualmente, pode ter evolução fatal precoce e não apresentar
histórico familiar (Sachatello-1974).
Nas outras formas clínicas, a polipose pode se iniciar em idade variada, sendo mais comum nas
primeira e segunda décadas e, em 15% dos casos, em adultos. Manifesta-se por sangramento retal, prolapso e

25
anemia. Os pólipos podem estar limitados ao intestino grosso ou ocorrer simultaneamente no estômago e no
intestino delgado. Pode haver histórico familiar, indicando mecanismo de herança autossômico dominante
(Desai-1995).
Descreveram-se alguns defeitos congênitos associados, como anormalidades cardíacas, do palato,
dentes supranumerários, macrocefalia, podactilia, alopecia e outros. Essas alterações são habituais em casos sem
histórico familiar da síndrome.
Havendo suspeita diagnóstica, devem-se realizar retossigmóidoscopia e colonoscopia para avaliação da
extensão da doença e exérese de alguns pólipos para exame histológico. Existe um numero variável de pólipos,
usualmente entre 50 e 200, distribuídos em todo trato gastrointestinal, mais no cólon e reto. Em 262 casos de PJ,
Hofting (1993) encontraram lesões colorretais em 98% dos casos; do estômago, em 13,6%; duodeno, em 2,3%
e jejuno / íleo, em 6,5%.
A PJ é uma síndrome rara, de caráter autossômico dominante, penetrância incompleta e
heterogeneidade genética. Existe histórico familiar em 20 a 50% dos pacientes. Foram identificadas mutações
germinativas no gene SMAD4 (MADH4)- também conhecido como DPCA4, localizado no cromossomo 18q
21.1 e no gene BMPR1A, que se localiza no cromossomo 10q 21-22. Estudos recentes indicam que pacientes
com mutações germinativas SMAD4 ou BMPR1A apresentam fenótipo mais proeminente que pacientes com
PJ sem a mutação; além disso, mutações SMAD4 predispõem à polipose no sistema digestivo superior.
Mutações germinativas do SMAD4 são responsáveis pela maioria dos casos de PJ, enquanto as
mutações BMPR1A são encontradas em 40 a 100%das famílias sem mutação SMAD4. O gene BMPR1A tem
11 éxons e se localiza perto do gene PTEN no cromossomo 10q 21-22, demonstrando que dois genes que
causam poliposes hamartomatosas estão fortemente ligados no braço longo do cromossomo 10.
Uma cuidadosa avaliação clinica e genética é necessária para o diagnostico diferencial de duas outras
síndromes hamartomatosas que apresentam mutações do gene PTEN (síndrome de Cowden e síndrome de
Bannayan-Riley-Ruvalcaba).
FIGURA 3: COLONOSCOPIA- INÚMEROS PÓLIPOS HAMARTOSOS

26
4.2.1.2.1. Potencial de malignidade
Os pólipos juvenis gastrointestinais são os mais encontrados na população pediátrica, classicamente
caracterizados como crescimento hamartomatosos ou proliferação inflamatória reativa. Entretanto, a associação
com carcinomas e displasia na PJ promoveu a caracterização dessa entidade como uma condição pré-maligna.
Carcinomas de diversas localizações (colorretais, de estômago, intestino delgado, pâncreas) têm sido
diagnosticados em associação com PJ em incidências significativas. Hofting et al. (1993) diagnosticaram 48
carcinomas em 272 pacientes (18%). Estima-se que o risco para câncer gástrico seja de 21% ( Dunlopp , 2002).
Em estudo do St. Mark’s Hospita,, Jass (1988) encontraram 15% de carcinomas em pacientes abaixo de 35
anos, estimando que o risco cumulativo de CCR seria de 68% aos 60 anos. Mas recentemente estimou-se que o
risco cumulativo de degeneração maligna na PJ ao longo da vida é de aproximadamente de 30 a 50%, para
cólon e reto e 10% , para o sistema digestivo superior. É possível que o risco diminuía com o aumento da idade,
uma vez que os pacientes vão sendo tratados e submetidos a colectomias, com diminuição do número de
pólipos.
Os carcinomas eventualmente associados à PJ ocorrem em idade precoce, em media entre 35 e 40
anos. A maioria dos tumores é do tipo mucinoso e/ou pouco diferenciados, configurando prognostico
desfavorável.
Serviços especializados mostram que 2 a 15% dos pacientes com PJ podem apresentar pólipos juvenis
com características adenomatosas ou adenomas puros. Subsequentemente numerosas alterações displásicas
foram documentadas em pólipos hamartomatosos ou nos adenomas associados à síndrome, achados que
surgerem um possível mecanismo histogênico para evolução de um câncer na PJ.
Não se sabe, porém, se tais adenomas são derivados da conversão total de um pólipo juvenil e se
constituem adenomas “de novo”. Revendo as características de 1032 pólipos juvenis, Jass (1988) encontraram
somente 21 adenomas (2%) sem qualquer característica de pólipo juvenil, a origem dos adenomas, a partir de
um pólipo juvenil, seja mais provável, estabelecendo o desenvolvimento em focos de displasia adenomatosa
localizados em pólipos juvenis.
4.2.1.2.2. Tratamento
O tratamento dos pólipos juvenis na criança é o mesmo realizado para pólipos em adulto. Quando
isolados podem ser completamente excisados cirúrgica ou endoscopicamente, dependendo de sua localização.
Embora tenha sido preconizada colectomia profilática em portadores de PJ, a realização regular dos
exames colonoscópio e endoscópio para polipectomia pode apresentar uma alternativa mais conservadora,

27
principalmente naqueles que aderem ao seguimento. Nesses casos, é necessária a atenção especial ao
aparecimento de pólipos juvenis com características adenomatosas, pelo maior risco de degeneração nesse
grupo de pacientes.
A tendência atual é fundamentar a abordagem do doente na gravidade dos sintomas e no numero de
pólipos, reservando-se o tratamento cirúrgico para os pacientes com diarréia, sangramentos recidivantes, com
mais de 20 pólipos, ou quando estes apresentarem crescimento acelerado ou displasia. As opções técnicas são
íleorretoanastomose ou proctocolectomia com bolsa ileal.
Existem aqueles que defendem o tratamento cirúrgico após os 20 anos de idade para prevenir o
desenvolvimento de câncer, enquanto outros defendem o acompanhamento endoscópico como alternativa
razoável. Parentes de primeiro grau devem ser rastreados por colonoscopia a partir da segunda década.
4.2.1.3. Síndrome de Cowden
A síndrome de Cowden é doença multissistêmica herdada por mecanismo autossômico dominante
com penetrância incompleta e expressão variável. É também conhecida como síndrome de múltiplos
hamartomas, tendo sido descrita por Lloyd e Dennis (1963) na família de Rachel Cowden, em 1963.
Caracteriza-se por uma combinação de alterações ectodérmicas, mesodérmicas e endodérmicas que podem
afetar pele, membranas mucosas, mamas, sistema digestivo e tireóide.
O defeito genético localiza-se no cromossomo 10q22-23 e implica o gene que codifica a proteína
PTEN. Hoje se reconhece que uma variação de alelos determina outra síndrome, a BRR, tendo-se descrita uma
família com duas mulheres portadoras de CS e dois homens com BRR. Mutações segmentativas foram
identificadas em até 80% dos portadores de CS. O achado de mutações somáticas do PTEN em vários tumores
esporádicos faz dessa síndrome um importante modelo clinico e genético de carcinogênese.
O diagnostico geralmente é feito na terceira década. As alterações mucocutâneas permitem o
reconhecimento precoce da doença e elas costumam estar presente antes do desenvolvimento de neoplasias
internas, o que facilita a identificação de lesões assintomáticas em outros órgãos. Manifestam-se em 80% dos
pacientes e são representadas por triquilemomas faciais múltiplos e, papilomatose da mucosa oral e ceratose
palmoplantar.
Além da polipose hamartomatosa gastrointestinal, tireóide, mamas e endométrio são os órgãos mais
afetados. Na tireóide aparecem bócio, adenomas e carcinoma folicular. Câncer de mama é a neoplasia mais
comum nesses pacientes (30 a 50%), ocorrendo em idade mais jovem que na população geral. Uma proporção
menor (10%) apresenta tumores do SNC, macrocefalia e discreto retardo mental.

28
Há pólipos em 35 a 65% dos pacientes. São sésseis, menores e menos exofíticos do que os encontrados
na síndrome de Peutz-Jeghers e assintomáticos na maioria dos pacientes. Embora ainda não tenha sido o risco de
câncer nessa afecção, recomenda-se colonoscopia a cada 3 a 5 anos. Devem-se também realizar exame clinico
anual das mamas e tireóide, mamografia anual (a partir dos 25 anos) e ultrassonografia de tireóide anual.
4.2.1.4. Síndrome de Bannayan-Riley-Ruvalcaba
A síndrome de Bannayan-Riley-Ruvalcaba também é denominada síndrome Bannayan-Zonana, tendo
sido primeiramente descrita por Riley e Smith, em 1961, a seguir por Bannayan em 1971 e posteriormente
caracterizada por Zonana em 1975.
Constitui síndrome autossômica dominante causada por mutação do gene PTEN no cromossomo
10q23,ocorrendo como resultado de uma variação alélica da síndrome de Cowden. Caracteriza-se por polipose
intestinal hamartomatosa associada a lesões dermatológicas típicas (aspecto lentiginoso de pênis e vulva,
verrugas, acantose nigricans e hiperpigmentação da pele peniana) e hamartomas na língua.
FIGURA 4: SÍNDROME DE COWDEN-PÁPULAS NA REGIAO ORAL
FIGURA 5: HIPERPIGMENTAÇÃO DA PELE PENIANA

29
Foram descritas ainda, manifestações extra intestinais como macrocefalia, lipomas subcutâneos e
viscerais, malformações vasculares e anormalidades esqueléticas. Alterações na retina são encontradas em ate
35% dos pacientes. Pelo menos 50% dos afetados apresentam anormalidades do SNC, como deficiência
mental, hipotonia e retardo do desenvolvimento psicomotor.
A polipose intestinal é diagnosticada em até 45% dos pacientes, no íleo distal e cólon. Como ainda não
foi descrita transformação maligna dos pólipos nesses pacientes, não há recomendações para rastreamento
gastrointestinal. Devido à estreita associação entre BRR e CS, pacientes com diagnóstico incerto devem fazer
exames de rastreamento para pulmão e tireóide.
4.2.1.5. Síndrome de Cronkhite-Canada
Essa síndrome extremamente rara foi descrita por Cronkhite e Canada, em 1955 (primeiros dois casos)
e caracteriza-se por polipose gastrointestinal não hereditária associada a alterações ectodérmicas. Sua etiologia é
desconhecida, não havendo dados que expliquem os distúrbios sincrônicos que ocorrem em dois epitélios.
Consequentemente, ainda não foram devidamente estabelecidos os fatores associados à sua progressão ou
remissão e as bases de tratamento. Admite-se que estresses mental e físico tenham alguma participação e não há
evidencias que sugiram base genética ou infecciosa.
Nos EUA, foram reportados apenas 15 casos, enquanto dois terços dos 150 casos relatados, foram
diagnosticados no Japão (sem justificativa aparente), onde a incidência estimada é de um caso por milhão.
Depois do Japão, a maioria dos relatos compreende indivíduos brancos da América do Norte e Europa
Ocidental.
Dados epidemiológicos mostram que a distribuição etária varia de 31 a 85 anos, com maior distribuição
da doença em pacientes entre 50 e 60 anos. No Japão, a afecção predomina no sexo masculino (2:1); em outros
países, o pequeno número de casos não demonstra predileção sexual.
As alterações gastrointestinais são representadas por lesões polipóides hamartomatosas generalizadas
interpostas por mucosa anormal. Os pólipos são encontrados no estômago, duodeno e cólon, aparecendo
também no intestino delgado esôfago. Já se observou regressão tanto dos pólipos gástricos como dos colônicos.
Esses pólipos apresentam risco significativo de malignidade. Já tendo sido descritos vários casos de câncer
colorretal.
A proliferação mucosa resulta em má absorção e enteropatia perdedora de proteínas, características
fisiopatológicas que determinam alterações hidroeletrolíticas, desnutrição, sangramento e complicações
cirúrgicas. Os sintomas mais comuns são diarréia aquosa, náuseas, anorexia, cólicas e eventual esteatorréia. A
doença usualmente evolui em alguns meses, período em que os sintomas gastrointestinais, inicialmente

30
moderados, podem progredir para perda de peso significativa e edema periférico. As consequências da
desnutrição podem ser potencialmente fatais. Exames bioquímicos revela anemia, hipoproteinemia e queda dos
níveis séricos de cálcio e potássio.
A diarréia é multifatorial e ocorre em 90% dos pacientes. Glândulas mucosas dilatadas liberam
secreções ricas em proteína na luz intestinal e a mucosa alterada é incapaz de digerir dissacarídeos e absorver
carboidratos e lipídeos. Muitos acreditam que os pólipos contribuam para a diarréia, embora algumas
modalidades terapêuticas e casos de remissão espontânea tenham obtido melhora do quadro diarreico sem afetar
o número de pólipos.
Apesar da maioria dos relatos associarem as alterações ectodérmicas à desnutrição, muitos sintomas e
sinais aparecem ou entram em remissão de maneira incompatível com essa teoria. As manifestações
epidérmicas podem se instalar antes ou depois do início do quadro diarréico. Caracterizam-se por alopecia,
hiperpigmentação cutânea, alterações do cabelo e atrofia das unhas. A alopecia inicialmente é irregular, com
rápida progressão para perda total do cabelo, podendo atingir sobrancelhas, face, axilas, região púbica e
extremidades. Nas unhas, podem surgir adelgaçamento, rachaduras e alterações da cor em mãos e pés. Máculas
e placas hiperpigmentadas acastanhadas distribuem-se difusamente., sendo mais comuns em mãos e braços.
A abordagem terapêutica inclui medidas gerais de suporte, terapia nutricional, antibióticos,
corticosteroides ou tratamento cirúrgico. Para atenuar a diarréia, empregam-se dietas de eliminação e agentes
antiperistálticos. Na vigência de distensão abdominal, restringe-se a ingestão de dissacarídeos, especialmente a
lactose. Corticóides são indicados para quadros de deterioração progressiva, podendo eventualmente induzir
remissão da doença. Dietas elementares e nutrição parenteral são empregadas para correção das deficiências
nutricionais.
O comprometimento das condições clinicas pré-operatórias contribui para as altas taxas de morbi-
mortalidade operatória. Assim, o tratamento cirúrgico é reservado para casos de câncer ou complicações como
sangramento intenso, perfuração ou obstrução (intussuscepção).
4.2.1.6. Síndrome Poliposa Mista Hereditária
Muitas síndromes hereditárias predispõem ao desenvolvimento de pólipos colônicos juvenis e câncer
colorretal, com importância potencial para compressão da carciogênese de tumores esporádicos. Na maioria dos
pacientes com polipose intestinal é possível diferenciar as síndromes PAF (Polipose adenomatosa familial), PJ e
SPJ. Entretanto, em raros casos, tal distinção não pode ser feita com base na histologia do pólipo, caracterizando-
se a chamada síndrome polioposa mista hereditária.
A HMPS distingue-se por pólipos juvenis atípicos, adenomas colônicos e carcinomas colorretais. A
síndrome é transmitida por mecanismo autossômico dominante. Pólipos adenomatosos e hiperplásicos podem

31
surgir em membros da família cuja lesão característica é um pólipo junvenil atípico. Outros indivíduos
desenvolvem mais de um tipo de pólipo, assim como pólipos individuais podem conter mais de uma
característica histológica. Os pólipos podem apresentar degeneração maligna. Tipicamente são encontrados
poucos pólipos à colonoscopia, além de não haver associações extra intestinais.
Descreveu-se, também, um caso de polipose juvenil em paciente cujos familiares eram portadores de
polipose adenomatosa familiar. No registro de poliposes do St. Mark’s Hospital identificou-se uma grande
família com HMPS em que a análise de 104 pólipos do pedigree revelou adenomas, pólipos juvenis,
hiperplásicos, pólipos Peutz-Jeghers e adenomas planos. Os autores sugerem que a HMPS pode ser uma
entidade genética não relacionada a outras síndromes hereditárias colorretais. Os dados de análise genética não
demonstraram ligação com lócus APC ou HNPCC.
4.2.2. Polipose adenomatosa
4.2.2.1.Polipose adenomatosa familiar e suas variações
Historicamente, denominações como polipose múltipla, polipose disseminada, adenomatose
hereditária, polipose colônica familiar e outras foram utilizadas para descrever a afecção, mas atualmente se
prefere o termo polipose adenomatosa familiar para destacar a origem dos pólipos e o caráter hereditário da
doença.
A descrição pioneira foi feita por Menzelio, em 1821, cabendo a Cripps chamar atenção a natureza
familiar da doença em 1882. Em seguida, Smith reconheceu seu potencial maligno, em 1887, mas somente
algumas décadas depois Lockhart-Mulmmery (1925) descreveu a enorme predisposição para o
desenvolvimento de carcinoma colorretal, observação também confirmada por outros importantes
pesquisadores como Dukes (1952) e Bussey (1975).
A PAF é afecção rara, responsável por cerca de 1% dos casos de CCR na população. Como é doença
autossômica dominante, os filhos de um individuo com PAF tem 50% de chance de herdar a mesma mutação.
É reconhecida a doença pré-cancerosa mais bem definida em toda a literatura médica, destacando-se a
penetrância virtual de 100%, que expressa a chance dos indivíduos não tratados por colectomia profilática
desenvolverem CCR. Segundo registros nacionais da doença, ocorre um caso para cada 6 a 20 mil novos
nascimentos, atingindo igualmente ambos os sexos.
Cerca de 20% dos pacientes não apresentam antecedentes familiares, sendo a afecção decorrente de
mutações genéticas. Os descendentes desses portadores sem histórico familiar tem o mesmo risco de
desenvolver a doença que os filhos com antecedentes, sendo a chance de transformação maligna semelhante nos

32
dois grupos. Em série da Clevelan Clinic 22% dos pacientes mostravam mutações espontâneas caracterizando-
se por manifestar doença mais grave, com maiores índices de câncer colônico (38% vs. 17%), tumores
desmóides (26% vs. 9%), pólipos duodenais (86% vs. 33%) e tumores extracolônicos (22% vs. 9%) em
comparação com pacientes que herdaram a doença.
O gene APC foi mapeado no cromossomo 5q21. Esse gene tem 8.538 pares, 15 éxons de codificação e
um produto proteico de 2843 aminoácidos. Regula a degradação de beta-catenina, agindo com um gene
supressor de tumor. Normalmente, cada pessoa possui duas cópias funcionais do APC em todas as células,
enquanto que indivíduos com PAF tem apenas uma cópia. Assim, a inativação somática ou perda do alelo
correspondente ao gene herdado resulta em inativação do gene, iniciando-se o processo neoplásico em que as
células epiteliais do cólon apresentam propensão a se proliferar, na puberdade ou na fase adulta. Outras
mutações somáticas em outros genes (oncogenes e outros genes supressores) facilitam o surgimento de
adenomas e carcinomas no tecido epitelial.
A mutação determina a formação dos chamados stop códons e de uma proteína truncada, identificada
pela realização do PTT ( protein truncation testing ou teste da proteína truncada). Quando se localiza a proteína
truncada, é possível encontrar a mutação em um seguimento especifico do gene e realizar o sequenciamento
genético para identificar um mais nucleotídeos mutantes.
Desde a identificação do gene APC, mais de 400 mutações foram identificadas, parecendo haver uma
relação entre local da mutação e expressões fenotípicas diferentes. Podem existir variações fenotípicas mesmo
em indivíduos com mutações idênticas. Genótipos individuais estão relacionados à expressão de algumas
manifestações extra intestinais, podendo também influir na gravidade e apresentação da doença colônica.
Dessa forma, mutações localizadas no final 5’ do códon 168 ou no final 3’ do códon 1.580 determinam
o aparecimento de fenótipo atenuado caracterizado por menor número de adenomas, progressão mais lente e
aparecimento tardio da doença (acima de 50 anos). Já mutações no códon 1.309 estão relacionadas a formas
mais agressivas, com início precoce (20 anos) e maior numero de pólipos. Enquanto mutações nos códons 1.061
e 1.369 predispõem à polipose típica, as localizadas no códon 1.465 estão associadas a expressões fenotípicas
variáveis no cólon e em outros órgãos.
As síndromes de Gardner e Turcot, assim como a forma atenuada da polipose adenomatosa familiar
são variações fenotípicas da PAF que merecem consideração especial. Em 1951, Eldon Gardner publicou um
artigo descrevendo polipose colônica em uma família de Utah com nove óbitos (idade média de 34 anos) por
câncer colônico em três gerações. A então chamada síndrome de Gardner passou a englobar pacientes com PAF
associada a manifestações extra intestinais como tumores de partes moles (fibromas, cistos epidermóides, cistos
sebáceos, lipomas, tumores desmóides), esteomas (mandíbula, maxila, crânio e ossos longos), adenomas do
sistema digestivo superior, carcinoma da tireóide e hepatoblastoma, entre outras. Apesar de expressões

33
fenotípicas distintas, a PAF clássica e a síndrome de Gardner se originam de mutações do mesmo gene, sem
diferença na localização ou natureza das mutações.
A associação de polipose com tumores do SNC é conhecida como síndrome de Turcot, descrita pela
primeira vez por Crail, em 1949. Dez anos mais tarde, Turcot et al. (1959) relataram a referida associação em
irmã e irmão cujos pais eram primos em terceiro grau. Desde então, pouco mais de 130 casos forma reportados
na literatura.
Ainda existe controvérsia quanto à sua diferenciação da PAF e quanto à forma de transmissão, havendo
discordância quanto à síndrome ser herdada por mecanismo autossômico recessivo ou dominante. Foram
descritas mutações do gene APC do cromossomo 5q21 (dando origem a tumores do tipo meduloblastoma -
mais comum – e astrocitomas) e nos genes de reparo associados ao HNPCC (dando origem a glioblastomas). A
formas atenuada da PAF caracteriza-se por menor número de pólipos colônicos (menos de 100), distribuição
mais proximal, maior frequência de adenomas planos, inicio mais tardio dos pólipos (depois dos 20 até os 30
anos) e do CCR ( depois dos 50 anos). Associa-se a mutações em regiões do gene APC diferentes da polipose
clássica (final 5’ ou final 3’). Sua frequência exata não é conhecida, pela dificuldade diagnóstica atribuída a suas
características tênues. A incidência de tumores desmóides depende da mutação. Alterações da retina e osteomas
são raros.
4.2.2.1.1. Aspectos Morfológicos, Clínicos e diagnósticos
Os múltiplos pólipos adenomatosos da PAF são indistintos daqueles encontrados em pacientes sem
polipose, diferindo apenas no numero e na época de aparecimento. Sua apresentação macroscópica varia de
pequenas áreas hiperplasia da mucosa até estruturas com vários centímetros, mas geralmente não ultrapassam 1
cm. Pólipos vilosos tem maior chance de degeneração, sendo o aparecimento de carcinomas (muitas vezes
múltiplos) indicados por maior tamanho, consistência, coloração mais escura e ulceração do pólipo.
Os pólipos colorretais predominam no cólon esquerdo e reto. A gravidade da polipose colônica é
atestada pelo número de pólipos observados à colonoscopia e na peça cirúrgica, na qual expressões como
“numerosos”, “incontáveis”, “milhares” ou “mucosa acarpetada” são indicativas de doença colônica grave. Por
outro lado, “poucos”, “esparsos” e “raros” são termos que refletem doença colônica branda.
Do ponto de vista clínico, a PAF costuma se manifestar na puberdade, com o aparecimento de pólipos
em diferentes estágios de evolução e grau de degeneração celular. Aproximadamente 15% dos pacientes
desenvolvem pólipos após os 10 anos de idade e 90% destes aparecem até os 30 anos. É menos comum
surgirem após os 40 anos de idade, situação em que se acredita haver uma baixa penetrância do defeito genético
básico. Nos primeiros anos da doença os sintomas são vagos ou, mesmo, ausentes. Sangramento nas fezes é a

34
manifestação inicial, tornando-se mais frequente e intenso com a evolução da doença. O aparecimento da
diarréia, sangue e muco nas fezes representa um alerta para o aparecimento de CCR, presente em mais de 60%
dos pacientes sintomáticos.
O intervalo entre o começo dos sintomas e o diagnostico de câncer diminui significativamente
conforme aumenta a idade dos pacientes, sugerindo uma fase pré-maligna mais curta em pacientes com mais
idade. A natureza adenomatosa e a enorme quantidade de pólipos tornam a possibilidade de degeneração
maligna uma preocupação constante em pacientes não tratados, situação em que o desenvolvimento de CCR é
uma regra, surgindo em média 10 a 15 anos após o desenvolvimento dos pólipos (ao redor dos 33 anos) e
levando a óbito no inicio da quarta década de vida.
A grande frequência de adenomas retais na PAF indicam que a avaliação inicial desses pacientes (e de
suspeitos) seja feita por retossigmóidoscopia a partir da adolescência. Esse exame permite avaliar a intensidade
do comprometimento retal, realizar a biópsia de estudos histológicos e, eventualmente, diagnosticar tumores
nesse segmento. Os achados à retoscopia tem sido valorizados na gravidade da doença e, por conseguinte,
auxiliam na escolha da melhor opção operatória em cada caso. Assim, estima-se que pacientes com mais de 20
pólipos retais ao exame proctológico tenham doença mais grave, enquanto a presença de menos de cinco
pólipos sugere doença mais branda. Para firmar o diagnostico da afecção e avaliar a extensão ou associação com
câncer, realiza-se a colonoscopia, cujo achado de mais de 1.000 adenomas também indica a gravidade da
doença.
A detecção precoce só é possível em parentes de portadores da afecção. Depois da identificação do
paciente chamado “índice”, um teste genético facilita o rastreamento dos familiares eventualmente acometidos e
a instituição de colectomia profilática. Os testes genéticos para análise da mutação devem ter o consentimento
prévio dos pacientes. Realizam-se o teste da proteína truncada e sequenciamento genético segundo normas já
descritas na literatura. Para facilitar o sequenciamento, faz-se o rastreamento prévio da mutação no éxon 15G do
FIGURA 6: ASPECTO DA MUCOSA COLÔNICA NA PAF

35
gene APC em casos de polipose grave. Naqueles pacientes com fenótipo acentuado, são rastreados os éxons 3 e
4.
A avaliação global dos pacientes com PAF deve incluir, também, exames que avaliem a eventual
associação com manifestações extra colônicas da doença, como endoscopia digestiva alta, ultrassonografia
abdominal, tomografia computadorizada, trânsito intestinal, radiografias ósseas e exame de fundo de olho, entre
outros.
4.2.2.1.2.Manifestações Extracolônicas
Desde a descrição original de polipose associada a cistos epidermóides e osteoma, a combinação de
PAF e manifestações extracolônicas (MEC) é comumente referida como síndrome de Gardner (1951).
Subsequentemente, reconheceu-se que a PAF é uma pan-polipose gastrointestinal que pode estar associada a
numerosas MEC, benignas e malignas.
Além do cólon e do reto, pólipos também podem ser encontrados no sistema digestivo superior
(estômago e duodeno), intestino delgado, tireóide, adrenais, pâncreas e hipófise. Outras MEC benignas incluem
cistos sebáceos, lipomas, osteomas, dedos hipocráticos, anormalidades dentárias (dentes supranumerários), lesão
da retina e tumores desmóides. Dentre as manifestações malignas, forma relatados tumores da região
periampular, entre ductos biliares, gástricos, no íleo (carcinoma e carcinóide), tireóide, supra renal e sistema
nervoso central.
Em pacientes com PAF, as principais causas de mortalidade são os tumores desmóides (TD) e as
neoplasias colorretais e periampulares. Assim, é preciso conhecer incidência, diagnosticar, prevenir e tratar tais
manifestações. As MEC mais diagnosticadas são as lesões da retina originalmente interpretadas como
congênitas, detectadas em até 90% de um portador de PAF. Embora achados histopatológicos indiquem que o
termo “hamartomas do epitélio pigmentar da retina” seja mais apropriado para designar essas lesões, o termo
CHRPE é o mais aceito e continua a ser usado. Sugeriu-se que a existência de quatro ou mais lesões CHRPE
distribuídas em ambos os olhos seria um marcador fenotípico da polipose, quando diagnosticado em uma
determinada família, também seria encontrado em pacientes do mesmo grupo familiar. Dessa forma, a CHRPE
poderia facilitar a detecção de mutações constitucionais do APC em parentes.
A CHRPE tem sido consistentemente associada a determinado domínio e é o único fenótipo sem
variação intrafamiliar. Assim a caracterização de CHRPE pode adicionar informação sobre a localização da
mutação genética. Atualmente, sugere-se que o achado de lesões CHRPE ao exame oftalmológico seja um
marcador clínico a mais para PAF em famílias CHRPE “positivas”. Em famílias CHRPE “negativas”, exames
oftálmicos negativos não tem valor predição e não devem eliminar a pessoa de rastreamento futuro.

36
Os tumores de partes moles (cistos epidermóides, lipomas e fibromas) podem ocorrer em qualquer
lugar da superfície cutânea, predominando nos membros, face e couro cabeludo. Leppard e Bussey (1975)
encontraram cistos em 53% de 70 portadores de PAF. Embora tenham apenas interesse cosmético, tem
importância por serem lesões raras na infância em indivíduos sem polipose. Quando aparecem antes da
puberdade, constituem verdadeiros marcadores da síndrome. Além deles, os osteomas podem preceder o
aparecimento de pólipos intestinais.
Os osteomas foram primeiro descritos por Gardner e Richards em 1953, podem ocorrer em qualquer
osso, conquanto sejam mais comuns na face (particularmente no ângulo da mandíbula) e menos frequentes em
ossos frontais e occiptais. São tumores benignos, ainda que possam causar sintomas por crescimento local.
Ocasionalmente, aparecem antes do diagnostico da polipose, podendo sugerir a herança do gene em
descendentes de indivíduos afetados. A incidência relatada (14 a 93%) é bastante variável, refletindo diferenças
na assiduidade com que se procura tais lesões, na interpretação das radiografias e na idade em que são
pesquisadas. Em nosso meio, diagnosticaram-se osteomas em 25% dos pacientes nos quais se investigou tal
manifestação.
FIGURA 8: OSTEOMAS DE MANDÍBULA E OSSO FRONTAL EM PACIENTE COM SINDROME DE GARDNER.
FIGURA 7: HIPERTROFIA CONGÊNITA DO EPITÉLIO PIGMENTAR DA RETINA DETECTADA AO EXAME DE
FUNDO DE OLHO (CHRPE)

37
Apesar de histologicamente benignos (lesões fibromatosas), os tumores desmóides podem exibir
comportamento biológico agressivo, com invasão local, mas não metastatizam. Formam tumores encapsulados,
de crescimento lento, podendo surgir em mesentério, parede abdominal, incisões, retoperitôneo, virilha e
nádegas; às vezes regridem espontaneamente e podem ser multifocais.
A incidência pós operatória desses tumores varia de 3,5 a 29% ( com media entre 10 a 18%), ocorrendo
cerca de dois anos após a cirurgia colorretal. Entretanto, o número fortuitos em pacientes assintomáticos sugere
que a incidência provavelmente seja maior do que a reportada. A ocorrência desses tumores está associada a
diferentes fatores de risco. Cerca de 80% dos casos aparecem em pacientes submetidos a operações abdominais
prévias.
Sexo feminino, histórico familiar de desmóides, presença de osteomas e mutações entre os códons
1.445 e 1.578 são considerados fatores de predição independentes. Histórico familiar de desmóides tem sido
reportado em mais de 50% dos portadores. Sua incidência também parece estar associada a mulher em pré
menopausa, gravidez e uso de contraceptivos. Essas observações indicam que a integração entre dados clínicos e
genéticos ajuda a definir subgrupos de pacientes com maior risco para desenvolver esses tumores.
Quando pequenos, os TD podem ser assintomáticos. À medida que crescem, podem determinar
sintomas relacionados a complicações como compressão ureteral, obstrução intestinal, infiltração de outros
órgãos, fístulas e oclusão vascular. A morte sobrevém em média, seis anos após o diagnóstico.
As opções para tratamento incluem cirurgia, radioterapia com drogas citotóxicas e não citotóxicas. O
manuseio dos tumores desmóides representa um grande desafio. A ressecção cirúrgica completa raramente é
possível TD mesentérico devido à extensão da ressecção e do alto recidiva. A cirurgia deve ser reservada para
alívio da obstrução, preferindo-se procedimentos bypass aos de ressecção.
Pólipos gastroduodenais são observados, com frequência na PAF e, mais raramente no intestino
delgado. As lesões gástricas encontradas são pólipos de glândulas fúndicas (dilatações císticas), hiperplásicos e,
menos habituais adenomas e carcinoma. De acordo com a literatura japonesa, a incidência de carcinoma
gástrico na PAF varia de 4,5 a 13,6%, enquanto fora do Japão essa é uma complicação rara. Estima-se que
pacientes com PAF tenham chance 300 vezes maior que a população geral para desenvolver câncer
gastroduodenal. Os adenomas doudenais são diagnosticados em ate 90% dos pacientes após 10 a 20 anos do
diagnostico dos pólipos colorretais, sendo os adenomas periampulares (muitas vezes microscópicos) as lesões
precursoras de carcinoma na região, embora essa progressão ocorra em menos de 5% dos casos.
Mesmo ainda não tendo sido definidos programas de vigilância endoscópica e tratamento efetivo para
essas lesões, sabe-se que o uso de antiinflamatórios não esteroides como sulindac não controla os pólipos.
Iwama et al. (1993) propuseram ressecção total da papila de Vater em pacientes acima de 35 anos, com
adenoma.

38
O carcinoma periampular é a forma mais comum de câncer extracolônico, estimando-se em 3 a 4% seu
risco de vida. Em pacientes submetidos à colectomia total, o carcinoma periampular é responsável por 22% das
mortes por câncer em média, 23 anos após o tratamento. Adenomas do intestino delgado foram reportados no
íleo terminal na mucosa ileal pós-ileostomia, anastomose ileorretal, bolsa ileal e bolsa de Koch. A transformação
maligna desses pólipos é rara. Já foram também relatados tumores hepatobiliares, pancreáticos, de bexiga, rins,
testículos, olhos e pulmões.
Esses autores observaram que carcinomas da tireóide desenvolvem em idade menor (32 anos) que
tumores do trato gastrointestinal (43 anos, em duodeno e 49 anos, no estômago) e que mulheres apresentam
maior propensão para desenvolver carcinoma de tireóide e TD. Estima-se que o risco de carcinoma da tireóide
associado à PAF seja 100 a 160 vezes superior ao da população geral em mulheres europeias , contra 25 vezes
no Japão.
Em 15 pacientes com carcinoma da tireóide associado à PAF, Cetta el al. (1998) observaram que a
mutação estava localizada entre códons 778 e 1.309 (éxon 15) em 13 pacientes, documentando manchas
oculares em 12 pacientes. Dessa maneira, as mutações agruparam-se na área genômica associada à CHRPE
(códons 463-1.387). Esses autores sugeriram que a incidência de câncer de tireóideo foi subestimada no passado
e que o rastreamento intensivo poderia detectar maior número desses tumores. Recomendam ainda pesquisa
sistemática em pacientes com manchas oculares e mutações genéticas no éxon 15, destacando que esses
tumores parecem ter excelente prognóstico.
A associação de tumores do SNC com polipose difusa colorretal caracteriza a síndrome de Turcot.
Esses tumores são representados por meduloblastomas e glioblastomas, surgindo habitualmente antes do
desenvolvimento da polipose.
Em trabalho recente, Bertario et al. (2003) identificaram associações entre manifestações especificas e
local da mutação em 953 com PAF entre 187 famílias, destacando que o conhecimento desses dados é útil para
vigilância e prevenção. As mutações do gene APC localizavam-se entre os códons 156 e 2.011. Encontraram-se
associações com CHRPE (códons 543 e 1.309), tumores desmóides (risco seis vezes maior entre códons 1.310 e
2.011 e risco menor entre 159 e 495), desenvolvimento precoce de CCR (códon 1.309) e adenomas duodenais
(risco 4 vezes maior entre códons 976 e 1.067). a frequência cumulativa de MEC foi maior entre mutações entre
códons 976 e 1.067.
Nossa experiência pessoal e a revisão da literatura sugerem que a incidência de manifestação
extracolônica ao longo da evolução da PAF é alta. Podendo ser detectadas em qualquer 40% dos pacientes
tratados. Deve-se ressaltar, provavelmente esse número possa ser maior, dependendo de pesquisa rotineira
dessas alterações e de seguimento prolongado. Verifica-se que algumas dessas manifestações ( especialmente as
neoplásicas) podem trazer graves consequências e gerar importantes complicações, afetando o tempo e a

39
qualidade de vida. Por tais motivos, torna-se necessário conhecer e investigar essas manifestações não só por
ocasião do diagnostico da PAF, como também no seguimento pós operatório dos pacientes.
4.2.2.1.3. Tratamento Cirúrgico
Todas as células do epitélio colunar em portadores da PAF possuem a mutação germinativa do gene
APC, podendo gerar múltiplos adenomas colorretais ao longo da vida. Por isso, esses doentes devem ser
submetidos à colectomia profilática para prevenir o desenvolvimento de CCR, a principal causa de morte nesses
pacientes. O melhor momento para operação depende do número de pólipos e do risco individual de cada um.
Nesse contexto, Heimann et al. (1985) reviram 69 casos operados em dois períodos entre 1947 e 1983,
no Mt. Sinai Hospital, observando diminuição de 50 para 20% na incidência de pacientes com CCR, atribuindo
o fato à intervenção cirúrgica com menos idade (40 e 25 anos, respectivamente) nesses períodos.
Para adolescentes ainda não há consenso quanto ao momento da indicação cirúrgica. Church et al.
(2002) conduziram um estudo para investigar o risco de câncer em adolescentes pertencentes a registros
afiliados ao Leeds Castle, identificando 14 pacientes abaixo de 20 anos com CCR invasivo (idade variou de 9 a
19 anos), três dos quais diagnosticados durante o tratamento cirúrgico e outro sete com sintomas associados. Os
autores concluíram que a incidência de CCR abaixo de 20 anos é rara (somente um caso tinha menos de 15
anos), sugerindo que a cirurgia pode ser protelada com segurança até pelo menos aos 15 anos, a menos que seja
encontrada alguma lesão suspeita.
Nas ultimas décadas, a introdução de novos procedimentos técnicos, a seleção mais apropriada dos
pacientes e o acúmulo de conhecimentos em biologia molecular propiciaram uma sensível evolução na
abordagem de pacientes com PAF. Apesar disso, a escolha da melhor opção cirúrgica ainda gera muitos
debates.
Além de prevenir o CCR, o tratamento cirúrgico deve permitir ao paciente preservar seu estilo de vida o
mais próximo do normal, livre de um estoma abdominal e com a função evacuatória preservada. Assim, a
decisão final deve cotejar não só a evolução a curto e longo prazos, como também os resultados funcionais e o
risco de câncer após a operação. As alternativas cirúrgicas incluem a realização de colectomia total com
anostomose ileorretal, proctolectomia restaurativa com confecção de bolsa ileal anastomosada ao canal anal ou
proctolectomia total com ileostomia definitiva.
Esta última determina profundas modificações na imagem corpórea e repercussões emocionais
significativas relacionadas ao estoma e a disfunções sexuais. Por esses motivos, sua indicação tem se restringido
a pacientes com câncer do reto baixo associado à polipose ou com disfunção enfincteriana importante. Assim, os
procedimentos mais indicados são a anastomose ileorretal ou bolsa ileal anastomosada ao canal anal.

40
A colostomia total com anastomose ileorretal é procedimento com baixa morbidade cirúrgica e que
preserva o reto como órgão reservatório, importante na continência, sendo indicada a pacientes com reto normal
ou pouco doente e que possam fazer seguimento pós operatório a longo prazo. Nesse segmento, os adenomas
do cólon retal podem ser ressecados ou cauterizados devendo-se examinar o resto em intervalos de quatro a seis
meses. A discrepância quanto à incidência de câncer no cólon retal pode ser explicada pela realização de
colectomias com diferentes extensões ou por tratamento cirúrgico em diferentes faixas etárias. Hoje se
reconhece que esse risco aumenta progressivamente com seguimento, variando de 5% após 10 anos.
Atualmente, a proctocolectomia restaurativa é o procedimento mais indicado, pois erradica toda a
mucosa colônica e retal doente, mantêm a musculatura esfincteriana e evita a ileostomia definitiva, embora seja
procedimento complexo associado a altos índices de morbidade pós operatória. Entretanto, a ascensão na curva
de aprendizado diminui o risco de perda definitiva da bolsa ileal e melhora os resultados funcionais. Dentre os
critérios de indicação cirúrgica, sabe-se que os resultados funcionais são piores em pacientes com função
esfincteriana deficiente e que a técnica deve ser seletivamente em doentes com câncer retal.
Ainda que talvez a premissa de que a confecção de bolsa ileal diminuía significativamente o risco de
câncer em comparação à anastomose ileorretal, não se conhecem os índices tardios de degeneração na bolsa ileal
e na zona de transição do epitélio anal. Desde seu advento, alguns trabalhos tem descrito o desenvolvimento de
pólipos adenomatosos em bolsas ileais em incidências variáveis de 4 a 50%.
Por isso, recomenda-se ressecar toda a mucosa retal a partir da linha pectínea, para evitar sua
regeneração e o desenvolvimento de novos pólipos. Mesmo que os adenocarcinomas descritos tenham se
originado de pequenas áreas de mucosa retal remanescentes, as vantagens desse procedimento devem ser
cotejadas com o risco duvidoso de degeneração, mesmo pequeno, que ressalta a necessidade de seguimento
desses pacientes. A ocorrência de pólipos na extremidade distal no íleo é pequena, não justificando ampliar a
extensão da ressecção do intestino delgado, pois esses pólipos apresentam pequeno potencial de degeneração e
podem ser tratados por fulguração no pós operatório.
Quanto à forma de anastomose de bolsa e canal anal, aceita-se que a sutura mecânica provê melhores
resultados funcionais comparada à anastomose manual, além de dispensar a necessidade de ileostomia
temporária em maior numero de casos. Em contra posição, pode-se associar à maior incidência de adenomas na
zona de transição.
Mais recentemente, tem-se sugerido testes genéticos moleculares para guiar o tratamento cirúrgico,
uma vez entre indivíduos da mesma família. Essa variação determina a existência de diferentes graus de
“gravidade” da doença, existindo indivíduos mais propensos a desenvolver numerosos pólipos e câncer retal
após anastomose ileal, casos em que a melhor opção terapêutica seria a confecção de bolsa ileal anastomosada
ao canal anal. Por outro lado, há outros que apresentam a doença intestinal mais branda, com menor número de

41
pólipos colorretais, candidatos ideias ao tratamento por anastomose ileorretal. Correlações entre genótipo e
fenótipo existem para número e localização dos adenomas, como a forma atenuada da PAF e mutações do gene
APC 3’ do códon 1250, que estão associadas a maior risco de câncer retal.
O risco de câncer retal após anastomose ileorretal depende de fatores clínicos (idade, tamanho do coto
retal), patológicos (presença de displasia, adenomas vilosos e associação de número, tamanho e forma dos
pólipos) e moleculares (localização da mutação do gene APC). Apesar disso muitos pacientes com seguimento
tardio desenvolvem câncer retal na ausência desses fatores.
A avaliação do risco associado a tais fatores evidencia-se resultados controversos na literatura, com
relatos em que não se demonstraram efeitos adversos quanto ao numero de pólipos, idade e câncer colônico e
outros em que o estudo de maior número de pacientes permitiu verificar maior risco relacionado à idade, a
câncer colônico (3,6 vezes), número de pólipos superior a 30 (4,6 vezes) e localização da mutação no gene APC.
A definição do numero de pólipos que determinaria maior risco no coto retal é variável na literatura.
Church et al. (2001) revificaram que pacientes com menos de cinco adenomas retais evoluira de maneira
satisfatória quando submetidos à anastomose ileorretal. Um dado importante a se considerar é a possibilidade
real de se estimar o numero de pólipos retais, uma vez que a contagem de pólipos colônicos à colonoscopia nem
sempre corresponde ao número de lesões encontradas nas peças ressecadas, conquanto a maioria dos pacientes
classificados como portadores da forma atenuada da doença à colonoscopia mantenha essa classificação após a
colectomia. Ainda mais a conceituação de menor gravidade de doença retal e colônica não garante que o câncer
não se desenvolverá, motivo que torna indispensável o seguimento prolongado dos pacientes.
A quimioprevenção com inibidores da cicloxigenase e ácido acetilsalicílico visando a alterar numero e
tamanho de pólipos retais e do sistema digestivo superior tendo obtido resultados conflitantes, razão pela qual
ainda não pode ser recomendada como medida padrão dos pacientes com PAF.
Uma perspectiva interessante refere-se ao emprego de técnicas laparoscópicas. Milsom et al. (1997)
publicaram resultados resultados animadores em pacientes jovens com polipose adenomatosa familiar tratados
com colectomia total e anastomose ileorretal, destacando o efeito cosmético e a rápida recuperação. Em elegante
estudo comparativo pareado, Marcello et al. (2000) relataram vantagens relacionadas ao retorno das funções
intestinais e o tempo de hospitalização em 40 pacientes com polipose e retocolite tratados com proctocolectomia
restaurativa laparoscópica quando comparada à via convencional.

42
5. Considerações finais
A polipose adenomatosa familiar (PAF) é doença hereditária de caráter autossômico
dominante, sendo responsável por apenas 1% dos casos de câncer colorretal (CCR) na
população. O defeito genético localiza-se no gene APC (adenomatous polyposis coli) situado
no braço longo do cromossomo 5q21. Em aproximadamente 20%, a doença pode ser
decorrente de mutações genéticas. Do ponto de vista clínico, a doença geralmente se
manifesta na puberdade, com o aparecimento de pólipos adenomatosos na mucosa
colorretal.Sintomatologia associada geralmente sucede o aparecimento dos adenomas na
segunda década de vida. A natureza adenomatosa e a enorme quantidade de pólipos tornam a
possibilidade de degeneração maligna preocupação importante em pacientes não tratados, em
que o desenvolvimento de CCR é regra, surgindo em média 10 anos após o desenvolvimento
dos pólipos.
Filhos de indivíduo afetado têm risco de 50% de herdar o gene. Após a identificação
do paciente chamado “índice”, a realização de teste genético facilita a identificação mais
acurada dos familiares eventualmente acometidos, permitindo focar o rastreamento e o
tratamento profilático somente naqueles com risco. Desta forma, assume fundamental
importância a detecção precoce de pacientes com esta afecção, seu adequado tratamento pela
colectomia profilática e o rastreamento de familiares para identificar eventuais portadores do
defeito genético.
Desde a descrição original de polipose associada a cistos epidermóides e osteoma, a
combinação de PAF e manifestações extracolônicas (MEC) é comumente referida como
síndrome de Gardner. Subsequentemente, reconheceu-se que a PAF é uma panpolipose
gastrointestinal que pode estar associada a numerosas MEC, benignas e malignas. Além do
cólon e reto, pólipos podem ser encontrados também no trato digestivo superior (estômago e
duodeno), intestino delgado, tireóide, adrenais, pâncreas e hipófise.

43
6. Referências
1Alves JB. Cirurgia Geral Especializada. Vol. 6. Belo Horizonte, 2004.
2.Hernegger GS, Moore HG, Guilem JG. Attenuated Familial Adenomatous Polyposis: An
evolving and poorly understoodentity.Dis Colon Rectum 2002; 45:127-136
3.Knudsen AL, Bisgaard ML, Bülow S. Attenuated Familial Adenomatous Polyposis
(AFAP).A review of the literature. Familial Cancer 2003; 2:43-55.
4. Contessini-Avesani E, Botti F, Negri C et al. Familial adenomatous polyposis. Surgical
treatment: when and how.
5.Moision A-L, Järvinen H, Peltomäki.Genetic and clinical characterisation of familial
adenomatous polyposis: a population based study.Gut 2002;50:845-850.
6. Neklason DW, Solomon CH, Dalton AL et al. Intron 4 mutation in APC gene results in
splice and attenuated FAP phenotype.Familial Cancer 2004;3:35-40.
7. Chetty R, Salahsho S, Bapat B et al. Intraductal papillary mucinous neoplasm of the
pancreas in a patient with attenuated familial adenomatous polyposis. J Clin Pathol
2005;58:97-101.



























![Trabalho ..[1] trabalho de soldadura](https://img.document.onl/doc/110x75/559b39b21a28ab113f8b473b/trabalho-1-trabalho-de-soldadura.jpg)

