Embed Size (px)
Citation preview

ANEMIA APLÁSTICA CARLOS EDUARDO PIZZINO

INTRODUÇÃO: -‐ A anemia aplás-ca é caracterizada pela acentuada redução ou ausência de células eritroides, mieloides e megacariocí-cas na medula óssea, com resultante pancitopenia -‐ Podem ser adquiridas ou hereditárias -‐ A incidência anual desta doença nos países ocidentais é de cerca de 2 a 4 por milhão -‐ Mais frequente em indivíduos de 10-‐ 25 anos ou maiores que 60 anos.

ETIOLOGIA:
ANEMIA APLÁSTICA HEREDITÁRIA
Anemia de Fanconi
Disceratose congênita
Outras e-ologias
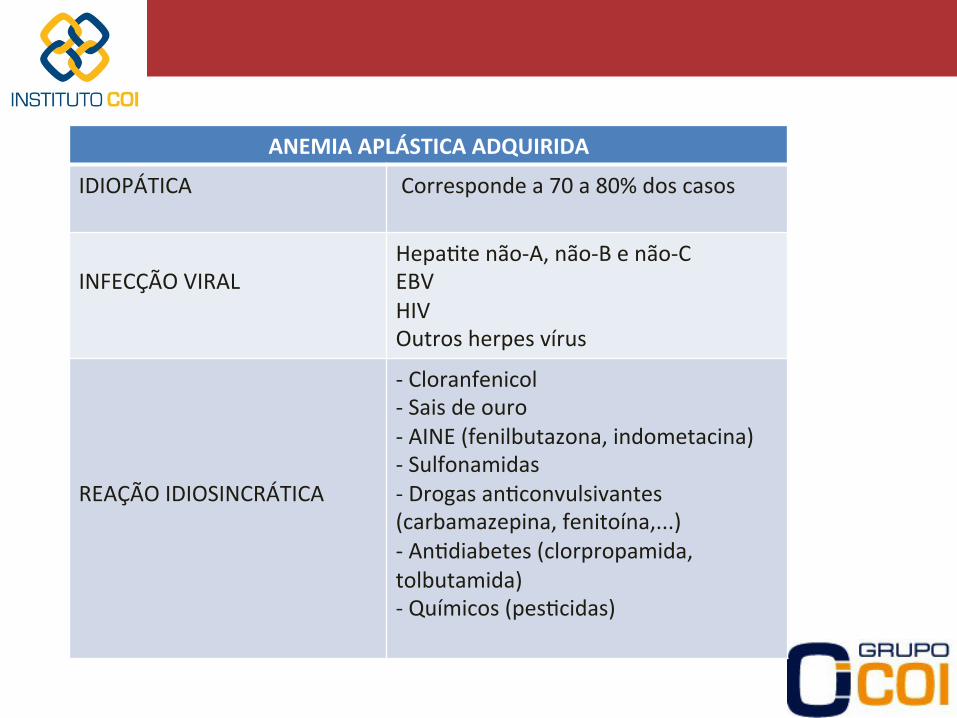
ANEMIA APLÁSTICA ADQUIRIDA
IDIOPÁTICA Corresponde a 70 a 80% dos casos
INFECÇÃO VIRAL
Hepa-te não-‐A, não-‐B e não-‐C EBV HIV Outros herpes vírus
REAÇÃO IDIOSINCRÁTICA
-‐ Cloranfenicol -‐ Sais de ouro -‐ AINE (fenilbutazona, indometacina) -‐ Sulfonamidas -‐ Drogas an-convulsivantes (carbamazepina, fenitoína,...) -‐ An-diabetes (clorpropamida, tolbutamida) -‐ Químicos (pes-cidas)

ANEMIA APLÁSTICA ADQUIRIDA
DOSE DEPENDENTE
Radiação ionizante Drogas citotóxicas Benzeno
DESORDENS AUTOIMUNES
Fasciíte eosinofilíca Lupus eritematoso sistêmico Doença enxerto versus hospedeiro
OUTRAS CAUSAS
Hemoglobinúria paroxís-ca noturna Gravidez Timoma, carcinoma gmico

DIAGNÓSTICO: -‐ Hemograma completo com re-culócitos • Pancitopenia • Re-culocitopenia -‐ Análise do sangue periférico (hematoscopia) • Hemácias normocí-cas, podem ser levemente macrocí-cas • Demais elementos reduzidos em número, porém de morfologia normal • Ausência de células anormais

-‐ Aspirado e biópsia de medula óssea • Medula óssea hipocelular (menos de 30% de celularidade) • Ausência de fibrose ou infiltração neoplásica • Células hematopoié-cas residuais com morfologia normal • Exclusão de síndrome hematofagocí-ca como causa da pancitopenia -‐ Citogené-ca da medula óssea • Se normal, a possibilidade de ser mielodisplasia hipoplásica diminui -‐ Imunofeno-pagem de marcadores eritrocitários • Ausência de CD55 e CD59: dado encontrado na hemoglobinúria paroxís-ca noturna
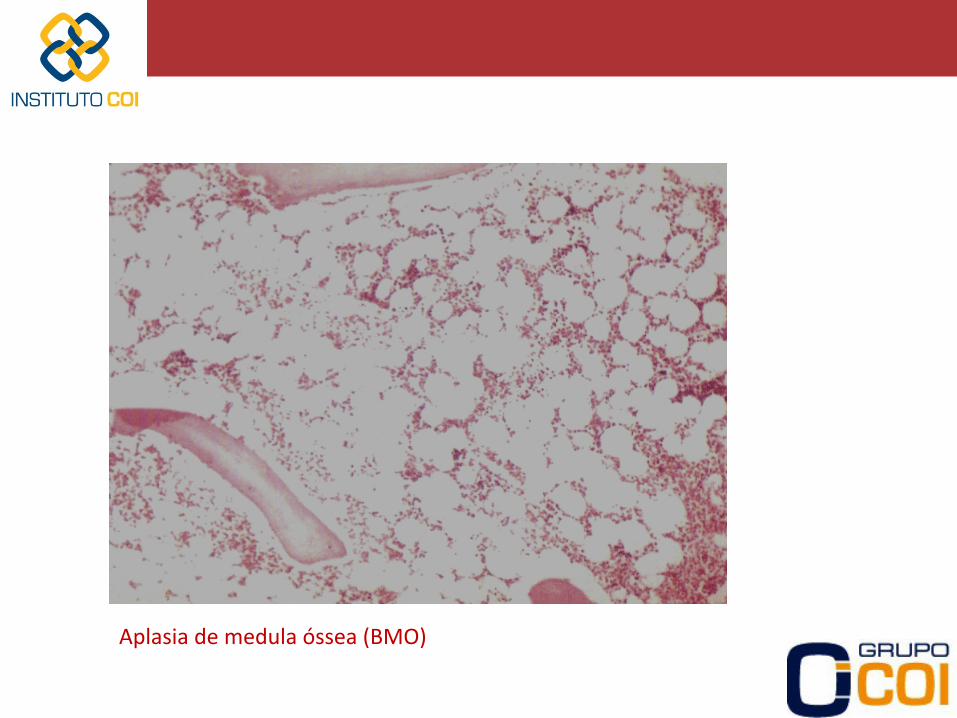
Aplasia de medula óssea (BMO)
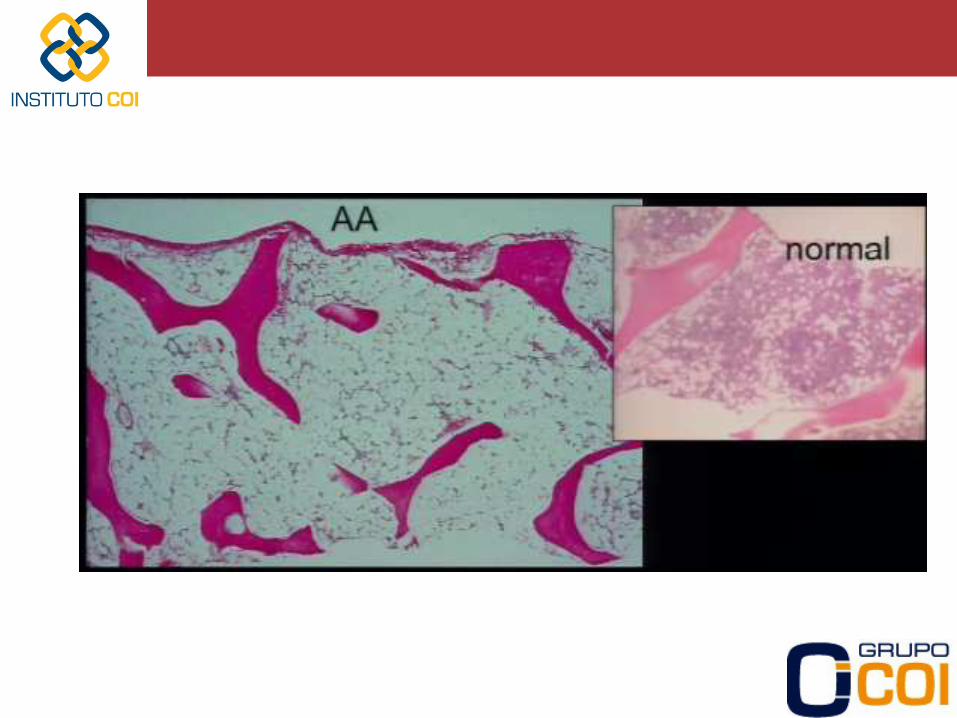

Diagnós>co de anemia de Fanconi: -‐ Presença de aumento na quebra cromossomial em culturas de linfócitos na presença de agentes ligadores de DNA (DEB -‐ diepoxibutano) -‐ Doença gené-ca caracterizada por: • Alterações ósseas • Baixa estatura • Manchas café-‐com-‐leite na pele • Anomalias renais (hipoplasia renal, rim pélvico) • Microcefalia • Retardo mental
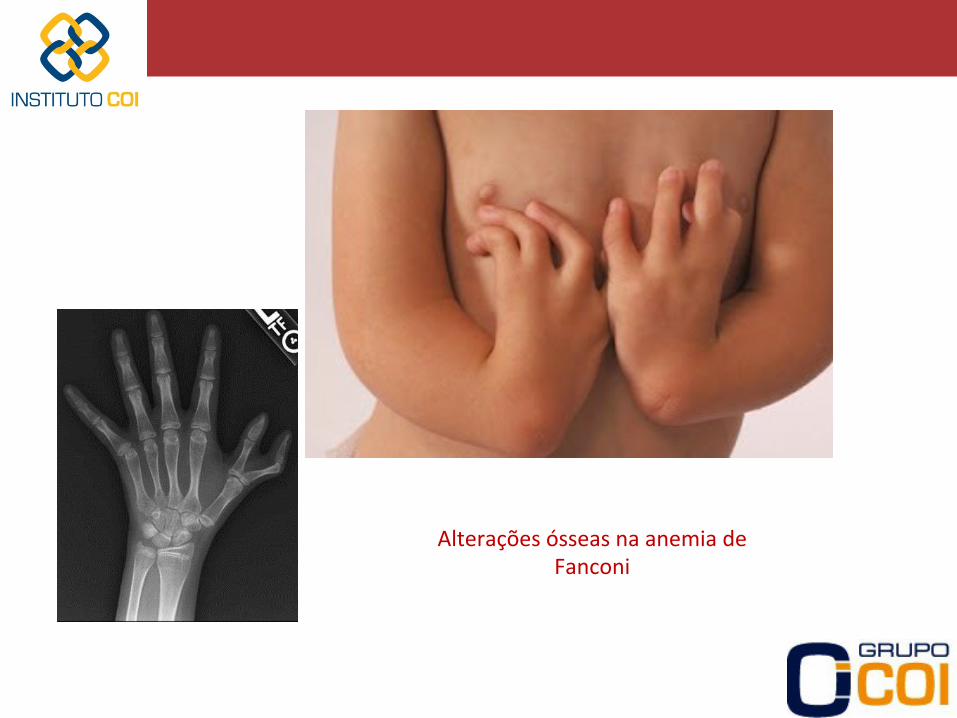
Alterações ósseas na anemia de Fanconi

Moderada
Celularidade medular menor que 30% Ausência de pancitopenia grave No mínimo 2 dos 3 seguintes critérios: Hb < 10g/dL; plaquetas <100.000/mL e neutrófilos < 1.500/mL
Grave
Celularidade medular inferior a 25% ou Celularidade de 25-‐50% com menos de 30% de células hematopoié>cas residuais com 2 das seguintes alterações: re>culócitos < 40.000/mcL, neutrófilos < 500/mcL ou plaquetas < 20.000/mcL
Muito Grave Critério para anemia aplás>ca grave + neutropenia < 200 mcL
CLASSIFICAÇÃO:

TRATAMENTO: O tratamento da anemia aplás-ca deve incluir: -‐ Re-rada de agentes potencialmente responsáveis pelo quadro clínico -‐ Terapia de suporte (uso de an-bió-cos e transfusões de hemocomponentes – tentar poupar o uso o máximo possível) -‐ Terapia defini-va específica (transplante alogênico x imunosupressão)

Modalidades de tratamento para anemia aplás-ca: -‐ Transplante alogênico de medula óssea (TMO): • Taxa de cura em torno de 60-‐70%, apesar de complicações como a doença enxerto versus hospedeiro (GVHD) • Rejeição do enxerto é ainda um problema em torno de 5-‐15% dos pacientes, principalmente naqueles subme-dos às inúmeras transfusões antes do TMO • Mortalidade relacionada ao tratamento aumenta com a idade

-‐ Imunosupressão: • U-lizado a combinação de globulina an--mócito (ATG), ciclosporina e
cor-coide com ou sem fator de crescimento de granulócitos (G-‐CSF) • Taxa de resposta (definida como independência transfusional) é em torno
de 60-‐70% em 6 meses nos pacientes com anemia aplás-ca grave • Risco de recaída em torno de 30-‐40%, com dependência da ciclosporina de
manutenção em torno de 26-‐62% dos pacientes • É uma opção terapêu-ca naqueles pacientes com anemia aplás-ca
moderada e dependência transfusional
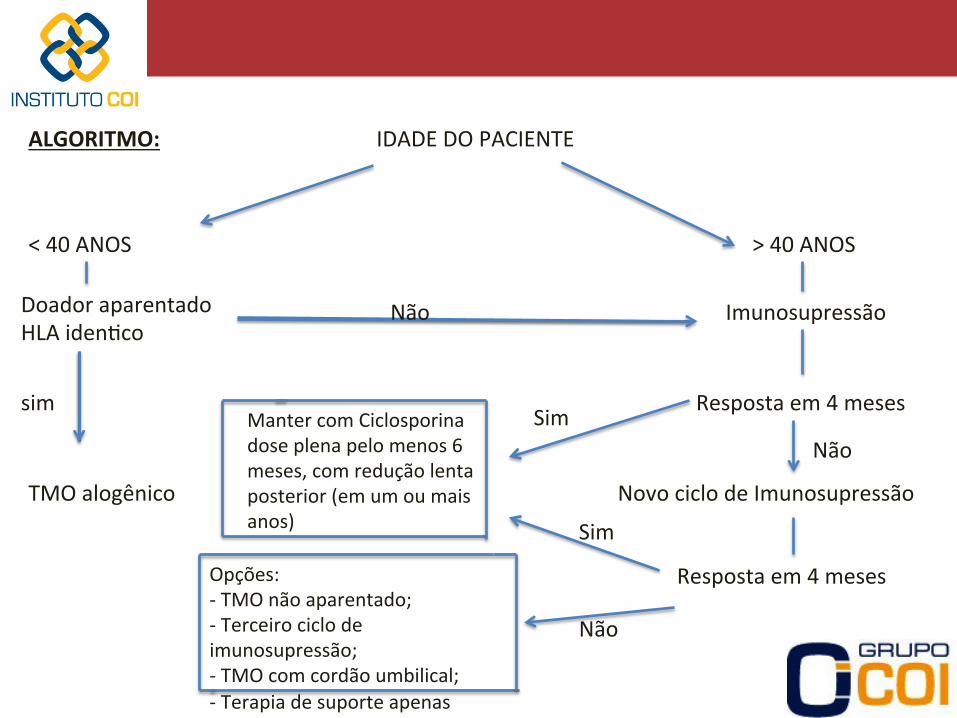
ALGORITMO: IDADE DO PACIENTE
< 40 ANOS > 40 ANOS
Doador aparentado HLA iden-co
sim
TMO alogênico
Imunosupressão Não
Resposta em 4 meses
Não Sim Manter com Ciclosporina
dose plena pelo menos 6 meses, com redução lenta posterior (em um ou mais anos)
Novo ciclo de Imunosupressão
Resposta em 4 meses
Sim
Não
Opções: -‐ TMO não aparentado; -‐ Terceiro ciclo de imunosupressão; -‐ TMO com cordão umbilical; -‐ Terapia de suporte apenas










![O significativo avanço na terapia medicamentosa da anemia ... - 09h00... · Zhao LP et al. Bone Marrow Transplant. 2019 Jan 22 25 [ahead of print] Lee JW et al. ... • Anemia Aplástica](https://img.document.onl/doc/110x75/5cca502f88c993cc428b97d0/o-significativo-avanco-na-terapia-medicamentosa-da-anemia-09h00-zhao.jpg)



















