Embed Size (px)
DESCRIPTION
metabolismo
Citation preview

Erros Inatos do Metabolismo
Abordagem Clínica
Prof Dra Ana Maria Martins
Departamento de Pediatria
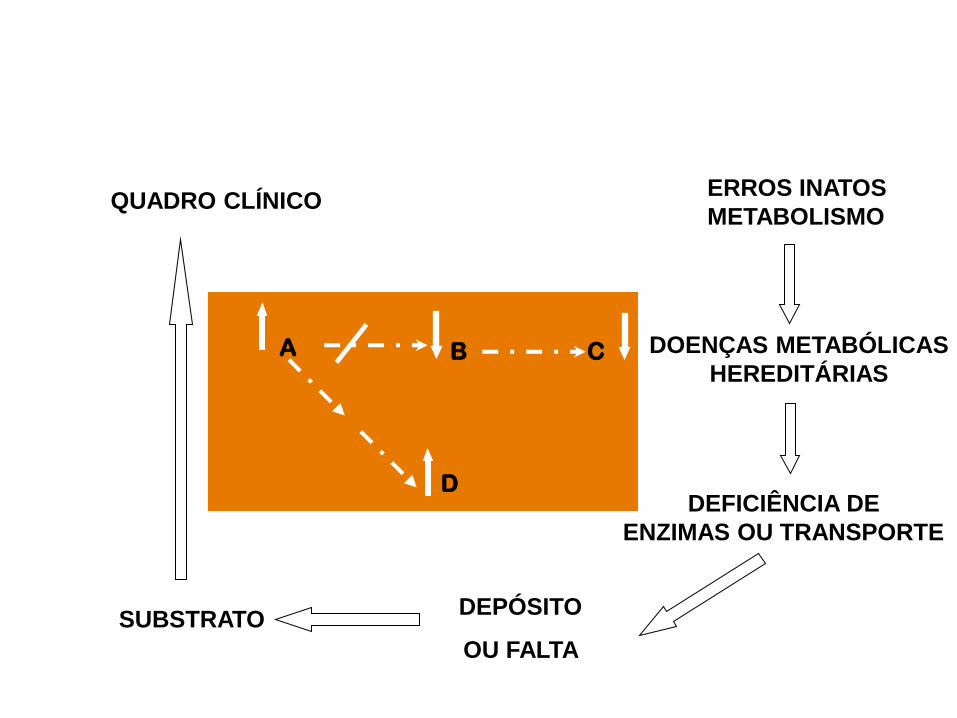
QUADRO CLÍNICO ERROS INATOS
METABOLISMO
DOENÇAS METABÓLICAS
HEREDITÁRIAS
DEPÓSITO
OU FALTA
DEFICIÊNCIA DE
ENZIMAS OU TRANSPORTE
SUBSTRATO
A B C
D

Erros Inatos do Metabolismo
• Incidência de EIM de 1:2.500 recém nascidos vivos1
– Brasil: 1200 novos casos por ano
• Incidência das DDL de 1:5.000 recém nascidos vivos2
• Maioria dos casos não chegam ao especialista pelo desconhecimento dos médicos em geral e carência de centros de referência no Brasil
1Applegarth DA et al, 2000, Pediatrics 105 (1): p. e10
2 Meikle PJ, Hopwood JJ, 2003, Eur J Pediatr, 162 (suppl 1): S34-7

Conceitos Gerais
• Idade de início do quadro clínico
• Gradação dos efeitos clínicos
• Tipo de herança
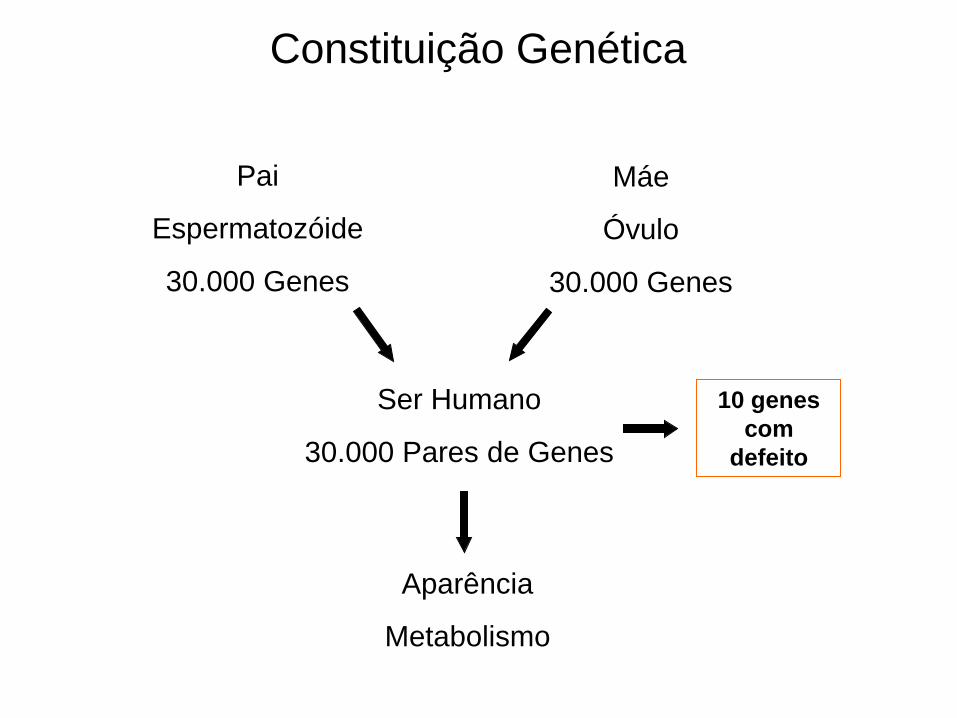
Aparência
Metabolismo
Constituição Genética
Pai
Espermatozóide
30.000 Genes
Máe
Óvulo
30.000 Genes
Ser Humano
30.000 Pares de Genes
10 genes
com
defeito
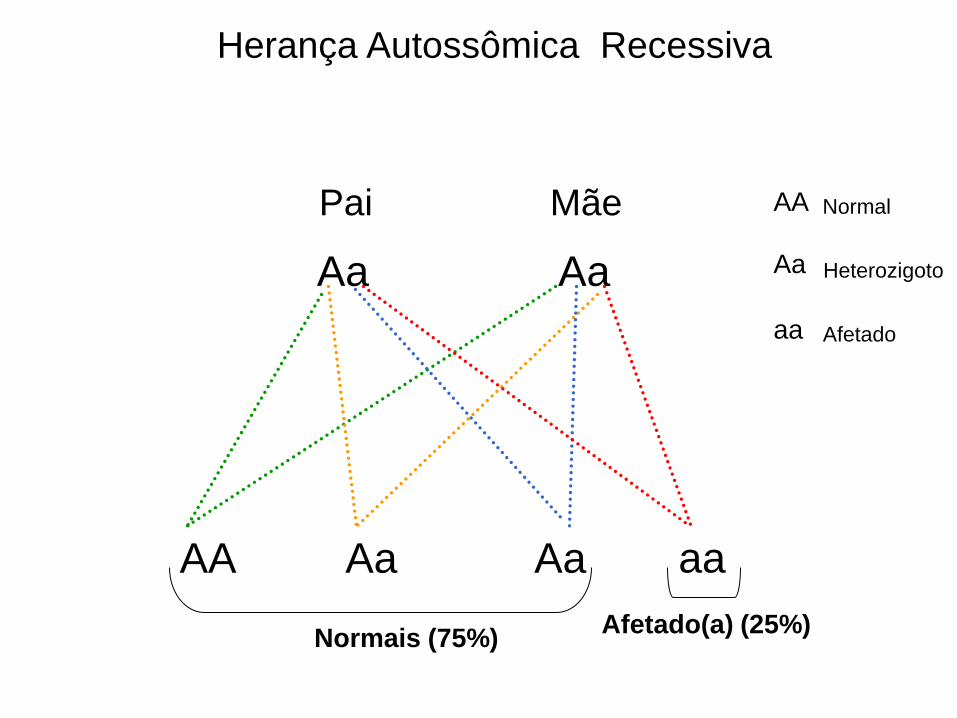
Herança Autossômica Recessiva
Aa Aa
AA Aa Aa aa
Pai Mãe Normal
Afetado
Heterozigoto
AA
Aa
aa
Normais (75%) Afetado(a) (25%)

XX XY XX XY
Normal
Alterado
X
X
XY
Pai
XX
Mãe
Normais (50%) mulher portadora
50%
homem afetado
50%
Herança Ligada ao X - Mulher portadora

XX XX XY XY
Normal
Alterado
X
X
XY
PAI
XX
MÃE
Mulheres Portadoras
100%
Homens Normais
100%
Herança Ligada ao X - Homem afetado
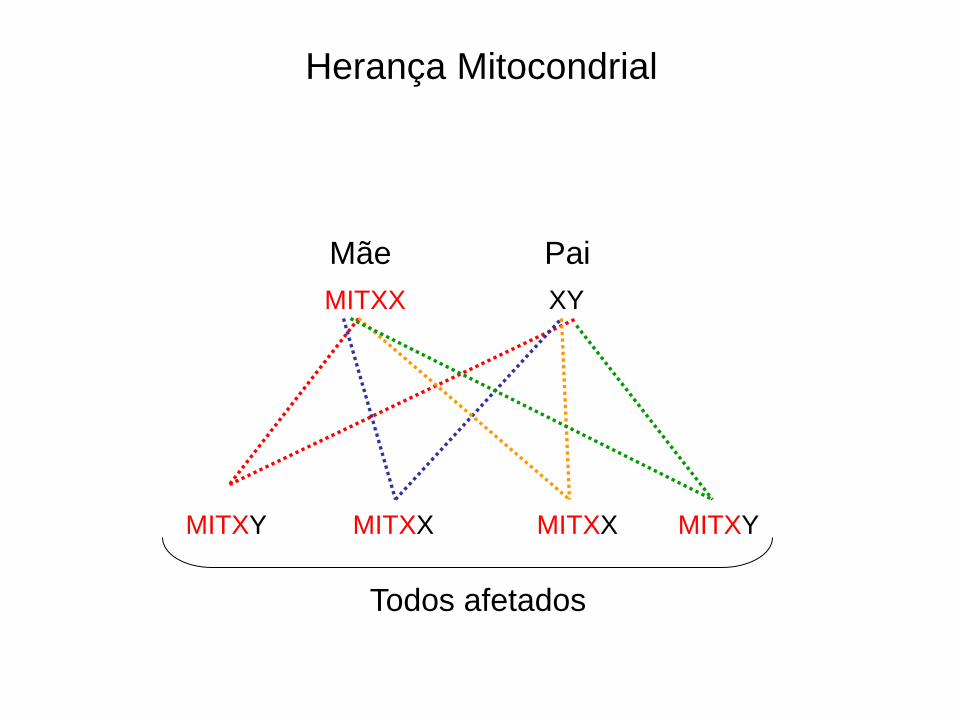
MITXX XY
MITXY MITXX MITXX MITXY
Herança Mitocondrial
Pai Mãe
Todos afetados

Abordagem do Paciente

- Atividade de fetal: época de início e intensidade, aleitamento - Problema de saúde materno-fetal - Retardo de crescimento intra-uterino - Apresentação fetal - Parto e condições de nascimento
História de Gestação

- Consangüinidade entre os pais
- Abortos múltiplos
- Saúde dos pais e irmãos
História - Familiar

Orientação e Conduta
- Orientação da família sobre os EIM e a investigação
do diagnóstico
- Aconselhamento genético provisório
- Atendimento multidisciplinar
- Investigação do diagnóstico específica
- Estudo individual do caso

Classificação
• Scriver CR, Beaudet L., Sly WS and Valle D - The Metabolic and
Molecular Bases of Inherited Disease, 2000.
• Fernandes J, Saudubray J-M, van den Berghe G, Walter JH – Inborn
Metabolic Diseases, 2006.

• Sinais de intoxicação aguda
• Sinais de intoxicação crônica
• Relação com ingestão alimentar e
intercorrências
• Intervalo livre de sintomas
Defeito no Metabolismo Intermediário
Grupo I

Aminoacidopatias Acidemias Orgânicas
Defeitos no ciclo da uréia Intolerância aos açúcares
Grupo I

Intoxicação Aguda
- Sépsis: alerta para EIM sem fator de risco
- Presença de Sepsis não afasta EIM, pode ser
concomitante
- Vômitos e desidratação recorrentes quadro
associado ou não a letargia
- Icterícia, hepatomegalia, insuficiência hepática

Intoxicação Aguda
- Odor anormal – urina e/ou suor
- Manifestações neurológicas agudas
- Estado neurológico flutuante- “intoxicação exógena”??
- Manifestações tromboembólicas
Fatores desencadeantes: infecções, estresse, transgressão
na dieta ou ingestão alimentar excessiva inadvertidamente

Intoxicação Crônica
- Atraso do DNPM, evoluindo para retardo mental
- Distúrbios do comportamento
- Eplepsia de difícil controle
- Alterações neurológicas: macro ou microcealia,
comprometimento do tonus
- Retardo de crescimento
- Alterações oculares: catarata, luxação do cristalino
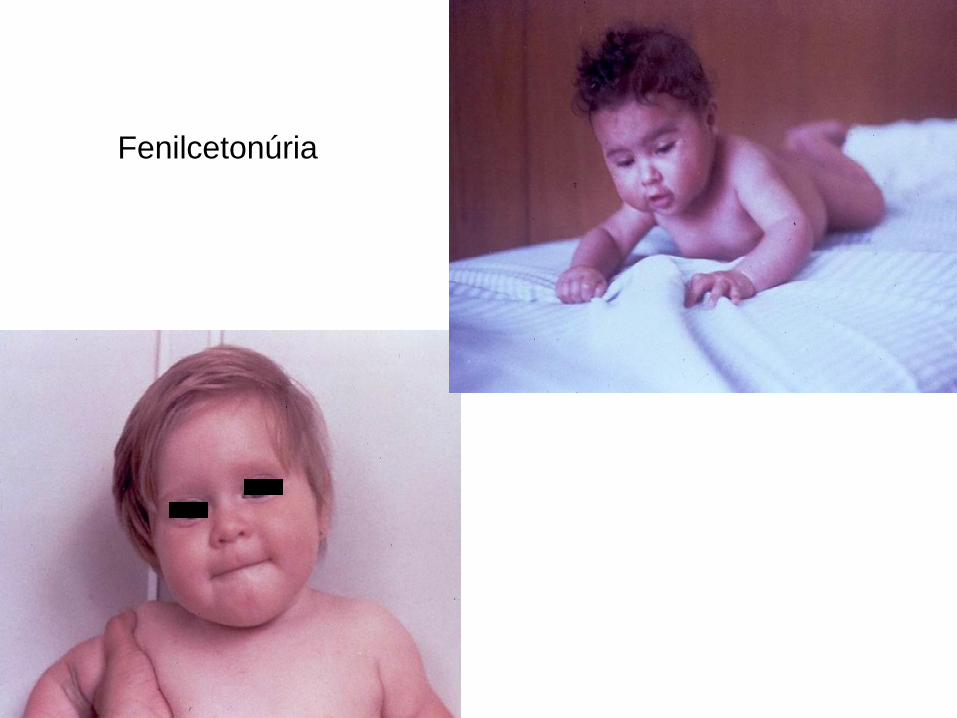
Fenilcetonúria


Homocistinúria
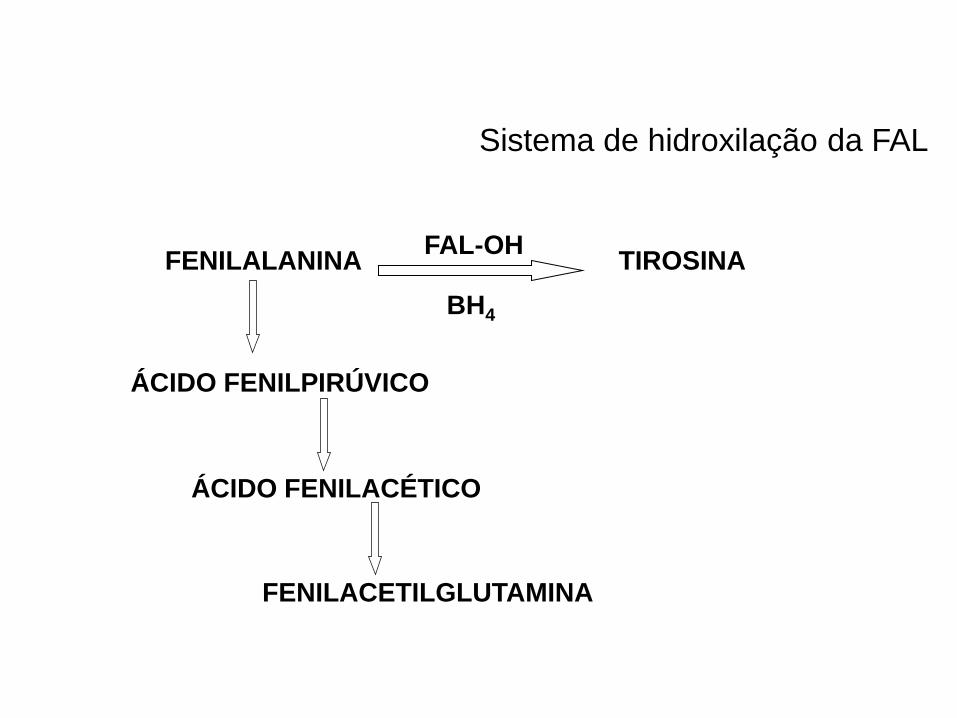
Sistema de hidroxilação da FAL
FENILALANINA TIROSINA
BH4
FAL-OH
ÁCIDO FENILPIRÚVICO
ÁCIDO FENILACÉTICO
FENILACETILGLUTAMINA
Clarke, JTR, 1996

Alimentos ricos em FAL
Carnes (boi, ave, peixe)
Leguminosas (feijão, lentilha, ervilha, etc...)
Ovos
Cereais (exceto arroz e farinha de mandioca)
Leite e derivados
Produtos industrializados
Produtos dietéticos (Aspartame)

Alimentos isentos ou com baixo teor de FAL
• Açúcar, gordura vegetal, chá, limão, vinagre, sal,
mel, pimenta, polvilho, tapioca, refrigerantes, sucos
artificiais, balas de frutas, geléias , algodão doce e
goiabada

0 - 4 meses
Divisão da dieta por faixa etária
4 - 7 meses
8 m - 1 ano
> 1 ano
Fórmula metabólica
leite materno / leite modificado
Fórmula metabólica Papa salgada (almoço) Frutas
Fórmula metabólica Papa salgada (almoço e jantar) Frutas Fórmula metabólica
Alimentos permitidos +
os livres

- Defeito no fígado, músculo, cérebro ou outros
- Sintomas decorrentes do depósito de
substâncias tóxicas e/ou déficit de energia
- Mitocondrial e Citoplasmático
Doenças Envolvendo Metabolismo de Energia
Grupo II

Mitocondrial
- Defeitos de Cadeia Respiratória
- Hiperlacticemias Congênitas
- Defeito de Oxidação de Ácidos Graxos
Citoplasmático
- Doenças de Depósito do Glicogênio
Grupo II

- Hipoglicemia, hiperlacticemia, acidose metabólica
- Hepatomegalia e/ou hepatopatia, cardiomiopatia, alterações
renais e oculares
- Alterações neurológicas: hipotonia, miopatia, convulsão,
malformação cerebral, "Acidente Vascular Encefálico”, alterações
evolutivas cerebrais
- Gerais: déficit de crescimento, morte súbita, apnéia, surdez,
diabetes e abortos de repetição
Manifestações Clínicas (Agudas)
Grupo II

Intoxicação Crônica
- Atraso do DNPM, evoluindo para retardo mental
- Involução do desenvolvimento
- Distúrbios do comportamento
- Eplepsia de difícil controle
- Alterações neurológicas: macro ou microcealia,
comprometimento do tonus, ataxia intermitente
- Retardo de crescimento
- Alterações oculares: retinite pigmentosa, cegueira
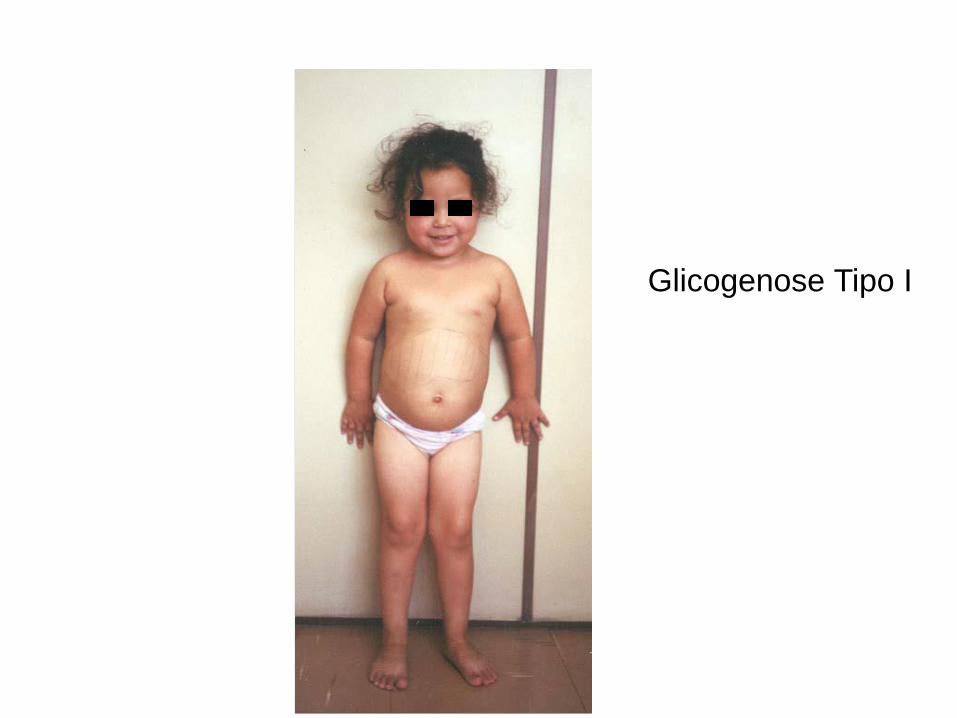
Glicogenose Tipo I

Grupo III
• Sintomas são permanentes e progressivos
• Quadro clínico independe de intercorrências
• Não tem relação com ingestão alimentar
Doenças que envolvem a Síntese ou
Catabolismo de Moléculas Complexas

Prevenção do jejum
Refeições freqüentes
Dietoterapia
Suplementação de vitaminas: L-carnitina,
riboflavina, tiamina, coenzima Q10, Vitamina C e E
Terapia com amido crú :
< 2 anos - 1.0 - 1.5 g/ Kg cada 4 h
> 2 anos - 1.75 - 2.0 g/Kg cada 6 h
Fernades J; Saudubray JM; Van Den Berghe G., 2000
Tratamento - Grupo II

Doenças
Lisossômicas Doenças dos
Peroxissomos
Defeito no Metabolismo
Defeitos na síntese do colesterol
Síntese de ácidos biliares
Purinas e pirimidinas
Porfirias
Transportes de metais
Deficiências de vitaminas
Defeitos nos neurotransmissores
Defeitos congênitos de glicosilação
Grupo III

Manifestações Clínicas
- Hidropsia fetal, ascite, achados dismórficos, hepato e/ou
esplenomegalia, discrasias sangüíneas
- Alterações neurológicas, psiquiátricas, irritabiliadade, cardiológicas,
gastrointestinais, esqueléticas, oculares, audiológicas, fácies e
tecido celular sub-cutâneo infiltrados.
- Alterações articulares, dores articulares, mãos, pés, coluna -
- Evolução: Atraso do DNPM, retardo mental, disfagia,
- Involução do DNPM

Grupo III - Manifestações Clínicas
Hidropsia Fetal
• Doenças de depósito lisossômico
- MPS VII, I e IVa
- Sialidose, mucolipidose II (I-cell disease)
- Esfingolipidoses (galactosialidose, Niemann-Pick A,
Gaucher, Farber, Gangliosidoses GM1
- Doenças de depósito lipídico
- Doenças de depósito do ácido siálico
• Defeitos da síntese do colesterol
- Smith-Lemli-Optiz
- Displasia esquelética de Greenberg
- Acidúria mevalônica
• Doenças dos peroxissomos (Zellweger)
• Doença de depósito do glicogênio tipo IV (Andersen)
• Defeitos congênitos de glicosilação (CDG)
• Deficiência primária de carnitina (Grupo III)
• Doenças mitocondriais, deficiência da fumarase(Grupo III)
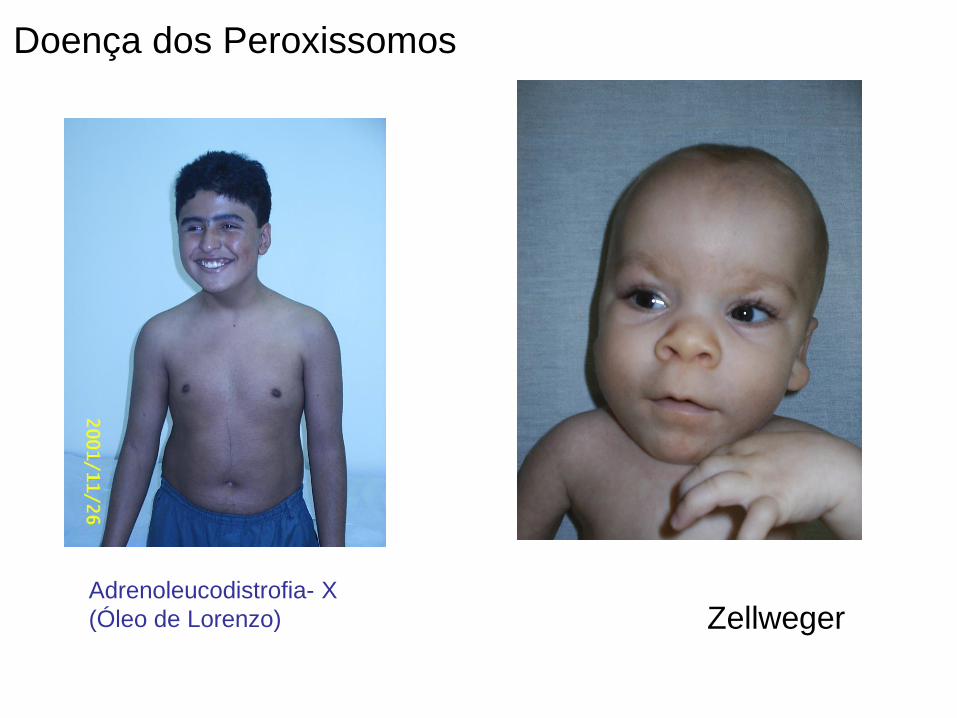
Zellweger
Doença dos Peroxissomos
Adrenoleucodistrofia- X
(Óleo de Lorenzo)

Doença de Gaucher
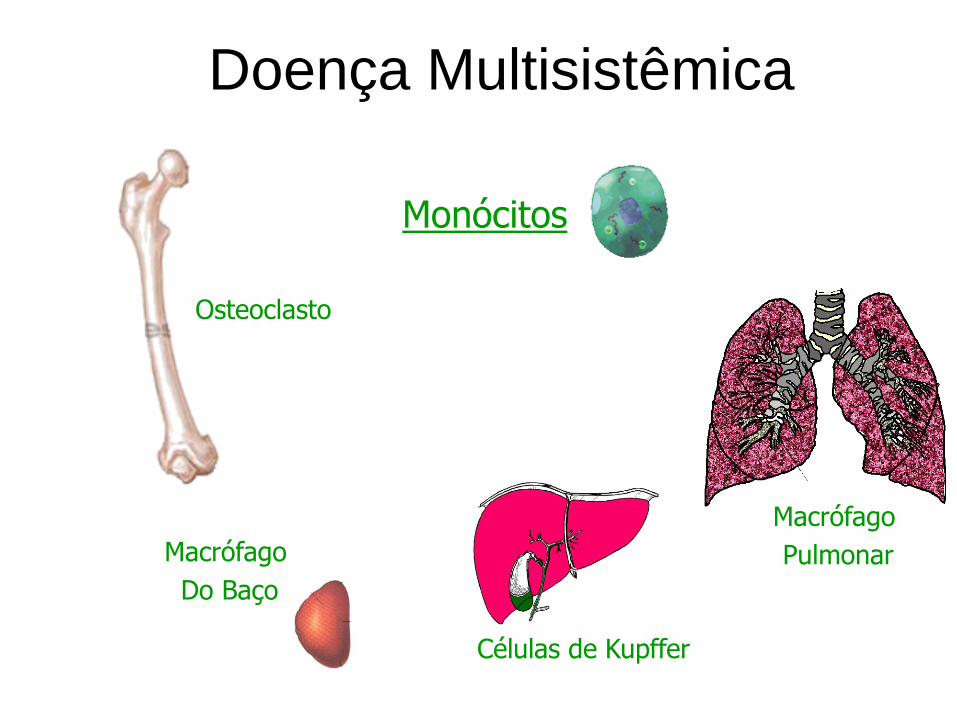
Doença Multisistêmica
Monócitos
Osteoclasto
Macrófago
Do Baço
Células de Kupffer
Macrófago
Pulmonar
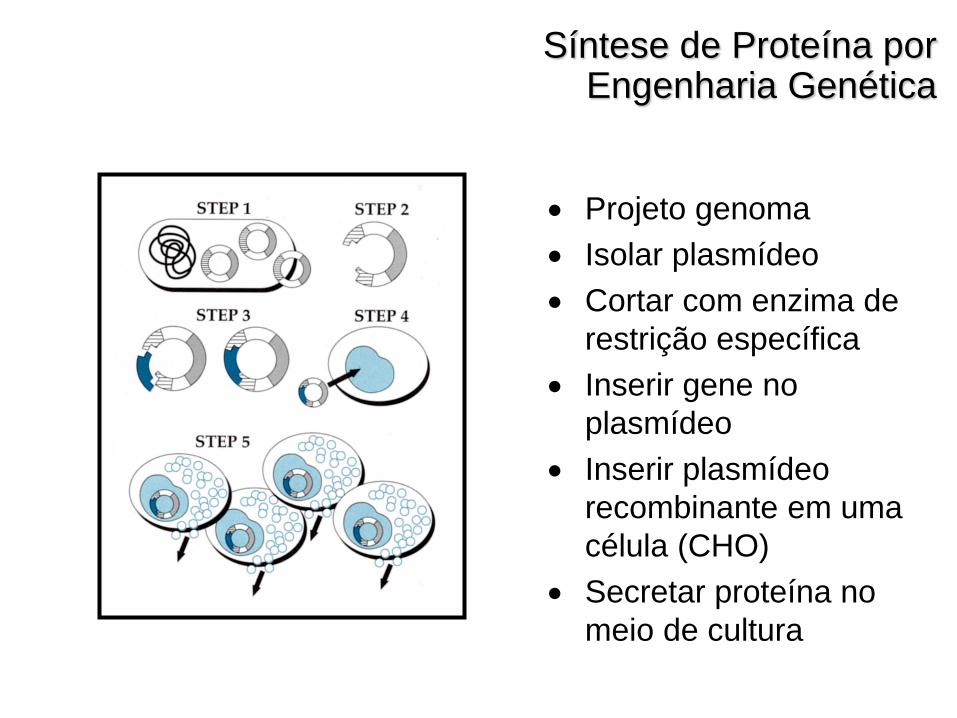
Síntese de Proteína por Engenharia Genética
Projeto genoma
Isolar plasmídeo
Cortar com enzima de
restrição específica
Inserir gene no
plasmídeo
Inserir plasmídeo
recombinante em uma
célula (CHO)
Secretar proteína no
meio de cultura

Doença de Gaucher

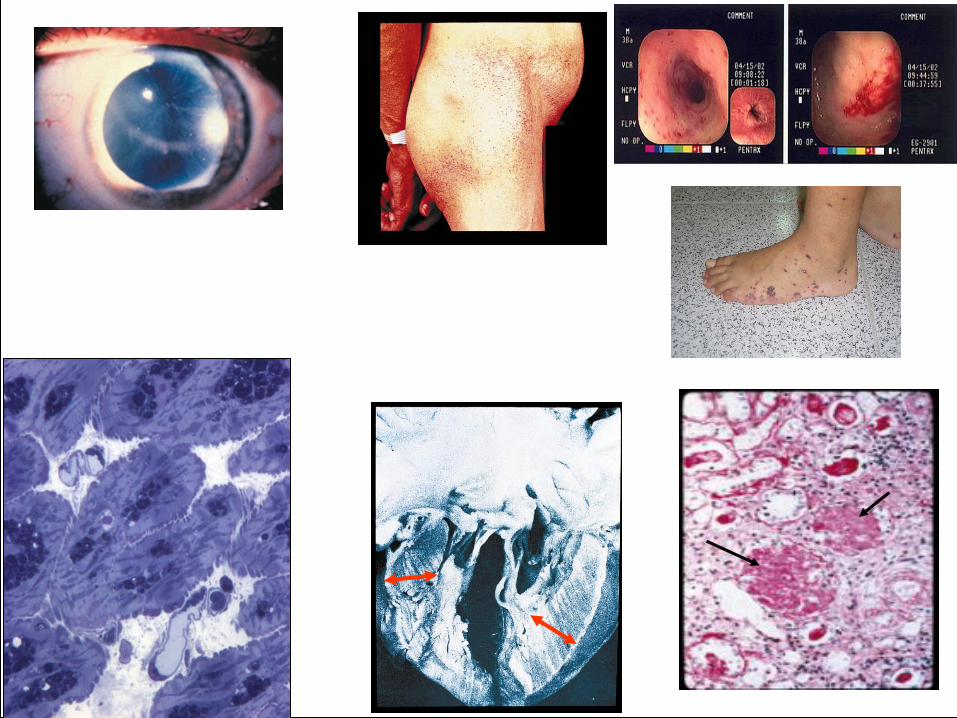
Doença de Fabry

acroparestesia crises episódicas de dor hipoidrose distrofia de córnea “febre” recorrente intolerância ao calor e ao frio distúrbios gastrointestinais angioqueratoma fadiga • complicações cerebrovasculares • complicações cardíacas • complicações renais
Quadro Clínico
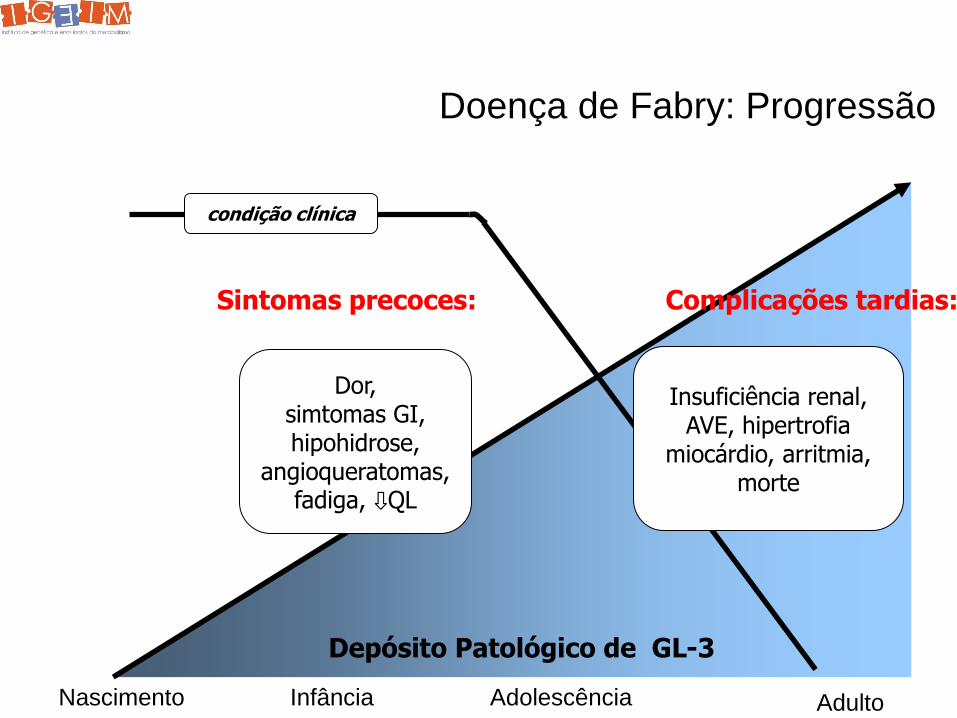
Doença de Fabry: Progressão
Insuficiência renal, AVE, hipertrofia
miocárdio, arritmia, morte
condição clínica
Sintomas precoces: Complicações tardias:
Depósito Patológico de GL-3
Dor, simtomas GI, hipohidrose,
angioqueratomas, fadiga, ⇩QL
Nascimento Infância Adolescência Adulto

Mucopolissacaridoses
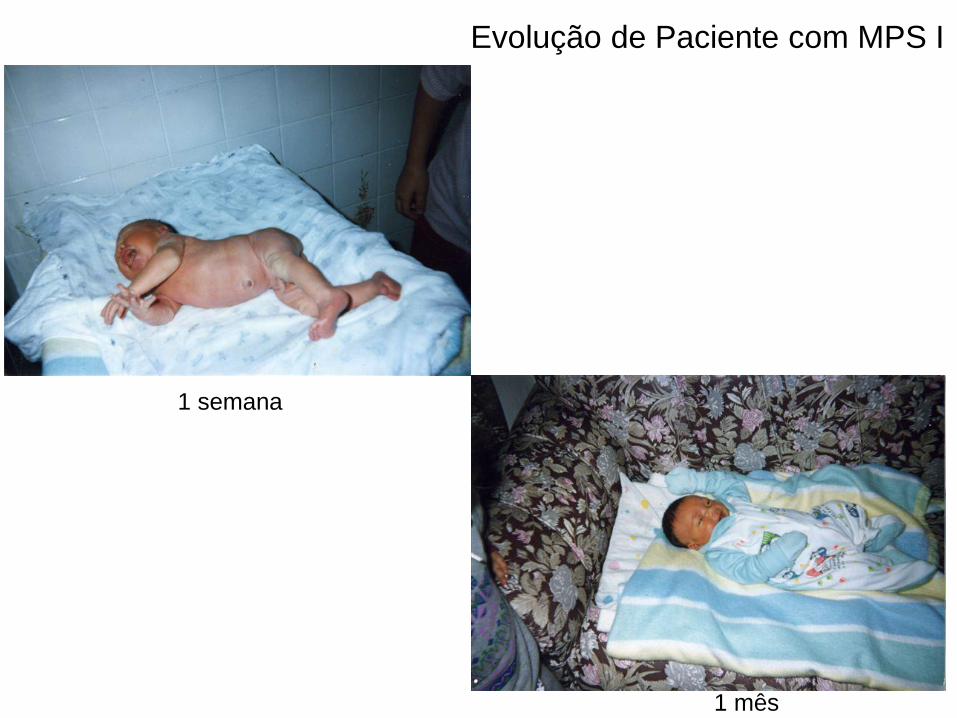
1 semana
1 mês
Evolução de Paciente com MPS I

9 meses
1 ano
Evolução de Paciente com MPS I
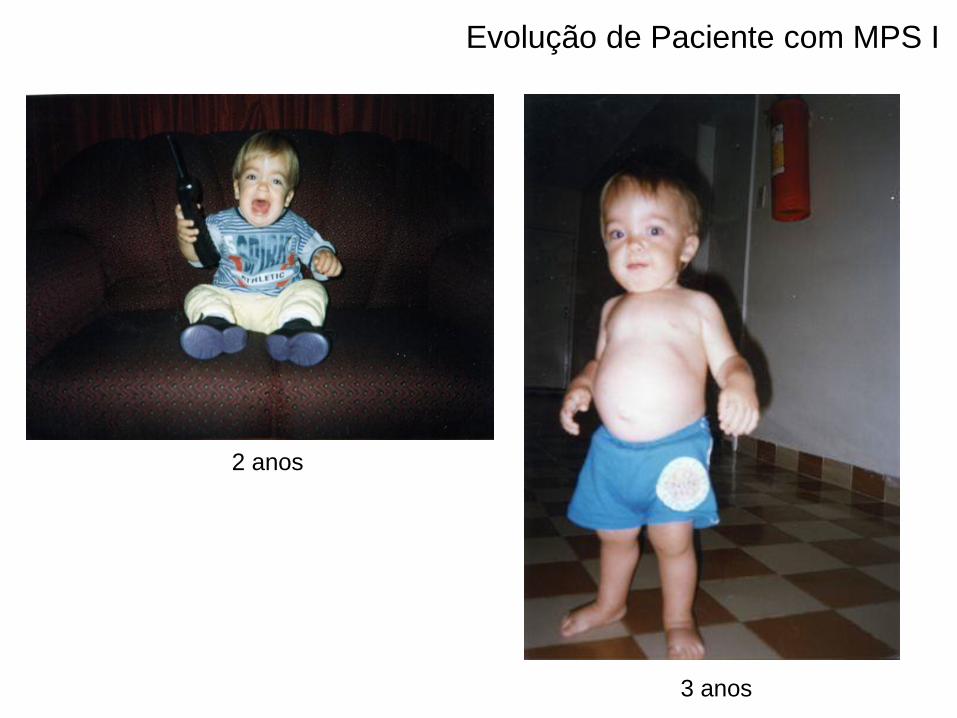
2 anos
3 anos
Evolução de Paciente com MPS I

4 anos 5 anos
Evolução de Paciente com MPS I

6 anos 7 anos
Evolução de Paciente com MPS I
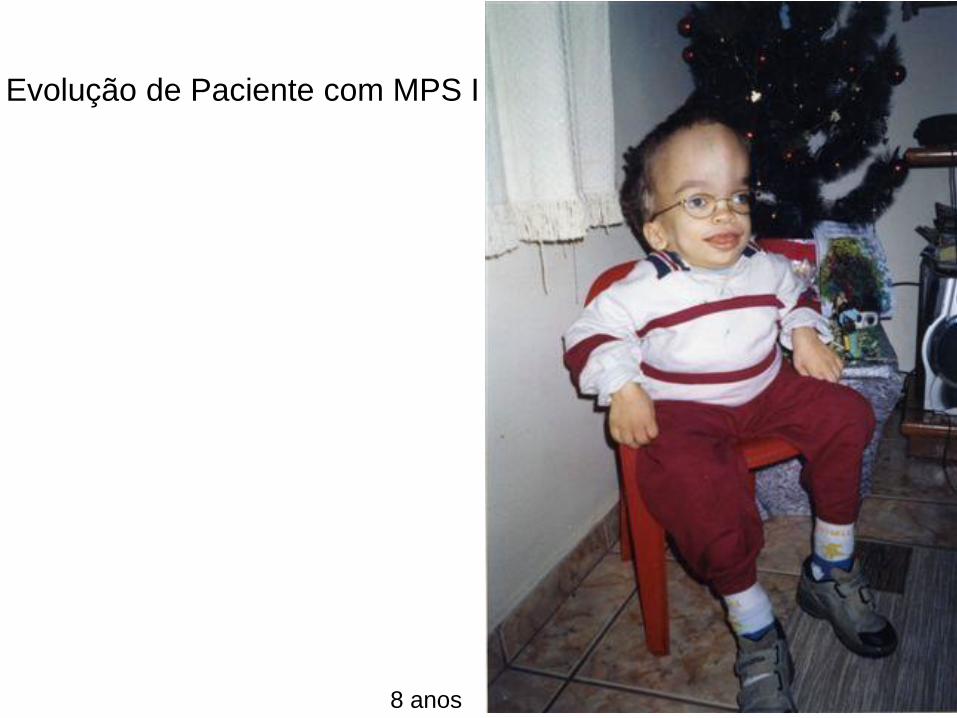
8 anos
Evolução de Paciente com MPS I

MPS I

MPS II

MPS VI

Doença de Pompe
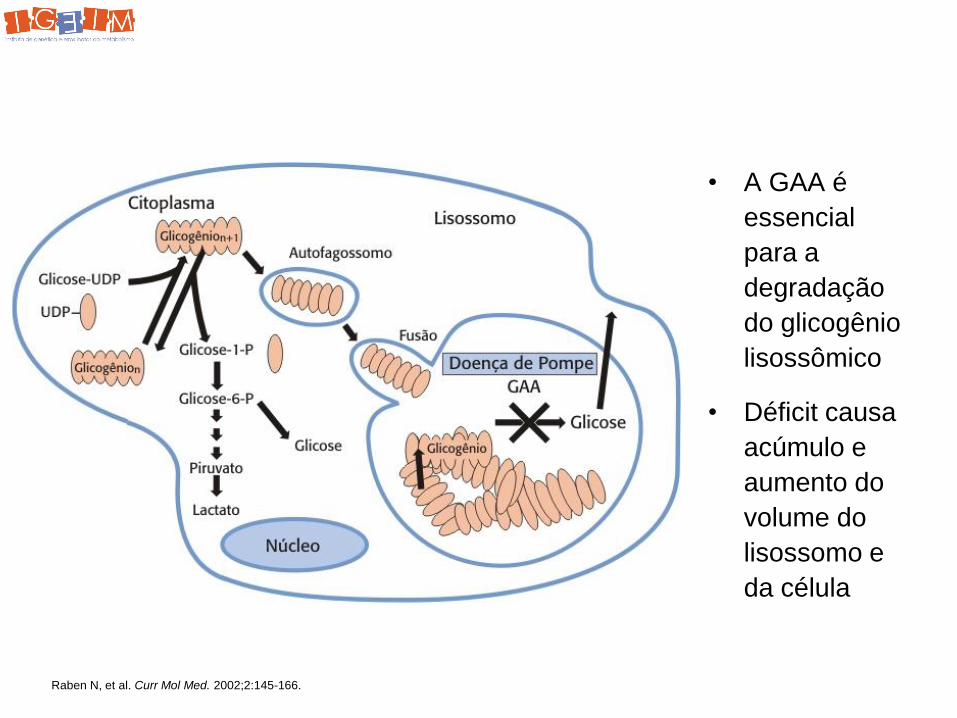
• A GAA é
essencial
para a
degradação
do glicogênio
lisossômico
• Déficit causa
acúmulo e
aumento do
volume do
lisossomo e
da célula
Raben N, et al. Curr Mol Med. 2002;2:145-166.
PATOLOGIA

• Miofibrilas substituídas
por glicogênio com
destruição do tecido
muscular
Raben N, et al. Curr Mol Med. 2002;2:145-166.
Dado de arquivo, Genzyme Corporation.
PATOLOGIA
Glicogênio
lisossômico entre
miofibrilas
Miofibrila Glicogênio lisossômico
se acumulando na
periferia da célula
Lisossomos com
membranas
rompidas e formação
de lago de glicogênio
DESTRUIÇÃO DOS TECIDOS

Estágios da progressão ultraestrutural
1. Glicogênio Lisossomal, Mitocôndria normal,
Miopatia leve
2. Glicogênio Lisossomal aumentado, Glicogênio
citoplasmático disperso, Mitocôndria anormal
3. Glicogênio lisossomal denso, Glicogênio
citoplasmático aumentado, Mitocôndria anormal,
Miopatia grave e dissolução fibrilar
4. Glicogênio Lisossomal reduzido, Glicogênio
citoplasmático aumentado, Mitocôndria reduzida
5. Glicogênio Lisossomal extensivo, Células
intumescidas com edema/influxo de água, Perda
total de fibrilas e estrutura sarcoplasmática
Adaptado de BL Thurberg, et al Laboratory Invest 2006 00, 1-13
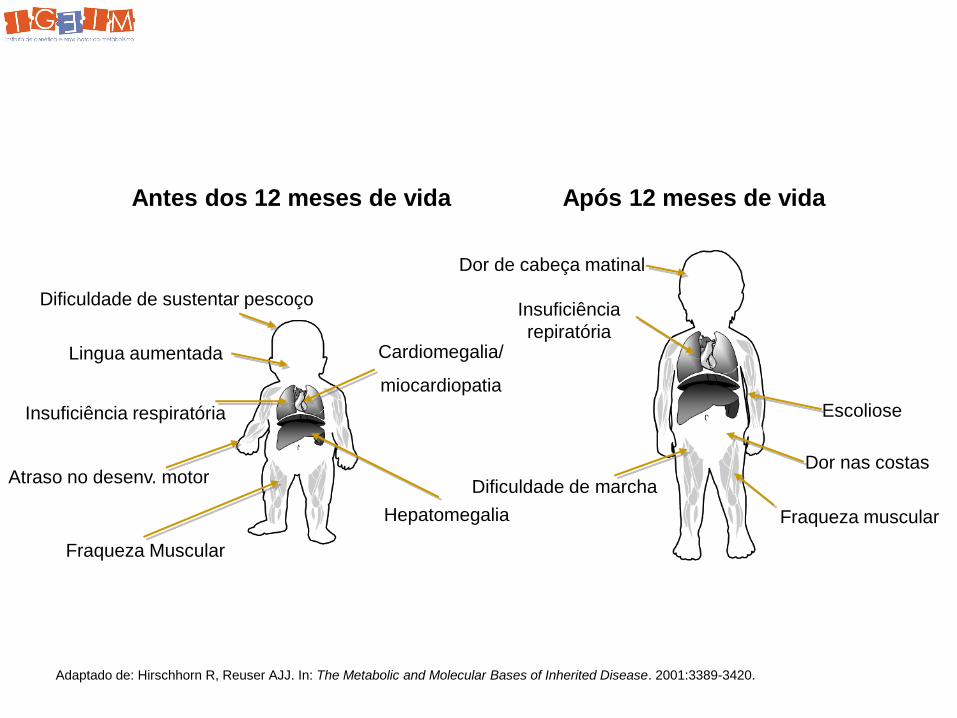
Antes dos 12 meses de vida Após 12 meses de vida
Dificuldade de sustentar pescoço
Lingua aumentada
Insuficiência respiratória
Atraso no desenv. motor
Fraqueza Muscular
Hepatomegalia
Cardiomegalia/
miocardiopatia
Dor de cabeça matinal
Escoliose
Dor nas costas
Fraqueza muscular
Adaptado de: Hirschhorn R, Reuser AJJ. In: The Metabolic and Molecular Bases of Inherited Disease. 2001:3389-3420.
Insuficiência
repiratória
Dificuldade de marcha
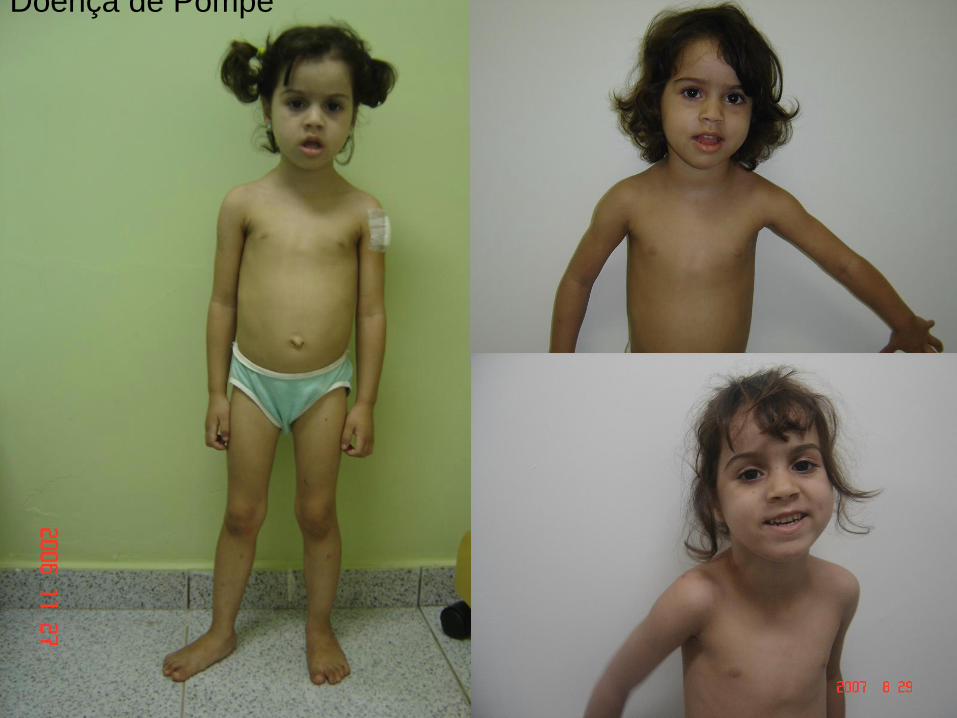
Doença de Pompe

Médico: Clínico, Pediatra, Geneticista, Neurologista,
Cardiologista, Oftalmologista, Endocrinologista,
Nefrologista, Gastroenterologista / Suporte Nutricional /
Hepatologista, Pneumologista /Distúrbios do sono,
Imunologista, Otorrinolaringologista, Acupuntura
Nutrição Enfermagem
Fonoaudio / Motricidade oral Psicologia
Fisioterapia/ Equoterapia Serviço Social
Terapia Ocupacional Musicoterapia
Suporte - Pacientes e Familiares

Quando Suspeitar de um Erro Inato do Metabolismo ?
- História familiar positiva
- Consangüinidade
- Involução do DNPM
- Hipoglicemia, hiperglicemia
- Acidose metabólica
- Discrasias sangüíneas
- Hepatomegalia e/ou esplenomegalia
- Letargia, coma
- Convulsões, ataxia, hipo ou hipertonia
- Estado neurológico flutuante
- Anormalidades oculares
- Anormalidades de cabelo
- Odor anormal - urina, suor
Waber, L, 1993; Lindor, NM & Karnes, PS; Martins, AM et al, 1999

www.igeim.org.br Fone/Fax: 5081-9620
LEIM/UNIFESP/IGEIM Triagem urinária
Cromatografia em papel (aa e açúcar)
Dosagem Enzimática e Genótipo de doenças de Gaucher, Fabry,
Pompe e MPS I, II, VI; Quitotriosidase; Homocisteína
Erros Inatos?
08007701006





























