Embed Size (px)
Citation preview

16
1 INTRODUÇÃO
O sistema sensorial trigeminal transmite informações mecânicas,
proprioceptivas (percepção espacial do próprio corpo), nociceptivas (dor), e
térmicas das regiões craniofaciais (MARTINO et al., 1996; BERETIER, 2000). As
vias neurais deste sistema atuam para manter a integridade dos tecidos
craniofaciais através de reflexos que protegem outros sistemas sensitivos (visão,
somestesia, olfação, gustação) e homeostáticos (tratos respiratório e digestório)
(BERETIER, 2000).
Considerando a importância do sistema trigeminal, na resposta a danos
orofaciais profundos e a possibilidade de eventos fisiopatológicos comuns, em
doenças neurodegenerativas agudas (isquemia e excitotoxicidade), também
ocorrerem neste sistema, este trabalho investigou comparativamente os padrões
inflamatórios, de lesão axonal e desmielinização, após a indução de lesão aguda
excitotóxica e isquêmica no núcleo trigeminal de ratos adultos, com ênfase nas
alterações na susbstância branca, do trato trigeminotalâmico.
As lesões foram induzidas, respectivamente, através de microinjeções de
N-Metil-D-Aspartato (NMDA) e Endotelina-1 (ET-1), no núcleo espinhal do sistema
trigeminal e avaliadas em um tempo precoce e outro mais tardio.
Um levantamento selecionado da literatura correspondente ao assunto
investigado foi realizado, servindo como base para o trabalho proposto e será
detalhado na seção seguinte.

17
2 REVISÃO DA LITERATURA
2.1 A NEUROANATOMIA DOS SISTEMAS TRIGEMINAIS
O nervo trigêmeo é assim chamado por possuir três ramos calibrosos
distribuídos por áreas extensas da face, tanto superficiais como profundas
(LAZAROV, 2002; RIZZOLO;MADEIRA, 2004). Estes ramos recebem
denominações conforme seus territórios de distribuição principais: nervo oftálmico
(V1), nervo maxilar (V2) e nervo mandibular (V3) (MARTINO et al., 1996;
MOORE;DALLEY, 2001; DI DIO, 2002). As duas primeiras divisões (V1 eV2) são
totalmente sensitivas e a terceira divisão (V3) é mista, contendo fibras sensitivas e
motoras (MARTINO et al., 1996; MOORE;DALLEY, 2001; DI DIO, 2002) (Figura
1). Cada raiz sensitiva inerva mucosas e pele de diferentes regiões da cabeça,
exceto a pele que reveste o ângulo da mandíbula, e a raiz motora inerva os
músculos da mastigação (SICHER;DU BRUL, 1977; MARTINO et al., 1996;
GO;KIM;ZEE, 2001; JAASKELAINEN, 2004). Os ramos periféricos destas
subdivisões levam sinais tácteis, térmicos e álgicos ao Sistema Nervoso Central
(SNC), enquanto os nervos facial, glossofaríngeo e vago inervam regiões
periféricas, como certas áreas da pele, do ouvido externo, faringe, laringe,
cavidade nasal, seios da face e ouvido médio (SICHER;DU BRUL, 1977;
MARTINO et al., 1996; GO;KIM;ZEE, 2001; JAASKELAINEN, 2004). Os
mecanorreceptores presentes nos tecidos periféricos mencionados transduzem a
informação tátil, enquanto que os nociceptores e termorreceptores veiculam as

18
sensibilidades dolorosa e térmica, respectivamente (BEAR;CONNORS;
PARADISO, 2002; LENT, 2002).
As células que dão origem aos aferentes sensoriais trigeminais primários
estão localizadas no gânglio sensorial periférico (Figura 1), de Gásser ou gânglio
semi-lunar, na fossa craniana média (SICHER;DU BRUL, 1977; MARTINO et al.,
1996).
O gânglio trigeminal é análogo ao gânglio da raiz dorsal do Sistema
Nervoso Periférico (SNP) (LAZAROV, 2002). Os neurônios deste gânglio são do
tipo pseudo-unipolar, no qual um único axônio possui ramos periférico e central
(LAZAROV, 2002). O complexo somestésico trigeminal é composto por três
núcleos que medeiam a sensibilidade craniana: núcleos mesencéfálico, principal e
espinhal (SICHER;DU BRUL, 1977; MARTINO et al., 1996; GRAY et al., 2003).
O núcleo mesencefálico, constituído dos corpos neuronais dos receptores
de estiramento presentes nos músculos da maxila, é formado por uma coluna de
células nervosas que se estendem superiormente da zona de entrada do nervo
trigêmeo, até o mesencéfalo. Este núcleo é importante para a percepção
proprioceptiva da maxila. O núcleo principal trigeminal está localizado próximo da
zona de entrada do nervo trigêmeo, seguido caudalmente pelo núcleo espinhal
que é composto por três sub-núcleos: oral, interpolar e caudal (MARTINO et al.,
1996; GRAY et al., 2003; RUB et al., 2003).
A raiz sensorial trigeminal emite um ramo ascendente e outro descendente,
através da face ventro-lateral da ponte. O ramo ascendente conecta as células
ganglionares trigeminais ao núcleo sensorial principal do nervo trigêmeo, através
de axônios de maior diâmetro, enquanto o ramo descendente chega ao trato

19
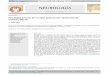
Figura 1. Divisões do nervo trigêmio. Nesta figura são ilustradas as divisões
oftálmica (V1), maxilar (V2) e mandibular (V3) da raiz do nervo trigêmio. Adaptado
de: http://www.umanitoba.ca/cranial_nerves/trigeminal_neuralgia/manuscript/
medications.html

20
trigeminal espinhal com fibras amielínicas e mielinizadas de menor diâmetro (LI et
al., 1992; MARTINO et al., 1996; GO;KIM;ZEE, 2001; DALLEL et al., 2003)
2.1.1 Via Trigeminal para Sensação Tátil
A habilidade de distinguir e interpretar informações térmicas e nociceptivas
é um importante componente da sobrevivência humana, pois possibilita
comportamentos motores adequados, de modo a evitar perda da integridade
tecidual (DASILVA et al., 2002). Os estudos neuroanatômicos clássicos da
organização funcional dos núcleos sensoriais trigeminais atribuiram ao núcleo
trigeminal principal, grande importância para a gênese da sensação tátil e
propriocepção (LI et al., 1992; MARTIN, 1998) e ao núcleo espinhal com seus
subnúcleos (oral, interpolar e caudal), um papel principal na transmissão da
informação nociceptiva das estruturas orofaciais (DALLEL et al., 1988).
Os axônios que veiculam a sensação tátil entram na região pontina ventral
e trafegam pela sua porção dorsal, principalmente em direção ascendente
terminando no núcleo sensorial trigeminal principal. A maioria dos neurônios deste
núcleo sensorial possui axônios que decussam na ponte e ascendem dorso-
medialmente até o núcleo da coluna dorsal, no lemnisco medial. Os axônios
ascendentes de segunda destes neurônios formam o lemnisco trigeminal e fazem
sinapse no tálamo, no núcleo ventral póstero-medial. Os axônios dos neurônios
talâmicos projetam-se via braço posterior da cápsula interna, para a parte lateral
do córtex somestésico primário (SI), no giro pós-central. O córtex somestésico
secundário (SII), localizado no opérculo parietal e córtex posterior, recebe

21
principalmente aferências de SI (MARTINO et al., 1996).
A segunda via, bem menor, origina-se dos neurônios da porção dorsal do
núcleo sensitivo principal e ascende ipsilateralmente (do mesmo lado), até chegar
no lemnisco trigeminal, terminando no núcleo ventral póstero-medial talâmico
(MARTINO et al., 1996) . Esta via menor é importante para o processamento de
estímulos mecânicos dos dentes e tecidos moles da cavidade oral (MARTINO et
al., 1996).
Portanto, o núcleo ventral póstero-medial do tálamo recebe aferências
bilaterais das estruturas intra-orais: projeção contralateral do lemnisco trigeminal e
projeção ipsilateral da via dorsal (MARTINO et al., 1996; LAZAROV, 2002). Do
tálamo, estas informações são direcionadas de forma topográfica para o giro pós-
central do córtex somestésico primário no córtex parietal. Estas informações
chegam veiculadas por axônios, que fazem sinapse em neurônios que estão
principalmente na área de representação da face, na porção lateral do giro pós-
central. SI possui conexões com áreas secundárias e terciárias (de associação) do
córtex somestésico. A atividade integrada destas áreas corticais, que são
conectadas através de sinapses, é que nos possibilita a percepção consciente das
informações sensoriais, incluindo tato, propriocepção e dor (MARTINO et al.,
1996; SHEPHERD, 1998; LENT, 2002).
2.1.2 Via Trigeminal Para Sensação Nociceptiva
Nesta via, as fibras amielínicas e mielinizadas de menor diâmetro entram na
ponte e descendem formando o trato trigeminal espinhal. Este trato extende-se da

22
porção média da ponte até a medula cervical (nível de C3 e C4), passando pelo
bulbo, e recebe aferências dos nervos facial, glossofaríngeo e vago somando-se
ao nervo trigêmeo (GO;KIM;ZEE, 2001; BOHOTIN et al., 2003). Colaterais axonais
do trato trigeminal espinhal fazem sinapses com neurônios presentes nos três
subnúcleos do núcleo espinhal: oral, interpolar e caudal. Os neurônios pós-
sinápticos, por sua vez, enviam projeções axonais que cruzam a linha média,
formando o trato trigeminotalâmico que veicula infomação nociceptiva até o tálamo
contralateral, no núcleo ventral póstero-medial e nos núcleos intra-laminares
(LAZAROV, 2002). O núcleo ventral póstero-medial projeta para as camadas III e
IV de SI, na porção lateral do giro pós-central. Os núcleos intra-laminares,
segundo local mais importante no tálamo a receber aferências trigeminais,
processam aspectos emocionais da dor e possuem uma projeção mais difusa, a
qual inclui aquela para o córtex insular anterior (MARTIN, 1998; GRAY et al.,
2003; IKEDA et al., 2003).
O núcleo caudal, a maior subdivisão do trato trigeminal espinhal, consiste
em uma porção laminada alongada que se funde sem limites claros, com o corno
dorsal cervical, enquanto a sua parte rostral forma uma zona de transição com a
extremidade caudal do núcleo interpolar (BERETIER, 2000). A zona de transição
entre os subnúcleos interpolar e caudal é uma região crítica para o processamento
da dor craniofacial, possui grande importância na resposta a danos orofaciais
profundos, configurando-se em uma região integradora de informações sensoriais
das regiões peri e intra-orais (BERETIER, 2000; IKEDA et al., 2003).
Anatomicamente, demonstrou-se que a projeção de neurônios trigeminais
primários, da polpa dentária do gato terminam em todas as sub-divisões do

23
complexo sensorial trigeminal (ARVIDSSON;GOBEL, 1981).
A representação somatotópica da face no núcleo trigeminal é semelhante a
uma casca de cebola : as áreas peri-orais são representadas rostralmente no
núcleo e áreas mais posteriores da face são representadas mais caudalmente no
núcleo trigeminal (DASILVA et al., 2002) .
Como vimos, diferenças anatômicas e funcionais distinguem fibras
aferentes individuais do nervo trigêmeo. Fibras de maior diâmetro veiculam a
sensibilidade tátil e fibras amielínicas, ou pouco mielinizadas veiculam sensações
álgicas e térmicas. Portanto, as informações álgicas, térmicas e proprioceptivas
são veiculadas através de vias neurais específicas até o tálamo, de modo análogo
ao que ocorre com as informações sensoriais, captadas em outros tecidos
periféricos e transmitidas a partir da medula espinhal. A principal via para
percepção tátil na face é semelhante ao sistema colunar dorsal-lemnisco medial,
enquanto que a organização da via ascendente, importante principalmente para a
percepção de sensações álgicas e térmicas da face, é similar ao sistema
anterolateral, para a transmissão de informações nociceptivas do resto do corpo.
2.1.3 A Fisiologia do Sistema Trigeminal
A transmissão e o processamento de informação no SNC envolvem a
participação de mediadores químicos, conhecidos como neurotransmissores.
Entre os neurotransmissores pode-se destacar o glutamato (principal
neurotransmissor excitatório do SNC) e o ácido gama amino butírico (GABA
principal neurotransmissor inibitório do SNC). Existe uma comunicação bi-

24
direcional, entre células gliais pré-sinápticas e elementos neuronais na sinapse. A
liberação de neurotransmissores do terminal pré-sináptico não apenas estimula os
neurônios pós-sinápticos, mas também ativa a glia peri-sináptica (BEZZI et al.,
1998). As células gliais ativadas, por sua vez, liberam gliotransmissores que
podem diretamente estimular neurônios pós-sinápticos e regular a liberação de
neurotransmissores (BEZZI et al., 1998; AULD;ROBITAILLE, 2003; NEWMAN,
2003).
Os neurônios trigeminais primários são quimicamente heterogêneos e
parecem utilizar vários neurotransmissores para a transmissão sináptica. Estes
neurotransmissores são liberados na fenda sináptica e, ao se ligarem em
receptores específicos na membrana do neurônio pós-sináptico, podem aumentar
(neurotransmissor excitatório), ou diminuir (neurotransmissor inibitório) a
excitabilidade do mesmo. Existe um grande número de neurotransmissores
excitatórios e inibitórios e esta ação, depende do local do sistema nervoso
(SHEPHERD, 1998). Dentre eles estão os aminoácidos excitatórios e inibitórios,
as monoaminas e outros como a histamina e a acetilcolina (LAZAROV, 2002). A
funcionalidade dos circuitos neurais depende da sintonia da comunicação entre
unidades sinápticas excitatórias e inibitórias, compostas por terminações nervosas
pré e pós-sinápticas, complexamente ligadas às células gliais (NEWMAN, 2003).
O glutamato e o aspartato são os principais neurotransmissores excitatórios
dos neurônios sensoriais primários (LAZAROV, 2002). A membrana dos neurônios
do gânglio trigeminal possui receptores ionotrópicos e metabotrópicos para os
diversos tipos de agonistas glutamatérgicos: ácido propiônico -amino-

25
hidroximetilsozazol (AMPA), cainato e NMDA.
Células imunopositivas para glutamato constituem 63,8% dos neurônios de
todos os tamanhos no gânglio trigeminal espinhal do gato (CLEMENTS et al.,
1991) e aferentes primários, que inervam a polpa dentária destes animais, contém
glutamato em seus terminais no núcleo caudal (CLEMENTS et al., 1991). O
glutamato e o aspartato, em menor expressão, são transmissores químicos de
informações proprioceptivas trigeminais, participam da transmissão química entre
neurônios aferentes primários e células do núcleo trigeminal principal (LAZAROV,
2002), bem como neurônios do núcleo trigeminal mesencefálico, também são
glutamatérgicos.
Os neurotransmissores inibitórios mais comumente encontrados no SNC
são a glicina e o GABA. A glicina não foi encontrada em neurônios do gânglio
trigeminal, mas junto com o GABA participa do controle do ritmo mastigatório
(LAZAROV, 2002). O GABA está presente em neurônios sensoriais no gânglio
trigeminal, e no núcleo trigeminal principal de ratos, porcos da índia, coelhos e
gatos (LAZAROV, 2002). Apesar do núcleo trigeminal caudal ser classicamente
visto como um equivalente anatômico do corno espinhal dorsal, que apresenta a
transmissão álgica bloqueada pelos neurônios gabaérgicos, neste núcleo os
neurônios parecem ter ação diferente. Viggiano et al. (2004), investigando o papel
da transmissão gabaérgica, na nocicepção dos núcleos do trato trigeminal
espinhal demonstraram o aumento de GABA, no núcleo trigeminal caudal, em um
modelo de dor inflamatória em ratos. O bloqueio de receptores GABAA inibiu a
expressão comportamental da percepção da dor, sugerindo que a transmissão
gabaérgica no núcleo trigeminal caudal sinaliza estímulos álgicos para centros

26
nervosos superiores.
As monoaminas dopamina, noradrenalina, adrenalina, serotonina, bem
como as catecolaminas, também estão associadas aos neurônios trigeminais
aferentes primários (KHODOROV, 2004). A histamina, sintetizada por neurônios
histaminérgicos, age no SNC como um neurotransmissor inibitório e/ou
neuromodulador e está presente no gânglio trigeminal (KHODOROV, 2004).
Os neurônios do gânglio trigeminal não sintetizam dopamina e não utilizam
catecolaminas, mas estão sob influência de fibras aferentes catecolaminérgicas.
Tallaksen-Greene et al. (1992) usaram autoradiografias quantitativas, para
examinar a densidade e distribuição de receptores para aminoácidos excitatórios,
nos núcleos do trato trigeminal espinhal e no núcleo trigeminal principal. A maior
densidade dos mediadores foi observada na zona marginal (lâmina I) e substância
gelatinosa (lâmina II), do sub-núcleo caudal, onde os receptores de AMPA
apresentaram maior densidade relativa, seguido dos receptores metabotrópicos,
cainato e NMDA. A densidade e distribuição observadas no núcleo sensorial
principal foram bastante similares, embora ligeiramente mais baixas. Portanto, os
receptores de aminoácidos excitatórios medeiam neurotransmissão somestésica
em terminais aferentes primários, estando envolvidos no processamento de
informações nociceptivas.
Um grande número de neuropeptídeos foi identificado no gânglio trigeminal:
substância P, neurocinina A, peptídeo relacionado ao gene da calcitonina,
colecistocinina, somatostatinas, peptídeos opióides (encefalinas
e endorfinas),
polipeptídeo intestinal vasoativo e galanina (KNYIHAR-CSILLIK et al., 1998;
LAZAROV, 2002; SHIN et al., 2003). Segundo Samsam et al. (2000), os

27
neurotransmissores neurocinina A, substância P, e peptídeo relacionado ao gene
da calcitonina atuam como moduladores da nocicepção, na primeira sinapse
central da via trigeminal durante a ativação deste sistema sensorial.
O óxido nítrico (NO), um importante mensageiro gasoso do sistema nervoso,
atua nos neurônios aferentes primários, difundindo-se do pericário das células que
o produzem para os neurônios adjacentes, onde ativa a guanilato ciclase solúvel
na membrana plasmática (LAZAROV, 2002).
2.2 DOENÇAS NEURODEGENERATIVAS AGUDAS: NOÇÕES GERAIS E
EPIDEMILOGIA
De acordo com a evolução temporal dos padrões lesivos, as doenças
neurodegenerativas podem ser classificadas em agudas (por exemplo, acidente
vascular cerebral, trauma cerebral e medular) e crônicas (por exemplo, doenças
de Alzheimer, Parkinson, Huntington, Esclerose Múltipla, Esclerose Lateral
Amiotrófica). Nas doenças neurodegenerativas agudas, a lesão neuronal ocorre
rapidamente, começando em minutos até poucas horas após a lesão primária.
Nas doenças crônicas, a morte celular ocorre durante um processo gradual mais
lento (meses a anos). Entre as doenças neurodegenerativas agudas mais comuns
destacam-se os traumas cerebral e da medula espinhal, além do Acidente
Vascular Cerebral (AVC) (GRAHAM et al., 1995; TATOR, 1995;
DIRNAGL;IADECOLA;MOSKOWITZ, 1999; LO, 2003; MERGENTHALER;DIRNAGL;MEISEL,
2004). Traumatismos cranioencefálicos (TCE) e medulares constituem importante
causa de morte, especialmente em adultos jovens, bem como uma grande causa

28
de incapacidade que afeta de maneira importante não só a vida do paciente, mas
também a de membros da família (KRAUS et al., 1985; KRAUS;NOURJAH, 1988;
KRAUS;ROCK;HEMYARI, 1990; GRAHAM et al., 1995; TAOKA;OKAJIMA, 1998;
MAYER;ROWLAND, 2002).
A lesão traumática no SNC inicia uma complexa cascata de eventos onde
ocorrem acúmulo patológico de aminoácidos excitatórios, edema, inflamação e
isquemia pós-traumática por comprometimento vascular, o que leva à morte
neuronal e glial e perda tecidual. (TATOR;FEHLINGS, 1991; FADEN, 1993;
HONMOU, 1995; SCHWAB;BARTHOLDI, 1996).
O AVC é a terceira causa de morte no mundo ocidental, sendo superado
apenas pelas doenças cardiovasculares e o câncer, e constitui a segunda causa
mais freqüente de morte em pessoas idosas nestas sociedades (DIRNAGL;
ADECOLA; MOSKOWITZ, 1999; LIPTON,1999; MERGENTHALER; DIRNAGL;
MEISEL, 2004). Pode ser classificado em isquêmico e hermorrágico, de acordo
com a patologia da lesão cerebral focal subjacente (SACCCO, 2002; LO, 2003).
No infarto cerebral, a anóxia causada pela oclusão de vasos por período superior
a cinco minutos dá início a uma cadeia de eventos, tais como: vasodilatação local,
estase da coluna sangúínea com segmentação das hemácias, edema e necrose
do tecido cerebral (SACCCO, 2002). Cerca de 75 % de todas as oclusões de
vasos cerebrais originam-se de embolismos de origem arterial ou cardíaca
(SACCCO, 2002). A Hemorragia Intracraniana decorre da ruptura de um vaso em
qualquer ponto, na cavidade craniana e possui incidência de aproximadamente
15%. De todos os casos, 20% são causados por tromboses e hialinoses de
arteríolas. As estenoses das arteríolas cerebrais correspondem a menos de 5 %

29
das ocorrëncias isquêmicas, sendo consideradas comparativamente raras
(DIRNAGL; IADECOLA; MOSKOWITZ, 1999; SACCCO, 2002; MERGENTHALER;
DIRNAGL; MEISEL, 2004).
De um modo geral, o acidente vascular isquêmico é três a quatro vezes
mais freqüente que o hemorrágico (SACCCO, 2002) e divide alguns fatores de
risco com as doenças cardiovasculares (diabetes, hipertensão e ateroesclerose)
(DIRNAGL; IADECOLA; MOSKOWITZ, 1999; MERGENTHALER; DIRNAGL;
MEISEL, 2004). Estudos estatísticos publicados pela American Heart Association
(2006) mostram que, em média, 700.000 pessoas/ano passam por alguma
experiência de AVC e a cada três minutos uma pessoa morre em decorrência dele
nos Estados Unidos. Estes dados impulsionaram a busca por terapias
neuroprotetoras com a finalidade de reduzir a morte celular e o volume do infarto
após episódios de AVC (CARMICHAEL, 2005), pois, dos pacientes que
sobrevivem, 50% sofrem de hemiparesias, 26 % dependem de algum auxílio para
realizar suas atividades diárias e 26 % permanecem restritos ao leito, implicando
em altos custos econômicos (MERGENTHALER; DIRNAGL; MEISEL, 2004).
2.2.1 Modelo de isquemia focal por injeção de Endotelina-1
Os peptídeos do tipo Endotelina (ET-1, ET-2 e ET-3), originalmente isolados
do sobrenadante da cultura de células endoteliais, são agentes vasoconstritores
potentes presentes em várias espécies, incluindo suínos e humanos
(YANAGISAWA et al., 1988; RUBANYI;POLOKOFF, 1994). A ET-1 age através de
receptores específicos, tipo A (ETA-R) e tipo B (ETB-R), amplamente distribuídos

30
pelo SNC no endotélio vascular, bem como em neurônios, astrócitos, microglia,
células epiteliais do plexo coreóide e células ependimais (RUBANYI;POLOKOFF,
1994). A ET-1 é principalmente sintetizada por células endoteliais, mas também
por outros tecidos, incluindo pulmão, coração, fígado, cérebro e algumas células
circulantes (MOTTE;MCENTEE;NAEIJE, 2005) e está envolvida em processos
celulares fundamentais como proliferação celular, fibrose e inflamação
(MASAKI;YANAGISAWA, 1992; MOTTE;MCENTEE;NAEIJE, 2005).
A aumentada liberação de ET-1, em AVC isquêmicos, sugere que estes
peptídeos endógenos podem ser um fator contribuinte na patogênese desta
doença, possivelmente devido a sua potente ação vasoconstritora (ZIV et al.,
1992; HUGHES et al., 2003; GUPTA et al., 2005).
No SNC de ratos, ETA-R é encontrado em células do músculo liso de
artérias do córtex cerebral (HAYNES et al., 1991; BHARDWAJ et al., 2000).
Receptores ETB-R foram identificados no córtex cerebral humano, hipocampo e
estriado de ratos.
O antagonismo seletivo de ETA possui efeitos neuroprotetores, em modelos
experimentais de isquemia (GUPTA et al., 2005). A ET-1 pode não ser
diretamente neurotóxica, mas, em regiões isquêmicas, pode exacerbar a morte
neuronal indiretamente, através da liberação excessiva de mediadores
potencialmente neurotóxicos, tais como o glutamato de astrócitos
(MOTTE;MCENTEE;NAEIJE, 2005).
A injeção direta de ET-1 no parênquima neural constitui um excelente
modelo experimental de isquemia focal (HUGHES et al., 2003). A injeção de 10
pmoles de ET-1, no estriado de ratos adultos induz isquemia focal transitória, com

31
redução de até 30% do fluxo sanguíneo. A injeção da quantidade mencionada de
ET-1, diluída em 1 ou 0.25 µl de solução salina estéril induz isquemia focal restrita
ao local de injeção, tanto em substância branca como cinzenta, corticais e ou o
estriado, sem o rompimento da barreira hematoencefálica (BHE) (HUGHES et al.,
2003).
2.3 DEGENERAÇÃO SECUNDÁRIA EM DOENÇAS NEURODEGENERATIVAS
AGUDAS: PRINCIPAIS MECANISMOS
Em doenças neurodegenerativas agudas, a morte neuronal e/ou de células
gliais pode ocorrer rapidamente, como conseqüência imediata de eventos
primários (por exemplo, o trauma), ou ocorrer tardiamente como conseqüência de
eventos secundários ativados, a partir do processo lesivo inicial. Na isquemia
cerebral, são comumente distinguidas duas regiões: o centro do infarto e uma
zona periférica, chamada de penumbra isquêmica. Uma redução significativa do
fluxo sanguíneo na área isquêmica central causa alterações nos processos
metabólicos, no suprimento energético da célula e na homeostase iônica, o que
gera perda da integridade celular, por necrose e apoptose em poucos minutos
(LIPTON, 1999; SACCCO, 2002; MERGENTHALER;DIRNAGL;MEISEL, 2004).
Este fenômeno é conhecido como degeneração secundária, é definido como um
grupo de eventos destrutivos que podem afetar células que não foram ou que
foram apenas marginalmente afetadas pela lesão inicial (TATOR;FEHLINGS,
1991; MCDONALD;SADOWSKY, 2002). No SNC, os neurônios e seus axônios
que não foram ou que foram apenas parcialmente afetados pela lesão primária,

32
podem degenerar tardiamente se não sofrerem intervenção terapêutica.
Eventos fisiopatológicos complexos e diversos são envolvidos nos
mecanismos de degeneração secundária (TATOR;FEHLINGS, 1991). No entanto,
sabe-se que excitotoxicidade e disfunção iônica, estresse oxidativo, resposta
inflamatória e apoptose são eventos cruciais neste importante fenômeno
patológico (PERRY et al., 1995; CHOI, 1998; LIPTON, 1999; POPOVICH et al.,
1999; CHOI, 2000; POPOVICH et al., 2002; LO, 2003; VILA, 2003).
2.3.1 Excitotoxicidade
2.3.1.1 Mecanismos Gerais
A liberação do glutamato na fenda sináptica possui um papel central na
comunicação neuronal e está envolvida nos vários aspectos da função cerebral
normal incluindo cognição, memória e aprendizado (KHALERT;REISER, 2004). Os
efeitos pós-sinápticos deste aminoácido excitatório endógeno são mediados por
receptores de membrana celular farmacológica e funcionalmente distintos
(SATTLER;TYMIANSKI, 2000). Os três receptores ionotrópicos principais são:
NMDA, cainato, e AMPA. Estes receptores, quando ionotrópicos, ao serem
ativados por seus agonistas, levam à abertura direta do canal iônico associado,
que pode ser permeável em menor ou maior quantidade para os íons Na+, K+ e
Ca++, dependendo do tipo de receptor (ATLANTE et al., 2001). Além disso, foi
posteriormente estabelecido que o glutamato pode se ligar aos chamados
receptores metabotrópicos, os quais atuam acoplados a um sistema envolvendo a

33
participação de proteinas G, que funciona através da liberação de segundos
mensageiros, os quais ativam canais iônicos presentes na membrana (CONN;PIN,
1997).
O cérebro contém grandes quantidades de glutamato, sendo o
neurotransmissor excitatório mais abundante no SNC (KHALERT;REISER, 2004;
HENDRIKS et al., 2005), porém só uma fração reduzida pode ser encontrada no
meio extracelular. As concentrações de glutamato, tanto no meio extracelular
como na fenda sináptica, são rigorosamente controladas por mecanismos
envolvendo enzimas e transportadores de glutamato em neurônios e células gliais
(DANBOLT, 2001). A capacidade funcional das sinapses é determinada pela
capacidade de biossíntese, liberação, degradação e transporte dos
neurotransmissores (SCHOUSBOE, 2003).
Excitotoxicidade envolve a ativação excessiva de receptores de glutamato
no SNC, resultando em morte neuronal (CHOI, 1988; 1992; ATLANTE et al., 2000;
MATUTE et al., 2001). Em certas condições patológicas como isquemia cerebral,
estado epiléptico, hipoglicemia, trauma mecânico e algumas doenças
neurodegenerativas crônicas, a excessiva liberação deste mediador por terminais
nervosos e seu acúmulo na fenda sináptica e no espaço extracelular, o torna uma
poderosa neurotoxina apta a induzir danos celulares irreversíveis (OLNEY, 1990;
CHOI, 1992; LI et al., 1999a), pois a íntima relação entre o metabolismo
energético anormal e a neurotransmissão glutamatérgica expõe neurônios à
excitotoxicidade mediada pelo glutamato (GREENE;GREENMYRE, 1996).
Durante a excitotoxicidade no SNC, células gliais (astrócitos e
oligodendrócitos), corpos neuronais e bainha de mielina sofrem danos estruturais

34
significativos, enquanto que o cilindro axonal é relativamente poupado durante a
lesão primária, pois alguns estudos imunohistoquímicos demonstraram a presença
de receptores glutamatérgicos, principalmente na membrana de oligodendrócitos,
na bainha de mielina, em astrócitos e, em menor quantidade, no cilindro axonal
(CHOI, 1992; LI;FIELD;RAISMAN, 1999; GALLO;GHINI, 2000; LI;JIANG;STYS,
2000; MATUTE et al., 2001; TEKKOK;GOLDBERG, 2001) .
2.3.1.2 Mecanismos moleculares e a participação do Ca++
O tecido neural possui um consumo relativamente alto de oxigênio e
glicose, depende quase que exclusivamente da fosforilação oxidativa para a
produção de energia (KHALERT;REISER, 2004). A diminuição do fluxo sangüíneo
restringe a chegada de substratos, em especial de oxigênio e glicose ao cérebro
(DIRNAGL;IADECOLA;MOSKOWITZ, 1999). Desta forma, com esta redução, as
reservas energéticas são perdidas com subsequente desequilíbrio iônico,
liberação de neurotransmissores e inibição da recaptação destes, especialmente
do glutamato (MELDRUM, 2000; LO, 2003; MERGENTHALER;DIRNAGL;MEISEL,
2004).
A despolarização de neurönios e células gliais em decorrência da carência
energética local resulta na ativação dos canais de Ca++ e leva a liberação de
aminoácidos excitotóxicos, especialmente glutamato, no meio extra-celular
(HERTZ;ZILKE, 2004; KANNURPATTI et al., 2004) . Este neurotransmissor, que
em condições normais sofre direta recaptação pré-sináptica ou astrocítica,
permanece no compartimento extra-celular e sofre acúmulo excessivo. A ativação

35
dos receptores de glutamato (NMDA, AMPA e metrabotrópicos) direta ou
indiretamente, mediado por prostaglandinas (BEZZI et al., 1998; CHOI, 2005), leva
a um aumento na concentração intra-celular de Ca++
(MERGENTHALER;DIRNAGL;MEISEL, 2004), através de sua liberação das
reservas internas, acidificação do meio e toxicidade (CHOI, 2005).
O aumento na concentração do Ca++ citoplasmático em neurônios do SNC
de mamíferos, induzido pela estimulação de receptores glutamatérgicos, ativa
mecanismos homeostáticos complexos, que proporcionam uma rápida
recuperação para níveis fisiológicos (SATTLER;TYMIANSKI, 2000; KHODOROV,
2004). Estes mecanismos consistem em interações complexas entre quatro tipos
de eventos: influxo de Ca++, tamponamento de Ca++ , estoque interno de Ca++, e
efluxo de Ca++ (SATTLER;TYMIANSKI, 2000). A extrapolação dos mecanismos
regulatórios fisiológicos, ativa inapropriadamente processos dependentes de Ca++
que tanto podem estar adormecidos como pouco ativados, induzindo
neurotoxicidade (CHOI, 1988; KANNURPATTI et al., 2004).
A neurotoxicidade do glutamato pode ser resumida em três fases, onde a
primeira é marcada pela tumefação neuronal imediata (CHOI, 1987), a segunda
pelo influxo excessivo de Ca++ (CHOI, 1988) e a terceira pela ativação excessiva
de proteases (lipases, fosfatases e endonucleases, caspases, calpaínas,
catepsinas) que tanto danificam diretamente a estrutura da célula, como induzem
a liberação de radicais livres oxidativos, que atuam como mediadores da morte
celular (CHOI, 1992; GOMES-LEAL, 2002b; ARUNDINE;TYMIANSKI, 2003;
BANO et al., 2005).
Em uma via bioquímica adicional, a sintase do óxido nítrico (SON), ao ser

36
ativada pelo Ca++, se liga à calmodulina e sintetiza NO (LIPTON;STAMLER,
1994; DAWSON;DAWSON, 1998; LIPTON, 2004). Quando sintetizado em
excesso, o NO combina com o ânion superóxido e forma peroxinitrito, que leva a
um estresse oxidativo e nitrosativo por disfunção mitocondrial
(LIPTON;SINGEL;STAMLER, 1994; LIPTON, 2004). Portanto, a disfunção
mitocondrial e o déficit energético resultante podem contribuir para a disfunção
neuronal através de alterações em canais iônicos, neurotransmissão e transportes
axonal e dendrítico (SATTLER;TYMIANSKI, 2000; BALES, 2004; BOSSY-
WETZEL;SCHWARZENBACHER;LIPTON, 2004; LIPTON, 2004; HOYER et al.,
2005).
Algumas evidências experimentais sugerem que a ativação de receptores do
tipo AMPA e cainato na substância branca causa morte de oligodendrócitos
maduros em cérebros isquêmicos, o que mostra a vulnerabilidade destas células à
excitotoxicidade mediada por receptores do tipo não-NMDA (MCDONALD et al.,
1998; LI;STYS, 2000; MATUTE et al., 2001; TEKKOK;GOLDBERG, 2001;
FOWLER et al., 2003). Este tópico será discutido posteriormente.
2.3.1.3 Estresse Oxidativo
A formação excessiva de radicais livres durante alterações patológicas do
SNC, tais como AVC e trauma, é um mecanismo fundamental de degeneração
secundária de doenças neurodegenerativas agudas e crônicas (LOVE, 1999;
LEWEN;MATZ;CHAN, 2000; LO, 2003). Os mecanismos de excitotoxicidade,
disfunção iônica e estresse oxidativo parecem agir em sinergismo durante o

37
desenrolar do processo neuropatológico (LOVE, 1999; LEWEN;MATZ;CHAN,
2000; ATLANTE et al., 2001; LO, 2003). Radicais livres parecem ser de extrema
importância para os mecanismos lesivos durante lesão excitotóxica mediada por
glutamato (SINGH et al., 1999; AGRAWAL;NASHMI;FEHLINGS, 2000). Espécies
reativas derivadas de oxigênio podem induzir peroxidação lipídica e promover
liberação de glutamato (AGRAWAL;NASHMI;FEHLINGS, 2000), tendo relação
direta com a excitotoxicidade (KANNURPATTI et al., 2004).
A ativação de receptores glutamatérgicos pode ter uma conexão com a
formação de radicais livres, através de pelo menos três maneiras: ativação pelo
Ca++ da fosfolipase A com a indução da liberação de ácido araquidônico e
formação de radicais livres; Indução pelo Ca++ da conversão de xantina
desidrogenase para xantina oxidase, o qual é fonte de radicais livres
(DYKENS;STERN;TRENKNER, 1987); estimulação de receptores de NMDA com
subsequente influxo de Ca++, ativação da SON com síntese de NO
(DAWSON;DAWSON, 1998). Estes autores acreditam que a produção de NO seja
um passo importante nos mecanimos de neurotoxicidade do glutamato. A inibição
da respiração mitocondrial pelo NO pode ser o principal mecanismo envolvido, em
desencadear a morte neuronal induzida pelo NO (BAL-PRICE;BROWN, 2001). O
metabolismo do ácido araquidônico pode ser a principal fonte de radicais livres,
pelo menos em cultura neuronal (CHOI, 1992, 1994).
Parece existir um elo de ligação entre estresse oxidativo e disfunção
mitocondrial (ATLANTE et al., 2000; ATLANTE et al., 2001). A liberação excessiva
de glutamato em condições patológicas pode gerar estresse oxidativo com
disfunção mitocondrial, por exemplo, diminuição dos suprimentos energéticos e do

38
transporte de metabólitos celulares (ATLANTE et al., 2000; ATLANTE et al., 2001).
A resposta mitocondrial à exposição ao glutamato, bem como a
excitotoxicidade são distintas entre animais jovens e adultos. A despolarização da
membrana mitocondrial de ratos adultos foi menor, e estes animais mostraram-se
menos sensíveis a citotoxicidade pelo glutamato (KANNURPATTI et al., 2004).
O trifosfato de adenosina (ATP) é essencial para a maioria dos processos
celulares e moleculares, tais como síntese proteica, manutenção do equilíbrio
iônico intra e extracelular, transporte e degradação de proteínas, funcionamento
do aparelho de Golgi e manutenção da transmissão sináptica. Uma redução no
ATP disponível, devido a falta de glicose ou alguma alteração metabólica pode
causar sérios distúrbios celulares, incluindo o metabolismo do precursor da
proteína amilóide (HOYER et al., 2005). Este fenômeno parece ocorrer in vivo em
algumas condições neurodegenerativas (ATLANTE et al., 2000; ATLANTE et al.,
2001).
Os mecanismos responsáveis pela indução de estresse oxidativo após
liberação excessiva de glutamato parecem envolver duas fases (ATLANTE et al.,
2001): produção inicial de xantina oxidase e uma fase tardia onde ocorre produção
de radicais livres, o que é relacionado à disfunção mitocondrial (HORAKOVA et
al., 1997; LEWEN;MATZ;CHAN, 2000). Além disso, a liberação de citocromo c
pode induzir mais formação de radicais livres e disfunção celular, incluindo lesão
mitocondrial (ATLANTE et al., 2000). Acredita-se que os mecanismos de defesa
celulares envolvendo anti-oxidantes naturais estejam reduzidos em tais
circunstâncias, levando a um considerável aumento do estresse oxidativo e lesão
celular generalizada, mediada por radicais livres (ATLANTE et al., 2001). Este

39
evento pode ser o destino final da cascata bioquímica responsável pela morte
neuronal mediada por ativação de receptores glutamatérgicos.
Experimentalmente, o uso de antioxidantes como o ebselen tem sido utilizado com
sucesso, para proteger as substâncias branca e cinzenta em modelos
experimentais de doenças neurodegenerativas agudas, incluindo o de isquemia
focal (REITER, 1998; RICHARDSON, 1999).
2.3.1.4 Modelos de excitotoxicidade por injeção de NMDA
A resposta excitotóxica obtida pela degeneração celular é mais complexa, do
que a obtida pela injeção de uma única citocina (BOLTON;PERRY, 1998).
Portanto, a injeção de NMDA vem sendo utilizada como modelo de lesão
excitotóxica.
Com a finalidade de documentar aspectos da resposta inflamatória
associada à degeneração neuronal induzida, a injeção intraestriatal de NMDA e
cainato mostrou resposta inflamatória aguda, com recrutamento de neutrófilos
seguidos de macrófagos. O infiltrado celular foi maior em animais jovens que em
adultos. Entretanto, a resposta inflamatória aumentada não pareceu ter um efeito
dramático no tamanho da lesão ou na resposta microglial e astrocítica, bem como
a morte celular ocorreu tanto por necrose como apoptose. Contrastando, a quebra
na BHE foi mais marcante em animais jovens após injeção de NMDA
(BOLTON;PERRY, 1998).
Em um modelo de lesão aguda da medula espinhal de ratos, demonstrou-
se recentemente a escala temporal dos padrões de ativação microglial e

40
astrocitária, após lesão excitotóxica (GOMES-LEAL, 2004). Células da microglia
são ativadas precocemente e podem contribuir para o início do processo lesivo na
substância cinzenta (GOMES-LEAL, 2004). A ativação astrocitária nas
substâncias branca e cinzenta da medula destes animais aumentou
consideravelmente entre 3 e 7 dias após a injeção de NMDA. A ativação dos
astrócitos começou mais rapidamente e foi mais intensa na substância branca,
que na substância cinzenta (GOMES-LEAL, 2004). Neste mesmo modelo
experimental, as células inflamatórias parecem contribuir para lesão axonal em
tempos de sobrevida mais tardios (GOMES-LEAL, 2005a).
O fator ativador de apoptose mostrou ser um importante mediador
excitotóxico, após a estimulação de receptores de NMDA. A exposição de uma
cultura de células neuronais ao NMDA resultou em excitotoxicidade pelo NMDA
independente de caspase. Portanto, a neurotoxicidade pelo NMDA não é afetada
pelos inibidores da caspase (WANG et al., 2004.
A habilidade de cabinóides em proteger neurônios contra lesões
excitotóxicas depende da inibição da SON neuronal. A morte celular induzida pelo
NMDA mostrou a participação do NO no efeito anti-excitotóxico de canabinóides
em neurônios cerebrais do córtex de ratos (KIM et al., 2005).
Sabendo que a neurogênese aumenta após lesão no encéfalo adulto, um
modelo de lesão excitotóxica usando NMDA foi empregado para avaliar a
dinâmica temporal e espacial, de proliferação nas zonas germinativas do cérebro
durante o desenvolvimento pós-natal. O dano excitotóxico no córtex motor de
ratos com 9 dias de idade causou degeneração neuronal, acompanhada de
resposta glial em todo o córtex ao nível do sítio de injeção, extendendo-se para o

41
septum, estriado e hipocampo rostral, sem comprometimento do hemisfério
contralateral. A lesão induziu diminuição significante do número de células
progenitoras da zona sub-ventricular (ipsilateralmente a lesão) e do giro dentato (3
primeiros dias) (FAIZ et al., 2005).
A expressão da enzima anti-oxidante superoxido dismutase do Cu e Zn em
encéfalos intactos é principalmente observada em neurônios (PELUFFO et al.,
2005). Em ratos recém-nascidos que sofreram lesão excitotóxica no SNC causada
por NMDA, observou-se diminuição na expressão desta enzima, seguida por uma
elevação de sua expressão por células astrogliais (PELUFFO et al., 2005).
2.3.2 Resposta Inflamatória
2.3.2.1 Inflamação no SNC
A inflamação é um componente chave do mecanismo de defesa contra
infecção, tanto no SNP como no SNC (FERRARI et al., 2004; MOYNAGH, 2005).
O encéfalo possui várias características peculiares que fazem com que a sua
resposta inflamatória seja diferente da que ocorre nos demais órgãos
(ALLAN;ROTHWELL, 2003). Uma barreira natural regula o fluxo de substâncias
que entram e saem do SNC, a BHE. Esta barreira protege o encéfalo de
substâncias lesivas, limitando a entrada de moléculas grandes e células
circulantes (PETTY;LO, 2002), protegendo-o de toxinas secretadas por bactérias e
outras originadas da atividade de células do sistema imune
(REICHEL;BEGLEY;ABBOTT, 2000).

42
A restrição imposta pela BHE às células do sistema imune faz com que a
resposta inflamatória no tecido nervoso, seja menos intensa do que em outros
tecidos (por exemplo, o tecido epitelial) (GOMES-LEAL, 2002a). Esta
característica peculiar levou o sistema nervoso a ser considerado um órgão de
privilégio imunológico (PERRY; ANDERSSON; GORDON, 1993; SCHWARTZ;
KIPNIS, 2001b; SCHWARTZ; MOALEM, 2001; ALLAN; ROTHWELL, 2003). Por
exemplo, estudos experimentais em roedores mostram que um estímulo
inflamatório com lipopolissacarídeos bacterianos (LPS) induz uma rápida e
elevada invasão de neutrófilos na pele, mas uma resposta limitada e tardia no
encéfalo (PERRY et al., 1995; MATYSZAK, 1998). O recrutamento de neutrófilos
em tecidos não neurais tem seu pico máximo em torno de 4-6 horas
(VILLARREAL;ZAGORSKI;WAHL, 2001), enquanto que no SNC ocorre em
24horas (BOLTON;PERRY, 1998; SCHNELL et al., 1999; GOMES-LEAL, 2002a,
2005b). Com relação aos monócitos sangüíneos, o recrutamento também é mais
lento no SNC (em torno de 72h) do que em tecidos não neurais (em torno de 24h).
A microinjeção de mediadores inflamatórios no parênquima do cérebro adulto
mostrou pouco recrutamento de neutrófilos, resposta vascular mínima e um atraso
no recrutamento de monócitos (ANDERSSON;PERRY;GORDON, 1992;
LAWSON;PERRY, 1995. Diferenças significativas são encontradas entre os
diversos compartimentos do SNC (SCHNELL et al., 1999).
A resposta às lesões no SNC tem característica multicelular que muda
continuamente com a evolução temporal e espacial do processo e é regulada por
múltiplos eventos moleculares intra e extracelulares (SOFRONIEW, 2005). Em
condições não patológicas, células do sistema nervoso podem mediar a resposta

43
inflamatória, mesmo que esta possua aspectos diferentes daquela de outros
tecidos (GOMES-LEAL, 2002a). O encéfalo responde a estímulos inflamatórios
periféricos (por resposta neural ou humoral), integra e regula vários aspectos da
resposta aguda e exibe muitas respostas inflamatórias locais, que parecem
contribuir com o aparecimento de doenças do SNC, tanto agudas como crônicas.
O estágio mais precoce da inflamação, que inicia poucas horas após a
isquemia, caracteriza-se pela liberação de moléculas de adesão tanto no endotélio
vascular como por leucócitos circulantes (ALLAN;ROTHWELL, 2003). Os
leucócitos aderem-se ao endotélio e transmigram do sangue para o parênquima
cerebral (STOLL;JANDER;SCHROETER, 1998). Esta adesão é de importância
decisiva na inflamação cerebral induzida por AVC
(MERGENTHALER;DIRNAGL;MEISEL, 2004). Neste caso, neutrófilos acumulam-
se em microvasos cerebrais localizados na área da penumbra isquêmica, levando
a um dano adicional na microcirculação. Macrófagos e monócitos seguem os
neutrófilos, migrando para o encéfalo isquêmico e tornam-se as células
predominantes 5 a 7 dias após a isquemia.
Leucócitos ativados (granulócitos, monócitos/macrófagos, linfócitos), bem
como neurônios e células gliais (astrócitos e microglia) produzem quimiocinas e
citocinas, principalmente citocinas pró-inflamatórias, tais como: Fator de necrose
tumoral
(FNT ) e as interleucinas-1 e 6 (IL-1 e IL-6), que exacerbam os danos
provocados pela lesão isquêmica (DIRNAGL;IADECOLA;MOSKOWITZ, 1999;
BECKER et al., 2001). Em modelos animais, o dano isquêmico é reduzido por
vários antagonistas de receptores de citocinas (ALLAN;ROTHWELL, 2003;

44
MERGENTHALER;DIRNAGL;MEISEL, 2004). Outras evidências mostram a
contribuição da inflamação pós-isquêmica na lesão do cérebro isquêmico, pois o
dano cerebral isquêmico reduz quando: a infiltração de neutrófilos é prevenida
pela indução de uma neutropenia sistêmica, moléculas de adesão ou seus
receptores são bloqueados por anticorpos neutralizantes, a ação de mediadores
da inflamação é bloqueada e no rato, inibição dos genes que coordenam a
liberação de genes relacionados com a inflamação
(DIRNAGL;IADECOLA;MOSKOWITZ, 1999). Além das citocinas inflamatórias,
algumas citocinas anti-inflamatórias também são sintetizadas, entre as quais fator
transformador de crescimento- 1 e a IL-10. Estas citocinas regulam a inflamação,
diminuindo-a e, por conseguinte, exercem efeito protetor no contexto da isquemia
cerebral (MERGENTHALER;DIRNAGL;MEISEL, 2004).
Duas enzimas também se mostraram essenciais para a inflamação: a
sintase do óxido nítrico imunológica e a ciclooxigenase tipo II (COX2)
(MERGENTHALER;DIRNAGL;MEISEL, 2004). Em modelos experimentais de
isquemia cerebral, a inibição da sintase do óxido nítrico imunológica reduziu o
tamanho do infarto em aproximadamente 30%, até mesmo quando o tratamento
começou apenas 24h após a indução da isquemia (IADECOLA;ZHANG;XU, 1995;
CHANG et al., 2004; PEREZ-ASENSIO et al., 2005). Além disso, esta enzima
produz uma grande quantidade de ON, o qual é altamente tóxico, devido a sua
participação na formação do peroxinitrito. A COX2 é mais facilmente detectada na
região da penumbra isquêmica, participa ativamente na produção de radicais
livres, o que a torna potencialmente lesiva para a área (NOGAWA et al., 1997;

45
HARA et al., 1998; NOGAWA et al., 1998). Dessa forma, tanto a sintase do óxido
nítrico imunológica quanto a COX2 apresentam-se como alvos de grande
interesse terapêutico, visto que em modelos experimentais de roedores, ambas
apresentaram efeito protetor por 6-24 h após a isquemia (CHOPP;ZHANG, 1996).
Existem diversas evidências na literatura que indicam que mecanismos
inflamatórios participam dos fenômenos de degeneração secundária em doenças
agudas e crônicas do SNC (BLIGHT, 1985,1994; PERRY et al., 1998; POPOVICH
et al., 1999; GOMES-LEAL, 2002a; POPOVICH et al., 2002; WYSS-CORAY,
2002). O uso de uma mistura de cloroquina e colchicina (GIULIAN, 1987) ou
injeção intraperitoneal de pó de sílica (BLIGHT, 1994) ou liposomas de clodronato
(POPOVICH et al., 1999), são procedimentos que inibem o recrutamento de
monócitos sangüíneos para parênquima do SNC lesado, diminuem
significativamente a área de lesão secundária, após lesão aguda da medula
espinhal de roedores. Além disso, foi demonstrado experimentalmente que o uso
de altas doses de metilpredinisolona, um anti-inflamatório esteróide, melhora a
capacidade de recuperação motora e diminui a área de lesão, em ratos
submetidos à lesão aguda da medula espinhal (BRACKEN, 2001a,2002).
Inúmeros testes demonstraram que este procedimento é eficaz e atualmente
utilizado como tratamento para lesão da medula espinhal de seres humanos
(BRACKEN, 2001a; BRACKEN;HOLFORD, 2002).
Acredita-se que componentes da resposta inflamatória também sejam
envolvidos nos eventos patológicos de doenças neurodegenerativas crônicas, tais
como as doenças de Parkinson, Huntington, Alzheimer, esclerose múltipla e
esclerose lateral amiotrófica (HIRSCH et al., 1998; MCGEER;MCGEER, 1998;

46
MCGEER;YASOJIMA;MCGEER, 2001).
2.3.2.3 O papel de células do sistema imune em doenças neurodegenerativas e
sua relação com a resposta inflamatória
A integridade da BHE depende da interação da matriz celular, composta por
células endoteliais e astrócitos (MERGENTHALER;DIRNAGL;MEISEL, 2004). A
isquemia cerebral pode danificar a interação entre estas estruturas. Proteases,
como metaloproteinases de matriz, são secretadas 1 a 3 horas após a isquemia
cerebral e atuam sobre as estruturas desta matriz
(MERGENTHALER;DIRNAGL;MEISEL, 2004). A destruição da lâmina basal, pelas
metaloproteinases de matriz permite que leucócitos atravessem a BHE e
contribuam para alterações patológicas nesta região (BOLTON;PERRY, 1998).
No SNC, assim como em tecidos não neurais, o sistema imune inato age
em sinergismo com o sistema imune específico, tanto contribuindo para a indução
de seus componentes como sendo estimulado por ele (MATYSZAK, 1998).
Acredita-se que alguns componentes da reposta inflamatória possam contribuir
para o aumento da lesão secundária, após lesão do SNC (BLIGHT, 1992;
POPOVICH et al., 1999; BLIGHT;ZIMBER, 2001). Abaixo sumarizamos a
participação destas células, nos mecanismos inflamatórios envolvidos na
fisiopatologia de doenças do SNC (GOMES-LEAL, 2002a, 2002b).
Neutrófilos

47
Os neutrófilos, juntamente com os eosinófilos e basófilos, formam o grupo de
leucócitos conhecidos como granulócitos (ABBAS;JANEWAY, 2000). Este termo é
empregado devido à aparência dos grânulos (lisossomas) encontrados no
citoplasma destas células. Os lisossomas possuem inúmeras enzimas,
principalmente proteases, que são liberadas durante a fagocitose e digestão
intracelular de patógenos (durante o processo de infecção), ou detritos celulares
(durante o processo de degeneração). Como já mencionado neste trabalho, são
as primeiras células inflamatórias a chegar ao sítio da inflamação aguda, tanto em
tecidos neurais como não neurais, sendo este fenômeno um pouco mais tardio no
SNC (STOLL;JANDER;SCHROETER, 1998; SCHNELL et al., 1999; GOMES-
LEAL, 2005b). São fisiologicamente células fagocitárias, mas acredita-se que os
mesmos podem contribuir como o processo lesivo, durante o trauma tecidual, nos
tecidos neurais e não neurais (TAOKA et al., 1997; TAOKA;OKAJIMA, 1998;
TAOKA et al., 1998).
Estas células podem ser encontradas no cérebro humano, após isquemia e
a quantidade delas no tecido tem correlação positiva com a presença de sangue
(TAOKA;OKAJIMA, 1998). Nestas circunstâncias, os neutrófilos podem liberar
proteases e radicais livres, que são lesivos para o parênquima tecidual
(CAMPBELL;CAMPBELL, 1988; OWEN;CAMPBELL, 1995). A diminuição do
número de neutrófilos circulantes ou administração de anticorpos anti-quimiocinas
tem mostrado redução significante na área do infarto, em modelos experimentais
de isquemia cerebral em ratos.
A expressão crônica de IL-1 leva ao recrutamento seletivo de neutrófilos, no
parênquima cerebral e induz ativação de microglia e astrócitos, lesão da BHE,

48
desmielinização reversível, sem neurodegeneração (FERRARI et al., 2004).
Macrófagos
Os macrófagos derivam de células tronco e de monoblastos da medula óssea
que transformam-se em monócitos, ao entrarem na corrente sangüínea
(PERRY;GORDON, 1991). Durante a inflamação aguda, estes monócitos são
recrutados para o parênquima tecidual, onde são ativados em situações
patológicas (GOMES-LEAL, 2002a). Os monócitos sangüíneos penetram no SNC
durante o seu desenvolvimento (PERRY;GORDON, 1991) e sua diferenciação em
macrófagos envolvem alterações morfológicas e o aumento da expressão de
diversos receptores de membrana de lisossomas (por exemplo, o antígeno
glicosilado citoplasmático que é marcado pelo anticorpo ED1) (DIJKSTRA et al.,
1985b) e o receptor C3 do complemento (REID et al., 1993). O fator de
estimulação de colônia de granulócitos-macrófagos é expresso em uma variedade
de células diferenciadas e não diferençadas, tipo: monócitos, macrófagos,
fibroclastos, células tipo T e células endoteliais e estimulam a proliferação e
maturação de progenitores mielóides, precursores de neutrófilos, monócitos,
macrófagos e eosinófilos (FRANZEN;BOUHY;SCHOENEN, 2004).
Os macrófagos são considerados as principais células efetoras da resposta
imune (GORDON, 2001), permanecendo por mais tempo e em maior número no
infiltrado inflamatório (POPOVICH et al., 1999). São fagócitos, portanto estão no
local da lesão para matar microorganismos invasores, remover restos e facilitar o
reparo e o retorno a homeostasia (PERRY;ANDERSSON;GORDON, 1993;

49
POPOVICH et al., 1999). Após lesão na medula espinhal o reparo precede a
infiltração de macrófagos hematogênicos, mas não a ativação da microglia
residente (POPOVICH et al., 1999). Desde que a matriz formada após a lesão não
seja suficiente para manter o crescimento axonal, é possível que o infiltrado de
macrófagos antagonize os esforços das células residentes para reparar o local da
lesão.
Uma ferramenta importante para eliminar células ou patógenos indesejados
é a produção de mediadores, causadores da morte celular ou de microorganismos
(HENDRIKS et al., 2005). A chegada destas células no local da inflamação está
relacionada com a ativação de plaquetas e cascatas de proteínas plasmáticas,
aumento da expressão de moléculas de adesão, nos vasos e aumento na
permeabilidade vascular (PERRY;ANDERSSON;GORDON, 1993).
Durante o processo de reparo tecidual, os macrófagos podem contribuir
para a exacerbação da lesão neural pela capacidade de liberar citocinas
inflamatórias
(FNT , FESGM; IL-1 e IL-6), proteases (MPM), radicais livres
(radicais hidroxila), NO (BLIGHT;SAITO;HEYES, 1993; BLIGHT, 1994;
FERGUSON et al., 1997; POPOVICH et al., 1999; GOMES-LEAL, 2005a) e
grandes quantidades de glutamato (HENDRIKS et al., 2005). Na lesão da medula
espinal ocorre grande resposta de macrófagos, sendo possível que estas células
atuem como mediadores da lesão traumática secundária e contribuam para o
insucesso da regeneração axonal no SNC (POPOVICH et al., 1999). Em
trabalhos experimentais, demonstrou-se que a depleção do recrutamento de
macrófagos com pó de sílica (BLIGHT, 1985, 1992, 1994), clodronato (POPOVICH
et al., 1999) ou metilpredinisolona (BRACKEN, 1990, 2000; BRACKEN et al.,

50
2000; BRACKEN, 2001a, 2001b, 2002; BRACKEN;HOLFORD, 2002) diminui a
área de lesão secundária e melhora o prognóstico de recuperação motora após
lesão medular em roedores.
Macrófagos são um alvo interessante para terapias que visam prevenir o
dano axonal na Esclerose Múltipla. A prevenção da migração de monócitos
através da BHE é um dos níveis de intervenção possível (HENDRIKS et al., 2005).
Microglia
As células microgliais são células gliais do SNC, possuem um importante
papel na resposta inflamatória e apresentam propriedades compatíveis com as
dos macrófagos hematogênicos (KHALERT;REISER, 2004;
MERGENTHALER;DIRNAGL;MEISEL, 2004). São ativadas por mediadores da
inflamação em uma gama de patologias no SNC, tais como inflamação cerebral,
trauma e AVC (BAL-PRICE;BROWN, 2001) e neste estado comportam-se como
macrófagos residentes do SNC (PERRY;ANDERSSON;GORDON, 1993; GOMES-
LEAL, 2002a), adotando inclusive uma morfologia ramificada típica
(STOLL;JANDER;SCHROETER, 1998). Quando não ativadas, possuem a função
geral de fazer a vigilância do sistema nervoso (GOMES-LEAL, 2002a). A microglia
parece ser responsável pela fagocitose mais generalizada envolvendo a ativação
de cascata complementar, onde astrócitos têm sido envolvidos em processos de
fagocitose, tais como a remoção de sinapses individuais (WYSS-CORAY, 2002;
MERGENTHALER;DIRNAGL;MEISEL, 2004). A microglia e outros fagócitos
participam não apenas da degradação de depósitos proteicos anormais, como na

51
Doença de Alzheimer, ou de micróbios invasores, mas também da remoção de
células do hospedeiro em degeneração (WYSS-CORAY, 2002).
É possível que haja um componente anti-inflamatório intrínseco no SNC
(PERRY;ANDERSSON;GORDON, 1993). Semelhante aos leucócitos, a microglia
ativada é capaz de produzir uma boa quantidade de citocinas inflamatórias, bem
como metabólitos tóxicos (radicais livres oxigenados: peroxinitrito e superóxido) e
enzimas (catepsinas) (MERGENTHALER;DIRNAGL;MEISEL, 2004). Durante o
fenômeno de ativação, a membrana de células microgliais aumenta a expressão
de moléculas do complexo principal de histocompatibilidade (CPH), do receptor C3
do complemento, receptores CD4, ED1 e moléculas de adesão
(PERRY;GORDON, 1991; STOLL;JANDER, 1999; STREIT, 2000; BAL-
PRICE;BROWN, 2001). Relatou-se que durante a isquemia cerebral, células da
microglia liberam NO e IL-1, os quais podem contribuir para o processo de
degeneração secundária (GEHRMANN et al., 1995;
STOLL;JANDER;SCHROETER, 1998; LOVE, 1999). Achados semelhantes foram
observados após lesão cerebral obtida através de modelos excitotóxicos
(GEHRMANN et al., 1995; STOLL;JANDER;SCHROETER, 1998; LOVE, 1999).
O fenômeno de ativação microglial foi dividido nas seguintes fases: estágio de
alerta (estágio 1), aderência (estágio 2), fagocitose (estágio3a), ativação de
células microgliais vizinhas (estágio 3b) (RAIVICH et al., 1999). O estágio 1 ocorre
nas primeiras 24 horas é caracterizado pelo aumento da expressão de algumas
moléculas relacionadas à função imune, tais como o componente Ci3b do
complemento e MAI-1 em camundongos. No estágio 2, as células tornam-se
menos ramificadas aderindo às estruturas lesadas, tais como neurônios sofrendo

52
degneração. Ao mesmo tempo, estas células apresentam diminuição da
expressão de algumas moléculas em suas membranas, tais como MAI-1 e
aumento da expressão de outras, como moléculas do CPH. No estágio 3a, as
moléculas microgliais tornam-se fagócitos na presença de neurônios sofrendo
degeneração. Neste momento, ocorre retração significativa das ramificações
microgliais, as células da microglia se tornam arredondadas, com morfologia de
macrófagos ativados. No estágio 3b, ocorre ativação generalizada da microglia em
sítios distais ao da lesão e de outras células da resposta inflamatória, tais como
linfócitos. O recrutamento de linfócitos é intenso nesta fase, sendo mediado pelo
aumento da expressão de moléculas da classe II do CPH, com subseqüente
apresentação de antígenos em seres humanos (GOMES-LEAL, 2002a).
As células microgliais parecem estar envolvidas nos mecanismos de lesão
autoimune (encefalite experimental alérgica) (NATHAN, 1987; NATHAN, 1992), de
lesão do SNC durante infecção pelo HIV (STOLL;JANDER, 1999).
Linfócitos
Em condições não patológicas, no parênquima do SNC são encontrados
poucos linfócitos ou mesmo sua total ausência. No entanto, em condições
patológicas, além de serem encontrados no parênquima cerebral existem
evidências de que podem causar dano tecidual adicional, após lesão primária
(BRADL;FLUGEL, 2002).
Existem evidências que sugerem que a atividade de células T pode ser
importante para regeneração neural (SCHWARTZ, 2001a, 2001b). O recrutamento

53
de linfócitos T é limitado durante a lesão do SNC, este fato pode contribuir para a
incapacidade de regeneração do tecido neural (SCHWARTZ;KIPNIS, 2001a).
Astrócitos
Nos últimos anos três funções principais dos astrócitos foram destacadas e
pesquisadas em detalhes: suporte nutricional a células neuronais, modulação do
nível glutamatérgico extra-celular e eliminação de radicais livres oxigenados. Os
astrócitos também são encarregados de manter o gradiente de íons dependentes
de energia (Ca++, N+ e K+) e o pH extracelular (KHALERT;REISER, 2004). Para
desempenhar suas funções, não só os astrócitos, mas as células gliais precisam
de quantidade considerável de energia (KHALERT;REISER, 2004). Ao contrário
dos neurônios, os astrócitos estão em contato com os vasos sangüíneos e
acessam o suporte de glicose do encéfalo pela circulação sangüínea
(KHALERT;REISER, 2004). Então, a maior quantidade de glicose utilizada pelo
encéfalo é entregue via astrócitos (KHALERT;REISER, 2004).
O glutamato, bem como o GABA, é removido da fenda sináptica pelos
astrócitos, através de um sistema de transporte altamente eficiente
(SCHOUSBOE, 2003). Os astrócitos estão localizados próximos à fenda sináptica,
por isso, são os primeiros candidatos a remover o glutamato, porém em situações
isquêmicas esta função é alterada e revertida (KHALERT;REISER, 2004).
A ativação de astrócitos ocasiona hipertrofia destas células, com aumento
do tamanho do corpo celular, encurtamento e aumento da espessura de suas
ramificações (astrocitose) (GOMES-LEAL, 2002a; SOFRONIEW, 2005). Estas

54
alterações morfológicas são reguladas por citocinas, tais como IL-6. A astrocitose
é considerada uma resposta geral do SNC, ao processo lesivo como uma tentativa
de preservar a integridade tecidual, mas a cicatriz glial formada, pode ser
prejudicial ao fenômeno de regeneração axonal (SYKOVA, 2001). A cicatriz glial,
composta por astrócitos e linhagens celulares de fibroblastos, demarca e envolve
o tecido lesado do tecido neural viável (SOFRONIEW, 2005). Os astrócitos podem
proliferar após lesões realmente severas (SOFRONIEW, 2005).
Astrócitos reativos exercem funções tanto pro, como anti-inflamatórias em
locais e períodos diferentes, como reposta a lesão e durante o reparo
(SOFRONIEW, 2005). Exercem papel importante na manutenção e reparo da
BHE, ajudam a regular os níveis de fluído tecidual, também regulam o edema
tóxico que pode ocorrer no parênquima cerebral após várias lesões ao SNC, bem
como ajudam a proteger neurônios, oligodendrócitos e a função neural
(SOFRONIEW, 2005). Entretanto, possuem a capacidade de gerar moléculas com
potencial citotóxico, tais como radicais de NO e radicais oxigenados
(SOFRONIEW, 2005).
2.3.3 Apoptose e Outros Tipos de Morte Celular
Durante o AVC, no centro do território de vascularização do vaso ocluído,
as células morrem principalmente por necrose e na penumbra isquêmica o
principal tipo de morte celular é apoptose (LO, 2003). Estudos experimentais estão
elucidando cada vez mais os mecanismos moleculares dos vários tipos de morte
celular, incluindo paraptose, necrose e apoptose (SYNTICHAKI, 2002a;

55
SYNTICHAKI, 2002b; LO, 2003; SYNTICHANKI, 2003; VILA, 2003). Estes
estudos, principalmente os realizados no nematódio C. elegans, consolidaram a
idéia que a necrose não é um tipo de morte celular caótica, mas que envolve a
ativação seqüencial de proteases, o que é expresso na hipótese calpaína-
catepsina (SYNTICHAKI, 2002b; SYNTICHANKI, 2003). Os mecanismos de
apoptose e necrose são extremamente complexos, e não serão aqui revistos.
Excelentes revisões estão disponíveis na literatura sobre o tema (HENGARTNER,
2000; YUAN, 2000; VILA, 2003).
Estabeleceu-se um papel essencial para a morte por apoptose, durante o
fenômeno de degeneração secundária em doenças neurodegenerativas agudas,
tais como trauma cerebral e da medula espinhal e AVC (LI et al., 1996; LIU et al.,
1997; LI et al., 1999a; BEATTIE;FAROOQUI;BRESNAHAN, 2000; LEE, 2002;
TIKKA et al., 2002; RUAN, 2003; TAKAGI et al., 2003; GOMES-LEAL, 2004;
STIRLING et al., 2004). Após lesão experimental da medula espinhal de ratos e
primatas demonstrou-se que oligodendrócitos morrem por apoptose, em um tempo
de sobrevida tardio (CROWE et al., 1997). Estudos na medula espinhal do rato,
demonstram claramente que a morte apoptótica de oligodendrócitos é um
componente importante da degeneração secundária (LI et al., 1996; LIU et al.,
1997; LI et al., 1999a; BEATTIE;FAROOQUI;BRESNAHAN, 2000; GOMES-LEAL,
2004). O tratamento com Minociclina, uma tetraciclina de segunda geração, reduz
a morte tardia de oligodendrócitos, impede lesão axonal retrógrada e melhora o
prognóstico neurológico de ratos submetidos à lesão aguda da medula espinhal
(LEE et al., 2003; STIRLING et al., 2004). A minociclina também possui efeito
neuroprotetor, em modelos de isquemia focal no cérebro de ratos (YRJANHEIKKI

56
et al., 1999).
2.3.4 Lesão Axonal em Doenças Neurodegenerativas
O SNC é composto pelo cérebro e medula espinhal. Estas duas regiões
do SNC possuem as substâncias cinzenta, contendo principalmente corpos
neuronais e células gliais, e substância branca que contém corpos celulares de
alguns grupos neuronais (por exemplo, neurônios diaforase positivos) e células
gliais, mas é principalmente formada por tratos de axônios mielinizados (MARTIN,
1998). Tanto a substância branca, como a substância cinzenta são lesadas
durante as doenças neurodegenerativas agudas, tais como trauma cerebral e
medular, AVC, doença neurológica devido à infecção pelo HIV, malária cerebral e
doenças neurodegenerativas crôncias como as doenças de Alzheimer, Parkinson,
Huntington e Esclerose mútipla (MEDANA;ESIRI, 2003). No entanto, os estudos
neuropatológicos clássicos foram mais centrados nos eventos patológicos que
acometem a substância cinzenta, os eventos que ocorriam na substância branca
foram geralmente negligenciados (COLEMAN;PERRY, 2002). Recentemente
percebeu-se que a lesão dos tratos axonais, presentes na substância branca do
SNC é uma das principais causas dos déficits funcionais subjacentes a estas
doenças, e que o desenrolar neuropatológico do processo lesivo depende da
localização destes tratos axonais (DE_KEYSER;SULTER;LUITEN, 1999;
DEWAR;YAM;MCCULLOCH, 1999; COLEMAN;PERRY, 2002; MEDANA;ESIRI,
2003).
Por exemplo, sabe-se que após um AVC, lesão da medula espinhal e

57
malária cerebral, o comprometimento da substância branca exacerba os déficits
funcionais em seres humanos (PETTY;WETTSTEIN, 1999;
MCDONALD;SADOWSKY, 2002; MEDANA et al., 2002; LO, 2003). Cerca de 25%
a 30% de todos os acidentes isquêmicos em humanos ocorrem na substância
branca (LO, 2003; STYS, 2004), uma região bastante vulnerável durante acidentes
isquêmicos (PANTONI;GARCIA;GUTIERREZ, 1996). Resultados experimentais
confirmaram que abordagens terapêuticas que privilegiam a preservação da
substância cinzenta e negligenciam a preservação da substância branca são
ineficazes após AVC e trauma (DE_KEYSER;SULTER;LUITEN, 1999;
DEWAR;YAM;MCCULLOCH, 1999).
A lesão da substância branca após AVC ou trauma, por exemplo, nos
tratos da medula espinhal e na cápsula interna, pode gerar déficits funcionais mais
significativos que a lesão de substância cinzenta (DEWAR;YAM;MCCULLOCH,
1999; MCDONALD;SADOWSKY, 2002; STYS, 2004). Um dos fatores importantes
para este fato é o comprometimento de axônios de passagem
tanto do cilindro
axonal como da bainha de mielina. As lesões da substância cinzenta induzem
déficits funcionais mais locais. Um exemplo claro deste fato é que uma lesão
restrita à substância cinzenta da sexta vértebra cervical em humanos, pode gerar
alterações funcionais das mãos sem afetar funções em segmentos caudais
(funções posturais, intestinais e da bexiga) (MCDONALD;SADOWSKY, 2002). No
entanto, uma lesão neste mesmo segmento vertebral, mesmo que não afete a
substância cinzenta, pode induzir tetraplegia e incontinência urinária
(MCDONALD;SADOWSKY, 2002). A lesão axonal é um fator importante para a
exacerbação dos déficits funcionais, principalmente por bloqueio total ou

58
disfunções no padrão de condução do potencial de ação (STYS, 2004). Lesão
axonal é um fenômeno freqüente em doenças neurodegenerativas agudas e
crônicas, tais como AVC (DE_KEYSER;SULTER;LUITEN, 1999;
DEWAR;YAM;MCCULLOCH, 1999; HUGHES et al., 2003), trauma cerebral e da
medula espinhal (GOMES-LEAL, 2002a; MCDONALD;SADOWSKY, 2002),
malária cerebral (MEDANA et al., 2002) e esclerose múltipla (FERGUSON et al.,
1997; TRAPP et al., 1998).
Os mecanismos de lesão axonal parecem envolver uma série complexa
de eventos fisiopatológicos, incluindo disfunções iônicas (influxo excessivo de Na+
e Ca++), alterações em bombas metabólicas do axônio e excitotoxicidade
mediada por receptores do tipo AMPA/cainato, afetando principalmente a bainha
de mielina, o corpo celular de oligodendrócitos e astrócitos (STYS, 2004).
Antagonistas de receptores de NMDA são ineficazes na proteção da substância
branca durante doenças neurodegenerativas agudas, indicando que estes
receptores não participam do processo lesivo nesta região do sistema nervoso
(YAM et al., 2000). Por outro lado, em uma série de estudos experimentais no
nervo óptico e na substância branca dorsal da medula espinhal, demonstraram de
forma irrefutável que a lesão da substância branca envolve a participação de
ativação excessiva de canais de Na+ e Ca++ e excitotoxicidade mediada pela
ativação de receptores AMPA/cainato, além de alterações em bombas
metabólicas transportadoras de glutamato (LI et al., 1999b; LI;JIANG;STYS, 2000;
LI;STYS, 2000, 2001). Em um modelo in vitro, estes autores infundiram os
agonistas glutamatérgicos AMPA e cainato em colunas dorsais isoladas da
medula espinhal de ratos (LI;STYS, 2000). Este procedimento resultou em

59
alterações estruturais em oligodendrócitos, astrócitos e particularmente da bainha
de mielina. Nestes estudos, não foram encontradas evidências para lesão do
cilindro axonal, como evidenciado pela imuncitoquímica para degradação da
espectrina por proteases, bem como pela presença de GluR2/3 e GluR4 em
células gliais.
Em um outro paradigma experimental utilizando o mesmo modelo in vitro,
a substância branca dorsal isolada de ratos foi submetida a 60 minutos de anóxia
ou à compressão durante 15 segundos (LI et al., 1999b). Tanto antagonistas
seletivos de receptores do tipo AMPA/cainato, como a inibição do transporte de
glutamato dependente de Na+, protegeram a substância branca dos mecanismos
lesivos induzidos por anóxia e trauma. Estudos posteriores pelo mesmo grupo
(LI;STYS, 2001) demonstraram que a liberação endógena de glutamato pode
ocorrer pela reversão dos gradientes de Na+ e K++ através do axolema. De
acordo com estes autores, a inibição da ATPase de Na+ e K++ e despolarização
induzem liberação de glutamato, através da reversão da atividade dos
transportadores de glutamato dependente de Na+ . O glutamato liberado de fontes
intracelulares agiria em receptores do tipo AMPA/cainato induzindo lesão
excitotóxica principalmente em oligodendrócitos e mielina. Nestes estudos, a
principal fonte de glutamato foi o cilindro axonal. Outros estudos sugerem que a
injeção do bloqueador de canal de Na+ -TTX- diminui significativamente a lesão
axonal, após contusão da medula espinhal, mostrando que influxo excessivo de
canais de Na+ pode ser um importante evento subjacente, aos mecanismos de
lesão, da substância branca após contusão da medula espinhal
(ROSENBERG;TENG;WRATHALL, 1999).

60
2.4 O PARADIGMA EXPERIMENTAL
O núcleo espinhal do sistema trigeminal, com seus subnúcleos, apresenta
importante atuação na transmissão da informação nociceptiva das estruturas
orofaciais (DALLEL et al., 1988). Dentre os sub-núcleos, destaca-se a zona de
transição entre os subnúcleos interpolar e caudal, como uma região crítica para o
processamento da dor craniofacial. Portanto, com grande importância na resposta
a danos orofaciais profundos, constituindo uma região integradora de informações
sensoriais das regiões peri e intra-orais (BERETIER, 2000; IKEDA et al., 2003).
Eventos isquêmicos e excitotóxicos nesta região certamente induzem
défices sensitivos em animais de experimentação e seres humanos
(VOS;STRASSMAN;MACIEWICZ, 1994), mas os eventos histopatológicos
relacionados aos padrões de lesão axonal, desmielinização e resposta inflamatória
não foram descritos de forma sistemática para esta região, principalmente
considerando-se os eventos que ocorrem nas substâncias branca e cinzenta da
região em questão. A comparação entre os padrões de resposta excitotóxica e
isquêmica no sistema trigeminal não está descrito na literatura.
Na presente dissertação, utilizamos modelo de isquemia focal por injeção
de ET-1 e de excitotoxicidade por injeção de NMDA para averiguarmos, em um
tempo de sobrevida agudo (1 dia) e em um tempo de sobrevida crônico (7 dias),
os padrões histopatológicos subseqüentes à isquemia focal e lesão excitotóxica
experimental induzidas na substância cinzenta do núcleo espinhal do sistema
trigeminal.

61
3 PROPOSIÇÃO
O propósito desta investigação foi avaliar comparativamente os padrões
inflamatórios, de lesão axonal e desmielinização após lesão aguda excitotóxica e
isquêmica, do núcleo espinhal do sistema trigeminal com ênfase nas alterações na
substância branca do trato trigeminotalâmico. Especificamente, induzir
degeneração neuronal aguda através da injeção focal de NMDA e ET-1 no núcleo
espinhal no sistema trigeminal, descrever os padrões de resposta inflamatória e
degeneração axonal, após lesão excitotóxica e isquêmica e por fim, comparar os
padrões de resposta inflamatória, lesão axonal e desmielinização obtidos após
lesão excitotóxica e isquêmica no tronco cerebral de ratos adultos.
Na presente dissertação, investigamos a hipótese de que eventos
excitotóxicos e isquêmicos, no núcleo espinhal do sistema trigeminal podem
induzir resposta inflamatória, com subsequente degeneração axonal secundária, e
comprometimento da bainha de mielina, dos tratos de substância branca do
sistema trigeminal.

62
4 MATERIAIS E MÉTODOS
4.1 PROCEDIMENTOS CIRÚRGICOS E INJEÇÃO DE NMDA E ENDOTELINA-1
Este estudo utilizou ratos machos adultos da raça Wistar (n= 4-5 por
tempo de sobrevida, sendo dois animais controle
Tabela 1), pesando em média
290 g. Após anestesia profunda mediante injeção intraperitoneal de uma mistura
de Vetanarcol ® (cloridrato de cetamina, 90 mg/Kg
Laboratórios Koning S.A.),
Kenzol ® (cloridrato de xilazina , 10 mg/Kg - Laboratórios Koning S.A.) e de sulfato
de atropina (0,125 mg/Kg), foi aplicada uma anestesia tópica no pavilhão auricular
com pomada de xilocaína (Cristália). De acordo com as normas internacionais, os
reflexos corneano e de retirada da pata foram testados, para garantir que o animal
estivesse devidamente anestesiado. Após este procedimento, os animais foram
mantidos em aparelho estereotáxico (David Kopff) para serem submetidos à
injeção de N-Metil-D-Aspartato (NMDA) ou Endotelina-1 (ET-1) no sub-núcleo
caudal do trato trigeminal.
Com o rato anestesiado e fixado ao aparelho estereotáxico, o campo
cirúrgico foi limpo entre os pavilhões auditivos e os olhos, através de tricotomia e
assepsia com solução de iodo à 2%. Em seguida, uma incisão foi realizada na
linha mediana, no sentido rostro-caudal, até que a calota craniana ficasse à
mostra, após a retirada de toda a gordura subcutânea e epicrânio, empregando
raspagem com uma espátula, até que os pontos bregma e lâmbda fossem
perfeitamente visualizados. O padrão de medidas estereotáxicas foi adotado a

63
partir do ponto brégma, de acordo com coordenadas pré-estabelecidas
(PAXINOS;WATSON, 1997).
Com a finalidade de demarcar o ponto da calota craniana, em que seria
realizada a abertura do crânio para a injeção de NMDA ou ET-1, foram utilizadas
as seguintes coordenadas estereotáxicas, considerando como referência o ponto
Brégma: 6 mm dorso-ventral , -13,8 mm posterior e 2,9 mm lateral. A abertura no
crânio, na região determinada pelas coordenadas estereotáxicas, foi realizada
com o auxílio de broca odontológica carbide PM No. 6 (Meisinger), em baixa
rotação, até a exposição da dura-máter. Esta meninge foi removida do campo
cirúrgico, com subseqüente injeção por pressão de 80 nmol de NMDA ou 40
pmoles de ET-1, durante 3 minutos, no subnúcleo caudal do trigêmio. A
micropipeta de vidro, (diâmetro interno 10-20 m), com a solução a ser injetada foi
mantida por cinco minutos no parênquima neural, antes de sua remoção lenta e
gradual, para evitar refluxo da solução de NMDA, durante a retirada da pipeta. Nos
animais controle foi injetado o mesmo volume de solução salina estéril (1µl),
(Tabela 1). As camadas musculares foram repostas e suturadas. A fim de
identificar o sítio de injeção, uma pequena quantidade de azul de colanil foi
adicionada à solução de NMDA, bem como à solução controle e de ET-1. Após a
sutura dos planos cirúrgicos, os animais foram mantidos com água e comida ad
libitum, durante os tempos de sobrevida de 1 e 7 dias. Este procedimento
experimental configura-se em um método não traumático de lesão do subnúcleo
caudal do trigêmio, onde o componente mecânico da lesão é desprezível. O
referido método foi estabelecido com sucesso em outras regiões do SNC

64
(HUGHES et al., 2003; FERRARI et al., 2004; GOMES-LEAL, 2004).
Todos os experimentos foram
de acordo com as normas sugeridas pela
Society for Neuroscience, National Institutes of Health (NIH, USA) e pelo Comitê
de Ética em Pesquisa Com Animais de Experimentação Animal da Universidade
Federal do Pará (CEPAE-UFPA).(Anexo A).
Tabela 1. Neurotóxicos injetados, concentração, volume e quantidade de animais
por grupo e tempo de sobrevida.
Neurotóxico Concentração Volume Animais de
Experimentação Animais Controle
NMDA 80 nmol 1 µl 4-5 animais 2 animais
ET-1 40 pmoles 1 µl 4-5 animais 2 animais
4.2 PERFUSÃO E ANÁLISE HISTOLÓGICA
Após os tempos de sobrevida de 1 e 7 dias, os animais foram
profundamente anestesiados com uma mistura de Vetanarcol ® (cloridrato de
catamina, 90 mg/Kg Laboratórios Koning S.A.) e Kenzol ® (cloridrato de xilazina,
10 mg/Kg - Laboratórios Koning S.A.), perfundidos através do ventrículo esquerdo
do coração, com solução salina a 0,9% heparinizada, seguida de solução de
Paraformaldeído (Sigma Company, USA) a 4% em tampão fosfato 0,2M (pH 7,4).
Após a dissecção do crânio, o tecido perfundido foi mantido em solução
de sacarose a 25% por 2 horas, em seguida em solução de sacarose a 50 % por
12 horas e finalmente, em solução de sacarose a 100% por 24 horas para
crioproteção. Os blocos de tecido para corte em criostato foram obtidos pela
imersão dos encéfalos, em gel especial para este fim (Jung Tissue Freezing

65
Médium - Leica Microsystems). O tecido imerso em gel foi congelado à 55º C, em
câmara de criostato (Microm HM 505 E), equipado com efeito Peltier. Secções
coronais e longitudinais (dos lados ipsilateral e contralateral ao sítio de injeção)
com 20 m de espessura foram obtidas com auxílio de criostato (Microm HM 505
E) a partir dos blocos feitos pelo procedimento descrito acima. Algumas secções
de 50 m foram coletadas e reagidas pela técnica da reação de NADPH-diaforase
e pela hematoxilina para a visualização da área de lesão. Todas as secções foram
montadas diretamente em lâminas gelatinizadas durante a microtomia. Para
aumento da aderência das secções, as lâminas permaneceram à temperatura
ambiente por no mínimo 24 horas, antes de qualquer outro procedimento
histológico. Após este período, as mesmas foram mantidas à temperatura de 20º
C, aguardando imunocitoquímica ou outra técnica de coloração.
4.3 ANÁLISE HISTOPATOLÓGICA E IMUNOCITOQUÍMICA
4.3.1 Visualização da Área de Lesão
Os sítios de injeção de NMDA e ET-1 foram reconhecidos pela presença
do azul de colanil no tecido, bem como pelo palor, vacuolização e perda de corpos
neuronais, como previamente descrito (DUSART;MARTY;PESCHANSKI, 1991;
GOMES-LEAL, 2002a, 2004). A área de lesão foi visualizada através da coloração
para hematoxilina (H). Como uma técnica adicional, a reação de NADPH-diaforase
foi realizada em algumas secções com espessura de 50 m para a visualização da

66
área de lesão. Segundo Gomes-Leal, com esta técnica de coloração, é possível
delimitar claramente a perda de reatividade da neurópila (azul devido á deposição
de formazan) reativa para NADAPH-diaforase após injeção de NMDA
(comunicação pessoal).
4.3.2 Imunocitoquímica
Com o intuito de avaliar a participação de células gliais e células da
resposta inflamatória nos mecanismos de lesão do trato trigeminal, bem como a
presença de lesão axonal e desmielinização, nos modelos de lesão excitotóxica e
isquêmica, uma série de estudos imunocitoquímicos foi realizada (Tabela 2).
Detalhes destes procedimentos podem ser encontrados nos anexos dete trabalho.
Tabela 2. Anticorpos, fabricantes e diluição utilizados na presente dissertação
Anticorpo Fabricante Diluição ED-1 (monoclonal) Serotec 1:500 MBS-1 (policlonal) CNS Inflammation Group 1:2000 ß-APP (monoclonal) Invitrogen 1:50 MBP (monoclonal) Chemicon international 1:500
4.3.2.1 Marcadores da Resposta Inflamatória
Para identificarmos a presença de macrófagos e microglia ativados, na
área de lesão e em regiões circundjacentes, utilizamos os anticorpos ED1 (1:500,
Serotec), que reconhece um epítopo na membrana de lisossomas no citoplasma
de macrófagos/microglia ativados (DIJKSTRA et al., 1985a).

67
Neutrófilos foram marcados utilizando o anticorpo policlonal MBS-1
(1:2000), o qual reconhece epítopos presentes na maioria da população de
neutrófilos. Este anticorpo foi desenvolvido por Martine Bernardes Silva do CNS,
inflammation Group da Universidade de Southampton e utilizados com sucesso
pelo nosso grupo, em um modelo de lesão aguda da medula espinhal (GOMES-
LEAL, 2002a, 2004, 2005a).
4.3.2.2 Marcadores de Lesão Axonal
Com o intuito de marcarmos lesão axonal, foi utilizada a imunocitoquímica
para a proteína precursora -amilóide ( -APP do inglês
amyloid precursor
protein 1:50, Zymed). Demonstrou-se que esta técnica é bastante sensível para
detectar lesão axonal (GENTLEMAN et al., 1993), tanto experimentalmente como
em estudos de distúrbios patológicos em seres humanos (AHLGREN;LI;OLSSON,
1996; MCKENZIE et al., 1996; AN et al., 1997).
4.3.2.3 Marcadores de Mielina
A fim de investigarmos se os modelos de degeneração neuronal aguda no
núcleo trigeminal caudal induziram alterações na bainha de mielina, utilizamos um
anticorpo monoclonal que reconhece a proteína básica de mielina (MBP, do inglês
myelin basic protein, 1:500, Chemicon International).

68
4.4 ANÁLISE QUALITATIVA
Todas as secções, coradas pelos diferentes métodos histológicos e
imunocitoquímicos, foram inspecionadas em microscópio óptico (ZEISS-AXI
OPHOT). Imagens de secções com campos mais ilustrativos, obtidas de animais
perfudidos 1 e 7 dias após a injeção de NMDA e ET-1, foram obtidas com o uso de
uma câmera digital acoplada ao microscópio óptico (Optiphot-Kikon) e ao
programa de computador Stereo Investigator (Microbrighfield, USA).
5. RESULTADOS
5.1 PADRÕES LESIVOS
No presente estudo, a lesão tecidual induzida pela injeção de 80 nmols
NMDA, ou 40 pmoles de ET-1 foi visualizada em secções coronais, ou
parassagitais coradas pela hematoxilina (Figura 2). A injeção destes neurotóxicos
induziu palor tecidual, lesão neuronal aguda, edema e resposta inflamatória no
local da injeção, qual seja o núcleo espinhal do sistema trigeminal (Figura 2).
Apesar da maior parte das injeções terem sido centradas no subnúcleo caudal,
algumas injeções atingiram os outros subnúcleos, principalmente a zona de
transição com o subnúcleo interpolar. Os animais controle, injetados com solução
salina estéril, não apresentaram este padrão lesivo, às vezes, apresentando lesão
mecânica desprezível (Figuras 2A-B). O sítio de injeção de NMDA ou ET-1 pôde

69
ser evidenciado pela presença do corante azul de colanil, co-injetado com os
neurotóxicos ou com a solução salina estéril (Figura 2).
A injeção de NMDA induziu clara perda tecidual, na substância cinzenta com
aspecto necrótico que pôde ser visualizada 1 dia após a injeção (Figura 2C). Esta
perda tecidual tornou-se mais intensa no tempo de sobrevida de 7dias (Figura 2E).
Não houve comprometimento da substância branca, após a injeção deste agonista
glutamatérgico (Figura 3). Houve pouco recrutamento de macrófagos e neutrófilos
(Figura 2C) nos animais injetados com NMDA, mas não nos animais controle
(Figura 2A), 1 dia após a injeção. No tempo de sobrevida de 7 dias, apesar do
aumento da área necrótica, a resposta inflamatória foi negligenciável, segundo
revelado pela coloração pela hematoxilina (Figura 2E).
O padrão de lesão isquêmica induzida por injeção do peptídeo vasconstritor
ET-1 apresentou aspectos diferenciais, quando comparada à lesão excitotóxica
induzida pela injeção de NMDA. Um dia após a injeção de endotelina-1, a perda
tecidual não foi tão intensa quanto a observada no mesmo tempo de sobrevida
para a injeção de NMDA (Figura 2B). No tempo de sobrevida de 7dias, houve
maior rarefação tecidual, sem a expansão necrótica observada na lesão
excitotóxica (Figura 2E). O centro isquêmico foi geralmente restrito à substância
cinzenta, mas o comprometimento da substância branca também foi encontrado, a
partir de 1 dia após a indução da isquemia. A região de penumbra isquêmica
englobava toda a substância branca, que neste caso correspondia ao trato
trigeminotalâmico.
O padrão de recrutamento de células inflamatórias marcadas pela
hematoxilina foi bem mais intenso nos animais injetados com ET-1 (Figuras 2D;F),

70
do que naqueles injetados com NMDA (Figuras 2C;E). Como confirmado pelos
estudos imunohistoquímicos descritos abaixo, apenas macrófagos foram
recrutados para o epicentro da lesão e regiões circundjacentes, após a injeção de
ET-1, nos dois tempos de sobrevida avaliados (Figuras 2D;F). Não houve
recrutamento de neutrófilos após a injeção de ET-1. Nos tempos de sobrevida
investigados, mas principalmente 7 dias após a injeção de ET-1, um grande
número de células picnóticas foi encontrado na substância branca, próxima ao
sítio de injeção no núcleo espinhal (Figura 4).
5.2 RESPOSTA INFLAMATÓRIA
Como mencionado anteriormente na descrição da histopatologia básica,
houve um padrão diferencial do recrutamento de neutrófilos e macrófagos, nos
modelos excitotóxico e isquêmico. Estes resultados foram confirmados através da
marcação imunohistoquímica com anticorpos específicos (anti-MBS-1 para
neutrófilos e anti-ED1 para macrófagos/microglia), para estas células do sistema
imune inato que participam da resposta inflamatória nos tecidos neurais e não
neurais.

71
Figura 2. Padrão histopatológico revelado pela coloração pela hematoxilina, em
animais controle injetados com solução salina estéril (A-B) e animais injetados com
NMDA (C;E) e endotelina (D;F), nos tempos de sobrevida de 1 e 7 dias. Notar a
presença do corante azul de colanil nos animais controle, sem perda tecidual
significativa (A-B). 1 dia após a injeção de NMDA (C) e endotelina-1 (D) a perda
tecidual torna-se evidente, juntamente com presença de células inflamatórias (setas
em C e D). Notar o maior número de células inflamatórias no subnúcleo caudal dos
animais injetados com ET-1, nos dois tempos de sobrevida (setas em D e F).
Escalas = 50 m.

72
Figura 3. Região necrótica (asterisco) induzida
pela injeção de NMDA na substância cinzenta
(SC) do subnúcleo caudal do núcleo espinhal, 7
dias após a injeção. A substância branca (SB)
está intacta, indicando que o NMDA é um
neurotóxico que lesa preferencialmente a
substância cinzenta. As setas apontam para
macrófagos no epicentro da lesão. Notar o azul
de colanil no centro necrótico. Escalas = 50 m.
Imunocitoquímica para macrófagos.

73
Figura 4. Picnose na substância branca do
tronco cerebral de ratos injetados com
endotelina-1. A região em B é uma ampliação
do campo delimitado pelo retângulo em A. As
setas apontam para células picnóticas em B.
Escalas: A = 50 m; B = 100
m.Hematoxilina.

74
A injeção de NMDA no núcleo espinhal do sistema trigeminal induziu
pouco recrutamento de neutrófilos no tempo de sobrevida de 1 dia (Figura 5C),
mas não induziu nos animais controle injetados com salina estéril (Figura 5A).
Estas células inflamatórias não foram encontradas no parênquima neural 7 dias
após a injeção (Figura 5E), o que está de acordo com o relato de investigações
prévias em outras regiões do SNC (BOLTON;PERRY, 1998; SCHNELL et al.,
1999; GOMES-LEAL, 2002b, 2005a). Também houve recrutamento de poucos
macrófagos, 1 (Figura 6C) e 7 (Figura 6E) dias após a injeção de NMDA no núcleo
espinhal de ratos. Ambos os tipos de leucócitos foram restritos às regiões de
perda tecidual na substância cinzenta. Estas células inflamatórias não foram
encontradas na substância branca (não ilustrado).
A lesão isquêmica no núcleo espinhal, induzida por injeção de ET-1 revelou
um padrão bem mais intenso de resposta inflamatória dominado pelo
recrutamento e ativação de macrófagos/microglia (células ED1 +), nos dois
tempos de sobrevida investigados (Figuras 2D;F, Figura 6D;F). Um grande
número de células ED1+ foi encontrado na substância cinzenta do núcleo espinhal
1 (Figura 6D) e 7 dias (Figura 6F) após a injeção de endotelina-1. As células
ED1+ também foram encontradas na substância branca, nos dois tempos de
sobrevida investigados, mas principalmente 7 dias após a indução da lesão
isquêmica (Figura 7). Não houve recrutamento de neutrófilos após a injeção de
endotelina-1 no núcleo espinhal do sistema trigeminal no presente estudo, com
exceção de um animal, em que alguns neutrófilos marcados foram encontrados
em uma região focal distante do sítio de injeção (não ilustrado).

75
Figura 5. Recrutamento de neutrófilos nos animais injetados com NMDA e
endotelina-1, 1 (C-D) e 7 dias (E-F) após a injeção. Nos animais controle
injetados com solução salina estéril não houve recrutamento de neutrófilos (A-B).
Poucos neutrófilos foram encontrados nos animais injetados com NMDA,
apenas 1 dia após a injeção (setas em C). Não houve recrutamento de
neutrófilos nos animais injetados com endotelina-1 em nenhum tempo de
sobrevida. Notar o azul de colanil em A e D. Escalas = 50 m.

76
Figura 6. Recrutamento de macrófagos/microglia ativados após injeção de NMDA
e endodelina-1 no núcleo espinhal de ratos adultos. Animais controle injetados com
solução salina estéril (A-B). Houve recrutamento mais intenso de
macrófagos/microglia ativados nos animais injetados com ET-1 (D;F) do que nos
animais injetados com NMDA (C;E). As setas apontam para macrófagos/microglia
ED1+. No o corante azul de colanil em E. Escalas = 50 m.

77
5.3 LESÃO AXONAL
Na presente dissertação, axônios lesados foram marcados através da
imunohistoquímica para a proteína precursora -amilóide ( -APP, do inglês -
amyloid precursor protein), um marcador clássico de lesão axonal. Microinjeções
de NMDA e ET-1 induziram o aparecimento de perfis axonais arredondados -
APP+ na substância cinzenta do núcleo espinhal, 1 e 7 dias após a injeção dos
neurotóxicos em questão (Figura 8), mas não nos animais controle (Figura 8-AB).
No tempo de sobrevida 1 dia, o número de perfis -APP+ foi comparável para os
animais injetados com NMDA e endotelina-1 (Figura 8C-D). No entanto, no tempo
de sobrevida de 7 dias, o número destes perfis -APP+ foi claramente maior no
núcleo espinhal de animais injetados com ET-1, do que naqueles injetados com
NMDA (Figura 8E-F).
Na substância branca dos animais injetados com NMDA, não houve
marcação significativa pela imunohistoquímica para o -APP 1 dia após a injeção
(Figura 9A-B). Sete dias após a injeção de NMDA, alguns perfis -APP+
arredondados foram encontrados na substância branca adjacente ao epicentro
necrótico da substância cinzenta (Figura 9C-D).
Na substância branca dos animais injetados com ET-1, os perfis
arredondados -APP+ foram mais freqüentes e encontrados a partir de 1 dia, após
a indução da isquemia (Figura 10C-D). Houve aumento do número e do tamanho
destas estruturas arredondadas positivas para o -APP no tempo de sobrevida de
7 dias (Figura 10D).

78
Figura 7. Presença de macrófagos/microglia ativados na substância branca do
sistema trigeminal dos animais injetados com endotelina-1, 1 dia (A-B) e 7 dias (C-D)
após a injeção. As setas apontam para macrófagos/microglia ativados ED1+. B e D
são ampliações de A e C. Escalas: A;C = 100 m; B;D = 20 m.

79
Figura 8. Lesão axonal na substância cinzenta do núcleo espinhal do sistema
trigeminal. Animais controle injetados com salina estéril (A-B). Animais injetados
com NMDA e endotelina-1, 1 (C-D) e 7 dias (E-F) após a injeção. As setas
apontam para axônios lesados marcados pela imunohistoquímica para o -APP.
Notar a maior quantidade de axônios lesados nos animais injetados com
endotelina-1 (D;F) do que nos animais injetados com NMDA (C;E),
principalmente no tempo de sobrevida de 7 dias. Escalas = 50 m.

80
Figura 9. Lesão axonal na substância branca do sistema trigeminal dos animais
injetados com NMDA. Perfis axonais marcados pela imunohistoquímica para o -
APP (setas em A-D), 1 (A-B) e 7 dias (C-D) após a injeção de NMDA. Escalas:
A;C = 100 m; B;D = 20 m.

81
5.4 ALTERAÇÕES NA BAINHA DE MIELINA
Injeção de NMDA e ET-1 no núcleo espinhal do sistema trigeminal induziu
alterações no padrão de marcação para MBP (MBP, do inglês myelin basic
protein) em escalas temporais diferentes. Um dia após a injeção de NMDA não
houve alteração aparente no padrão de marcação revelado pelo anticorpo anti-
MBP (Figura 11C), o qual foi comparável ao dos animais controle (Figura 11A-B).
Para este mesmo tempo de sobrevida, houve perda significativa da marcação para
MBP nos animais experimentais injetados com endotelina-1 (Figura 11D). Nestes
mesmos animais, a substância branca contralateral apresentou um padrão de
marcação normal (Figura 11A).
Sete dias após a injeção de NMDA, em torno do epicentro necrótico na
substância cinzenta, houve intensa perda da marcação para MBP, sem
vacuolização significativa da substância branca (Figura 11E). Nos animais
injetados com ET-1, a perda de marcação para MBP foi evidente, o que foi
concomitante com intensa vacuolização da substância branca (Figuras 11F e 12).

82
Figura 10. Lesão axonal na substância branca do sistema trigeminal dos animais
injetados com ET-1. Perfis axonais marcados pela imunohistoquímica para o -
APP (setas em A-D), 1 (A-B) e 7 dias (C-D) após a injeção de endotelina-1. B e D
são ampliações de A e C. SC = substância cinzenta; SB = substância branca.
Escalas: A;C = 100 m; B;D = 20 m.

83
Figura 11. Desmielinização na substância branca do sistema trigeminal após
a injeção de NMDA e endotelina-1 no núcleo espinhal e ratos adultos. Animais
controle: lado contralateral do animal injetado com ET-1 ilustrado em D (A);
animal injetado com salina estéril (B). Notar conspícua desmielinização no trato
trigeminotalâmico 1 dia após a injeção de endotelina-1 (D), mas não nos
animais injetados com NMDA (C). Os animais injetados com NMDA
apresentam desmielinização no tempo de sobrevida de 7 dias (E). Neste
tempo de sobrevida, ocorre degradação da bainha de mielina nos animais
injetados com endotelina-1 (F). A seta aponta para um microcisto em F. SC =
substância cinzenta; SB = substância branca. Escalas = 50 m.

84
Figura 12. Formação de microcistos na substância
branca dos animais injetados com ET-1, 7 dias após a
injeção. As setas apontam para microcistos na
substância branca. Escalas: A = 50 m; B = 20 m.

85
6 DISCUSSÃO
6.1 CONSIDERAÇÕES TÉCNICAS
No presente estudo, injetou-se ET-1 e NMDA, no núcleo espinhal do
sistema trigeminal de ratos adultos, para induzir degeneração neuronal aguda e
resposta inflamatória nesta região a fim de avaliar os padrões de degeneração
secundária da substância branca no sistema neural em questão. Um aspecto
original da presente dissertação, foi a comparação implementada entre os eventos
histopatológicos induzidos por eventos isquêmicos e excitotóxicos, em um mesmo
paradigma experimental, o que era inexistente da literatura. Este fato torna-se
mais relevante, considerando-se que eventos isquêmicos e excitotóxicos são
eventos fisiopatológicos importantes de doenças neurodegenerativas agudas, tais como AVC e
trauma (SCHWAB;BARTHOLDI, 1996; DIRNAGL;IADECOLA;MOSKOWITZ, 1999; LO, 2003;
MERGENTHALER;DIRNAGL;MEISEL, 2004).
O principal argumento para o uso da injeção de NMDA para induzir
degeneração neuronal aguda experimental encontra-se no fato de que a injeção
deste neurotóxico induz alterações histopatológicas similares às encontradas
durante trauma do cérebro e da medula espinhal de seres humanos
(KAO;CHANG;BLOODWORTH, 1977; WOZNIEWICZ et al., 1983; FOX et al., 1988;
TATOR;FEHLINGS, 1991; DAVIS;KHANGURE, 1994; SCHWAB;BARTHOLDI, 1996;
TATOR;KOYANAGI, 1997). Estas alterações patológicas incluem necrose progressiva
no eixo rostro-caudal com cavitação hemorrágica e seringomielia
(KAO;CHANG;BLOODWORTH, 1977; WOZNIEWICZ et al., 1983; FOX et al.,
1988; DAVIS;KHANGURE, 1994; TATOR;KOYANAGI, 1997), ativação glial e

86
lesão axonal (SCHWAB;BARTHOLDI, 1996; LOPACHIN;LEHNING, 1997; STYS,
1998; CORNISH et al., 2000). Inexistem estudos que tenham investigado os
padrões histopatológicos induzidos por injeção de NMDA no sistema trigeminal.
Demonstrou-se experimentalmente que microinjeções de ET-1 constituem-se
em um excelente modelo de isquemia focal transitória (CLARKE et al., 1989;
FUXE et al., 1989; AGNATI et al., 1991; HUGHES et al., 2003). Injeções de
pequenas quantidades deste neurotóxico induzem redução de mais de 60 % do
fluxo sanguíneo na região injetada (HUGHES et al., 2003). Esta técnica de
indução de lesão isquêmica focal é bem menos trabalhosa do que os métodos
clássicos de clampeamento de vasos sanguíneos específicos, como a artéria
cerebral média, onde a cirurgia possui considerável grau de complexidade. Além
disso, a injeção de ET-1 e NMDA através de microcapilares de vidro com diâmetro
interno de ponta variando entre 20-30 m faz com que os compomenentes
mecânicos sejam desprezíveis, o que permite diferenciar claramente eventos
lesivos primários e secundários.
No presente modelo experimental, microinjeções de NMDA e ET-1 induziram
eventos histopatológicos diferenciais, no que concerne aos padrões necróticos,
inflamatórios e de lesão do cilindro axonal e da bainha de mielina. A injeção do
agonista glutamatérgico NMDA induziu necrose mais intensa da substância
cinzenta, sem indução de perda tecidual na substância branca. A resposta
inflamatória induzida pela injeção deste neurotóxico foi moderada, com pouco
recrutamento de neutrófilos e macrófagos para o parênquima neural. Houve lesão
axonal na substância cinzenta após injeção de NMDA, com pouco

87
comprometimento axonal na substância branca. A mielina foi afetada no tempo de
sobrevida mais tardio (sete dias) no modelo de lesão excitotóxica. Por outro lado,
apesar da injeção de ET-1 induzir perda tecidual menos intensa do que a induzida
pela injeção de NMDA, este peptídeo vasoconstritor induziu resposta inflamatória
intensa dominada por recrutamento de macrófagos, mais lesão axonal e perda de
mielina, a partir de tempos de sobrevida mais precoces. Além disso, a lesão
isquêmica induziu o aparecimento de um grande número de células picnóticas, o
que sugere apoptose.
6.2 PERDA TECIDUAL EM DOENÇAS NEURODEGENERATIVAS AGUDAS
Perda tecidual com necrose progressiva é um evento final, após lesão
experimental do cérebro e medula espinhal, independente do modelo lesivo
utilizado (SCHWAB;BARTHOLDI, 1996; LIU et al., 1997;
GUTH;ZHANG;STEWARD, 1999). Achados histopatológicos semelhantes são
encontrados no SNC de seres humanos, após trauma e AVC
(KAO;CHANG;BLOODWORTH, 1977; WOZNIEWICZ et al., 1983; FOX et al.,
1988; DAVIS;KHANGURE, 1994; TATOR;KOYANAGI, 1997;
MERGENTHALER;DIRNAGL;MEISEL, 2004). Estas alterações patológicas
incluem necrose na direção rostro caudal com cavitação hemorrágica e
seringomielia (KAO;CHANG;BLOODWORTH, 1977; WOZNIEWICZ et al., 1983;
FOX et al., 1988; DAVIS;KHANGURE, 1994; TATOR;KOYANAGI, 1997), ativação
glial e lesão axonal (SCHWAB;BARTHOLDI, 1996; LOPACHIN;LEHNING, 1997;
STYS, 1998; CORNISH et al., 2000).

88
O tratamento da necrose hemorrágica primária não influencia no prognóstico
da lesão, no entanto, em doenças neurodegenerativas agudas do SNC, a lesão
primária, seja na substância branca ou na substância cinzenta, expande-se com o
tempo, o que aumenta o déficit funcional principalmente através do aumento do
número de axônios lesados ou degeneração de oligodendrócitos
(TATOR;FEHLINGS, 1991; SCHWAB;BARTHOLDI, 1996; LIU et al., 1997;
TATOR;KOYANAGI, 1997). Este fenômeno é conhecido como degeneração
neuronal secundária.
Sabe-se que na medula espinhal, a lesão necrótica induzida por contusão
ou compressão, que é inicialmente restrita à substância cinzenta, pode atingir a
substância branca em torno de 8h (SCHWAB;BARTHOLDI, 1996). Na presente
dissertação, apesar da maioria das injeções de ET-1 e NMDA terem sido restritas
aos subnúcleos do núcleo espinhal, principalmente o subnúcleo caudal, observou-
se comprometimento da substância branca, incluindo lesão axonal, perda da
reatividade para a proteína básica de mielina e vacuolização. Como previamente
relatado, estes efeitos foram mais intensos no modelo isquêmico do que no
modelo excitotóxico. Estes resultados sugerem que a substância branca é afetada
por mecanismos secundários, nos dois modelos experimentais, mas
principalmente no modelo isquêmico. Estes achados estão de acordo com outras
investigações que sugerem que a substância branca do SNC é altamente
vulnerável à isquemia (PANTONI;GARCIA;GUTIERREZ, 1996;
DEWAR;YAM;MCCULLOCH, 1999; PETTY;WETTSTEIN, 1999;
KANELLOPOULOS et al., 2000). Como o acúmulo de aminoácidos excitatórios é
um evento secundário ao processo isquêmico (LO, 2003;

89
MERGENTHALER;DIRNAGL;MEISEL, 2004), e, considerando-se que o modelo
excitotóxico pela injeção de NMDA, usado na presente dissertação simula este
evento patológico, os resultados da presente dissertação mostram que outros
eventos patológicos além da excitotoxicidade, são responsáveis pelos eventos
lesivos durante a degeneração neuronal secundária subjacentes à isquemia.
O fenômeno de degeneração neuronal secundária foi descrito originalmente
por Allen em 1911, em seus estudos na medula espinhal do gato (apud Tator &
Felings, 1991) e consiste de um grupo de eventos destrutivos, que podem afetar
células e axônios que foram apenas marginalmente afetados pelo evento lesivo
primário, ou mesmo que não foram afetados (TATOR;FEHLINGS, 1991;
SCHWAB;BARTHOLDI, 1996; YAMAURA et al., 2002; LO, 2003;
MERGENTHALER;DIRNAGL;MEISEL, 2004). Durante este fenômeno, neurônios
e axônios não afetados pelo estímulo lesivo primário podem degenerar por
mecanimos ditos secundários se não forem protegidos por intervenção terapêutica
(TATOR;FEHLINGS, 1991; SCHWAB;BARTHOLDI, 1996). Acredita-se que
inúmeros mecanimos sejam envolvidos no fenômeno de degeneração secundária
no SNC, mas a inflamação aguda, lesão vascular, excitotoxicidade mediada pelo
glutamato, estresse oxidativo, acidose metabólica, despolarização peri-infarto
parecem ser os principais fatores envolvidos (TATOR;FEHLINGS, 1991;
SCHWAB;BARTHOLDI, 1996; YAMAURA et al., 2002; LO, 2003;
MERGENTHALER;DIRNAGL;MEISEL, 2004).

90
6.3 PADRÃO DIFERENCIAL DA RESPOSTA INFLAMATÓRIA EM MODELOS
EXCITOTÓXICOS E ISQUÊMICOS
No presente estudo, avaliou-se os padrões de recrutamento de neutrófilos e
macrófagos, em um modelo excitotóxico (injeção de NMDA) e um modelo
isquêmico (injeção de ET-1), em um tempo precoce (1dia) e no tempo de
sobrevida mais tardio (7dias). Os resultados mostraram um padrão diferencial de
recrutamento destes leucócitos. No modelo excitotóxico, houve recrutamento de
poucos neutrófilos, no tempo de sobrevida de 1 dia e de pouquíssimos
macrófagos no tempo de sobrevida de 7 dias. Estas células inflamatórias foram
basicamente restritas à substância cinzenta do núcleo espinhal. No modelo
isquêmico induzido por injeção focal de ET-1, a resposta inflamatória foi
caracterizada por recrutamento intenso de macrófagos, tanto para a substância
cinzenta como para a substância branca. Não houve recrutamento de neutrófilos
para o parênquima neural induzido por injeção de endotelina-1, no modelo
experimental proposto. O aparecimento de células ED1 +, na substância branca
ocorreu no tempo de sobrevida de 7 dias.
A escala temporal do recrutamento de células inflamatórias nos modelos
experimentais investigados segue o já relatado na literatura, em modelos
traumáticos e não traumáticos de lesão do cérebro (BOLTON;PERRY, 1998) e
medula espinhal (SCHNELL et al., 1999; GOMES-LEAL, 2002b, 2004, 2005a). No
cérebro e na medula espinhal foram estabelecidos que o pico para o recrutamento
de neutrófilos ocorre 1 dia após a injeção de NMDA, enquanto o pico para o
recrutamento de macrofágos ocorre a partir de 3 dias após a injeção deste

91
neurotóxico (BOLTON;PERRY, 1998; SCHNELL et al., 1999; GOMES-LEAL,
2002b, 2004, 2005a). Sabe-se também que a intensidade da resposta inflamatória
depende do compartimento do SNC analisado, por exemplo, sendo mais intensa
na medula espinhal do que no cérebro (SCHNELL et al., 1999). No tronco
cerebral, de acordo com os resultados da presente dissertação para o modelo
excitotóxico, a quantidade de células inflamatórias recrutadas foi aparentemente
menor do que as relatada para outras regiões do SNC, por exemplo, medula
espinhal e núcleos da base (BOLTON;PERRY, 1998; SCHNELL et al., 1999;
GOMES-LEAL, 2002b, 2004, 2005a). Isto pode significar que, dependendo do
compartimento do SNC, a regulação da resposta inflamatória pode ser mais ou
menos intensa, provavelmente para a preservação da integridade do tecido neural.
Isto seria oportuno no tronco cerebral, onde existem núcleos neuronais que
constituem-se em centros respiratórios. Lesão nestes centros neurais vitais podem
induzir coma e morte rapidamente (MEDANA et al., 2002; MEDANA;ESIRI, 2003).
O fato de ter havido recrutamento de neutrófilos, mesmo que moderado,
apenas no modelo excitotóxico, mas não no modelo isquêmico, pode ter relação
com o comprometimento ou não da barreira hematoencefálica. Hughes et al.,
(2003) relataram que microinjeções ET-1 no estriato, substância cinzenta e
substância branca cortical do cérebro de ratos adultos induzem resposta
inflamatória caracterizada por recrutamento de macrófagos, principalmente 3 dias
após a indução isquêmica. Estes autores também relataram ausência de
recrutamento de neutrófilos, no modelo experimental utilizado. Portanto,
neutrófilos não são recrutados no modelo de isquemia focal induzido por injeção
de ET-1. No modelo utilizado por Hughes et al. (2003), não houve

92
comprometimento da barreira hematoencefálica, como avaliado por um ensaio
histoquímico específico para estes fins utilizando injeção da peroxidase de raiz
forte (HRP). Portanto, o comprometimento da barreira hematoencefálica é um
evento crucial para que neutrófilos, mas não monócitos, cheguem ao parênquima
neural. Isto foi demonstrado claramente por Schnell et al., (1999). Neste estudo, o
recrutamento mais intenso de neutrófilos foi concomitante com um
comprometimento mais conspícuo da barreira hematoencefálica. A ausência de
recrutamento de neutrófilos no modelo isquêmico utilizado no presente estudo
mostra que estas células não participam do processo lesivo, por exemplo,
desmielinização, vacuolização e lesão axonal.
Monócitos têm livre acesso ao SNC, mesmo através de uma barreira
hematoencefálica intacta (PERRY;GORDON, 1991). Estas células transformam-se
nos macrófagos residentes do SNC na vida adulta, as células microgliais
(PERRY;GORDON, 1991). Durante lesão aguda do SNC, tais como durante
trauma e AVC, monócitos derivados da corrente sanguínea alcançam o
parênquima neural lesado e transformam-se em macrófagos ativados (POPOVICH
et al., 1999; POPOVICH;HICKEY, 2001; POPOVICH et al., 2002; GOMES-LEAL,
2005a). Na presente dissertação, a resposta inflamatória do modelo isquêmico foi
dominada por um intenso recrutamento de macrófagos/microglia para o
parênquima lesado. Estes resultados mostram que os mecanismos moleculares,
que induzem a ativação de macrófagos/microglia no modelo isquêmico diferem
daqueles presentes no modelo excitotóxico, onde o recrutamento de macrófagos
foi escasso. Após lesão mecânica e excitotóxica do cérebro e da medula espinhal,
macrófagos são recrutados para o tecido neural lesado, principalmente nos

93
tempos de sobrevida mais tardios (BOLTON;PERRY, 1998; SCHNELL et al.,
1999; GOMES-LEAL, 2002a, 2005a). Os resultados da presente dissertação
mostraram o aumento do número de células ED1+ (macrófagos/microglia
ativados), principalmente no tempo de sobrevida de 7 dias, o que está de acordo
com os estudos anteriormente relatados. A presente investigação também
mostrou, entre as duas técnicas comparadas, um recrutamento diferencial de
células ED1+, para a substância branca no tempo de sobrevida mais tardio, após
a indução da lesão isquêmica em relação a trabalhos citados na literatura. A
imunohistoquímica utilizando o anticorpo anti-ED1 marca macrófagos/microglia
ativados, mas não é capaz de diferenciar os macrófagos derivados da microglia,
daqueles macrófagos hematogênicos oriundos de monócitos derivados da
corrente sanguínea. Estes dois subgrupos de macrófagos podem contribuir
diferencialmente para o processo lesivo (POPOVICH;HICKEY, 2001). Popovich e
Hickey, (2001) utilizaram ratos quiméricos de diferentes linhagens, que tiveram a
medula óssea destruída por radiação para a caracterização da participação dos
dois subgrupos de macrófagos mencionados após contusão experimental da
medula espinhal de ratos. Os animais quiméricos tiveram seus macrófagos
derivados da corrente sanguínea, marcados por anticorpos específicos que os
diferenciavam dos macrófagos ativados derivados da microglia residente. Estes
autores demonstraram que a cena lesiva é inicialmente dominada por macrófagos
derivados de células microgliais, que depois são suplantados por macrófagos
hematogênicos. Ainda segundo estes autores, os macrófagos hematogênicos
ficavam geralmente restritos à substância cinzenta da medula espinhal, enquanto
que os macrófagos derivados da microglia residente possuíam uma distribuição

94
mais ampla, inclusive na substância branca. No presente estudo, o aparecimento
de células ED1+ na substância branca em um tempo de sobrevida mais tardio
pode significar o aumento da atividade microglial, ou mesmo a maior infiltração de
macrófagos hematogênicos na substância branca.
Existem fortes evidências na literatura de que células da resposta
inflamatória ao desempenharem seus papéis fisiológicos como células
fagocitárias, removendo detritos celulares e outros produtos da lesão, podem
também agravar o fenômeno lesivo (BLIGHT, 1985; BLIGHT;DECRESCITO, 1986;
BLIGHT, 1991, 1992, 1993; GIULIAN et al., 1993; BLIGHT, 1994; POPOVICH et
al., 1994; BLIGHT et al., 1995; POPOVICH et al., 1996;
POPOVICH;STOKES;WHITACRE, 1996; POPOVICH;WEI;STOKES, 1997;
POPOVICH;YU;WHITACRE, 1997; POPOVICH et al., 1999; POPOVICH, 2000;
BLIGHT;ZIMBER, 2001; HERMANN et al., 2001; POPOVICH;HICKEY, 2001;
POPOVICH et al., 2001). Os mecanismos específicos pelos quais células da
resposta inflamatória podem agravar o processo lesivo após lesão do SNC não
são totalmente estabelecidos, mas inúmeras possibilidades têm sido aventadas na
literatura. Acredita-se que macrófagos e microglia ativados podem contribuir para
exacerbação da lesão no SNC, como uma espécie de efeito colateral de suas
atividades fisiológicas (BLIGHT, 1985; GIULIAN et al., 1989;
GIULIAN;VACA;NOONAN, 1990; BLIGHT, 1992, 1994; POPOVICH;WEI;STOKES,
1997; POPOVICH et al., 1999). A depleção do recrutamento de macrófagos com
pó de sílica (BLIGHT, 1985, 1992, 1994) ou clodronato (POPOVICH et al., 1999)
diminui a área de lesão secundária e melhora o prognóstico de recuperação
motora, após lesão da medula espinhal em roedores. A Injeção intraperitoneal de

95
pó de sílica induz uma forte resposta inflamatória local, tornando monócitos menos
disponíveis em outros tecidos (BLIGHT, 1994; FISCHER;REHN;BRUCH, 1996;
IYER et al., 1996). Blight propôs inicialmente, que a atividade de macrófagos
poderia ser importante para a exacerbação de lesão neuronal, após lesão da
medula espinhal (BLIGHT, 1985;1992). Blight (1994) testou esta hipótese através
de injeções intraperitoneais de pó de sílica, em porquinhos da índia submetidos a
contusão da ME. Este autor relatou que o referido procedimento resultou em
redução da perda de axônios mielinizados e diminuição da hipervascularização da
area lesada. Blight concluiu que macrófagos poderiam causar lesão direta a
axônios ou vasos sanguíneos nas condições experimais analisadas.
Popovich et al., (1999) relataram outra forte evidência para a participação
de macrófagos em mecanismos lesivos. Estes autores efetuaram injeção
intravenosa de lipossomas de clodronato, em ratos submetidos à lesão aguda da
medula espinhal. Animais que receberam injeção de clodronato apresentaram
melhora das funções motoras, acompanhada por diminuição da cavitação no eixo
rostrocaudal, além de regeneração axonal. Além disso, tem sido relatado que a
injeção de altas doses do anti-inflamatório metilprednisolona produz diminuição da
área de lesão secundária e induz melhora da recuperação motora em ratos
submetidos a LME (BRACKEN, 1990, 2000; BRACKEN et al., 2000; BRACKEN,
2001b; 2001a, 2002; BRACKEN;HOLFORD, 2002). Esta droga é atualmente
usada para o tratamento de seres humanos que sofreram lesão aguda da ME
(BRACKEN et al., 2000; BRACKEN, 2002; BRACKEN;HOLFORD, 2002). Os
mecanismos pelos quais a metilpredinisolona diminue a área de lesão secundária
podem ser relacionados aos seus efeitos anti-inflamatórios, por exemplo, a

96
diminuição da liberação de FNT-
(XU et al., 1992; XU et al., 1998) ou mesmo a
uma função anti-oxidante (KOC et al., 1999; BLIGHT;ZIMBER, 2001). O TNF-
é
uma citocina pro-inflamatória considerada importante para os mecanismos lesivos,
relacionados à resposta inflamatória e degeneração secundária, após lesão do
cérebro e da medula espinhal (HERMANN et al., 2001). Hermann et al., (2001)
relataram que injeções de pequenas doses de FNT-
(60pg) ou cainato não
induzem ou induzem lesões diminutas na SC da ME, mas que injeções
concomitantes de TNF-
(60pg) e cainato induzem lesões significativamente
maiores. Estes autores sugeriram que o TNF-
poderia potencializar os efeitos da
excitotoxicidade mediada por receptores AMPA, talvez induzindo liberação de
glutamato por microglia ou astrócitos.
Finalmente, existem evidências in vitro a favor da participação do NO
sintetizado pela SON imunológica (NOSi), presente em células gliais na expansão
da lesão necrótica durante lesão do SNC (BAL-PRICE;BROWN, 2001). Estes
estudos mostram que o NO liberado por células gliais, durante a inflamação aguda
pode ser responsável por cerca de 70-75 % da morte celular, por necrose que
ocorre no meio de cultura.
Em outro estudo, Popovich et al., (2002) realizaram microinjeções do ativador
microglial zymosan, com esta abordagem demonstraram lesão axonal e
desmielinização seletiva no sítio de injeção.
Todos os estudos, relatados acima mostram o potencial para células
inflamatórias causarem perda tecidual após lesão aguda do SNC, como a induzida
no presente modelo experimental. No entanto, é válido mencionar que existem

97
evidências experimentais de que o processo inflamatório pode possuir efeito
benéfico, após lesão do SNC (LAZAROV-SPIEGLER et al., 1998; RAPALINO et
al., 1998; KNOLLER, 2005; SCHWARTZ;YOLES, 2005). Recentemente, relatou-
se um ensaio clínico de fase I, no qual o transplante autólogo de macrófagos foi
bem tolerado, por alguns pacientes humanos com lesão aguda da medula
espinhal. Estas pessoas apresentaram melhor prognóstico clínico, do que o grupo
que não foi submetido ao tratamento (KNOLLER, 2005).
6.4 INJEÇÃO DE ENDOTELINA-1 INDUZ MAIS LESÃO AXONAL DO QUE
INJEÇÃO DE NMDA
No presente modelo experimental, o modelo de isquemia focal por injeção de
ET-1 induziu mais lesão axonal, tanto na substância branca como na substância
cinzenta, do que a injeção de NMDA. Sabe-se que os déficits energéticos,
concomitantemente com mecanismos inflamatórios, excitotóxicos e cidose
metabólica contribuem para a perda tecidual após isquemia (DIRNAGL;
IADECOLA; MOSKOWITZ,1999; MERGENTHALER; DIRNAGL; MEISEL, 2004).
Poucos estudos investigaram os padrões de lesão axonal após lesão aguda do
SNC. Gomes-Leal et al., (2005) relataram lesão axonal na substância cinzenta
ventral e dorsal, após microinjeções de NMDA no corno ventral da medula
espinhal de ratos. Neste estudo, houve correlação entre a ativação de
macrófagos/microglia e o número de axônios lesados, três dias após a indução da
lesão primária.
No presente estudo, a injeção de NMDA no núcleo espinhal do sistema

98
trigeminal induziu lesão axonal, principalmente na substância cinzenta. Sabe-se
que a lesão neuronal aguda excitotóxica induz degeneração de corpos neuronais,
com subsequente ativação astrocitária, microglial, inflamação, apoptose e
formação de radicais livres (GOMES-LEAL, 2002a, 2004, 2005a). Estes eventos
podem induzir lesão axonal direta. Existem evidências experimentais que ativação
de macrófagos e microglia podem contribuir diretamente para lesão axonal,
através da liberação de citocinas inflamatórias (por exemplo, TNF- , IL-1, IL-6),
proteases e NO (POPOVICH et al., 2002; GOMES-LEAL, 2005a; HENDRIKS et
al., 2005).
Um papel direto do NMDA nos mecanismos de lesão axonal, na substância
cinzenta no presente modelo experimental seria muito improvável, quando se
considera que não existem receptores de NMDA em células gliais, mielina e
axônios (AGRAWAL;FEHLINGS, 1996; 1997; GARCIA_BARCINA;MATUTE, 1998;
MATUTE, 1998). O TNF- pode contribuir para a lesão axonal, através de efeitos
lesivos primários em oligodendrócitos (HENDRIKS et al., 2005). O glutamato
liberado de neurônios ou células gliais sofrendo degeneração na substância
cinzenta ou de outras fontes, também poderia contribuir para lesão axonal.
No presente modelo de lesão excitotóxica, os efeitos lesivos na substância
branca foram desprezíveis, quando comparados aos induzidos por injeção de ET-
1. Estes resultados diferem dos relatados por Gomes-Leal et al., (2005), que
encontraram quantidade significativa de axônios lesados nos tratos de substância
branca da medula espinhal, após lesão excitotóxica induzida por injeção de NMDA
no corno ventral da medula espinhal de ratos. A razão destes resultados
discrepantes não é clara, mas pode estar relacionada a fatores anatômicos e

99
fisiológicos inerentes à constituição destes diferentes compartimentos do SNC.
Por exemplo, a medula espinhal possui tratos de substância branca mais
exuberantes do que os encontrados no tronco cerebral, que certamente são mais
vulneráveis a processos lesivos. Na medula espinhal, diferentes fascículos de
substância branca podem ser lesados por diferentes mecanismos lesivos
(ZHANG;KREBS;GUTH, 1997). Estes mesmos mecanismos podem não ser
atuantes em outras regiões do SNC, como no trato trigeminotalâmico do sistema
trigeminal.
A lesão isquêmica induzida por injeção de ET-1 induziu, comparativamente,
mais lesão axonal do que a lesão excitotóxica induzida pela injeção de NMDA,
tanto na substância branca, como na substância cinzenta. Sabe-se que o tecido
neural é altamente vulnerável à isquemia, principalmente a substância branca
(PANTONI;GARCIA;GUTIERREZ, 1996; DE_KEYSER;SULTER;LUITEN, 1999;
DEWAR;YAM;MCCULLOCH, 1999; PETTY; WETTSTEIN,
1999;KANELLOPOULOS et al., 2000).
Existem diversos possíveis mecanismos que poderiam ser responsáveis pela
lesão axonal durante doenças neurodegenerativas agudas (STYS, 2005).
Recentemente, Stys e colabaoradores (LI et al., 1999b; LI;JIANG;STYS, 2000;
LI;STYS, 2000), usando substância branca isolada da medula espinhal in vitro,
relataram evidências para um papel de receptores do tipo não-NMDA, nos
mecanismos lesivos após trauma e isquemia. No primeiro paradigma experimental
estudado (LI;STYS, 2000), a infusão dos agonistas glutamatérgicos AMPA e
cainato na substância branca dorsal isolada da medula espinhal resultou em
alterações de oligodendrócitos, astrócitos e particularmente da bainha de mielina.

100
Nestes estudos, não foram encontradas evidências de lesão do cilindro axonal,
como evidenciado pela imunocitoquímica para degradação da espectrina por
proteases, bem como pela presença de GluR2/3 e GluR4 em células gliais. Em
um outro paradigma experimental utilizando o mesmo modelo in vitro, a
substância branca isolada de ratos foi submetida a 60 minutos de anóxia ou à
compressão durante 15 segundos (LI et al., 1999b). Tanto antagonistas seletivos
de receptores do tipo AMPA/cainato, como a inibição to transporte de glutamato
dependente de Na+, protegeram a SB medular dos mecanismos lesivos induzidos
por anóxia e trauma. Estudos posteriores pelo mesmo grupo (LI;STYS, 2001)
demonstraram que a liberação endógena de glutamato pode ocorrer pela reversão
dos gradientes de Na+ e K++, através do axolema. De acordo com estes autores, a
inibição da ATPase de Na+ e K++ e despolarização induzem liberação de
glutamato, através da reversão da atividade dos transportadores de glutamato
dependente de Na+. O glutamato liberado de fontes intracelulares agiria em
receptores do tipo AMPA/cainato induzindo lesão excitotóxica, principalmente em
oligodendrócitos e mielina. Nestes estudos, a principal fonte de glutatamo foi o
cilindro axonal. Outros estudos sugerem que a injeção do bloqueador de canal de
Na+ -TTX- diminui significativamente a lesão axonal, após contusão da ME,
mostrando que ativação de canal de Na+ pode ser um importante evento
subjacente aos mecanismos de lesão da SB após contusão da ME. No presente
modelo in vivo, é possível que mecanismos similares possam ocorrer. Nos tempos
de sobrevida mais tardios, encontramos maior índice de lesão axonal, tanto na
substância branca como na substância cinzenta.
Apesar de sua imensa importância fisiológica para a homeostase tecidual,

101
em condições patológicas incluindo isquemia, trauma e doenças
neurodegenerativas, a ativação de macrófagos/microglia parece estar envolvida
no aumento da área de lesão. Entre os diversos mecanismos em potencial,
macrófagos/microglia ativados poderiam exacerbar o processo lesivo através da
liberação de fatores tóxicos incluindo NO, enzimas proteolíticas, metabólitos do
ácido araquidônico, TNF-
e IL-1 (NATHAN, 1987; NATHAN, 1992; BANATI et al.,
1993; MINGHETTI;LEVI, 1998). Durante isquemia cerebral, células da microglia
liberam NO e IL-1, os quais podem contribuir para o fenômeno de degeneração
neuronal secundária (GEHRMANN et al., 1995; STOLL;JANDER;SCHROETER,
1998; LOVE, 1999). Achados similares foram relatados após lesão cerebral
isquêmica (GEHRMANN et al., 1995; STOLL;JANDER;SCHROETER, 1998;
LOVE, 1999; STOLL;JANDER, 1999).
Recentemente, Hermann et al., (2001) sugeriram que o FNT-
pode
potenciar os efeitos da excitotoxicidade mediada por receptors AMPA durante
lesão da ME em ratos. De acordo com esta hipótese, FNT-
poderia agir em
receptores AMPA, presentes na membrana de células microgliais induzindo-as a
liberarem mais glutamato e FNT- , portanto exacerbando o processo lesivo.
Outros estudos sugerem que células da microglia e/ou astrócitos podem liberar
glutamato ou ácido quinolínico em condições patológicas (PIANI et al., 1992;
BARGER;HARMON, 1997; HERMANN et al., 2001). É possível que através de um
dos mecanismos descritos, ou mesmo através da associação destes, a ativação
de macrófagos/microglia pode contribuir para a exacerbação da lesão na
substância branca no presente estudo.

102
6.5 ALTERAÇÃO DIFERENCIAL NA BAINHA DE MIELINA NOS MODELOS
EXCITOTÓXICO E ISQUÊMICO
No presente estudo, encontramos perda da reatividade para proteína básica
de mielina a partir de 24h, após a injeção de ET-1 no subnúcleo caudal, do
sistema trigeminal e 7 dias após a injeção de NMDA na mesma região. Nos
animais injetados com ET-1, a substância branca apresentou microcistos no
tempo de sobrevida de 7 dias.
Outros estudos relataram achados similares em outras regiões do SNC,
tanto no modelo de lesão excitotóxica por injeção de NMDA (GOMES-LEAL,
2002a), como no modelo de isquemia focal por injeção de ET-1 (HUGHES et al.,
2003). Gomes-Leal, (2002) relatou diminuição da reatividade para a proteína
básica de mielina 3 dias após a lesão excitotóxica. Esta alteração na bainha de
mielina foi confirmada por microscopia eletrônica (GOMES-LEAL, 2002a, 2005a) e
foi concomitante com a formação de microcistos na substância branca.
Hughes et al., (2003) relataram perda da reatividade para proteína básica
de mielina, 24h após injeção de ET-1 na substância branca do córtex motor de
ratos adultos.
No modelo de desmielinização induzida por cuprizona (LUDWIN, 1978), a
desmielinização ocorre como um evento secundário às alterações patológicas
primárias em oligodendrócitos. Nestas circunstâncias, alterações osmóticas em
oligodendrócitos poderiam induzir a formação de vacúolos intramielínicos
apresentando periodicidade espacial. No entanto, em condições patológicas, tais

103
como a EEA e toxicidade induzida pelo hexaclorofenol, a formação de microcistos
parece ser o evento patológico primário (WINCHELL;STERNBERGER;WEBSTER,
1982).
Na presente investigação, as alterações precoces na bainha de mielina
podem estar relacionadas à resposta inflamatória, pois grande número de
macrófagos/microglia foi encontrado na substância branca, a partir deste tempo de
sobrevida. Em outras condições patológicas, como a esclerose múltipla, ou no seu
modelo experimental, a encefalomielite alérgica experimental, a perda da mielina é
sabidamente devida às ações deletérias de células inflamatórias como macrófagos
(FERGUSON et al., 1997; TRAPP et al., 1998; TRAPP et al., 1999;
TRAPP;RANSOHOFF;RUDICK, 1999). Este fenômeno patológico pode ser
mediado por citocinas e proteases liberadas por macrófagos (HENDRIKS et al.,
2005).
Formação de microcistos na substância branca do trato trigeminotalâmico
foi observada 7 dias após a injeção de endotelina-1, no subnúcleo caudal. Este
fenômeno foi relatado em outros modelos de isquemia (KANELLOPOULOS et al.,
2000) e lesão por impacto da ME (LI et al., 1995). Li et al., (1995) relataram
intensa formação de microcistos nos tratos longitudinais da substância branca da
medula espinhal de ratos, após lesão por impacto severa. Após lesão excitotóxica
por injeção de NMDA na medula espinhal de ratos, Gomes-Leal et al., (2004,2005)
relataram formação de microcistos entre 3 e 7 dias após injeção de NMDA, no
corno ventral da medula espinhal de ratos adultos.
Os mecanismos responsáveis pela formação de microcistos não são
totalmente estabelecidos, mas podem estar relacionados à tumefação de axônios

104
e/ou ramificações astrocitárias, bem como formação de espaços entre a bainha de
mielina e o axolema (PANTONI;GARCIA;GUTIERREZ, 1996; YAM et al., 1998).
A formação de microcistos pode estar relacionada ao fenômeno de
desmielinização. Todas as doenças desmielinizantes apresentam duas
caracerísticas patológicas principais: perda e/ou vacuolização (microcistos) da
bainha de mielina (LUDWIN, 1987). A formação de microcistos pode ser uma
consequência direta de lesão à bainha de mielina, ou aparecer como um evento
secundário à lesão de oligodendrócitos, ou mesmo ocorrer devido a ambos os
fatores, em condições patológicas que incluem doenças imunes, tóxicas e
desmielinização viral (LUDWIN, 1987). Nestas condições patológicas, geralmente
a formação de microcistos representa a separação da mielina do axolema,
gerando acumúlo de fluído entre a mielina e o cilindro axonal, com periodicidade
espacial no tecido edemaciado. É possível que mecanismos semelhantes sejam
os responsáveis pela formação de microcistos, na substância branca dos animais
submetidos à lesão isquêmica. Resultados semelhantes foram obtidos com o
modelo de intoxicação pelo hexaclorofenol (ANDREAS, 1993;
HANTZSCHEL;ANDREAS, 1998; , 2000). Durante a excitotoxicidade induzida pelo
hexaclorofenol edema e vacuolização da substância branca cerebral são achados
comuns, o que é concomitante com o edema de oligodendrócitos (ANDREAS,
1993; HANTZSCHEL;ANDREAS, 1998, 2000). Nestas circunstâncias,
mecanismos excitotóxicos mediados por receptores do tipo não-NMDA parecem
estar envolvidos, à medida que o uso de antagonistas para estes receptores
induzem neuroproteção (HANTZSCHEL;ANDREAS, 2000). Na presente
investigação, não investigamos o comprometimento patológico de

105
oligodendrócitos, o que deve ser implementado em estudos futuros.
No presente estudo, claramente a formação de microcistos ocorreu
posteriormente à desmielinização, o que sugere que o comprometimento da
bainha de mielina e/ou oligodendrócitos é um evento crucial para esta condição
patológica.
6.6 PICNOSE NA SUBSTÂNCIA BRANCA APÓS LESÃO ISQUÊMICA
Sete dias após a injeção de ET-1 no núcleo espinhal do sistema trigeminal,
um grande número de células picnóticas foi observado na substância branca.
Outros estudos relataram a presença de picnose, na substância branca após lesão
aguda da medula espinhal (GOMES-LEAL, 2004). Gomes-Leal et al., (2004)
relataram grande quantidade de células picnóticas em todos os tratos de
substância da medula espinhal, principalmente nos tempos de sobrevida mais
tardios após a injeção de NMDA no corno ventral da medula espinhal de ratos.
Estes autores correlacionaram os perfis picnóticos com a degeneração tardia de
oligodendrócitos, como sugerido por outros autores
(SHUMAN;BRESNAHAN;BEATTIE, 1997; LI et al., 1999a; CASHA;YU;FEHLINGS,
2001; TAKAGI et al., 2003). Na presente investigação, os perfis picnóticos
encontrados na substância branca do sistema trigeminal podem ser
oligodendrócitos apoptóticos, mas estudos com dupla marcação utilizando
técnicas específicas para a marcação de apoptose e oligodendrócitos devem ser
utilizados para a averiguação desta hipótese.

106
7 CONCLUSÃO
Na presente dissertação, investigamos os padrões inflamatórios de lesão
axonal e da bainha de mielina, após lesão excitotóxica e isquêmica no núcleo
espinhal do sistema trigeminal. A análise comparativa dos padrões
histopatológicos demomonstraram padrões lesivos diferenciais, com resposta
inflamatória mais intensa caracterizada por recrutamento de macrófagos, mas não
de neutrófilos no modelo de lesão isquêmica e resposta inflamatória discreta
caracterizada por recrutamento escasso dos dois tipos de leucócitos. O modelo
isquêmico induziu mais lesão axonal, sugerindo que a substância branca é mais
vulnerável ao modelo isquêmico, do que ao modelo excitotóxico. Alterações na
bainha de mielina ocorreram mais precocemente, na substância branca dos
animais submetidos à isquemia, do que nos submetidos à excitotoxicidade
induzida pela injeção de NMDA. Futuros estudos, utilizando marcadores de
oligodendrócitos, microglia, de células apoptóticas e técnicas de microscopia
eletrônica devem contribuir para a elucidação das diferenças observadas nos dois
modelos experimentais.

107
8 REFERÊNCIAS BILIOGRÁFICAS
Abbas AK, Janeway CA, Jr. Immunology: improving on nature in the twenty-first century. Cell 2000; 100(1):129-38.
Agnati LF, et al. A new model of focal brain ischemia based on the intracerebral injection of endothelin-1. Ital J Neurol Sci 1991; 12(3 Suppl 11):49-53.
Agrawal SK, Fehlings MG. Mechanisms of secondary injury to spinal cord axons in vitro: role of Na+, Na(+)-K(+)-ATPase, the Na(+)-H+ exchanger, and the Na(+)-Ca2+ exchanger. J Neurosci 1996; 16(2):545-52.
Agrawal SK, Fehlings MG. Role of NMDA and non-NMDA ionotropic glutamate receptors in traumatic spinal cord axonal injury. J Neurosci 1997; 17(3):1055-63.
Agrawal SK, Nashmi R, Fehlings MG. Role of L- and N-type calcium channels in the pathophysiology of traumatic spinal cord white matter injury. Neuroscience 2000; 99(1):179-88.
AHA. American Heart Association, Heart Disease and Stroke Statistics Update 2006. 2006. American Heart Association, Heart Disease and Stroke Statistics Update 2006.
Ahlgren S, Li GL, Olsson Y. Accumulation of beta-amyloid precursor protein and ubiquitin in axons after spinal cord trauma in humans: immunohistochemical observations on autopsy material. Acta Neuropathologica 1996; 92(1):49-55.
Allan SM, Rothwell NJ. Inflammation in central nervous system injury. Philos Trans R Soc Lond B Biol Sci 2003; 358(1438):1669-77.
An SF, et al. Axonal damage revealed by accumulation of beta-APP in HIV-positive individuals without AIDS. J Neuropathol Exp Neurol 1997; 56(11):1262-8.
Andersson PB, Perry VH, Gordon S. The acute inflammatory response to lipopolysaccharide in CNS parenchyma differs from that in other body tissues. Neuroscience 1992; 48(1):169-86.
Andreas K. Efficacy of cerebroprotective substances in the management of functional disorders induced by the cytotoxic brain oedema-producing substance hexachlorophene. Naunyn Schmiedebergs Archives of Pharmacology 1993; 347(1):79-83.
Arundine M, Tymianski M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 2003; 34(4-5):325-37.

108
Arvidsson J, Gobel S. An HRP study of the central projections of primary trigeminal neurons qhich innervate tooth pulps in the cat. Brain Res 1981; 210:1-16.
Atlante A, Calissano P, Bobba A, Azzariti A, Marra E, Passarella S. Cytochrome c is released from mitochondria in a reactive oxygen species (ROS)-dependent fashion and can operate as a ROS scavenger and as a respiratory substrate in cerebellar neurons undergoing excitotoxic death. J Biol Chem 2000; 275(47):37159-66.
Atlante A, Calissano P, Bobba A, Giannattasio S, Marra E, Passarella S. Glutamate neurotoxicity, oxidative stress and mitochondria. FEBS Lett 2001; 497(1):1-5.
Auld DS, Robitaille R. Glial cells and neurotransmission: an inclusive view of synaptic function. Neuron 2003; 40(2):389-400.
Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci 2001; 21(17):6480-91.
Bales KR. Neurodegenerative disease research in the 21st century. Drug Discovery Today 2004; 9(13):553-6.
Banati RB, Gehrmann J, Schubert P, Kreutzberg GW. Cytotoxicity of microglia. Glia 1993; 7(1):111-8.
Bano D, et al. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell 2005; 120(2):275-85.
Barger SW, Harmon AD. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 1997; 388(6645):878-81.
Bear M, Connors BW, Paradiso MA. Neurociências-Desvendando o sistema nervoso. Porto Alegre: Artemed; 2002.
Beattie MS, Farooqui AA, Bresnahan JC. Review of current evidence for apoptosis after spinal cord injury. J Neurotrauma 2000; 17(10):915-25.
Becker K, Kindrick D, Relton J, Harlan J, Winn R. Antibody to the alpha4 integrin decreases infarct size in transient focal cerebral ischemia in rats. Stroke 2001; 32(1):206-11.
Beretier DH, H.; Hu, J. Trigeminal subnucleus caudalis: beyond homologies with the spinal dorsal horn. Pain 2000; 88:221-4.
Bezzi P, et al. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature 1998; 391(6664):281-5.

109
Bhardwaj A, Wu Y, Hurn PD, Kirsch JR, Traystman RJ. Administration of selective endothelin receptor type A antagonist Ro 61-1790 does not improve outcome in focal cerebral ischemia in cat. Journal of Cerebral Blood Flow and Metabolism : Official Journal of the International Society of Cerebral Blood Flow and Metabolism 2000; 20(3):499-504.
Blight AR. Delayed demyelination and macrophage invasion: a candidate for secondary cell damage in spinal cord injury. Cent Nerv Syst Trauma 1985; 2(4):299-315.
Blight AR. Effects of silica on the outcome from experimental spinal cord injury: implication of macrophages in secondary tissue damage. Neuroscience 1994; 60(1):263-73.
Blight AR. Macrophages and inflammatory damage in spinal cord injury. J Neurotrauma 1992; 9(1):S83-91.
Blight AR. Morphometric analysis of blood vessels in chronic experimental spinal cord injury: hypervascularity and recovery of function. J Neurol Sci 1991; 106(2):158-74.
Blight AR. Remyelination, revascularization, and recovery of function in experimental spinal cord injury. Adv Neurol 1993; 59:91-104.
Blight AR, Cohen TI, Saito K, Heyes MP. Quinolinic acid accumulation and functional deficits following experimental spinal cord injury. Brain 1995; 118(Pt 3):735-52.
Blight AR, Decrescito V. Morphometric analysis of experimental spinal cord injury in the cat: the relation of injury intensity to survival of myelinated axons. Neuroscience 1986; 19(1):321-41.
Blight AR, Saito K, Heyes MP. Increased levels of the excitotoxin quinolinic acid in spinal cord following contusion injury. Brain Res 1993; 632(1-2):314-6.
Blight AR, Zimber MP. Acute spinal cord injury: pharmacotherapy and drug development perspectives. Curr Opin Investig Drugs 2001; 2(6):801-8.
Bohotin C, Scholsem M, Bohotin V, Franzen R, Schoenen J. Vagus nerve stimulation attenuates heat- and formalin-induced pain in rats. Neurosci Lett 2003; 351(2):79-82.
Bolton SJ, Perry VH. Differential blood-brain barrier breakdown and leucocyte recruitment following excitotoxic lesions in juvenile and adult rats. Exp Neurol 1998; 154(1):231-40.

110
Bossy-Wetzel E, Schwarzenbacher R, Lipton SA. Molecular pathways to neurodegeneration. Nat Med 2004; 10 Suppl:S2-9.
Bracken MB. High dose methylprednisolone must be given for 24 or 48 hours after acute spinal cord injury. Bmj 2001a; 322(7290):862-3.
Bracken MB. Methylprednisolone and acute spinal cord injury: an update of the randomized evidence. Spine 2001b; 26(24 Suppl):S47-54.
Bracken MB. Methylprednisolone and spinal cord injury. J Neurosurg 2002; 96(1 Suppl):140-1; discussion 2.
Bracken MB. Methylprednisolone in the management of acute spinal cord injuries. Med J Aust 1990; 153(6):368.
Bracken MB. The use of methylprednisolone. J Neurosurg 2000; 93(2 Suppl):340-1.
Bracken MB, et al. Clinical measurement, statistical analysis, and risk-benefit: controversies from trials of spinal injury. J Trauma 2000; 48(3):558-61.
Bracken MB, Holford TR. Neurological and functional status 1 year after acute spinal cord injury: estimates of functional recovery in National Acute Spinal Cord Injury Study II from results modeled in National Acute Spinal Cord Injury Study III. J Neurosurg 2002; 96(3 Suppl):259-66.
Bradl M, Flugel A. The role of T cells in brain pathology. Curr Top Microbiol Immunol 2002; 265:141-62.
Campbell EJ, Campbell MA. Pericellular proteolysis by neutrophils in the presence of proteinase inhibitors: effects of substrate opsonization. J Cell Biol 1988; 106(3):667-76.
Carmichael ST. Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx 2005; 2(3):396-409.
Cartier L, Hartley O, Dubois-Dauphin M, Krause K-H. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev 2005; 48(1):16-42.
Casha S, Yu WR, Fehlings MG. Oligodendroglial apoptosis occurs along degenerating axons and is associated with FAS and p75 expression following spinal cord injury in the rat. Neuroscience 2001; 103(1):203-18.
Chang CP, Lee CC, Chen SH, Lin MT. Aminoguanidine protects against intracranial hypertension and cerebral ischemic injury in experimental heatstroke. J Pharmacol Sci 2004; 95(1):56-64.

111
Chavarria A, Alcocer-Varela J. Is damage in central nervous system due to inflammation. Autoimmunity Reviews 2004; 3:251-60.
Choi D. Antagonizing excitotoxicity: a therapeutic strategy for stroke? Mt Sinai J Med 1998; 65(2):133-8.
Choi D. Stroke. Neurobiol Dis 2000; 7(5):552-8.
Choi DW. Excitotoxic cell death. J Neurobiol 1992; 23(9):1261-76.
Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988; 1(8):623-34.
Choi DW. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res 1994; 100:47-51.
Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci 1987; 7(2):369-79.
Choi DW. Neurodegeneration: cellular defences destroyed. Nature 2005; 433(7027):696-8.
Chopp M, Zhang ZG. Anti-adhesion molecule and nitric oxide protection strategies in ischemic stroke. Curr Opin Neurol 1996; 9(1):68-72.
Clarke JG, et al. Endothelin-1 is a potent long-lasting vasoconstrictor in dog peripheral vasculature in vivo. J Cardiovasc Pharmacol 1989; 13 Suppl 5:S211-2.
Clarkson AN, Rahman R, Appleton I. Inflammation and autoimmunity as a central theme in neurodegenerative disorders: fact or fiction? Curr Opin Investig Drugs 2004; 5(7):706-13.
Clements JR, Magnusson K, R., Hautman J, Beitz AJ. Rat tooth pulp projections to spinal trigeminal subnucleus caudalis are glutamate-like immunoreactive. J Comp Neurol 1991; 15:2-9.
Coleman M, Perry V. Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci 2002; 25(10):532.
Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 1997; 37:205-37.
Cornish R, Blumbergs PC, Manavis J, Scott G, Jones NR, Reilly PL. Topography and severity of axonal injury in human spinal cord trauma using amyloid precursor protein as a marker of axonal injury. Spine 2000; 25(10):1227-33.

112
Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS. Apoptosis and delayed degeneration after spinal cord injury in rats and. Nat Med 1997 Jan;3(1):73-6. 1997; 3(1):73-6.
Dallel R, Raboisson P, Auroy P, Woda A. The rostral part of the trigeminal sensory complex is involved in orofacial nociception. Brain Res 1988; 448(1):7-19.
Dallel R, Villanueva L, Woda A, Voisin D. [Neurobiology of trigeminal pain]. Med Sci (Paris) 2003; 19(5):567-74.
Danbolt NC. Glutamate uptake. Prog Neurobiol 2001; 65(1):1-105.
DaSilva AF, et al. Somatotopic activation in the human trigeminal pain pathway. J Neurosci 2002; 22(18):8183-92.
Davis SJ, Khangure MS. A review of magnetic resonance imaging in spinal trauma. Australas Radiol 1994; 38(4):241-53.
Dawson VL, Dawson TM. Nitric oxide in neurodegeneration. Prog Brain Res 1998; 118:215-29.
De_Keyser J, Sulter G, Luiten PG. Clinical trials with neuroprotective drugs in acute ischaemic stroke: are we doing the right thing? Trends Neurosci 1999; 22(12):535-40.
Dewar D, Yam P, McCulloch J. Drug development for stroke: importance of protecting cerebral white matter. Eur J Pharmacol 1999; 375(1-3):41-50.
Di Dio LJA. Tratado de Anatomia Sistêmica Aplicada. 2a. ed. São Paulo: Atheneu; 2002. 2 vols. 600 p.
Dijkstra CD, Dopp EA, Joling P, Kraal G. The heterogeneity of mononuclear phagocytes in lymphoid organs: distinct macrophage subpopulations in rat recognized by monoclonal antibodies ED1, ED2 and ED3. Adv Exp Med Biol 1985a; 186:409-19.
Dijkstra CD, Van Vliet E, Dopp EA, van der Lelij AA, Kraal G. Marginal zone macrophages identified by a monoclonal antibody: characterization of immuno- and enzyme-histochemical properties and functional capacities. Immunology 1985b; 55(1):23-30.
Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 1999; 22(9):391-7.
Dusart I, Marty S, Peschanski M. Glial changes following an excitotoxic lesion in the CNS--II. Astrocytes. Neuroscience 1991; 45(3):541-9.

113
Dykens JA, Stern A, Trenkner E. Mechanism of kainate toxicity to cerebellar neurons in vitro is analogous to reperfusion tissue injury. J Neurochem 1987; 49(4):1222-8.
Faden AI. Experimental neurobiology of central nervous system trauma. Crit Rev Neurobiol 1993; 7(3-4):175-86.
Faiz M, Acarin L, Castellano B, Gonzalez B. Proliferation dynamics of germinative zone cells in the intact and excitotoxically lesioned postnatal rat brain. BMC Neurosci 2005; 6(1):26.
Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain 1997; 120 ( Pt 3):393-9.
Ferrari C, et al. Reversible Demielination, Blood-Brain Barrier Breakdown, and Pronounced Neutrophil Reccruitment Induced by Chronic IL-1 Expression in the Brain. Am J Pathol 2004; 165(5):1827-37.
Fischer R, Rehn B, Bruch J. Dust specific effects on the macrophage pneumocyte type II cell system; comparing in vitro and in vivo data. Exp Toxicol Pathol 1996; 48(6):520-2.
Fowler JH, McCracken E, Dewar D, McCulloch J. Intracerebral injection of AMPA causes axonal damage in vivo. Brain Res 2003; 991(1-2):104-12.
Fox JL, Wener L, Drennan DC, Manz HJ, Won DJ, Al_Mefty O. Central spinal cord injury: magnetic resonance imaging confirmation and operative considerations. Neurosurgery 1988; 22(2):340-7.
Franzen R, Bouhy D, Schoenen J. Nervous system injury: focus on the inflamatory cytocine "granolocyte-macrophage colony stimulating factor". Neurosci Lett 2004; 361:76-78.
Fuxe K, Cintra A, Andbjer B, Anggard E, Goldstein M, Agnati LF. Centrally administered endothelin-1 produces lesions in the brain of the male rat. Acta Physiol Scand 1989; 137(1):155-6.
Gallo V, Ghini CA. Glutamate receptors in gliaa New cells, New inputs and new functions. TiPS 2000; 21:252-8.
Garcia_Barcina JM, Matute C. AMPA-selective glutamate receptor subunits in glial cells of the adult bovine white matter. Molecular Brain Research 1998; 53(1-2):270-6.
Gehrmann J, Banati RB, Wiessner C, Hossmann KA, Kreutzberg GW. Reactive microglia in cerebral ischaemia: an early mediator of tissue damage? Neuropathol Appl Neurobiol 1995; 21(4):277-89.

114
Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett 1993; 160(2):139-44.
Giulian D. Ameboid microglia as effectors of inflammation in the central nervous system. J Neurosci Res 1987; 18(1):155-71, 32-3.
Giulian D, Chen J, Ingeman JE, George JK, Noponen M. The role of mononuclear phagocytes in wound healing after traumatic injury to adult mammalian brain. J Neurosci 1989; 9(12):4416-29.
Giulian D, Corpuz M, Chapman S, Mansouri M, Robertson C. Reactive mononuclear phagocytes release neurotoxins after ischemic and traumatic injury to the central nervous system. J Neurosci Res 1993; 36(6):681-93.
Giulian D, Vaca K, Noonan CA. Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science 1990; 250(4987):1593-6.
Go JL, Kim PE, Zee CS. The trigeminal nerve. Semin Ultrasound CT MR 2001; 22(6):502-20.
Gomes-Leal W. Inflamação aguda, resposta glial e degeneração axonal em um modelo de excitotoxicidade na medula espinhal [Doutorado]. Belém: Universidade Federal do Pará; 2002a. 197 p.
Gomes-Leal W, Corkill, D.J., Freire, M.A.M, Picanço-Diniz, C.W.P., Perry, V.H. Astrocytosis, microglia activation, oligodendrocyte degeneration and pyknosis following acute spinal cord injury. Exp Neurol 2004; 190(456-67.
Gomes-Leal W, Corkill, D.J., Picanço-Diniz, C.W., Perry, V.H. Systematic analysis of axonal damage and its possible correlation with inflammatory response in different white matter tracts of acutely injured rat spinal cord. Brain Res 2005a; 1066(1-2):57-70.
Gomes-Leal W, Corkill, D.J., Picanço-Diniz, C.W.P., Perry, V.H. Acute inflammation, glial activation and axonal damage in an excitotoxic model of acute spinal cord injury. International Spinal Cord Annual Meeting abstracts. 2002b.
Gomes-Leal W, Martins, L.C, Diniz, J.A.P., Dos Santos, Z.A., Borges, J.A., Macedo, C.A.C., et al. Differential neurotropism and neuropathological effects of selected rhabdoviruses on the brains of intranasally-infected newborn mice (In press). Acta Tropica 2005b.
Gordon S. Macrophage function disorders. Encyclopedia of Life Sciences 2001:1-11.

115
Graham DI, Adams JH, Nicoll JA, Maxwell WL, Gennarelli TA. The nature, distribution and causes of traumatic brain injury. Brain Pathol 1995; 5(4):397-406.
Gray H, Bannister L, Berry M, Williams P. Gray´s Anatomy: The Anatomical Basis of Medicine & Surgery. 38 ed: Churchill Livinstone; 2003.
Greene JG, Greenmyre JT. Bioenergetics and Glutamate Excitotoxicity. Prog Neurobiol 1996; 48:613-34.
Gupta YK, Briyal S, Sharma U, Jagannathan NR, Gulati A. Effect of endothelin antagonist (TAK-044) on cerebral ischemic volume, oxidative stress markers and neurobehavioral parameters in the middle cerebral artery occlusion model of stroke in rats. Life Sci 2005; 77(1):15-27.
Guth L, Zhang Z, Steward O. The unique histopathological responses of the injured spinal cord. Implications for neuroprotective therapy. Ann N Y Acad Sci 1999; 890:366-84.
Hantzschel A, Andreas K. Efficacy of glutamate receptor antagonists in the management of functional disorders in cytotoxic brain oedema induced by hexachlorophene. Pharmacology & Toxicology 1998; 82(2):80-8.
Hantzschel A, Andreas K. Non-N-methyl-D-aspartate (NMDA) receptor antagonist 1,2,3, 4-tetrahydro-6-nitro-2,3-dioxo-benzo(f)quinoxaline-7-sulphonamide (NBQX) decreases functional disorders in cytotoxic brain oedema. Arch Toxicol 2000; 73(10-11):581-7.
Hara K, Kong DL, Sharp FR, Weinstein PR. Effect of selective inhibition of cyclooxygenase 2 on temporary focal cerebral ischemia in rats. Neurosci Lett 1998; 256(1):53-6.
Haynes WG, Clarke JG, Cockcroft JR, Webb DJ. Pharmacology of endothelin-1 in vivo in humans. J Cardiovasc Pharmacol 1991; 17 Suppl 7:S284-6.
Hendriks JJA, Teunissen CE, Vries HE, Dijkstra CD. Macrophages and neurodegeneration. Brain Research Reviews 2005; 48:185-95.
Hengartner M. The biochemistry of apoptosis. Nature 2000; 407(12):770-6.
Hermann GE, Rogers RC, Bresnahan JC, Beattie MS. Tumor necrosis factor-alpha induces cFOS and strongly potentiates glutamate-mediated cell death in the rat spinal cord. Neurobiol Dis 2001; 8(4):590-9.
Hertz L, Zilke R. Astrocytic control of glutamatergic activityÇ astrocytes as stars of the show. Trends Neurosci 2004; 27(12):735-43.

116
Hirsch EC, Hunot S, Damier P, Faucheux B. Glial cells and inflammation in Parkinson's disease: a role in neurodegeneration? Ann Neurol 1998; 44(3 Suppl 1):S115-20.
Honmou OaY, W. Traumatic injury to spinal cord. In: SGK Waxman, J.D; Stys, P.K, editor The axon:Structure, Function and Pathophysiology. New York: Oxford University Press; 1995.
Horakova L, Stolc S, Chromikova Z, Pekarova A, Derkova L. Mechanisms of hippocampal reoxygenation injury. Treatment with antioxidants. Neuropharmacology 1997; 36(2):177-84.
Hoyer A, Bardenheuer J, Martin E, Plaschke K. Amyloid precursor protein (APP) and its derivatives change after cellular energy depletion. An in vitro-study. J Neural Transm 2005; 112(239-53.
Hughes PM, et al. Focal lesions in the rat central nervous system induced by endothelin-1. J Neuropathol Exp Neurol 2003; 62(12):1276-86.
Iadecola C, Zhang F, Xu X. Inhibition of inducible nitric oxide synthase ameliorates cerebral ischemic damage. Am J Physiol 1995; 268(1 Pt 2):R286-92.
Ikeda T, Terayama R, Jue SS, Sugiyo S, Dubner R, Ren K. Differential rostral projections of caudal brainstem neurons receiving trigeminal input after masseter inflammation. J Comp Neurol 2003; 465(2):220-33.
Iyer R, Hamilton RF, Li L, Holian A. Silica-induced apoptosis mediated via scavenger receptor in human alveolar macrophages. Toxicol Appl Pharmacol 1996; 141(1):84-92.
Jaaskelainen SK. Clinical neurophysiology and quantitative sensory testing in the investigation of orofacial pain and sensory function. J Orofac Pain 2004; 18(2):85-107.
Kanellopoulos GK, Xu XM, Hsu CY, Lu X, Sundt TM, Kouchoukos NT. White matter injury in spinal cord ischemia: protection by AMPA/kainate glutamate receptor antagonism. Stroke 2000; 31(8):1945-52.
Kannurpatti BG, Sanganahalli BG, Mishra S, Joshi P, Joshi NB. Glutamate-induced differencial mithocondrial response in young and adult rats. Neurochemistry International 2004; 44:361-9.
Kao CC, Chang LW, Bloodworth JM. The mechanism of spinal cord cavitation following spinal cord transection. Part 3: Delayed grafting with and without spinal cord retransection. J Neurosurg 1977; 46(6):757-66.

117
Khalert S, Reiser R. Glial perspectives of metabolic stages during cerebral hypoxia-calcium regulation and metabolic energy. Cell Calcium 2004; 36:295-302.
Khodorov B. Glutamate-induced deregulation of calcium homeostasis and mitochondrial dysfunction in mammalian central neurones. Prog Biophys Mol Biol 2004; 86(2):279-351.
Kim SH, Won SJ, Mao XO, Jin K, Greenberg DA. Molecular mechanisms of cannabinoid protection from neuronal excitotoxicity. Mol Pharmacol 2005.
Knoller N AG, Fulga V, Zelig G, Attias J, Bakimer R, Marder JB, Yoles E, Belkin M, Schwartz M, Hadani M. Clinical experience using incubated autologous macrophages as a treatment for complete spinal cord injury: phase I study results. J Neurosurg Spine 2005; 3(3):173-81.
Knyihar-Csillik E, Tajti J, Samsam M, Sary G, Buzas P, Vecsei L. Depletion of calcitonin gene-related peptide from the caudal trigeminal nucleus of the rat after electrical stimulation of the Gasserian ganglion. Exp Brain Res 1998; 118(1):111-4.
Koc RK, Akdemir H, Karakucuk EI, Oktem IS, Menku A. Effect of methylprednisolone, tirilazad mesylate and vitamin E on lipid peroxidation after experimental spinal cord injury. Spinal Cord 1999; 37(1):29-32.
Kraus J, Conroy C, Cox P, Ramstein K, Fife D. Survival times and case fatality rates of brain-injured persons. J Neurosurg 1985; 63(4):537-43.
Kraus JF, Nourjah P. The epidemiology of mild, uncomplicated brain injury. J Trauma 1988; 28(12):1637-43.
Kraus JF, Rock A, Hemyari P. Brain injuries among infants, children, adolescents, and young adults. Am J Dis Child 1990; 144(6):684-91.
Lawson LJ, Perry VH. The unique characteristics of inflammatory responses in mouse brain are acquired during postnatal development. Eur J Neurosci 1995; 7(7):1584-95.
Lazarov-Spiegler O, Rapalino O, Agranov G, Schwartz M. Restricted inflammatory reaction in the CNS: a key impediment to axonal regeneration? Mol Med Today 1998; 4(8):337-42.
Lazarov NE. Comparative analysis of the chemical neuroanatomy of the mammalian trigeminal ganglion and mesencephalic trigeminal nucleus. Prog Neurobiol 2002; 66(1):19-59.
Lee SH, Kim, M., Kim, Y.J., Kim, Y.A., Chi, J.G.,Roh, J.K., Yoon, B.W. Ischemic intensity influences the distribution of delayed infarction and apoptotic cell death following transient focal cerebral ischemia in rats. Brain Res 2002; 956(14-23.

118
Lee SM, et al. Minocycline reduces cell death and improves functional recovery after traumatic spinal cord injury in the rat. J Neurotrauma 2003; 20(10):1017-27. Lent R. Cem bilhões de neurônios; 2002.
Lewen A, Matz P, Chan PH. Free radical pathways in CNS injury. J Neurotrauma 2000; 17(10):871-90.
Li GL, et al. Apoptosis and expression of Bcl-2 after compression trauma to rat spinal cord. J Neuropathol Exp Neurol 1996; 55(3):280-9.
Li GL, Farooque M, Holtz A, Olsson Y. Apoptosis of oligodendrocytes occurs for long distances away from the primary injury after compression trauma to rat spinal cord. 1999a; 98(5):473-80.
Li GL, Farooque M, Holtz A, Olsson Y. Changes of beta-amyloid precursor protein after compression trauma to the spinal cord: an experimental study in the rat using immunohistochemistry. J Neurotrauma 1995; 12(3):269-77.
Li S, Jiang Q, Stys PK. Important role of reverse Na(+)-Ca(2+) exchange in spinal cord white matter injury at physiological temperature. J Neurophysiol 2000; 84(2):1116-9.
Li S, Mealing GA, Morley P, Stys PK. Novel injury mechanism in anoxia and trauma of spinal cord white matter: glutamate release via reverse Na+-dependent glutamate transport. J Neurosci 1999b; 19(14):RC16.
Li S, Stys PK. Mechanisms of ionotropic glutamate receptor-mediated excitotoxicity in isolated spinal cord white matter. J Neurosci 2000; 20(3):1190-8.
Li S, Stys PK. Na(+)-K(+)-ATPase inhibition and depolarization induce glutamate release via reverse Na(+)-dependent transport in spinal cord white matter. Neuroscience 2001; 107(4):675-83.
Li Y, Field PM, Raisman G. Death of oligodendrocytes and microglial phagocytosis of myelin precede immigration of Schwann cells into the spinal cord. J Neurocytol 1999; 28(4-5):417-27.
Li YQ, Takada M, Ohishi H, Shinonaga Y, Mizuno N. Trigeminal ganglion neurons which project by way of axon collaterals to both the caudal spinal trigeminal and the principal sensory trigeminal nuclei. Brain Res 1992; 594(1):155-9.
Lipton P. Ischemic cell death in brain neurons. Physiol Rev 1999; 79(4):1432-568.
Lipton SA. Turning down, but not off. Nature 2004; 428(6982):473.

119
Lipton SA, Singel DJ, Stamler JS. Nitric oxide in the central nervous system. Prog Brain Res 1994; 103:359-64.
Lipton SA, Stamler JS. Actions of redox-related congeners of nitric oxide at the NMDA receptor. Neuropharmacology 1994; 33(11):1229-33.
Liu XZ, et al. Neuronal and glial apoptosis after traumatic spinal cord injury. The Journal of Neuroscience : the Official Journal of the Society For Neuroscience 1997; 17(14):5395-406.
Lo EH, Dalkara, T., Moskowitz, M.A. Mechanisms, challenges and opportunities in stroke. Nature reviews neuroscience 2003; 4:399-415.
LoPachin RM, Lehning EJ. Mechanism of calcium entry during axon injury and degeneration. Toxicology and Applied Pharmacology 1997; 143(2):233-44.
Love S. Oxidative stress in brain ischemia. Brain Pathol 1999; 9(1):119-31.
Ludwin SK. Central nervous system demyelination and remyelination in the mouse: an ultrastructural study of cuprizone toxicity. Laboratory Investigation; a Journal of Technical Methods and Pathology 1978; 39(6):597-612.
Ludwin SK. Remyelination in demyelinating diseases of the central nervous system. Crit Rev Neurobiol 1987; 3(1):1-28.
Martin JH. Neuroanatomia funcional. 2a. ed. São Paulo: Artes médicas; 1998.
Martino G, Grimaldi LM, Bazzigaluppi E, Passini N, Sinigaglia F, Rogge L. The insulin-dependent diabetes mellitus-associated ICA 105 autoantigen in stiff-man syndrome patients. J Neuroimmunol 1996; 69(1-2):129-34.
Masaki T, Yanagisawa M. Endothelins. Essays Biochem 1992; 27:79-89.
Matute C. Characteristics of acute and chronic kainate excitotoxic damage to the optic nerve. Proc Natl Acad Sci U S A 1998; 95(17):10229-34.
Matute C, Alberdi E, Domercq M, Perez_Cerda F, Perez_Samartin A, Sanchez_Gomez MV. The link between excitotoxic oligodendroglial death and demyelinating diseases. Trends Neurosci 2001; 24(4):224-30.
Matyszak MK. Inflammation in the CNS: balance between immunological privilege and immune responses. Prog Neurobiol 1998; 56(1):19-35.
Mayer SA, Rowland LP. Traumetismo Cranioencefálico. In: LP Rowland, editor Merrit. Tratado de Neurologia. Rio de Janeiro: Guanabara-Koogan; 2002.

120
McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nature Medicine 1998; 4(3):291-7.
McDonald JW, Sadowsky C. Spinal-cord injury. Lancet 2002; 359(9304):417-25. McGeer EG, McGeer PL. The importance of inflammatory mechanisms in Alzheimer disease. Exp Gerontol 1998; 33(5):371-8.
McGeer PL, Yasojima K, McGeer EG. Inflammation in Parkinson's disease. Adv Neurol 2001; 86:83-9.
McKenzie KJ, McLellan DR, Gentleman SM, Maxwell WL, Gennarelli TA, Graham DI. Is beta-APP a marker of axonal damage in short-surviving head injury? Acta Neuropathologica 1996; 92(6):608-13.
Medana IM, et al. Axonal injury in cerebral malaria. Am J Pathol 2002; 160(2):655-66.
Medana IM, Esiri MM. Axonal damage: a key predictor of outcome in human CNS diseases. Brain 2003; 126(Pt 3):515-30.
Meldrum B. Amino acids as dietary excitotoxins: a contribution to understanding neurodegenerative disorders. Brain Res Brain Res Rev 2000; 18(3):293-314.
Mergenthaler P, Dirnagl U, Meisel A. Pathophysiology of stroke: lessons from animal models. Metab Brain Dis 2004; 19(3-4):151-67.
Minghetti L, Levi G. Microglia as effector cells in brain damage and repair: focus on prostanoids and nitric oxide. Prog Neurobiol 1998; 54(1):99-125.
Moore KL, Dalley AF. Anatomia orientada para a clínica. 4a. ed. Rio de janeiro: Guanabara Koogan; 2001. 1023 p.
Motte S, McEntee K, Naeije R. Endothelin receptor antagonists. Pharmacol Ther 2005.
Moynagh PN. The interleukin-1 signalling pathway in astrocytes: a key contributor to inflammation in the brain. J Anat 2005; 207:265-9.
Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB J 1992; 6(12):3051-64.
Nathan CF. Secretory products of macrophages. J Clin Invest 1987; 79(2):319-26.
Newman EA. New roles for astrocytes: regulation of synaptic transmission. Trends Neurosci 2003; 26(10):536-42.

121
Nogawa S, Forster C, Zhang F, Nagayama M, Ross ME, Iadecola C. Interaction between inducible nitric oxide synthase and cyclooxygenase-2 after cerebral ischemia. Proc Natl Acad Sci U S A 1998; 95(18):10966-71.
Nogawa S, Zhang F, Ross ME, Iadecola C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci 1997; 17(8):2746-55.
Olney JW. Excitotoxicity: an overview. Can Dis Wkly Rep 1990; 16 Suppl 1E(47-57; discussion -8.
Owen CA, Campbell EJ. Neutrophil proteinases and matrix degradation. The cell biology of pericellular proteolysis. Semin Cell Biol 1995; 6(6):367-76.
Pantoni L, Garcia JH, Gutierrez JA. Cerebral white matter is highly vulnerable to ischemia. Stroke 1996; 27(9):1641-6; discussion 7.
Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 4a. ed. San Diego: Academic Press; 1997.
Peluffo H, Acarin L, Faiz M, Castellano B, Gonzalez B. Cu/Zn superoxide dismutase expression in the postnatal rat brain following an excitotoxic injury. J Neuroinflammation 2005; 2(1):12.
Perez-Asensio FJ, et al. Inhibition of iNOS activity by 1400W decreases glutamate release and ameliorates stroke outcome after experimental ischemia. Neurobiol Dis 2005; 18(2):375-84.
Perry VH, Andersson PB, Gordon S. Macrophages and inflammation in the central nervous system. Trends Neurosci 1993; 16(7):268-73.
Perry VH, Bell MD, Brown HC, Matyszak MK. Inflammation in the nervous system. Curr Opin Neurobiol 1995; 5(5):636-41.
Perry VH, Bolton SJ, Anthony DC, Betmouni S. The contribution of inflammation to acute and chronic neurodegeneration. Res Immunol 1998; 149(7-8):721-5.
Perry VH, Gordon S. Macrophages and the nervous system. Int Rev Cytol 1991; 125:203-44.
Petty MA, Lo EH. Junctional complexes of the blood-brain barrier, permeability changes in neuroinflamation. Prog Neurobiol 2002; 68:311-23.
Petty MA, Wettstein JG. White matter ischaemia. Brain Res Brain Res Rev 1999; 31(1):58-64.
Piani D, Spranger M, Frei K, Schaffner A, Fontana A. Macrophage-induced cytotoxicity of N-methyl-D-aspartate receptor positive neurons involves excitatory

122
amino acids rather than reactive oxygen intermediates and cytokines. Eur J Immunol 1992; 22(9):2429-36.
Popovich PG. Immunological regulation of neuronal degeneration and regeneration in the injured spinal cord. Prog Brain Res 2000; 128:43-58.
Popovich PG, Guan Z, McGaughy V, Fisher L, Hickey WF, Basso DM. The neuropathological and behavioral consequences of intraspinal microglial/macrophage activation. J Neuropathol Exp Neurol 2002; 61(7):623-33.
Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes BT. Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol 1999; 158(2):351-65.
Popovich PG, Hickey WF. Bone marrow chimeric rats reveal the unique distribution of resident and recruited macrophages in the contused rat spinal cord. J Neuropathol Exp Neurol 2001; 60(7):676-85.
Popovich PG, Horner PJ, Mullin BB, Stokes BT. A quantitative spatial analysis of the blood-spinal cord barrier. I. Permeability changes after experimental spinal contusion injury. Exp Neurol 1996; 142(2):258-75.
Popovich PG, Reinhard JF, Jr., Flanagan EM, Stokes BT. Elevation of the neurotoxin quinolinic acid occurs following spinal cord trauma. Brain Res 1994; 633(1-2):348-52.
Popovich PG, Stokes BT, Whitacre CC. Concept of autoimmunity following spinal cord injury: possible roles for T lymphocytes in the traumatized central nervous system. J Neurosci Res 1996; 45(4):349-63.
Popovich PG, Stuckman S, Gienapp IE, Whitacre CC. Alterations in immune cell phenotype and function after experimental spinal cord injury. J Neurotrauma 2001; 18(9):957-66.
Popovich PG, Wei P, Stokes BT. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol 1997; 377(3):443-64.
Popovich PG, Yu JY, Whitacre CC. Spinal cord neuropathology in rat experimental autoimmune encephalomyelitis: modulation by oral administration of myelin basic protein. J Neuropathol Exp Neurol 1997; 56(12):1323-38.
Raivich G, Bohatschek M, Kloss CU, Werner A, Jones LL, Kreutzberg GW. Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Brain Res Rev 1999; 30(1):77-105.

123
Rapalino O, et al. Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat Med 1998; 4(7):814-21.
Reichel A, Begley DJ, Abbott NJ. Carrier-mediated delivery of metabotrophic glutamate receptor ligands to the central nervous system: structural tolerance and potential of the L-system amino acid transporter at the blood-brain barrier. J Cereb Blood Flow Metab 2000; 20(1):168-74.
Reid DM, Perry VH, Andersson PB, Gordon S. Mitosis and apoptosis of microglia in vivo induced by an anti-CR3 antibody which crosses the blood-brain barrier. Neuroscience 1993; 56(3):529-33.
Reiter RJ. Oxidative damage in the central nervous system: protection by melatonin. Prog Neurobiol 1998; 56(3):359-84.
Richardson JS. Neuroprotective agents. Phys Med Rehabil Clin N Am 1999; 10(2):447-61.
Rizzolo RJC, Madeira MC. Anatomia Facial com Fundamentos de Anatomia Sistëmica Geral 2004.
Rosenberg LJ, Teng YD, Wrathall JR. Effects of the sodium channel blocker tetrodotoxin on acute white matter pathology after experimental contusive spinal cord injury. J Neurosci 1999; 19(14):6122-33.
Ruan YW, Ling, G.Y., Zhang, J.L., Xu, Z.C. A poptosis in the adult striatum after transient forebrain ischemia and the effects of ischemic severity. Brain Res 2003; 982(228-40.
Rub U, et al. Anatomically based guidelines for systematic investigation of the central somatosensory system and their application to a spinocerebellar ataxia type 2 (SCA2) patient. Neuropathol Appl Neurobiol 2003; 29(5):418-33.
Rubanyi GM, Polokoff MA. Endothelins: molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharmacol Rev 1994; 46(3):325-415.
Saccco RL. Patogênese, classificação e Epidemiologia das doenças vasculares cerebrais. In: LP Rowland, editor Merrit. Tratado de Neurologia. Rio de Janeiro: Guanabara Koogan; 2002; p. 184-95.
Samsam M, Covenas R, Ahangari R, Yajeya J, Narvaez JA, Tramu G. Simultaneous depletion of neurokinin A, substance P and calcitonin gene-related peptide from the caudal trigeminal nucleus of the rat during electrical stimulation of the trigeminal ganglion. Pain 2000; 84(2-3):389-95.

124
Sattler R, Tymianski M. Molecular mechanisms of calcium-dependent excitotoxicity. J Mol Med 2000; 78(1):3-13.
Schnell L, Fearn S, Klassen H, Schwab ME, Perry VH. Acute inflammatory responses to mechanical lesions in the CNS: differences between brain and spinal cord. Eur J Neurosci 1999; 11(10):3648-58.
Schousboe A. Role os astrocytes in the maintenance and modulation of glutamatergic and GABAergic neurotransmission. Neurochem Res 2003; 28(347-52.
Schwab ME, Bartholdi D. Degeneration and regeneration of axons in the lesioned spinal cord. Physiol Rev 1996; 76(2):319-70.
Schwartz M. Harnessing the immune system for neuroprotection: therapeutic vaccines for acute and chronic neurodegenerative disorders. Cell Mol Neurobiol 2001a; 21(6):617-27.
Schwartz M. Immunological approaches to the treatment of spinal cord injury. BioDrugs 2001b; 15(9):585-93.
Schwartz M, Kipnis J. Protective autoimmunity: regulation and prospects for vaccination after brain and spinal cord injuries. Trends Mol Med 2001a; 7(6):252-8.
Schwartz M, Kipnis J. Protective autoimmunity: regulation and prospects for vaccination after brain and spinal cord injuries. Trends in Molecular Medicine 2001b; 7(6):252-8.
Schwartz M, Moalem G. Beneficial immune activity after CNS injury: prospects for vaccination. J Neuroimmunol 2001; 113(2):185-92.
Schwartz M, Yoles E. Macrophages and dendritic cells treatment of spinal cord injury: from the bench to the clinic. Acta Neurochir Suppl 2005; 93:147-50.
Shepherd G. Synaptic organization of the brain. New york: Oxford university press; 1998.
Shin NS, Lee IS, Yoon YS, Lee HS. Immunohistochemical localization of substance P, calcitonin gene-related peptide, galanin and calcium-binding proteins in trigeminal ganglia of goat (Capra hircus). Anat Histol Embryol 2003; 32(5):310-5.
Shuman SL, Bresnahan JC, Beattie MS. Apoptosis of microglia and oligodendrocytes after spinal cord contusion in rats. J Neurosci Res 1997; 50(5):798-808.
Sicher H, Du Brul EL. Anatomia Bucal. 6a. ed. Rio de Janeiro: Guanabara Koogan; 1977. 511 p.

125
Singh RJ, Hogg N, Goss SP, Antholine WE, Kalyanaraman B. Mechanism of superoxide dismutase/H(2)O(2)-mediated nitric oxide release from S-nitrosoglutathione--role of glutamate. Arch Biochem Biophys 1999; 372(1):8-15.
Sofroniew MV. Reactive Astrocytes in Neural Repair and Protection. Neuroscientist 2005; 11(5):400-7.
Stirling DP, et al. Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. The Journal of Neuroscience 2004; 24(9):2182-90.
Stoll G, Jander S. The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol 1999; 58(3):233-47.
Stoll G, Jander S, Schroeter M. Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol 1998; 56(2):149-71.
Streit WJ. Microglial response to brain injury: a brief synopsis. Toxicol Pathol 2000; 28(1):28-30.
Stys PK. Anoxic and ischemic injury of myelinated axons in CNS white matter: from mechanistic concepts to therapeutics. J Cereb Blood Flow Metab 1998; 18(1):2-25.
Stys PK. General mechanisms of axonal damage and its prevention. Journal of Neurological Sciences 2005; 233:3-13.
Stys PK. White matter injury mechanisms. Current molecular medicine 2004; 4:113-30.
Sykova E. Glial diffusion barriers during aging and pathological states. Prog Brain Res 2001; 132:339-63.
Syntichaki P, Xu, K., Driscoll, M., Tavernarakis, N. Speficic aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature 2002a; 419:939-44.
Syntichaki T, N. Death by necrosis:uncontrollabel catastrophe, or is there order behind the chaos ? EMBO report 2002b; 3:604-9.
Syntichanki P, Tavernarakis, N. The biochemistry of neuronal necrosis: rogue biology ? Nature reviews neuroscience 2003; 4:672-84.
Takagi T, Takayasu M, Mizuno M, Yoshimoto M, Yoshida J. Caspase activation in neuronal and glial apoptosis following spinal cord injury in mice. 2003; 43(1):20-9; discussion 9-30.

126
Tallaksen-Greene SJ, Young AB, Penney JB, Beitz AJ. Excitatory amino acid binding sites in the trigeminal principal sensory and spinal trigeminal nuclei of the rat. Neurosci Lett 1992; 141(1):79-83. Taoka Y, Okajima K. Spinal cord injury in the rat. Prog Neurobiol 1998; 56(3):341-58.
Taoka Y, Okajima K, Murakami K, Johno M, Naruo M. Role of neutrophil elastase in compression-induced spinal cord injury in rats. Brain Res 1998; 799(2):264-9.
Taoka Y, et al. Role of neutrophils in spinal cord injury in the rat. Neuroscience 1997; 79(4):1177-82.
Tator CH. Update on the pathophysiology and pathology of acute spinal cord injury. Brain Pathology (Zurich, Switzerland) 1995; 5(4):407-13.
Tator CH, Fehlings MG. Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J Neurosurg 1991; 75(1):15-26.
Tator CH, Koyanagi I. Vascular mechanisms in the pathophysiology of human spinal cord injury. J Neurosurg 1997; 86(3):483-92.
Tekkok SB, Goldberg MP. Ampa/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J Neurosci 2001; 21(12):4237-48.
Tikka TM, et al. Minocycline prevents neurotoxicity induced by cerebrospinal fluid from patients with motor neurone disease. Brain 2002; 125(Pt 4):722-31.
Trapp BD, Bo L, Mork S, Chang A. Pathogenesis of tissue injury in MS lesions. J Neuroimmunol 1999; 98(1):49-56.
Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998; 338(5):278-85.
Trapp BD, Ransohoff R, Rudick R. Axonal pathology in multiple sclerosis: relationship to neurologic disability. Curr Opin Neurol 1999; 12(3):295-302.
Viggiano A, Monda M, Chiefari M, Aurilio C, De Luca B. Evidence that GABAergic neurons in the spinal trigeminal nucleus are involved in the transmission of inflammatory pain in the rat: a microdialysis and pharmacological study. Eur J Pharmacol 2004; 496(1-3):87-92.
Vila M, Przedborski. targeting programmed cell death in neurodegenerative diseases. Nature reviews neuroscience 2003; 4:1-11.
Villarreal G, Zagorski J, Wahl SMT, F. Inflammation:Acute. Encyclopedia of Life Sciences 2001:1-8.

127
Vos BP, Strassman AM, Maciewicz RJ. Behavioral evidence of trigeminal neuropathic pain following chronic constriction injury to the rat's infraorbital nerve. J Neurosci 1994; 14(5 Pt 1):2708-23.
Wakefield D, Kumar RK. Inflammation:Chronic. Encyclopedia of Life Sciences 2001.
Wang H, et al. Apoptosis-Inducing Factor Substitutes for Caspase Executioners in NMDA-Triggered Excitotoxic Neuronal Death. Neurobiol Dis 2004; 24(48):10964-73.
Weiss SJ. Tissue destruction by neutrophils. New Eng J Med 1989; 320:365-76.
Winchell KH, Sternberger NH, Webster HD. Myelin-associated glycoprotein localized immunocytochemically in periaxonal regions of oligodendroglia during hexachlorophene intoxication. Brain Res 1982; 239(2):679-84.
Wozniewicz B, Filipowicz K, Swiderska SK, Deraka K. Pathophysiological mechanism of traumatic cavitation of the spinal cord. Paraplegia 1983; 21(5):312-7.
Wyss-Coray T, Mucke, L. Inflammation in neurodegenerative disese - a double edged sword. Neuron 2002; 35(1):419-32.
Xu J, Fan G, Chen S, Wu Y, Xu XM, Hsu CY. Methylprednisolone inhibition of TNF-alpha expression and NF-kB activation after spinal cord injury in rats. Brain Res Mol Brain Res 1998; 59(2):135-42.
Xu J, Qu ZX, Hogan EL, Perot PL, Jr. Protective effect of methylprednisolone on vascular injury in rat spinal cord injury. J Neurotrauma 1992; 9(3):245-53.
Yam PS, Dunn LT, Graham DI, Dewar D, McCulloch J. NMDA receptor blockade fails to alter axonal injury in focal cerebral ischemia. J Cereb Blood Flow Metab 2000; 20(5):772-9.
Yam PS, Patterson J, Graham DI, Takasago T, Dewar D, McCulloch J. Topographical and quantitative assessment of white matter injury following a focal ischaemic lesion in the rat brain. Brain Res 1998; 2(4):315-22.
Yamaura I, et al. Mechanism of destructive pathologic changes in the spinal cord under chronic mechanical compression. Spine 2002; 27(1):21-6.
Yanagisawa M, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988; 332(6163):411-5.

128
Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A 1999; 96(23):13496-500.
Yuan J, Yankner, B.A. Apoptosis in the nervous system. Nature 2000; 407(12):802-9.
Zhang Z, Krebs CJ, Guth L. Experimental analysis of progressive necrosis after spinal cord trauma in the rat: etiological role of the inflammatory response. Exp Neurol 1997; 143(1):141-52.
Ziv I, Fleminger G, Djaldetti R, Achiron A, Melamed E, Sokolovsky M. Increased plasma endothelin-1 in acute ischemic stroke. Stroke 1992; 23(7):1014-6.

129
9 ANEXOS
Anexo A - Parecer do Comitê de Ética

130
Anexo B - Protocolo de Imunohistoquímica
Pré Tratamento das Secções
Realizamos pré tratamento das secções a fim de melhorar o padrão de coloração.
a. Pré Tratamento Com Tampão Citrato ou Tampão Borato
As secções foram imersas em tampão borato (pH 9.0) aquecido a 60 0C em
banho maria por 30 minutos. A temperatura foi mantida constante neste período.
Após este período, permitiu-se que as seções resfriassem por 25 minutos, tempo
após o qual as secções foram submetidas ao protocolo normal para
imunocitoquímica.
Imunocitoquímica
a. secções de 20 m já montadas em lâminas foram retiradas do freezer e
deixadas na estufa por 30 minutos e em seguidas submetidas ao pré tratamento
com tamão borato.
b. peróxido de hidrogênio a 1% (100ml de H2O
2/250ml de metanol) - 20 minutos
c. lavagem em tampão fosfato salino (TFS)/Tween - 3 vezes de 3 minutos
d. Bloqueio em soro normal do animal que produziu o anticorpo secundário por 30
minutos. No caso dos anticorpos ED1, APP, e MBP o soro normal de cavalo a
10% foi utilizado. No caso dos anticorpos anti MBS-1 o soro nomal de cabra a
10% foi utilizado .
e. Após o bloqueio em soro normal, sem retirá-lo das secões incubou-se as
mesmas no anticorpo primário, diluído em TFS por 1.5 a 2h (cerca de 50 l de
solução por secção) .

131
f. Lavagem TFS/Tween 3 vezes durante 3 minutos
g. Anticorpo secundário por uma hora com as seguintes diluições: para os
anticorpos ED1, APP, MBP utilizou-se o anticorpo secundário biotinilado anti-
cavalo feito em camundongo (1:100, Vector). Para os anticorpos anti MBS-1
utilizou-se o anticorpo secundário biotinilado feito em cabra anti coelho.
h. Lavagem TFS/Tween 3 vezes durante 3 minutos
i. Incubação no complexo avidina-biotina-peroxidase (Kit ABC, vector) por 45
minutos (uma gota da solução A + uma gota da solução B/5ml de TFS (peletes,
Sigma Aldrich)
j. Lavagem TFS Tween 3 vezes durante 3 minutos
k. Reação em diamino benzidina (DAB ) para revelação da peroxidase do
antígeno. Aliquotas de DAB foram descongeladas e diluídas em 250 ml de
tampão fosfato 0.1M em uma cubeta. Antes do início da reação, acrescentou-
se à solução de DAB 100 l de H2O
2 . As secções montadas em lâminas
dispostas em uma cesta histológica foram imersas na solução de DAB/H2O
2 e
monitoradas com auxílio de um microscópio até que o padrão da reação fosse
satisfatório. Em seguida, as secções foram lavadas em tampão fosfato 0.1M
por cerca de 2 vezes durante 3 minutos, contracoradas com Hematoxilina e,
desidratadas e cobertas com lamînula em DPX.

This document was created with Win2PDF available at http://www.daneprairie.com.The unregistered version of Win2PDF is for evaluation or non-commercial use only.