Embed Size (px)
Citation preview

Técnicas de análise de DNA e RNA

Fundamento e aplicação das técnicas de análise de DNA
• Extracção, purificação, quantificação e detecção de ácidos nucleicos
• Electroforese convencional em gel de agarose
• Electroforese em gel de campo pulsado (PFGE)
• Hibridação Southern, hibridação dot-blot e hibridação in situ
• Sequenciação de DNA
• Footprinting de DNA
• Retardação em gel
• Mutagénese de DNA

Extracção, purificação, quantificação e detecção de ácidos nucleicos
Extracção e purificação de ácidos nucleicos – Utilização de Kits comerciais
Quantificação de ácidos nucleicos Método espectrofotométrico (A concentração de DNA é medida pela absorção a 260 nm, sendo uma absorvância de 1,0 equivalente a 50 µg/ml de DNA de cadeia dupla.)Método baseado na fluorescência do brometo de etídio (Comparação com a fluorescência de amostras de DNA de concentração conhecida.)
Detecção de ácidos nucleicosColoração com brometo de etídioColoração com nitrato de prataMétodos de detecção não radioactivos na hibridação de ácidos nucleicos
Marcação das sondas in vitro com:Corantes fluorescentes ou fluoróforos (fluoresceína e rodamina)Biotina ou digoxigenina (através de ligandos de afinidade – avidina, estreptavidina ou anticorpo anti-digoxigenina - acoplados a uma enzima de sinalização que converte os substratos em produtos corados ou quimioluminescentes)Enzimas (fosfatase alcalina – AP ou peroxidase de rábano silvestre – HRP)
Métodos de detecção radioactivos na hibridação de ácidos nucleicosMarcação das sondas in vitro com 32PMarcação interna (Nick translation, Random primers, PCR)Marcação terminal 5´ou 3’

Electroforese em gel convencional (1)

Electroforese em gel convencional (2)A técnica de electroforese em gel permite a separação de fragmentos de DNA de
dimensões diferentes
A matriz de gel (ovais laranja) consiste em longas cadeias de polímeros, entrelaçadas. A dimensão das cadeias interligadas, ou poros, depende da concentração de agarose ou de acrilamida utilizada no gel.
Os poros nos géis de agarose são maiores do que nos géis de acrilamida. Os primeiros são utilizados para separar fragmentos grandes de DNA (≈500 pb a ≈20 kb) e os últimos para separar fragmentos pequenos de DNA (1 nucleótido a ≈2 kb).
Sob a acção da corrente eléctrica que passa através do gel os fragmentos são separados, movendo-se para o pólo positivo a uma taxa inversamente proporcional ao logaritmo dasua dimensão, formando bandas que são visualizadas por auto-radiografia (fragmentos radioactivos) ou pela adição de um corante fluorescente, como por exemplo o brometo de etídio.
Os géis de agarose são horizontais, mas os de acrilamida são verticais (preparados entre duas placas de vidro com um afastamento de alguns milímetros apenas) porque o oxigénio do ar inibe a polimerização da acrilamida.

Electroforese em gel de campo pulsado (PFGE) (1)
A técnica de electroforese em gel de campo pulsado – PFGE é utilizada para separar cromossomas diferentes, como por exemplo de Saccharomyces cerevisiae, cujas dimensões variam entre 220 000 a 2,5 milhões de pares de nucleótidos, ou moléculas de DNA com 107 pares de nucleótidos. O DNA écorado com brometo de etídio.

Electroforese em gel de campo pulsado (PFGE) (2)
Definição das condições: Nº de fases; duração dos pulsos; duração de cada fase; amperagem.Exemplo: Fase 1; pulso 1 segundo; duração da fase 36 minutos; 180 mA
Fase 2; pulso 2 segundos; duração da fase 36 minutos; 180 mA

Hibridação Southern e hibridação Northern (1)
As técnicas de hibridação Southern e hibridação Northern permitem, respectivamente, a detecção de DNAs ou de RNAs específicos por hibridação com sondas apropriadas, de DNA e RNA.
Neste exemplo, a sonda de DNA é detectada pela sua radioactividade, mas as sondas também podem ser detectadas por métodos não radioactivos.

Hibridação Southern e hibridação Northern (2)
Estas técnicas envolvem as seguintes etapas: (A) Misturas de moléculas de RNA de cadeia simples (Hibridação Northern) ou de moléculas de DNA de cadeia dupla obtidas por digestão com enzimas de restrição (Hibridação Southern)são separadas por electroforese com base na dimensão.
(B) As moléculas de RNA ou os fragmentos de DNA desnaturados por exposição do DNA a condições alcalinas desnaturantes, são transferidos para membranas de nitrocelulose ou de nylon.
(C) A membrana, contendo os ácidos nucleicos ligados, é cuidadosamente removida do gel.
(D) A membrana é depois colocada num saco de plástico selado ou numa garrafa de hibridação, juntamente com a solução de hibridação (uma solução salina tamponada) e a sonda de DNA ou de RNA, radioactivamente marcada e na forma de cadeia simples, por um período de tempo prolongado em condições que favorecem a hibridação.
(E) A membrana é em seguida removida do saco ou da garrafa de hibridação e lavada vigorosamente, de modo que apenas as moléculas de sonda que hibridaram com o RNA ou o DNA imobilizado na membrana permanecem ligadas. Após auto-radiografia, o DNA ou o RNA que hibridou com a sonda marcada é visualizado como uma banda.

Hibridação dot-blot

Estratégias de sequenciação shotgun do genoma humano
Digestão parcialClonagem em vectores BAC
ClonagemVector M13 ou
Fagemídio
Sonicação e reparação das extremidades dos fragmentos
Estratégia da empresa pública Estratégia da empresa privada
Celera
A sequenciação shotgun significa que o DNA genómico original é aleatoriamente clivado em pequenos fragmentos.

Sequenciação de insertos grandes
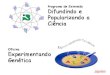
Pesquisa de regiões codificantes numa sequência de DNA (1)

Pesquisa de regiões codificantes numa sequência de DNA (2)
(A) Qualquer região da sequência de DNA, em princípio, codifica seis sequências diferentes de aminoácidos, porque as três grelhas diferentes de leitura são utilizadas para interpretar a sequência de nucleótidos em cada cadeia.
A sequência de nucleótidos é sempre lida na direcção 5’→3’ da cadeia e codifica um polipéptido a partir da extremidade amínica (N) para a extremidade carboxílica (C).
Na leitura de uma sequência nucleotídica aleatória numa grelha particular é encontrado, em média, um sinal de paragem da síntese proteica em 21 aminoácidos (um em 63 nucleótidos).
Na sequência de 48 pb, os codões stop estão representados a verde, e apenas a grelha de leitura 2 não tem codão stop.
(B) Pesquisa na sequência de DNA de 1700 pb de uma possível sequência codificante de uma proteína. A informação está apresentada como em (A), com cada codão stop representado a verde.
Todas as regiões entre possíveis codões de iniciação e terminação da síntese proteica estão representadas como barras vermelhas. Apenas a grelha de leitura 1 codifica realmente uma proteína, de 475 aminoácidos.

Aplicações da sequenciação de genomas
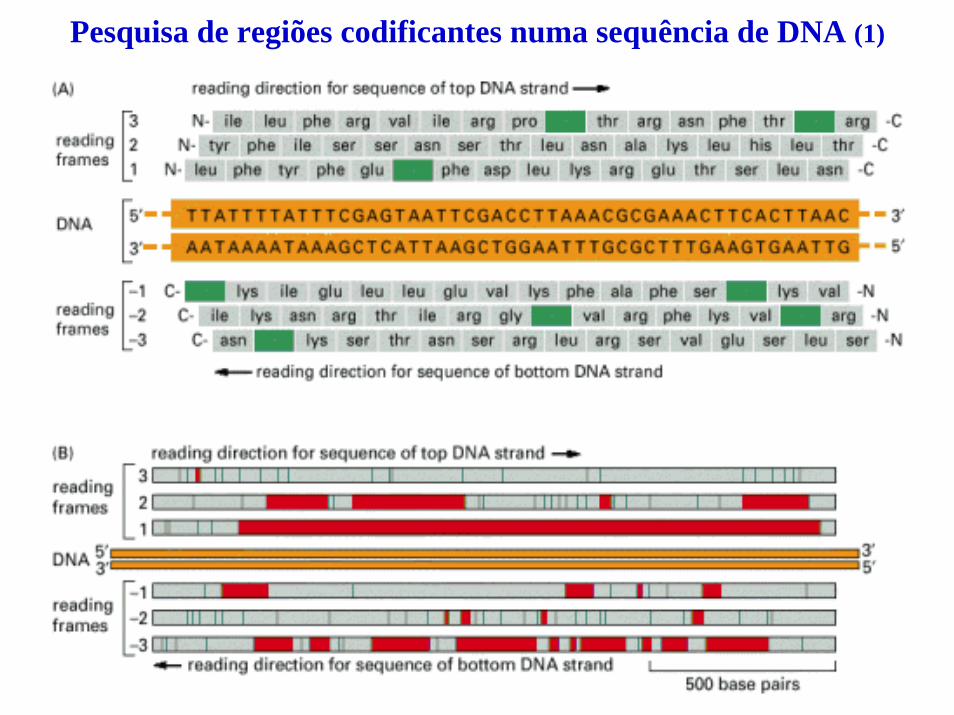
Técnica de footprinting de DNA (1)
Esta técnica permite detectar interacções DNA-proteínaFigure 8-54. © 2002 by Bruce Alberts, Alexander Johnson, Julian Lewis, Martin Raff, Keith Roberts, and Peter Walter.

Técnica de footprinting de DNA (2)
(A) A técnica de footprinting de DNA requer uma molécula de DNA previamente marcada numa das extremidades.
A proteína representada na figura tem uma ligação forte com sete nucleótidos da sequência de DNA específica, protegendo esses nucleótidos do agente de clivagem, a enzima DNase I.
Na reacção paralela de clivagem com DNase I realizada sem a proteína de ligação a DNA, é visualizado no gel de acrilamida desnaturante o conjunto completo de bandas (não estárepresentado).
Os resultados são analisados, em paralelo, com as quatro reacções de sequenciação pelo método de Sanger do fragmento em estudo, o que permite determinar a sequência de DNA reconhecida pela proteína. A identificação dos nucleótidos envolvidos na interacção com a proteina é obtida por mutagénese in vitro.
(B) O footprint foi realizado para determinar o local de ligação de uma proteína humana que estimula a transcrição de genes eucarióticos específicos. Estes resultados indicaram que o local de ligação se situa a cerca de 60 nucleótidos a montante do local de início da transcrição.

Retardação em gel (1)
Esta técnica permite detectar interacções DNA-proteína

Retardação em gel (2)
Este método permite determinar, após sequenciação, a sequência de DNA reconhecida por uma proteína reguladora de um gene. A identificação dos nucleótidos envolvidos na interacção com a proteína é obtida por mutagénese in vitro.
A proteína relevante é purificada e misturada com milhões de diferentes fragmentos curtos de DNA, cada um com uma sequência diferente de nucleótidos. Um conjunto destes fragmentos pode ser produzido programando um sintetizador de DNA, aparelho que sintetiza quimicamente DNA com a sequência pretendida. Por exemplo, há 411, ou aproximadamente 42 milhões de sequências possíveis para um fragmento de DNA de 11 nucleótidos.
Os fragmentos de DNA de cadeia dupla que se ligam fortemente à proteína reguladora do gene são depois separados dos fragmentos de DNA que não se ligam por retardação em gel.
Após separação dos complexos proteína-DNA do DNA livre, os fragmentos de DNA são removidos da proteína, e são realizados vários ciclos adicionais do mesmo processo de selecção. As sequências de nucleótidos dos fragmentos de DNA que permanecem através de múltiplos ciclos de selecção são determinadas, e uma sequência consenso de DNA reconhecida pela proteína pode ser obtida.

Retardação em gel (3)
Esta técnica tem diferentes designações: Electroforetic mobility shift assay (EMSA); Gelshift; Gel retardation; Bandshift assay.Figure 7-29. © 2002 by Bruce Alberts, Alexander Johnson, Julian Lewis, Martin Raff, Keith Roberts, and Peter Walter.

Retardação em gel (4)
O fundamento da técnica de retardação em gel está esquematizado em (A).
Neste exemplo, o extracto proteico de uma linha celular produtora de anticorpo é misturado com o fragmento de DNA radioactivo contendo cerca de 160 nucleótidos da sequência de DNA reguladora do gene que codifica a cadeia leve do anticorpo produzido pela linha celular.
O efeito das proteínas do extracto na mobilidade do fragmento de DNA é analisado por electroforese em gel de poliacrilamida, seguida de auto-radiografia, comparando com a mobilidade do fragmento de DNA sem adição do extracto.
Os fragmentos de DNA livres migram rapidamente para o fundo do gel, enquanto os fragmentos ligados a proteínas são retardados; as seis bandas retardadas sugerem que o extracto contém seis proteínas diferentes de ligação a sequências específicas de DNA (C1 a C6), que se ligam a este fragmento. (Para simplificar, os fragmentos de DNA com mais de uma proteína ligada foram omitidos da figura).
Em (B), o extracto foi fraccionado por uma técnica padrão de cromatografia (em cima), e cada fracção foi misturada com o fragmento de DNA radioactivo, aplicada num poço do gel de poliacrilamida e analisada como em (A).

Purificação de proteínas de ligação a DNA por cromatografia de afinidade (1)
Figure 7-30. © 2002 by Bruce Alberts, Alexander Johnson, Julian Lewis, Martin Raff, Keith Roberts, and Peter Walter.

Purificação de proteínas de ligação a DNA por cromatografia de afinidade (2)
Na etapa 1, as proteínas de ligação a DNA são separadas das restantes proteínas celulares numa coluna com diferentes sequências de DNA.
A fraca afinidade por DNA não específico resulta de atracções iónicas, sendo as proteínas removidas por uma solução com uma concentração moderada de sal.
Na etapa 2, a coluna contém apenas uma sequência particular de DNA, e as proteínas retidas devido a interacções não específicas são eluídas por soluções de concentração moderada de sal, deixando na coluna as proteínas (em geral uma) fortemente ligadas à sequência de DNA. Estas proteínas são eluídas da coluna por soluções contendo uma concentração muito elevada de sal.
Um método alternativo de purificação de uma proteína de ligação a DNA consiste na triagem de uma biblioteca de cDNA com uma sonda de DNA que contém o local de ligação à proteína.

Mutagénese sítio-específica com primer mutagénico
Apesar da falta de complementaridade dos nucleótidos internos, o emparelhamento do primer mutagénico é possível, sendo a segunda cadeia sintetizada pela polimerase de DNA, e as suas extremidades ligadas pela ligase de DNA.Os homodúplices são identificados por hibridação molecular, utilizando o primer mutagénico como sonda oligonucleotídica alelo-específica, ou por PCR alelo-específico.

Mutagénese sítio-dirigida por PCR
• Deve ser utilizada uma polimerase termoestável com capacidade de revisão de provas para minimizar eventuais erros.
• O DNA molde, isolado de estirpes dam+, émetilado e susceptível àdigestão com DpnI, uma endonuclease específica de DNA metilado e hemimetilado.

Fundamento e aplicação das técnicas de análise de RNA
• Hibridação Northern
• Mapeamento de extremidades 5’ e 3’ do mRNA com nuclease S1
• Mapeamento de extremidades 5’ do mRNA por extensão do primer
• Microarrays de DNA

Hibridação Northern
A hibridação Northern permite:
• Detectar a presença de mRNA específico
• Estimar a dimensão do mRNA, relativamente a RNAs marcadores
• Estimar a quantidade relativa do mRNA (por comparação do sinal de hibridação com o de outros genes que são expressos em níveis conhecidos).
• Detectar a presença de transcritos diferentes de um gene específico (splicing alternativo, promotores alternativos).Para além da hibridação Northern, as técnicas de RT-PCR e
hibridação in situ permitem analisar em que tecidos ou condições experimentais os genes se expressam.

Mapeamento de extremidades 5’ do mRNATécnica de protecção da nuclease S1 Técnica de extensão do primer
A extremidade 5’ do mRNA corresponde ao local de início da transcrição.
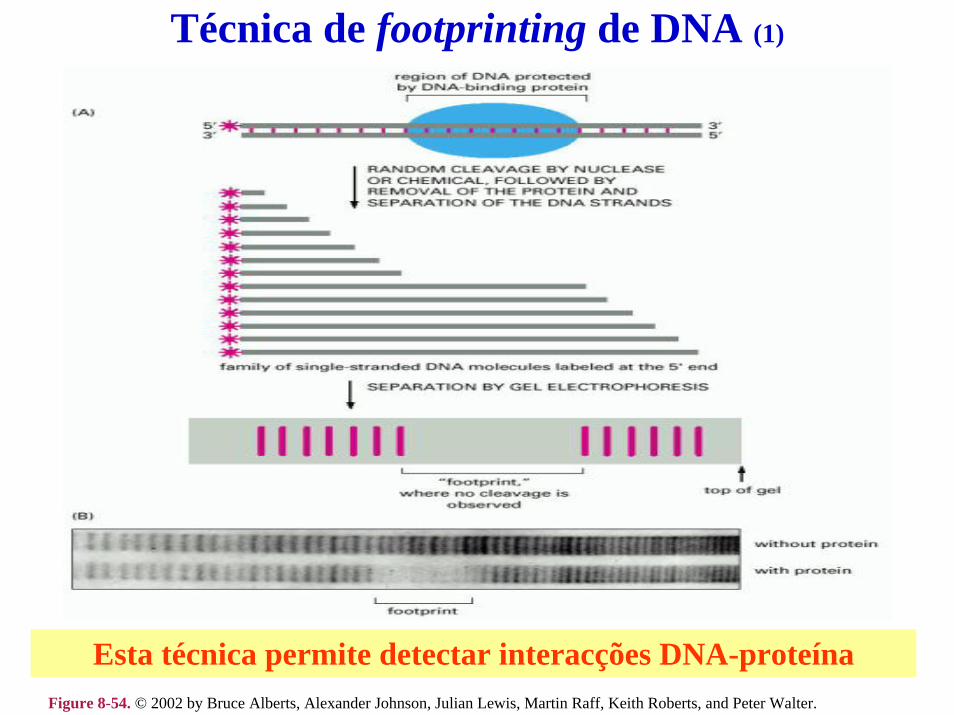
Análise da expressão génica com microarrays de DNA (1)
c
Cada spot contém 106-109 cópias da mesma sequência de cDNA de várias centenas de pares de bases.
Cada gene está representado por 20 pares de oligonucleótidos (PM e MM) diferentes de cadeia simples com 20-25 nt.
Perfect matchMismatch

Análise da expressão génica com microarrays de DNA (2)
Transcritase reversacDNA
cDNA marcado
Avidina conjugadacom um fluoróforo
cDNA marcado
Nível de expressãoX - elevado em 1Y – elevado em 2 Z – idêntico em 1 e 2

Análise da expressão génica com microarrays de DNA (3)
Os oligos MM contêm uma única base mismatch e servem para controlar a hibridação inespecífica.
O controlo positivo é o gene da actina, gene expresso constitutivamente.
O controlo negativo incluído nos microarrays de cDNA serve para normalizar o background ou hibridação não específica.
Para determinar o sinal de um gene particular, os sinais dos 20 oligos PM são somados e os sinais dos 20 oligos MM são subtraídos do total.
Em (C) um dos nucleótidos está conjugado com um fluoróforo.
X – representa genes hipotéticos presentes em níveis elevados na amostra 1; Y – níveis elevados na amostra 2; Z – níveis idênticos nas amostras 1 e 2.
Para este tipo de análise da expressão baseada na hibridação multiplex muitas empresas comercializam (A), mas (B) é comercializado como GeneChips apenas pela Companhia de Biotecnologia US Affymetrix Inc..